Chapter 11 Electrodiagnostic Medicine III
Case Studies
The electromyographer tailors the EDX study based on the history and physical examination. A differential diagnosis is developed before the study that considers disease processes involving multiple levels of the central and peripheral nervous system. A deductive process is used in which each NCS and muscle examined with needle EMG should assist with narrowing the differential diagnosis until a conclusion is reached. This dynamic (rather than protocol or rote) process for the selection of nerves and muscles to study is more likely to ultimately provide an accurate diagnosis, helps limit the number of studies to the minimum required for the diagnosis, is required by the American Association of Neuromuscular and Electrodiagnostic Medicine guidelines, is required by the Current Procedural Terminology code used for billing these studies, and is the standard of practice.3
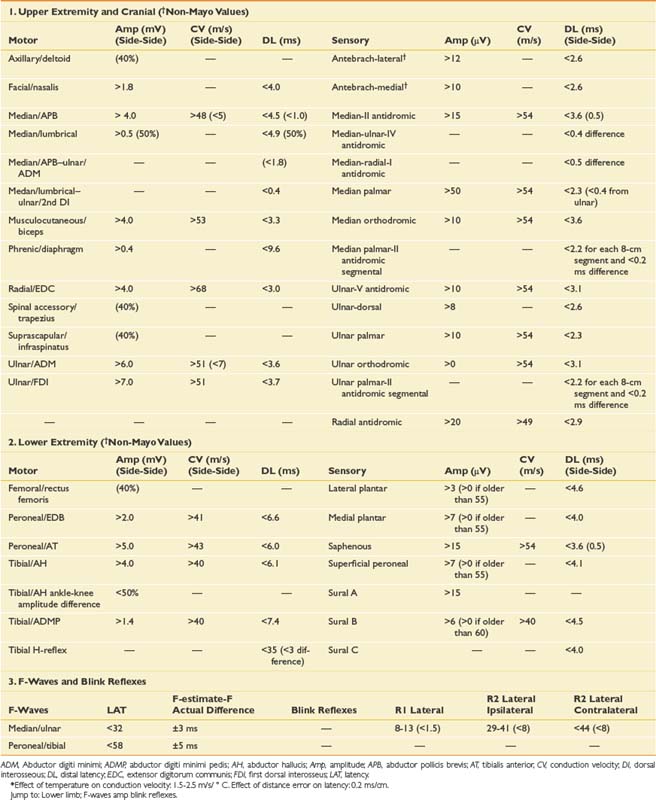
This chapter reviews a series of cases to illustrate a logical approach to the neurophysiologic workup of various clinical complaints. In clinical practice each case is unique, however, and will vary from patient to patient and clinician to clinician. The specific testing must be individualized based initially on the presenting complaint; however, during the study, the procedure changes based on the data obtained from initial NCS or needle EMG findings. Specific testing performed also depends on patient tolerance and other factors such as anticoagulation status; the presence of lymphedema, central lines, or pacemakers; and patient positioning. The cases reported below demonstrate how to apply the basic science and techniques described in Chapters 9 and 10 to a clinical situation. The EMG findings in the following cases are graded using the Mayo Clinic rating scale (Box 11-1).9 Insertional activity is described as increased if there is anything more than minimal electrical noise generated by the electrode movement itself. This can include positive waves, fibrillation potentials, myotonic discharges, myokymia, neuromyotonia, or the “snap-crackle-pop” insertional activity seen in some normal subjects. Fibrillation potentials and positive sharp waves are interpreted the same in our laboratory, both representing active or uncompensated denervation, which is reflected on the tables in a single column referred to as fibrillation potentials. The grading of fibrillation potentials, fasciculation potentials, and voluntary MUPs is otherwise defined in Box 11-1. Normative data for NCSs (based on the Mayo Clinic EMG laboratory database) are provided in Table 11-1.
BOX 11-1 EMG Grading Guidelines (Mayo Clinic EMG Reports)
Spontaneous Activity
Polyphasic (graded as % of all MUPs assessed that have >4 phases, rounded to the nearest whole number of the following choices: 15%, 25%, 50%, 75%, 100%)
Case 1: Upper Limb Paresthesia and Pain
A 45-year-old, right-handed female secretary presents with pain and aching in her right hand that wakes her from sleep and prevents her from typing for more than 5 to 10 minutes. She describes a pins-and-needles sensation in all five digits of her right hand, but on specific questioning admits there are also mild symptoms in the left hand. When the symptoms are severe, she feels pain throughout her entire hand, forearm, and arm as far as the shoulder. These symptoms have been present and getting gradually worse for at least 6 months. She also feels her right hand is weak, as she is dropping objects and is more clumsy than usual. She reports chronic neck pain that seems to be most problematic toward the end of the workday.
Differential Diagnosis
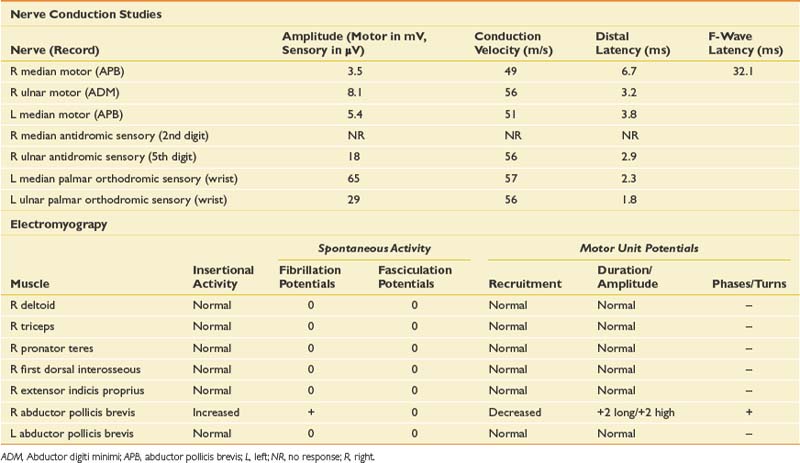
When evaluating for a mononeuropathy, it is helpful to consider the potential etiologies because this will determine which EDX studies should be performed. Although focal compression, such as carpal tunnel syndrome, is by far the most common etiology for a mononeuropathy, a more generalized process involving multiple nerves (e.g., mononeuritis multiplex, hereditary neuropathy with tendency to pressure palsies, multifocal motor neuropathy with conduction block) needs to be considered. Cervical radiculopathy is also high on the list of differential diagnoses, and as she reports paresthesias throughout the entire hand, this could reflect involvement of the C6, C7, and/or C8 nerve roots. The NCS and needle EMG findings for this case are shown in Table 11-2.
Discussion
Given the differential diagnosis outlined above, it is important to perform median and ulnar motor and sensory NCSs. A median F-wave was also performed because this may be prolonged where proximal slowing is present, such as in radiculopathy. When median neuropathy at the wrist (carpal tunnel syndrome) is a clinical consideration, we perform the median motor study first and then determine the most appropriate median sensory study based on the motor results. If the motor study is normal, as it was on the left, there are a number of different orthodromic or antidromic sensory studies that can be performed. We chose the orthodromic palmar sensory study on the left because it is one of the most sensitive techniques available and can be abnormal in cases of very mild median neuropathy.29 Other options include radial-to-median distal latency comparison recording from the thumb, ulnar-to-median distal latency comparison recording from the fourth digit, or antidromic median sensory studies recorded from the most symptomatic digit.29 If the motor distal latency is prolonged, a less sensitive but technically less challenging study such as a median antidromic sensory study would be appropriate, and that was the choice on the right. The ulnar sensory response must also be obtained to ensure there is not evidence for a more generalized process. In this case, the NCSs show evidence of a median neuropathy at the wrist, bilaterally, with focal slowing demonstrated by prolonged median motor and antidromic sensory latencies on the right, and relative prolongation of the orthodromic median palmar sensory distal latency on the left, compared with the ulnar response (with a difference greater than 0.3 ms considered abnormal).29
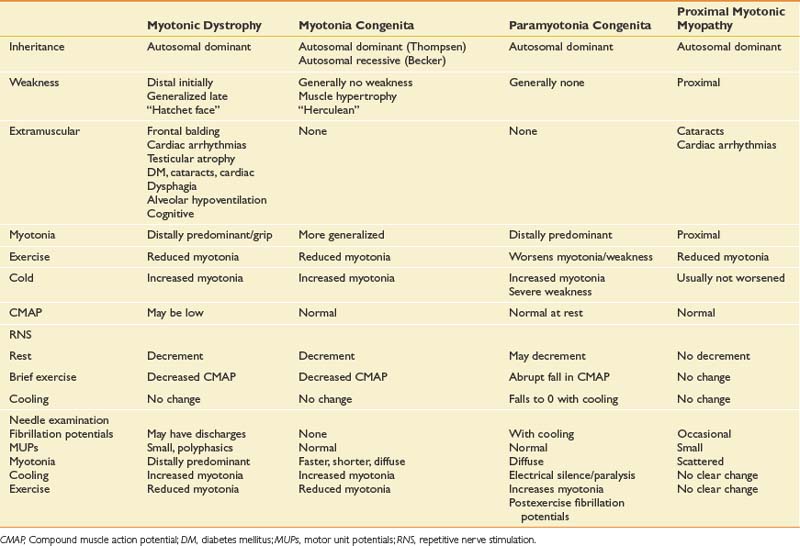
Since the NCSs have confirmed the presence of a median neuropathy at the wrist, the purpose of the needle examination in this case is threefold: (1) to confirm the level of the median nerve lesion, (2) to provide additional information regarding severity, and (3) to exclude a superimposed process that could be contributing to the presenting symptoms, such as cervical radiculopathy. Examining a median-innervated thenar muscle provides the information on severity; in this case the finding of long-duration MUPs and some fibrillation potentials provides further evidence that this is a relatively severe lesion. Examining the pronator teres is helpful because this is a more proximal median-innervated muscle, and if normal, helps exclude a more proximal median neuropathy. Additionally, as the pronator teres is innervated by the C6 and C7 roots, a normal examination helps exclude a cervical radiculopathy at those levels. The deltoid also assesses for C5 and C6 root involvement, which can lead to similar symptoms. Examining the first dorsal interosseous helps exclude an ulnar neuropathy (unlikely given the normal ulnar NCS), but also excludes a C8 radiculopathy or lower trunk plexopathy as a cause for the abnormal findings in the abductor pollicis brevis muscle. Cervical paraspinal muscles were not examined in this case because all the findings on both NCSs and needle EMG pointed to carpal tunnel syndrome. However, if there is a strong clinical suspicion for recent-onset cervical radiculopathy, the paraspinals should be examined because they are often the first muscle to show changes on needle EMG.
