[level-membership-for-hematology-oncology-and-palliative-medicine-category]Endocrine complications and paraneoplastic syndromes
Sai-Ching Jim Yeung, MD, PhD, FACP  Robert F. Gagel, MD
Robert F. Gagel, MD
Overview
Transformation of normal cells results in activation and/or suppression of a number of hormonally active genes. This chapter outlines the several clinical endocrine paraneoplastic syndromes and their appropriate management. Treatment of cancer results in a number of endocrine or metabolic manifestations, most of which are related to hormone deficiency or drug-related toxicity. The introduction of targeted therapy that disrupts signaling pathways and immunotherapy, contrary to expectations, has actually increased the number of endocrine manifestations. This chapter will chronicle the major new endocrine toxicities in addition to those observed with older cytotoxic chemotherapies and radiation.
Introduction
This chapter is divided into two major sections. The first focuses on endocrine complications in cancer patients and the second on endocrine paraneoplastic syndromes. Cancer and its treatment can lead to endocrine dysfunction or clinical and laboratory abnormalities that obscure or mimic endocrine diseases. Paraneoplastic syndromes are a group of diverse clinical syndromes seen in cancer patients caused by circulating biologic/humoral factors that include hormones, immunoglobulins, cytokines, and other agents.
Endocrine complications
Hypothalamic–pituitary dysfunction
Radiotherapy is a common cause of hypothalamic–pituitary dysfunction in cancer patients. There is no strong direct evidence to implicate chemotherapy as a cause of permanent dysfunction of the anterior pituitary, although some of the newer targeted therapies may affect pituitary function. Metastasis to the hypothalamic region or the pituitary gland is uncommon,1 and clinical manifestations of endocrine dysfunction because of metastatic disease in this region are rare. However, benign tumors such as pituitary tumors and craniopharyngiomas frequently affect this anatomic region and cause endocrine dysfunction. The introduction of immunotherapy by immune checkpoint blockade [anti-CTLA4 antibodies (ipilimumab and tremelimumab) and anti-PD-1 antibody (nivolumab)] has been associated with development of autoimmune hypophysitis that requires hormonal replacement (corticosteroids or thyroid hormone) in 2–3% of treated patients.
Development of radiation-induced hypothalamic dysfunction is insidious; hormonal deficiency can manifest years after radiation. In general, the rapidity of onset and severity of dysfunction depend on the total dose of radiation and the rate of delivery. The sequence and frequency of dysfunction among the axes of hypothalamic–pituitary functions vary. The somatotropic axis is the most susceptible, while the thyrotropic axis is the least susceptible (Figure 1).2–5 The diagnosis of hypothalamic–pituitary dysfunction requires vigilance of the physician because most presenting symptoms (e.g., fatigue and weakness) are nonspecific and attributable to other causes common among cancer patients. A diagnostic screen for hypothalamic/pituitary dysfunction may include serum growth hormone (GH) and insulin-like growth factor-1 (IGF-1) measurement and evaluation for gonadal failure. Signs of overt hypopituitarism include hypoglycemia, hypotension, and hypothermia.
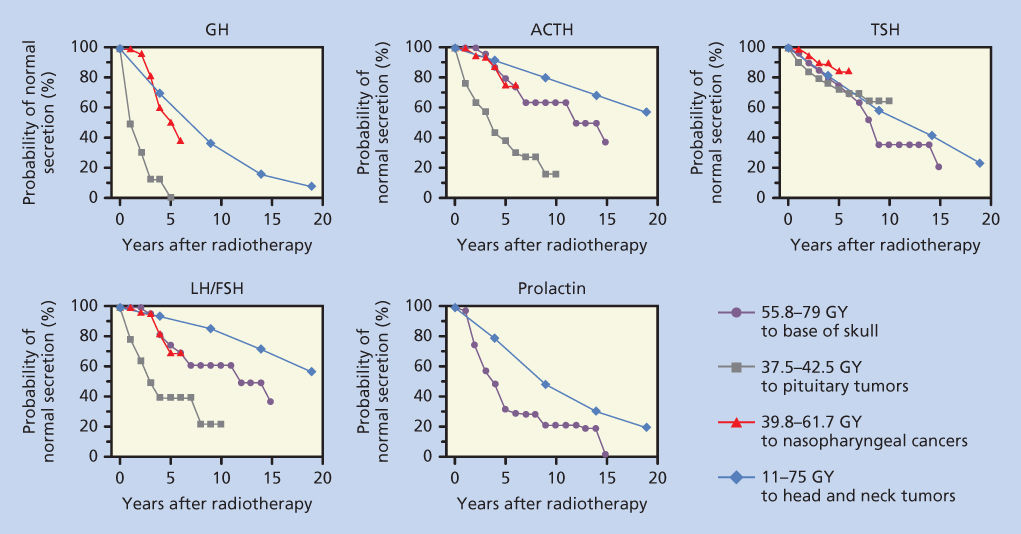
Figure 1 Probability of normal pituitary hormone secretion over time after radiation exposure to the hypothalamic–pituitary areas. Data from four studies were replotted on this single figure. The first set of values (closed circle) are from Pai et al.,3 where the patient received 55.8–79 Gy to the base of the skull. The second set of values (solid square) are from Shalet et al.,83 where patients with pituitary tumors were treated with 37.5–42.5 Gy. The third series (open triangle), from Lam et al.,2 shows the effect of radiation treatment for nasopharyngeal carcinoma with 39.8–61.7 Gy. The final series (open diamond) represents data from Samaan et al.,4 in which 11–75 Gy was administered to treat head and neck tumors.
In children and adolescents, evaluation of sexual development is a useful diagnostic tool. Staging sexual development according to Tanner’s criteria, menstrual history in girls and penile/testicular size in boys should be evaluated. In children who have had cranial irradiation, height and weight should be measured every 6 months. In children treated with spinal and craniospinal irradiation, local rather than general growth abnormalities may be present and, if so, require specific evaluation. Foot size is a reliable indicator of growth that can be easily measured. Deviation from normal growth curves should be evaluated for GH deficiency, hypothyroidism, and adrenal insufficiency. If the initial evaluation of GH, IGF-1, thyrotropin (TSH) and free thyroxine (T4) levels, and radiographic bone age reveal abnormality, then detailed dynamic testing to evaluate the hypothalamic/pituitary axes should be performed (Table 1).
Table 1 Dynamic testing of the hypothalamic/pituitary axes
| Test | Dose/sampling | Contraindications |
| Growth hormone axis | ||
| Insulin hypoglycemia | 0.075–0.1 U regular insulin/kg IV to achieve glucose ≤40 mg/dL. Sample for glucose and GH at 0, 30, 45, 60, and 90 min | Coronary heart disease or seizures |
| Arginine | 0.5 g/kg (up to 30 g) IV over 30 min. Sample for GH at 0, 30, 60, 90, and 120 min | Liver disease or renal disease |
| L-Dopa | 500 mg by mouth. Sample for GH at 0, 30, 60, 90, and 120 min | Systolic blood pressure <100 mm Hg or age > 60 years |
| Arginine and GHRH | Arginine dose as above. GHRH 1 µg/kg IV push. Sample for GH at 0, 30, 60, 90, and 120 min | Liver disease or renal disease |
| Clonidine stimulation protocol | Clonidine 0.15 mg/m2 by mouth. Collect GH samples at baseline, 30, 60, 90, and 120 min | |
| Growth hormone-releasing hormone (GHRH) stimulation protocol | GHRH at 1.0 µg/kg body weight IV push. Collect GH samples at baseline, 15, 30, 45, 60, 90, and 120 min | |
| Growth hormone suppression test | The test should be performed after an overnight fast with the patient maintained at bed rest. The patient should drink a solution of 100 g glucose. Collect GH samples at baseline, 60, and 120 min | |
| Adrenal axis | ||
| ACTH stimulation test, 1-h | Synthetic ACTH 1–24 1 or 250 µg IM or IV. Draw blood for cortisol at 30 and 60 min after injection | |
| ACTH stimulation test, 48-h | Beginning at 9 a.m., obtain baseline 24-h urine for 17-hydroxycorticosteroids (17-OHCS) and creatinine. Collect 24-h urine as on day 1. Beginning at 9 a.m., start IV and give 250 µg synthetic ACTH 1–24 in 250 mL normal saline over 8 h every 8 h for 48 h. Alternatively, 40 IU of depot formulation of purified bovine ACTH in gelatin IM every 12 h for 48 h. Repeat 24-h urine as on days 1 and 2 Days 4 and 5: Collect 24-h urine as on previous days |
|
| Corticotropin-releasing hormone (CRH) stimulation test | Fast for at least 4 h before the test. Human CRH at 1.0 µg/kg IV bolus over 30 s. Blood samples should be collected at 15 and 1 min before CRH administration and at 15, 30, 45, 60, 90, and 120 min after for measurements of cortisol and ACTH | |
| Low-dose dexamethasone test, overnight | Dexamethasone 1.0 mg (adult) or 20 µg/kg (children) PO between 11 p.m. and midnight. Serum cortisol is collected at 8–9 a.m. the next morning. A cortisol level <1.8 µg/dL essentially excludes Cushing syndrome | |
| Low-dose dexamethasone test, 48-h | Serum cortisol is collected at 8–9 a.m. Dexamethasone 0.5 mg (adult) or 10 µg/kg (children) PO immediately after the cortisol is drawn and again every 6 h for 48 h. A second plasma cortisol is drawn at 9 a.m., 6 h after the last dexamethasone dose. Serum cortisol concentrations <1.8 µg/dL exclude Cushing syndrome | |
| High-dose dexamethasone test, 48-h | Serum cortisol is collected at 9 a.m. Dexamethasone is administered (2.0 mg; 50 µg/kg in children) every 6 h for 48 h. A second plasma cortisol is drawn at 9 a.m., 6 h after the last dexamethasone dose. Patients with functional adrenal adenomas show no suppression of cortisol levels in the 48-h sample relative to the initial (baseline) sample. Seventy-eight percent of patients with pituitary source of excess ACTH showed >50% suppression of plasma cortisol, while only 11% of patients with an ectopic source of excess ACTH had a >50% suppression | |
| Comprehensive, 6-day, low-/high-dose dexamethasone test | This protocol incorporates the low- and high-dose dexamethasone tests in succession. 24-h urinary free cortisol and/or 17-hydroxycorticosteroid (17-OHCS) measurement can help verify the results of serum cortisol and ACTH | |
| Gonadotropin-releasing hormone (GnRH) stimulation test | GnRH 100 µg IV. A sample for serum LH should be collected at baseline and 40 min after GnRH administration | |
| Metyrapone stimulation (overnight) test | At 11 p.m., metyrapone 30 mg/kg (maximum 3 g) PO with a snack. On the following morning, at 8 a.m., measure serum cortisol and 11-deoxycortisol | |
GH = growth hormone; GNRH = growth hormone-releasing hormone; IV = intravenous; IM = intramuscular; PO = by mouth.
In adults who have received cranial or head and neck irradiation, detection of hypothalamic–pituitary abnormalities is more challenging. One strategy to detect hypothalamic–pituitary abnormalities in adults consists of routine screening for GH deficiency and gonadal failure. It is recommended that measurements of IGF-1 and testosterone levels in males and documentation of menstrual history in females be obtained annually for 5 years, and then at 5-year intervals for another 10 years. Any abnormalities noted on the screening tests should be pursued with further dynamic testing to evaluate all the axes of hypothalamic–pituitary functions.
Immunotherapy-induced hypophysitis
The development of immunotherapeutic approaches to treat melanoma and a growing list of other malignancies has resulted in a number of bystander effects. Prominent among these side effects is immunotherapy-induced hypophysitis (IH). In the phase III clinical trial of ipilimumab, 2.3% of patients developed symptoms of IH and evidence of hormonal deficiency including glucocorticoid, thyroid, gonadal, and GH deficiency, necessitating long-term replacement therapy. The pituitary-related events were grade 3 or 4 in 1.9% of these patients, necessitating urgent treatment or hospitalization. The onset of pituitary failure ranged from 11 to 19 weeks, following initiation of ipilimumab therapy. Common presenting complaints include headache, mental status alterations, abdominal pain, modified bowel habits, and hypotension; it is often difficult to distinguish IH from brain metastasis or symptoms associated with widespread metastatic disease. This difficulty suggests that clinicians initiating therapy with immunotherapy agents should obtain baseline thyroid (free T4 and TSH), adrenal (ACTH, adrenocorticotropic hormone and 8 a.m. serum cortisol), gonadal [luteinizing hormone (LH), follicle-stimulating hormone, and estradiol in females or testosterone in males], and growth-related hormones (GH and IGF-1) as baseline before initiating therapy. These measurements should be repeated if a patient develops symptoms suggestive of endocrine deficiency or acute symptoms of hypophysitis (headache and visual disturbance). In patients with symptoms or physical findings consistent with IH, an MRI (magnetic resonance imaging) of the pituitary gland and repeat hormone testing as described above should be performed together with dynamic testing of the adrenal endocrine axis (e.g., low-dose cosyntropin stimulation test). A recent report highlighting a larger experience in a single center indicates that IH is observed not only with anti-CTLA4 agents (ipilimumab and tremelimumab),6 but also with an anti-PD1 agent (nivolumab). In a report describing 968 patients treated with these agents for melanoma, prostate cancer, and renal cell carcinoma at a single center, 2.7% (27 subjects) had one or two hormonal deficiencies together with either pituitary enlargement in MRI scans or headache.6 In these 27 patients, central adrenal deficiency was identified in 77%, central hypothyroidism in 89%, and central hypogonadism in 79%. An abnormality of the pituitary gland was found by MRI in 85%.6 After a median follow-up period of 17 months (range: 1–76), none of the patients recovered normal adrenocortical function, whereas 12–13% recovered thyroid or gonadal function. Clearly, this is a significant and evolving issue; it is unclear at this time whether combinatorial therapy with two or more immunotherapeutic agents, currently under development, will be associated with higher incidence and/or severity of toxicity.
Thyroid disorders
Thyroid disorders and abnormalities in thyroid function are commonly associated with cancer and its therapy.
Serum thyroid hormone-binding protein abnormalities
The levels of thyroid hormone-binding proteins [thyroxine-binding globulin (TBG), prealbumin, and albumin] can be modified by sex hormone levels and nutritional factors; abnormalities of both are encountered frequently in cancer patients. Several chemotherapy drugs affect thyroid function test results. L-Asparaginase appears to reversibly inhibit synthesis of albumin and TBG, resulting in low total thyroxine (T4), but normal free T4 levels.7 The combination of podophyllin and alkylating agents has also been reported to decrease TBG.8 Both 5-fluorouracil9 and mitotane10 increase the total T4 and triiodothyronine (T3) levels without suppressing TSH, suggesting that these drugs increase thyroid hormone-binding capacity in the serum.
Euthyroid sick syndrome
Alterations in thyroid hormone metabolism occur in patients with cancer and other serious systemic illnesses.11 Low serum T3 levels, which may be found in up to 70% of moderately to seriously ill cancer patients, are caused by a decrease in the extrathyroidal conversion of T4 to T3. Serum concentrations of free T4 are usually normal or high, while concentrations of free T3 are below normal or low. The patients are clinically euthyroid, and serum TSH level and TRH stimulation test results are normal.
In most patients with euthyroid sick syndrome, T3, T4, and TSH levels are normal. Clinical manifestations of hypothyroidism are usually absent, but assessment may be confounded by obtundation, edema, and hypothermia that may accompany severe illness. Low free T4 levels in the context of euthyroid sick syndrome usually indicate a grave prognosis, with a mortality rate of more than 50%. Although it is generally accepted that thyroid hormone therapy has no benefit, in practice it is sometimes difficult to differentiate between the euthyroid sick syndrome and secondary hypothyroidism. Judicious replacement of T4 at physiologic levels in these uncommon patients may be appropriate if there are no contraindications (e.g., active ischemic heart disease).
Hypothyroidism
Thyroidectomy
Thyroidectomy may be performed for a variety of oncologic reasons in the management of thyroid cancer, head and neck cancer, or thyroid metastasis. Thyroid replacement is needed in this group of patients. In thyroid cancer patients, supraphysiologic doses of thyroid hormone are adjusted to suppress TSH without overt hyperthyroid symptoms. In others, the dose of thyroid hormone should be adjusted to keep TSH in the normal range.
Radiation
Irradiation is an important cause of hypothyroidism [primary (thyroid), secondary (pituitary), and tertiary (hypothalamic)]. Radiation-induced primary hypothyroidism is caused by thyroid cell destruction, inhibition of cell division, vascular damage, and possibly an immune-mediated phenomenon. Factors that increase the risk of developing primary hypothyroidism include a high radiation dose to the vicinity of the thyroid gland, duration since therapy, lack of shielding of the thyroid during therapy, and combined irradiation and surgical treatments.12
The incidences of hypothyroidism after radiation therapy for various cancers and conditions are tabulated in Table 2.4, 12–22 A relationship between radiation dose and the prevalence of hypothyroidism is based on studies of patients with Hodgkin disease.15, 18 Long-term follow-up of patients treated with low-dose radiotherapy suggests that the threshold for causing clinically evident hypothyroidism is approximately 10 Gy. For Hodgkin-disease patients who received >30 Gy, the actuarial risk of hypothyroidism was up to 45%, 20 years after irradiation.15 Patients with frank or subclinical hypothyroidism should receive thyroid hormone-replacement therapy.
Table 2 Incidence of hypothyroidism (including compensated hypothyroidism) after radiotherapy.a
| Type of malignancy or conditions | Radiation dose | % with hypothyroidism |
| Hodgkin disease | 30–60 Gy | 30–50 |
| Head and neck cancer | 40–72 Gy | 25–50 |
| Lymphoma | 20–40 Gy (median 36 Gy) | 30–42 |
| Breast carcinoma | ? | 15–21 |
| Total-body irradiation in BMT | 13.75–15 Gy | 15–43 |
BMT = bone marrow transplantation.
a Data based on Refs 4 and 1213141516171819202122.
Chemotherapy
The diagnosis of hypothyroidism in 14% of BMT (bone marrow transplantation) patients who received chemotherapy but did not receive total-body irradiation23 suggests a causal relation between hypothyroidism and high-dose combination cytotoxic chemotherapy. This notion is also supported by studies that showed an increased incidence of primary hypothyroidism in patients treated with multiple combination drug regimens24, 25 with or without radiation.24 L-Asparaginase, in addition to inhibition of TBG synthesis discussed above, may also inhibit TSH synthesis reversibly and lead to temporary hypothyroidism with decreased free T4 levels.26
Thyroid dysfunction is a recognized side effect of cytokine treatments. Treatment with interleukin-2 produces thyroid dysfunction in approximately 20–35% of patients.27 These patients have hypothyroidism, hyperthyroidism, or hyperthyroidism followed by hypothyroidism.28 Approximately 10% of interferon-treated patients develop primary hypothyroidism.29 Pituitary enlargement secondary to interferon-induced hypothyroidism has also been reported.30 Patients with antithyroid antibodies before therapy are at higher risk of cytokine-induced thyroid dysfunction.
