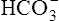Chapter 22
Electrolyte and Acid-Base Regulation
After reading this chapter you will be able to:
• Explain why sodium reabsorption tends to take priority over reabsorption of other ions, and why this may lead to acid-base and electrolyte imbalances under certain conditions
• Describe how the kidney reabsorbs sodium through primary and secondary active transport
• Explain why plasma chloride ion imbalances lead to acid-base and potassium ion imbalances
• Explain why acid-base imbalances cause potassium ion imbalances
• Explain why potassium imbalances cause acid-base imbalances
• Describe how chloride and potassium ion reabsorption is tied to sodium ion reabsorption
• Describe how the kidney either reabsorbs or excretes bicarbonate to compensate for respiratory acid-base disturbances
• Explain why urinary buffers must be present for the kidney to secrete hydrogen ions in conditions of acidemia
• Explain why diuretics that block sodium reabsorption tend to cause potassium and chloride depletion
• Explain why dehydration leads to alkalemia
• Explain why individuals who are chronically hypercapnic tend to be hypochloremic
• Explain how renal failure affects acid-base, fluid, and electrolyte balance
Renal Electrolyte Regulation
Sodium, potassium, and chloride regulation are intimately related to acid-base regulation. The regulation of these electrolytes is also important for maintaining normal fluid volume, nerve-impulse transmission, and muscle contraction. For the kidney to reabsorb water and other substances from the tubular lumen into the peritubular capillary blood, the substance first must be transported across the tubular epithelium into the renal interstitial fluid and then through the peritubular capillary membrane into the blood. Substances cross the tubular epithelial cells either by active transport through the cell membranes themselves (transcellular pathway) or by passive diffusion through the junctional spaces between the cells (paracellular pathway).1 Active transport requires adenosine triphosphate (ATP) for an energy source and can move a substance “uphill” across a cell membrane, against its concentration gradient. Such transport is called primary active transport if it is coupled directly to the ATP energy source; it is called secondary active transport if it is indirectly linked to the energy source, for example, if the process of primary active transport of one ion creates an electrical gradient that affects the movement of an oppositely charged ion.
Sodium and Chloride Regulation
Sodium is the major osmotically active substance in the extracellular fluid, which means wherever sodium goes, water follows. Na+ along with Cl−, the most abundant anion, determine the extracellular fluid volume.2 About 26,000 mEq of Na+ passes through the glomerular membrane into the tubular filtrate daily. However, daily Na+ intake averages only about 150 mEq.1 Therefore the kidney’s main job is to reabsorb Na+, not to excrete it. Because of the major role of Na+ in maintaining fluid balance, the body places a priority on Na+ reabsorption, even at the expense of imbalances in other electrolytes. As noted in Chapter 21, greater than 99% of the Na+ in the filtrate is reabsorbed. Na+ is reabsorbed from the tubular filtrate by two mechanisms: (1) primary active transport and (2) secondary active secretion of H+ and K+ ions.
Primary Active Transport of Sodium
Primary active transport accounts for most Na+ reabsorption from the tubular filtrate and occurs in all tubules except the descending loop of Henle.1 On the nonluminal side of the tubular epithelial cell (Figure 22-1), the sodium-potassium-adenosine triphosphatase (Na+-K+-ATPase) pump actively transports Na+ ions out of the cell, into the interstitial fluid, and ultimately into the peritubular capillary blood. The membrane protein forming this pump hydrolyzes ATP molecules to generate the energy necessary to actively transport Na+ ions out of the cell. K+ ions are simultaneously transported into the cell from the interstitial fluid, but more Na+ ions are pumped out of the cell than K+ ions are pumped in. This process keeps Na+ concentration in the tubule cell very low and creates a negatively charged intracellular environment.
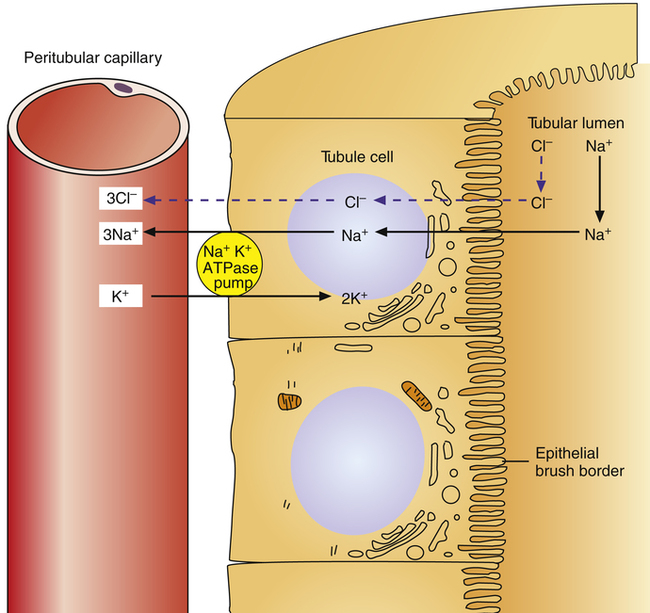
Because [Na+] is comparatively much higher in the tubular filtrate, and Na+ is a positively charged ion, both chemical diffusion and electrostatic forces favor its movement from the tubular lumen into the cell, down its concentration and electrostatic gradients.1 Specialized sodium-carrier proteins in the luminal membrane of the tubule cell facilitate this process by binding with Na+ ions and releasing them inside of the tubule cell. Cl− ions passively follow Na+ ions, which maintains the filtrate’s electrical neutrality. That is, the transport of the positively charged Na+ ion out of the filtrate into the tubular cell leaves the tubular lumen negatively charged with respect to the extracellular fluid and the blood; this electrostatic gradient causes Cl− (the most abundant anion in the filtrate) to diffuse passively through paracellular pathways (between the tubule cells) into the interstitial space and ultimately into the capillary blood.1 Cl− diffusion also occurs because of a concentration gradient; that is, Na+ transport out of the filtrate creates an osmotic gradient for water reabsorption from the tubular lumen, which concentrates Cl− ions in the filtrate. Cl− thus passively diffuses out of the filtrate in response to both electrostatic and chemical diffusion forces.1
