[level-membership-for-emergency-medicine-category]Chapter 125
Electrolyte Disorders
Hyperkalemia
Hyperkalemia, defined as serum potassium level greater than 5.0 mEq/L, is the most dangerous acute electrolyte abnormality, potentially leading to life-threatening arrhythmias and death. Although hyperkalemia may have vague and varied symptoms, it is usually totally asymptomatic, with cardiac arrest as its first “symptom.”1 Thus the diagnosis of hyperkalemia depends on paying specific attention to risk factors for impaired potassium excretion, such as dehydration and renal failure, along with an awareness of medications that cause potassium retention. Evaluation of the electrocardiogram (ECG) of patients at risk for this electrolyte disturbance is critical. Hyperkalemia can be rapidly progressive, and lifesaving interventions must be instituted at the earliest suspicion of toxicity.
Upwards of 98% of potassium in the body is contained intracellularly, whereas less than 2% remains circulating in the blood. Serum potassium concentration is normally between 3.5 and 5.0 mEq/L and is tightly regulated by the kidney. In the healthy state, at least 90% of potassium excretion occurs through the kidney; in the renally impaired state, the gastrointestinal tract may account for roughly 25% of excretion. Hyperkalemia usually develops from impaired renal excretion or increased release from cells; however, in advanced chronic kidney disease or end-stage renal disease, dietary intake of potassium may be a significant factor in its development.2
The most common cause of hyperkalemia is spurious elevation due to hemolysis during or after the blood draw. Thus an ECG should be used to assess for true hyperkalemia while another sample is analyzed. Most causes of true hyperkalemia are due to release from cells or renal insufficiency. Renal failure is the most common cause of confirmed hyperkalemia and is often compounded by medications that further impair renal potassium handling. Box 125-1 organizes the most common causes of hyperkalemia. The presence of one of these conditions may be the lone historical clue in hyperkalemia.3
Clinical Features
Hyperkalemia remains a difficult clinical diagnosis to make on clinical grounds alone. It is not uncommon for a patient with mild to moderate hyperkalemia to be identified during routine blood sampling for an unrelated condition. Patients with moderate to severe hyperkalemia may have gastrointestinal effects such as nausea, vomiting, and diarrhea often in association with their underlying disease. Neuromuscular findings, including muscle cramps, generalized weakness, paresthesias, tetany, and focal or global paralysis, may be seen in patients with severe hyperkalemia. The signs and symptoms of progressive muscle weakness, paresthesias, dyspnea, and depressed deep tendon reflexes are neither sensitive nor specific, nor do they appear reliably with a particular serum potassium level.3 Patients with severe hypokalemia may present with hemodynamic instability and cardiac arrhythmias requiring immediate intervention.
Diagnostic Strategies
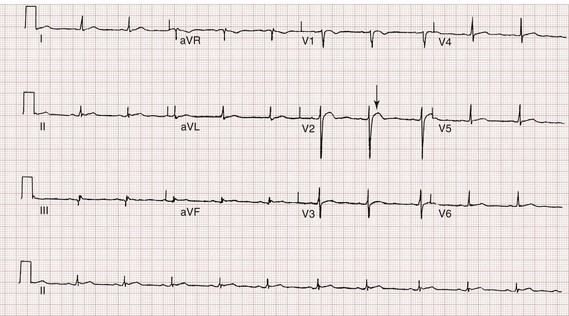
The ECG is helpful in making the diagnosis of hyperkalemia and can be used in unstable patients to initiate treatment (Figs. 125-1 to 125-3). Classic electrocardiographic changes—the peaked T wave, flattened p wave with prolonged PR interval or a totally absent P wave, wide QRS, and sine wave pattern, portending imminent cardiac arrest—have been well described as appearing sequentially with rising serum potassium levels.4,5 Peaked T waves usually appear as serum potassium levels exceed 5.5 to 6.5 mEq/L; P wave disappearance and PR prolongation are common with levels above 6.5 to 7.5 mEq/L; and QRS prolongation is seen with potassium levels above 7.0 to 8.0 mEq/L. Although these changes may occur in only half the patients, recognition of these patterns when they are present is vital to rapid diagnosis and initiation of lifesaving treatment.6 A serum potassium level above 5.0 mEq/L is diagnostic of hyperkalemia, but the value itself does not always predict electrocardiographic changes or the degree of cardiotoxicity. Subtle electrocardiographic changes consistent with hyperkalemia should not be the sole reason to treat a stable patient not likely to have an elevated potassium concentration until serum levels have returned.3
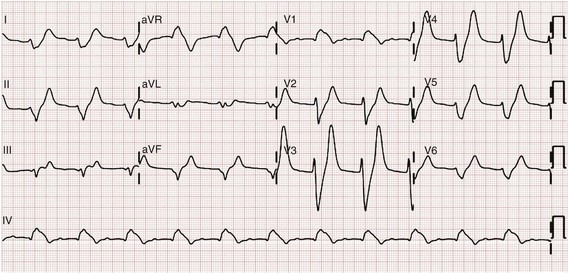
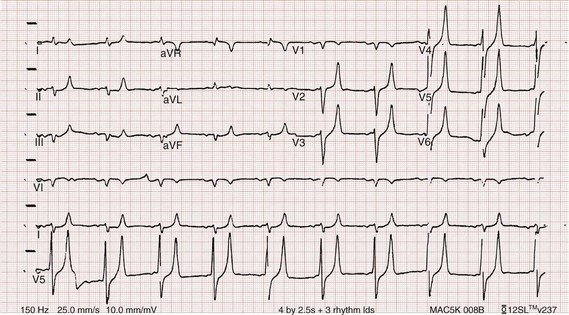
Figure 125-2 Hyperkalemia in the same patient as in Figure 125-1 after potassium-lowering therapy has begun. Tall peaked T waves, decreased P wave.
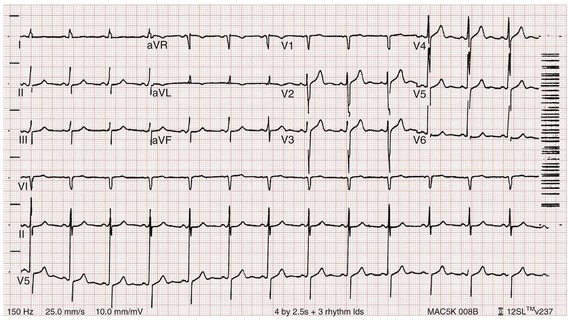
Figure 125-3 The same patient as in Figures 125-1 and 125-2 after dialysis. The electrocardiogram is now normal.
Management
Patients with suspected or known hyperkalemia require intravenous access and continuous cardiac monitoring. The treatment of hyperkalemia is based on the clinical scenario combined with the 12-lead ECG and the laboratory potassium value. The treatment strategy consists of three main steps: stabilization of the cardiac membrane, shifting of potassium into the cells, and then removal of potassium from the body. In patients who do not require urgent treatment, lowering of total body potassium may be the only step necessary. A variety of treatment options are considered for the acute management of hyperkalemia, including calcium, insulin, beta2-adrenergic agonists, sodium bicarbonate, resins, and dialysis (Table 125-1).
Table 125-1
| TREATMENT | MEDICATION | FEATURES |
| Stabilize cardiac membrane | Calcium chloride (10 mL, maximum 20 mL) or calcium gluconate (10-30 mL), IV push | For wide QRS, restores the electrical gradient; does not decrease serum potassium |
| Shift potassium into cells | Insulin, 10 units, IV push, combined with 100 mL of 50% dextrose, IV push High-dose nebulized albuterol by face mask (15-25 mg by continuous inhalation) Bicarbonate 50-100 mL Normal saline 100-250 mL |
If severely acidotic In conjunction with nephrologist if dialysis dependent |
| Remove potassium from the body | Hemodialysis Normal saline and furosemide Ion exchange resin |
Emergently in cardiac arrest, urgently in renal failure; may delay if renal function is normal In patients with rhabdomyolysis or tumor lysis syndrome with intact urine output Not effective acutely |
Intravenous calcium is used to stabilize the cardiac membrane by restoring the electrical gradient. Calcium increases the depolarization threshold and the calcium gradient across the cardiac membrane, quieting myocyte excitability and increasing cardiac conduction speed, thus narrowing the QRS. Calcium does not decrease serum potassium levels, and its effect is rapid but transient. The dose is one ampule, or 10 mL of 10% calcium chloride solution. Some authors prefer calcium gluconate rather than calcium chloride on the basis of the reduced risk of tissue necrosis should it extravasate at the injection site.4,5 More than 10 mL of calcium gluconate will often be required as it contains only one third the calcium contained in calcium chloride. Calcium gluconate is also preferred in pediatric cases as well as in more chronic, less emergent hyperkalemic patients when a slow infusion is desired.
Nebulized albuterol by face mask is effective in shifting potassium into cells by stimulation of the Na+,K+-ATPase pump.6 Nebulized albuterol begins to take measurable effect after 15 minutes and lowers the serum potassium level by 0.5 to 1 mEq/L, depending on the dose.7 The effective dose is at least four times higher than that typically used for bronchodilation.8 Mild tachycardia is the main side effect but is generally well tolerated. The combination of nebulized albuterol and insulin with glucose appears to be additive, lowering serum potassium by a mean of 1.2 mEq/L.9
Saline infusions also stimulate the Na+,K+-ATPase pump, and only a few hundred milliliters is required for beneficial effects. Saline infusions are given judiciously in anuric patients and usually in consultation with the nephrologist. Sodium bicarbonate is effective only in hyperkalemic patients who are acidotic and has no benefit when it is used for hyperkalemia in nonacidotic patients.10 Sodium bicarbonate buffers hydrogen ions extracellularly while shifting potassium intracellularly but should be reserved for patients with confirmed acidosis. Although intravenous magnesium also can drive potassium intracellularly, it should never be used in hyperkalemia as most patients with elevated potassium levels are also at risk for concomitant hypermagnesemia.
Potassium can best be removed from the body acutely by hemodialysis. Hemodialysis effectively and reliably decreases serum potassium levels by at least 1 mEq/L in the first hour and another 1 mEq/L during the next 2 hours.7,11 It is the only reliable method of potassium removal that has been experimentally studied and should be instituted early in the treatment of life-threatening hyperkalemia in patients with renal failure. In patients with intact renal function, medical management alone is usually sufficient, even in extreme cases, and hemodialysis may not be necessary unless multiple medical modalities fail. There are no randomized trials addressing the use of diuretics such as furosemide in the emergent management of hyperkalemia, but in cases such as rhabdomyolysis or tumor lysis syndrome, it may be appropriate to use a normal saline infusion supplemented by furosemide to enhance diuresis and potassium excretion in the urine.