Retinoid X receptor (RXR) ligands may be used in the treatment of certain malignancies such as cutaneous T-cell lymphoma. Bexarotene (a RXR-selective ligand) caused secondary hypothyroidism dose-dependently.31 A single dose can rapidly suppress TSH in healthy subjects.32 In addition to suppressing transcription of TSH by an RXR-mediated thyroid hormone-independent mechanism, bexarotene also increases metabolic clearance of thyroid hormones by a nondeiodinase-mediated pathway.33
Tyrosine kinase inhibitors
The rapid introduction of small organic molecules that inhibit kinase activity (tyrosine kinase inhibitors, TKIs) into cancer therapy over the past decade has led to the recognition that many of these agents affect thyroid function in profound ways. The first mechanism is a direct effect of these agents on thyroid function. A number of TKIs target receptor tyrosine kinases with well-defined functions in the thyroid gland including the vascular endothelial growth factor receptor, the epidermal growth factor receptor, RET, KIT, and MET. Other agents target downstream signaling pathways such as RAF, PI3K, and mTOR. These signaling pathways are operative in the thyroid and important for normal thyroid cell growth and function. It is not surprising that disruption of these pathways has effects on thyroid cell growth and death and thyroid hormone synthesis. Indeed hypothyroidism occurs at significant incidence rates with a number of agents: sunitinib, 7–85%; pazopanib, 12%; nilotinib, 22%; axitinib, 20–100%; cabozantinib, 15%; sorafenib, 8–39%; dasatinib, 50%; and imatinib, 0–25%.34 Routine measurement of thyroid function (free T4 and TSH) at regular intervals during the first year of therapy with one of these agents seems prudent as it is often difficult to differentiate nonspecific side effects of TKI therapy from primary hypothyroidism or effects of metastatic cancer.
There is a second mechanism for development of hypothyroidism in patients treated with specific TKIs for metastatic thyroid carcinoma who have also undergone total thyroidectomy. Specific agents approved (vandetanib, cabozantinib, sorafenib, and lenvatinib) or studied (pazopanib, motesanib, and sunitinib) for treatment of differentiated or medullary thyroid carcinoma cause an elevation of the serum TSH concentration and low serum T4 levels with high percent incidences: vandetanib, 49%; cabozantinib, 57%; sorafenib, 41%; and lenvatinib, 57%. As each of these agents causes diarrhea and malabsorption, one presumed mechanism for this effect is reduced absorption of thyroid hormone, although effects on thyroid hormone metabolism have not been excluded. Increasing the thyroid hormone dosages by as much as one-third generally resolves the problem. It is important to note that there may also be malabsorption of calcium, magnesium, and vitamin D, causing worsening hypocalcemia or hypomagnesemia in patients with mild thyroidectomy-associated hypoparathyroidism.35 Routine measurement of free T4 and TSH should be incorporated into management as more than 50% of these patient will require a dose modification of thyroid hormone (as well as magnesium, calcium, and vitamin D supplementation). It is important to note that primary hypothyroidism, hypocalcemia, or hypomagnesemia can be readily treated, and their appearance, if addressed promptly, should not limit the use of the TKIs. However, in patients with hypothyroidism, hypocalcemia, or hypomagnesemia, all of which can prolong the QT interval, it may be necessary to withhold TKIs that prolong QT interval, that is, vandetanib,35 lenvatinib,36 and sorafenib in patients with QT prolongation until the QT abnormalities are resolved.
131I-containing compounds
The use of 131I for treatment of thyroid cancer requires a high serum TSH level. High TSH level is achieved by either withholding thyroid hormone replacement or increasingly by administration of recombinant human TSH. The use of 131I-containing compounds in the treatment of other tumors may result in hypothyroidism. For instance, using high-dose (100–1000 mCi) [131I]-metaiodobenzylguanidine to treat unresectable pheochromocytoma may result in primary hypothyroidism.37
Metastasis to the thyroid
Hypothyroidism secondary to metastatic infiltration and replacement of the thyroid by cancer is extremely rare.
Screening
Children who have received either head and neck or cranial irradiation should have a free T4 and a TSH measurement annually for 5 years, and every 2 years thereafter. Early detection of abnormal T4 and TSH levels will permit medical intervention before hypothyroidism adversely affects physical and intellectual development and growth. In adults, neck irradiation for treatment of lymphoma and various head and neck tumors is associated with a high incidence of primary hypothyroidism. Patients who have received irradiation should have free T4 and TSH levels measured annually for 5 years, and then every other year for 10 years, and thereafter every 5 years for another 10 years. Once hypothyroidism is diagnosed, the patient should receive thyroid hormone-replacement therapy.
Hyperthyroidism
Radiation-induced painless thyroiditis with hyperthyroxinemia is an uncommon side effect of external-beam radiotherapy to the head and neck area. Transient hyperthyroidism may occur as a result of inflammation and destruction of thyroid tissue with release of thyroglobulin (containing T4 and T3), and is usually followed by hypothyroidism. Transient hyperthyroidism has been reported after mantle radiotherapy in Hodgkin disease patients, and occurs usually within 18 months of treatment.38 A low uptake of radioiodine in most of these cases suggests a diagnosis of silent thyroiditis, but some have Graves’ disease. In one series of Hodgkin disease patient treated with radiation, the risk of Graves’ disease in these patients was estimated to be at least 7.2 times that in a healthy population.15
Ophthalmopathy similar to that in Graves’ disease has been reported within 18–84 months of high-dose radiotherapy to the neck for lymphoma, breast cancer, and nasopharyngeal or laryngeal cancer. Ophthalmopathy may occur without hyperthyroidism and in the absence of the human leukocyte antigen-B8.39 This suggests that radiation-induced thyroid injury may induce an autoimmune process that is similar to Graves’ disease.
There have also been examples of TKI-associated hyperthyroidism, including two deaths (sorafenib and sunitinib associated).40 The presumed mechanism of hyperthyroidism is thyroiditis. Patients with severe hyperthyroidism should be treated aggressively for thyroid storm; consideration should be given to holding TKI therapy until the patient is stabilized in such cases.34
Thyroid nodules and cancers
Low-dose radiation increases the risk of thyroid nodules and cancer. The association between thyroid cancer and low-dose irradiation has been extensively examined,41 and is discussed in the chapter about thyroid cancer (see 81).
Energy balance and glucose metabolism
Obesity and metabolic syndrome
Cancer treatments may lead to obesity and the metabolic syndrome.42–44 The metabolic syndrome is a cluster of abnormalities consisting of central obesity, dyslipidemia, hyperglycemia, and hypertension that increases the risk of type 2 diabetes and cardiovascular disease. Obesity is a modifiable risk factor for carcinogenesis as well as cancer progression. The mechanisms by which obesity promotes cancer include: hyperinsulinemia due to insulin resistance, high IGF-1, adipokines, low adiponectin, increased production of estrogens by adipose tissue, and increased inflammation. Obesity in cancer survivors may place them at increased risk for poor disease outcomes.42, 45, 46 Obesity increases the risk of colorectal and genitourinary second primary cancers.47 Although low levels of physical activity in cancer survivors can contribute to obesity, the pathophysiologic basis of the weight gain is unclear. As obesity is an adverse prognostic factor for many cancers and is a modifiable risk factor, secondary obesity after cancer treatment needs to be addressed.
Diabetes mellitus
Diabetes mellitus type 2 (DM2) is associated with an elevated risk of pancreatic, liver, colon, gastric, breast, and endometrial cancer.48–53 Extensive epidemiologic data suggest important roles of diabetes in carcinogenesis48–53 and cancer survival.54 The strongest association is perhaps with pancreatic cancer.55–59 Apart from the frequently coexisting obesity, the mechanisms by which diabetes promotes cancer include: hyperinsulinemia, high IGF-1, and hyperglycemia. Hyperglycemia per se has a promoting effect on cancer proliferation. In male, cancer survivors with a fasting serum glucose concentration ≥126 mg/dL had a higher relative risk for hepatopancreatobiliary second primary cancer.47 Evidence-based guidelines for the management of DM2 in cancer patients to optimize patient survival are lacking.
The administration of glucocorticoids (e.g., in combination therapy regimens, for edema of brain metastasis, for prevention of transplant rejection, for graft-versus-host disease in BMT, and for nausea/vomiting) is probably the most common cause of diabetes mellitus in cancer patients. Therefore, patients who receive glucocorticoids must be periodically screened for diabetes with evaluation of fasting glucose levels during therapy. Treatment with streptozocin60 or L-asparaginase61 may result in insulin-deficient diabetes mellitus. Although there is no evidence of a delayed onset of diabetes mellitus following treatment with streptozocin, follow-up has been limited and short term. For long-term survivors treated with streptozocin, periodic screening for delayed development of diabetes mellitus may be indicated. Diabetes mellitus may also develop as a consequence of serious pancreatitis secondary to treatment with L-asparaginase. Immunotherapy for cancer using cytokines such as interleukin-2 and interferons may cause toxicity to pancreatic β cells and lead to insulin-dependent diabetes.62 Tacrolimus, an immunosuppressive agent used to prevent graft-versus-host disease in BMT, also increases the incidence of diabetes, perhaps by damaging pancreatic β cells.63 Patients who received allogenic BMT are likely to be receiving both glucocorticoids, cyclosporine A and tacrolimus, and are particularly at risk for developing diabetes mellitus.64 Management of the blood glucose levels would depend on the severity of the blood glucose level abnormality and on the underlying pathophysiologic mechanism of the increase in blood sugar. In general, insulin will be needed in patients who are insulin deficient.
Metabolic bone diseases
Osteoporosis
Four groups of adult patients are at particular risk for accelerated bone loss and osteoporosis: (i) patients with lymphoma, myeloma, or leukemia; (ii) women with breast cancer treated with cytotoxic chemotherapy frequently undergo an early menopause65 and cannot receive estrogen-replacement therapy; (iii) postmenopausal women with estrogen-receptor positive breast cancer; and (iv) men with prostate cancer who are on antiandrogenic therapy and made hypogonadal. Normal bone remodeling involves a delicate balance between bone formation by osteoblasts and bone resorption by osteoclasts. Antineoplastic therapy is toxic to osteoblast function and decreases bone formation. Production by the tumor of hormonally active substances [e.g., parathyroid hormone-related protein (PTHrP), lymphotoxin, interleukin-1, and interleukin-6] may contribute to bone loss. In most cases, it is not clear whether bone loss is caused by antineoplastic therapy or by the underlying disease process and its effects (including cachexia, malnutrition, poor calcium and vitamin D intake, or a combination of these). In patients with breast or prostate cancer, sex steroid hormone deficiency induced by therapy is the most important cause of bone loss. Bone loss is prominent in patients with several disorders (myeloma, leukemia, and lymphoma), affecting the hematopoietic cells, perhaps because of cytokine production and an intimate relationship of hematopoietic cells with bone-forming cells or the use of high-dose or prolonged therapy with glucocorticoids.
A number of drugs can induce osteoporosis.66 In cancer patients, glucocorticoids, methotrexate, and cytotoxic drugs that cause renal loss of calcium, magnesium, or phosphorus (e.g., platinum compounds, cyclophosphamide, and ifosfamide) have significant impact on bone density. Osteoporosis (generalized or localized) is observed in children receiving methotrexate therapy for acute lymphoblastic leukemia (ALL).67 The osteoporosis improves significantly after cessation of methotrexate therapy. Longitudinal study of ALL patients showed that the leukemic process, high-dose glucocorticoids, and hypomagnesemia (owing to renal wastage following cyclical glucocorticoid and nephrotoxic chemotherapy or antiinfective agents) contributed to the impairment of calcium and vitamin D metabolism and decrease in bone mass at different stages of the treatment process.68 Adjuvant chemotherapy for breast cancer (usually involving 5-fluorouracil, cyclophosphamide, and doxorubicin or methotrexate) is associated with low bone mass in premenopausal patients.69 Bone loss during chemotherapy is substantial and may lead to increased risk of fracture. Posttreatment hypogonadism appears to be a major factor in these adult women with osteoporosis. While tamoxifen has a slight protective effect on bone loss, the opposite is true for aromatase inhibitors. BMT usually involves treatment with high-dose cytotoxic drug, glucocorticoids, and immunosuppressive agents. In 24 patients who underwent BMT with high-dose chemotherapy, profound effects on bone biomarkers were observed.70
Prompt investigation of gonadal dysfunction in cancer survivors and prompt replacement of gonadal steroids (in the absence of contraindications) in young hypogonadal men or women are recommended to decrease the risk of future bone fractures. The bone mass of long-term cancer survivors should be assessed when the patient is about 30 years old, the age at which most people have attained peak bone mass.71 If bone mass is normal, no further evaluation is needed beyond the usual recommendations for prevention of osteoporosis. If it is abnormal (more than 2 standard deviations below normal), the patient should be referred for evaluation of the multiple reversible causes of osteoporosis.
A key point in the management of the osteoporosis syndrome in cancer patients is the use of bone mineral density measurement (e.g., by dual-energy X-ray absorptiometry) to assess fracture risk and to monitor the effects of therapy. This measurement should be performed early in the course of management of the malignancy so that appropriate preventive measures can be implemented. The oncologist who is prescribing medications that are likely to decrease bone mass should consider active use of bisphosphonates (e.g., alendronate, risedronate, ibandronate, or zoledronate), calcitonin, selective estrogen-receptor modulators (SERMs), or denosumab, in addition to a daily intake of 1200–1500 mg elemental calcium and vitamin D supplementation. Bisphosphonates and denosumab are effective therapies for prevention of bone loss. While osteoporosis in children with leukemia will frequently reverse because the children are in the formative years of bone development, in adults more active measures such as bisphosphonate or teriparatide therapy to prevent bone loss should be considered, rather than waiting for the development of a fracture syndrome.
Another key point is correction of abnormal mineral and vitamin D metabolism by dietary supplements. Nutritional deficiency in teenagers and young adults results in lower bone mass. Treatment of hypocalcemia, hypomagnesemia, and vitamin D deficiency is integral to the successful therapy of osteoporosis in cancer patients. Recent studies document that clinically relevant vitamin D deficiency is present in 50% of the normal population; the percentage is almost certainly higher in patients undergoing cancer therapy.
Osteomalacia
Osteomalacia, a condition characterized by unmineralized bone matrix, is a rare complication of chemotherapy, but should be considered in osteopenic patients and those with osteomalacic clinical syndrome (bone pain and proximal myopathy). The most common cause is a decrease in the serum calcium and/or phosphorus concentrations caused by nutritional deficiency and renal wasting of phosphorus and calcium. Patients who have received chemotherapeutic agents that cause hypophosphatemia, hypomagnesemia, or hypocalcemia are particularly at risk. Investigation of the levels of serum ionized calcium, phosphorus, magnesium, and vitamin D metabolites should be included in the initial evaluation. Appropriate replacement therapy of these vitamins and minerals should be instituted once deficiencies have been identified. Other contributing factors include systemic acidosis and drugs such as anticonvulsants and aluminum.66 Tumor-induced osteomalacia will be addressed in the section that follows discussing paraneoplastic syndromes.
Ifosfamide causes tubular damage leading to renal phosphate wasting, hypophosphatemia, and rickets/osteomalacia.72 The toxic effects of ifosfamide on renal tubular function include Fanconi syndrome in adults and children. Tubular damage is seen most commonly when ifosfamide is administered in doses of 50 g/m2 or more, or when it is used in combination with cisplatin.73 Rickets is reported most commonly in children. Estramustine, used in the treatment of prostate cancer, has been reported to increase bone resorption and at the same time cause hypocalcemia, hypophosphatemia, and secondary hyperparathyroidism.74
Adrenal diseases
Adrenal metastasis
Hematogenous metastasis to the adrenal glands is common, exceeded in frequency only by hematogenous metastasis to the lung, liver, and bone.75 Autopsies have documented that 9–27% of patients who died from malignant illness had adrenal metastasis, with bilateral involvement in one-half to two-thirds of patients with adrenal metastasis.
The presence of adrenal metastasis may have important implications for diagnostic and therapeutic planning. When patients with cancer have an adrenal mass but no evidence of metastasis elsewhere, it is important to determine whether this mass represents a metastatic tumor or a separate, unrelated adrenal lesion. Recent advances in imaging techniques have allowed the identification of adrenal lesions antemortem as part of the tumor-staging evaluation. The location of the adrenal glands in the perinephric fat allows the detection of almost all normal glands and contour-deforming masses as small as 5–10 mm. Computed tomography (CT) has a sensitivity and specificity in the detection of adrenal masses. Characteristics on CT examination that suggest adrenal metastasis rather than primary adrenal disease include heterogeneity, contrast enhancement, bilaterality, and size >3 cm.76
Without other evidence of metastatic disease, whether the adrenal mass is actually a metastatic tumor is critical information in determining the appropriate therapy for the cancer. Evaluation of a patient who has a malignant adrenal mass should include a history and physical examination to elicit evidence of adrenal insufficiency, Cushing syndrome, mineralocorticoid excess, or pheochromocytoma. Biochemical assessment should include a short ACTH stimulation test with measurements of serum cortisol to rule out adrenal insufficiency. A 24-h urine collection should be obtained to measure urinary free cortisol, aldosterone, catecholamines, and metanephrines. Pheochromocytoma must be excluded, especially if there is hypertension, or an operative procedure of any type is contemplated. It has been reported that one-half of the patients who had a clinically unsuspected pheochromocytoma had clinical deterioration or even death immediately following a non-adrenal-related surgical procedure.77
If the biochemical assessment for pheochromocytoma is negative, CT-guided fine-needle aspiration should be considered. This procedure has a sensitivity of 85% in detecting cancer.78 MRI may be helpful in the diagnosis of pheochromocytoma. Functional scintigraphy using 131I-6-iodomethyl-19-nor-cholesterol (NP-59) may be used in conjunction with CT and MRI to aid in the diagnosis of a unilateral adrenal mass >2 cm.79
Adrenal insufficiency
Despite the relatively high prevalence of adrenal infiltration by many common cancers, clinically evident adrenal hypofunction occurs infrequently, except when both adrenal glands are affected by metastatic disease.80 It is estimated that more that 80% of adrenal tissue must be destroyed before corticosteroid production, under both basal and stress conditions, is impaired.81 Because the clinical manifestations of adrenal insufficiency are nonspecific and overlap findings in cancer patients, a high index of suspicion is required to detect this treatable condition. The cachexia and weakness seen in patients with adrenal insufficiency can mimic the general wasting seen in patients with extensive metastatic disease. Electrolyte abnormalities can easily be explained by poor intake, malnutrition, side effects of chemotherapeutic agents, or paraneoplastic syndromes. Adrenal insufficiency may develop gradually and therefore be confused with cancer-associated cachexia.
