133 Ehlers–Danlos syndrome
Salient features
History
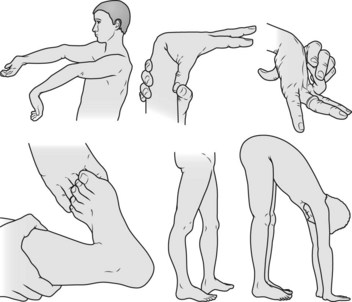
Examination
Skin
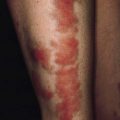
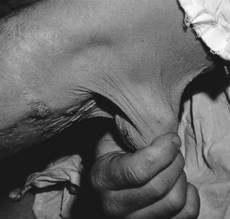
• The skin over the neck, axillae and groin is smooth and elastic (Fig. 133.2). It can be stretched and, when released, it returns immediately to its normal position. Late in the disease it becomes lax, wrinkled and hangs in folds.

Diagnosis
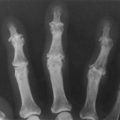
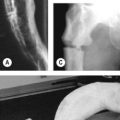
This patient has hypermobile joints (lesion) caused by Ehlers–Danlos syndrome (aetiology).
Advanced-level questions
What do you know about Ehlers–Danlos syndrome?
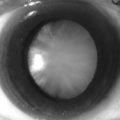
Type VI (kyphoscoliotic type). The most common and autosomal recessive form of inheritance. There is a defect in the enzyme lysyl hydroxylase, which is necessary for hydroxylation of lysyl residues during collagen synthesis. Only collagen types 1 and 3 are affected, and collagen types 2, 4 and 6 are normal. The predominant clinical features are ocular fragility with rupture of the cornea and retinal detachment. Some of these patients respond to ascorbic acid.
Type IV (‘vascular type’). This is caused by a defect in the structural gene for collagen type 3 (COL3A1) and is inherited as an autosomal dominant trait. As type 3 collagen is present largely in blood vessels and intestines, the typical features of this syndrome include easy bruising; thin skin with visible veins; characteristic facial features; rupture of the large arteries, uterus or colon; and reduced life expectancy. This is the most severe form of Ehlers–Danlos syndrome (N Engl J Med 2000;342:673–80). Loeys–Dietz syndrome overlaps with this type.
Type VII. A defect in the conversion of procollagen to collagen, caused by the mutation that affects one of the two type 1 collagen genes (COL1A1 and COL1A2). Although in the single mutant allele, only 50% of type 1 collagen chains are affected, heterozygotes manifest the syndrome because these abnormal chains interfere with the formation of normal collagen helices.
Type IX. This is caused by a defect in copper metabolism, with high levels of copper in the cells but low levels of serum copper and caeruloplasmin. Because the gene influencing copper metabolism is on the X chromosome, it is inherited as an X-linked recessive trait. This disease illustrates how copper metabolism can affect connective tissue.
Classical or types I and II. Null alleles of the COL5A1 gene encoding type V collagen are a cause of the classical form. A single base mutation in COL5A2 causes type II. Mutations in a gene that encodes tenascin-X, which is neither a collagen nor a collagen-modifying protein, can result in a variation of the classic form of the Ehlers–Danlos syndrome.
Asociación Sindromes de Ehlers-Danlos e Hiperlaxitud (www.asedh.org): Spanish patient support group.
Association Francaise des Syndromes d’Ehlers-Danlos (www.afsed.com): French patient support group.
Ehlers-Danlos Foundation of New Zealand (www.edfnz.org.nz): New Zealand patient support group.
Ehlers-Danlos National Foundation (www.ednf.org): US patient support group.
Ehlers-Danlos Support Group (www.ehlers-danlos.org): UK patient support group.