Cation exchange resins, such as sodium polystyrene sulfonate (Kayexalate), have not been shown to decrease the serum potassium level within the first 4 hours of treatment and should not be used in the acute management of hyperkalemia.12
Hyperkalemia is seen in diabetic ketoacidosis (DKA), although most hyperkalemic patients with DKA are actually total body deficient of potassium. In this insulin-deficient and acidotic state, serum potassium levels rise because of cellular shifts. Simply treating the patient’s underlying DKA will also treat the hyperkalemia. In fact, the mainstay of treatment of DKA—fluids and insulin—closely mirrors the treatment of hyperkalemia itself.13
As hyperkalemia progresses, the end result will be cardiopulmonary arrest due to ventricular fibrillation, pulseless electrical activity, or asystole. In a known or suspected hyperkalemic arrest, an approach beginning with standard advanced cardiac life support combined with the use of multiple potassium-lowering medications is rational.14,15 Epinephrine has been shown to lower potassium by 0.25 mEq/L.16,17 Calcium chloride is given immediately by intravenous push, followed by insulin and glucose. Bicarbonate should be given by bolus dose if the patient is believed to be acidotic. Emergent hemodialysis is recommended if it is readily available. Hemodialysis through central venous access can be used during ongoing cardiopulmonary resuscitation to acutely lower the serum potassium level and may result in return of spontaneous circulation with intact neurologic status despite prolonged resuscitative efforts and failure of conventional medications and defibrillation.18
Hypokalemia
Hypokalemia is the most common electrolyte abnormality encountered in clinical practice. When it is defined as a value of less than 3.5 mEq/L, hypokalemia is found in more than 20% of hospitalized patients and in 10 to 40% of patients treated with thiazide diuretics in the outpatient setting.19 Although hypokalemia is usually asymptomatic, severe cardiac dysrhythmias and rhabdomyolysis can occur secondary to potassium’s effect on the heart and muscle.20 The five most common causes of hypokalemia are renal losses, increased nonrenal losses, decreased potassium intake, intracellular shift, and endocrine etiologies (Box 125-2). Hypokalemia is often seen in association with hypomagnesemia, and patients with low serum potassium levels should be assumed to be hypomagnesemic also.20–22
Increased excretion of potassium, especially coupled with poor intake, is the most common cause of hypokalemia, and patients receiving diuretics represent the single most common patient group encountered in clinical practice. Hypokalemia from thiazide diuretics occurs through increases in distal sodium delivery in the nephron and by activation of the renin-angiotensin-aldosterone system. Thiazide diuretics are more likely than loop or osmotic diuretics to cause hypokalemia, but both the thiazide and loop diuretics block chloride-associated sodium and increase delivery of sodium to the collecting tubules. Hypokalemia is a common adverse effect of treatment with diuretics and may cause fatal arrhythmias and increase the risk of digitalis toxicity.23 In addition to diuretics, other drugs and disorders can cause significant renal potassium losses, including hyperaldosteronism, steroid excess, metabolic acidosis, DKA, renal tubular acidosis, and alcohol consumption. Penicillin and its synthetic derivatives, when they are given in large doses, promote renal potassium excretion by increasing sodium delivery to the distal nephron.
Administration of insulin may cause a reduction in serum potassium because of insulin’s ability to stimulate the Na+,K+-ATPase pump and move potassium intracellularly; hypokalemia can be a dangerous complication with intentional overdoses of insulin and during treatment of DKA. Although most patients with DKA present with high-normal or mildly elevated serum potassium levels, patients are usually 2 to 3 mEq/kg body weight deficient in total body potassium.24 Failure to appreciate this total body deficit—and to not begin potassium infusion once significant hyperkalemia has been ruled out and intact renal function confirmed—may lead to otherwise unexplained arrhythmias or cardiac arrest in patients hours after their initial therapy has begun.
Dietary potassium deficiency should be considered in the severely malnourished patient and the chronic alcoholic.25 When poor potassium intake is combined with increased nonrenal losses, severe hypokalemia can result. Hypokalemia can also result from an acute shift of potassium from the extracellular compartment into cells. This is most commonly seen in patients with metabolic alkalosis, in patients with hyperventilation, and in those patients taking medications such as beta-agonists or decongestants. Beta receptor stimulation can lead to hypokalemia, especially in patients using repetitive and high doses of beta-agonists for chronic obstructive pulmonary disease or asthma. Albuterol-induced hypokalemia can occur even at normal therapeutic doses.26 A standard dose of nebulized albuterol reduces serum potassium by 0.2 to 0.4 mEq/L, and a second dose taken within 1 hour has the potential to reduce it by almost 1 mEq/L. Patients with starvation or near-starvation may suffer from hypokalemia when they are fed because insulin secretion and increased cellular uptake can cause an acute intracellular migration of potassium.
Clinical Features
Hypokalemia is usually asymptomatic but can be manifested with nonspecific complaints, including palpitations, skeletal muscle weakness, easy fatigability, depression, and muscle pain. Although short periods of mild potassium depletion are typically well tolerated in healthy individuals, severe potassium depletion can result in serious cardiovascular instability, neurologic dysfunction, glucose intolerance, gastrointestinal symptoms, and renal failure as well as affect the acid-base balance in the body.27 The likelihood of symptoms appears to correlate with the rapidity of the decrease in serum potassium. In patients without underlying heart disease, abnormalities in cardiac conduction are extremely unusual, even when the serum potassium concentration is below 3.0 mEq/L. Paresthesias, depressed deep tendon reflexes, fasciculations, muscle weakness, and confusion can occur when the serum potassium level is less than 2.5 mEq/L. However, in patients with cardiac ischemia or heart failure, even mild to moderate hypokalemia increases the likelihood of cardiac arrhythmias secondary to potassium’s effect on the action potential. The data linking hypokalemia with arrhythmias and cardiac arrest in acute myocardial infarction are fairly strong, but the direct myocardial stimulatory effects of increased circulating epinephrine is a possible confounder.20 Hypokalemia is an independent risk factor contributing to reduced survival of cardiac patients and increased incidence of arrhythmic death. On the basis of available evidence, it appears best to attempt to maintain a serum potassium concentration above 4.5 mEq/L in patients having an acute myocardial infarction. Hypokalemic patients can demonstrate first- and second-degree heart block, atrial fibrillation, ventricular fibrillation, and asystole. Hypokalemia can also promote metabolic acidosis.
Hypokalemic periodic paralysis is a rare disorder characterized by potentially fatal episodes of muscle weakness through the involvement of the respiratory muscles. Life-threatening cardiac arrhythmias are managed by restoration of serum potassium levels into the normal range.28
Diagnostic Strategies
Hypokalemia is rarely suspected on the basis of clinical presentation, and the diagnosis is typically made by measurement of the serum potassium concentration during routine laboratory study. If there is any suspicion for hypokalemia or a patient presents with generalized weakness, palpitations, or arrhythmias, an ECG should be obtained. Just as a tall-peaked T wave is characteristic of hyperkalemia, a flattened T wave can be seen in hypokalemia. U waves, which are small deflections after the T wave, may also be seen (Figs. 125-4 and 125-5). The real danger of hypokalemia is that it may also cause a prolonged QT interval. Once the QT interval becomes longer than 500 milliseconds, the risk of malignant ventricular arrhythmias and torsades de pointes increases dramatically.28 Hypokalemia is also notorious for causing nonspecific ST and T wave changes. In addition, prolonged potassium depletion of even modest proportion can provoke or exacerbate kidney injury or hypertension. A severe degree of hypokalemia with paralysis is a potentially life-threatening medical emergency; measurement of relative urinary potassium excretion and an assessment of the acid-base status might help narrow the differential diagnosis in the emergency setting.
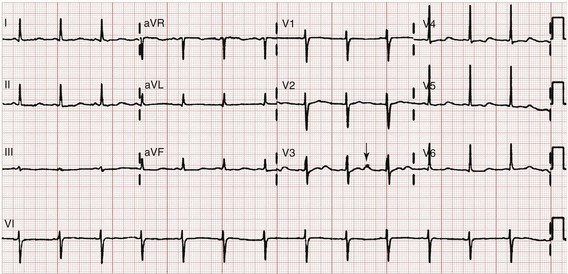
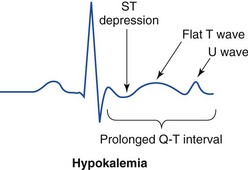
Figure 125-5 Electrocardiographic changes in hypokalemia.
Management
Potassium is an intracellular cation, so a low serum potassium level almost always reflects a significant total potassium deficit. When treating hypokalemia, one should remember that each 0.3 mEq potassium drop below normal correlates with an approximately 100 mEq total body deficit.29 Patients who have mild or moderate hypokalemia are usually asymptomatic or present with minor symptoms. These patients may only need oral potassium replacement therapy if they do not have nausea or vomiting as the cause of their hypokalemia. Oral replacement is available in liquid, powder, and tablet form. Potassium chloride is the most commonly used supplementation, and 40 to 60 mEq orally every 2 to 4 hours is typically well tolerated. If the cause of hypokalemia is not clear or the hypokalemia is severe and associated with profound weakness, obtain a spot urine potassium level before starting therapy to assess whether the patient’s kidneys are inappropriately wasting potassium from a renal or endocrine cause. Patients in whom severe hypokalemia is suspected should be immediately placed on a cardiac monitor and intravenous access secured.
Treatment of hypokalemia is essential in multiple populations of patients. Hypokalemia is arrhythmogenic, especially in the settings of acute myocardial infarction, high catecholamine states, and hypertrophied or dilated ventricles. Hypokalemia is an important independent risk factor for morbidity and mortality in patients with heart failure. Correction of serum potassium levels to between 4.0 and 5.0 mEq/L is important in these patients.30
Hypokalemia is associated with hypomagnesemia, and the severity of the hypokalemia correlates with a similar degree of hypomagnesemia.31 Magnesium replacement should usually accompany potassium repletion. Unless the patient receives at least 0.5 g/hr of magnesium sulfate along with potassium replacement, potassium will not move intracellularly and the patient will lose potassium through excretion.22 Correction of large potassium deficits may require several days, and oral and intravenous replacement can occur simultaneously.
Hypernatremia
Hypernatremia is defined as a serum sodium concentration above 145 mEq/ L and is usually associated with a poor prognosis. It is uncommon in previously normal patients, and in adults it is almost exclusively due to a total body water deficit.32 Most hypernatremic patients have either an impaired sense of thirst or no access to water. Thus elders, infants, patients in coma or with mental impairment, and those who are intubated and paralyzed are at highest risk for this disorder.33,34 Hypernatremia can be divided into three physiologic pairings (Box 125-3). Diabetes insipidus, a condition that results in insufficient production of or lack of response to antidiuretic hormone, can lead to life-threatening hypernatremia35 (Box 125-4).

Clinical Features
Hypernatremia is a disease seen predominantly in elders, but it can also be seen in patients who depend on others to provide them with water, including infants, intubated patients, and persons with mental debilitation.36,37 In addition, patients will also have multifactorial causes leading to severe hypernatremia.38 Patients may complain of polyuria or polydipsia or have obvious causes of extrarenal fluid losses; others may have no complaints at all.
Diagnostic Strategies
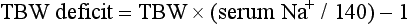
A patient’s total body water is usually calculated by multiplying the patient’s body weight in kilograms times 0.6. However, because of percentage body fat differences based on the age and sex of the patient, it is more accurate to use the correction factors listed in Table 125-2.
Table 125-2
| POPULATION | TOTAL BODY WATER |
| Children and adult men | Body weight (kg) × 0.6 |
| Adult women | Body weight (kg) × 0.5 |
| Elderly men | Body weight (kg) × 0.5 |
| Elderly women | Body weight (kg) × 0.45 |
Management
The treatment of hypernatremia has three interdependent goals: first, to quickly correct underlying shock, hypoperfusion, or significant hypovolemia with normal saline; second, to treat the underlying cause of hypernatremia, such as fever, vomiting, or diabetes insipidus; and third, to carefully lower the serum sodium level, usually by replacement of the body’s total water deficit.39 Until hypoperfusion and hypovolemia are corrected, homeostatic mechanisms for sodium balance will promote sodium resorption to maintain intravascular volume, even at the expense of the serum sodium concentration.
The rate of correction in hypernatremia is extremely important to minimize morbidity and mortality. In adult patients who have had hypernatremia during a short time as a result of sodium loading, “rapid correction” at 1 to 2 mEq/hr lowering of serum sodium appears relatively safe.39,40 However, most adult patients have hypernatremia during days to weeks. In this group of patients, serum sodium concentration should be slowly corrected at no more than 0.5 mEq/hr or 10 to 12 mEq/day.