Approximately 20–30% of the patients with bilateral adrenal metastasis will develop adrenal insufficiency.80 These patients should all be evaluated by the ACTH stimulation test with serum cortisol measurements and should receive glucocorticoid- and mineralocorticoid-replacement therapy when adrenal insufficiency is suspected and until normal adrenal function is documented. Patients who are stable should receive 20 mg of hydrocortisone in the morning and 10 mg in the early afternoon. In the event of circulatory instability, sepsis, emergency surgery, or other major complications, stress dosages of parenteral glucocorticoid should be given (e.g., hydrocortisone succinate 100 mg intravenously every 8 h).
Other causes of primary adrenal insufficiency in cancer patients include autoimmune adrenalitis, adrenal hemorrhage, and granulomatous diseases. Many cancer patients may be immunocompromised. For example, patients with leukemia or lymphoma or patients who have undergone BMT are immunosuppressed. In these patients, infection of the adrenal glands by cytomegalovirus, mycobacteria, or fungi may lead to adrenal insufficiency.
Adrenal insufficiency may be drug induced. Etomidate,80 a common intravenous anesthetic, and ketoconazole, an antifungal drug, both inhibit the production of cytochrome P450-dependent enzymes in the glucocorticoid synthetic pathway. Aminoglutethimide and metyrapone are drugs that inhibit enzymes in steroidogenesis, and may cause adrenal insufficiency when used in the treatment of prostate, breast, and adrenocortical cancers. Mitotane, structurally related to the insecticide dichlorodiphenyltrichloroethane (DDT), has selective toxicity for adrenocortical cells. Adrenal insufficiency is commonly observed when mitotane is administered in doses necessary to treat adrenocortical cancer; glucocorticoid replacement therapy is mandatory in such patients.10 Increased protein binding is observed in mitotane-treated patients, and may lead to an increased daily requirement of glucocorticoids during replacement therapy. Suramin, recently proposed as an anticancer agent based on its activity against the tumor growth factors, may also cause adrenal insufficiency.
Secondary adrenal insufficiency because of metastasis to the pituitary or hypothalamus may also occur. The most common cause of secondary adrenal hypofunction, however, is exogenous glucocorticoid therapy that suppresses hypothalamic–pituitary adrenal function. A prolonged course of therapy may lead to hypothalamic–pituitary suppression lasting for many months. Short periods of steroid therapy (i.e., 1, 2, or 4 weeks) in patients with leukemia and lymphoma suppress adrenal function for 2–4 days in most patients, and for longer in some patients. In patients who have received glucocorticoids for more than 2 weeks, a tapering period of 10–14 days should be considered. This is especially true for chemotherapy regimens that included high-dose glucocorticoids such as those used in the treatment of acute leukemia and lymphoma. In addition, patients who have been treated within the past year with prolonged glucocorticoid courses should receive stress dosages of glucocorticoid if acute medical or surgical complications occur (e.g., neutropenic fever with hypotension and acute typhlitis). Irradiation of the hypothalamic–pituitary region causes ACTH deficiency and secondary adrenal insufficiency in 19–42% of treated patients (Figure 1). Several diagnostic approaches have been used to evaluate secondary adrenal insufficiency, including basal 8 a.m. serum cortisol measurements and dynamic tests with 1 µg of synthetic ACTH(1–24), insulin-induced hypoglycemia, or metyrapone. The effects of immunotherapy on pituitary ACTH production were discussed earlier in this chapter.
Disorders of growth hormone secretion and growth
Childhood cancer or its treatment commonly impairs growth. Medulloblastoma and ALL, common childhood malignancies, are frequently treated with cranial or craniospinal irradiation and/or chemotherapy. Close to 40% of adult survivors of childhood brain cancer are below the tenth population percentile for height, and the risk factors for short stature are young age at diagnosis and radiation treatment affecting the hypothalamic–pituitary axis.82 GH deficiency and damage to the osseous growth plates are two common mechanisms of growth retardation.
Cranial irradiation may cause hypothalamic or pituitary dysfunction. The hypothalamus appears to be more radiosensitive than does the pituitary gland and may be damaged by lower radiation doses (<40 Gy). Higher doses (>40 Gy) are likely to damage both hypothalamic and pituitary function. Deficiency of one or more pituitary hormones following irradiation of the hypothalamic/pituitary area occurs in almost 100% of patients 5 years after irradiation (Figure 1).
GH deficiency is the most frequently noted and often the first pituitary manifestation to arise after cranial irradiation. Isolated GH deficiency following irradiation is common, and the effects are dose related. At lower doses (20–24 Gy), the only effect may be an altered GH secretory pattern and subnormal response to insulin-induced hypoglycemia. With intermediate and higher doses, the GH response to the stimulatory effect of arginine is impaired, and the frequency and amplitude of pulsatile GH secretion are decreased.5 At doses up to 30 Gy, abnormal GH secretion and growth retardation are observed in more than 35% of patients, necessitating GH treatment.83
In addition to growth retardation caused by GH deficiency, craniospinal or spinal irradiation for hematologic malignancy or central nervous system tumors and total-body irradiation before BMT may cause two other effects. First, irradiation affects the growth plates in vertebral bodies and in the pelvis and decreases vertebral growth. Second, irradiation causes resistance to GH or IGFs.
Children treated with chemotherapy for malignancy frequently have a period of reduced growth velocity, followed by a “catch-up” growth phase. Systemic illness seems to be the most important component of growth retardation in these children, although chemotherapy may play a significant role. Both growth velocity and height are lower in children who are treated with higher doses of chemotherapy and for a longer duration with combination chemotherapy than in those who receive regular therapy or less-intensive chemotherapy. If there is no catch-up growth after 1.5–2 years, it is important to exclude GH deficiency.
In adults, GH deficiency is thought to cause decreased bone and muscle mass, lower exercise capacity, increased adipose tissue, fatigue, a poor sense of well-being, impaired myocardial function, and increased cardiovascular risks. GH replacement may be indicated to improve the patients’ quality of life and sense of well being,84 but the concern over IGF-1-induced reactivation of malignant disease should be factored into the decision.
Disorders of electrolyte/mineral metabolism
Hyponatremia
Risk factors for hyponatremia include treatment-induced nausea and vomiting, certain chemotherapy agents, hydration with hypotonic fluid, pain, opiates, and stress (both physical and psychological). In a prospective study of in-patient cancer patients, the incidence of hyponatremia is 3.7% with sodium depletion and syndrome of inappropriate antidiuretic hormone (SIADH), each accounting for about one-third of all causes.85 SIADH is characterized by low serum osmolality and inappropriately high urine osmolality in the absence of diuretics, heart failure, cirrhosis, adrenal insufficiency, and hypothyroidism. In cancer patients, SIADH may be caused by vasopressin secreted by tumors [e.g., up to 15% of small-cell lung cancers (SCLCs)], abnormal secretory stimuli (e.g., intrathoracic infection and positive pressure ventilation), or cytotoxicity affecting paraventricular and supraoptic neurons. It is also possible that chemotherapy-induced lysis of vasopressin-containing cancer cells leads to or worsens SIADH. Drug-induced renal salt wasting or tumor-induced salt wasting (mediated by atrial natriuretic peptide)86 can also cause hyponatremia, hypoosmolality, elevated urinary sodium, and urinary osmolality. These SIADH-like syndromes are difficult to distinguish from SIADH when signs and symptoms of fluid volume depletion are subtle or absent. Nonetheless, there are convincing reports that provide evidence of chemotherapy-induced hypothalamic or pituitary damage in the context of SIADH. There are at least seven reports associating vincristine with SIADH, and some of these reports document inappropriately high serum levels of vasopressin.87 Vinblastine has also been reported to cause severe hyponatremia and SIADH.87 The presumed mechanism of vinca alkaloid-induced SIADH is paraventricular or supraoptic cell microtubular damage.
Cyclophosphamide therapy has been associated with hyponatremia and SIADH. Autopsy findings in a case of fatal hyponatremia induced by cyclophosphamide (1800 mg/m2) suggest that cyclophosphamide directly affects the hypothalamus.88 Those findings included infundibular necrosis, decreased intraaxonal secretory granules, and depletion of posterior pituitary vasopressin. Patients treated with lower doses of cyclophosphamide also develop hyponatremia, hypotonicity, urinary hypertonicity, and increased plasma vasopressin levels. Damage to the renal tubules and resulting defects in salt and water transport may be the major cause of hyponatremia associated with low-dose cyclophosphamide therapy.89
There are many reports of cisplatin-induced hyponatremia caused by renal salt wasting.90 Several reports claim that cisplatin induces SIADH. The mechanism of cisplatin-induced hyponatremia is unclear, but it has been suggested that renal toxic effects of cisplatin, that is, decreased papillary solute content and maximal urinary osmolarity are the major factors, rather than a direct effect of cisplatin on vasopressin secretion. In a majority of the patients who have elevated vasopressin levels, the vasopressin levels became suppressed after correction of hypovolemia.90 Therefore, the stimulus for vasopressin release in these patients was probably hypovolemia caused by renal salt wasting.
Figure 2 outlines the algorithm for evaluation and treatment of hyponatremia. For hypovolemia and sodium loss, fluid and sodium replacement is the primary treatment. The AVP-receptor antagonists directly block the binding of AVP with its receptors. In clinical trials, conivaptan, lixivaptan, tolvaptan, and satavaptan effectively correct hyponatremia associated with SIADH, cirrhosis, or congestive heart failure, and this new class of drugs are likely to be useful for cancer-related hyponatremia.91
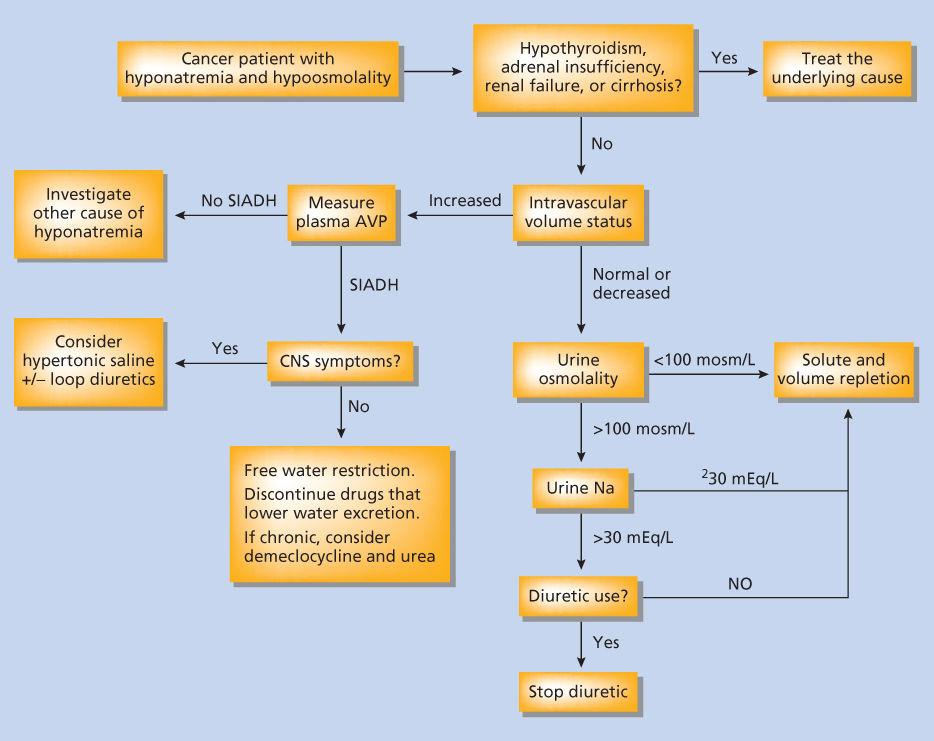
Figure 2 Approach to evaluation of hyponatremia in a cancer patient.
Hypernatremia
Hypernatremia secondary to central diabetes insipidus occurs frequently as a complication of neurosurgery or destruction by the tumor of the anterior pituitary or the related hypothalamic nuclei. Nephrogenic diabetes insipidus can result from the effects of ifosfamide or streptozocin on tubular reabsorption of water. Ifosfamide has broad nephrotoxic effects, although tubular damage predominates. Distal tubular defects develop in about half of patients treated with ifosfamide. However, frank nephrogenic diabetes insipidus leading to hypernatremia is not common.92 Streptozocin is another nephrotoxic drug; in addition to causing glomerular defects (proteinuria) and tubular defects (Fanconi syndrome), streptozocin therapy has been reported to cause nephrogenic diabetes insipidus.93
Hypocalcemia
Hypocalcemia may be one of the features of tumor lysis syndrome. Hypocalcemia can also be caused by primary hypoparathyroidism after surgical procedures in the neck that sacrificed or damaged the parathyroid glands (e.g., total laryngectomy and total thyroidectomy). Hypocalcemia is also a common complication of chemotherapy.87 Hypocalcemia has been reported in 6–20% of cisplatin-treated patients. Effects of cisplatin on renal tubular function, magnesium metabolism, bone resorption, and vitamin D metabolism may explain the hypocalcemia. Profound hypomagnesemia causes a decrease in the secretion of parathyroid hormone and a reduction in the calcium-mobilizing effects of parathyroid hormone. Hypomagnesemia also inhibits formation of 1,25-dihydroxy vitamin D3 (1,25-dihydroxycholecalciferol). Cisplatin may inhibit the mitochondrial function in the kidneys and thereby inhibits conversion of 25-hydroxycholecalciferol to 1,25-dihydroxy cholecalciferol by the enzyme 1-alpha-hydroxylase. In addition, cisplatin may have a direct inhibitory effect on bone resorption. Carboplatin therapy, similar to cisplatin therapy, is associated with a 16–31% incidence of hypocalcemia. Dactinomycin is another antitumor antibiotic that blocks DNA-directed RNA synthesis, causing hypocalcemia in animals. Dactinomycin also abolishes the calcium-mobilizing effect of thyroid hormone, presumably by interfering with osteoclast-mediated bone resorption. Asymptomatic hypomagnesemia, hypocalcemia, and hypoparathyroidism have also been reported in patients treated with a combination of doxorubicin and cytarabine.
Hypercalcemia
The incidence of hypercalcemia in cancer patients is approximately 1%.94 Hypercalcemia in cancer patients is a poor prognostic sign associated with a shortened survival. The paraneoplastic syndrome of hypercalcemia of malignancy is discussed under the section titled “Endocrine Paraneoplastic Syndromes (‘Ectopic’ Hormone Production).” No chemotherapy has been identified as a cause of hypercalcemia. However, there is a clear association between low-dose (usually 2–7.5 Gy) external-beam irradiation of the head and neck area and subsequent development of primary hyperparathyroidism. There is a 2.5- to 3-fold increase in the incidence of primary hyperparathyroidism after low-dose irradiation of the neck.95 Among patients who developed primary hyperparathyroidism, 14–30% had prior exposure to radiation. The interval from irradiation to development of hyperparathyroidism ranges from 29 to 47 years. Primary hyperparathyroidism also develop in the context of multiple endocrine neoplasia, types 1 and 2. Surgery is the principal treatment for primary hyperparathyroidism. Removal of adenoma is usually curative, but in the context of MEN1, the surgical procedure of choice is 3.5-gland parathyroidectomy. 96
Hypomagnesemia
Cisplatin causes morphologic changes and necrosis in the proximal tubule, an important site of magnesium reabsorption. Hypomagnesemia occurs in approximately 90% of patients treated with cisplatin,97 and 10% of the hypomagnesemic patients have symptoms of muscle weakness, tremors, and dizziness. Vigorous hydration and the use of osmotic diuretics such as mannitol may prevent renal failure, but has little effect on renal magnesium wasting. Hypomagnesemia may persist long after cessation of cisplatin therapy. There are no large series in the literature addressing the incidence of hypomagnesemia, but the information from the manufacturer indicates that 60% of those taking cisplatin may be affected. Hypomagnesemia also occurs in patients who receive cyclophosphamide and carboplatin.
Disorders of lipid metabolism
Short-term lipid abnormalities caused by cancer therapy are generally of little clinical significance. However, major abnormalities can lead to acute complications. Interferons and vitamin A derivatives can cause significant increases in triglycerides that can lead to pancreatitis. Interferons cause hypertriglyceridemia by increasing hepatic and peripheral fatty acid production98 and by suppressing hepatic triglyceride lipase.99 Long-term treatment with interferon-α2 causes hypertriglyceridemia in approximately one-third of patients, most of whom had previous serum lipid abnormalities. Serum triglyceride levels of more than 1000 mg/dL are not unusual. In a case report, a therapeutic effect of diet and gemfibrozil was observed in the presence of continued interferon-α therapy.100 All-trans-retinoic acid (tretinoin) and other derivatives, for example, 13-cis-retinoic acid (isotretinoin), have been used in the treatment of several malignancies, most notably head and neck cancers and acute promyelocytic leukemia. The effects on lipid metabolism are well characterized, although the mechanism of development of lipid abnormalities is less clear. These abnormalities include hypertriglyceridemia caused by elevated very low-density lipoprotein levels, and hypercholesterolemia caused by increased low-density lipoprotein level. Retinoid-induced hypertriglyceridemia can cause stroke and pancreatitis. Hyperlipidemia associated with retinoid therapy has been treated with gemfibrozil or fish oil.
Sexual dysfunction
Radiation treatment to the head may cause a broad spectrum of hypothalamic–pituitary abnormalities (Figure 1). The resultant thyroid, GH, or adrenal deficiency may indirectly affect reproductive function. Sexual function is directly affected by hyperprolactinemia or gonadotropin deficiency, commonly observed in patients treated with <40 Gy of cranial irradiation.
Hyperprolactinemia occurs commonly (up to 50% incidence within 2 years) following head and neck irradiation with a median hypothalamic–pituitary radiation exposure of 50–57 Gy.4 Radiation damage to the hypothalamus leading to a loss of the normal inhibition of prolactin secretion is the proposed mechanism of hyperprolactinemia. Hyperprolactinemia inhibits the secretion of gonadotropin by the pituitary and decreases the responsiveness of the pituitary to gonadotropin-releasing hormone (GnRH), thereby causing secondary hypogonadism. Treatment with dopamine agonists (bromocriptine and cabergoline) inhibits prolactin secretion, and it may be reasonable to proceed with a therapeutic trial if other anterior pituitary functions are normal.
