[level-membership-for-cardiovascular-category]
Chapter 12 Effects of high altitude
ALTITUDE AND OXYGEN TRANSPORT
Effects of altitude on plasma oxygen uptake
The standard atmospheric pressure of 760 mmHg (101 kPa) at sea level reflects the weight exerted by the gas molecules that make up the air, under gravitational force. As one ascends from sea level, the air becomes progressively less compressed and so the constituent gas molecules become less tightly packed. In consequence, a given volume of inspired air contains fewer molecules of all gases, including oxygen. The relationship between altitude and atmospheric pressure is not a strictly linear one because the air volume increases in 3 dimensions but, in rough terms, pressure falls by around 100 mmHg for every 1000 m (3300 ft) of ascent up to 3000 m (10 000 ft) (Fig. 12.1).
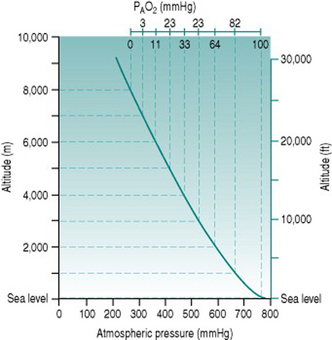
The absolute change in oxygen availability imposed by a given ascent can be calculated easily. Since oxygen represents 21% of normal air, the partial pressure of oxygen (PO2) at sea level is (21%.760) or 160 mmHg. Once inspired, the air becomes saturated with water vapour (partial pressure 47 mmHg) so that the total gas pressure is reduced to 713 mmHg and PO2 falls to (21%.713) or 150 mmHg. In the alveoli, the oxygen is diluted further by approximately 50 mmHg, due primarily to the presence of carbon dioxide, resulting in a local PO2 (PAO2) of around 100 mmHg: at equilibrium with the plasma, arterial PO2 (PaO2) is, therefore, usually also around 100 mmHg.
Effects of altitude on oxygen carriage
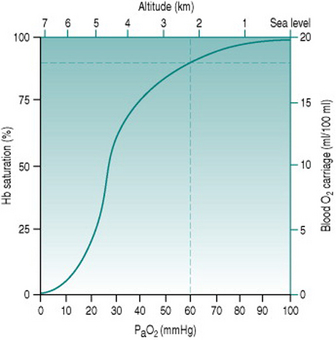
Because of the sigmoid shape of the haemoglobin dissociation curve, the falls in PaO2 associated with acute exposure to altitudes up to around 2000 m (6700 ft) cause only a slight reduction in haemoglobin saturation (Fig. 12.2) and so do not reduce oxygen delivery at rest. During exercise, however, the combination of reduced binding and reduced pulmonary capillary transit time leads to a greater degree of desaturation that is proportional both to altitude and to cardiac output. Thus, the threshold altitude for oxygen limitation of maximum exercise in sedentary individuals is typically around 1500 m (5000 ft), but, in trained athletes with substantially greater cardiac outputs, maximum work capacity begins to fall at much lower altitudes (Johnson et al 1994). In the only Summer Olympics held at a significant altitude, in Mexico City in 1968, winning times for all track events longer than 800 m were well above the existing records.
COMPENSATION FOR HYPOXIA
Respiratory stimulation
The reduced oxygen carriage associated with moderate altitude results in more rapid fatigue during exercise, but no respiratory compensation occurs because ventilation is still driven by the central chemoreceptors. These respond to rises in local proton concentration (that is, reduced pH) secondary to arterial carbon dioxide diffusing into the hindbrain, but are insensitive to hypoxia. The peripheral chemoreceptors responsible for monitoring arterial oxygen status are triggered only when PaO2 falls to around 60 mmHg which, as we saw earlier (p. 145), corresponds to an approximate altitude of 2300 m or 7600 ft. At or above this height, chemoreceptor stimulation initiates increased minute ventilation the magnitude of which depends both on the initial PaO2 and on whether increased ventilation is able to restore PaO2 to a value above 60 mmHg.
Haematological changes
Over the first few days of exposure to hypoxia, haematological changes increase the capacity of the red blood cell pool to deliver oxygen. The alkalosis produced by hypocapnia increases red cell expression of 2,3-diphosphglycerate (DPG), which shifts the haemoglobin dissociation curve to the right and increases oxygen unloading in the systemic capillaries (Fig. 12.3). Simultaneously, erythropoietin release from the renal medulla in response to hypoxia stimulates red cell production, so that haematocrit rises progressively over the ensuing several weeks.