Secondary Active Secretion of Hydrogen and Potassium
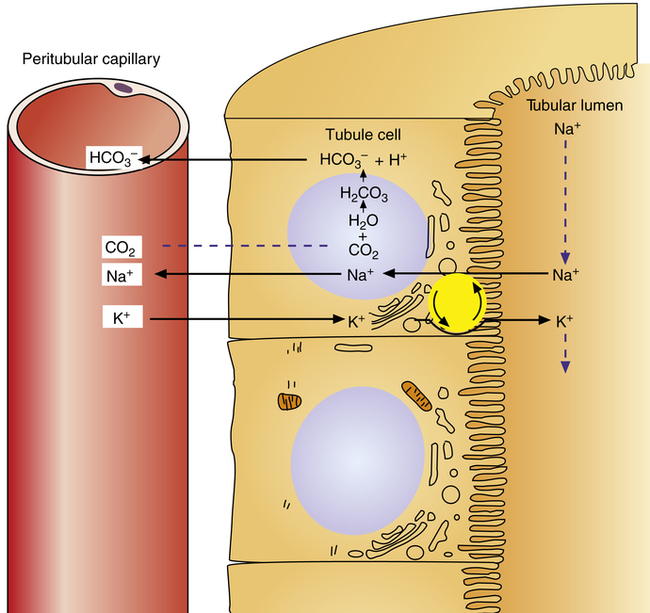
Reabsorption of Na+ by way of the active secretion of H+ and K+ ions is a more complex process. For H+ secretion (Figure 22-2), the process proceeds in the following manner: First, carbon dioxide (CO2) from the peritubular capillary blood diffuses into the tubular cells, where it reacts with water (in the presence of carbonic anhydrase) and forms H+ ions. The H+ ion in the tubule cell and Na+ ion in the filtrate simultaneously combine with opposite ends of a protein-carrier molecule in the luminal border of the cell membrane. Na+ diffusion into the cell, down its concentration and electrostatic gradients (described previously), provides the energy for H+ transport into the filtrate (see Figure 22-2). This process is called countertransport because the transported ions move in opposite directions. Figure 22-2 shows that the < ?xml:namespace prefix = "mml" />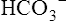 ion generated by the reaction between CO2 and water accompanies Na+ transport out of the cell and into the peritubular capillaries. In this way, Na+ reabsorption from the filtrate occurs without Cl− reabsorption. A similar countertransport mechanism is involved in the secondary active secretion of K+ into the tubules in exchange for Na+ (Figure 22-3). This mechanism of potassium secretion is more likely to occur in the presence of alkalemia when H+ is scarce; this is the reason alkalemia tends to cause K+ depletion (hypokalemia).
ion generated by the reaction between CO2 and water accompanies Na+ transport out of the cell and into the peritubular capillaries. In this way, Na+ reabsorption from the filtrate occurs without Cl− reabsorption. A similar countertransport mechanism is involved in the secondary active secretion of K+ into the tubules in exchange for Na+ (Figure 22-3). This mechanism of potassium secretion is more likely to occur in the presence of alkalemia when H+ is scarce; this is the reason alkalemia tends to cause K+ depletion (hypokalemia).
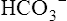
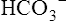
Secondary Active Transport of Chloride
In most of the tubular segments, Cl− ions are reabsorbed with Na+ ions by passive diffusion as described earlier. In the thick segment of the loop of Henle, Cl− ions are transported in a secondary active transport process also known as cotransport. In this mechanism, the same carrier protein referred to previously that combines with Na+ in the luminal tubular membrane simultaneously combines with Cl−. As Na+ diffuses down its electrochemical gradient into the tubule cell, it pulls Cl− with it. This secondary active transport of Cl− requires no ATP energy source; it simply uses the force of the Na+ ions’ “downhill” diffusion into the cell to energize the process. In addition to Cl−, a significant amount of K+ is reabsorbed with Na+ through the same mechanism; for each Na+ ion, two Cl− ions and one K+ ion are cotransported by a membrane carrier protein known as the 1 sodium, 2 chloride, 1 potassium cotransporter.1
Potassium Regulation
Precise control of extracellular [K+] is extremely important because cardiac muscle cells are very sensitive to slight concentration changes; an elevation of only 3 to 4 mEq/L in the plasma [K+] can cause lethal arrhythmias.1 The maintenance of K+ balance depends mainly on renal excretion, which must adapt quickly to large variations in K+ intake to prevent lethal hyperkalemia. Because more than 98% of total body K+ is in the cells, the intracellular compartment is a K+ reservoir in hyperkalemia and a source of K+ in hypokalemia; redistribution of K+ between intracellular and extracellular fluid compartments is an important part of controlling extracellular K+ levels.1 After a normal meal, a person’s K+ level would increase to lethal levels if most of the K+ did not rapidly move to the intracellular
CLINICAL FOCUS 22-1 Effect of Hypochloremia on Acid-Base Status and Potassium Balance
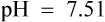
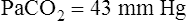
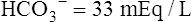
Her plasma electrolyte results are as follows:
| Measured | Normal Range | |
| Na+ | 140 mEq/L | 137-147 mEq/L |
| K+ | 3.0 mEq/L | 3.5-4.8 mEq/L |
| Cl− | 89 mEq/L | 98-105 mEq/L |
| CO2 | 34 mEq/L | 25-33 mEq/L |
What are the mechanisms contributing to this patient’s metabolic alkalosis?