Gonadotropin deficiency occurs commonly (up to 61%) in patients treated with irradiation for brain tumors.101 In children, delayed puberty, absent menarche, and inadequate sexual development are significant problems related to gonadotropin deficiency. Early or even precocious puberty has been reported in patients treated with combined chemotherapy and cranial irradiation for ALL102 or brain tumor.103 This phenomenon occurs more frequently in female patients. Concomitant GH deficiency is frequently noted, although its role in the development of precocious puberty is unclear.
In adults, gonadotropin deficiency may cause sex hormone deficiency and sexual dysfunction. Sex hormone deficiency may alter libido and adversely affect bone and lipid metabolism. Sexual dysfunction and impotence need to be evaluated and appropriately treated. Figure 3 outlines a diagnostic algorithm for the evaluation of hypogonadism.
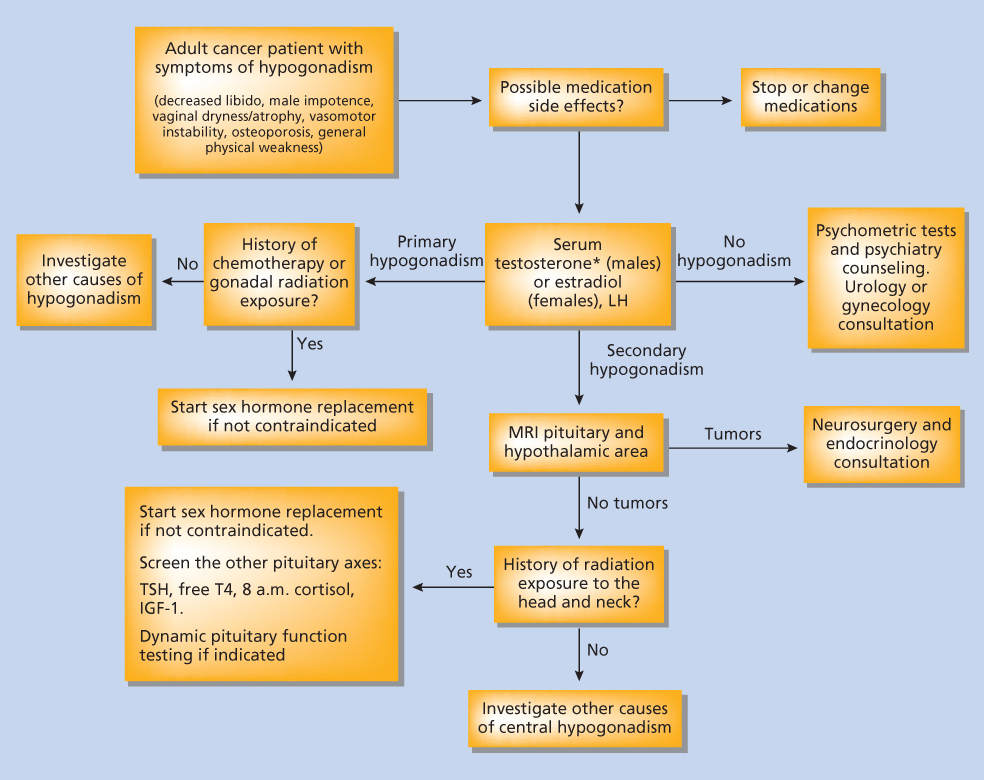
Figure 3 Approach to evaluation of hypogonadism in a cancer patient.
Gonadal complications and dysfunction caused by anticancer therapy have been reviewed.87
Endocrine paraneoplastic syndromes (“ectopic” hormone production)
Among the more interesting and protean manifestations of cancer is the production of hormonal substances that produce unique clinical syndromes. These syndromes can be classified broadly into several different types. The first is the production of a hormonal substance by a cell type that normally produces the hormone. Examples include parathyroid hormone production by a parathyroid cancer, production of calcitonin by medullary thyroid carcinoma, and serotonin by carcinoid tumors. In each of these examples a malignancy of a differentiated cell type continues to produce its normal product, but does so in a manner that is largely independent of the normal regulatory processes. These clinical syndromes are discussed in the relevant chapters in this text that discuss these malignancies. The second type, to be discussed in detail here, is the “ectopic” production of a hormone by a cell type that does not normally produce the hormonal substance or produces it normally at very low levels. In some examples, the cell may have produced the hormonal product at an earlier stage in its development. An example of this is PTHrP production by a squamous cell carcinoma. PTHrP is normally expressed in differentiating squamous cells, but is not expressed or is expressed at low levels in differentiated squamous epithelium.104 Another example of “ectopic” hormone production occurs in a hormone-producing cell whose machinery has been co-opted to produce another hormone. An example of this is the production of ACTH by a wide spectrum of neuroendocrine tumor types, small-cell carcinoma of the lung being one example.
Production of peptides by neuroendocrine tumors comprises the most common of the “ectopic” hormone syndromes. Neuroendocrine cells are dispersed throughout nearly all organs. Prominent are lung, gastrointestinal tract, pancreas, thyroid gland, adrenal medulla, breast, prostate, and skin. The list of hormones produced by tumors derived from members of this group of neuroendocrine cells includes ACTH, calcitonin, vasoactive intestinal peptide (VIP), bombesin, growth hormone-releasing hormone (GHRH), pancreatic polypeptide, corticotropin-releasing hormone (CRH), neurotensin, somatostatin (SRIH), and other small peptides.
Defined clinical syndromes
There are clearly defined clinical ectopic hormone syndromes that occur with some frequency. Their recognition may help in the definition of the cancer type and lead to appropriate management approaches. In addition, these syndromes are a major cause of morbidity and mortality; treatment approaches are available for many of these syndromes and can improve both quality and duration of survival.
Ectopic ACTH production
Inappropriate secretion of ACTH, although uncommon, is an important cause of morbidity and mortality in certain types of malignancies. There are at least two different mechanisms: ectopic ACTH production or ectopic production of CRH, the hypothalamic peptide that normally stimulates ACTH synthesis and release.
The most common cause of ectopic ACTH production is the expression of proopiomelanocortin (POMC) by a tumor. Posttranslational processing of POMC normally proceeds down one of two mutually exclusive pathways.105 The POMC precursor can be cleaved in two different ways to produce peptides with very different biologic activities. The pathway that leads to ACTH production and Cushing syndrome is one in which POMC is cleaved to produce big melanocyte-stimulating hormone and ACTH. Fortunately for cancer patients, the enzymes that cleave POMC to produce ACTH are expressed uncommonly outside of the normal pituitary gland. Most malignant tumors expressing the POMC gene produce peptides that cause no identifiable clinical syndrome. The most common tumor type that overexpresses POMC and the proteolytic enzymes that cleave it to produce ACTH is SCLC, although a broader spectrum of tumors including pulmonary carcinoid, medullary thyroid carcinoma, islet cell malignancy, pheochromocytoma, and occasional ganglioneuromas will produce this hormone. Cushing syndrome produced by “ectopic” production of ACTH is characterized by adrenal cortical hyperplasia and hypercortisolism.106
The second cause of excessive ACTH production is tumor production of CRH.107 Ectopic production of this peptide causes a clinical syndrome characterized by pituitary corticotrope hyperplasia leading to adrenal cortical hyperplasia and Cushing syndrome. Identification of excessive CRH production requires that the clinician consider this possibility and measure CRH in blood. Neoplasms that can produce CRH include medullary thyroid carcinoma, paragangliomas, prostate cancer, and islet cell neoplasms. There are examples of cancers that produce both ACTH and CRH.
Patients with ectopic ACTH syndrome may present with clinical features of Cushing syndrome—easy bruising, centripetal obesity, muscle wasting, hypertension, diabetes, and metabolic alkalosis predominate. Alternatively, patients with rapidly growing SCLC may present with a clinical syndrome characterized by cachexia, muscle atrophy, profound hypokalemic metabolic alkalosis, and hypertension without the other clinical findings of Cushing syndrome.
The hallmark of ectopic ACTH syndrome is the finding of an elevated plasma ACTH concentration. However, in the differential diagnosis of hypercortisolism in a cancer patient with an elevated plasma ACTH concentration, one should also consider an ACTH producing pituitary tumor.108 Differentiation between pituitary ACTH production (a primary pituitary tumor) and ectopic tumor production of ACTH or ectopic CRH production, mimicking a pituitary tumor, is among the most difficult diagnostic workups in the discipline of endocrinology. There are numerous examples where failure to correctly differentiate between ectopic ACTH production and a pituitary tumor producing ACTH has led to incorrect treatment.109 In some cases, such as ectopic ACTH production by a SCLC, the clinical syndrome (hypokalemia, metabolic alkalosis, hypertension, exceedingly high plasma cortisol levels, and a rapidly growing tumor) will lead to a straightforward diagnosis.110 In other cases, particularly those with tumors that have not been associated with frequent ectopic ACTH production, the diagnosis can be challenging and require the assistance of an endocrinologist experienced in this area and with full access to the spectrum of interventional radiologic techniques used to differentiate pituitary from nonpituitary sources of ACTH.
The diagnostic evaluation begins with the measurement of plasma ACTH in a patient with hypercorticism (Figure 4). A plasma ACTH concentration >100 pg/mL should prompt investigation for an ectopic source of ACTH.111 In patients with an ACTH value below 10 pg/mL (collected under appropriate conditions to prevent degradation of ACTH), a primary adrenocortical source of cortisol (adrenal adenoma or carcinoma) should be considered. The majority of patients will fall into the range of ACTH values between 10 and 100 pg/mL. The major differential diagnostic possibilities in these patients include a central (pituitary) or peripheral (ectopic) source of ACTH, with production of ectopic CRH by a tumor a rare possibility. Differentiation between central and peripheral sources of ACTH is accomplished by several approaches. The identification of a pituitary tumor by MRI provides a presumptive diagnosis of a pituitary source, although pituitary tumors that are nonfunctional or produce another hormonal product occur with some frequency. For this reason, petrosal venous sinus sampling is often employed and permits differentiation between a central (pituitary) and peripheral (ectopic) source of ACTH.112 The diagnosis of ectopic CRH production can only be made by suspecting this diagnosis and measuring the plasma concentration of this peptide. It is also possible to use other approaches to diagnose ectopic ACTH syndrome. ACTH production from a tumor is not generally suppressed by a high dose of dexamethasone. In patients with an ACTH >10 pg/mL, administration of a single 8 mg oral dose of dexamethasone at 11:00 p.m. followed with measurement of serum cortisol level at 8:00 a.m. permits differentiation between central and peripheral sources.106 In pituitary Cushing syndrome, dexamethasone will generally suppress ACTH by 50%, whereas ACTH production by an ectopic source will generally not be suppressed. False-positive or false-negative results occur with each of these testing procedures.
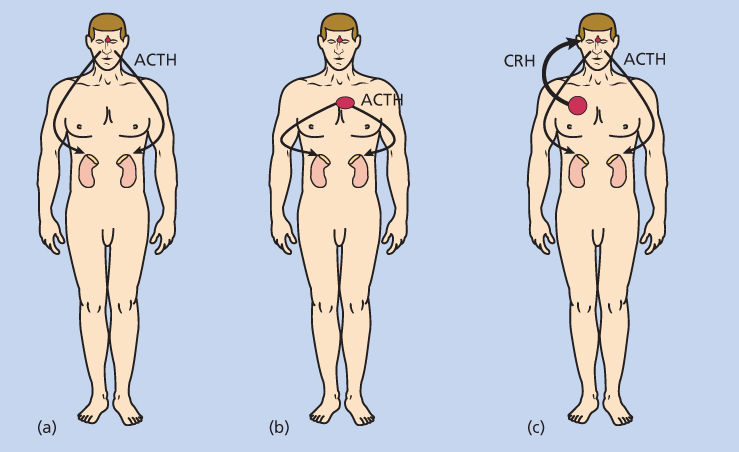
Figure 4 Differential diagnosis of adrenocorticotropin (ACTH)-dependent Cushing syndrome. It is difficult to differentiate between a pituitary (a) and ectopic (b) source of ACTH, the most common causes of ACTH-dependent Cushing syndrome. To differentiate with certainty, placement of catheters in veins draining the pituitary gland (inferior petrosal sinuses) combined with stimulation by exogenous corticotropin-releasing hormone permits differentiation with certainty (discussed in text). Ectopic corticotropin-releasing hormone (CRH) production by a tumor results in increased ACTH production by the pituitary gland (c). It is difficult to differentiate between ectopic CRH production and pituitary-dependent Cushing syndrome, necessitating the measurement of CRH in peripheral blood to make the diagnosis with certainty. Malignant tumors occasionally produce both CRH and ACTH, further complicating the diagnostic evaluation (not shown).
Surgical removal or treatment of a malignant tumor with chemotherapeutic agents is the primary therapy for an ACTH- or CRH-producing tumor. Any electrolyte abnormalities, diabetes mellitus, or hypertension should be corrected before a planned surgical procedure. Patients with long-standing Cushing syndrome and elevated plasma cortisol values have higher morbidity and mortality postoperatively. Preoperative treatment options include metyrapone (1–4 g/day orally), aminoglutethimide (250 mg orally four times per day with upward titration), or ketoconazole (200–400 mg twice a day orally).113, 114 Parenteral etomidate, used for sedation and induction of anesthesia, rapidly inhibits cortisol synthesis at subhypnotic concentrations.115 It is titrated from 0.3 to 4 mg/kg/h to normalize serum cortisol measurements and has been used to rapidly reverse hypercortisolism in a small number of patients. Replacement glucocorticoid therapy is needed when pharmacologic inhibitors of cortisol production are used to prevent adrenal insufficiency. If surgical removal of an ACTH- or CRH-producing tumor is not possible or inadvisable, chronic therapy with inhibitors of cortisol synthesis may be required. Alternatively, laparoscopic adrenalectomy with subsequent replacement of corticosteroids provides a rapid and generally safe approach to management of hypercortisolism. The development of retroperitoneal laparoscopic adrenalectomy during the past decade provides a rapid and safe technique to remove the adrenal glands. This technique is probably underutilized as a therapeutic strategy for management of ectopic ACTH syndrome.
Patients with rapidly progressive SCLC and ectopic ACTH syndrome form a unique subset of patients because of the need to initiate chemotherapy on a timely basis. Unfortunately, these patients are also highly susceptible to opportunistic infections, and initiation of therapy will often lead to death or serious morbidity related to infection.116 The central dilemma in these patients is that a period of 1–2 weeks may be required for pharmacologic inhibitors of cortisol synthesis to normalize the serum cortisol (and a longer period may be required for normalization of immunity), a delay in the initiation of chemotherapy that is generally deemed unacceptable from an oncologic perspective. Retroperitoneal laparoscopic adrenalectomy, a straightforward and well-tolerated technique following normalization of electrolyte abnormalities and hypertension, may provide a strategy for rapid normalization of excessive cortisol secretion. Prophylactic therapy for opportunistic infections caused by pneumocystis carinii or fungi should be considered if chemotherapy is initiated shortly after normalization of the serum cortisol.
Hypercalcemia caused by malignancy
Hypercalcemia is a common and serious cause of morbidity and mortality in cancer patients.
The most common causes of hypercalcemia in patients with cancer are PTHrP-mediated hypercalcemia, increased production of the active metabolite of vitamin D, calcitriol or 1,25 dihydroxy vitamin D3, and localized osteolytic hypercalcemia.117, 118 Ectopic production of PTH is a rare cause of cancer-associated hypercalcemia. Other common causes of hypercalcemia, most notably primary hyperparathyroidism, should be considered in the cancer patient with hypercalcemia. Measurement of serum intact parathyroid hormone (iPTH) permits differentiation between hyperparathyroidism and a number of other causes of hypercalcemia. The combination of hypercalcemia and an elevated parathyroid hormone level combined with increased urinary calcium excretion provides reasonable evidence for primary hyperparathyroidism. Suppression of the iPTH below the normal range is found in PTHrp or calcitriol-mediated hypercalcemia. Other less-common causes of hypercalcemia in cancer will be discussed below.
Parathyroid hormone-related protein
PTHrP is a small peptide in which 8 of the first 16 amino acids are identical to PTH. This small peptide causes hypercalcemia by binding to the PTH receptor and activating the expression of an osteoblast-specific cell surface protein, RANK ligand (RANKL). Interaction between RANKL and the RANK receptor on the osteoclast precursor causes increased osteoclast differentiation, bone resorption, and hypercalcemia. Other PTH-like actions of PTHrp include hypophosphatemia and increased urinary calcium excretion. PTHrp-mediated hypercalcemia is characterized by a suppressed iPTH level and a low or normal calcitriol level. This contrasts with the finding of elevated iPTH and calcitriol levels in primary hyperparathyroidism. PTHrP production is found commonly in squamous cell carcinomas; other tumors that produce it include: breast, neuroendocrine, renal, melanoma, and prostate tumors.
Calcitriol production by malignant tumors
Lymphoma commonly produces calcitriol, leading to increased gastrointestinal absorption of calcium. Lymphomatous tissue, such as granulomatous tissue seen in sarcoid, berylliosis, silicone-induced granulomatous, tuberculosis, and fungal infection, expresses 1α-hydroxylase, the enzyme that converts 25-hydroxy vitamin D3 to calcitriol. Clinical studies show that a high percentage of lymphoma patients have hypercalciuria at the time of diagnosis; a smaller percentage have frank hypercalcemia.119 The characteristic clinical features of hypercalcemia in the context of lymphoma include a suppressed serum iPTH, a normal or slightly increased phosphorus level (caused by the suppression of PTH), hypercalciuria, absence of bone metastasis, and an elevated serum calcitriol level in approximately one-half of hypercalcemic patients.119
Localized osteolytic bone resorption causing hypercalcemia
Certain malignancies metastasize to bone frequently, and some cause hypercalcemia. Hypercalcemia associated with breast cancer and myeloma is common. In contrast, prostate cancer, despite its more frequent presence in bone, rarely produces hypercalcemia. Malignancies that cause hypercalcemia produce cytokines, PTHrP, or other factors that stimulate increased bone resorption.117, 118 Perhaps, the best characterized is the production of PTHrP by breast carcinoma cells. There is compelling evidence in animal models that PTHrP production by breast cancer cells stimulates osteoclastic resorption and release of transforming growth factor (TGF)-β from normal bone. In this animal model, TGF-β release from bone stimulates proliferation of breast carcinoma growth, setting up a loop in which PTHrP production not only stimulates increased osteoclastic resorption, but also accelerates growth of adjacent breast cancer.120, 121 Indeed, this thought process led to an examination of and subsequent approval of bisphosphonates and denosumab, classes of drugs that inhibit osteoclast bone resorption, for prevention of cancer metastasis.122, 123
In multiple myeloma, several factors contribute to localized osteolysis. Increased expression of RANKL causing localized osteoclast proliferation appears to be the most important cause. Other factors that may contribute to osteoclast proliferation in myeloma are interleukin-6 and macrophage inflammatory protein 1α.118
Impact of malignancy-related hypercalcemia
Severe hypercalcemia in the context of hypercalcemia is associated with a shortened life span. The average survival for patients with severe and unresponsive hypercalcemia can be measured in weeks to months. The causes of death include complications of hypercalcemia (coma and renal failure) and progression of tumor. The development of hypercalcemia is often, although not always, an indicator of tumor progression in the face of adequate therapy. As it is not always possible to predict which patients will respond to oncologic therapy, it is important to treat hypercalcemia in all newly diagnosed patients with cancer. Whether to continue to treat recurrent and/or refractory hypercalcemia is a decision that should be based on response of the causative tumor to oncologic therapy and the overall prognosis of the patient. Severe hypercalcemia frequently causes depression of cerebral function or coma, a clinical situation that may reduce suffering in a dying patient.