Although the diagnosis in this case is bilateral median neuropathy at the wrist, consistent with carpal tunnel syndrome, the presentation highlights that many patients do not describe classic symptoms, and one must consider a broad differential diagnosis in such cases. In carpal tunnel syndrome, more than 50% of patients report sensory symptoms outside the median nerve distribution,30 and this is a reason that EDX examination is critical in refining the clinical impression.
Case 2: Aching and Numbness of the Left Thumb, Index Finger, and Middle Finger
Differential Diagnosis
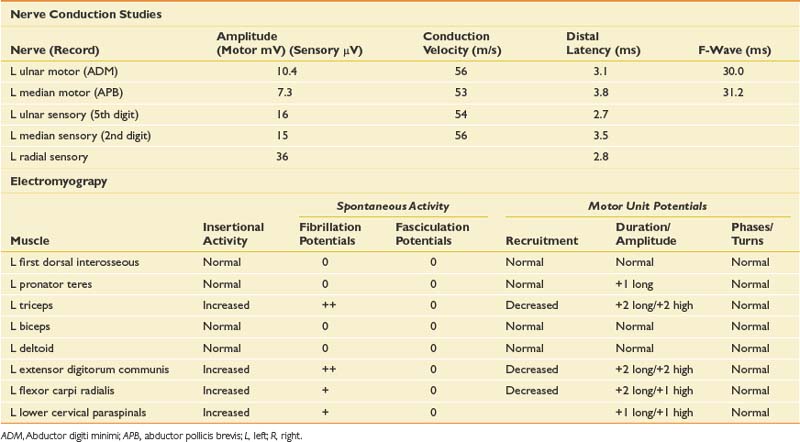
This is a very common referral to the electromyographer with the primary differential diagnosis being a cervical radiculopathy, median neuropathy at the wrist or myofascial syndrome. A brachial plexopathy is possible but unlikely in the absence of trauma or a history of carcinoma. Although an idiopathic brachial plexopathy (Parsonage-Turner syndrome) could present spontaneously, the pattern of pain and weakness would be unusual. The sensory findings confirmed on clinical examination would argue against a myofascial process and narrow the diagnosis. The pattern of sensory loss could be seen with either a median neuropathy or cervical radiculopathy, but the mild triceps weakness and reflex changes would certainly argue for a radiculopathy, most likely in the C7 distribution. Given the triceps and finger extensor weakness, a radial neuropathy would also need to be excluded with the EDX evaluation. NCSs and needle EMG were performed (Table 11-3).
Case 3: Shoulder Pain and Weakness
A 54-year-old woman with a history of breast cancer, status post right modified mastectomy, radiation, and chemotherapy 5 years previously, presents with a 4-week history of right shoulder pain and weakness as well as numbness in the right hand and forearm. She is otherwise healthy and has not been taking any medications except tamoxifen since completing chemotherapy. She denies previous episodes of similar symptoms, nor any history of neck or shoulder trauma. She had recently increased her activity with involvement in a weightlifting program, and had noted some generalized muscle aching in association with that. She states the pain came on suddenly during the night approximately 6 weeks previously with a severe burning and at times deep aching sensation, preventing her from sleeping. The severe pain lasted about 10 days and has now significantly improved, but as the pain improved, she noticed significant weakness that has persisted, such that she can barely lift her arm from her side. She still has good use of her hand.
Differential Diagnosis
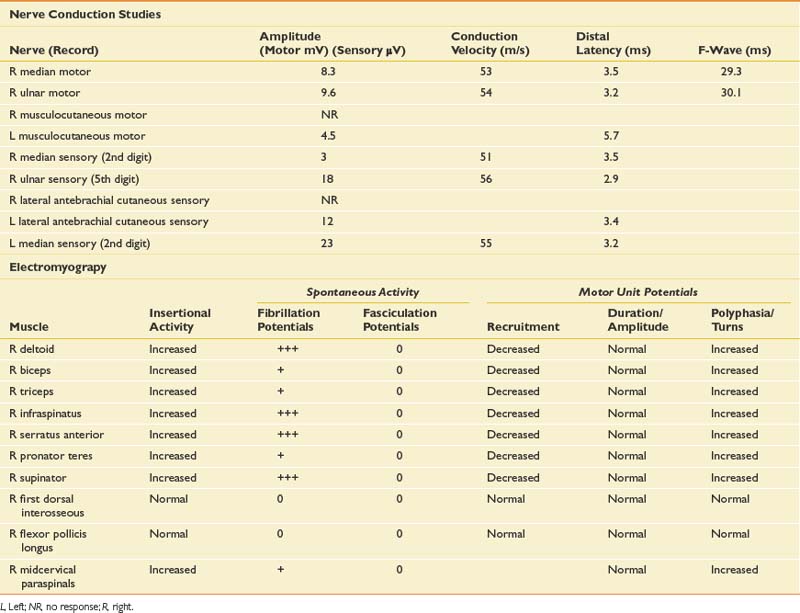
Given the history, an infiltrative plexopathy caused by breast carcinoma would be of primary concern; however, the sudden onset of symptoms would argue for an inflammatory/idiopathic brachial plexopathy. In this case a radiation-induced injury should also be considered, but the acute and painful onset would also make this less likely. The finding of myokymia on needle examination is very helpful in such cases, because this is much more frequently found in radiation plexopathy than in cases of metastatic infiltration of the plexus.15,19 The NCSs and needle EMG for this patient are listed in Table 11-4.
Discussion
NCSs in this case show low-amplitude median antidromic and absent lateral antebrachial cutaneous sensory responses, which implies that the lesion is distal to the dorsal root ganglion at the level of the plexus or peripheral nerve. The normal median motor response (C8, T1, lower trunk) in the presence of a low-amplitude median sensory response (C6, C7, middle trunk) suggests the lesion is proximal to the median nerve and at the level of the plexus. The median sensory response on the left is mildly low amplitude with a prolonged distal latency, most likely representing a preexisting median neuropathy at the wrist of the type seen in carpal tunnel syndrome. The musculocutaneous compound muscle action potential (CMAP) is absent on the right but easily elicited on the left. This study was performed because the more easily obtained median and ulnar motor studies are both derived from C8–T1 axons and therefore are spared in this case.
The needle EMG findings, when taken in combination with the NCS results, are most consistent with a severe right upper, and to a lesser extent, middle trunk brachial plexopathy, with evidence to suggest mild cervical root involvement also. Given the somewhat patchy nature of the needle EMG findings, the absence of trauma, and the classic history of severe pain at onset followed by weakness that persists, the cause is almost certainly inflammatory and is typical of neuralgic amyotrophy or Parsonage-Turner syndrome.32 Often a particular peripheral nerve will be severely involved. For example, patients can present with sudden onset of severe dyspnea that is worse when recumbent, because of involvement of the phrenic nerve. They can also present with a classic anterior interosseous neuropathy with weakness only involving the flexor pollicis longus, flexor digitorum profundus to the second and third digits, and the pronator quadratus. Such cases are frequently misdiagnosed as entrapment neuropathy. Therefore it is critical when evaluating such cases to perform a careful needle examination, looking for patchy involvement outside the muscles that are clearly involved clinically. This includes a search for subclinical contralateral limb involvement, to avoid unnecessary surgery in such cases. A careful history should quickly alert the electromyographer to the likely diagnosis in such cases. Imaging of the cervical spine in this setting can be helpful in excluding a compressive process, but care should be taken to not assign clinical significance to mild spondylotic changes. Imaging of the brachial plexus can also reveal signal abnormalities, and in this case it would also be indicated to exclude an infiltrative process.