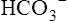
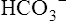
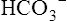
compartment. K+ uptake by the cells, and thus its lowered concentration in extracellular fluid, is stimulated by insulin, aldosterone, and beta-adrenergic drugs, all of which activate the Na+,K+-ATPase pump present in all cell membranes.1 For this reason, albuterol (a beta2 agonist) is sometimes administered to individuals with life-threatening hyperkalemia.
Potassium Reabsorption
About 65% of K+ in the filtrate is reabsorbed into the blood by cotransport with Na+ and Cl− in the proximal tubules (by way of the 1 sodium, 2 chloride, 1 potassium cotransporter).1 Anything that blocks Na+ reabsorption, such as a loop diuretic, impairs K+ (and Cl−) reabsorption; overuse of loop diuretics can cause hypokalemia and hypochloremia. Approximately another 25% of the filtrate’s K+ is reabsorbed by the same cotransport mechanism in the ascending limb of the loop of Henle. The small amount of K+ remaining in the filtrate after it leaves the loop of Henle is almost completely reabsorbed by the distal tubule and cortical segments of the collecting ducts. However, the kidneys must excrete a quantity of K+ in the urine at least equal to the daily K+ intake to avoid hyperkalemia.
Control of Potassium Excretion
Because it greatly influences cardiac impulse conduction (see Chapter 18 and nervous system function, extracellular fluid [K+] regulation is especially important. K+ ion excretion is controlled by (1) extracellular fluid [K+] and (2) aldosterone secretion by the adrenal gland cortex.2
A rising extracellular [K+] directly stimulates tubular K+ secretion, especially when [K+] is already above the normal range. It does so by stimulating the Na+,K+-ATPase pump, increasing K+ uptake through the nonluminal tubule cell membrane. Conversely, low extracellular [K+] depresses the rate of K+ secretion. Extracellular fluid [K+] also directly affects the rate of aldosterone secretion by the adrenal cortex.1 Aldosterone also activates the Na+,K+-ATPase pump, pumping more K+ into the tubular cell as it pumps more Na+ out of the cell. A functioning aldosterone system is extremely important in regulating extracellular [K+]. Diseases that abnormally increase aldosterone secretion, such as primary aldosteronism, greatly increase K+ secretion into the filtrate, causing severe hypokalemia. Conversely, diseases that destroy the adrenal glands, such as Addison’s disease, stop aldosterone production and produce severe hyperkalemia.
Hydrogen Ion Regulation
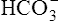
Tubular Hydrogen Ion Secretion
All parts of the nephron’s tubular system actively secrete H+ except the thin segments of the loop of Henle. About 95% of H+ secretion occurs via secondary active transport; the remaining 5% occurs via primary active transport.1
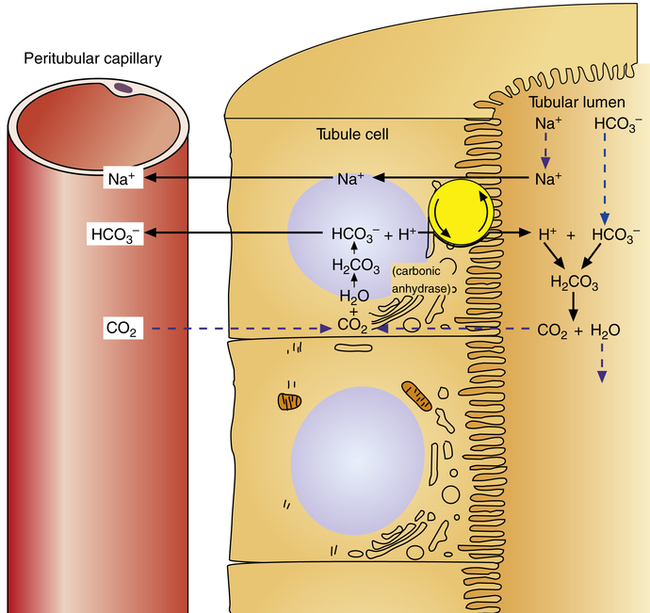
Secondary active secretion of H+ occurs mainly in the proximal tubules, loop of Henle, and early distal tubules. This process is the same as that described for the Na+ reabsorption mechanism (see Figure 22-2). First, CO2 in the peritubular blood plasma freely diffuses into the tubular cell, where it reacts instantly with water under the influence of carbonic anhydrase. As a result, carbonic acid forms and dissociates into 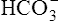 and H+ ions. The H+ ions are secreted into the tubular filtrate by the Na+-H+ countertransport mechanism described previously (see Figure 22-2). The luminal brush border of the tubule cell is impermeable to
and H+ ions. The H+ ions are secreted into the tubular filtrate by the Na+-H+ countertransport mechanism described previously (see Figure 22-2). The luminal brush border of the tubule cell is impermeable to 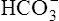 ; therefore
; therefore 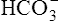 cannot follow H+, and intracellular [
cannot follow H+, and intracellular [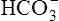 ] increases. In this way, a concentration gradient for
] increases. In this way, a concentration gradient for 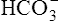 diffusion is formed, and it diffuses out of the cell along with newly reabsorbed Na+ into the interstitial fluid and peritubular capillary blood. Generally, for every H+ ion secreted, one
diffusion is formed, and it diffuses out of the cell along with newly reabsorbed Na+ into the interstitial fluid and peritubular capillary blood. Generally, for every H+ ion secreted, one 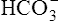 ion enters the blood.
ion enters the blood.
