[level-membership-for-neurology-category]
Chapter 4 Electrodiagnostic Findings in Neuromuscular Disorders
Details of the electrodiagnostic findings in various neuromuscular disorders are outlined within the case studies in this book. The following is a brief summary of these findings in the most common neuromuscular disorders.
FOCAL MONONEUROPATHIES
Demyelinative Mononeuropathy
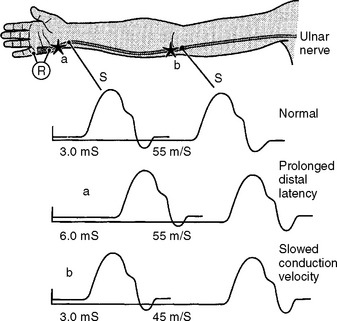
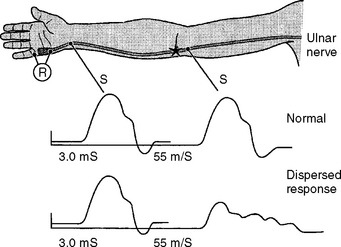
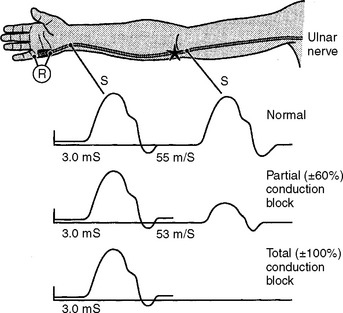

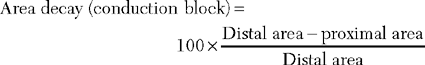
There are several limitations to the definitive diagnosis of demyelinative conduction block:
| Definite in Any Nerve* |
* Caution should be taken in evaluating the tibial nerve, where stimulation at the knee can be submaximal, resulting in 50% or at times greater than 50% drop in amplitude and area, especially in overweight and very tall patients.
Axon-Loss Mononeuropathy
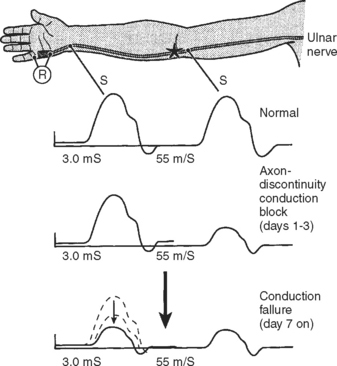
Following acute focal axonal damage, the distal nerve segment undergoes wallerian degeneration. However, early after axonal transaction, the distal axon remains excitable. Hence, stimulation distal to the lesion elicits a normal CMAP, whereas proximal stimulation elicits an absent response (complete conduction block) when the lesion is total and reduced CMAP amplitude (partial conduction block) when the lesion is incomplete (Figure 4-4). In an attempt to distinguish this pattern from a demyelinative conduction block, some refer to this pattern as an axonal noncontinuity, early axon loss, or axon discontinuity conduction block.
Wallerian degeneration of the axons distal to the nerve lesions is completed in 7–11 days. In the first 1–2 days, the distal CMAP and SNAP are normal. The distal CMAP amplitude then decreases and reaches its nadir in 5–6 days, while the distal SNAP amplitude lags slightly behind. It starts declining in amplitude after 4–5 days and reaches its nadir in 10–11 days (Figure 4-5). The earlier decline of the CMAP amplitude comparing to the SNAP amplitude following axon-loss nerve lesion is likely related to the early neuromuscular transmission failure that affects the recording of the CMAP amplitudes only. This is supported by the fact that MNAPs, recorded directly from nerve trunks, follow the time course of SNAPs.
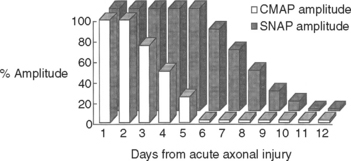
Figure 4-5 Effect of wallerian degeneration on distal CMAP and SNAP amplitudes following acute nerve lesion.
On motor NCS, a conduction block is present soon after axonal injury. However, as the distal axons undergo wallerian degeneration, this is replaced by unelicitable or low CMAP amplitudes with both distal and proximal stimulations corresponding to complete or partial motor axonal loss lesions respectively (see Figure 4-4). At this time, the distal CMAP amplitude is a reliable semiquantitative estimate of the amount of axonal loss in peripheral nerve lesions. In the chronic phases of partial axonal nerve lesions with reinnervation via collateral sprouting, the CMAP may improve to reach normal or near normal values giving a false indication of a milder degree of original axonal loss.