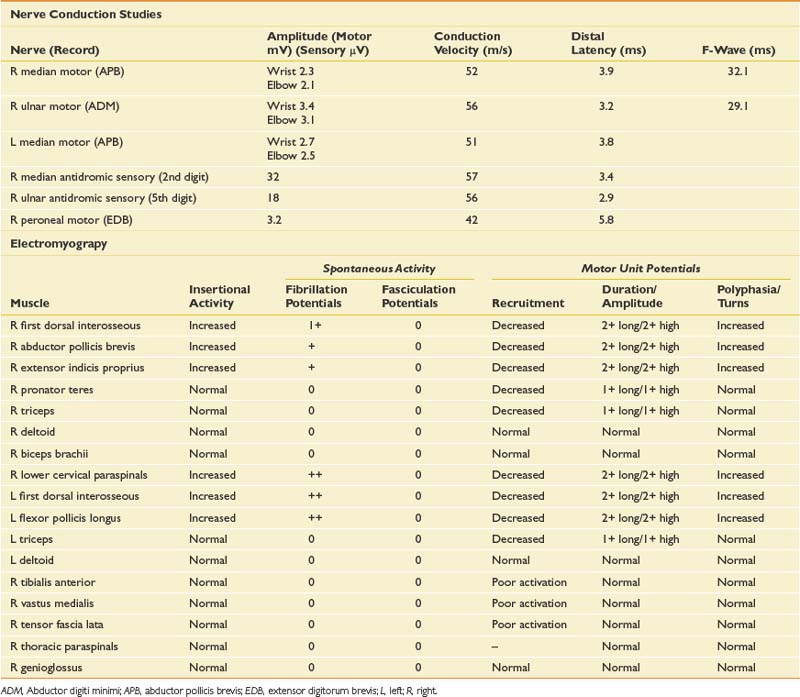
Case 4: Hand Numbness and Weakness
Differential Diagnosis
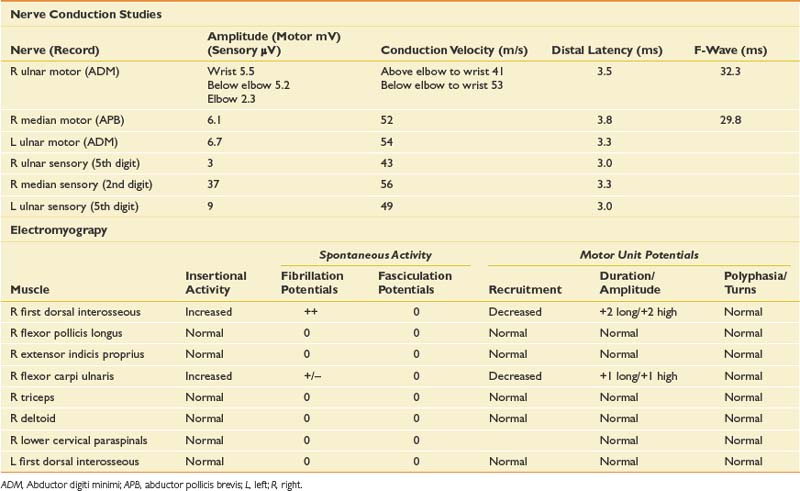
The patient presents with numbness and tingling in a right ulnar nerve distribution and weakness of intrinsic hand muscles. Given the history of compression, the most likely cause for these symptoms would be mononeuropathy involving the ulnar nerve at the wrist or elbow. Such symptoms could also represent a more generalized process such as a hereditary neuropathy with a tendency to pressure palsies, or mononeuritis multiplex (typically the latter would be painful). Other considerations should include lower trunk or medial cord brachial plexopathy and C8/T1 radiculopathy. Anterior horn cell disease involving the lower cervical cord is unlikely given the sensory symptoms. Rarely a spinal cord lesion such as a syrinx or a cortical infarct might present in this way. NCS and needle EMG findings for this case are shown in Table 11-5.
Discussion
NCSs showed a low-amplitude ulnar motor response on the right, with slowed conduction velocity and borderline distal latency. There was a significant reduction in the CMAP amplitude between the elbow and wrist stimulation sites suggesting a conduction block. This was confirmed with stimulation at the below-elbow site, followed by short-segment stimulation (inching) between the elbow and below-elbow sites (Figure 11-1). There was also significant slowing localized to the elbow segment (evident as a conduction velocity in the above-elbow to wrist segment that was more than 10 ms slower than the conduction velocity of the below-elbow to wrist segment). The left ulnar motor response was normal but the ulnar sensory response was low amplitude with a slowed conduction velocity. With normal median motor and sensory studies on the right, these findings are most suggestive of bilateral ulnar neuropathy at the elbow, more pronounced on the right.
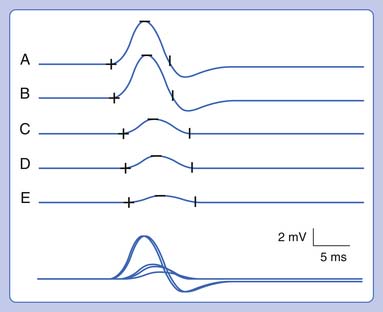
Ulnar neuropathies are relatively common, generally presenting with varying degrees of sensory symptoms in the fourth and fifth digits as well as weakness of ulnar-innervated hand and sometimes forearm muscles. Sensory fibers are usually the most vulnerable and can show abnormalities in very mild neuropathies. Motor responses can be recorded from both the abductor digiti minimi and first dorsal interosseous muscles to improve sensitivity, as a normal response can be obtained from one of the muscles or the other because of the fascicular nature of nerve topography. The diagnosis of a focal ulnar neuropathy at the elbow is suspected with slowing across the elbow segment of greater than 10 m/s compared with the below-elbow segment, of if there is an amplitude drop of 20% over a 10-cm segment. If a question remains, sequential 2-cm stimulation across the elbow can confirm an ulnar neuropathy if there is a 10% drop over a 2-cm segment or a latency difference of greater than 0.7 ms.4,5 An incidental median-to-ulnar crossover in the forearm can simulate an ulnar neuropathy and should always be excluded, particularly if there appears to be a conduction block in the forearm.
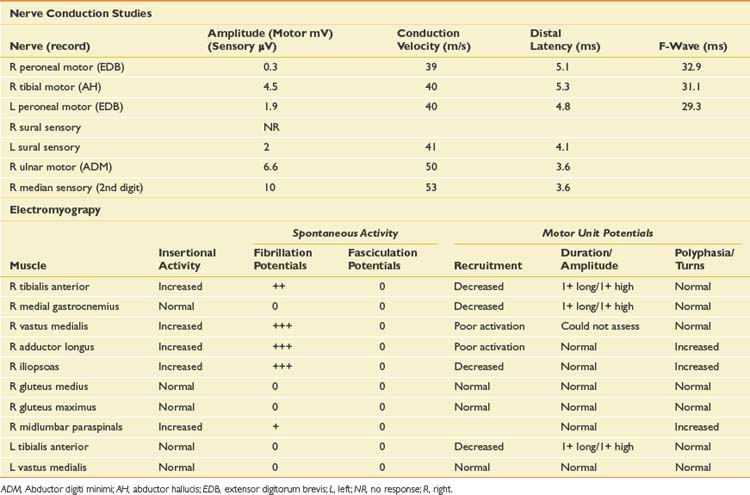
Case 5: Painless Weakness
On examination, he has an obvious foot drop when ambulating, atrophy of the left calf, and some fasciculations noted at rest in the gastrocnemius muscle, as well as rare fasciculation potentials in the triceps. Strength testing reveals moderate weakness of the left ankle dorsiflexors, plantar flexors, toe flexors and extensors, and mild weakness of the proximal left lower limb muscles. In the right lower limb, he has mild weakness of the ankle dorsiflexors and toe extensors. Strength is normal in the upper limbs, and sensation is intact throughout. Reflexes are diffusely brisk but symmetric. Plantar responses are downgoing on the right and equivocal on the left. Cranial nerve examination is normal.
Differential Diagnosis
When a patient presents with progressive, painless weakness, one must always consider the possibility of amyotrophic lateral sclerosis (ALS), particularly when muscle stretch reflexes are normal or hyperreflexic in the presence of lower motor neuron weakness. Other causes for painless weakness include multifocal motor neuropathy with conduction block (MMNCB), which initially presents with weakness in a single peripheral nerve distribution, but progresses to involve multiple nerves. In early MMNCB, muscle bulk is typically preserved in the face of significant weakness, but with time patients develop axon loss with associated muscle atrophy and loss of reflexes. Reflexes would generally be reduced in the affected distribution as well. A lumbar plexopathy could explain the left lower limb weakness particularly given the history of mild sensory symptoms. In some cases of infiltrative or inflammatory etiology, bilateral involvement of the plexus can be seen. Although unusual in the absence of pain, multiple lumbosacral radiculopathies should be considered, particularly in the setting of diabetes, or as in this case with a background history of low back pain. When hyperreflexia is present, cervical or thoracic stenosis or intramedullary lesions are a possible explanation for brisk reflexes in an otherwise lower motor neuron problem at the level of the lumbar spine. Some myopathies can present with predominantly lower limb weakness; side-to-side asymmetry is common in inclusion body myositis, in which there is typically early involvement of the quadriceps and ankle dorsiflexors. His young age would generally argue against this. Inherited myopathies such as limb girdle dystrophy could also present with lower limb weakness and some asymmetry. Neuromuscular junction disease such as Lambert-Eaton myasthenic syndrome should be considered, but there is typically prominent proximal weakness with a fatigable component, depressed muscle stretch reflexes, and autonomic symptoms such as dry mouth. The NCS and EMG findings are presented in Table 11-6.
Discussion
ALS is a disorder of the anterior horn cell that presents with progressive, painless weakness combined with upper motor neuron findings and the absence of sensory symptoms. It is an almost universally fatal disease, with an average life span from the time of diagnosis of 3 to 5 years.28 Other disease processes that could potentially masquerade as ALS, such as polyradiculopathy, polyradiculoneuropathy, or MMNCB, must be entirely excluded because they can have treatment implications. EDX testing is helpful to confirm the diagnosis when it is clinically apparent, and to diagnose the condition when it is clinically suspected but too early to diagnose on clinical grounds alone (abnormal findings are usually present on needle examination long before they become clinically evident).