Normal saline can typically be started for volume replacement until the patient is hemodynamically stable and then changed to half-normal saline at 100 mL/hr once vital signs have normalized. The treatment of central diabetes insipidus with desmopressin (DDAVP) is an effective means of improving polyuria and hypernatremia; initial doses in the acute setting range from 1 to 2 µg.33
Hyponatremia
Hyponatremia, defined as serum sodium concentration of less than 135 mEq/L, is the second most common electrolyte abnormality encountered in clinical practice.37 It is important to recognize hyponatremia because of its potential morbidity and also because it can be a marker of underlying disease. The most common causes of severe hyponatremia in adults are therapy with thiazides, the postoperative state including transurethral prostatectomy, the syndrome of inappropriate secretion of antidiuretic hormone (SIADH), polydipsia in psychiatric patients, and unintentional water intoxication. Gastrointestinal fluid loss, ingestion of overly dilute formula, accidental ingestion of excessive water, and receipt of multiple tap-water enemas are the main causes of severe hyponatremia in infants and children. Most patients presenting to the emergency department (ED) with hyponatremia are asymptomatic and do not require emergent therapy. If symptoms are present, they are typically based on the degree of hyponatremia and how acutely the hyponatremia developed. Symptoms range from headache, nausea, and vomiting to confusion, seizures, and coma. There are two groups of hyponatremic patients that will require treatment with either normal saline or hypertonic saline: (1) severe but asymptomatic hyponatremia with a sodium level of 110 mEq/L or less and (2) acute symptomatic hyponatremia with a sodium level below 120 mEq/L.
Central nervous system (CNS) damage due to hyponatremia may be caused by cerebral edema and increased intracranial pressure, by osmotic fluid shifts during overly aggressive treatment, or by both. When they are subjected to a hyponatremic environment, neurons become depleted of sodium and potassium in an attempt to limit their own osmolarity to prevent intracellular fluid shifts that would lead to cerebral edema. If fluid therapy raises extracellular sodium levels too quickly, fluids shift out of neurons and diffuse demyelination may occur, leading to flaccid paralysis and often death due to a syndrome most commonly referred to as central pontine myelinolysis, although it is more accurately labeled the osmotic demyelinating syndrome.41
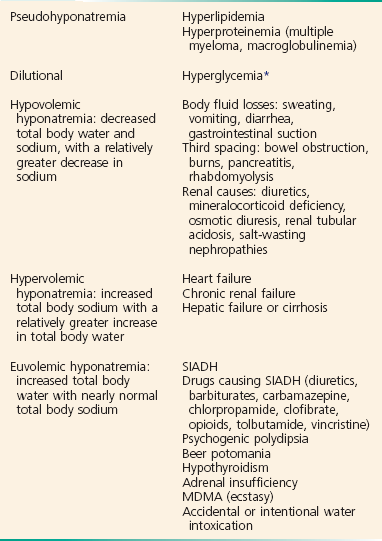
Although hyponatremia has many causes, they fall into four general categories: pseudohyponatremia, hyponatremia with dehydration and decreased extracellular volume, hyponatremia with increased extracellular volume, and euvolemic hyponatremia with increased total body water (Box 125-5).
BOX 125-5 Causes of Hyponatremia
Pseudohyponatremia
Pseudohyponatremia is a falsely low sodium reading caused by the presence of other osmolar particles in the serum. The phenomenon of pseudohyponatremia is explained by the increased percentage of large molecular particles relative to sodium. These large molecules do not contribute to plasma osmolality, resulting in a state in which the relative sodium concentration is decreased but the overall osmolality remains unchanged. Severe hypertriglyceridemia and hyperproteinemia are two common causes of this condition.42 Blood draw or laboratory error should also be considered a possible cause of a patient’s hyponatremia, especially if the blood sample was drawn near an infusion site using 5% dextrose in water (D5W) or 5% dextrose in half-normal saline or when a very abnormal sodium level is reported in an otherwise healthy patient.
Hyperglycemia is sometimes considered a cause of pseudohyponatremia; however, it actually causes a dilutional hyponatremia by pulling water into the vascular space by osmosis. Two different formulas based on the degree of a patient’s hyperglycemia are currently used to correct serum sodium levels. One commonly used formula advocates the addition of 1.6 mEq/L to the measured sodium for every 100 mg/dL of glucose above 100. Another formula recommends use of 2.4 mEq as the correction factor because glucose values above 400 mg/dL may lower sodium values by 4 mEq/L per each 100 mg/dL of glucose rise.40
Euvolemic Hyponatremia
The final category of hyponatremia is one in which patients are euvolemic but have increased total body water. Causes of this type of hyponatremia include SIADH, psychogenic polydipsia,43 beer potomania, hypothyroidism, diuretic use in patients with mild CHF, and accidental or intentional water intoxication. These patients do not present with edema because most of the increased body water is intracellular and not intravascular. Hyponatremia without edema has also been described in patients after the use of the recreational drug N-methyl-3,4-methylenedioxyamphetamine (MDMA or ecstasy). MDMA-induced hyponatremia is multifactorial and is related to increased free water intake to avoid dehydration and rhabdomyolysis, along with the tendency to be very active while using the drug, leading to sweating and antidiuretic hormone secretion.41 In addition, there are extensive case reports of significant hyponatremia in endurance athletes.44
SIADH is an important cause of hyponatremia that occurs when normal antidiuretic hormone secretion is lost and antidiuretic hormone is secreted independently of the body’s need to conserve water. The process results from excess antidiuretic hormone production that causes total body water to increase, diluting the body’s sodium and causing the serum sodium to decrease. Patients with SIADH have inappropriately concentrated urine despite a low serum osmolality and normal circulating blood volume. Patients with SIADH have excess total body water but no signs of edema, ascites, or heart failure because most of the increased body water is intracellular, not intravascular. The three most common causes of SIADH are pulmonary lung masses and infections, CNS disorders, and drugs (Box 125-6). Lung cancers (especially small cell cancer), pneumonia, and tuberculosis can lead to SIADH. CNS infections, masses, and psychosis can also cause SIADH. A large number of medications are associated with SIADH, the most common of which are thiazide diuretics, narcotics, lithium, oral hypoglycemics, barbiturates, and antineoplastics. The mainstay of treatment of most patients with SIADH and other causes of euvolemic hyponatremia is free water restriction.
Management
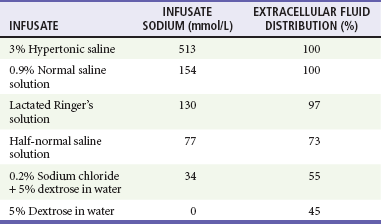
Treatment of hyponatremia is guided by the patient’s clinical presentation, severity of symptoms, estimated duration of illness, fluid status, and underlying cause of the sodium disturbance. Typically, sodium should be corrected during a time course of 48 to 72 hours. Central pontine myelinolysis can occur with too rapid correction of sodium.43 Most cases of central pontine myelinolysis occur in the alcoholic, malnourished, and elder population, although this devastating side effect can occur in healthy, young patients as well. Patients with central pontine myelinolysis have a flaccid paralysis, dysarthria, dysphagia, and hypotension. If a patient have these symptoms during therapy, stop all sodium-containing fluids and administer D5W immediately to lower sodium values temporarily.44 Most patients presenting to the ED with hyponatremia are stable and require no emergent therapy. However, patients who have serum sodium levels of significantly less than 120 mEq/L and those who have acute alterations in mental status, seizures, or new focal findings due to hyponatremia need immediate intervention. Table 125-3 presents the sodium concentration of various infusates, and the following equation is helpful to estimate the effect of 1 liter of any infusate on serum sodium:

In more severe cases when the sodium value is 120 mEq/L or less and the patient has alterations in mental status, has focal findings, or is seizing, hypertonic saline is indicated.45–47 Correction of hyponatremia by 4 to 6 mEq/L within 6 hours, with bolus infusions of 3% saline if necessary, is sufficient to manage the most severe manifestations of hyponatremia.45
The serum sodium level should be below 120 mEq/L when administration of hypertonic saline is being considered. It is recommended that critically ill hyponatremic patients with seizures, focal findings, or coma receive 100 mL of 3% hypertonic saline during 10 minutes. If a second bolus is required, an additional 100 mL of the 3% solution (513 mEq/L of sodium) may be administered during the next 50 minutes. Increased neurologic stability is highly likely once a symptomatic hyponatremic patient’s serum sodium concentration has been raised by about 4 to 6 mEq acutely by the hypertonic saline. To minimize the likelihood of central pontine myelinolysis, it is essential that symptomatic patients with severe hyponatremia not have serum sodium levels rise by any more than a total of 10 to 12 mEq within the first 24 hours.46,47 Potassium deficits should be replaced aggressively in the treatment of hyponatremic patients with a sodium disorder. If patients are retaining volume and diuresis is not adequate, furosemide can be used; D5W is infused if the sodium level is rising too quickly.46 Patients may be able to make full neurologic recoveries from central pontine myelinolysis with the reinduction of hyponatremia in these extreme cases.44 Demeclocycline in a dosage of 600 to 1200 mg daily is effective in patients with refractory hyponatremia.48
Hypovolemic Hyponatremia
Treatment of hypovolemic hyponatremia begins with rehydration. Hypotensive, dehydrated patients are volume resuscitated with normal saline. Once the patient is hemodynamically stable, the infusion rate is slowed. Typically, the normal saline is started at 500 to 1000 mL/hr until the blood pressure is stable and then slowed to 200 mL/hr with frequent sodium checks. If the sodium value is below 120 mEq/L, the sodium concentration should be allowed to rise only by an average of 0.5 mEq/hr or 10 to 12 mEq/day.47 It is essential to treat the underlying cause of hyponatremia.
Hypervolemic Hyponatremia
Normal saline and hypertonic saline can cause pulmonary edema in the hypervolemic hyponatremic patient. Restriction of fluid and sodium is the preferred treatment of patients with hypervolemic hyponatremia, although loop diuretics can be used in severe cases. Hemodialysis is an alternative in patients with renal impairment and will be required in significantly hyponatremic renal failure patients with volume overload. Patients with CHF will usually benefit from diuretics that will increase water excretion and cause vasodilation to improve cardiac output.49 In those patients with liver failure, albumin is a consideration, along with diuretics and possibly paracentesis to improve the underlying pathologic process. Water restriction may make the largest impact on the long-term care of these patients.
Euvolemic Hyponatremia
The mainstay of treatment of euvolemic hyponatremia is free water restriction. As the hypo-osmolality in SIADH results from a relative abundance of water in the intracellular and extracellular volumes, maintained by a reduced ability to excrete water, the restriction of free oral water intake is the first recommendation. The use of water restriction is insufficient to treat acute severe hyponatremia and is not recommended as a sole intervention in severely symptomatic hyponatremia, in which a more rapid correction rate is necessary. The only definitive treatment of SIADH is elimination of its underlying cause. Most cases of SIADH caused by malignant disease resolve with effective antineoplastic therapy, and most due to medication resolve promptly when the offending agent is discontinued.50
In patients with SIADH, normal saline may cause the serum sodium concentration to decrease even more as free water is retained and hypertonic urine is excreted. If a patient is symptomatic because of a rapid decrease in serum sodium concentration, treatment with hypertonic saline is recommended. Demeclocycline and lithium are rarely used in the treatment of SIADH because of their many side effects and nephrotoxicity. Rapid correction of hyponatremia may occur during hemodialysis. To minimize the risks of central pontine myelinolysis, hemodialysis is reserved for patients with documented renal failure and used in a very careful manner.50 Vaptans, which are oral agents that inhibit the effects of vasopressin, have been studied for treatment of patients with hyponatremia due to SIADH but need further evaluation before becoming standard of care.46,51
Hypercalcemia
There are five major causes of hypercalcemia (Box 125-7). Primary hyperparathyroidism is the most common cause of hypercalcemia in outpatients, whereas malignant disease is the most common cause in hospitalized patients. Mild hypercalcemia, in an otherwise normal person, may be due to thiazide diuretics with minimal dehydration. Other less common causes of elevated calcium concentration are usually not considered until malignant disease and parathyroid disease are ruled out. Malignancy-associated hypercalcemia occurs in up to 10% of all patients with advanced cancer and generally conveys a poor prognosis. Other causes of hypercalcemia include granulomatous disease, such as sarcoidosis and tuberculosis; medications and pharmacologic agents; and a number of diverse conditions, such as rhabdomyolysis and prolonged immobilization.