Therapy of hypercalcemia
Dehydration is a common finding in hypercalcemic patients. Increased urine excretion of calcium causes a concentrating defect, leading to increased fluid loss. Initial management should focus on the reversal of dehydration by infusion of a solution of normal saline at rates between 100 and 300 mL/h. Hydration will commonly lower the serum calcium by 10–40% over a period of 6–12 h. Patients with severe hypercalcemia, defined as a serum calcium concentration >13 mg/dL (3.25 mmol/L), an alteration of mental status, or evidence of renal dysfunction attributable to hypercalcemia should be treated with either intravenous pamidronate (60–90 mg over 4 h) or zoledronate (4 mg over 30 min),124 glucocorticoids (40–60 mg/day prednisone equivalent), or gallium nitrate (200 mg/m2/day, infused daily for 7 days).125, 126 Salmon calcitonin may lower the serum calcium concentration by 1–2 mg/dL early in the treatment course, but is rarely effective long term. These drugs are sometimes used in combination or sequentially in a patient who is poorly responsive. Glucocorticoids, which inhibit calcium absorption, are most commonly used as primary therapy for lymphoma, whereas bisphosphonate therapy is more likely to be effective in hypercalcemia associated with solid tumors. Zoledronate is generally more effective than pamidronate because of its increased potency.127 Use of bisphosphonates for long-term treatment of bone metastasis has been associated with development of osteonecrosis of the jaw in 1–2.5% of treated patients. This has not been an issue in patients treated short term for hypercalcemia associated with malignancy.
A monoclonal antibody directed against RANKL (denosumab) prevents its interaction with the RANK receptor on osteoclast precursors, thereby reducing osteoclast-mediated bone resorption.128 Denosumab is an effective inhibitor of bone resorption, and recent clinical studies have established its efficacy in patients with hypercalcemia and bone metastasis, particularly those patients who have become unresponsive to bisphosphonate therapy.128
Human chorionic gonadotropin
Human chorionic gonadotropin (HCG) is formed from two different protein subunits encoded by separate genes. The first is the α subunit that is shared by all members of the pituitary class of glycoprotein hormones including HCG, LH, follicle-stimulating hormone (FSH), and thyroid-stimulating hormone (TSH). The second, the β subunit, is unique for each of these hormones. Production of HCG is found in trophoblastic tumors (choriocarcinomas, testicular embryonal carcinomas, and seminomas) and, uncommonly, in tumors of the lung and pancreas. In younger children, precocious puberty, caused by HCG stimulation of ovarian function, is seen. In adult males, gynecomastia is a common occurrence. Hyperthyroidism may develop from an interaction of HCG with the thyroid-stimulating hormone receptor (TSHR), particularly when β-HCG is expressed at high levels.
Removal or effective therapy for the underlying tumor is the most effective therapy for clinical syndromes caused by excessive β-HCG production. Hyperthyroidism can be treated short term with thionamide therapy if there is belief that chemotherapy or other strategies to treat the underlying malignancy are likely to be effective. In patients with less-responsive tumors, thyroidectomy or radioactive iodine may be required.
Hypoglycemia
Tumor-induced hypoglycemia is an uncommon but challenging cause of morbidity for cancer patients. Three different clinical syndromes have been identified. First, insulin can be produced by islet cell malignancy. Islet cell tumors commonly produce low levels of insulin that are clinically insignificant until large tumor burdens, most commonly in the form of hepatic metastasis, develop. A second cause is insufficient gluconeogenesis, seen in patients with near complete replacement of hepatic parenchyma by tumor, interfering with or eliminating glucose production. The third form is caused by increased concentrations of insulin-like growth factor II (IGF-II), a peptide that activates the insulin receptor. This syndrome is most commonly seen in patients with fibrosarcomas, hemangiopericytomas, or hepatomas. In these patients IGF-II levels are most commonly elevated, the result of a failure of insulin-like growth factor binding protein 3 (IGFBP3) and acid labile subunit to form a complex capable of binding IGF-II efficiently. The elevated circulating IGF-II activates the insulin receptor and causes hypoglycemia.129–131
The most common presentation for each of these clinical syndromes is fasting hypoglycemia, and patients are most likely to develop symptoms during normal periods of fasting, particularly during nocturnal hours. Measurement of a plasma insulin, proinsulin, and C-peptide during a period of hypoglycemia is the most important diagnostic tool for separating the first clinical type (insulin production) from the second (replacement of liver by tumor) and third (IGF-II) types. The findings of elevated insulin, proinsulin, and C-peptide in the face of hypoglycemia (and the absence of any drugs that might stimulate insulin release from normal pancreas) make a compelling case for unregulated insulin production as a cause of the hypoglycemia. In contrast, insulin, proinsulin, and C-peptide levels will be low in tumor replacement of the liver or IGF-II-mediated hypoglycemia. Laboratory findings in IGF-II-mediated hypoglycemia include an elevated serum IGF-II, low or normal insulin, proinsulin, and C-peptide measurements, low IGF-I levels, and generally normal IGFBP3 or acid labile subunit measurements in the context of a large sarcoma or retroperitoneal tumor.
Surgical excision or antineoplastic therapy to reduce tumor mass is effective in insulin or IGF-II-mediated hypoglycemia; there is little effective therapy for hepatic replacement by tumor other than providing glucose. Hypoglycemia is treated with frequent meals. Patients may remain symptom-free by being awakened for caloric intake during nocturnal hours. A continuous infusion of 20% dextrose through a central venous line may be required to maintain normal blood glucose in patients, particularly those with hepatic replacement by tumors. Glucagon infusion (0.5–2 mg/h) to stimulate hepatic gluconeogenesis is also an effective therapy for patients with insulin-producing tumors or those with IGF-II-mediated hypoglycemia. It is important to document a response to glucagon (1 mg subcutaneously with measurement of plasma glucose at 30 and 60 min following injection) before trying this therapeutic approach. Glucagon can be administered in small volumes (1–5 mL over 24 h), making it possible to use small infusion pumps.132 Patients treated with glucagon may develop the characteristic rash associated with glucagonoma, necessitating discontinuance of this treatment modality. Other therapies that have been applied with periodic success include recombinant GH (3–6 µg/kg subcutaneously daily) or glucocorticoids (20–40 mg prednisone equivalents per day). Octreotide or lanreotide have been used in patients with insulin-producing islet cell tumors, generally without success. The lack of success may relate to the fact that SRIH analogs currently available are more effective for inhibiting glucagon than insulin secretion. Diazoxide (3–8 mg/kg/day in 2–3 divided doses) has been used successfully to inhibit insulin secretion, but causes fluid retention, thereby limiting its usefulness at effective doses.
Hypoglycemia may also occur in patients with lactic acidosis in the context of end-stage leukemia or lymphoma. This clinical syndrome occurs in patients with end-stage or extensive disease and leukemic/lymphomatous involvement of the liver. It is hypothesized that lactic acid production by tumor cells exceeds the ability of the liver to clear it. The etiology of the hypoglycemia is unclear, but may result from impaired hepatic gluconeogenesis.133
Syndrome of inappropriate antidiuretic hormone
SIADH was discussed earlier (see the section titled “Disorders of Electrolyte/Mineral Metabolism: Hyponatremia”) as a side effect of cancer therapy. In addition, approximately 15% of SCLC, 1% of other lung cancers, and 3% of squamous cell head and neck cancers produce vasopressin in an unregulated manner, leading to hyponatremia, hypoosmolality, increased urine sodium excretion, and an inappropriately high urine osmolality relative to the plasma tonicity.134 In addition, other benign or malignant neoplasms that include primary brain tumors, hematologic neoplasms, skin tumors, and gastrointestinal, gynecologic, breast, and prostate cancers, and sarcomas can produce this clinical syndrome.
Most patients who develop this syndrome are asymptomatic. In cases where the serum sodium concentration falls <120 mEq/L, altered mental status and seizures may develop. In particular, women of reproductive age who develop hyponatremia may develop profound cerebral degeneration. Fluid restriction can be used effectively for short-term management, but treatment with demeclocycline (150–300 mg/day), an agent that inhibits the effects of vasopressin on the kidney, is preferable for long-term treatment. Vasopressin receptor antagonists, conivaptan, lixivaptan, tolvaptan, and satavaptan, have shown efficacy in clinical trials and at least one, conivaptan, is currently FDA-approved in the United States in an intravenous form (20 mg intravenously over 30 min, followed by 20 mg over 24 h). Tolvaptan (15–60 mg/day) is approved for oral use. Both of these agents are highly effective and it is important to monitor the serum sodium concentration carefully after initiation of therapy.
A consideration in the long-term use of demeclocycline and the V2 receptor antagonists is the cost of therapy. In a patient with a serum sodium concentration <120 mEq/L, use of intravenous conivaptan results in improvement or normalization of the serum sodium over a 24–48 h period and may be most appropriate in this emergent situation; demeclocycline effects are seen over several days. For longer-term therapy, particularly when it is not possible to reduce tumor mass, consideration should be given to the less-expensive demeclocycline, which is generally well tolerated and effective.
Other ectopic hormone syndromes
Tumor-induced osteomalacia
Severe hypophosphatemia caused by renal phosphate wasting is the hallmark of tumor-induced osteomalacia. This clinical syndrome is characterized by osteomalacia, caused by inadequate mineralization of osteoid, and moderate to severe proximal myopathy.135 Tumors that produce this clinical syndrome include mesenchymal tumors (osteoblastomas, giant cell osteosarcomas, hemangiopericytomas, hemangiomas, and nonossifying fibromas)136 and, rarely, malignant tumors such as prostate or lung cancer. There is compelling evidence that fibroblast growth factor-23, a member of the fibroblast growth factor family that is mutated in autosomal dominant osteomalacia,137 is overexpressed by some neoplasms causing tumor-induced osteomalacia.138 Oral or intravenous supplementation of phosphate combined with vitamin D therapy is generally effective for eradicating or improving clinical symptoms. Complete surgical removal of the tumor is generally curative.
Erythropoietin, thrombopoietin, leukopoietin, or colony-stimulating factor production
Polycythemia caused by ectopic erythropoietin production is a rare clinical syndrome. It is found in cerebellar hemangioblastoma, uterine fibroids, pheochromocytomas, and renal cell, ovarian, and hepatic cancers.139, 140 Treatment can include surgical or chemotherapy reduction of tumor mass or phlebotomy. Other less well-defined syndromes include production of thrombopoietin, leukopoietin, or colony-stimulating factor by some tumors. These conditions are treated by appropriate chemotherapy to reduce its size or by surgical removal.
Renin production
Production of renin by renal (Wilms tumor, renal cell carcinoma, or hemangiopericytoma), lung (SCLC and adenocarcinoma), hepatic, pancreatic, or ovarian carcinomas can produce a clinical syndrome characterized by hypertension, hypokalemia, and evidence of increased aldosterone production.141 Therapy with spironolactone, angiotensin-converting enzyme inhibitors, or angiotensin receptor antagonists may lower the blood pressure and normalize electrolyte abnormalities in patients in whom the tumor cannot be resected.
Growth hormone and prolactin
Acromegaly is a condition characterized by elevated GH and IGF-1 values, most commonly caused by a pituitary tumor. There are uncommon examples of GH production by lung and gastric adenocarcinomas. Ectopic production of GHRH, the hypothalamic peptide that normally regulates GH production by the pituitary,142 has been demonstrated for islet cell tumors, bronchogenic carcinoids, and SCLC. It is unlikely that treatment with SRIH analogs, one of the mainstay therapies for pituitary tumors producing GH, will be effective. However, the GH antagonist, pegvisomant, is likely to be effective for treatment of GH excess. Ectopic prolactin production is found rarely in gonadoblastoma,143 lymphoma,141 leukemia,144 and colorectal cancer.145 The clinical syndrome includes galactorrhea and amenorrhea in women and hypogonadism and gynecomastia in men. Dopamine agonists (bromocriptine, quinagolide, or cabergoline), effective for treatment of pituitary prolactinomas, are generally ineffective for treatment of ectopic prolactin production.
References
- 1 Fassett DR, Couldwell WT. Metastases to the pituitary gland. Neurosurg Focus. 2004;16:E8.
- 2 Lam KS, Tse VK, Wang C, Yeung RT, Ho JH. Effects of cranial irradiation on hypothalamic-pituitary function—a 5-year longitudinal study in patients with nasopharyngeal carcinoma. Q J Med. 1991;78:165–176.
- 3 Pai HH, Thornton A, Katznelson L, et al. Hypothalamic/pituitary function following high-dose conformal radiotherapy to the base of skull: demonstration of a dose-effect relationship using dose-volume histogram analysis. Int J Radiat Oncol Biol Phys. 2001;49:1079–1092.
- 4 Samaan NA, Schultz PN, Yang KP, et al. Endocrine complications after radiotherapy for tumors of the head and neck. J Lab Clin Med. 1987;109:364–372.
- 5 Shalet SM. Disorders of the endocrine system due to radiation and cytotoxic chemotherapy. Clin Endocrinol (Oxf). 1983;19:637–659.
- 6 Pitteloud MJ, Dadu R, Cabanillas ME, et al. Hypophysitis in the Age of Cancer Immunotherapy: Experience in a Large Cancer Center. Annual Meeting of the Endocrine Society 2015; pp. OR30–OR35.
- 7 Garnick MB, Larsen PR. Acute deficiency of thyroxine-binding globulin during L-asparaginase therapy. N Engl J Med. 1979;301:252–253.
- 8 Djurica SN, Plecas V, Milojevic Z, et al. Direct effects of cytostatic therapy on the functional state of the thyroid gland and TBG in serum of patients. Exp Clin Endocrinol. 1990;96:57–63.
- 9 Beex L, Ross A, Smals A, Kloppenborg P. 5-fluorouracil-induced increase of total serum thyroxine and triiodothyronine. Cancer Treat Rep. 1977;61:1291–1295.
- 10 van Seters AP, Moolenaar AJ. Mitotane increases the blood levels of hormone-binding proteins. Acta Endocrinol (Copenh). 1991;124:526–533.
- 11 Chopra IJ. Clinical review 86: euthyroid sick syndrome: is it a misnomer? J Clin Endocrinol Metabol. 1997;82:329–334.
- 12 Tami TA, Gomez P, Parker GS, Gupta MB, Frassica DA. Thyroid dysfunction after radiation therapy in head and neck cancer patients. Am J Otolaryngol. 1992;13:357–362.
- 13 Grande C. Hypothyroidism following radiotherapy for head and neck cancer: multivariate analysis of risk factors. Radiother Oncol. 1992;25:31–36.
- 14 Tamura K, Shimaoka K, Friedman M. Thyroid abnormalities associated with treatment of malignant lymphoma. Cancer. 1981;47:2704–2711.
- 15 Hancock SL, Cox RS, McDougall IR. Thyroid diseases after treatment of Hodgkin’s disease. N Engl J Med. 1991;325:599–605.
- 16 Constine LS, Donaldson SS, McDougall IR, et al. Thyroid dysfunction after radiotherapy in children with Hodgkin’s disease. Cancer. 1984;53:878–883.
- 17 Devney RB, Sklar CA, Nesbit ME Jr et al. Serial thyroid function measurements in children with Hodgkin disease. J Pediatr. 1984;105:223–227.
- 18 Schimpff SC, Diggs CH, Wiswell JG, Salvatore PC, Wiernik PH. Radiation-related thyroid dysfunction: implications for the treatment of Hodgkin’s disease. Ann Intern Med. 1980;92:91–98.
- 19 Fuks Z, Glatstein E, Marsa GW, Bagshaw MA, Kaplan HS. Long-term effects on external radiation on the pituitary and thyroid glands. Cancer. 1976;37:1152–1161.
- 20 Joensuu H, Viikari J. Thyroid function after postoperative radiation therapy in patients with breast cancer. Acta Radiolog Oncol. 1986;25:167–170.
- 21 Sklar CA, Kim TH, Ramsay NK. Thyroid dysfunction among long-term survivors of bone marrow transplantation. Am J Med. 1982;73:688–694.
- 22 Boulad F, Bromley M, Black P, et al. Thyroid dysfunction following bone marrow transplantation using hyperfractionated radiation. Bone Marrow Transplant. 1995;15:71–76.
- 23 Toubert ME, Socie G, Gluckman E, et al. Short- and long-term follow-up of thyroid dysfunction after allogeneic bone marrow transplantation without the use of preparative total body irradiation. Br J Haematol. 1997;98:453–457.
- 24 Sutcliffe SB, Chapman R, Wrigley PF. Cyclical combination chemotherapy and thyroid function in patients with advanced Hodgkin’s disease. Med Pediatr Oncol. 1981;9:439–448.
- 25 Ogilvy-Stuart AL, Shalet SM, Gattamaneni HR. Thyroid function after treatment of brain tumors in children. J Pediatr. 1991;119:733–737.
- 26 Heidemann PH, Stubbe P, Beck W. Transient secondary hypothyroidism and thyroxine binding globulin deficiency in leukemic children during polychemotherapy: an effect of L-asparaginase. Eur J Pediatr. 1981;136:291–295.
- 27 Krouse RS, Royal RE, Heywood G, et al. Thyroid dysfunction in 281 patients with metastatic melanoma or renal carcinoma treated with interleukin-2 alone. J Immunother Emphasis Tumor Immunol. 1995;18:272–278.
- 28 Vassilopoulou-Sellin R, Sella A, Dexeus FH, Theriault RL, Pololoff DA. Acute thyroid dysfunction (thyroiditis) after therapy with interleukin-2. Horm Metab Res. 1992;24:434–438.
- 29 Baudin E, Marcellin P, Pouteau M, et al. Reversibility of thyroid dysfunction induced by recombinant alpha interferon in chronic hepatitis C. Clin Endocrinol (Oxf). 1993;39:657–661.