In ALS, the needle examination typically shows profuse fibrillation and fasciculation potentials, and very complex (polyphasic), unstable MUPs. These findings must be present in at least three different spinal segments (e.g., an upper and lower limb, and thoracic paraspinal muscles), or two spinal segments in combination with bulbar muscles. Within those segments, two or more muscles with different segmental and peripheral nerve innervation must be affected before one can confidently conclude that a progressive motor neuron disease is present (this grading scale is based on the El Escorial criteria for possible or probable ALS).7
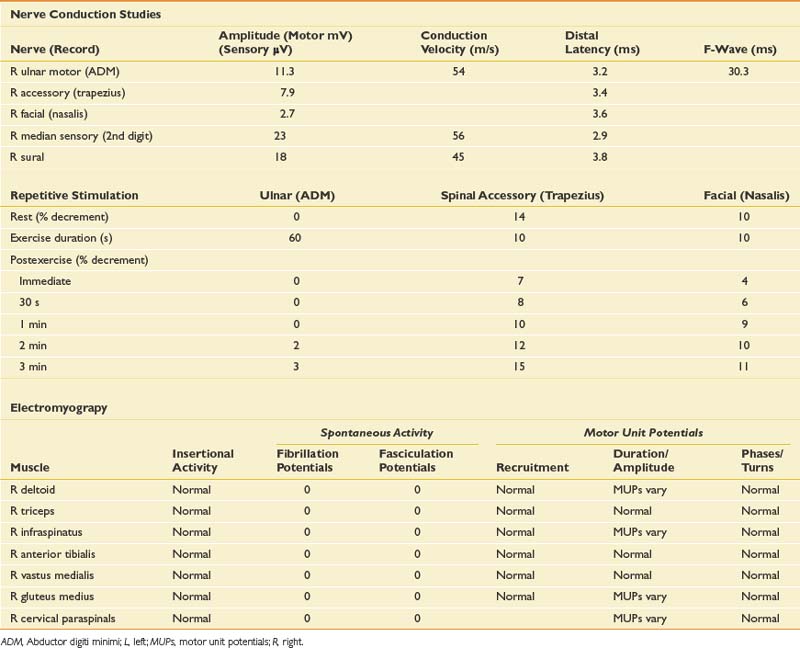
Case 6: Failure to Wean From the Ventilator
Differential Diagnosis
In the ICU setting, critical illness myopathy (CIM) and critical illness polyneuropathy are common causes of unexplained weakness.14 Other considerations should include neuromuscular junction disorders such as botulism or a myasthenic crisis, which can present with predominantly respiratory symptoms. Respiratory failure can be the initial presentation of ALS, as well as some myopathies such as acid maltase disease. Other myopathies that might present in the ICU should also be considered, including acute necrotizing myopathy and inflammatory myopathies (polymyositis, dermatomyositis), which can occasionally present fulminantly. A polyradiculoneuropathy should be considered, either acute inflammatory demyelinating polyradiculoneuropathy (Guillain-Barré syndrome) or in this case, diabetic polyradiculoneuropathy combined with severe deconditioning, given the history of 2 weeks of immobility in the ICU.
EDX testing in the ICU is fraught with technical challenges related to patient positioning, inaccessibility of some nerves or muscles because of line placement, inability of the patient to cooperate with needle examination because of altered mental status or severe weakness, anticoagulant medications, and electrical interference from equipment, which can make sensory NCSs very difficult to obtain. Sometimes a study has to be limited because of these issues. The NCS and needle EMG findings for this case are presented in Table 11-7.
Discussion
Phrenic CMAPs were also low amplitude bilaterally. Several techniques for phrenic NCSs have been described, but the CMAP can be technically difficult to obtain. The most frequent cause of unreliable findings is the misinterpretation of volume-conducted responses from nearby chest wall muscles.25 It is important to stimulate the nerve in between inspiration and expiration because this results in the biggest response and minimizes movement artifact, although some patients are unable to comply with this instruction.
Based on the NCSs, the findings were most suggestive of CIM, with low-amplitude motor responses but relative preservation of sensory responses. The long-duration CMAPs were also very suggestive of CIM.1 Needle EMG was limited by the inability of the patient to activate most muscles. Low-grade fibrillation potentials, however, were seen in most muscles examined, and when motor units could be briefly activated, they were noted to be of short duration, some were complex, and recruitment was markedly increased. The diaphragm was also studied, and the findings were consistent with the generalized myopathic process noted elsewhere. Needle examination of the diaphragm can be very helpful in determining the underlying pathophysiology of neuromuscular disorders, but does carry inherent risks because of the proximity of vital organs. Several techniques have been described, including an ultrasound-guided technique.6 One must weigh the potential risks against the importance of the additional diagnostic information the EMG examination can provide. Evaluation of MUPs can differentiate between neuropathic and myopathic processes, and evaluation for fibrillation potentials can provide information on the degree of axon loss or severity of the myopathic process. One must take care not to interpret low-amplitude, very complex, nascent MUPs as myopathic (nascent motor units are seen after severe nerve injury during early reinnervation), and the recruitment pattern is critical in differentiating the two.
Case 7: Foot and Hand Numbness and Pain
Differential Diagnosis
The patient presents with symptoms suggestive of a rather rapid-onset, length-dependent peripheral neuropathy. There is a wide differential diagnosis to consider in cases of length-dependent peripheral neuropathy, although most are inherited, metabolic, toxic, or idiopathic.11 In this case, the two most likely causative agents would be recent chemotherapy and the history of heavy alcohol intake. The relative subacute onset and the presence of positive sensory symptoms would argue for an acquired process. Although the presentation is characteristic of a length-dependent peripheral neuropathy, other possibilities should also be considered, including polyradiculoneuropathy, which can present with predominantly length-dependent symptoms (e.g., acute inflammatory demyelinating polyradiculoneuropathy, chronic inflammatory demyelinating polyradiculoneuropathy, diabetic polyradiculoneuropathy). Multiple mononeuropathies would not typically present in such a symmetric fashion. The prominent sensory symptoms make muscle, neuromuscular junction, and anterior horn cell disease unlikely. The NCS and needle EMG findings are presented in Table 11-8.
Discussion
The findings on NCSs in this case were most consistent with an axonal peripheral neuropathy, with mild generalized slowing of conduction velocities, borderline distal latencies, and significant loss of amplitude (with sensory fibers affected more than motor). The sural response was still within normal limits for age, and therefore the medial plantar study was performed, because it is a more sensitive test for sensory neuropathy in younger patients. The electrophysiologic confirmation of length-dependent peripheral neuropathy is based on the distribution of findings on needle examination. In this case, distal muscles were abnormal with large MUPs and fibrillation potentials, and more proximal muscles had normal findings. It is important to examine a proximal muscle with the same myotomal innervation to confirm that the process is length dependent (e.g., if the medial gastrocnemius is abnormal, the gluteus maximus should be examined because both distal and proximal muscles are predominantly S1 innervated). Paraspinal muscles should also be normal. Changes are typically more profound in the lower limbs, but it is often helpful to document the length-dependent pattern in the upper limbs by showing abnormalities in a hand intrinsic muscle and normal findings in a more proximal muscle such as the triceps or deltoid. Although alcohol is a very common cause of peripheral neuropathy, the abruptness of onset and severity of the symptoms and EDX findings argued against that as the primary etiology in this case. The likely etiology was thought to be vincristine toxicity, a component of his chemotherapy regimen. When the vincristine was discontinued, his symptoms gradually improved over the following months.
Case 8: Bilateral Hand Weakness and Numbness
Physical examination shows a spastic, scissoring gait, increased muscle stretch reflexes in the upper and lower limbs, upgoing plantar response on the right, normal sensation to pinprick and joint position sense in the lower extremities, and decreased sensation to pinprick in the ulnar aspect of both hands. Strength is normal in both lower limbs and in the proximal upper limb muscles. There is moderate weakness in the intrinsic hand muscles, with some atrophy, and in the extensor digitorum communis bilaterally. Cranial nerve examination is normal.
Differential Diagnosis
The clinical presentation is most notable for upper motor neuron findings with diffuse hyperreflexia and lower limb spasticity. There is some mild atrophy suggesting lower motor neuron involvement, but this is less clear and a clear indication for EMG. The upper motor neuron findings should alert one to the possibility of ALS, but could certainly be seen with a cervical myelopathy or a bilateral cortical process. ALS would be less likely in this case given the sensory symptoms, although a coexisting peripheral neuropathy or superimposed mononeuropathies would be possible. Bilateral median and/or ulnar neuropathies are a relatively common cause of bilateral hand weakness and numbness but would not explain the upper motor neuron signs. NCS and EMG findings are shown in Table 11-9.
Discussion
The primary purpose of the EDX studies in this setting is to determine whether there is lower motor neuron involvement, and if present, whether it is diffuse or limited to a specific localizing region. NCSs showed low-amplitude median and ulnar motor responses with preservation of the sensory responses. This implies that the lesion is either proximal to the dorsal root ganglion or at the level of the muscle itself (in this case the latter is unlikely given the prominent sensory symptoms). Lesions proximal to the dorsal root ganglion can involve the nerve root or the anterior horn cell. No motor dispersion and no conduction blocks were noted. A lower limb motor study was normal, arguing against a more diffuse process. Needle examination showed fibrillation potentials and neurogenic MUPs in bilateral C8–T1-innervated muscles, including the paraspinals, indicating pathology at the level of the nerve roots or anterior horn cell. Neurogenic MUP changes were also seen in C7-innervated muscles, without fibrillation potentials, suggesting mild, chronic C7 involvement. Given the presentation and potential concern for ALS, muscles in the lower limb as well as the thoracic paraspinal and genioglossus (tongue) muscles were examined, and the only finding of note was poor voluntary motor unit activation, which can be seen in spasticity or when the patient is giving poor effort. Also, no fasciculation potentials were identified, making ALS unlikely.
These findings are most consistent with multiple bilateral cervical radiculopathies or focal anterior horn cell disease, with uncompensated denervation evident at the C8–T1 levels bilaterally. Focal anterior horn cell disease can be seen with intrinsic spinal cord pathology or in focal motor neuron disease (diplegic amyotrophy).17 Such changes could also be seen in early ALS; however, subclinical involvement would typically be noted in the form of unstable, complex MUPs and fasciculations in a more widespread distribution. The most likely explanation for these findings in this case, given the spasticity in the lower limbs and the numbness in the hands despite normal sensory studies, would be cervical stenosis with associated cord compression. Because one cannot differentiate between nerve root and anterior horn cell disease on the basis of electrophysiologic findings, it is important to take into account the clinical presentation and any other available diagnostic testing such as imaging, when reporting the final impression. In this case, magnetic resonance imaging of the cervical cord showed severe stenosis with cord signal change at the C7–8 level.