Primary active secretion accounts for a small amount of H+ secretion, occurring in the late distal tubule and the entire length of the collecting duct. In this process, a specific transport protein in the tubule cell luminal membrane, hydrogen-transporting ATPase, pumps the H+ formed by the reaction between CO2 and water into the filtrate. The only difference between this process and secondary active secretion is that H+ ions are transported across the luminal membrane by an active hydrogen pump instead of by countertransport with Na+ ions. Although this mechanism accounts for only about 5% of the total H+ secreted, it is much more powerful than the secondary active secretion mechanism. Primary active transport can concentrate H+ ions in the filtrate 900-fold compared with a maximum of about 3-fold to 4-fold for the secondary process.1 This extreme H+ ion concentrating ability can reduce urine pH to a maximum of about 4.5. All tubular H+ secretion stops below this pH level.1
Hydrogen Ion Secretion and Bicarbonate Ion Reabsorption
The H+ ion secreted into the filtrate immediately reacts with the 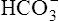 ion (see Figure 22-2). (Remember that numerous
ion (see Figure 22-2). (Remember that numerous 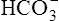 ions are filtered out of the blood at the glomerulus.) When the
ions are filtered out of the blood at the glomerulus.) When the 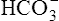 ion buffers the secreted H+ ion, H2CO3 and ultimately CO2 and water are formed. All cell membranes are freely permeable to CO2; CO2 immediately diffuses into the tubular cell where it instantly reacts with water under the influence of carbonic anhydrase. Again, the CO2 hydrolysis reaction produces a
ion buffers the secreted H+ ion, H2CO3 and ultimately CO2 and water are formed. All cell membranes are freely permeable to CO2; CO2 immediately diffuses into the tubular cell where it instantly reacts with water under the influence of carbonic anhydrase. Again, the CO2 hydrolysis reaction produces a 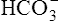 ion; the intracellular concentration of
ion; the intracellular concentration of 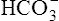 thus increases, and it diffuses into the interstitial fluid. In this way, the filtrate’s
thus increases, and it diffuses into the interstitial fluid. In this way, the filtrate’s 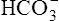 ions are “reabsorbed” into the blood, although the reabsorbed
ions are “reabsorbed” into the blood, although the reabsorbed 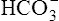 ion is not the same ion that was in the filtrate.
ion is not the same ion that was in the filtrate.
Normally, H+ secretion slightly exceeds the rate of 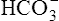 filtration at the glomerulus. This means that the titration of
filtration at the glomerulus. This means that the titration of 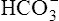 against H+ in the tubular filtrate is incomplete, leaving a slight excess of H+. Thus, the excreted urine is usually slightly acidic.1
against H+ in the tubular filtrate is incomplete, leaving a slight excess of H+. Thus, the excreted urine is usually slightly acidic.1
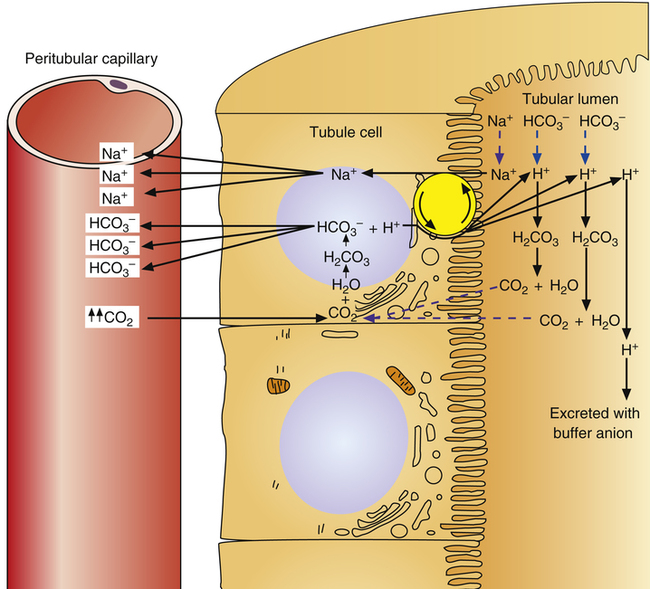
Renal Compensation for Respiratory Acidosis
In respiratory acidosis, the ratio of dissolved CO2 to 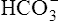 ions increases (see Chapter 10). As a result, the rate of H+ secretion increases relative to the rate of
ions increases (see Chapter 10). As a result, the rate of H+ secretion increases relative to the rate of 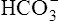 filtration into the tubules (Figure 22-4). When arterial blood PCO2 is elevated, relatively more CO2 diffuses into the tubule cell from the blood, generating more H+ ions, which are secreted into the filtrate. All available
filtration into the tubules (Figure 22-4). When arterial blood PCO2 is elevated, relatively more CO2 diffuses into the tubule cell from the blood, generating more H+ ions, which are secreted into the filtrate. All available 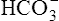 ions react with these H+ ions and are reabsorbed, as described previously. The remaining H+ ions are excreted in the urine, carrying with them a different urinary buffer anion from the filtrate (discussed in a later section). In this way, for every H+ secreted, one
ions react with these H+ ions and are reabsorbed, as described previously. The remaining H+ ions are excreted in the urine, carrying with them a different urinary buffer anion from the filtrate (discussed in a later section). In this way, for every H+ secreted, one 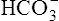 ion is gained by the blood, which is the desired response to acidosis (i.e., elimination of acid and retention of base). In this way the normal kidney compensates for respiratory acidosis.
ion is gained by the blood, which is the desired response to acidosis (i.e., elimination of acid and retention of base). In this way the normal kidney compensates for respiratory acidosis.