- 30 Vecil GG, Papadopoulos NV, Vassilopoulou-Sellin R, McCutcheon IE. Interferon-induced hypothyroidism causing reversible pituitary enlargement. Endocr Pract. 2008;14:219–223.
- 31 Sherman SI. Etiology, diagnosis, and treatment recommendations for central hypothyroidism associated with bexarotene therapy for cutaneous T-cell lymphoma. Clin Lymphoma. 2003;3:249–252.
- 32 Golden WM, Weber KB, Hernandez TL, et al. Single-dose rexinoid rapidly and specifically suppresses serum thyrotropin in normal subjects. J Clin Endocrinol Metab. 2007;92:124–130.
- 33 Smit JW, Stokkel MP, Pereira AM, Romijn JA, Visser TJ. Bexarotene-induced hypothyroidism: bexarotene stimulates the peripheral metabolism of thyroid hormones. J Clin Endocrinol Metab. 2007;92:2496–2499.
- 34 Illouz F, Braun D, Briet C, Schweizer U, Rodien P. Endocrine side-effects of anti-cancer drugs: thyroid effects of tyrosine kinase inhibitors. Eur J Endocrinol. 2014;171:R91–R99.
- 35 Wells SA Jr Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28:767–772.
- 36 Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372:621–630.
- 37 Quach A, Ji L, Mishra V, et al. Thyroid and hepatic function after high-dose 131 I-metaiodobenzylguanidine (131 I-MIBG) therapy for neuroblastoma. Pediatr Blood Cancer. 2011;56:191–201.
- 38 Petersen M, Keeling CA, McDougall IR. Hyperthyroidism with low radioiodine uptake after head and neck irradiation for Hodgkin’s disease. J Nucl Med. 1989;30:255–257.
- 39 Wasnich RD, Grumet FC, Payne RO, Kriss JP. Graves’ ophthalmopathy following external neck irradiation for nonthyroidal neoplastic disease. J Clin Endocrinol Metab. 1973;37:703–713.
- 40 Haraldsdottir S, Li Q, Villalona-Calero MA, et al. Case of sorafenib-induced thyroid storm. J Clin Oncol. 2013;31:e262–e264.
- 41 Ron E, Lubin JH, Shore RE, et al. Thyroid cancer after exposure to external radiation: a pooled analysis of seven studies. Radiat Res. 1995;141:259–277.
- 42 Camoriano JK, Loprinzi CL, Ingle JN, et al. Weight change in women treated with adjuvant therapy or observed following mastectomy for node-positive breast cancer. J Clin Oncol. 1990;8:1327–1334.
- 43 Makari-Judson G, Judson CH, Mertens WC. Longitudinal patterns of weight gain after breast cancer diagnosis: observations beyond the first year. Breast J. 2007;13:258–265.
- 44 Saquib N, Flatt SW, Natarajan L, et al. Weight gain and recovery of pre-cancer weight after breast cancer treatments: evidence from the women’s healthy eating and living (WHEL) study. Breast Cancer Res Treat. 2007;105:177–186.
- 45 Courneya KS, Katzmarzyk PT, Bacon E. Physical activity and obesity in Canadian cancer survivors: population-based estimates from the 2005 Canadian Community Health Survey. Cancer. 2008;112:2475–2482.
- 46 Kroenke CH, Chen WY, Rosner B, Holmes MD. Weight, weight gain, and survival after breast cancer diagnosis. J Clin Oncol. 2005;23:1370–1378.
- 47 Park SM, Lim MK, Jung KW, et al. Prediagnosis smoking, obesity, insulin resistance, and second primary cancer risk in male cancer survivors: National Health Insurance Corporation Study. J Clin Oncol. 2007;25:4835–4843.
- 48 Nilsen TI, Vatten LJ. Prospective study of colorectal cancer risk and physical activity, diabetes, blood glucose and BMI: exploring the hyperinsulinaemia hypothesis. Br J Cancer. 2001;84:417–422.
- 49 Muti P, Quattrin T, Grant BJ, et al. Fasting glucose is a risk factor for breast cancer: a prospective study. Cancer Epidemiol Biomarkers Prev. 2002;11:1361–1368.
- 50 Verlato G, Zoppini G, Bonora E, Muggeo M. Mortality from site-specific malignancies in type 2 diabetic patients from Verona. Diabetes Care. 2003;26:1047–1051.
- 51 Richardson LC, Pollack LA. Therapy insight: influence of type 2 diabetes on the development, treatment and outcomes of cancer. Nat Clin Pract Oncol. 2005;2:48–53.
- 52 Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol. 2004;159:1160–1167.
- 53 Yamagata H, Kiyohara Y, Nakamura S, et al. Impact of fasting plasma glucose levels on gastric cancer incidence in a general Japanese population: the Hisayama study. Diabetes Care. 2005;28:789–794.
- 54 van de Poll-Franse LV, Houterman S, Janssen-Heijnen ML, et al. Less aggressive treatment and worse overall survival in cancer patients with diabetes: a large population based analysis. Int J Cancer. 2007.
- 55 Lu XH, Wang L, Li H, et al. Establishment of risk model for pancreatic cancer in Chinese Han population. World J Gastroenterol. 2006;12:2229–2234.
- 56 Kuriki K, Hirose K, Tajima K. Diabetes and cancer risk for all and specific sites among Japanese men and women. Eur J Cancer Prev. 2007;16:83–89.
- 57 Fisher WE. Diabetes: risk factor for the development of pancreatic cancer or manifestation of the disease? World J Surg. 2001;25:503–508.
- 58 Stolzenberg-Solomon RZ, Graubard BI, Chari S, et al. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA. 2005;294:2872–2878.
- 59 Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273:1605–1609.
- 60 Schein PS, O’Connell MJ, Blom J, et al. Clinical antitumor activity and toxicity of streptozotocin (NSC-85998). Cancer. 1974;34:993–1000.
- 61 Gillette PC, Hill LL, Starling KA, Fernbach DJ. Transient diabetes mellitus secondary to L-asparaginase therapy in acute leukemia. J Pediatr. 1972;81:109–111.
- 62 Almawi WY, Tamim H, Azar ST. Clinical review 103: T helper type 1 and 2 cytokines mediate the onset and progression of type I (insulin-dependent) diabetes. J Clin Endocrinol Metab. 1999;84:1497–1502.
- 63 Drachenberg CB, Klassen DK, Weir MR, et al. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation. 1999;68:396–402.
- 64 Jindal RM, Sidner RA, Milgrom ML. Post-transplant diabetes mellitus. The role of immunosuppression. Drug Saf. 1997;16:242–257.
- 65 Headley JA, Theriault RL, LeBlanc AD, Vassilopoulou-Sellin R, Hortobagyi GN. Pilot study of bone mineral density in breast cancer patients treated with adjuvant chemotherapy. Cancer Invest. 1998;16:6–11.
- 66 Jones G, Sambrook PN. Drug-induced disorders of bone metabolism. Incidence, management and avoidance. Drug Saf. 1994;10:480–489.
- 67 Schwartz AM, Leonidas JC. Methotrexate osteopathy. Skeletal Radiol. 1984;11:13–16.
- 68 Atkinson SA, Halton JM, Bradley C, Wu B, Barr RD. Bone and mineral abnormalities in childhood acute lymphoblastic leukemia: influence of disease, drugs and nutrition. Int J Cancer Suppl. 1998;11:35–39.
- 69 Bruning PF, Pit MJ, de Jong-Bakker M, et al. Bone mineral density after adjuvant chemotherapy for premenopausal breast cancer. Br J Cancer. 1990;61:308–310.
- 70 Carlson K, Simonsson B, Ljunghall S. Acute effects of high-dose chemotherapy followed by bone marrow transplantation on serum markers of bone metabolism. Calcif Tissue Int. 1994;55:408–411.
- 71 Vassilopoulou-Sellin R, Brosnan P, Delpassand A, et al. Osteopenia in young adult survivors of childhood cancer. Med Pediatr Oncol. 1999;32:272–278.
- 72 Brade WP, Herdrich K, Kachel-Fischer U, Araujo CE. Dosing and side-effects of ifosfamide plus mesna. J Cancer Res Clin Oncol. 1991;117(Suppl 4):S164–S186.
- 73 Moncrieff M, Foot A. Fanconi syndrome after ifosfamide. Cancer Chemother Pharmacol. 1989;23:121–122.
- 74 Citrin DL, Wallemark CB, Nadler R, et al. Estramustine affects bone mineral metabolism in metastatic prostate cancer. Cancer. 1986;58:2208–2213.
- 75 Abrams H, Spiro R, Goldstein N. Metastasis in carcinoma—one thousand autopsied cases. Cancer. 1950;3:74.
- 76 Hussain S, Belldegrun A, Seltzer SE, Richie JP, Abrams HL. CT diagnosis of adrenal abnormalities in patients with primary non-adrenal malignancies. Eur J Radiol. 1986;6:127–131.
- 77 Platts JK, Drew PJ, Harvey JN. Death from phaeochromocytoma: lessons from a post-mortem survey. J R Coll Physicians Lond. 1995;29:299–306.
- 78 Katz RL, Patel S, Mackay B, Zornoza J. Fine needle aspiration cytology of the adrenal gland. Acta Cytol. 1984;28:269–282.
- 79 Francis IR, Smid A, Gross MD, et al. Adrenal masses in oncologic patients: functional and morphologic evaluation. Radiology. 1988;166:353–356.
- 80 Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer. 1987;60:103–107.
- 81 Cedermark BJ, Sjoberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet. 1981;152:607–610.
- 82 Gurney JG, Ness KK, Stovall M, et al. Final height and body mass index among adult survivors of childhood brain cancer: childhood cancer survivor study. J Clin Endocrinol Metab. 2003;88:4731–4739.
- 83 Shalet SM, Clayton PE, Price DA. Growth and pituitary function in children treated for brain tumours or acute lymphoblastic leukaemia. Horm Res. 1988;30:53–61.
- 84 Burman P, Broman JE, Hetta J, et al. Quality of life in adults with growth hormone (GH) deficiency: response to treatment with recombinant human GH in a placebo-controlled 21-month trial. J Clin Endocrinol Metab. 1995;80:3585–3590.
- 85 Berghmans T, Paesmans M, Body JJ. A prospective study on hyponatraemia in medical cancer patients: epidemiology, aetiology and differential diagnosis. Support Care Cancer. 2000;8:192–197.
- 86 Johnson BE, Damodaran A, Rushin J, et al. Ectopic production and processing of atrial natriuretic peptide in a small cell lung carcinoma cell line and tumor from a patient with hyponatremia. Cancer. 1997;79:35–44.
- 87 Yeung SC, Chiu AC, Vassilopoulou-Sellin R, Gagel RF. The endocrine effects of nonhormonal antineoplastic therapy. Endocr Rev. 1998;19:144–172.
- 88 Harlow PJ, DeClerck YA, Shore NA, et al. A fatal case of inappropriate ADH secretion induced by cyclophosphamide therapy. Cancer. 1979;44:896–898.
- 89 Bode U, Seif SM, Levine AS. Studies on the antidiuretic effect of cyclophosphamide: vasopressin release and sodium excretion. Med Pediatr Oncol. 1980;8:295–303.
- 90 Anand AJ, Bashey B. Newer insights into cisplatin nephrotoxicity. Ann Pharmacother. 1993;27:1519–1525.
- 91 Raftopoulos H. Diagnosis and management of hyponatremia in cancer patients. Support Care Cancer. 2007;15:1341–1347.
- 92 Skinner R, Pearson AD, Price L, Coulthard MG, Craft AW. Nephrotoxicity after ifosfamide. Arch Dis Child. 1990;65:732–738.
- 93 Delaney V, de Pertuz Y, Nixon D, Bourke E. Indomethacin in streptozocin-induced nephrogenic diabetes insipidus. Am J Kidney Dis. 1987;9:79–83.
- 94 Vassilopoulou-Sellin R, Newman BM, Taylor SH, Guinee VF. Incidence of hypercalcemia in patients with malignancy referred to a comprehensive cancer center. Cancer. 1993;71:1309–1312.
- 95 Cohen J, Gierlowski TC, Schneider AB. A prospective study of hyperparathyroidism in individuals exposed to radiation in childhood. JAMA. 1990;264:581–584.
- 96 Norton JA, Venzon DJ, Berna MJ, et al. Prospective study of surgery for primary hyperparathyroidism (HPT) in multiple endocrine neoplasia-type 1 and Zollinger-Ellison syndrome: long-term outcome of a more virulent form of HPT. Ann Surg. 2008;247:501–510.
- 97 Stewart AF, Keating T, Schwartz PE. Magnesium homeostasis following chemotherapy with cisplatin: a prospective study. Am J Obstet Gynecol. 1985;153:660–665.
- 98 Ehnholm C, Aho K, Huttunen JK, et al. Effect of interferon on plasma lipoproteins and on the activity of postheparin plasma lipases. Arteriosclerosis. 1982;2:68–73.
- 99 Yamagishi S, Abe T, Sawada T. Human recombinant interferon alpha-2a (r IFN alpha-2a) therapy suppresses hepatic triglyceride lipase, leading to severe hypertriglyceridemia in a diabetic patient. Am J Gastroenterol. 1994;89:2280.
- 100 Berruti A, Gorzegno G, Vitetta G, Tampellini M, Dogliotti L. Hypertriglyceridemia during long-term interferon-alpha therapy: efficacy of diet and gemfibrosil treatment. A case report. Tumori. 1992;78:353–355.
- 101 Constine LS, Woolf PD, Cann D, et al. Hypothalamic-pituitary dysfunction after radiation for brain tumors. N Engl J Med. 1993;328:87–94.
- 102 Quigley C, Cowell C, Jimenez M, et al. Normal or early development of puberty despite gonadal damage in children treated for acute lymphoblastic leukemia. N Engl J Med. 1989;321:143–151.
- 103 Brauner R, Rappaport R. Precocious puberty secondary to cranial irradiation for tumors distant from the hypothalamo-pituitary area. Horm Res. 1985;22:78–82.
- 104 Maioli E, Fortino V. The complexity of parathyroid hormone-related protein signalling. Cell Mol Life Sci. 2004;61:257–262.
- 105 Eipper BA, Mains RE. Structure and biosynthesis of pro-adrenocorticotropin/endorphin and related peptides. Endocr Rev. 1980;1:1–27.
- 106 Findling JW, Raff H. Diagnosis and differential diagnosis of Cushing’s syndrome. Endocrinol Metab Clin North Am. 2001;30:729–747.
- 107 O’Brien T, Young WF Jr Davila DG, et al. Cushing’s syndrome associated with ectopic production of corticotrophin-releasing hormone, corticotrophin and vasopressin by a phaeochromocytoma. Clin Endocrinol (Oxf). 1992;37:460–467.
- 108 Newell-Price J. Cushing’s syndrome. Clin Med. 2008;8:204–208.
- 109 Shrager JB, Wright CD, Wain JC, et al. Bronchopulmonary carcinoid tumors associated with Cushing’s syndrome: a more aggressive variant of typical carcinoid. J Thorac Cardiovasc Surg. 1997;114:367–375.
- 110 Islam M, Paul RV. Correction of metabolic alkalosis by potassium chloride in ectopic adrenocorticotropic hormone syndrome. Am J Kidney Dis. 1996;28:610–613.
- 111 Cook DM, Loriaux DL. Cushing’s: medical approach. Curr Ther Endocrinol Metab. 1997;6:59–62.
- 112 Doppman JL, Oldfield EH, Nieman LK. Bilateral sampling of the internal jugular vein to distinguish between mechanisms of adrenocorticotropic hormone-dependent Cushing syndrome. Ann Intern Med. 1998;128:33–36.
- 113 Winquist EW, Laskey J, Crump M, Khamsi F, Shepherd FA. Ketoconazole in the management of paraneoplastic Cushing’s syndrome secondary to ectopic adrenocorticotropin production. J Clin Oncol. 1995;13:157–164.
- 114 Nieman LK, Ilias I. Evaluation and treatment of Cushing’s syndrome. Am J Med. 2005;118:1340–1346.
- 115 Johnson TN, Canada TW. Etomidate use for Cushing’s syndrome caused by an ectopic adrenocorticotropic hormone-producing tumor. Ann Pharmacother. 2007;41:350–353.
- 116 Dimopoulos MA, Fernandez JF, Samaan NA, Holoye PY, Vassilopoulou-Sellin R. Paraneoplastic Cushing’s syndrome as an adverse prognostic factor in patients who die early with small cell lung cancer. Cancer. 1992;69:66–71.
- 117 Stewart AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med. 2005;352:373–379.
- 118 Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664.
- 119 Seymour JF, Gagel RF, Hagemeister FB, Dimopoulos MA, Cabanillas F. Calcitriol production in hypercalcemic and normocalcemic patients with non-Hodgkin lymphoma. Ann Intern Med. 1994;121:633–640.
- 120 Guise TA, Yin JJ, Thomas RJ, et al. Parathyroid hormone-related protein (PTHrP)-(1-139) isoform is efficiently secreted in vitro and enhances breast cancer metastasis to bone in vivo. Bone. 2002;30:670–676.
- 121 Kakonen SM, Selander KS, Chirgwin JM, et al. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J Biol Chem. 2002;277:24571–24578.
- 122 Wang Z, Qiao D, Lu Y, et al. Systematic literature review and network meta-analysis comparing bone-targeted agents for the prevention of skeletal-related events in cancer patients with bone metastasis. Oncologist. 2015;20:440–449.
- 123 Dougall WC, Holen I, Gonzalez SE. Targeting RANKL in metastasis. Bonekey Rep. 2014;3:519.
- 124 Body JJ, Bartl R, Burckhardt P, et al. Current use of bisphosphonates in oncology. International Bone and Cancer Study Group. J Clin Oncol. 1998;16:3890–3899.
- 125 Chisholm MA, Mulloy AL, Taylor AT. Acute management of cancer-related hypercalcemia. Ann Pharmacother. 1996;30:507–513.
- 126 Apseloff G. Therapeutic uses of gallium nitrate: past, present, and future. Am J Ther. 1999;6:327–339.
- 127 Berenson JR, Rosen LS, Howell A, et al. Zoledronic acid reduces skeletal-related events in patients with osteolytic metastases. Cancer. 2001;91:1191–1200.
- 128 Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab. 2014;99:3144–3152.
- 129 Baxter RC, Holman SR, Corbould A, et al. Regulation of the insulin-like growth factors and their binding proteins by glucocorticoid and growth hormone in nonislet cell tumor hypoglycemia. J Clin Endocrinol Metab. 1995;80:2700–2708.
