CHAPTER 35 DYSTONIA
The dystonias are an unusual group of movement disorders whose main feature is involuntary muscle contraction or spasm. The term dystonia was originally introduced by Hermann Oppenheim in 1911 to describe alterations in muscle tone and postural abnormalities that are seen in this condition. The concept of dystonia can be confusing because the term has been used to describe a symptom (e.g., a dystonic arm posture), a disease (primary torsion dystonia), or a syndrome. The dystonias constitute a relatively common group of movement disorders that encompass a wide range of conditions from those in which the only manifestation is dystonic muscle spasms to those in which dystonia is one part of a more severe neurological condition.
DEFINITION AND CLASSIFICATION
Dystonia is characterized by involuntary sustained muscle contractions affecting one or more sites of the body, frequently causing twisting and repetitive movements or abnormal postures.1 The movements range from slower twisting athetosis to rapid, shocklike jerky movements. They are repetitive and sometimes rhythmic and can be accompanied by tremor. Dystonic movements can be aggravated by movement (action dystonia) that can be nonspecific or task-specific (e.g., writing). Over time the dystonia can occur with less specific movements and eventually can be present at rest, leading to sustained abnormal postures.
Three basic approaches are used to classify dystonia: age at onset, distribution of affected body parts, and etiology. The categories of age at onset and affected body distribution (Table 35-1) are important in describing clinical signs and have clinical implications for prognosis and treatment.
±, with or without.
EPIDEMIOLOGY
The true population incidence and prevalence of dystonia are unknown. The prevalence data available are usually based on studies of diagnosed cases only and therefore are underestimates of the real numbers. This is particularly the case with dystonia, which can manifest in a variety of ways, and a significant number of cases of focal dystonia are undiagnosed or even misdiagnosed. In an early study in the United States that was based on case note review, the prevalence for PTD was estimated to be 329 per million population. In more recent studies of diagnosed cases in Japan and Europe, the prevalence was estimated to be between 101 and 150 per million. The most reliable estimate is from an ongoing study in the northeast of England, where ascertainment was more complete, and some previously undiagnosed cases were identified. This finding has implied a prevalence rate of 485 per million. The prevalence of secondary dystonia is unknown, although it is estimated from case series that it may be less than 20% to 25% the rate for PTD.
In a study in South Tyrol in Austria, a random sample of the population older than 50 years was examined.2 Primary dystonia was diagnosed in 6 of the 707 individuals studied, implying a prevalence of 7320 per million in this age-selected population, although 95% confidence intervals were very wide, at 3190 to 15,640, because of the small sample. However, this indicates that in the aging population, dystonia is a relatively common neurological disorder.
CLINICAL FEATURES
The diagnosis of dystonia is based on clinical findings. Primary dystonia has no other neurological features apart from dystonia and tremor.3 Features suggestive of a secondary cause of dystonia are listed in Table 35-2. Investigations are usually performed to help rule out a secondary cause of dystonia.
TABLE 35-2 Clinical Features Suggestive of Secondary Dystonia
Primary Dystonia
Early-Onset Primary Torsion Dystonia (Dystonia Musculorum Deformans, Oppenheim’s Dystonia)
Most forms of early-onset PTD are genetic in origin, and Table 35-3 lists the genetic forms that have been identified to date. Most are autosomal dominant, some reported only in single families. The existence of autosomal recessive forms (DYT2) is controversial.

Focal Primary Torsion Dystonia
These are by far the commonest forms of dystonia. Usually sporadic, they have onset in adult life and remain focal in distribution. Families with autosomal dominant forms have been described, and it is believed that a proportion of the apparently sporadic cases may represent manifestation of a dominant gene with very low penetrance (estimated at 12% to 15%). The individual types are discussed as follows.