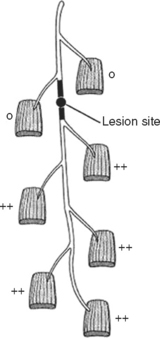
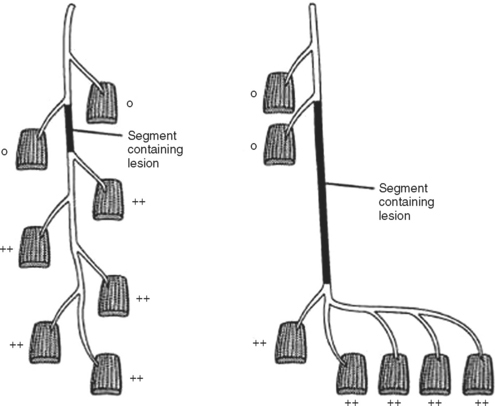
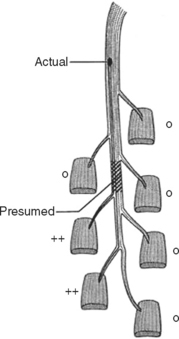
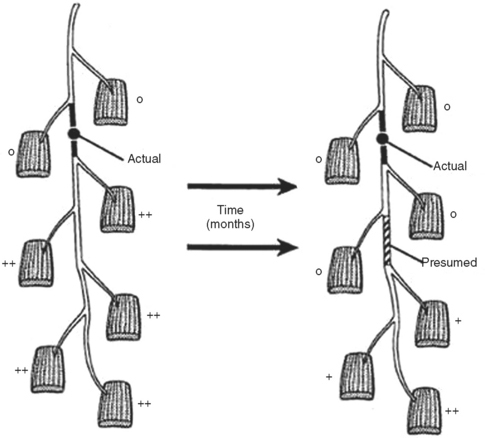
In contrast to demyelinating or mixed mononeuropathies, pure axon-loss peripheral nerve lesions cannot be localized by NCSs when studied after the completion of wallerian degeneration, since they are not associated with focal conduction slowing or block. The identification of conduction block in the early days of axonal loss is extremely helpful in localizing a peripheral nerve injury. Waiting for the completion of wallerian degeneration results in diffusely low or unevoked CMAPs (regardless of stimulation site), which does not allow for accurate localization of the injury site. Localizing a purely axon-loss mononeuropathy after the completion of wallerian degeneration depends on needle EMG, with principles that are similar to manual muscle strength testing used during the neurological examination. Typically, the needle EMG reveals neurogenic changes (fibrillation potentials, reduced MUAP recruitment, chronic neurogenic MUAP morphology changes) that are limited to muscles innervated by the injured nerve distal to the site of the lesion (Figure 4-6). In contrast, muscles innervated proximal to the lesion remain normal. Unfortunately, attempting to localize axon loss lesions solely by needle EMG has several shortcomings that may result in poor localization or, sometimes, mislocalization of the site of the nerve lesion. These include the following scenarios:
RADICULOPATHIES AND PLEXOPATHIES
The dorsal root ganglia contain unipolar sensory neurons with a peripheral and a central axon. In radiculopathies associated with axonal loss due to lesions of the proximal sensory axons, the distal sensory axons do not degenerate since the dorsal root neurons usually escape injury. Hence, the SNAP remains normal despite sensory loss and degeneration of proximal sensory axons. When the motor axons within the ventral roots are also injured, radiculopathies exhibits signs of motor axon degeneration including abnormal needle EMG and, when severe, low-amplitude CMAPs.
GENERALIZED POLYNEUROPATHIES
Demyelinating Polyneuropathies
With distal stimulation, the CMAP amplitude is mildly or moderately reduced because of abnormal temporal dispersion and phase cancellation, and the distal latency is delayed because of demyelination. With more proximal stimulation, the CMAP amplitude is lower due to temporal dispersion and conduction block along some fibers. The proximal conduction velocity is markedly slowed because of increased probability for the nerve action potentials to pass through demyelinated segments (Figure 4-10C).

Axonal Polyneuropathies
Axonal polyneuropathies produce length-dependent dying-back degeneration of axons. The major change on NCS is a decrease of the CMAP and SNAP amplitudes, more marked in the lower extremities (see Figure 4-10B). In contrast, conduction velocities and distal latencies are normal. As with axon loss mononeuropathies, selective loss of many fast-conducting fibers associated with more than a 50% reduction in mean CMAP amplitude can slow conduction velocity to 70–80% of normal value.
In axon-loss polyneuropathy, the needle EMG is most useful in depicting the temporal profile of the illness. Fibrillation potentials typically develop within 2–3 weeks of an acute neuropathy, and reinnervated MUAPs become apparent within 1–2 months. In acute polyneuropathies, needle EMG during the first few weeks of illness may show only reduced recruitment of MUAPs in weak muscles with normal MUAP morphology and no spontaneous activity. In relatively active or progressive axon-loss polyneuropathies, a combination of fibrillation potentials with reduced recruitment of reinnervated MUAPs is most prominent distally. In chronic and very slowly progressive polyneuropathies, reinnervation may completely keep pace with active denervation yielding little or no fibrillation potentials but reduced recruitment of reinnervated MUAPs.