- 130 Baxter RC. Insulin-like growth factor binding proteins as glucoregulators. Metabolism. 1995;44:12–17.
- 131 Baxter RC. The role of insulin-like growth factors and their binding proteins in tumor hypoglycemia. Horm Res. 1996;46:195–201.
- 132 Hoff AO, Vassilopoulou-Sellin R. The role of glucagon administration in the diagnosis and treatment of patients with tumor hypoglycemia. Cancer. 1998;82:1585–1592.
- 133 Sillos EM, Shenep JL, Burghen GA, et al. Lactic acidosis: a metabolic complication of hematologic malignancies: case report and review of the literature. Cancer. 2001;92:2237–2246.
- 134 Flombaum CD. Metabolic emergencies in the cancer patient. Semin Oncol. 2000;27:322–334.
- 135 Kumar R. Tumor-induced osteomalacia and the regulation of phosphate homeostasis. Bone. 2000;27:333–338.
- 136 Nuovo MA, Dorfman HD, Sun CC, Chalew SA. Tumor-induced osteomalacia and rickets. Am J Surg Pathol. 1989;13:588–599.
- 137 Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–6505.
- 138 Jonsson KB, Zahradnik R, Larsson T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663.
- 139 Suzuki M, Takamizawa S, Nomaguchi K, et al. Erythropoietin synthesis by tumour tissues in a patient with uterine myoma and erythrocytosis. Br J Haematol. 2001;113:49–51.
- 140 Gold PJ, Fefer A, Thompson JA. Paraneoplastic manifestations of renal cell carcinoma. Semin Urol Oncol. 1996;14:216–222.
- 141 Arbaiza D, Noriega K, Marcial J, et al. Ectopic production of prolactin in an infant with non-Hodgkin lymphoma. Med Pediatr Oncol. 1999;32:311–312.
- 142 Doga M, Bonadonna S, Burattin A, Giustina A. Ectopic secretion of growth hormone-releasing hormone (GHRH) in neuroendocrine tumors: relevant clinical aspects. Ann Oncol. 2001;12(Suppl 2):S89–S94.
- 143 Hoffman WH, Gala RR, Kovacs K, Subramanian MG. Ectopic prolactin secretion from a gonadoblastoma. Cancer. 1987;60:2690–2695.
- 144 Hatfill SJ, Kirby R, Hanley M, Rybicki E, Bohm L. Hyperprolactinemia in acute myeloid leukemia and indication of ectopic expression of human prolactin in blast cells of a patient of subtype M4. Leuk Res. 1990;14:57–62.
- 145 Bhatavdekar JM, Patel DD, Chikhlikar PR, et al. Ectopic production of prolactin by colorectal adenocarcinoma. Dis Colon Rectum. 2001;44:119–127.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]Endocrine complications and paraneoplastic syndromes
Sai-Ching Jim Yeung, MD, PhD, FACP Robert F. Gagel, MD
Overview
Transformation of normal cells results in activation and/or suppression of a number of hormonally active genes. This chapter outlines the several clinical endocrine paraneoplastic syndromes and their appropriate management. Treatment of cancer results in a number of endocrine or metabolic manifestations, most of which are related to hormone deficiency or drug-related toxicity. The introduction of targeted therapy that disrupts signaling pathways and immunotherapy, contrary to expectations, has actually increased the number of endocrine manifestations. This chapter will chronicle the major new endocrine toxicities in addition to those observed with older cytotoxic chemotherapies and radiation.
Introduction
This chapter is divided into two major sections. The first focuses on endocrine complications in cancer patients and the second on endocrine paraneoplastic syndromes. Cancer and its treatment can lead to endocrine dysfunction or clinical and laboratory abnormalities that obscure or mimic endocrine diseases. Paraneoplastic syndromes are a group of diverse clinical syndromes seen in cancer patients caused by circulating biologic/humoral factors that include hormones, immunoglobulins, cytokines, and other agents.
Endocrine complications
Hypothalamic–pituitary dysfunction
Radiotherapy is a common cause of hypothalamic–pituitary dysfunction in cancer patients. There is no strong direct evidence to implicate chemotherapy as a cause of permanent dysfunction of the anterior pituitary, although some of the newer targeted therapies may affect pituitary function. Metastasis to the hypothalamic region or the pituitary gland is uncommon,1 and clinical manifestations of endocrine dysfunction because of metastatic disease in this region are rare. However, benign tumors such as pituitary tumors and craniopharyngiomas frequently affect this anatomic region and cause endocrine dysfunction. The introduction of immunotherapy by immune checkpoint blockade [anti-CTLA4 antibodies (ipilimumab and tremelimumab) and anti-PD-1 antibody (nivolumab)] has been associated with development of autoimmune hypophysitis that requires hormonal replacement (corticosteroids or thyroid hormone) in 2–3% of treated patients.
Development of radiation-induced hypothalamic dysfunction is insidious; hormonal deficiency can manifest years after radiation. In general, the rapidity of onset and severity of dysfunction depend on the total dose of radiation and the rate of delivery. The sequence and frequency of dysfunction among the axes of hypothalamic–pituitary functions vary. The somatotropic axis is the most susceptible, while the thyrotropic axis is the least susceptible (Figure 1).2–5 The diagnosis of hypothalamic–pituitary dysfunction requires vigilance of the physician because most presenting symptoms (e.g., fatigue and weakness) are nonspecific and attributable to other causes common among cancer patients. A diagnostic screen for hypothalamic/pituitary dysfunction may include serum growth hormone (GH) and insulin-like growth factor-1 (IGF-1) measurement and evaluation for gonadal failure. Signs of overt hypopituitarism include hypoglycemia, hypotension, and hypothermia.
Figure 1 Probability of normal pituitary hormone secretion over time after radiation exposure to the hypothalamic–pituitary areas. Data from four studies were replotted on this single figure. The first set of values (closed circle) are from Pai et al.,3 where the patient received 55.8–79 Gy to the base of the skull. The second set of values (solid square) are from Shalet et al.,83 where patients with pituitary tumors were treated with 37.5–42.5 Gy. The third series (open triangle), from Lam et al.,2 shows the effect of radiation treatment for nasopharyngeal carcinoma with 39.8–61.7 Gy. The final series (open diamond) represents data from Samaan et al.,4 in which 11–75 Gy was administered to treat head and neck tumors.
In children and adolescents, evaluation of sexual development is a useful diagnostic tool. Staging sexual development according to Tanner’s criteria, menstrual history in girls and penile/testicular size in boys should be evaluated. In children who have had cranial irradiation, height and weight should be measured every 6 months. In children treated with spinal and craniospinal irradiation, local rather than general growth abnormalities may be present and, if so, require specific evaluation. Foot size is a reliable indicator of growth that can be easily measured. Deviation from normal growth curves should be evaluated for GH deficiency, hypothyroidism, and adrenal insufficiency. If the initial evaluation of GH, IGF-1, thyrotropin (TSH) and free thyroxine (T4) levels, and radiographic bone age reveal abnormality, then detailed dynamic testing to evaluate the hypothalamic/pituitary axes should be performed (Table 1).
Table 1 Dynamic testing of the hypothalamic/pituitary axes
| Test | Dose/sampling | Contraindications |
| Growth hormone axis | ||
| Insulin hypoglycemia | 0.075–0.1 U regular insulin/kg IV to achieve glucose ≤40 mg/dL. Sample for glucose and GH at 0, 30, 45, 60, and 90 min | Coronary heart disease or seizures |
| Arginine | 0.5 g/kg (up to 30 g) IV over 30 min. Sample for GH at 0, 30, 60, 90, and 120 min | Liver disease or renal disease |
| L-Dopa | 500 mg by mouth. Sample for GH at 0, 30, 60, 90, and 120 min | Systolic blood pressure <100 mm Hg or age > 60 years |
| Arginine and GHRH | Arginine dose as above. GHRH 1 µg/kg IV push. Sample for GH at 0, 30, 60, 90, and 120 min | Liver disease or renal disease |
| Clonidine stimulation protocol | Clonidine 0.15 mg/m2 by mouth. Collect GH samples at baseline, 30, 60, 90, and 120 min | |
| Growth hormone-releasing hormone (GHRH) stimulation protocol | GHRH at 1.0 µg/kg body weight IV push. Collect GH samples at baseline, 15, 30, 45, 60, 90, and 120 min | |
| Growth hormone suppression test | The test should be performed after an overnight fast with the patient maintained at bed rest. The patient should drink a solution of 100 g glucose. Collect GH samples at baseline, 60, and 120 min | |
| Adrenal axis | ||
| ACTH stimulation test, 1-h | Synthetic ACTH 1–24 1 or 250 µg IM or IV. Draw blood for cortisol at 30 and 60 min after injection | |
| ACTH stimulation test, 48-h | Beginning at 9 a.m., obtain baseline 24-h urine for 17-hydroxycorticosteroids (17-OHCS) and creatinine. Collect 24-h urine as on day 1. Beginning at 9 a.m., start IV and give 250 µg synthetic ACTH 1–24 in 250 mL normal saline over 8 h every 8 h for 48 h. Alternatively, 40 IU of depot formulation of purified bovine ACTH in gelatin IM every 12 h for 48 h. Repeat 24-h urine as on days 1 and 2 Days 4 and 5: Collect 24-h urine as on previous days |
|
| Corticotropin-releasing hormone (CRH) stimulation test | Fast for at least 4 h before the test. Human CRH at 1.0 µg/kg IV bolus over 30 s. Blood samples should be collected at 15 and 1 min before CRH administration and at 15, 30, 45, 60, 90, and 120 min after for measurements of cortisol and ACTH | |
| Low-dose dexamethasone test, overnight | Dexamethasone 1.0 mg (adult) or 20 µg/kg (children) PO between 11 p.m. and midnight. Serum cortisol is collected at 8–9 a.m. the next morning. A cortisol level <1.8 µg/dL essentially excludes Cushing syndrome | |
| Low-dose dexamethasone test, 48-h | Serum cortisol is collected at 8–9 a.m. Dexamethasone 0.5 mg (adult) or 10 µg/kg (children) PO immediately after the cortisol is drawn and again every 6 h for 48 h. A second plasma cortisol is drawn at 9 a.m., 6 h after the last dexamethasone dose. Serum cortisol concentrations <1.8 µg/dL exclude Cushing syndrome | |
| High-dose dexamethasone test, 48-h | Serum cortisol is collected at 9 a.m. Dexamethasone is administered (2.0 mg; 50 µg/kg in children) every 6 h for 48 h. A second plasma cortisol is drawn at 9 a.m., 6 h after the last dexamethasone dose. Patients with functional adrenal adenomas show no suppression of cortisol levels in the 48-h sample relative to the initial (baseline) sample. Seventy-eight percent of patients with pituitary source of excess ACTH showed >50% suppression of plasma cortisol, while only 11% of patients with an ectopic source of excess ACTH had a >50% suppression | |
| Comprehensive, 6-day, low-/high-dose dexamethasone test | This protocol incorporates the low- and high-dose dexamethasone tests in succession. 24-h urinary free cortisol and/or 17-hydroxycorticosteroid (17-OHCS) measurement can help verify the results of serum cortisol and ACTH | |
| Gonadotropin-releasing hormone (GnRH) stimulation test | GnRH 100 µg IV. A sample for serum LH should be collected at baseline and 40 min after GnRH administration | |
| Metyrapone stimulation (overnight) test | At 11 p.m., metyrapone 30 mg/kg (maximum 3 g) PO with a snack. On the following morning, at 8 a.m., measure serum cortisol and 11-deoxycortisol | |
GH = growth hormone; GNRH = growth hormone-releasing hormone; IV = intravenous; IM = intramuscular; PO = by mouth.
In adults who have received cranial or head and neck irradiation, detection of hypothalamic–pituitary abnormalities is more challenging. One strategy to detect hypothalamic–pituitary abnormalities in adults consists of routine screening for GH deficiency and gonadal failure. It is recommended that measurements of IGF-1 and testosterone levels in males and documentation of menstrual history in females be obtained annually for 5 years, and then at 5-year intervals for another 10 years. Any abnormalities noted on the screening tests should be pursued with further dynamic testing to evaluate all the axes of hypothalamic–pituitary functions.
Immunotherapy-induced hypophysitis
The development of immunotherapeutic approaches to treat melanoma and a growing list of other malignancies has resulted in a number of bystander effects. Prominent among these side effects is immunotherapy-induced hypophysitis (IH). In the phase III clinical trial of ipilimumab, 2.3% of patients developed symptoms of IH and evidence of hormonal deficiency including glucocorticoid, thyroid, gonadal, and GH deficiency, necessitating long-term replacement therapy. The pituitary-related events were grade 3 or 4 in 1.9% of these patients, necessitating urgent treatment or hospitalization. The onset of pituitary failure ranged from 11 to 19 weeks, following initiation of ipilimumab therapy. Common presenting complaints include headache, mental status alterations, abdominal pain, modified bowel habits, and hypotension; it is often difficult to distinguish IH from brain metastasis or symptoms associated with widespread metastatic disease. This difficulty suggests that clinicians initiating therapy with immunotherapy agents should obtain baseline thyroid (free T4 and TSH), adrenal (ACTH, adrenocorticotropic hormone and 8 a.m. serum cortisol), gonadal [luteinizing hormone (LH), follicle-stimulating hormone, and estradiol in females or testosterone in males], and growth-related hormones (GH and IGF-1) as baseline before initiating therapy. These measurements should be repeated if a patient develops symptoms suggestive of endocrine deficiency or acute symptoms of hypophysitis (headache and visual disturbance). In patients with symptoms or physical findings consistent with IH, an MRI (magnetic resonance imaging) of the pituitary gland and repeat hormone testing as described above should be performed together with dynamic testing of the adrenal endocrine axis (e.g., low-dose cosyntropin stimulation test). A recent report highlighting a larger experience in a single center indicates that IH is observed not only with anti-CTLA4 agents (ipilimumab and tremelimumab),6 but also with an anti-PD1 agent (nivolumab). In a report describing 968 patients treated with these agents for melanoma, prostate cancer, and renal cell carcinoma at a single center, 2.7% (27 subjects) had one or two hormonal deficiencies together with either pituitary enlargement in MRI scans or headache.6 In these 27 patients, central adrenal deficiency was identified in 77%, central hypothyroidism in 89%, and central hypogonadism in 79%. An abnormality of the pituitary gland was found by MRI in 85%.6 After a median follow-up period of 17 months (range: 1–76), none of the patients recovered normal adrenocortical function, whereas 12–13% recovered thyroid or gonadal function. Clearly, this is a significant and evolving issue; it is unclear at this time whether combinatorial therapy with two or more immunotherapeutic agents, currently under development, will be associated with higher incidence and/or severity of toxicity.
Thyroid disorders
Thyroid disorders and abnormalities in thyroid function are commonly associated with cancer and its therapy.
Serum thyroid hormone-binding protein abnormalities
The levels of thyroid hormone-binding proteins [thyroxine-binding globulin (TBG), prealbumin, and albumin] can be modified by sex hormone levels and nutritional factors; abnormalities of both are encountered frequently in cancer patients. Several chemotherapy drugs affect thyroid function test results. L-Asparaginase appears to reversibly inhibit synthesis of albumin and TBG, resulting in low total thyroxine (T4), but normal free T4 levels.7 The combination of podophyllin and alkylating agents has also been reported to decrease TBG.8 Both 5-fluorouracil9 and mitotane10 increase the total T4 and triiodothyronine (T3) levels without suppressing TSH, suggesting that these drugs increase thyroid hormone-binding capacity in the serum.
Euthyroid sick syndrome
Alterations in thyroid hormone metabolism occur in patients with cancer and other serious systemic illnesses.11 Low serum T3 levels, which may be found in up to 70% of moderately to seriously ill cancer patients, are caused by a decrease in the extrathyroidal conversion of T4 to T3. Serum concentrations of free T4 are usually normal or high, while concentrations of free T3 are below normal or low. The patients are clinically euthyroid, and serum TSH level and TRH stimulation test results are normal.
In most patients with euthyroid sick syndrome, T3, T4, and TSH levels are normal. Clinical manifestations of hypothyroidism are usually absent, but assessment may be confounded by obtundation, edema, and hypothermia that may accompany severe illness. Low free T4 levels in the context of euthyroid sick syndrome usually indicate a grave prognosis, with a mortality rate of more than 50%. Although it is generally accepted that thyroid hormone therapy has no benefit, in practice it is sometimes difficult to differentiate between the euthyroid sick syndrome and secondary hypothyroidism. Judicious replacement of T4 at physiologic levels in these uncommon patients may be appropriate if there are no contraindications (e.g., active ischemic heart disease).
Hypothyroidism
Thyroidectomy
Thyroidectomy may be performed for a variety of oncologic reasons in the management of thyroid cancer, head and neck cancer, or thyroid metastasis. Thyroid replacement is needed in this group of patients. In thyroid cancer patients, supraphysiologic doses of thyroid hormone are adjusted to suppress TSH without overt hyperthyroid symptoms. In others, the dose of thyroid hormone should be adjusted to keep TSH in the normal range.
Radiation
Irradiation is an important cause of hypothyroidism [primary (thyroid), secondary (pituitary), and tertiary (hypothalamic)]. Radiation-induced primary hypothyroidism is caused by thyroid cell destruction, inhibition of cell division, vascular damage, and possibly an immune-mediated phenomenon. Factors that increase the risk of developing primary hypothyroidism include a high radiation dose to the vicinity of the thyroid gland, duration since therapy, lack of shielding of the thyroid during therapy, and combined irradiation and surgical treatments.12
The incidences of hypothyroidism after radiation therapy for various cancers and conditions are tabulated in Table 2.4, 12–22 A relationship between radiation dose and the prevalence of hypothyroidism is based on studies of patients with Hodgkin disease.15, 18 Long-term follow-up of patients treated with low-dose radiotherapy suggests that the threshold for causing clinically evident hypothyroidism is approximately 10 Gy. For Hodgkin-disease patients who received >30 Gy, the actuarial risk of hypothyroidism was up to 45%, 20 years after irradiation.15 Patients with frank or subclinical hypothyroidism should receive thyroid hormone-replacement therapy.
Table 2 Incidence of hypothyroidism (including compensated hypothyroidism) after radiotherapy.a
| Type of malignancy or conditions | Radiation dose | % with hypothyroidism |
| Hodgkin disease | 30–60 Gy | 30–50 |
| Head and neck cancer | 40–72 Gy | 25–50 |
| Lymphoma | 20–40 Gy (median 36 Gy) | 30–42 |
| Breast carcinoma | ? | 15–21 |
| Total-body irradiation in BMT | 13.75–15 Gy | 15–43 |
BMT = bone marrow transplantation.
a Data based on Refs 4 and 1213141516171819202122.