Writer’s cramp and task-specific limb dystonias
Writer’s cramp is the most common form of task-specific dystonia and, in contrast to craniocervical dystonia, is more common in men than in women. Onset usually occurs between the ages of 30 and 50 years and often starts with a feeling of tension in fingers and forearms that interferes with writing fluency. The pen is held abnormally forcefully as a result of dystonic contraction of hand and/or forearm muscles. This commonly involves excessive flexion of the thumb and index finger with pronation of the hand and ulnar deviation of the wrist. Affected individuals may also experience lifting of the thumb or index finger off the pen or isolated extension of fingers. Up to 50% of patients also experience upper limb tremor, either on writing or a postural tremor. Strain and aching, particularly in affected forearm muscles, is common on writing, but pain is an uncommon feature.
Secondary/Symptomatic Dystonias
Dystonia-Plus Syndromes
Myoclonus-dystonia syndrome
MDS usually has onset in childhood or early adolescence, with myoclonic jerks affecting the upper limbs and axial muscles (trunk and neck). The myoclonus can occur on rest and also be precipitated by action. Dystonia occurs in approximately two thirds of patients; cervical dystonia and writer’s cramp are the most common forms. On occasion, dystonia affects the legs. Several reports have identified psychiatric features associated with MDS, including obsessive-compulsive disorder, panic attacks, and anxiety. Most affected patients note significant relief of symptoms with alcohol or benzodiazepines and can have marked rebound of symptoms after administration of these drugs. This can often lead to abuse of these substances.
Secondary Dystonia
The term secondary implies that an identifiable cause for the dystonia can be found. Many of these directly involve the basal ganglia and lead to contralateral hemidystonia. Table 35-4 summarizes the most common causes of secondary dystonia. Strokes, tumors, vascular malformations, and traumatic injuries to the basal ganglia are well-described causes of dystonia, but dystonia also, more rarely, occurs after injury to cortical or brainstem structures, the spinal cord, and even peripheral nerves. Perinatal injury may cause dystonia at the time of brain injury (dystonic or choreoathetoid cerebral palsy) or can lead to delayed-onset dystonia, which can begin years after the injury and progress. Infectious, postinfectious, and inflammatory syndromes associated with dystonia usually manifest in combination with other movement disorders, such as parkinsonism, chorea, athetosis, and tics. Drugs may also cause transient or chronic (tardive) dystonia. Acute dystonic reactions occur shortly after the introduction of a drug and have been described for a number of drugs, including dopamine receptor-blocking (DRB) agents, antidepressants (selective serotonin reuptake inhibitors, monoamine oxidase inhibitors), calcium antagonists, general anesthetic agents, anticonvulsants (carbamazepine, phenytoin), L-dopa, ranitidine, 3,4-methylenedioxymethamphetamine (“ecstasy”), and cocaine. Tardive dystonia is usually seen with use of dopamine receptor-blocking drugs and is defined as dystonia present for at least 1 month and occurring either during or within 3 months of discontinuation of a dopamine receptor-blocking drug. It most commonly affects the face and neck but can spread and even generalize in some cases.
| Cause | Examples |
|---|---|
| CNS lesion | Brain tumor, stroke, hypoxia, intracranial hemorrhage, CNS trauma, congenital malformations, cervical cord lesions |
| Perinatal cerebral injury | Cerebral palsy, delayed-onset dystonia, perinatal hypoxia, kernicterus |
| Infectious, postinfectious, and inflammatory | Subacute sclerosing panencephalopathy, Reye’s syndrome, viral encephalitis, Creutzfeld-Jakob disease, systemic lupus erythematosus, antiphospholipid syndrome, Sjögren’s syndrome |
| Peripheral nerve injury | |
| Drug-induced | Dopaminergic agents (L-dopa, dopamine agonists), dopamine receptor–blocking drugs (neuroleptics, prochlorperazine, metoclopramide), selective serotonin reuptake inhibitors, MAO inhibitors, antiepileptic drugs, ergots, flecainide, cocaine, ranitidine, calcium antagonists, anesthetic agents |
| Toxin-induced | Manganese, carbon monoxide, carbon disulfide, cyanide, methanol, disulfiram, wasp sting venom |
| Metabolic | Hypoparathyroidism |
CNS, central nervous system; L-dopa, levodopa; MAO, monoamine oxidase.