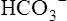
Renal Compensation for Respiratory Alkalosis
In respiratory alkalosis, the rate of 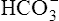 filtration into the tubules exceeds the rate of tubular H+ secretion. Because of their high numbers, not all
filtration into the tubules exceeds the rate of tubular H+ secretion. Because of their high numbers, not all 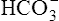 ions in the filtrate have a H+ ion with which to react (Figure 22-5). Because of the impermeability of the luminal tubular membrane to
ions in the filtrate have a H+ ion with which to react (Figure 22-5). Because of the impermeability of the luminal tubular membrane to 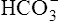 , these ions are trapped in the filtrate and pass into the urine, carrying with them positive ions such as Na+ or K+. This is the desired response to alkalosis (i.e., elimination of base and reduced secretion of acid) and is the mechanism whereby the kidney compensates for respiratory alkalosis.
, these ions are trapped in the filtrate and pass into the urine, carrying with them positive ions such as Na+ or K+. This is the desired response to alkalosis (i.e., elimination of base and reduced secretion of acid) and is the mechanism whereby the kidney compensates for respiratory alkalosis.
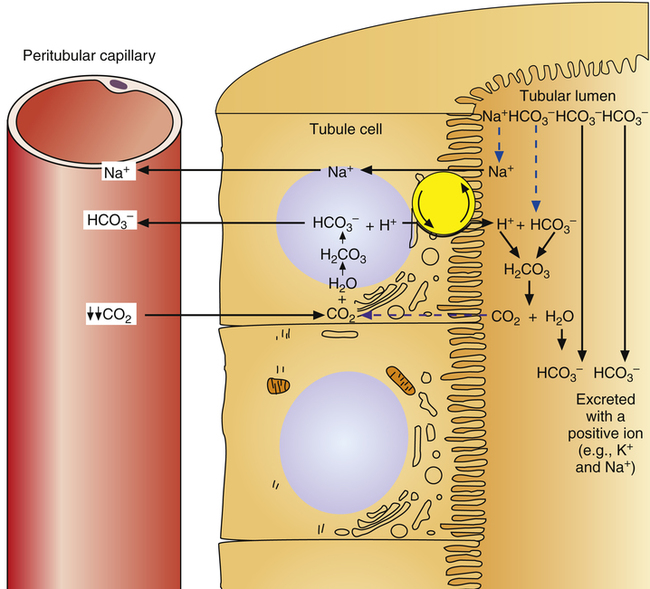
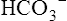
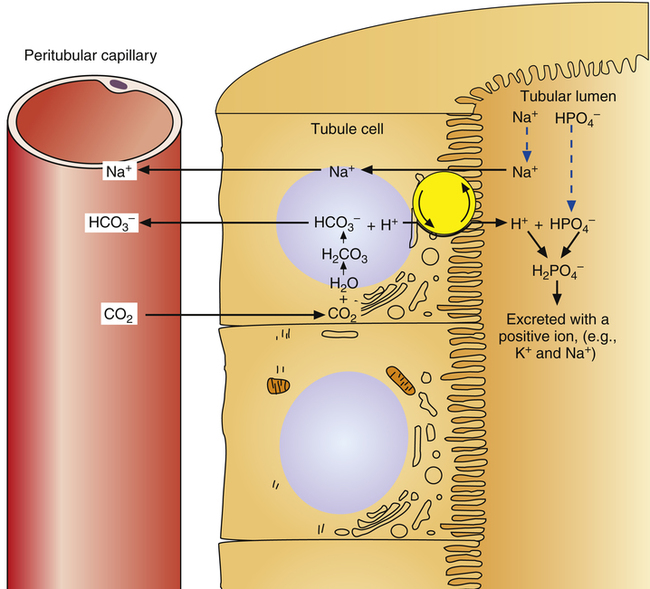
Role of Urinary Buffers
Not many H+ ions can be excreted in the urine in the free form. Free H+ ions rapidly reduce the filtrate pH to 4.5, a level below which H+ secretion mechanisms cannot function.1 Therefore, filtrate buffers are necessary for normal H+ ion excretion. The primary urinary buffer is 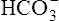 as already described. The two other major buffer systems that transport excess H+ ions in the urine are the phosphate buffer system and ammonia buffer system.
as already described. The two other major buffer systems that transport excess H+ ions in the urine are the phosphate buffer system and ammonia buffer system.
Phosphate Buffers
Phosphate buffers in the tubular filtrate include 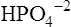 and
and 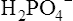 . Because these buffer anions are poorly reabsorbed from the filtrate, they become concentrated and powerful as water is reabsorbed. The phosphate buffer system’s pKa is 6.8, a pH level close to that of the tubular filtrate.1 This means the phosphate buffer system operates in its most effective range in the tubular filtrate (see Chapter 10). The function of the phosphate buffer system is illustrated in Figure 22-6.
. Because these buffer anions are poorly reabsorbed from the filtrate, they become concentrated and powerful as water is reabsorbed. The phosphate buffer system’s pKa is 6.8, a pH level close to that of the tubular filtrate.1 This means the phosphate buffer system operates in its most effective range in the tubular filtrate (see Chapter 10). The function of the phosphate buffer system is illustrated in Figure 22-6.