Chemotherapy
The diagnosis of hypothyroidism in 14% of BMT (bone marrow transplantation) patients who received chemotherapy but did not receive total-body irradiation23 suggests a causal relation between hypothyroidism and high-dose combination cytotoxic chemotherapy. This notion is also supported by studies that showed an increased incidence of primary hypothyroidism in patients treated with multiple combination drug regimens24, 25 with or without radiation.24 L-Asparaginase, in addition to inhibition of TBG synthesis discussed above, may also inhibit TSH synthesis reversibly and lead to temporary hypothyroidism with decreased free T4 levels.26
Thyroid dysfunction is a recognized side effect of cytokine treatments. Treatment with interleukin-2 produces thyroid dysfunction in approximately 20–35% of patients.27 These patients have hypothyroidism, hyperthyroidism, or hyperthyroidism followed by hypothyroidism.28 Approximately 10% of interferon-treated patients develop primary hypothyroidism.29 Pituitary enlargement secondary to interferon-induced hypothyroidism has also been reported.30 Patients with antithyroid antibodies before therapy are at higher risk of cytokine-induced thyroid dysfunction.
Retinoid X receptor (RXR) ligands may be used in the treatment of certain malignancies such as cutaneous T-cell lymphoma. Bexarotene (a RXR-selective ligand) caused secondary hypothyroidism dose-dependently.31 A single dose can rapidly suppress TSH in healthy subjects.32 In addition to suppressing transcription of TSH by an RXR-mediated thyroid hormone-independent mechanism, bexarotene also increases metabolic clearance of thyroid hormones by a nondeiodinase-mediated pathway.33
Tyrosine kinase inhibitors
The rapid introduction of small organic molecules that inhibit kinase activity (tyrosine kinase inhibitors, TKIs) into cancer therapy over the past decade has led to the recognition that many of these agents affect thyroid function in profound ways. The first mechanism is a direct effect of these agents on thyroid function. A number of TKIs target receptor tyrosine kinases with well-defined functions in the thyroid gland including the vascular endothelial growth factor receptor, the epidermal growth factor receptor, RET, KIT, and MET. Other agents target downstream signaling pathways such as RAF, PI3K, and mTOR. These signaling pathways are operative in the thyroid and important for normal thyroid cell growth and function. It is not surprising that disruption of these pathways has effects on thyroid cell growth and death and thyroid hormone synthesis. Indeed hypothyroidism occurs at significant incidence rates with a number of agents: sunitinib, 7–85%; pazopanib, 12%; nilotinib, 22%; axitinib, 20–100%; cabozantinib, 15%; sorafenib, 8–39%; dasatinib, 50%; and imatinib, 0–25%.34 Routine measurement of thyroid function (free T4 and TSH) at regular intervals during the first year of therapy with one of these agents seems prudent as it is often difficult to differentiate nonspecific side effects of TKI therapy from primary hypothyroidism or effects of metastatic cancer.
There is a second mechanism for development of hypothyroidism in patients treated with specific TKIs for metastatic thyroid carcinoma who have also undergone total thyroidectomy. Specific agents approved (vandetanib, cabozantinib, sorafenib, and lenvatinib) or studied (pazopanib, motesanib, and sunitinib) for treatment of differentiated or medullary thyroid carcinoma cause an elevation of the serum TSH concentration and low serum T4 levels with high percent incidences: vandetanib, 49%; cabozantinib, 57%; sorafenib, 41%; and lenvatinib, 57%. As each of these agents causes diarrhea and malabsorption, one presumed mechanism for this effect is reduced absorption of thyroid hormone, although effects on thyroid hormone metabolism have not been excluded. Increasing the thyroid hormone dosages by as much as one-third generally resolves the problem. It is important to note that there may also be malabsorption of calcium, magnesium, and vitamin D, causing worsening hypocalcemia or hypomagnesemia in patients with mild thyroidectomy-associated hypoparathyroidism.35 Routine measurement of free T4 and TSH should be incorporated into management as more than 50% of these patient will require a dose modification of thyroid hormone (as well as magnesium, calcium, and vitamin D supplementation). It is important to note that primary hypothyroidism, hypocalcemia, or hypomagnesemia can be readily treated, and their appearance, if addressed promptly, should not limit the use of the TKIs. However, in patients with hypothyroidism, hypocalcemia, or hypomagnesemia, all of which can prolong the QT interval, it may be necessary to withhold TKIs that prolong QT interval, that is, vandetanib,35 lenvatinib,36 and sorafenib in patients with QT prolongation until the QT abnormalities are resolved.
131I-containing compounds
The use of 131I for treatment of thyroid cancer requires a high serum TSH level. High TSH level is achieved by either withholding thyroid hormone replacement or increasingly by administration of recombinant human TSH. The use of 131I-containing compounds in the treatment of other tumors may result in hypothyroidism. For instance, using high-dose (100–1000 mCi) [131I]-metaiodobenzylguanidine to treat unresectable pheochromocytoma may result in primary hypothyroidism.37
Metastasis to the thyroid
Hypothyroidism secondary to metastatic infiltration and replacement of the thyroid by cancer is extremely rare.
Screening
Children who have received either head and neck or cranial irradiation should have a free T4 and a TSH measurement annually for 5 years, and every 2 years thereafter. Early detection of abnormal T4 and TSH levels will permit medical intervention before hypothyroidism adversely affects physical and intellectual development and growth. In adults, neck irradiation for treatment of lymphoma and various head and neck tumors is associated with a high incidence of primary hypothyroidism. Patients who have received irradiation should have free T4 and TSH levels measured annually for 5 years, and then every other year for 10 years, and thereafter every 5 years for another 10 years. Once hypothyroidism is diagnosed, the patient should receive thyroid hormone-replacement therapy.
Hyperthyroidism
Radiation-induced painless thyroiditis with hyperthyroxinemia is an uncommon side effect of external-beam radiotherapy to the head and neck area. Transient hyperthyroidism may occur as a result of inflammation and destruction of thyroid tissue with release of thyroglobulin (containing T4 and T3), and is usually followed by hypothyroidism. Transient hyperthyroidism has been reported after mantle radiotherapy in Hodgkin disease patients, and occurs usually within 18 months of treatment.38 A low uptake of radioiodine in most of these cases suggests a diagnosis of silent thyroiditis, but some have Graves’ disease. In one series of Hodgkin disease patient treated with radiation, the risk of Graves’ disease in these patients was estimated to be at least 7.2 times that in a healthy population.15
Ophthalmopathy similar to that in Graves’ disease has been reported within 18–84 months of high-dose radiotherapy to the neck for lymphoma, breast cancer, and nasopharyngeal or laryngeal cancer. Ophthalmopathy may occur without hyperthyroidism and in the absence of the human leukocyte antigen-B8.39 This suggests that radiation-induced thyroid injury may induce an autoimmune process that is similar to Graves’ disease.
There have also been examples of TKI-associated hyperthyroidism, including two deaths (sorafenib and sunitinib associated).40 The presumed mechanism of hyperthyroidism is thyroiditis. Patients with severe hyperthyroidism should be treated aggressively for thyroid storm; consideration should be given to holding TKI therapy until the patient is stabilized in such cases.34
Thyroid nodules and cancers
Low-dose radiation increases the risk of thyroid nodules and cancer. The association between thyroid cancer and low-dose irradiation has been extensively examined,41 and is discussed in the chapter about thyroid cancer (see 81).
Energy balance and glucose metabolism
Obesity and metabolic syndrome
Cancer treatments may lead to obesity and the metabolic syndrome.42–44 The metabolic syndrome is a cluster of abnormalities consisting of central obesity, dyslipidemia, hyperglycemia, and hypertension that increases the risk of type 2 diabetes and cardiovascular disease. Obesity is a modifiable risk factor for carcinogenesis as well as cancer progression. The mechanisms by which obesity promotes cancer include: hyperinsulinemia due to insulin resistance, high IGF-1, adipokines, low adiponectin, increased production of estrogens by adipose tissue, and increased inflammation. Obesity in cancer survivors may place them at increased risk for poor disease outcomes.42, 45, 46 Obesity increases the risk of colorectal and genitourinary second primary cancers.47 Although low levels of physical activity in cancer survivors can contribute to obesity, the pathophysiologic basis of the weight gain is unclear. As obesity is an adverse prognostic factor for many cancers and is a modifiable risk factor, secondary obesity after cancer treatment needs to be addressed.
Diabetes mellitus
Diabetes mellitus type 2 (DM2) is associated with an elevated risk of pancreatic, liver, colon, gastric, breast, and endometrial cancer.48–53 Extensive epidemiologic data suggest important roles of diabetes in carcinogenesis48–53 and cancer survival.54 The strongest association is perhaps with pancreatic cancer.55–59 Apart from the frequently coexisting obesity, the mechanisms by which diabetes promotes cancer include: hyperinsulinemia, high IGF-1, and hyperglycemia. Hyperglycemia per se has a promoting effect on cancer proliferation. In male, cancer survivors with a fasting serum glucose concentration ≥126 mg/dL had a higher relative risk for hepatopancreatobiliary second primary cancer.47 Evidence-based guidelines for the management of DM2 in cancer patients to optimize patient survival are lacking.
The administration of glucocorticoids (e.g., in combination therapy regimens, for edema of brain metastasis, for prevention of transplant rejection, for graft-versus-host disease in BMT, and for nausea/vomiting) is probably the most common cause of diabetes mellitus in cancer patients. Therefore, patients who receive glucocorticoids must be periodically screened for diabetes with evaluation of fasting glucose levels during therapy. Treatment with streptozocin60 or L-asparaginase61 may result in insulin-deficient diabetes mellitus. Although there is no evidence of a delayed onset of diabetes mellitus following treatment with streptozocin, follow-up has been limited and short term. For long-term survivors treated with streptozocin, periodic screening for delayed development of diabetes mellitus may be indicated. Diabetes mellitus may also develop as a consequence of serious pancreatitis secondary to treatment with L-asparaginase. Immunotherapy for cancer using cytokines such as interleukin-2 and interferons may cause toxicity to pancreatic β cells and lead to insulin-dependent diabetes.62 Tacrolimus, an immunosuppressive agent used to prevent graft-versus-host disease in BMT, also increases the incidence of diabetes, perhaps by damaging pancreatic β cells.63 Patients who received allogenic BMT are likely to be receiving both glucocorticoids, cyclosporine A and tacrolimus, and are particularly at risk for developing diabetes mellitus.64 Management of the blood glucose levels would depend on the severity of the blood glucose level abnormality and on the underlying pathophysiologic mechanism of the increase in blood sugar. In general, insulin will be needed in patients who are insulin deficient.
Metabolic bone diseases
Osteoporosis
Four groups of adult patients are at particular risk for accelerated bone loss and osteoporosis: (i) patients with lymphoma, myeloma, or leukemia; (ii) women with breast cancer treated with cytotoxic chemotherapy frequently undergo an early menopause65 and cannot receive estrogen-replacement therapy; (iii) postmenopausal women with estrogen-receptor positive breast cancer; and (iv) men with prostate cancer who are on antiandrogenic therapy and made hypogonadal. Normal bone remodeling involves a delicate balance between bone formation by osteoblasts and bone resorption by osteoclasts. Antineoplastic therapy is toxic to osteoblast function and decreases bone formation. Production by the tumor of hormonally active substances [e.g., parathyroid hormone-related protein (PTHrP), lymphotoxin, interleukin-1, and interleukin-6] may contribute to bone loss. In most cases, it is not clear whether bone loss is caused by antineoplastic therapy or by the underlying disease process and its effects (including cachexia, malnutrition, poor calcium and vitamin D intake, or a combination of these). In patients with breast or prostate cancer, sex steroid hormone deficiency induced by therapy is the most important cause of bone loss. Bone loss is prominent in patients with several disorders (myeloma, leukemia, and lymphoma), affecting the hematopoietic cells, perhaps because of cytokine production and an intimate relationship of hematopoietic cells with bone-forming cells or the use of high-dose or prolonged therapy with glucocorticoids.
A number of drugs can induce osteoporosis.66 In cancer patients, glucocorticoids, methotrexate, and cytotoxic drugs that cause renal loss of calcium, magnesium, or phosphorus (e.g., platinum compounds, cyclophosphamide, and ifosfamide) have significant impact on bone density. Osteoporosis (generalized or localized) is observed in children receiving methotrexate therapy for acute lymphoblastic leukemia (ALL).67 The osteoporosis improves significantly after cessation of methotrexate therapy. Longitudinal study of ALL patients showed that the leukemic process, high-dose glucocorticoids, and hypomagnesemia (owing to renal wastage following cyclical glucocorticoid and nephrotoxic chemotherapy or antiinfective agents) contributed to the impairment of calcium and vitamin D metabolism and decrease in bone mass at different stages of the treatment process.68 Adjuvant chemotherapy for breast cancer (usually involving 5-fluorouracil, cyclophosphamide, and doxorubicin or methotrexate) is associated with low bone mass in premenopausal patients.69 Bone loss during chemotherapy is substantial and may lead to increased risk of fracture. Posttreatment hypogonadism appears to be a major factor in these adult women with osteoporosis. While tamoxifen has a slight protective effect on bone loss, the opposite is true for aromatase inhibitors. BMT usually involves treatment with high-dose cytotoxic drug, glucocorticoids, and immunosuppressive agents. In 24 patients who underwent BMT with high-dose chemotherapy, profound effects on bone biomarkers were observed.70
Prompt investigation of gonadal dysfunction in cancer survivors and prompt replacement of gonadal steroids (in the absence of contraindications) in young hypogonadal men or women are recommended to decrease the risk of future bone fractures. The bone mass of long-term cancer survivors should be assessed when the patient is about 30 years old, the age at which most people have attained peak bone mass.71 If bone mass is normal, no further evaluation is needed beyond the usual recommendations for prevention of osteoporosis. If it is abnormal (more than 2 standard deviations below normal), the patient should be referred for evaluation of the multiple reversible causes of osteoporosis.
A key point in the management of the osteoporosis syndrome in cancer patients is the use of bone mineral density measurement (e.g., by dual-energy X-ray absorptiometry) to assess fracture risk and to monitor the effects of therapy. This measurement should be performed early in the course of management of the malignancy so that appropriate preventive measures can be implemented. The oncologist who is prescribing medications that are likely to decrease bone mass should consider active use of bisphosphonates (e.g., alendronate, risedronate, ibandronate, or zoledronate), calcitonin, selective estrogen-receptor modulators (SERMs), or denosumab, in addition to a daily intake of 1200–1500 mg elemental calcium and vitamin D supplementation. Bisphosphonates and denosumab are effective therapies for prevention of bone loss. While osteoporosis in children with leukemia will frequently reverse because the children are in the formative years of bone development, in adults more active measures such as bisphosphonate or teriparatide therapy to prevent bone loss should be considered, rather than waiting for the development of a fracture syndrome.
Another key point is correction of abnormal mineral and vitamin D metabolism by dietary supplements. Nutritional deficiency in teenagers and young adults results in lower bone mass. Treatment of hypocalcemia, hypomagnesemia, and vitamin D deficiency is integral to the successful therapy of osteoporosis in cancer patients. Recent studies document that clinically relevant vitamin D deficiency is present in 50% of the normal population; the percentage is almost certainly higher in patients undergoing cancer therapy.
Osteomalacia
Osteomalacia, a condition characterized by unmineralized bone matrix, is a rare complication of chemotherapy, but should be considered in osteopenic patients and those with osteomalacic clinical syndrome (bone pain and proximal myopathy). The most common cause is a decrease in the serum calcium and/or phosphorus concentrations caused by nutritional deficiency and renal wasting of phosphorus and calcium. Patients who have received chemotherapeutic agents that cause hypophosphatemia, hypomagnesemia, or hypocalcemia are particularly at risk. Investigation of the levels of serum ionized calcium, phosphorus, magnesium, and vitamin D metabolites should be included in the initial evaluation. Appropriate replacement therapy of these vitamins and minerals should be instituted once deficiencies have been identified. Other contributing factors include systemic acidosis and drugs such as anticonvulsants and aluminum.66 Tumor-induced osteomalacia will be addressed in the section that follows discussing paraneoplastic syndromes.
Ifosfamide causes tubular damage leading to renal phosphate wasting, hypophosphatemia, and rickets/osteomalacia.72 The toxic effects of ifosfamide on renal tubular function include Fanconi syndrome in adults and children. Tubular damage is seen most commonly when ifosfamide is administered in doses of 50 g/m2 or more, or when it is used in combination with cisplatin.73 Rickets is reported most commonly in children. Estramustine, used in the treatment of prostate cancer, has been reported to increase bone resorption and at the same time cause hypocalcemia, hypophosphatemia, and secondary hyperparathyroidism.74
Adrenal diseases
Adrenal metastasis
Hematogenous metastasis to the adrenal glands is common, exceeded in frequency only by hematogenous metastasis to the lung, liver, and bone.75 Autopsies have documented that 9–27% of patients who died from malignant illness had adrenal metastasis, with bilateral involvement in one-half to two-thirds of patients with adrenal metastasis.
The presence of adrenal metastasis may have important implications for diagnostic and therapeutic planning. When patients with cancer have an adrenal mass but no evidence of metastasis elsewhere, it is important to determine whether this mass represents a metastatic tumor or a separate, unrelated adrenal lesion. Recent advances in imaging techniques have allowed the identification of adrenal lesions antemortem as part of the tumor-staging evaluation. The location of the adrenal glands in the perinephric fat allows the detection of almost all normal glands and contour-deforming masses as small as 5–10 mm. Computed tomography (CT) has a sensitivity and specificity in the detection of adrenal masses. Characteristics on CT examination that suggest adrenal metastasis rather than primary adrenal disease include heterogeneity, contrast enhancement, bilaterality, and size >3 cm.76
Without other evidence of metastatic disease, whether the adrenal mass is actually a metastatic tumor is critical information in determining the appropriate therapy for the cancer. Evaluation of a patient who has a malignant adrenal mass should include a history and physical examination to elicit evidence of adrenal insufficiency, Cushing syndrome, mineralocorticoid excess, or pheochromocytoma. Biochemical assessment should include a short ACTH stimulation test with measurements of serum cortisol to rule out adrenal insufficiency. A 24-h urine collection should be obtained to measure urinary free cortisol, aldosterone, catecholamines, and metanephrines. Pheochromocytoma must be excluded, especially if there is hypertension, or an operative procedure of any type is contemplated. It has been reported that one-half of the patients who had a clinically unsuspected pheochromocytoma had clinical deterioration or even death immediately following a non-adrenal-related surgical procedure.77
If the biochemical assessment for pheochromocytoma is negative, CT-guided fine-needle aspiration should be considered. This procedure has a sensitivity of 85% in detecting cancer.78
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