Clinical Features
Unfortunately, the clinical presentation of hypercalcemia is often vague and nonspecific. Symptoms include nonfocal abdominal pain, constipation, fatigue, diffuse body aches, anorexia, nausea, and vomiting. Symptom severity depends on the degree of hypercalcemia, the rapidity of onset, and the patient’s baseline neurologic and renal function.52 The diagnosis should be considered in a large number of differential diagnoses. In addition, some patients complain of polyuria or polydipsia. Neuropsychiatric disturbances include anxiety, depression, confusion, and hallucinations. The CNS manifestations that often predominate in more severe cases include lethargy, altered mental status, seizures, and coma. Death due to hypercalcemia is usually related to complications caused by coma, dehydration, or electrolyte disturbances. Cardiac conduction abnormalities may occur; bradydysrhythmias are the most common.53 Severe hypercalcemia has also been associated with sinus arrest, atrioventricular block, atrial fibrillation, and ventricular tachycardia.
Diagnostic Strategies
The diagnostic evaluation of a patient with suspected hypercalcemia begins with obtaining of electrolyte and renal function tests and an ECG. Calcium is measured by determination of either a total serum calcium level or an ionized calcium level. Ionized calcium is the active form of the total calcium level. It is more accurate in the diagnosis and treatment of hypocalcemia, but it does need to be routinely evaluated in hypercalcemia.54 The serum total calcium level represents both bound and unbound calcium and thus should be corrected on the basis of the albumin concentration. The adjustment to serum albumin is accomplished by adding or subtracting 0.08 mg/dL to the measured total serum calcium for every 1.0 g/L of albumin below or above 4 g/L albumin, respectively.
A short QT interval can be seen in hypercalcemia and is considered a classic finding. However, although the incidence and duration of QT shortening appear to be correlated with the degree of hypercalcemia, it is not a reliable finding and is not routinely seen in most patients (Fig. 125-6). ST segment elevation may be the least well documented but most consistent electrocardiographic finding, and hypercalcemia should be considered in the differential diagnosis of ST segment elevation caused by conditions other than myocardial infarction.55,56 In severe cases of hypercalcemia, sinus bradycardia, bundle branch block, and high-degree atrioventricular block may also been seen.
Management
Patients in hypercalcemic crisis are usually dehydrated, often obtunded, and also predisposed to arrhythmias as a result of concomitant electrolyte disturbances; thus they require intravenous access with a normal saline infusion and close monitoring. Normal saline will inhibit proximal tubule reabsorption of calcium and also correct the patient’s volume depletion. Normal saline is infused “wide open” until blood pressure and perfusion are normalized. After the initial bolus, the saline infusion is adjusted to a rate of approximately 200 to 300 mL/hr, depending on the patient’s age and renal function. Patients with underlying cardiac or renal disease may require lower infusion rates that are carefully titrated. Although the administration of higher volumes of saline may further augment calcium excretion, it is much more likely to result in increased morbidity and mortality from volume overload, pulmonary edema, and myocardial ischemia. The routine use of furosemide in the management of hypercalcemia is no longer recommended. Furosemide was once thought to block the distal reabsorption of calcium, thus complementing saline’s proximal tubule effects. However, no modern-day studies have shown furosemide to have significant calcium reabsorption blocking effects. The use of furosemide is reserved to augment saline diuresis and to avoid volume overload during the treatment of hypercalcemia.57 If it is given to patients who are not yet volume replete, this loop diuretic may worsen the hypercalcemia because of its volume depleting effects and also adversely affect the patient’s hemodynamics and renal status. Once calcium excretion by saline infusion has begun, other electrolyte values should be carefully monitored with attention to serum potassium levels.
Osteoclast-inhibiting therapies for severe hypercalcemia are generally considered in consultation with the patient’s primary physician or oncologist. Drugs that inhibit osteoclast-mediated bone resorption include the bisphosphonates, mithramycin, calcitonin, and glucocorticoids. Intravenous bisphosphonates are the most extensively studied and most efficacious agents for the treatment of malignancy-associated hypercalcemia.58 Their calcium-lowering effect is achieved predominantly by inhibition of osteoclast function and survival. Zoledronic acid is the bisphosphonate of choice in hypercalcemia of malignancy.59 The infusion takes 15 minutes, and zoledronic acid may be more effective than other bisphosphonates at keeping the calcium level down over time. The use of intravenous bisphosphonates is restricted to the treatment of acute hypercalcemia associated with serum calcium concentrations above 15 mg/dL and rapid deterioration of CNS, cardiac, gastrointestinal, and renal function.
In the rare case in which a patient has a life-threatening hypercalcemic arrhythmia or heart block, phosphates and hemodialysis are considered.60,61 In cases of hypercalcemic crisis resulting from primary hyperparathyroidism, urgent parathyroidectomy is potentially curative.
Hypocalcemia
There are multiple causes of hypocalcemia, of which hypoalbuminemia is the most common (Box 125-8). Because calcium is bound to albumin and other serum proteins, hypoalbuminemia will cause a fall in the measured serum calcium by about 0.8 mg/dL for every 1 g/dL reduction in serum albumin. The active form of calcium is the ionized calcium, which is not affected by changes in albumin.
Hypoparathyroidism is a common cause of hypocalcemia and often develops because of surgery for head and neck cancers. It develops in 1 to 2% of patients after total thyroidectomy. Patients with vitamin D deficiency, including those with malabsorption syndromes, liver disease, malnutrition, and very little sunlight exposure, are at high risk for development of hypocalcemia.62 Derangements in magnesium and phosphate can also lead to hypocalcemia. Hyperphosphatemic patients often have hypocalcemia because of phosphate’s affinity to bind calcium, whereas hypomagnesemia causes end-organ resistance to parathyroid hormone and inhibits the hypocalcemic feedback loop. Patients with sepsis demonstrate hypocalcemia usually associated with hypoalbuminemia.
The most common causes of symptomatic hypocalcemia are massive blood transfusions, toxins, pancreatitis, tumor lysis syndrome, and chronic malnutrition (Box 125-9). Patients receiving massive blood transfusions are at risk for development of hypocalcemia because of citrate toxicity. Rapid blood transfusions and radiocontrast dyes containing citrate should be monitored closely in patients with hepatic failure, CHF, or other low-output states to avoid hypocalcemia.63
Diagnostic Strategies
Most cases of hypocalcemia are discovered by clinical suspicion followed by appropriate laboratory testing. A serum calcium level less than 8.5 mg/dL or an ionized calcium level less than 2.0 mEq/L is considered diagnostic. Total serum calcium is approximately 50% free (ionized) and 50% bound, primarily to albumin; thus the serum level must be “corrected” when hypoalbuminemia exists. The ionized calcium level, which is not affected by the albumin level, is more accurate. It is best to perform the whole blood ionized calcium determination rapidly to avoid changes in chelation and pH.64 In select cases, a parathyroid hormone level may be sent to assist the admitting or consulting physician. Electrocardiography and cardiac monitoring are recommended in suspected hypocalcemia patients to evaluate the QT interval and to provide continuous monitoring for potential dysrhythmias.
Management
Most asymptomatic patients and those with mild symptoms can be treated with oral calcium supplementation, such as calcium carbonate. Intravenous calcium is administered, either as calcium chloride or calcium gluconate, to patients with moderate to severe symptoms; 100 to 300 mg of elemental calcium given during 5 to 30 minutes will raise the ionized calcium level 0.5 to 1.5 mEq. Calcium chloride contains 272 mg of elemental calcium but can be caustic to veins and thus should be given only to critically ill hypocalcemic patients who require calcium urgently. It is best given through a central line. Calcium gluconate contains 92 mg of elemental calcium. Although this is one-third the amount contained in calcium chloride, it is safer to administer and can be given peripherally.62 Most patients requiring intravenous calcium should be admitted to the hospital for monitoring and treatment of nausea, vomiting, hypertension, and bradycardia. Patients taking digoxin have increased cardiac sensitivity to fluctuations in serum calcium, so intravenous calcium administration is accompanied by continuous electrocardiographic monitoring.65
Hypermagnesemia
Hypermagnesemia is a relatively rare electrolyte abnormality defined as a serum magnesium concentration above 2.2 mg/dL. Hypermagnesemia is most often seen in patients with renal insufficiency who cannot optimally regulate magnesium excretion, especially as their magnesium load increases. There have been reports of fatal and near-fatal cases involving hypermagnesemia in patients receiving magnesium with unrecognized renal failure.66 Hypermagnesemia can also be caused iatrogenically in patients receiving intravenous magnesium for medical treatment or in patients taking over-the-counter laxatives and antacids.67 Even though most patients at risk for hypermagnesemia have underlying renal impairment, hypermagnesemia has been reported in patients with normal renal function, especially in elders. Box 125-10 lists the most common causes of increased serum magnesium levels.
In patients with normal renal function, large amounts of magnesium can be excreted daily in the stool and urine. However, in patients with impaired renal function, hypermagnesemia can be seen even with therapeutic doses of magnesium-containing products. For example, a patient with renal insufficiency should not use magnesium citrate for treatment of constipation. An adult dose of 10 ounces of laxative syrup results in consumption of approximately 2.0 g of elemental magnesium per single dose. A healthy adult can excrete more than 6.0 g of magnesium daily, but renally impaired patients may not be able to tolerate a single dose of laxative syrup. Hypermagnesemia resulting from Epsom salt gargles and salt enemas has been reported.68,69
Clinical Features
Patients with hypermagnesemia may present with flushing, nausea, vomiting, headache, and diminished deep tendon reflexes. Typically, symptoms begin to be manifested around magnesium levels of 4 mg/dL (Table 125-4). Magnesium is a CNS and neuromuscular depressant and can cause cardiac instability. Magnesium acts as a calcium channel blocker and also blocks potassium channels needed for repolarization. As magnesium levels rise, hypotension and electrocardiographic changes, including QRS widening and QT and PR prolongation, begin to occur. When serum magnesium levels rise above 7 mg/dL, patients can have signs and symptoms of hypotension, respiratory insufficiency, and heart block. Cardiac arrest and death have been reported in patients with serum magnesium levels above 10 mg/dL.70 Finally, hypermagnesemia causes suppression of parathyroid hormone secretion and can be associated with hypocalcemia.
Table 125-4
Clinical Effects of Hypermagnesemia
| EFFECT | LEVEL (mg/dL) |
| Decreased deep tendon reflexes | 4-5 |
| Hypotension | 5-7 |
| Respiratory insufficiency | 10 |
| Heart block | 10-15 |
| Cardiac arrest | 10-24 |
Diagnostic Strategies
Measured plasma magnesium levels often do not reflect total magnesium content, making it difficult to consistently correlate symptoms to specific magnesium levels. Although there is some question of the role of measuring ionized magnesium in patients with hypomagnesemia, only total body magnesium needs to be followed in hypermagnesemic patients.71
Management
In patients with higher levels of serum magnesium or more severe symptoms, renal consultation should be initiated immediately to arrange for dialysis. Intravenous calcium therapy to reverse magnesium toxicity should be reserved for patients with life-threatening symptoms while dialysis is being arranged. Calcium directly antagonizes the neuromuscular and cardiovascular effects of magnesium and is recommended in hypotensive patients with respiratory depression and cardiac instability. In treating life-threatening hypermagnesemia, initially administer 100 to 200 mg of intravenous calcium as either calcium chloride or calcium gluconate (1-2 mL of 10% calcium chloride or 5 mL of 1% calcium gluconate during 2-5 minutes) and then titrate to effect.72 One can then consider a continuous infusion at 2 to 4 mg/kg/hr if it is needed while dialysis is being arranged.