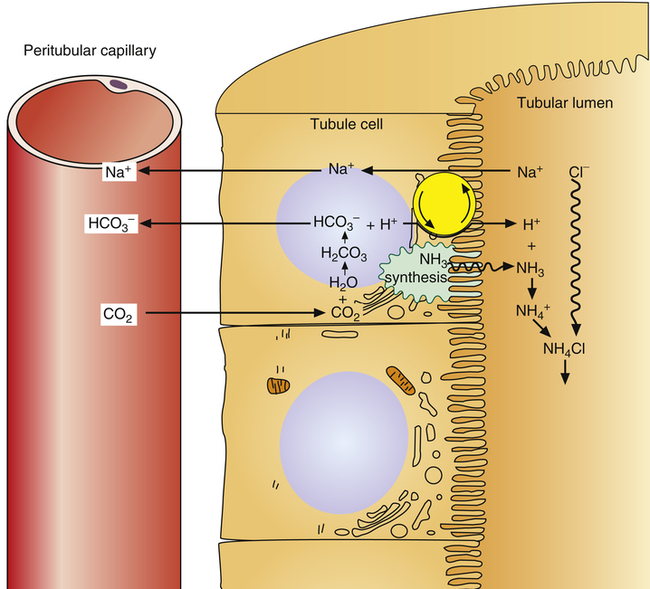
Ammonia Buffer System
The ammonia buffer system consists of the ammonia molecule (NH3) and ammonium ion (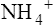 ). Most of the tubular epithelium synthesizes NH3, which diffuses into the filtrate (Figure 22-7). The NH3 molecule buffers the H+ ion in the filtrate, forming NH4+. Every time this process occurs, the filtrate NH3 concentration decreases, causing more NH3 to diffuse out of the tubule cell, which stimulates more NH3 synthesis. In this way, the amount of free H+ in the filtrate controls the rate of NH3 synthesis.
). Most of the tubular epithelium synthesizes NH3, which diffuses into the filtrate (Figure 22-7). The NH3 molecule buffers the H+ ion in the filtrate, forming NH4+. Every time this process occurs, the filtrate NH3 concentration decreases, causing more NH3 to diffuse out of the tubule cell, which stimulates more NH3 synthesis. In this way, the amount of free H+ in the filtrate controls the rate of NH3 synthesis.
Potassium and Acid-Base Balance
Hypokalemia Caused by Alkalosis
The kidney’s main task in Na+ ion regulation is to reabsorb it from the filtrate. (Greater than 99% of it is reabsorbed.) The kidneys reabsorb sodium even if this causes other electrolyte imbalances. For example, in a state of alkalosis, the tubule cells rely more heavily on the potassium-secreting mechanism to reabsorb Na+ (see Figure 22-3). In other words, the relative shortage of H+ ions forces the kidney to secrete more K+ ions; this is a major reason alkalosis tends to cause hypokalemia. Also, in alkalosis, the glomerulus filters more  ions into the filtrate than can be matched by tubular H+ ion secretion (see Figure 22-5). Because H+ is scarce and Na+ reabsorption is a high priority, the tubules secrete more K+ than normal to accompany the excess
ions into the filtrate than can be matched by tubular H+ ion secretion (see Figure 22-5). Because H+ is scarce and Na+ reabsorption is a high priority, the tubules secrete more K+ than normal to accompany the excess  in the urine.
in the urine.
Alkalosis Caused by Hypokalemia
Similarly, a state of hypokalemia also can cause alkalosis. For example, diuretic therapy or lack of dietary K+ intake may cause hypokalemia. The tubules must then secrete inappropriately large amounts of H+ ions to reabsorb Na+ via the countertransport mechanism (see Figure 22-2), which normally accounts for a relatively small percentage of Na+ reabsorption. Consequently, alkalosis occurs. Hypokalemic alkalosis is compounded by a coexisting state of dehydration.3 Dehydration produces a profound stimulus for Na+ reabsorption, which (coupled with hypokalemia) intensifies the kidney’s need to secrete H+. Overuse of diuretic drugs can lead to such a condition.
Intracellular Mechanisms Affecting Plasma Hydrogen Ion Concentration and Potassium Ion Concentration
The relationship of [K+] and plasma pH has a further explanation at the cellular level in the body.4 These interrelationships are illustrated in red blood cells in Figure 22-8 (the same relationships are also present in all of the body’s cells). Plasma hypokalemia creates a concentration gradient that favors intracellular K+ diffusion out of the cell (Figure 22-8, A). The resulting intracellular negativity causes a net movement of plasma H+ into the cell. The reduction in plasma [H+] produces alkalemia. Conversely, in alkalemia (Figure 22-8, B), intracellular H+ ions diffuse out of the cell in response to a H+ ion concentration gradient, and plasma K+ diffuses into the cell. The movement of K+ into the cell causes hypokalemia.
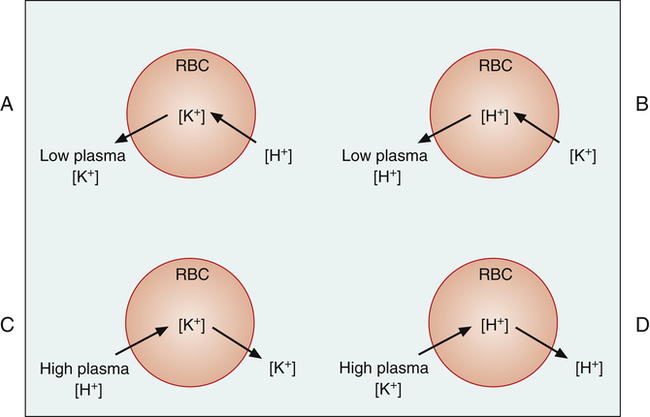
Similarly, in acidemia (Figure 22-8, C), the plentiful plasma H+ diffuses into the cell, and K+ diffuses out of the cell, producing hyperkalemia. Conversely, hyperkalemia causes K+ to move into the cell and H+ to move out of the cell, causing acidemia (Figure 22-8, D).