Case 9: Leg Pain and Weakness
Differential Diagnosis
The diagnosis that should come to mind immediately with this history is diabetic lumbar radiculoplexus neuropathy (diabetic amyotrophy).13 The history of sudden onset of severe pain, prominent weakness, recently diagnosed diabetes, and significant weight loss are quite typical of this entity. Other causes of lumbar plexopathy, as well as an acute severe lumbar radiculopathy, would also be obvious considerations. The distribution of weakness suggests multilevel root involvement, L2–L4 predominantly. It would be important to know whether he had undergone any recent interventions related to the peripheral arterial disease, because a compressive hematoma in the region of the femoral nerve or lumbar plexus can present with thigh pain and weakness. An acute necrotizing myopathy could rarely present with focal pain and weakness such as this, although the sensory symptoms make that unlikely in this case.
The goals of EDX testing in this case are to identify as accurately as possible the level of pathology to help direct the diagnostic workup, and to evaluate the severity of axonal loss to provide prognostic information. The NCS and EMG findings are presented in Table 11-10.
Discussion
Needle EMG is used to map out the involved myotomes, with severe denervation and poor activation as well as markedly reduced recruitment in all L2–L4 innervated muscles examined on the right. There is also moderate active denervation noted in the tibialis anterior muscle, which is supplied by the L4 and L5 nerve roots and lumbar plexus. Mild fibrillation potentials were also present in the lumbar paraspinals, but not to the same degree as in the right thigh muscles. Some longstanding neurogenic changes were noted in distal L5- and S1-innervated muscles (indicated by long-duration, high-amplitude MUPs, but no increase in turns or phases), without corresponding changes in the gluteus medius or gluteus maximus muscles. These findings confirm the length-dependent peripheral neuropathy evident on NCSs.
In this case there is also mild denervation in the right lumbar paraspinal muscles, which is consistent with a radiculoplexus neuropathy as is typically seen with diabetic and nondiabetic inflammatory plexopathies.13 It is helpful to check one or two of the most severely affected muscles in the contralateral limb to evaluate for subclinical involvement, which is also very suggestive of an inflammatory etiology. Imaging of both the lumbar spine and plexus is recommended to rule out a compressive or infiltrative lesion, even in cases such as this with a history typical of diabetic radiculoplexus neuropathy (diabetic amyotrophy). This can be diagnostically challenging because the electrophysiologic findings could easily reflect multiple compressive radiculopathies, and in this age-group, imaging may show significant spondylosis and stenosis. The key to diagnosis involves carefully obtaining a clinical history. In this setting, the acute onset in conjunction with the prominent weight loss would lead one down the correct path to diagnosis.
Case 10: Weakness and Fatigue
A 45-year-old woman presents with weakness and fatigue. Although relatively nonspecific, she reports fatigue especially as the day progresses and worsening functional limitations (such as going up stairs). There is no complaint of pain or sensory symptoms. She denies dysphagia or weight loss, but does report intermittent diplopia. Physical examination shows normal strength in limb and trunk muscles, normal cranial nerve examination, normal muscle stretch reflexes, and normal sensory examination. With repetitive strength testing there did appear to be a slight degree of fatigue, but this was equivocal. There were no fasciculations noted.
Differential Diagnosis
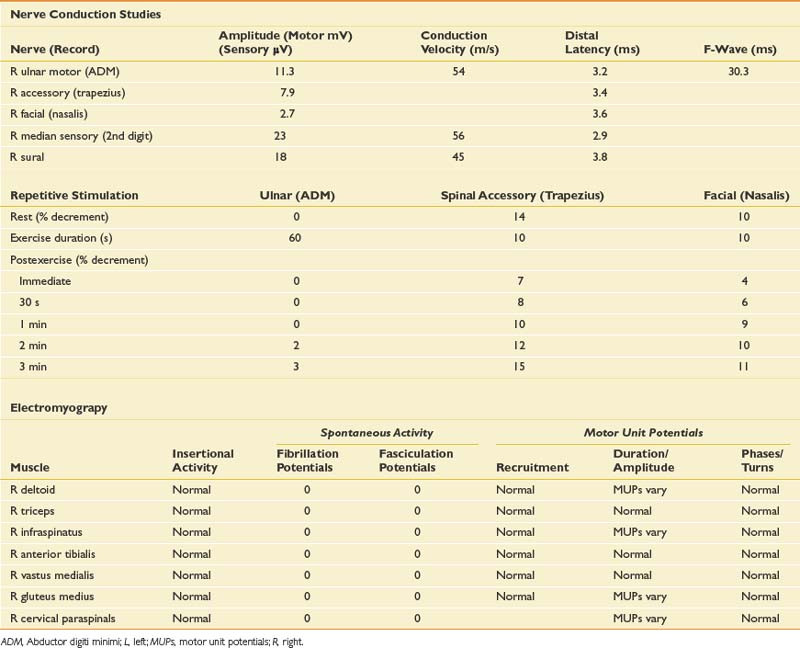
The EDX evaluation in this case includes standard NCSs in addition to needle EMG. The NCSs include repetitive stimulation to distal and proximal muscles at rest and after exercise (Table 11-11). Needle examination was performed on proximal and distal muscles of the upper and lower limbs.
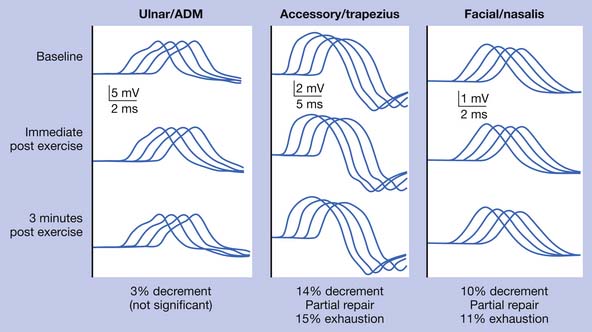
Discussion
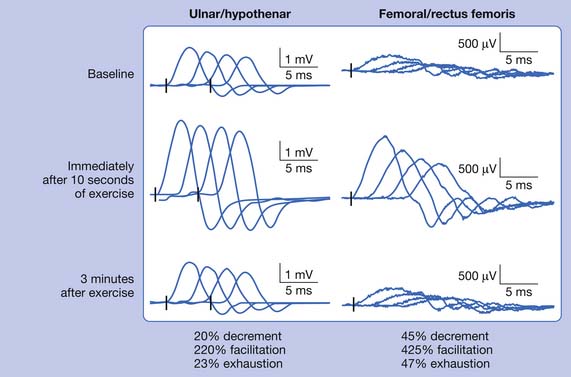
Routine NCSs showed normal CMAP and SNAP amplitudes. In this case there was no significant decrement to 2-Hz repetitive stimulation when recorded over the abductor digiti minimi, but there was a consistent pattern of decrement with at least partial repair when recorded over the trapezius and a facial muscle (Figure 11-2). The EMG revealed no abnormal spontaneous activity and varying MUP morphology of the same MUP without other morphologic changes in primarily proximal muscles. The EDX testing was critical to confirm the diagnosis of a neuromuscular junction disorder. The degree of decrement was mild but reproducible, with the expected greater involvement of proximal muscles. This has been shown by Özdemir and Young,22 who found abnormalities in the abductor digiti minimi in 59% of patients, but in greater than 80% when proximal repetitive stimulation was performed. Typically this would be performed on the spinal accessory or facial nerves because these are technically less challenging. Recent reports also describe this technique recording over the masseter for those who present with dysphagia or jaw weakness.26 Proximal lower limb repetitive stimulation can be performed on the femoral nerve with percutaneous near-needle stimulation if necessary, otherwise a decrement might be noted in the tibialis anterior. The typical amplitude decrement is a smooth pattern of decline, which becomes maximal at the fourth or fifth response but has the greatest relative reduction between the first and second responses (Figure 11-3). If stimulation were continued past the fifth response there would be stabilization, presumably associated with mobilization of acetylcholine stores. If the decrement is not smooth or the major decrement does not occur between the first and second responses, this should be attributed to technical factors until proven otherwise (Figure 11-4). Immediately after brief exercise, there should be at least a partial repair of the decrement, characterized by return of the CMAP to the baseline amplitude of the first response in the train. During the 2 to 4 minutes after exercise, there is postactivation exhaustion that will again show a smooth decrement that can exceed that at rest. A consistent decrement of greater than 10% in two nerves with typical repair and postexercise exhaustion is necessary to support the diagnosis.31 In myasthenia gravis, there can be a decrement at rest if more severe disease, or a decrement after exercise (generally 1 minute) in more mild disease.


Case 11: Progressive Lower Greater Than Upper Limb Weakness
Differential Diagnosis
The primary symptoms and clinical findings are related to proximal limb weakness, which in this case appears to be most severe in the quadriceps where there is clear atrophy. There is also weakness in the finger flexors. Although the clinical history of hand weakness certainly raises the more common suspicion of median neuropathy at the wrist, the findings on this examination are more consistent with the long finger flexors. A unifying diagnosis for the motor-greater-than-sensory symptoms would be a polyradiculopathy or polyradiculoneuropathy. The mild sensory symptoms without significant clinical sensory findings can be relatively nonspecific. The proximal pattern of lower limb involvement would suggest a myopathy, and the specific pattern of quadriceps and finger flexor involvement would be relatively typical of inclusion body myositis (IBM). Other potential dystrophies such as proximal myotonic dystrophy (DM2) or inflammatory myopathies such as polymyositis or dermatomyositis are also possible. A motor neuron disease is possible but less likely in the absence of long tract signs. A disorder of neuromuscular transmission is possible and will need to be excluded with the EDX studies. NCSs and needle EMG were performed (Table 11-12).