Hypomagnesemia
Hypomagnesemia is a common electrolyte abnormality that often goes undetected. Normal serum magnesium levels range from 1.5 to 3.0 mEq/L. Symptoms of hypomagnesemia typically begin to be manifested at serum levels below 1.2 mEq/L, although symptoms are often not well correlated with the patient’s serum level. This is because most of the body’s magnesium is intracellular, and thus a single blood sample with a low serum magnesium level may not accurately reflect total body magnesium or the extent of true hypomagnesemia. Magnesium exists in three states: ionized magnesium, protein bound, and complexed to serum anions. Even though studies show the importance of measuring ionized calcium, most research shows that ionized magnesium can be inferred from total magnesium. Currently, the clinical role of measurement of ionized magnesium is unclear, and measurement of ionized magnesium is not standard practice in the ED; there may be a role for measurement of ionized magnesium in the intensive care setting.73
There are many causes of hypomagnesemia (Box 125-11). The following are the five most common ED presentations of hypomagnesemia.
Patients Maintained with Diuretics.: Patients using either loop or thiazide diuretics are at increased risk for hypomagnesemia. Both types of diuretics can inhibit magnesium reabsorption. Conversely, potassium-sparing diuretics are also magnesium sparing because they enhance magnesium reabsorption and decrease magnesium excretion. The degree of hypomagnesemia induced by the loop and thiazide diuretics is generally mild, partly because the associated volume contraction will tend to increase proximal sodium, water, and magnesium reabsorption.
Malnourished and Alcoholic Patients.: Healthy patients consume enough magnesium in green vegetables, legumes, fruits, shellfish, fresh meat, and cocoa on a regular basis to maintain normal total body magnesium stores. However, hypomagnesemia is common in patients with chronic protein-calorie malnutrition because of an associated lack of essential minerals and vitamins including magnesium. This is especially true in chronic alcoholics, who may not eat foods rich in magnesium.74 Magnesium losses are further increased in chronic alcoholics because of alcohol’s diuretic effects. Hypomagnesemia may also be seen in patients with malabsorption disorders (celiac sprue and short bowel syndrome) and in patients with increased magnesium excretion (chronic diarrhea or inflammatory bowel conditions).
Patients with Hypokalemia.: Both potassium and magnesium are critical to help stabilize the membrane potential, to decrease cell excitability, and for function of the Na+,K+-ATPase pump. Approximately 50% of patients with hypokalemia also have concomitant magnesium deficiency. Increasing degrees of hypokalemia are correlated with an increasing likelihood of a magnesium deficit.21 Hypokalemic patients who are refractory to potassium replacement are likely to also be hypomagnesemic.
Patients with Acute Coronary Artery Disease and Ventricular Arrhythmias.: There appears to be a relationship between low serum magnesium levels and the subsequent development of coronary heart disease.75 Patients who have a myocardial infarction are more likely than controls to be hypomagnesemic. At present, magnesium supplementation is recommended only for those acute coronary syndrome patients who have evidence of hypokalemia, prolonged QT, or known hypomagnesemia. Similarly, patients with acute myocardial infarction who have mild hypomagnesemia appear to have a twofold to threefold increase in the frequency of ventricular arrhythmias in the first 24 hours compared with those with normal magnesium levels.76 There is controversy about whether magnesium should be administered empirically after acute myocardial infarction.77 Dysrhythmia is the most common cardiovascular manifestation of hypomagnesemia. Magnesium affects the duration of phase 2 of the action potential, and hypomagnesemia can prolong the QT interval. Magnesium also has effects on phase 4, the resting membrane potential, when it serves to keep the cell more negative by stimulating the sodium-potassium pump. The exact mechanism underlying a possible association between hypomagnesemia and arrhythmias is at present unknown. Arrhythmias are likely to be due to concurrent hypokalemia, hypomagnesemia, or both, resulting in a prolonged QT interval and increases in spontaneous depolarization.
Patients Receiving Specific Medications.: In addition to diuretics, many medications are associated with hypomagnesemia. Many nephrotoxic drugs, including aminoglycosides, amphotericin B, cisplatin, and pentamidine, can produce magnesium wasting. The mechanism responsible for hypomagnesemia associated with long-term proton pump inhibitor use is unknown; however, long-term use of proton pump inhibitors may be associated with changes in intestinal absorption of magnesium.78 Severe hypomagnesemia can also be seen in patients preparing for colonoscopy with polyethylene glycol–based bowel preparations.
Clinical Features
Determination of the clinical consequences of isolated hypomagnesemia is often difficult because patients with hypomagnesemia typically have hypokalemia, hypocalcemia, and hyponatremia. However, many signs and symptoms are reported to correlate with hypomagnesemia, including muscle cramping, diffuse weakness, palpitations, vertigo, ataxia, depression, and seizures. The clinical manifestations most likely seen in the ED typically involve the neuromuscular and cardiovascular systems. Patients may present with hyperactive deep tendon reflexes, muscle cramps, Trousseau’s and Chvostek’s signs, and dysarthria and dysphagia from esophageal dysmotility. Cardiac conduction abnormalities secondary to magnesium depletion, and often coexisting hypokalemia, can result in PR as well as QT interval prolongation. Dysrhythmias including atrial fibrillation, multifocal atrial tachycardia, premature ventricular complexes, ventricular tachycardia, torsades de pointes, and ventricular fibrillation are the most common cardiovascular manifestations of hypomagnesemia. Women with adequate intakes of magnesium are less likely to be affected by preeclampsia than are those with an inadequate intake.79
Hyperphosphatemia
Hyperphosphatemia is defined as a serum level above 2.5 mg/dL, but it is usually clinically significant only when levels are greater than 5 mg/dL. Although it is rare in the general population, hyperphosphatemia is extremely common in patients with renal insufficiency or renal failure.80 Almost all patients with renal failure experience hyperphosphatemia at some time during the course of their disease. Hyperphosphatemia can occur because of four major pathways: decreased phosphate excretion, excessive phosphate intake, increased renal tubular reabsorption, and shift of phosphate from intracellular to extracellular space. In addition, physicians must be aware of spurious elevations in phosphate (Box 125-12).
Decreased excretion of phosphate combined with excessive intake is the most common mechanism for the development of hyperphosphatemia. Excessive phosphate intake alone is an uncommon cause of hyperphosphatemia in patients with normal renal function. When patients have glomerular filtration rates below 30 mL/min, the kidneys do not allow excretion of the full amount of ingested phosphate to maintain homeostasis. Exogenous phosphate, including intravenous or oral phosphate administration and phosphate enemas and laxatives can cause a large burden on the kidneys if they do not have normal baseline function.19
Hyperphosphatemia can be a spurious finding in cases of hyperproteinemia, such as multiple myeloma, hyperlipidemia, hemolysis, or hyperbilirubinemia. Drawing of a blood sample from a line containing heparin is another cause of a falsely elevated phosphate level.81
Management
Dietary restriction alone may suffice for control of hyperphosphatemia in persons with mild renal insufficiency, but it is inadequate for control in those with overt renal failure. Because most patients presenting with severe hyperphosphatemia also have hypocalcemia, treatment focuses on the correction of both electrolytes. In patients with normal renal function, phosphate excretion can be increased by saline infusion coupled with loop diuretics. Hyperphosphatemia usually resolves in 6 to 12 hours in patients with normal renal function.82 In patients with hyperphosphatemia with renal failure, hemodialysis or peritoneal dialysis should be considered early in the management. Currently, phosphate control is initiated only when hyperphosphatemia occurs, but a potentially beneficial and simple approach may be to intervene earlier in patients with chronic kidney disease.83 The optimal method for control of serum phosphate in patients undergoing dialysis is unknown and may involve combinations of dietary modification and enhancement of phosphate clearance through longer dialysis sessions.84,85
Hypophosphatemia
Symptoms of hypophosphatemia typically begin to be manifested at serum levels below 1.0 mg/dL. Acute hypophosphatemia is most commonly due to a rapid intracellular shift, but there are many other causes of clinical manifestations as well.86 Hyperventilation, glucose, insulin, volume, and resolving acidosis lead to hypophosphatemia by rapid intracellular shift. The many causes of hypophosphatemia include decreased phosphate intake or increased absorptive states, hyperventilatory states, hormonal and endocrine effects, medications, and disease states (Box 125-13). The ED patients most likely to have hypophosphatemia are those who are malnourished with alcohol withdrawal, acute hyperventilation, or sepsis and patients with DKA or alcohol ketoacidosis in whom reintroduction of insulin and glucose causes phosphate uptake into cells.87,88

Clinical Features
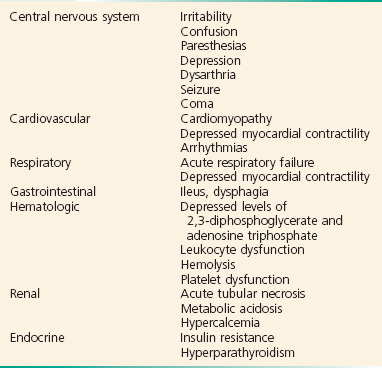
Mild to moderate hypophosphatemia is usually asymptomatic, but major clinical manifestations can occur with severe hypophosphatemia. Hypophosphatemia can affect a variety of organ systems and cause a wide variety of symptoms because phosphate is an essential component to adenosine triphosphate (Box 125-14). Patients with hypophosphatemia may present with nonspecific complaints including joint pain, myalgias, irritability, and depression. Severe hypophosphatemia can be manifested as seizures, arrhythmias, cardiomyopathy, insulin resistance, acute tubular necrosis, rhabdomyolysis, and acute respiratory failure.
Management
Therapy for hypophosphatemia is recommended as levels drop below 1.5 to 2.0 mg/dL, and therapy is essential as levels fall below 1.0 mg/dL. Because hypophosphatemia often is manifested with hypokalemia, phosphate repletion is considered in conjunction with potassium administration. Oral phosphorous, 250 to 500 mg twice daily, can be given to stable or asymptomatic patients. Intravenous preparations are available as sodium phosphate (Na2PO4 and NaPO4) or potassium phosphate (K2PO4 and KPO4), and rate of infusion and choice of initial dosage should be based on severity of hypophosphatemia and presence of symptoms. If the serum phosphorus concentration is less than 1.5 mg/dL (0.48 mmol/L), 1.3 mmol/kg of elemental phosphorous (up to a maximum of 100 mmol) can be given in three or four divided doses in a 24-hour period.89 For routine replacement, give 0.5 mL/hr K2PO4; this may be increased to 1 mL/hr in severe symptomatic patients.87 Each milliliter of K2PO4 contains 3 mmol of phosphorus and 4.4 mEq of potassium. Typical replacement therapy provides approximately 1 g of phosphorus per day. Monitoring of patients for the development of hypocalcemia, hyperkalemia, and hyperphosphatemia is required while intravenous phosphate is administered, especially in patients with renal insufficiency. Patients with DKA are initially hypophosphatemic. However, no studies have shown significant benefit to routine phosphate therapy in DKA.91 Risks of routine treatment with phosphate include hyperphosphatemia, renal failure, hypocalcemia, and hypomagnesemia. In patients with severe malnutrition or significant hypophosphatemia, replacement can be considered, but never administer more than 60 mmol/day without reason.






References
1. Glasziou, P. Practice corner: The first symptom of hyperkalemia is death. ACP J Club. 2004;140:A13.
2. Whang, R, et al. Predictors of clinical hypomagnesemia. Hypokalemia, hypophosphatemia, hyponatremia, and hypocalcemia. Arch Intern Med. 1984;144:1794–1796.
3. Wrenn, KD, Slovis, CM, Slovis, BS. The ability of physicians to predict hyperkalemia from the ECG. Ann Emerg Med. 1991;20:1229–1232.
4. Ahmed, J, Weisberg, LS. Hyperkalemia in dialysis patients. Semin Dial. 2001;14:348–356.
5. Semple, P, Booth, C. Calcium chloride; a reminder. Anaesthesia. 1996;51:93.
6. Singh, BS, et al. Efficacy of albuterol inhalation in treatment of hyperkalemia in premature neonates. J Pediatr. 2002;141:16–20.