Because of these intracellular-extracellular K+ shifts, the plasma [K+] on a clinical laboratory report must be interpreted carefully. In the examples in Figure 22-8, total body stores of K+ do not change; K+ merely shifts between fluid compartments. However, consider the acidotic condition in Figure 22-8, C, in which plasma [K+] increases. In response to the increasing [K+], the kidneys excrete more K+, causing more K+ to diffuse out of the cells into the plasma. Thus, plasma [K+] may be normal or high, but total body K+ stores may actually be depleted. Similarly, low [K+] in alkalemia (see Figure 22-8, B) does not always mean the body’s K+ stores are depleted.
Speed of Renal Acid-Base Correction
The blood buffers can readjust pH in seconds, whereas the lungs can readjust pH in minutes. In contrast, the renal mechanisms require hours to days to readjust pH. Although the kidneys are slow to act, they are much more powerful and complete in their ability to restore a normal pH. In contrast to the blood buffers and respiratory system, the kidneys can continue to act until pH is readjusted almost exactly to normal rather than a certain percentage of the way.1 The kidneys are important, not for their speed but for their ability to compensate for acid-base disturbances more completely.
Harmful Effects of Fluid, Electrolyte, and Acid-Base Imbalances
Volume Depletion
Chloride and Potassium Imbalances
Causes of hypochloremia and hypokalemia are listed in Boxes 22-1 and 22-2. With regard to hypochloremia, patients with congestive heart failure are often on low-salt diets and taking diuretic drugs to prevent fluid retention and edema; this the body’s Cl− stores. Critically ill patients on ventilators usually have nasogastric suction tubes placed that empty the stomach to prevent regurgitation and aspiration of vomitus; this causes loss of gastric HCL. In another example, a patient with COPD and chronic hypercapnia may have a paradoxically alkalotic pH, which would appear to be “overcompensation.” However, this condition is not overcompensation for respiratory acidosis; instead, it reflects chronic Cl− depletion as a result of increased NH3 buffering activity (loss of NH4Cl in the urine). Alkalosis occurs because the lack of Cl− ions causes the kidney to rely more heavily on H+ ion secretion to reabsorb Na+.
Severe diarrhea (see Box 22-2) may cause hypokalemia and acidosis because the lower bowel is rich in K+ and 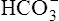 ions. Respiratory alkalosis is sometimes a complication of inappropriate ventilator therapy; regardless of its cause, alkalosis causes hypokalemia because the kidneys respond to low [H+] by secreting more K+ (see Figure 22-5).
ions. Respiratory alkalosis is sometimes a complication of inappropriate ventilator therapy; regardless of its cause, alkalosis causes hypokalemia because the kidneys respond to low [H+] by secreting more K+ (see Figure 22-5).
Harmful Effects of Potassium Imbalances, Acidosis, and Alkalosis
Box 22-3 lists harmful effects of K+ imbalances, and Box 22-4 lists adverse effects of acidosis and alkalosis. The main clinical effect of acidosis is central nervous system depression, whereas the major effect of alkalosis is overexcitability of the central and peripheral nervous systems.
Effects of Renal Failure
Renal failure falls into two general physiological categories: (1) acute renal failure and (2) chronic renal failure.1 Renal failure usually interferes with electrolyte and water balance, causing sodium and fluid retention. In severe instances, the kidney fails to excrete adequate amounts of fixed acids and nitrogenous waste products. In complete renal failure without treatment, patients soon die.
Acute Renal Failure
Acute renal failure falls into three categories:1 (1) decreased blood supply, as might occur in heart failure or hemorrhage; (2) intrarenal failure, or abnormalities within the kidney itself; and (3) postrenal failure, which refers to obstruction to urine outflow from the kidney.
Intrarenal Failure
Intrarenal failure includes conditions that injure the glomerular capillaries, the tubular epithelium, and the renal interstitium. Acute glomerulonephritis is an example of a disease that is usually caused by an abnormal immune reaction in which the body develops antibodies to group A beta streptococci microorganisms.1 Renal manifestations of the disease usually follow streptococcal infections elsewhere in the body, such as the throat or skin. The streptococcal antigen-antibody complex lodges in the basement membrane of the glomerulus; this causes a general inflammatory glomerular reaction, blocking the glomerular capillaries or making them excessively permeable. High glomerular permeability allows plasma proteins and red blood cells to leak into the filtrate. As a result, plasma oncotic pressure decreases, which accounts for pleural effusions in the chest that often accompany acute glomerulonephritis.5 (That is, plasma oncotic pressure may become so low that it can no longer keep fluids from leaking out of the vascular space into the pleural cavity.) In severe instances, total renal shutdown occurs. The acute inflammation usually disappears in a few weeks to a few months.1
Another form of intrarenal failure is tubular necrosis. Certain poisons and severe renal ischemia can cause tubular necrosis (death of tubular epithelial cells).1 Renal poisons include carbon tetrachloride and mercury compounds. In addition, tetracycline antibiotics and some anticancer drugs are toxic to the nephron.1 Dead epithelial cells slough away from their attachments and plug the tubules, preventing the normal flow of filtrate. Severe renal ischemia, usually caused by acute cardiovascular failure, also can kill tubular epithelial cells and plug the tubular lumen.