Discussion
Motor NCSs were performed on distal motor nerves as well as the spinal accessory nerve. The primary purpose for spinal accessory stimulation was to perform repetitive stimulation, which in this case was negative. This would argue against but not entirely exclude a disorder of neuromuscular transmission. CMAP amplitudes were borderline or mildly reduced, with sparing of the conduction velocities and distal latencies. No motor dispersion or conduction blocks were noted. The sural response was absent suggesting some degree of sensory axon loss, whether part of a unifying diagnosis or as a separate peripheral neuropathy. The differential diagnosis has narrowed slightly, but EMG examination is critical for the diagnosis. The EMG provided many clues regarding the diagnosis, most notable of which were mixed MUPs in several muscles, primarily short duration and low amplitude, suggestive of a myopathy. Some large MUPs were also seen, which could indicate an underlying peripheral neuropathy or a chronic myopathy. Associated with this were fibrillation potentials, which in the setting of short MUPs would generally reflect some degree of myonecrosis, fiber splitting, or vacuolar changes. These findings would be consistent with an inflammatory myopathy, but the specific pattern again gives clues to differentiate polymyositis from IBM. The particular involvement of the quadriceps in the lower limbs is typical of IBM,24 and greater involvement of the forearm finger flexors in the upper limb is relatively common. In polymyositis the findings can be relatively diffuse but tend to be more proximally predominant in the upper and lower limbs. The clinical history also provides some clues in that the patient had previously attempted treatment with prednisone. Corticosteroids are not generally effective in IBM, and there is generally no clear pharmacologic treatment to suppress the process. Neurogenic abnormalities characterized by some large MUPs are not uncommonly seen in IBM. These large MUPs could represent either collateral sprouting to split fibers that have lost nerve supply over a period of time (as in a chronic myopathy), or in this case a probable underlying mild peripheral neuropathy. The sensory response can be helpful to separate these possibilities. IBM can be misdiagnosed as a progressive motor neuron disease if one focuses on the large MUPs and does not recognize the underlying short-duration, low-amplitude MUPs that can be present at lower levels of activation. In this case a biopsy was performed of the vastus lateralis, which was conclusive for IBM. Prednisone was discontinued because it can be a factor in type II fiber atrophy and the potential for increased weakness.16
Case 12: Acute Right Foot Drop
Differential Diagnosis
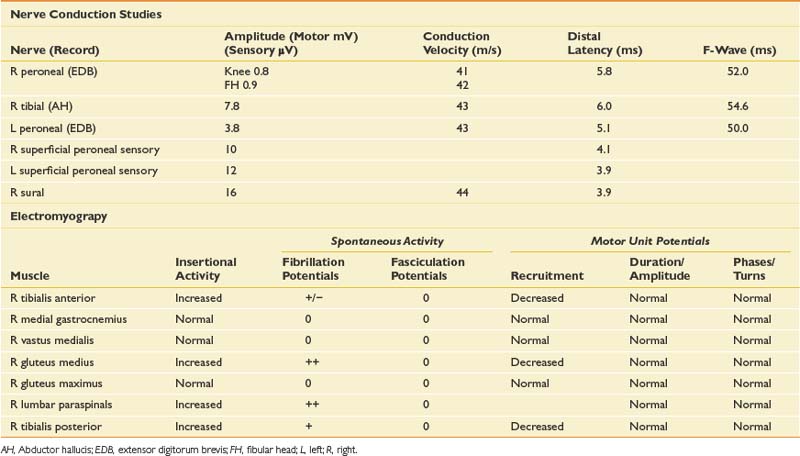
A very limited physical examination is reported because the design of the EDX study will mirror the findings that should be present on the physical examination. The most obvious diagnostic possibilities would be a peroneal neuropathy versus an L5 radiculopathy. The most common site of a peroneal neuropathy would be compressive at the fibular head, possibly in association with the weight loss. A sciatic involvement or less likely, a plexopathy, needs to be considered. With the smoking history and weight loss, a mass lesion such as a metastatic lung carcinoma leading to a cauda equina lesion is possible, but in this case the findings were purely unilateral and without sphincter disturbance. NCSs and needle examination were performed (Table 11-13).
Discussion
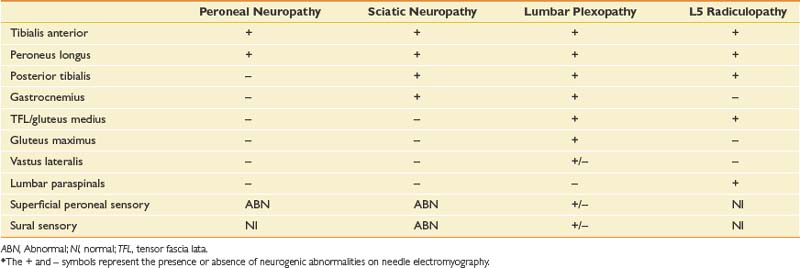
Table 11-14 describes the typical electrophysiologic findings for various causes of foot drop. The abnormalities in this case confirm an L5 radiculopathy. The NCSs, particularly the normal superficial peroneal sensory response, and the paraspinal abnormalities also argue for a radiculopathy rather than a peripheral nerve injury. It is important to perform a sensory study of a nerve in the root distribution for the clinically weak muscle or a corresponding sensory study for the reduced CMAP, or both. In this case the peroneal CMAP was reduced, and the normal superficial sensory response, in comparison with the contralateral limb, gave a clue to a presynaptic process. If the tibial CMAP amplitude had been reduced, then the sural study would have been more appropriate (because they both arise primarily from the S1 root). The other critical feature for the diagnosis of a radiculopathy is the presence of neurogenic abnormalities, preferably fibrillation potentials, in two or more limb muscles innervated by the same root but by different peripheral nerves. In this case there was involvement of distal L5 peroneal- and tibial-innervated muscles as well as a proximal superior gluteal–innervated L5 muscle. A sciatic neuropathy is excluded by the gluteus medius findings, the abnormal paraspinal findings, and the normal sensory responses.
Case 13: Hand Weakness, Worsening Gait, and Progressive Foot Drop
A 28-year-old man presents with a history of hand weakness, worsening gait over the past 3 to 4 years, and progressive foot drop with subjective reports of “cramping.” He denies any other pain or sensory symptoms. Occasionally he chokes on liquids but has no diplopia or dysarthria. The symptoms are relatively constant, although he does tend to trip more when fatigued. The medical history is significant for borderline diabetes. Family history is notable for some family members with “muscular dystrophy,” but the patient offers little detail. The symptoms have progressed to the point where he is falling weekly and is having difficulty with fine motor control of the hands, with “cramping” when grasping objects. This is especially severe during cold weather. The physical examination shows a well-nourished gentleman with some premature frontal balding. There is mild atrophy of the anterior compartment of the lower leg. Gait evaluation reveals moderate foot drop bilaterally, slightly worse on the right, with compensation by circumduction. He is unable to walk on heels and has difficulty walking on toes. Strength testing shows distally predominant weakness, particularly in the tibialis anterior. Grip strength is mildly reduced, but more notable is difficulty releasing the grasp consistent with grip myotonia. Percussion myotonia is noted in the thenar muscles. There are no sensory deficits, and muscle stretch reflexes are normal.
Differential Diagnosis
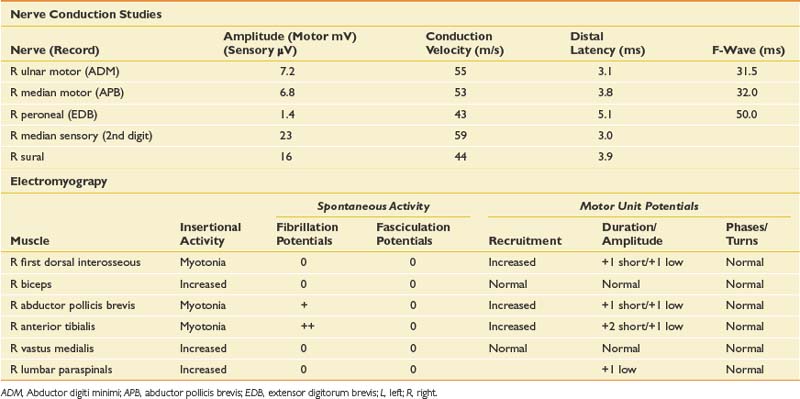
The primary feature of the clinical history and examination is that of distally predominant weakness without sensory deficits. Although a peripheral neuropathy or multiple compressive neuropathies are possible, the lack of sensory symptoms would make these less likely. Multifocal processes such as a hereditary neuropathy with liability to pressure palsies can present with multiple mononeuropathies and minimal sensory symptoms, but the history of gradual progression in this case would make this less likely. Multifocal motor neuropathy with conduction block is also possible. A motor neuron disease is within the differential, but a relatively rapidly progressive process such as ALS would be unlikely given the prolonged history. The history of difficulty with grasp relaxation is characteristic of a myotonic disorder, and the presence of clinical weakness would suggest, in this case, a disorder with both myotonia and a myopathy. NCSs and needle EMG were performed (Table 11-15).
Discussion
The primary finding on this study is the myotonia, which appears to be more predominant distally. In association with this are myopathic MUPs, again in a distal pattern. The potential etiology of myotonia is relatively broad, but with other clinical features such as weakness, and more importantly the pattern of weakness, the differential diagnosis can be narrowed (Boxes 11-2 and 11-3).