7. Allon, M, Dunlay, R, Copkney, C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426–429.
8. Nair, S, et al. A randomized controlled trial to assess the optimal dose and effect of nebulized albuterol in acute exacerbations of COPD. Chest. 2005;128:48–54.
9. Allon, M, Copkney, C. Albuterol and insulin for treatment of hyperkalemia in hemodialysis patients. Kidney Int. 1990;38:869–872.
10. Allon, M, Shanklin, N. Effect of bicarbonate administration on plasma potassium in dialysis patients: Interactions with insulin and albuterol. Am J Kidney Dis. 1996;28:508–514.
11. Mahoney, A, et al. Emergency interventions for hyperkalaemia. Cochrane Database Syst Rev. (2):2005.
12. Gruy-Kapral, C, et al. Effect of single dose resin-cathartic therapy on serum potassium concentration in patients with end-stage renal disease. J Am Soc Nephrol. 1998;9:1924–1930.
13. Kitabchi, AE, et al. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32:1335–1343.
14. Alfonzo, AV, et al. Potassium disorders—clinical spectrum and emergency management. Resuscitation. 2006;70:10–25.
15. Vanden Hoek, TL, et al. Part 12: Cardiac arrest in special situations: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122(Suppl 3):S829–S861.
16. Blumberg, A, et al. Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure. Am J Med. 1988;85:507–512.
17. Roffey, P, et al. Implication of epinephrine-induced hypokalemia during cardiac arrest. Resuscitation. 2003;58:231.
18. Lin, JL, et al. Outcomes of severe hyperkalemia in cardiopulmonary resuscitation with concomitant hemodialysis. Intensive Care Med. 1994;20:287–290.
19. Buckley, MS, Leblanc, JM, Cawley, MJ. Electrolyte disturbances associated with commonly prescribed medications in the intensive care unit. Crit Care Med. 2010;38(Suppl):S253–S264.
20. Osadchii, OE. Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin Pharmacol. 2010;24:547–559.
21. Huang, CL, Kuo, E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18:2649–2652.
22. Whang, R, Whang, DD, Ryan, MP. Refractory potassium repletion. A consequence of magnesium deficiency. Arch Intern Med. 1992;152:40–45.
23. Hunt, SA, et al. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: Developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:e391–e479.
24. Umpierrez, GE, et al. Treatment of diabetic ketoacidosis with subcutaneous insulin aspart. Diabetes Care. 2004;27:1873–1878.
25. Finsterer, J, et al. Malnutrition-induced hypokalemic myopathy in chronic alcoholism. J Toxicol Clin Toxicol. 1998;36:369–373.
26. Udezue, E, D’Souza, L, Mahajan, M. Hypokalemia after normal doses of nebulized albuterol (salbutamol). Am J Emerg Med. 1995;13:168–171.
27. Greenlee, M, et al. Narrative review: Evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med. 2009;150:619–625.
28. Krahn, LE, et al. Hypokalemia leading to torsades de pointes. Munchausen’s disorder or bulimia nervosa? Gen Hosp Psychiatry. 1997;19:370–377.
29. Gennari, FJ. Hypokalemia. N Engl J Med. 1998;339:451–458.
30. Coca, SG, Perazella, MA, Buller, GK. The cardiovascular implications of hypokalemia. Am J Kidney Dis. 2005;45:233–247.
31. Hamill-Ruth, RJ, McGory, R. Magnesium repletion and its effect on potassium homeostasis in critically ill adults: Results of a double-blind, randomized, controlled trial. Crit Care Med. 1996;24:38–45.
32. Huang, WY, et al. Central pontine and extrapontine myelinolysis after rapid correction of hyponatremia by hemodialysis in a uremic patient. Ren Fail. 2007;29:635–638.
33. Adler, SM, Verbalis, JG. Disorders of body water homeostasis in critical illness. Endocrinol Metab Clin North Am. 2006;35:873–894.
34. Jacobson, J, Bohn, D. Severe hypernatremic dehydration and hyperkalemia in an infant with gastroenteritis secondary to rotavirus. Ann Emerg Med. 1993;22:1630–1632.
35. Verbalis, JG. Diabetes insipidus. Rev Endocr Metab Disord. 2003;4:177–185.
36. Palevsky, PM, Bhagrath, R, Greenberg, A. Hypernatremia in hospitalized patients. Ann Intern Med. 1996;124:197–203.
37. Price, TG, Kallenborn, JC. Infant hypernatremia: A case report. J Emerg Med. 2000;19:153–157.
38. Holley, AD, Green, S, Davoren, P. Extreme hypernatraemia: A case report and brief review. Crit Care Resusc. 2007;9:55–58.
39. Adrogue, HJ, Madias, NE. Hyponatremia. N Engl J Med. 2000;342:1581–1589.
40. Kaplan, LJ, Kellum, JA. Fluids, pH, ions and electrolytes. Curr Opin Crit Care. 2010;16:323–331.
41. Pirzada, NA, Ali, II. Central pontine myelinolysis. Mayo Clin Proc. 2001;76:559–562.
42. Turchin, A, Seifter, JL, Seely, EW. Clinical problem-solving. Mind the gap. N Engl J Med. 2003;349:1465–1469.
43. Cheng, JC, et al. Long-term neurologic outcome in psychogenic water drinkers with severe symptomatic hyponatremia: The effect of rapid correction. Am J Med. 1990;88:561–566.
44. Almond, CS, et al. Hyponatremia among runners in the Boston Marathon. N Engl J Med. 2005;352:1550–1556.
45. Lien, YHH, Shapiro, JI. Hyponatremia: Clinical diagnosis and management. Am J Med. 2007;120:653–658.
46. Sterns, RH, Hix, JK, Silver, S. Treatment of hyponatremia. Curr Opin Nephrol Hypertens. 2010;19:493–498.
47. Verbalis, JG, et al. Hyponatremia treatment guidelines 2007: Expert panel recommendations. Am J Med. 2007;120(Suppl 1):S1–S21.
48. Janicic, N, Verbalis, JG. Evaluation and management of hypo-osmolality in hospitalized patients. Endocrinol Metab Clin North Am. 2003;32:459–481.
49. Oren, RM. Hyponatremia in congestive heart failure. Am J Cardiol. 2005;95:2B–7B.
50. Ellison, DH, Berl, T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064–2072.
51. Vaidya, C, Ho, W, Freda, BJ. Management of hyponatremia: Providing treatment and avoiding harm. Cleve Clin J Med. 2010;77:715–726.
52. Stewart, AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med. 2005;352:373–379.
53. Ziegler, R. Hypercalcemic crisis. J Am Soc Nephrol. 2001;12(Suppl 17):S3–S9.
54. Ariyan, CE, Sosa, JA. Assessment and management of patients with abnormal calcium. Crit Care Med. 2004;32(4 Suppl):S146–S154.
55. Nishi, SP, Barbagelata, NA, Atar, S, Birnbaum, Y, Tuero, E. Hypercalcemia-induced ST-segment elevation mimicking acute myocardial infarction. J Electrocardiol. 2006;39:298–300.
56. Littmann, L, Taylor, L, Brearley, WD. ST-segment elevation: A common finding in severe hypercalcemia. J Electrocardiol. 2007;40:60–62.
57. LeGrand, SB, Leskuski, D, Zama, I. Narrative review: Furosemide for hypercalcemia: An unproven yet common practice. Ann Intern Med. 2008;149:259–263.
58. Drake, MT, Clarke, BL, Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clinic Proc. 2008;83:1032–1045.
59. Major, P, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: A pooled analysis of two randomized, controlled clinical trials. J Clin Oncol. 2001;19:558–567.
60. Stewart, AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med. 2005;352:373–379.
61. Koo, WS, et al. Calcium-free hemodialysis for the management of hypercalcemia. Nephron. 1996;72:424–428.
62. Cooper, MS, Gittoes, NJ. Diagnosis and management of hypocalcaemia. BMJ. 2008;336:1298–1302.
63. Aguilera, M, Vaughan, RS. Calcium and the anaesthetist. Anaesthesia. 2000;55:779–790.
64. Bosworth, M, et al. Clinical inquiries: What is the best workup for hypocalcemia? J Fam Pract. 2008;57:677–679.
65. Levine, M, Nikkanen, H, Pallin, DJ. The effects of intravenous calcium in patients with digoxin toxicity. J Emerg Med. 2011;40:41–46.
66. Schelling, JR. Fatal hypermagnesemia. Clin Nephrol. 2000;53:61–65.
67. Qureshi, T, Melonakos, TK. Acute hypermagnesemia after laxative use. Ann Emerg Med. 1996;28:552–555.
68. Birrer, RB, Shallash, AJ, Totten, V. Hypermagnesemia-induced fatality following Epsom salt gargles. J Emerg Med. 2002;22:185–188.
69. Tofil, NM, et al. Fatal hypermagnesemia caused by an Epsom salt enema: A case illustration. South Med J. 2005;98:253–256.
70. Slovis, CM, Meehan, PM. Electrolyte abnormalities. In: Roppolo LP, ed. Emergency Medicine Handbook: Critical Concepts for Clinical Practice. Philadelphia: Mosby/Elsevier; 2007:1166–1184.
71. Topf, JM, Murray, PT. Hypomagnesemia and hypermagnesemia. Rev Endocr Metab Disord. 2003;4:195–206.
72. Vanden Hoek, TL, et al. Cardiac arrest in special situations: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S829–S861.
73. Soliman, HM. Development of ionized hypomagnesemia is associated with higher mortality rates. Crit Care Med. 2003;31:1082–1087.
74. Poikolainen, K, Alho, H. Magnesium treatment in alcoholics: A randomized clinical trial. Subst Abuse Treat Prev Policy. 2008;3:1.
75. Liao, F, Folsom, AR, Brancati, FL. Is low magnesium concentration a risk factor for coronary heart disease? The Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 1998;136:480–490.
76. Agus, ZS. Hypomagnesemia. J Am Soc Nephrol. 1999;10:1616–1622.
77. ISIS-4. A randomized factorial trial assessing early captopril, oral mononitrate, and intravenous magnesium sulfphate in 58,050 patients with suspected acute myocardial infarction. ISIS-4 Collaborative Group. Lancet. 1995;345:669.
78. Broeren, MA, Geerdink, EA, Vader, HL, van den Wall Bake, AW. Hypomagnesemia induced by several proton-pump inhibitors. Ann Intern Med. 2009;151:755–756.
79. Champagne, CM. Magnesium in hypertension, cardiovascular disease, metabolic syndrome, and other conditions: A review. Nutr Clin Pract. 2008;23:142–151.
80. Kapoor, M, Chan, G. Fluid and electrolyte abnormalities. Crit Care Clin. 2001;17:503–529.
81. Ball, CL, Tobler, K, Ross, BC, Connors, MR, Lyon, ME. Spurious hyperphosphatemia due to sample contamination with heparinized saline from an indwelling catheter. Clin Chem Lab Med. 2004;42:107–108.
82. Slovis, CM, Meehan, PM. Electrolyte abnormalities. In: Roppolo LP, ed. Emergency Medicine Handbook: Critical Concepts for Clinical Practice. Philadelphia: Mosby/Elsevier; 2007:1166–1184.
83. Martin, KJ, Gonzalez, EA. Prevention and control of phosphate retention/hyperphosphatemia in CKD-MBD: What is normal, when to start, and how to treat? Clin J Am Soc Nephrol. 2011;6:440–446.
84. Tonelli, M, Pannu, N, Manns, B. Oral phosphate binders in patients with kidney failure. N Engl J Med. 2010;362:1312–1324.
85. Culleton, BF, et al. Effect of frequent nocturnal hemodialysis vs conventional hemodialysis on left ventricular mass and quality of life: A randomized controlled trial. JAMA. 2007;298:1291–1299.
86. Subramanian, R, Khardori, R. Severe hypophosphatemia. Pathophysiologic implications, clinical presentations, and treatment. Medicine (Baltimore). 2000;79:1–8.