ANTERIOR HORN CELL DISORDERS
Needle EMG is the most important electrodiagnostic study for providing evidence of generalized lower motor neuron degeneration. Early in the course of the illness, denervation in clinically normal muscles and limbs is most useful in establishing early dissemination of denervation. Needle EMG in amyotrophic lateral sclerosis often shows signs of active denervation (fibrillation and fasciculation potentials), chronic denervation (reinnervated and unstable MUAPs), and reduced MUAP recruitment. Lambert’s original criteria for diagnosis include detecting fibrillation and fasciculation potentials in muscles of the lower as well as the upper extremities or in the extremities as well as the head. These criteria evolved over the years into denervation at least three extremities or two extremities and cranial muscles (the head and neck considered an extremity). Although lower motor neuron degeneration ultimately affects almost the entire neuraxis (brainstem and cervical, thoracic, or lumbosacral segments of spinal cord), the early phases of the illness are often characterized by limited and more focal weakness. The revised El Escorial criteria recommend that needle EMG signs of lower motor neuron degeneration should be present in at least two of the four central nervous system regions, i.e., the brainstem, cervical, thoracic, or lumbosacral regions.
Brooks BR, Miller RG, Swash M, Munsat TL, for the World Federation of Neurology Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293-299.
Campbell WW, Pridgeon RM, Sahni KS. Short segment incremental studies in the evaluation of ulnar neuropathy at the elbow. Muscle Nerve. 1992;15:1050-1054.
Gordon PH, Wilbourn AJ. Early electrodiagnostic findings in Guillain-Barré syndrome. Arch Neurol. 2001;58:913-917.
Katirji MB, Agrawal R, Kantra TA. The human cervical myotomes. An anatomical correlation between electromyography and CT/myelography. Muscle Nerve. 1988;11:1070-1073.
Kimura J. The carpal tunnel syndrome: localization of conduction abnormalities within the distal segment of the median nerve. Brain. 1979;102:619-635.
Lacomis D. Electrodiagnostic approach to the patient with suspected myopathy. Neurol Clin N Am. 2002;20:587-603.
Lambert EH, Mulder DW. Electromyographic studies in amyotrophic lateral sclerosis. Mayo Clin Proc. 1957;32:441-446.
McIntosh KA, Preston DC, Logigian EL. Short segment incremental studies to localize ulnar entrapments at the wrist. Neurology. 1998;50:303-306.
Lambert EH. Electromyography in amyotrophic lateral sclerosis. In: Norris FHJr, Kurland LT, editors. Motor neuron diseases. New York: Grune and Stratton; 1969:135-153.
Chad DA. Electrodiagnostic approach to the patient with suspected motor neuron disease. Neurol Clin N Am. 2002;20:527-555.
Rhee RK, England JD, Sumner AJ. Computer simulation of conduction block: effects produced by actual block versus interphase cancellation. Ann Neurol. 1990;28:146-159.
Wilbourn AJ. The electrodiagnostic examination in myopathies. J Clin Neurophysiol. 1993;10:132-148.
Wilbourn AJ, Aminoff MJ. The electrodiagnostic examination in patients with radiculopathies. Muscle Nerve. 1998;21:1612-1631.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 4 Electrodiagnostic Findings in Neuromuscular Disorders
Details of the electrodiagnostic findings in various neuromuscular disorders are outlined within the case studies in this book. The following is a brief summary of these findings in the most common neuromuscular disorders.
FOCAL MONONEUROPATHIES
Demyelinative Mononeuropathy
There are several limitations to the definitive diagnosis of demyelinative conduction block:
| Definite in Any Nerve* |
* Caution should be taken in evaluating the tibial nerve, where stimulation at the knee can be submaximal, resulting in 50% or at times greater than 50% drop in amplitude and area, especially in overweight and very tall patients.
Axon-Loss Mononeuropathy
Following acute focal axonal damage, the distal nerve segment undergoes wallerian degeneration. However, early after axonal transaction, the distal axon remains excitable. Hence, stimulation distal to the lesion elicits a normal CMAP, whereas proximal stimulation elicits an absent response (complete conduction block) when the lesion is total and reduced CMAP amplitude (partial conduction block) when the lesion is incomplete (Figure 4-4). In an attempt to distinguish this pattern from a demyelinative conduction block, some refer to this pattern as an axonal noncontinuity, early axon loss, or axon discontinuity conduction block.
Wallerian degeneration of the axons distal to the nerve lesions is completed in 7–11 days. In the first 1–2 days, the distal CMAP and SNAP are normal. The distal CMAP amplitude then decreases and reaches its nadir in 5–6 days, while the distal SNAP amplitude lags slightly behind. It starts declining in amplitude after 4–5 days and reaches its nadir in 10–11 days (Figure 4-5). The earlier decline of the CMAP amplitude comparing to the SNAP amplitude following axon-loss nerve lesion is likely related to the early neuromuscular transmission failure that affects the recording of the CMAP amplitudes only. This is supported by the fact that MNAPs, recorded directly from nerve trunks, follow the time course of SNAPs.
Figure 4-5 Effect of wallerian degeneration on distal CMAP and SNAP amplitudes following acute nerve lesion.
On motor NCS, a conduction block is present soon after axonal injury. However, as the distal axons undergo wallerian degeneration, this is replaced by unelicitable or low CMAP amplitudes with both distal and proximal stimulations corresponding to complete or partial motor axonal loss lesions respectively (see Figure 4-4
[/not-level-membership-for-neurology-category]