In this case, there is clear weakness in addition to the myotonia, which reinforces the suspicion of a distally predominant myopathy, most likely myotonic dystrophy (see Table 11-15). EDX testing is important to better define the degree and pattern of myotonia, as well as to define whether there is an associated myopathy. Specialized techniques such as repetitive stimulation, response to cooling, or response to exercise will generally confirm the diagnosis. Table 11-16 outlines the typical responses to these techniques with various myotonic disorders. Of particular note is the response to repetitive stimulation that occurs in many myotonic disorders. This is characterized by a decremental response at rest, which is very similar to that seen in neuromuscular junction disorders. It differs physiologically, however, in that it is likely caused by membrane inactivation.2,18 There is also a repair after brief exercise, but distinguishing it from neuromuscular junction disorders is a clear reduction of the CMAP amplitude immediately after exercise. This phenomenon is seen in most myotonic disorders, except in proximal myotonic myopathy. The absence of neuromuscular junction abnormalities and the proximal pattern of weakness are helpful characteristics that can be used to distinguish this disorder.27 This case is relatively typical of myotonic dystrophy, and this was confirmed with genetic testing.
Case 14: Progressive Lower Limb Weakness and Numbness
A 52-year-old woman presents with a 2-year history of progressive numbness and weakness involving the distal upper and lower limbs bilaterally. The symptoms initially started with numbness in the left side of her face, but then progressed to involve her feet and then her hands. She denies any pain but is finding it increasingly difficult to perform her activities of daily living; she trips and falls quite often when ambulating, and now uses a cane. She has also noted difficulty walking up stairs or getting up from a low chair. She is unable to perform fine motor activities with her hands, such as using buttons or holding her utensils. More recently she has developed dizziness if she gets up too quickly. She denies any family history of neurologic symptoms. She reports that the symptoms had been gradually progressive for several months but then seemed to progress quite suddenly (prompting her to seek medical evaluation). Her medical history is significant for hypertension and mild type 2 diabetes, which has been managed with dietary modification with good blood sugar control.
Differential Diagnosis
With unequivocal sensory symptoms and signs, anterior horn cell, muscle, and neuromuscular junction disease are all essentially excluded unless part of a coexisting process. Plexopathy is also unlikely because of the involvement of all four limbs. The clinical presentation is most suggestive of a peripheral neuropathy that could be length dependent. Given the examination findings of proximal weakness, however, a polyradiculoneuropathy or a multifocal neuropathy should also be considered. With a 2-year history of progressive symptoms, an inherited neuropathy and various causes of acquired neuropathy (including toxic, metabolic, infectious, paraneoplastic, immune mediated, or inflammatory) are all potential etiologies. The history of stepwise progression and the possible dysautonomia make chronic inflammatory demyelinating neuropathy (CIDP) a strong consideration. The NCS and EMG findings are shown in Table 11-17.
Discussion
NCSs in this case were diffusely abnormal, with prolonged distal latencies, markedly slowed conduction velocities, and low CMAP and SNAP amplitudes. The median motor study showed evidence of conduction block in the forearm, and the ulnar motor study showed conduction block between the below-elbow and wrist sites of stimulation. When evaluating for conduction block, it is important to rule out technical explanations for the drop in amplitude, such as understimulation at the proximal site or overstimulation at the distal site resulting in spread of current to other nerves and a volume-conducted response. F-waves were markedly prolonged, also suggesting proximal conduction block or slowing. Sensory responses were notable for preservation of the sural response with low median and ulnar sensory responses. This pattern of sural sparing is relatively typical for CIDP but not specific.12 Blink responses were also performed to evaluate for very proximal slowing, and were abnormal, with prolongation of both the R1 and R2 responses.
The classic electrophysiologic picture in CIDP is that of demyelination, evident on both motor and sensory NCSs. Focal demyelination is seen as conduction block or focal slowing, and often distal latencies are markedly prolonged and conduction velocities severely slowed with relative preservation of amplitude. Proximal responses are dispersed in comparison with responses obtained with distal stimulation (temporal dispersion), and this is the hallmark of demyelination (Figure 11-5). Prolonged F-waves may be one of the earliest signs of more proximal slowing, which can alert the clinician that this is in fact a polyradiculoneuropathy, as opposed to a length-dependent process. Often there is associated axonal loss (and in rare cases this is the predominant picture). This is evident as loss of amplitude on NCSs, and fibrillation potentials and polyphasic large MUPs on EMG examination. Usually changes are more severe distally, although paraspinal muscles are also affected, indicating nerve root involvement.
One of the most important diagnoses to exclude when evaluating a patient with possible CIDP is inherited demyelinating sensorimotor neuropathy (Charcot-Marie-Tooth [CMT] disease type I, III, or IV), because this has important treatment implications. Many patients with mild forms of CMT disease are relatively asymptomatic and can go undiagnosed, making the family history less obvious. The history is slowly progressive, often dating back to childhood when a patient might recall being somewhat clumsy or unable to keep up with other children. This is different from CIDP, in which the onset is relatively abrupt, and has a progressive, stepwise or recurrent course with a positive response to immunotherapy. As in this case, autonomic dysfunction can be seen with CIDP. Conduction block, significant temporal dispersion, and evidence of proximal slowing (prolonged F-waves or prolonged blink reflexes) are key findings on electrophysiologic testing that help differentiate acquired forms of demyelinating neuropathies from CMT disease. Similarly, on needle examination, markedly reduced recruitment in the absence of significant axonal loss (indicative of conduction block) is more suggestive of an acquired process. The clinical and EDX criteria for CIDP are outlined in Box 11-4.
Clinical Diagnostic Criteria
Inclusion Criteria
Electrodiagnostic Criteria
Case 15: Progressive Weakness
A 65-year-old man presents with a 6-month history of progressive weakness characterized by difficulty rising from a chair and ascending stairs. He denies pain but does report mild “numbness” of the toes. There are no visual or bulbar symptoms. Bladder and bowel function are normal. The medical history is significant for well-controlled diabetes mellitus and chronic obstructive pulmonary disease. He has a 40–pack-year smoking history. The patient also reports a 25-lb weight loss during this period. Physical examination reveals mild proximal weakness in hip flexion, knee extension, and hip abduction (Medical Research Council 4/5) with difficulty rising from a squat position. There is also mild weakness in shoulder internal and external rotation bilaterally. Cranial nerve examination is normal. Sensory examination is grossly intact to light touch, pinprick, and proprioception. Muscle stretch reflexes are absent throughout.
Differential Diagnosis
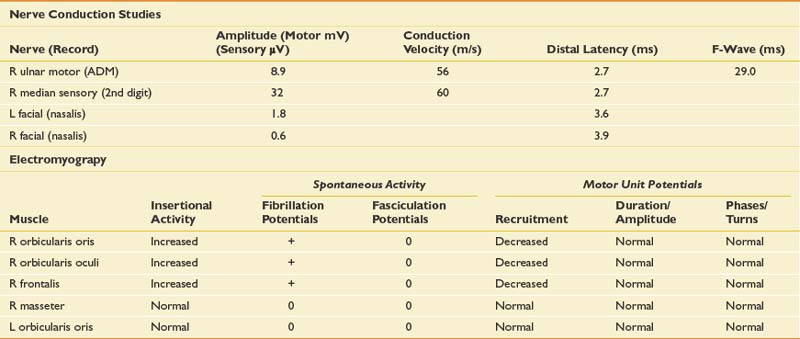
The primary finding on clinical examination is proximal muscle weakness, with the leading diagnostic possibilities being myopathy, neuromuscular junction disorder, motor neuron disease, polyradiculoneuropathy, or debility. The relatively symmetric proximal findings with loss of reflexes make motor neuron disease less likely. The mild sensory symptoms and loss of muscle stretch reflexes raise the possibility of a polyradiculoneuropathy, which can be seen with diabetes in association with prominent weight loss versus coexisting peripheral neuropathy and myopathy. An inflammatory myopathy such as inclusion body myositis or polymyositis remains in the differential. Given the prominent weight loss, the possibility of a paraneoplastic process involving any of these muscles or nerves or a presynaptic neuromuscular junction disorder must be considered. Motor and sensory NCSs, repetitive stimulation studies, and needle EMG were performed (Table 11-18).
Discussion
NCSs showed markedly reduced CMAP amplitudes as well as borderline sensory amplitudes. The conduction velocities were relatively spared, and there was no evidence for motor conduction blocks or dispersion. Needle examination showed varying MUPs throughout without abnormal spontaneous activity. MUP morphology revealed an increased proportion of short-duration, low-amplitude MUPs in some proximal muscles. If the electromyographer were to quit at this point without performing repetitive stimulation, the conclusion could be that the patient has a proximally predominant myopathy in addition to a mild underlying peripheral neuropathy. This conclusion would be incorrect, and some inconsistencies in this regard need to be recognized. The degree of weakness was relatively mild, but the CMAPs were markedly reduced, clearly out of proportion to the clinical examination. In addition, there was prominent MUP variation that would not be expected to this degree in a myopathy. The examiner performed repetitive stimulation on distal upper and lower limb muscles as well as the weak quadriceps. As noted in Figure 11-6, there was a marked increase of the CMAP amplitude immediately after brief exercise, with greater than doubling of the baseline response (greater than 200% facilitation). This was associated with partial repair of the subsequent responses of the train and further decrement out to 3 minutes after exercise. This pattern of low-amplitude baseline CMAPs, decrement to repetitive stimulation, and greater than 200% facilitation after brief exercise is consistent with a presynaptic neuromuscular junction disorder, specifically Lambert-Eaton myasthenic syndrome (LEMS).8 A mild underlying peripheral neuropathy is also probable. Further questioning of the patient revealed prominent small-fiber symptoms such as dry mouth, dry eyes, and erectile dysfunction, which may be seen either from the LEMS or a small-fiber peripheral neuropathy. On reexamination, the muscle stretch reflexes were relatively normal after brief muscle contraction, again consistent with a presynaptic neuromuscular junction disorder. P/Q voltage-gated calcium channel antibodies were positive in this patient. They are found in approximately 90% of patients with LEMS.20 The patient was found to have small cell lung carcinoma.