87. Miller, DW, Slovis, C. Hypophosphatemia in the emergency department therapeutics. Am J Emerg Med. 2000;18:457–461.
88. Shor, R, et al. Severe hypophosphatemia in sepsis as a mortality predictor. Ann Clin Lab Sci. 2006;36:67–72.
89. Kraft, MD, Btaiche, IF, Sacks, GS, Kudsk, KA. Treatment of electrolyte disorders in adult patients in the intensive care unit. Am J Health Syst Pharm. 2005;62:1663.
90. Reference deleted in proofs.
91. Chiasson, JL, et al. Diagnosis and treatment of diabetic ketoacidosis and the hyperglycemic hyperosmolar state. CMAJ. 2003;168:859–866.
92. Casavant, MJ, Fitch, JA. Fatal hypernatremia from saltwater used as an emetic. J Toxicol Clin Toxicol. 2003;41:861–863.
93. Trepiccione, F, Christensen, BM. Lithium-induced nephrogenic diabetes insipidus: New clinical and experimental findings. J Nephrol. 2010;23(Suppl 16):S43–S48.
94. Porath, A, et al. Dead Sea water poisoning. Ann Emerg Med. 1989;18:187–191.
95. Liamis, G, et al. Medication-induced hypophosphatemia: A review. QJM. 2010;103:449–459.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]Chapter 125
Electrolyte Disorders
Hyperkalemia
Hyperkalemia, defined as serum potassium level greater than 5.0 mEq/L, is the most dangerous acute electrolyte abnormality, potentially leading to life-threatening arrhythmias and death. Although hyperkalemia may have vague and varied symptoms, it is usually totally asymptomatic, with cardiac arrest as its first “symptom.”1 Thus the diagnosis of hyperkalemia depends on paying specific attention to risk factors for impaired potassium excretion, such as dehydration and renal failure, along with an awareness of medications that cause potassium retention. Evaluation of the electrocardiogram (ECG) of patients at risk for this electrolyte disturbance is critical. Hyperkalemia can be rapidly progressive, and lifesaving interventions must be instituted at the earliest suspicion of toxicity.
Upwards of 98% of potassium in the body is contained intracellularly, whereas less than 2% remains circulating in the blood. Serum potassium concentration is normally between 3.5 and 5.0 mEq/L and is tightly regulated by the kidney. In the healthy state, at least 90% of potassium excretion occurs through the kidney; in the renally impaired state, the gastrointestinal tract may account for roughly 25% of excretion. Hyperkalemia usually develops from impaired renal excretion or increased release from cells; however, in advanced chronic kidney disease or end-stage renal disease, dietary intake of potassium may be a significant factor in its development.2
The most common cause of hyperkalemia is spurious elevation due to hemolysis during or after the blood draw. Thus an ECG should be used to assess for true hyperkalemia while another sample is analyzed. Most causes of true hyperkalemia are due to release from cells or renal insufficiency. Renal failure is the most common cause of confirmed hyperkalemia and is often compounded by medications that further impair renal potassium handling. Box 125-1 organizes the most common causes of hyperkalemia. The presence of one of these conditions may be the lone historical clue in hyperkalemia.3
Clinical Features
Hyperkalemia remains a difficult clinical diagnosis to make on clinical grounds alone. It is not uncommon for a patient with mild to moderate hyperkalemia to be identified during routine blood sampling for an unrelated condition. Patients with moderate to severe hyperkalemia may have gastrointestinal effects such as nausea, vomiting, and diarrhea often in association with their underlying disease. Neuromuscular findings, including muscle cramps, generalized weakness, paresthesias, tetany, and focal or global paralysis, may be seen in patients with severe hyperkalemia. The signs and symptoms of progressive muscle weakness, paresthesias, dyspnea, and depressed deep tendon reflexes are neither sensitive nor specific, nor do they appear reliably with a particular serum potassium level.3 Patients with severe hypokalemia may present with hemodynamic instability and cardiac arrhythmias requiring immediate intervention.
Diagnostic Strategies
The ECG is helpful in making the diagnosis of hyperkalemia and can be used in unstable patients to initiate treatment (Figs. 125-1 to 125-3). Classic electrocardiographic changes—the peaked T wave, flattened p wave with prolonged PR interval or a totally absent P wave, wide QRS, and sine wave pattern, portending imminent cardiac arrest—have been well described as appearing sequentially with rising serum potassium levels.4,5 Peaked T waves usually appear as serum potassium levels exceed 5.5 to 6.5 mEq/L; P wave disappearance and PR prolongation are common with levels above 6.5 to 7.5 mEq/L; and QRS prolongation is seen with potassium levels above 7.0 to 8.0 mEq/L. Although these changes may occur in only half the patients, recognition of these patterns when they are present is vital to rapid diagnosis and initiation of lifesaving treatment.6 A serum potassium level above 5.0 mEq/L is diagnostic of hyperkalemia, but the value itself does not always predict electrocardiographic changes or the degree of cardiotoxicity. Subtle electrocardiographic changes consistent with hyperkalemia should not be the sole reason to treat a stable patient not likely to have an elevated potassium concentration until serum levels have returned.3
Figure 125-2 Hyperkalemia in the same patient as in Figure 125-1 after potassium-lowering therapy has begun. Tall peaked T waves, decreased P wave.
Figure 125-3 The same patient as in Figures 125-1 and 125-2 after dialysis. The electrocardiogram is now normal.
Management
Patients with suspected or known hyperkalemia require intravenous access and continuous cardiac monitoring. The treatment of hyperkalemia is based on the clinical scenario combined with the 12-lead ECG and the laboratory potassium value. The treatment strategy consists of three main steps: stabilization of the cardiac membrane, shifting of potassium into the cells, and then removal of potassium from the body. In patients who do not require urgent treatment, lowering of total body potassium may be the only step necessary. A variety of treatment options are considered for the acute management of hyperkalemia, including calcium, insulin, beta2-adrenergic agonists, sodium bicarbonate, resins, and dialysis (Table 125-1).
Table 125-1
| TREATMENT | MEDICATION | FEATURES |
| Stabilize cardiac membrane | Calcium chloride (10 mL, maximum 20 mL) or calcium gluconate (10-30 mL), IV push | For wide QRS, restores the electrical gradient; does not decrease serum potassium |
| Shift potassium into cells | Insulin, 10 units, IV push, combined with 100 mL of 50% dextrose, IV push High-dose nebulized albuterol by face mask (15-25 mg by continuous inhalation) Bicarbonate 50-100 mL Normal saline 100-250 mL |
If severely acidotic In conjunction with nephrologist if dialysis dependent |
| Remove potassium from the body | Hemodialysis Normal saline and furosemide Ion exchange resin |
Emergently in cardiac arrest, urgently in renal failure; may delay if renal function is normal In patients with rhabdomyolysis or tumor lysis syndrome with intact urine output Not effective acutely |
Intravenous calcium is used to stabilize the cardiac membrane by restoring the electrical gradient. Calcium increases the depolarization threshold and the calcium gradient across the cardiac membrane, quieting myocyte excitability and increasing cardiac conduction speed, thus narrowing the QRS. Calcium does not decrease serum potassium levels, and its effect is rapid but transient. The dose is one ampule, or 10 mL of 10% calcium chloride solution. Some authors prefer calcium gluconate rather than calcium chloride on the basis of the reduced risk of tissue necrosis should it extravasate at the injection site.4,5 More than 10 mL of calcium gluconate will often be required as it contains only one third the calcium contained in calcium chloride. Calcium gluconate is also preferred in pediatric cases as well as in more chronic, less emergent hyperkalemic patients when a slow infusion is desired.
Nebulized albuterol by face mask is effective in shifting potassium into cells by stimulation of the Na+,K+-ATPase pump.6 Nebulized albuterol begins to take measurable effect after 15 minutes and lowers the serum potassium level by 0.5 to 1 mEq/L, depending on the dose.7 The effective dose is at least four times higher than that typically used for bronchodilation.8 Mild tachycardia is the main side effect but is generally well tolerated. The combination of nebulized albuterol and insulin with glucose appears to be additive, lowering serum potassium by a mean of 1.2 mEq/L.9
Saline infusions also stimulate the Na+,K+-ATPase pump, and only a few hundred milliliters is required for beneficial effects. Saline infusions are given judiciously in anuric patients and usually in consultation with the nephrologist. Sodium bicarbonate is effective only in hyperkalemic patients who are acidotic and has no benefit when it is used for hyperkalemia in nonacidotic patients.10 Sodium bicarbonate buffers hydrogen ions extracellularly while shifting potassium intracellularly but should be reserved for patients with confirmed acidosis. Although intravenous magnesium also can drive potassium intracellularly, it should never be used in hyperkalemia as most patients with elevated potassium levels are also at risk for concomitant hypermagnesemia.
Potassium can best be removed from the body acutely by hemodialysis. Hemodialysis effectively and reliably decreases serum potassium levels by at least 1 mEq/L in the first hour and another 1 mEq/L during the next 2 hours.7,11 It is the only reliable method of potassium removal that has been experimentally studied and should be instituted early in the treatment of life-threatening hyperkalemia in patients with renal failure. In patients with intact renal function, medical management alone is usually sufficient, even in extreme cases, and hemodialysis may not be necessary unless multiple medical modalities fail. There are no randomized trials addressing the use of diuretics such as furosemide in the emergent management of hyperkalemia, but in cases such as rhabdomyolysis or tumor lysis syndrome, it may be appropriate to use a normal saline infusion supplemented by furosemide to enhance diuresis and potassium excretion in the urine.
Cation exchange resins, such as sodium polystyrene sulfonate (Kayexalate), have not been shown to decrease the serum potassium level within the first 4 hours of treatment and should not be used in the acute management of hyperkalemia.12
Hyperkalemia is seen in diabetic ketoacidosis (DKA), although most hyperkalemic patients with DKA are actually total body deficient of potassium. In this insulin-deficient and acidotic state, serum potassium levels rise because of cellular shifts. Simply treating the patient’s underlying DKA will also treat the hyperkalemia. In fact, the mainstay of treatment of DKA—fluids and insulin—closely mirrors the treatment of hyperkalemia itself.13
As hyperkalemia progresses, the end result will be cardiopulmonary arrest due to ventricular fibrillation, pulseless electrical activity, or asystole. In a known or suspected hyperkalemic arrest, an approach beginning with standard advanced cardiac life support combined with the use of multiple potassium-lowering medications is rational.14,15 Epinephrine has been shown to lower potassium by 0.25 mEq/L.16,17 Calcium chloride is given immediately by intravenous push, followed by insulin and glucose. Bicarbonate should be given by bolus dose if the patient is believed to be acidotic. Emergent hemodialysis is recommended if it is readily available. Hemodialysis through central venous access can be used during ongoing cardiopulmonary resuscitation to acutely lower the serum potassium level and may result in return of spontaneous circulation with intact neurologic status despite prolonged resuscitative efforts and failure of conventional medications and defibrillation.18
Hypokalemia
Hypokalemia is the most common electrolyte abnormality encountered in clinical practice. When it is defined as a value of less than 3.5 mEq/L, hypokalemia is found in more than 20% of hospitalized patients and in 10 to 40% of patients treated with thiazide diuretics in the outpatient setting.19 Although hypokalemia is usually asymptomatic, severe cardiac dysrhythmias and rhabdomyolysis can occur secondary to potassium’s effect on the heart and muscle.20 The five most common causes of hypokalemia are renal losses, increased nonrenal losses, decreased potassium intake, intracellular shift, and endocrine etiologies (Box 125-2). Hypokalemia is often seen in association with hypomagnesemia, and patients with low serum potassium levels should be assumed to be hypomagnesemic also.20–22
Increased excretion of potassium, especially coupled with poor intake, is the most common cause of hypokalemia, and patients receiving diuretics represent the single most common patient group encountered in clinical practice. Hypokalemia from thiazide diuretics occurs through increases in distal sodium delivery in the nephron and by activation of the renin-angiotensin-aldosterone system. Thiazide diuretics are more likely than loop or osmotic diuretics to cause hypokalemia, but both the thiazide and loop diuretics block chloride-associated sodium and increase delivery of sodium to the collecting tubules. Hypokalemia is a common adverse effect of treatment with diuretics and may cause fatal arrhythmias and increase the risk of digitalis toxicity.23 In addition to diuretics, other drugs and disorders can cause significant renal potassium losses, including hyperaldosteronism, steroid excess, metabolic acidosis, DKA, renal tubular acidosis, and alcohol consumption. Penicillin and its synthetic derivatives, when they are given in large doses, promote renal potassium excretion by increasing sodium delivery to the distal nephron.