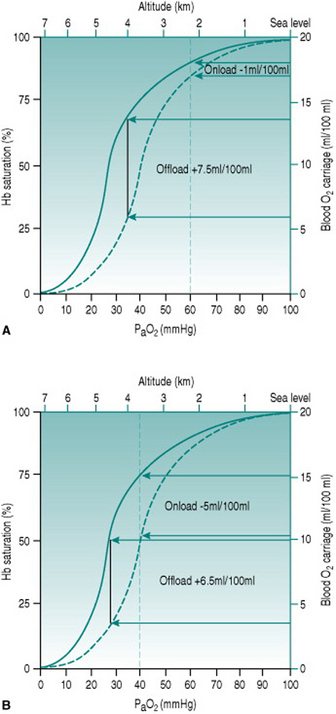
Figure 12.3 Effect of altitude on the efficiency of DPG expression as an aid to oxygen delivery. Note that the values for PaO2 represent those occurring at the respective altitudes before any compensation. (a) At altitude 2200 m (PaO2 60 mmHg) there is a doubling of oxygen offloading, while onloading is virtually unaffected. The net tissue gain is 6.5 ml oxygen/100 ml blood. (b) At altitude 3500 m (PaO2 40 mmHg), by contrast, the improved offloading is almost obviated by reduced onloading. The net tissue gain now is only 1.5 ml oxygen/100 ml blood.
ALTITUDE AND WORK CAPACITY
Benefits of adaptation for exercise capacity at high altitude
The spectrum of compensatory processes means that fully acclimated individuals have vastly better capacities for work than when they first arrive at altitude. They are, however, still not able to achieve workloads identical to those that were possible at sea level. The compensations that occur cannot overcome fully the reduced haemoglobin saturation imposed by a low PaO2 and, in addition, the adaptive processes themselves impose some limits of circulatory efficiency. One problem is that increased haematocrit increases blood viscosity and so increases cardiac workload for a given cardiac output. In addition, the rightwards shift of the haemoglobin dissociation curve induced by DPG has the effect of, under moderate-to-severely hypoxic conditions, reducing pulmonary loading of oxygen as well as increasing the unloading process (Fig. 12.3). Finally, since even slight alkalosis shifts the dissociation curve to the left, the profound hypocapnia seen in fully acclimated individuals may actually reverse the effect of DPG and reduce capillary oxygen unloading.
Benefits of adaptation for exercise capacity at low altitude
The greatest significance for athletic performance of adaptations to high altitude is its potential effect on performance at low altitude. Here, the positive effects on oxygen delivery of DPG and raised haematocrit can be exploited without interference from reduced ambient oxygen availability or hypocapnia. This scenario has led to training at altitude being adopted as a routine component of preparation for competition of many athletes. Unfortunately, the value of this strategy is limited by the reduced intensity of training that is possible under hypoxic conditions. More convincing results have been obtained by allowing training to be carried out at low altitude and stimulating the haematological adaptations by exposure to a hypoxic environment overnight (Hendricksenn & Meeuwsen 2003).
The potential advantages for performance of adaptations to hypoxia have led to the administration of synthetic erythropoietin in order to increase haematocrit. Leaving aside the illegality, ‘blood doping’ with erythropoietin carries with it very substantial risks. The normal response to hypoxia incorporates both increased red cell mass and increased oxygen carriage efficiency through DPG expression. The red cells produced in response to erythropoietin do not have up-regulated DPG and so, in order to achieve a given increment in tissue oxygen delivery, there needs to be a much greater increment in red cell mass. In consequence, athletes who have taken erythropoietin may have haematocrits of the order of 70%, with massively elevated cardiac workload and a significant risk of cardiac events during exercise, as well as increased risk of venous thrombosis due to red cell clumping (see Chapter 5, p. 55).
PROBLEMS WITH EXPOSURE TO ALTITUDE
Acute mountain sickness
Individuals who ascend rapidly to altitudes around 3000 m (9800 ft) frequently suffer from a syndrome termed acute mountain sickness, which involves nausea, headache and fatigue, among other symptoms. These symptoms usually appear within 1 or 2 h of ascent and usually disappear over the next several days as acclimation occurs. Several factors may be involved in genesis of acute mountain sickness. Significant dehydration often occurs, because hypoxia and the alkalosis that follows respiratory stimulation appear to inhibit thirst and so exaggerate the increased respiratory water loss associated with altitude. This dehydration probably plays a part in the headache by tightening the connective tissue strands that support the brain. Alkalosis seems also to be a key factor in the other symptoms, since most people can prevent acute mountain sickness except at extreme altitudes by dosing themselves with a carbonic anhydrase inhibitor before ascent.