Postrenal Failure
Postrenal failure occurs when obstructions to renal outflow occur. If this involves only one kidney, the other kidney can usually maintain normal fluid and electrolyte balance. If obstruction affects both kidneys and persists for several days, it can lead to irreversible kidney damage.1 Causes of obstruction include kidney stones and blood clots of the ureters, which also may block the bladder opening or the urethra.1
Blood Transfusion Reactions
Severe immune reactions to blood transfusion can cause acute renal failure. Transfusion reactions are characterized by the destruction of many red blood cells, releasing hemoglobin into the plasma. The hemoglobin molecule is small enough to pass through the glomerular membrane but may not be adequately reabsorbed by the proximal tubules. Excess hemoglobin in the filtrate precipitates and blocks the tubules.1
All of the foregoing forms of acute renal failure prevent the normal flow of filtrate through the tubules. Consequently, the kidneys retain sodium and fluid because they cannot excrete these substances. As a result, body tissues become edematous, and blood pressure increases.1
Chronic Renal Failure
Chronic renal failure is characterized by a decrease in the number of functional nephrons. In chronic glomerulonephritis, the glomerular filtration membrane progressively thickens and becomes fibrotic, rendering the affected glomeruli functionless.1 This condition reduces the kidney’s filtering capacity.
In pyelonephritis, the renal pelvis becomes inflamed, eventually extending back into the renal tubules. Bacteria commonly cause pyelonephritis, especially fecal bacteria that contaminate the urinary tract.1 If unchecked, the resulting infection progressively destroys many nephrons.
Arteriosclerotic disease reduces blood flow through the renal arteries and may lead to the ischemic death of many nephrons. The kidney tissue becomes fibrotic and greatly reduced in size. This process occurs to some extent in the aging kidney, decreasing renal blood flow to about 45% of normal by the age of 80.1
Physiological Effects of Chronic Renal Failure
Generally, the kidneys can maintain adequate function with only one third of the normal number of nephrons. Further reduction in functioning nephrons increases the body’s retention of waste products, especially urea and creatinine. The tubular filtrate load of solutes in the remaining functional nephrons increases greatly, exceeding the kidney’s reabsorption capacity. Consequently, the solutes remaining in the filtrate act as osmotic diuretics, increasing each nephron’s filtrate flow and urine output. Despite significant renal impairment, total urine output often paradoxically increases.1 In other words, the increase in each nephron’s urine output outweighs the effect of decreased nephron numbers. The high flow rate of filtrate through the remaining functional tubules impairs the kidney’s urine-concentrating ability. Flow is too rapid through the collecting ducts for adequate water reabsorption to occur. Consequently, the kidneys excrete dilute urine, manifested by a decrease in the urine’s specific gravity.1
If a person with complete renal shutdown continues to ingest food and water, extracellular concentrations of nitrogenous waste products, sodium, potassium, and fixed acids rapidly increase. The most important effects of retaining these substances are the following:1 (1) general edema secondary to salt and water retention; (2) metabolic acidosis caused by the kidney’s failure to excrete fixed acids; (3) high blood concentrations of nitrogenous compounds such as urea, creatinine, and uric acid, which are end products of protein breakdown; and (4) increased blood concentration of phenols, sulfates, phosphates, and potassium. This condition is called uremia because of the high urea concentrations in all body fluids. After about one week of renal failure, patients develop a confused mental state that may progress to a condition known as uremic coma. Acidosis, which depresses the central nervous system, is apparently responsible for the coma.1
Nephrotic Syndrome
Nephrotic syndrome is characterized by increased glomerular membrane permeability and subsequent loss of plasma proteins into the urine. This syndrome may occur in the absence of other renal abnormalities but is usually associated with some degree of renal failure.1 Any condition that increases glomerular permeability can cause nephrotic syndrome. Low plasma protein concentration lowers plasma osmotic pressure, causing generalized interstitial edema and fluid collection in body cavities such as the abdomen (ascites), pleural space (pleural effusion), and pericardial space (pericardial effusion).
Goodpasture’s Syndrome
Goodpasture’s syndrome is an autoimmune disease affecting glomerular and pulmonary alveolar basement membranes.5 The body forms circulating antibodies against these structures, causing hemorrhage and fibrosis. Patients with this disease develop hemoptysis (coughing up of blood) and hematuria (blood in the urine). Goodpasture’s syndrome targets primarily the kidney; the lung’s alveoli become involved because of a cross-reaction.5
Goodpasture’s syndrome affects mainly young adult males over the age of 16 years. This rare syndrome causes renal and respiratory failure. The prognosis is quite poor; average survival after the appearance of the first symptom is about six months.5
Renal Function Tests
Renal Clearance Tests
Renal clearance tests are used to assess the ability of the kidneys to clear the plasma of a specific substance. Intravenous pyelography (IVP) involves the injection of an iodine-containing substance intravenously and watching the subsequent rise in urinary iodine concentration over the next several minutes.1 Since iodine is opaque to x-rays, urinary iodine outlines the renal pelvis on x-ray pictures. Lack of renal pelvis shadows on the x-ray film (pyelogram) indicates that the patient has poor renal clearance.
Blood Urea Nitrogen and Creatinine
Common blood tests of renal function are blood urea nitrogen (BUN) and creatinine tests. Normal BUN concentration ranges from 8 to 20 mg/dL; blood creatinine concentration ranges from 0.6 to 1.2 mg/dL.6 Urea is synthesized in the liver from ammonia, a by-product of protein (amino acid) catabolism. Urea is constantly formed and must be excreted by the kidneys. BUN increases in kidney failure as a result of decreased tubular filtration capacity. In extreme instances, BUN may increase to 300 mg/dL.1