Administration of insulin may cause a reduction in serum potassium because of insulin’s ability to stimulate the Na+,K+-ATPase pump and move potassium intracellularly; hypokalemia can be a dangerous complication with intentional overdoses of insulin and during treatment of DKA. Although most patients with DKA present with high-normal or mildly elevated serum potassium levels, patients are usually 2 to 3 mEq/kg body weight deficient in total body potassium.24 Failure to appreciate this total body deficit—and to not begin potassium infusion once significant hyperkalemia has been ruled out and intact renal function confirmed—may lead to otherwise unexplained arrhythmias or cardiac arrest in patients hours after their initial therapy has begun.
Dietary potassium deficiency should be considered in the severely malnourished patient and the chronic alcoholic.25 When poor potassium intake is combined with increased nonrenal losses, severe hypokalemia can result. Hypokalemia can also result from an acute shift of potassium from the extracellular compartment into cells. This is most commonly seen in patients with metabolic alkalosis, in patients with hyperventilation, and in those patients taking medications such as beta-agonists or decongestants. Beta receptor stimulation can lead to hypokalemia, especially in patients using repetitive and high doses of beta-agonists for chronic obstructive pulmonary disease or asthma. Albuterol-induced hypokalemia can occur even at normal therapeutic doses.26 A standard dose of nebulized albuterol reduces serum potassium by 0.2 to 0.4 mEq/L, and a second dose taken within 1 hour has the potential to reduce it by almost 1 mEq/L. Patients with starvation or near-starvation may suffer from hypokalemia when they are fed because insulin secretion and increased cellular uptake can cause an acute intracellular migration of potassium.
Clinical Features
Hypokalemia is usually asymptomatic but can be manifested with nonspecific complaints, including palpitations, skeletal muscle weakness, easy fatigability, depression, and muscle pain. Although short periods of mild potassium depletion are typically well tolerated in healthy individuals, severe potassium depletion can result in serious cardiovascular instability, neurologic dysfunction, glucose intolerance, gastrointestinal symptoms, and renal failure as well as affect the acid-base balance in the body.27 The likelihood of symptoms appears to correlate with the rapidity of the decrease in serum potassium. In patients without underlying heart disease, abnormalities in cardiac conduction are extremely unusual, even when the serum potassium concentration is below 3.0 mEq/L. Paresthesias, depressed deep tendon reflexes, fasciculations, muscle weakness, and confusion can occur when the serum potassium level is less than 2.5 mEq/L. However, in patients with cardiac ischemia or heart failure, even mild to moderate hypokalemia increases the likelihood of cardiac arrhythmias secondary to potassium’s effect on the action potential. The data linking hypokalemia with arrhythmias and cardiac arrest in acute myocardial infarction are fairly strong, but the direct myocardial stimulatory effects of increased circulating epinephrine is a possible confounder.20 Hypokalemia is an independent risk factor contributing to reduced survival of cardiac patients and increased incidence of arrhythmic death. On the basis of available evidence, it appears best to attempt to maintain a serum potassium concentration above 4.5 mEq/L in patients having an acute myocardial infarction. Hypokalemic patients can demonstrate first- and second-degree heart block, atrial fibrillation, ventricular fibrillation, and asystole. Hypokalemia can also promote metabolic acidosis.
Hypokalemic periodic paralysis is a rare disorder characterized by potentially fatal episodes of muscle weakness through the involvement of the respiratory muscles. Life-threatening cardiac arrhythmias are managed by restoration of serum potassium levels into the normal range.28
Diagnostic Strategies
Hypokalemia is rarely suspected on the basis of clinical presentation, and the diagnosis is typically made by measurement of the serum potassium concentration during routine laboratory study. If there is any suspicion for hypokalemia or a patient presents with generalized weakness, palpitations, or arrhythmias, an ECG should be obtained. Just as a tall-peaked T wave is characteristic of hyperkalemia, a flattened T wave can be seen in hypokalemia. U waves, which are small deflections after the T wave, may also be seen (Figs. 125-4 and 125-5). The real danger of hypokalemia is that it may also cause a prolonged QT interval. Once the QT interval becomes longer than 500 milliseconds, the risk of malignant ventricular arrhythmias and torsades de pointes increases dramatically.28 Hypokalemia is also notorious for causing nonspecific ST and T wave changes. In addition, prolonged potassium depletion of even modest proportion can provoke or exacerbate kidney injury or hypertension. A severe degree of hypokalemia with paralysis is a potentially life-threatening medical emergency; measurement of relative urinary potassium excretion and an assessment of the acid-base status might help narrow the differential diagnosis in the emergency setting.
Figure 125-5 Electrocardiographic changes in hypokalemia.
Management
Potassium is an intracellular cation, so a low serum potassium level almost always reflects a significant total potassium deficit. When treating hypokalemia, one should remember that each 0.3 mEq potassium drop below normal correlates with an approximately 100 mEq total body deficit.29 Patients who have mild or moderate hypokalemia are usually asymptomatic or present with minor symptoms. These patients may only need oral potassium replacement therapy if they do not have nausea or vomiting as the cause of their hypokalemia. Oral replacement is available in liquid, powder, and tablet form. Potassium chloride is the most commonly used supplementation, and 40 to 60 mEq orally every 2 to 4 hours is typically well tolerated. If the cause of hypokalemia is not clear or the hypokalemia is severe and associated with profound weakness, obtain a spot urine potassium level before starting therapy to assess whether the patient’s kidneys are inappropriately wasting potassium from a renal or endocrine cause. Patients in whom severe hypokalemia is suspected should be immediately placed on a cardiac monitor and intravenous access secured.
Treatment of hypokalemia is essential in multiple populations of patients. Hypokalemia is arrhythmogenic, especially in the settings of acute myocardial infarction, high catecholamine states, and hypertrophied or dilated ventricles. Hypokalemia is an important independent risk factor for morbidity and mortality in patients with heart failure. Correction of serum potassium levels to between 4.0 and 5.0 mEq/L is important in these patients.30
Hypokalemia is associated with hypomagnesemia, and the severity of the hypokalemia correlates with a similar degree of hypomagnesemia.31 Magnesium replacement should usually accompany potassium repletion. Unless the patient receives at least 0.5 g/hr of magnesium sulfate along with potassium replacement, potassium will not move intracellularly and the patient will lose potassium through excretion.22 Correction of large potassium deficits may require several days, and oral and intravenous replacement can occur simultaneously.
Hypernatremia
Hypernatremia is defined as a serum sodium concentration above 145 mEq/ L and is usually associated with a poor prognosis. It is uncommon in previously normal patients, and in adults it is almost exclusively due to a total body water deficit.32 Most hypernatremic patients have either an impaired sense of thirst or no access to water. Thus elders, infants, patients in coma or with mental impairment, and those who are intubated and paralyzed are at highest risk for this disorder.33,34 Hypernatremia can be divided into three physiologic pairings (Box 125-3). Diabetes insipidus, a condition that results in insufficient production of or lack of response to antidiuretic hormone, can lead to life-threatening hypernatremia35 (Box 125-4).
Clinical Features
Hypernatremia is a disease seen predominantly in elders, but it can also be seen in patients who depend on others to provide them with water, including infants, intubated patients, and persons with mental debilitation.36,37 In addition, patients will also have multifactorial causes leading to severe hypernatremia.38 Patients may complain of polyuria or polydipsia or have obvious causes of extrarenal fluid losses; others may have no complaints at all.
Diagnostic Strategies
A patient’s total body water is usually calculated by multiplying the patient’s body weight in kilograms times 0.6. However, because of percentage body fat differences based on the age and sex of the patient, it is more accurate to use the correction factors listed in Table 125-2.
Table 125-2
| POPULATION | TOTAL BODY WATER |
| Children and adult men | Body weight (kg) × 0.6 |
| Adult women | Body weight (kg) × 0.5 |
| Elderly men | Body weight (kg) × 0.5 |
| Elderly women | Body weight (kg) × 0.45 |
Management
The treatment of hypernatremia has three interdependent goals: first, to quickly correct underlying shock, hypoperfusion, or significant hypovolemia with normal saline; second, to treat the underlying cause of hypernatremia, such as fever, vomiting, or diabetes insipidus; and third, to carefully lower the serum sodium level, usually by replacement of the body’s total water deficit.39 Until hypoperfusion and hypovolemia are corrected, homeostatic mechanisms for sodium balance will promote sodium resorption to maintain intravascular volume, even at the expense of the serum sodium concentration.
The rate of correction in hypernatremia is extremely important to minimize morbidity and mortality. In adult patients who have had hypernatremia during a short time as a result of sodium loading, “rapid correction” at 1 to 2 mEq/hr lowering of serum sodium appears relatively safe.39,40 However, most adult patients have hypernatremia during days to weeks. In this group of patients, serum sodium concentration should be slowly corrected at no more than 0.5 mEq/hr or 10 to 12 mEq/day.
Normal saline can typically be started for volume replacement until the patient is hemodynamically stable and then changed to half-normal saline at 100 mL/hr once vital signs have normalized. The treatment of central diabetes insipidus with desmopressin (DDAVP) is an effective means of improving polyuria and hypernatremia; initial doses in the acute setting range from 1 to 2 µg.33
Hyponatremia
Hyponatremia, defined as serum sodium concentration of less than 135 mEq/L, is the second most common electrolyte abnormality encountered in clinical practice.37 It is important to recognize hyponatremia because of its potential morbidity and also because it can be a marker of underlying disease. The most common causes of severe hyponatremia in adults are therapy with thiazides, the postoperative state including transurethral prostatectomy, the syndrome of inappropriate secretion of antidiuretic hormone (SIADH), polydipsia in psychiatric patients, and unintentional water intoxication. Gastrointestinal fluid loss, ingestion of overly dilute formula, accidental ingestion of excessive water, and receipt of multiple tap-water enemas are the main causes of severe hyponatremia in infants and children. Most patients presenting to the emergency department (ED) with hyponatremia are asymptomatic and do not require emergent therapy. If symptoms are present, they are typically based on the degree of hyponatremia and how acutely the hyponatremia developed. Symptoms range from headache, nausea, and vomiting to confusion, seizures, and coma. There are two groups of hyponatremic patients that will require treatment with either normal saline or hypertonic saline: (1) severe but asymptomatic hyponatremia with a sodium level of 110 mEq/L or less and (2) acute symptomatic hyponatremia with a sodium level below 120 mEq/L.
Central nervous system (CNS) damage due to hyponatremia may be caused by cerebral edema and increased intracranial pressure, by osmotic fluid shifts during overly aggressive treatment, or by both. When they are subjected to a hyponatremic environment, neurons become depleted of sodium and potassium in an attempt to limit their own osmolarity to prevent intracellular fluid shifts that would lead to cerebral edema. If fluid therapy raises extracellular sodium levels too quickly, fluids shift out of neurons and diffuse demyelination may occur, leading to flaccid paralysis and often death due to a syndrome most commonly referred to as central pontine myelinolysis, although it is more accurately labeled the osmotic demyelinating syndrome.41
Although hyponatremia has many causes, they fall into four general categories: pseudohyponatremia, hyponatremia with dehydration and decreased extracellular volume, hyponatremia with increased extracellular volume, and euvolemic hyponatremia with increased total body water (Box 125-5).