Pulmonary oedema
A small number of individuals show a more serious acute response to the same levels of ascent that induce acute mountain sickness. In these people, there is damage to the pulmonary capillary wall so that plasma leaks into the lung interstitium and may enter the alveoli. The precise mechanisms that underlie this high-altitude pulmonary oedema are not fully understood, but it almost certainly involves generation of high intracapillary pressures in response to hypoxia. Superficially, this seems counterintuitive. You will remember from Chapter 8 (pp. 94) that capillary hydrostatic pressure in the lung is normally well below the plasma oncotic pressure, so that there is a good safety margin to ensure no fluid extravasation. We also saw in Chapter 8 (pp. 96) that the pulmonary arteriolar smooth muscle possesses hypoxia receptors that induce vasoconstriction in response to reduced alveolar PO2. In theory, therefore, breathing hypoxic air should cause generalized pulmonary vasoconstriction which, while it will elevate pulmonary arterial pressure, will reduce capillary hydrostatic pressure.
In practice, the likely explanation is that, in the susceptible individuals that suffer high altitude pulmonary oedema, not all areas of the pulmonary arteriolar bed respond equally strongly to the vasoconstrictor effect of hypoxia. Under these circumstances, a disproportionate volume of right cardiac output is directed through the least constricted vessels exposing the capillaries in these areas to an intravascular pressure close to pulmonary arterial pressure (West 2004).
Discussion
Breathlessness, or dyspnoea, is a subjective feeling caused by inadequate pulmonary gas exchange activating chemoreceptors. These might be central CO2 receptors responding to hypercapnia or peripheral hypoxia receptors responding to inadequate oxygenation of arterial blood or to plasma-borne protons. In either case, the sensation of breathlessness is due partly to inputs from these receptors and partly to the respiratory muscle fatigue that results from increased respiratory work. Breathlessness associated with acute ascent to a higher altitude automatically suggests hypoxia rather than acidosis or hypercapnia and this is consistent with the fact that Kevin’s lips were bluish. Blue colouration of the mucous membranes or skin, known as cyanosis, reflects the presence in arterial blood of more than 5 g/100 ml of deoxygenated haemoglobin. Thus, cyanosis usually indicates inefficient pulmonary uptake of oxygen. In individuals who have very high haematocrits, on the other hand, it is sometimes possible to see cyanosis in the presence of normal pulmonary oxygenation, because at high rates of pulmonary blood flow there is not enough time to saturate the extra haemoglobin molecules.
Many people have detectable heart murmurs and the cause of these is not always obvious without investigations. From the discussions in Chapter 3 (pp. 24) you will remember that the only criterion for generation of a murmur is the presence of turbulent flow at some time during the cardiac cycle. In most cases, the structural distortion that produces this turbulent flow is a minor one that does not interfere with exercise but, unless the extent of the abnormality has been clarified, you cannot be sure of this. Possible causes of turbulence are valvular stenosis, valvular incompetence or a shunt that allows a proportion of stroke volume to be ejected through a narrow orifice at high velocity.
Pulmonary hypertension
Chronic hypoxia also has other disadvantageous consequences that are seen in long-term residents and, in particular, in individuals who have been born at high altitude. The pulmonary vasoconstrictor effect of hypoxia generates an increased afterload for the right ventricle leading to myocardial hypertrophy and the possibility of right heart failure because of inadequate coronary vascularization (see Chapter 8, p. 95). In addition, the relatively low PaO2 results in reduced efficiency of ductus arteriosus closure after birth (see Chapter 8, p. 102), so that a relatively large number of native highland children have some residual shunting between pulmonary and systemic circulations, which further increases pulmonary afterload.
 in sedentary individuals is around 1500 m (5000 ft), but a considerably lower ascent is needed to impair performance in trained athletes with higher cardiac outputs.
in sedentary individuals is around 1500 m (5000 ft), but a considerably lower ascent is needed to impair performance in trained athletes with higher cardiac outputs.Hendriksenn IJ, Meeuwsen T. The effect of intermittent training in hypobaric hypoxia on sea-level exercise: a cross-over study in humans. European Journal of Applied Physiology. 2003;88:396-403.
Johnson RL, Grover RF, DeGraff AC. Effects of high altitude and training on oxygen transport and exercise performance. In: Fletcher GF, editor. Cardiovascular response to exercise. Mt Kisco, NY: Futura Publishing Co; 1994:223-252.
West JB. The physiologic basis of high-altitude diseases. Annals of Internal Medicine. 2004;141:789-800.