Case 16: “Drooping” of the Right Face
A 54-year-old otherwise healthy man awoke and was noted by his wife to have “drooping” of the right face. He noted the inability to close the right eye completely and was drooling out of the right side of the mouth while attempting to brush his teeth. He was brought to the emergency department, where a comprehensive neurologic examination showed right facial weakness involving all peripheral facial branches including the frontalis. There was trace movement of the orbicularis oculi and oris. The remainder of the cranial nerve examination including ocular movements, jaw strength, facial sensory, and palatal and tongue movements was normal. Mild hyperacusis was reported. The patient did report a moderate headache, and a computed tomography scan was performed that was normal. The patient received a diagnosis of right facial neuropathy and was told to contact his primary physician for follow-up the next day. The patient was referred to the EMG laboratory 13 days after onset to determine the prognosis for recovery and exclude other potential causes.
Differential Diagnosis
The clinical history and physical examination are consistent with a lower motor neuron process involving the right facial nerve. The most likely cause would be idiopathic facial neuropathy (Bell’s palsy). Other potential causes for acute-onset facial weakness include an infectious process such as herpes zoster or Lyme disease, granulomatous disorders such as sarcoid, or other inflammatory- or immune-mediated processes such as acute inflammatory demyelinating polyneuropathy or mononeuritis multiplex. Subacute or chronic facial paralysis could be seen with a tumor of the cerebellar pontine angle or structural process around the ear or parotid gland (cholesteatoma, salivary tumor). An upper motor neuron process is unlikely because the frontalis would typically be relatively spared in such cases. NCSs were performed as in Table 11-19. Blink reflex results are noted in Table 11-20.

Discussion
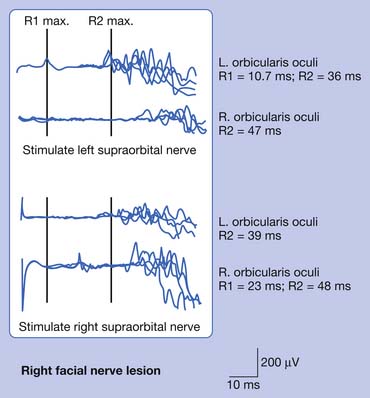
Bilateral facial NCSs were performed, showing markedly reduced right facial CMAP amplitude. This alone would support a lower motor neuron process and exclude central causes. The left facial response was normal, and limited upper limb studies were performed to exclude a more diffuse polyradiculoneuropathy. Blink reflexes (Figure 11-7) revealed marked prolongation of the right R1 and ipsilateral R2 components (with normal contralateral R2 latency) when stimulating the right supraorbital nerve. When stimulating the left supraorbital nerve, there was prolongation of the contralateral R2 response. These findings are consistent with left efferent pathway involvement, specifically of the facial nerve. The needle examination revealed sparse fibrillation potentials with markedly reduced recruitment in right facial–innervated muscles. The masseter was examined to exclude more diffuse cranial neuropathies, and was normal.
Recovery of facial function can be reasonably well predicted by physical examination and EDX abnormalities. In a study of the natural history of Bell’s palsy, Peitersen23 found complete recovery in 71% of patients overall, and in those with incomplete paresis at presentation, 94% recovered fully (as opposed to only 61% with complete paresis at presentation). Two thirds of patients recovered function within 3 months, but after 6 months no additional patients regained normal mimical function. EDX findings, particularly the CMAP amplitude, can also assist with prediction of recovery. An amplitude of 10% or less after 7 days in comparison with the contralateral response would generally predict poor recovery, whereas an amplitude of greater than 30% predicted full recovery within 3 months.21 Those with amplitudes of 10% to 30% had incomplete recovery over 6 to 8 months with some residual weakness or additional symptoms such as synkinesis or “crocodile tears.” Some have reported the utilization of stimulus thresholds within the first 72 hours.10 In this study, those with a stimulation threshold of greater than 10 mA did more poorly. In this case, the patient had some sparing of motor function as well as a facial CMAP amplitude of greater than 30%, arguing for complete recovery.
1. Allen D.C., Arunachalam R., Mills K.R. Critical illness myopathy: further evidence from muscle-fiber excitability studies of an acquired channelopathy. Muscle Nerve. 2008;37:14-22.
2. Aminoff M.J., Layzer R.B., Satya-Murti S., et al. The declining electrical response of muscle to repetitive nerve stimulation in myotonia. Neurology. 1977;27:812-816.
3. Anonymous. Guidelines in electrodiagnostic medicine. The electrodiagnostic medicine consultation. Muscle Nerve Suppl. 1999;8:S73-S90.
4. Anonymous. Practice parameter: electrodiagnostic studies in ulnar neuropathy at the elbow: American Association of Electrodiagnostic Medicine, American Academy of Neurology, and American Academy of Physical Medicine and Rehabilitation. Neurology. 1999;52:688-690.
5. Anonymous. Practice parameter for electrodiagnostic studies in ulnar neuropathy at the elbow: summary statement: American Association of Electrodiagnostic Medicine, American Academy of Neurology, American Academy of Physical Medicine and Rehabilitation. Muscle Nerve. 1999;22:408-411.
6. Boon A.J., Alsharif K.I., Harper C.M., et al. Ultrasound-guided needle EMG of the diaphragm: technique description and case report. Muscle Nerve. 2008;38:1623-1626.
7. Brooks B.R. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124(suppl):96-107.
8. Chiou-Tan F.Y., Tim R.W., Gilchrist J.M., et al. Literature review of the usefulness of repetitive nerve stimulation and single fiber EMG in the electrodiagnostic evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2001;24:1239-1247.
9. Daube J., Rubin D. Clinical neurophysiology, New York. Oxford University Press; 2009.
10. Devi S., Challenor Y., Duarte N., et al. Prognostic value of minimal excitability of facial nerve in Bell’s palsy. J Neurol Neurosurg Psychiatry. 1978;41:649-652.
11. Dyck P.J., Dyck P.J., Grant I.A., et al. Ten steps in characterizing and diagnosing patients with peripheral neuropathy. Neurology. 1996;47:10-17.
12. Dyck P.J., Lais A.C., Ohta M., et al. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975;50:621-637.
13. Dyck P.J., Windebank A.J. Diabetic and nondiabetic lumbosacral radiculoplexus neuropathies: new insights into pathophysiology and treatment. Muscle Nerve. 2002;25:477-491.
14. Goodman B.P., Boon A.J. Critical illness neuromyopathy. Phys Med Rehabil Clin N Am. 2008;19:97-110.
15. Harper C.M.Jr., Thomas J.E., Cascino T.L., et al. Distinction between neoplastic and radiation-induced brachial plexopathy, with emphasis on the role of EMG. Neurology. 1989;39:502-506.
16. Kaminski H.J., Ruff R.R. Endocrine myopathies. New York: McGraw-Hill; 1994.
17. Katz J.S., Wolfe G.I., Andersson P.B., et al. Brachial amyotrophic diplegia: a slowly progressive motor neuron disorder. Neurology. 1999;53:1071-1076.
18. Kuntzer T., Flocard F., Vial C., et al. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000;23:1089-1094.
19. Lederman R.J., Wilbourn A.J. Brachial plexopathy: recurrent cancer or radiation? Neurology. 1984;34:1331-1335.
20. Lennon V.A., Lambert E.H. Autoantibodies bind solubilized calcium channel-omega-conotoxin complexes from small cell lung carcinoma: a diagnostic aid for Lambert-Eaton myasthenic syndrome. Mayo Clin Proc. 1989;64:1498-1504.
21. Olsen P.Z. Prediction of recovery in Bell’s palsy. Acta Neurol Scand Suppl. 1975;61:1-121.
22. Özdemir C., Young R.R. The results to be expected from electrical testing in the diagnosis of myasthenia gravis. Ann N Y Acad Sci. 1976;274:203-222.
23. Peitersen E. Bell’s palsy: the spontaneous course of 2,500 peripheral facial nerve palsies of different etiologies. Acta Otolaryngol Suppl 549. 2002:4-30.
24. Phillips B.A., Cala L.A., Thickbroom G.W., et al. Patterns of muscle involvement in inclusion body myositis: clinical and magnetic resonance imaging study. Muscle Nerve. 2001;24:1526-1534.
25. Resman-Gaspersc A., Podnar S. Phrenic nerve conduction studies: technical aspects and normative data. Muscle Nerve. 2008;37:36-41.
26. Rubin D.I., Harper C.M., Auger R.G. Trigeminal nerve repetitive stimulation in myasthenia gravis. Muscle Nerve. 2004;29:591-596.
27. Sanders H.W., Scelsa S.N., Conigliari M.F., et al. The short exercise test is normal in proximal myotonic myopathy. Clin Neurophysiol. 2000;111:362-366.
28. Sorenson E.J., Stalker A.P., Kurland L.T., et al. Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925 to 1998 [see comment]. Neurology. 2002;59:280-282.
29. Stevens J.C. AAEM minimonograph no. 26: The electrodiagnosis of carpal tunnel syndrome: American Association of Electrodiagnostic Medicine. Muscle Nerve. 1997;20:1477-1486.
30. Stevens J.C., Smith B.E., Weaver A.L., et al. Symptoms of 100 patients with electromyographically verified carpal tunnel syndrome. Muscle Nerve. 1999;22:1448-1456.
31. Strommen J. Electrodiagnosis in neuromuscular junction disorders. Phys Med Rehabil. 1999;13:281-306.
32. Turner J.W., Parsonage M.J. Neuralgic amyotrophy (paralytic brachial neuritis); with special reference to prognosis. Lancet. 1957;273:209-212.