Sallis R, Chassay CM. Recognizing and treating common cold-induced injury in outdoor sports. Medicine and Science in Sports and Exercise. 1999;31:1367-1373.
Schmidt W, Heinicke K, Rojas J, et al. Blood volume and hemoglobin mass in endurance athletes from moderate altitude. Medicine and Science in Sports and Exercise. 2002;34:1934-1940.
Questions for revision
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 12 Effects of high altitude
ALTITUDE AND OXYGEN TRANSPORT
Effects of altitude on plasma oxygen uptake
The standard atmospheric pressure of 760 mmHg (101 kPa) at sea level reflects the weight exerted by the gas molecules that make up the air, under gravitational force. As one ascends from sea level, the air becomes progressively less compressed and so the constituent gas molecules become less tightly packed. In consequence, a given volume of inspired air contains fewer molecules of all gases, including oxygen. The relationship between altitude and atmospheric pressure is not a strictly linear one because the air volume increases in 3 dimensions but, in rough terms, pressure falls by around 100 mmHg for every 1000 m (3300 ft) of ascent up to 3000 m (10 000 ft) (Fig. 12.1).
The absolute change in oxygen availability imposed by a given ascent can be calculated easily. Since oxygen represents 21% of normal air, the partial pressure of oxygen (PO2) at sea level is (21%.760) or 160 mmHg. Once inspired, the air becomes saturated with water vapour (partial pressure 47 mmHg) so that the total gas pressure is reduced to 713 mmHg and PO2 falls to (21%.713) or 150 mmHg. In the alveoli, the oxygen is diluted further by approximately 50 mmHg, due primarily to the presence of carbon dioxide, resulting in a local PO2 (PAO2) of around 100 mmHg: at equilibrium with the plasma, arterial PO2 (PaO2) is, therefore, usually also around 100 mmHg.
Effects of altitude on oxygen carriage
Because of the sigmoid shape of the haemoglobin dissociation curve, the falls in PaO2 associated with acute exposure to altitudes up to around 2000 m (6700 ft) cause only a slight reduction in haemoglobin saturation (Fig. 12.2) and so do not reduce oxygen delivery at rest. During exercise, however, the combination of reduced binding and reduced pulmonary capillary transit time leads to a greater degree of desaturation that is proportional both to altitude and to cardiac output. Thus, the threshold altitude for oxygen limitation of maximum exercise in sedentary individuals is typically around 1500 m (5000 ft), but, in trained athletes with substantially greater cardiac outputs, maximum work capacity begins to fall at much lower altitudes (Johnson et al 1994). In the only Summer Olympics held at a significant altitude, in Mexico City in 1968, winning times for all track events longer than 800 m were well above the existing records.
COMPENSATION FOR HYPOXIA
Respiratory stimulation
The reduced oxygen carriage associated with moderate altitude results in more rapid fatigue during exercise, but no respiratory compensation occurs because ventilation is still driven by the central chemoreceptors. These respond to rises in local proton concentration (that is, reduced pH) secondary to arterial carbon dioxide diffusing into the hindbrain, but are insensitive to hypoxia. The peripheral chemoreceptors responsible for monitoring arterial oxygen status are triggered only when PaO2 falls to around 60 mmHg which, as we saw earlier (p. 145), corresponds to an approximate altitude of 2300 m or 7600 ft. At or above this height, chemoreceptor stimulation initiates increased minute ventilation the magnitude of which depends both on the initial PaO2 and on whether increased ventilation is able to restore PaO2 to a value above 60 mmHg.
Haematological changes
Over the first few days of exposure to hypoxia, haematological changes increase the capacity of the red blood cell pool to deliver oxygen. The alkalosis produced by hypocapnia increases red cell expression of 2,3-diphosphglycerate (DPG), which shifts the haemoglobin dissociation curve to the right and increases oxygen unloading in the systemic capillaries (Fig. 12.3). Simultaneously, erythropoietin release from the renal medulla in response to hypoxia stimulates red cell production, so that haematocrit rises progressively over the ensuing several weeks.
Figure 12.3 Effect of altitude on the efficiency of DPG expression as an aid to oxygen delivery. Note that the values for PaO2 represent those occurring at the respective altitudes before any compensation. (a) At altitude 2200 m (PaO2 60 mmHg) there is a doubling of oxygen offloading, while onloading is virtually unaffected. The net tissue gain is 6.5 ml oxygen/100 ml blood. (b) At altitude 3500 m (PaO2 40 mmHg), by contrast, the improved offloading is almost obviated by reduced onloading. The net tissue gain now is only 1.5 ml oxygen/100 ml blood.