Heredodegenerative Disorders
Dystonia also occurs in a wide range of heredodegenerative disorders in which there is progressive neuronal loss with a mixture of neurological symptoms and signs, sometimes with systemic involvement. Table 35-5 lists the various heredodegenerative disorders that can be divided into those with disorders of metabolism, mitochondrial disease, trinucleotide repeat diseases, parkinsonian disorders, and other degenerative processes without defined causes.
TABLE 35-5 Heredodegenerative Disorders That Can Cause Dystonia
| Metabolic Disorders | |
| Metal and mineral metabolism | Wilson’s disease, neurodegeneration with brain iron accumulation type I, neuroferritinopathy, idiopathic basal ganglia calcification (Fahr’s disease) |
| Lysosomal storage disorders | Niemann-Pick disease type C, GM1 and GM2 gangliosidoses, metachromatic leukodystrophy, Krabbe’s disease, Pelizaeus-Merzbacher disease, fucosidosis |
| Inborn errors of metabolism | Lesch-Nyhan syndrome, triosephosphate isomerase deficiency, glucose transport defects |
| Amino and organic acidurias | Glutaric aciduria type I, homocystinuria, propionic acidemia, methylmalonic aciduria, 4-hydroxybutyric aciduria, 3-methylglutaconic aciduria, 2-oxoglutaric aciduria, Hartnup’s disease |
| Mitochondrial Disorders | |
| Leigh’s disease | |
| Leber’s hereditary optic neuropathy | |
| Mohr-Tranebjaerg syndrome (dystonia/deafness) | |
| Trinucleotide Repeat Disorders | |
| Huntington’s disease | |
| Spinocerebellar ataxias | |
| Parkinsonian Disorders | |
| Parkinson’s disease (especially familial young-onset forms) | |
| Progressive supranuclear palsy | |
| Multiple-system atrophy | |
| Corticobasal ganglionic degeneration | |
| X-linked dystonia–parkinsonism (“Lubag”) | |
| Others | |
| Ataxia-telangiectasia | |
| Chorea-acanthocytosis | |
| Rett’s syndrome | |
| Infantile bilateral striatal necrosis | |
| Ataxia with vitamin E deficiency | |
| Progressive pallidal degeneration | |
| Sjögren-Larsson syndrome | |
| Ataxia–amyotrophy–mental retardation–dystonia syndrome | |
Psychogenic Dystonia
The concept of psychogenic dystonia is difficult because in the first half of the 20th century many cases of organic dystonia were thought to be psychiatric in origin as a result of the unusual and variable nature of the symptoms. Psychogenic dystonia does exist, often resulting in profound disability, but is a rare form of dystonia. As with organic dystonias, it is difficult to diagnose with certainty, and the assessment should be undertaken only by a neurologist with considerable experience with organic dystonias. One reason for this is the absence of a specific diagnostic test for dystonia. The diagnosis is based on the presence of clinical inconsistencies and incongruities with organic dystonia. The pathophysiology is poorly understood. Prompt diagnosis and treatment are necessary, in view of the poor prognosis of conversion disorders when there has been considerable delay between symptom onset and diagnosis, and treatment often involves a combination of psychotherapy, physical therapy, and psychopharmacologic therapy.
INVESTIGATIONS
Table 35-6 list potential investigations. However, the clinical presentation determines which of these are appropriate. For instance, a woman aged 60 presenting with blepharospasm alone may need no investigation, whereas a child with dystonia and other neurological features would need extensive tests to identify a potential secondary cause. A history of birth injury, a family history of other neurological disorders, and exposure to dystonia-inducing drugs are important.
| Dystonia Phenotype | Investigation |
|---|---|
| Primary torsion dystonia | |
| Early onset (<28 years) | Copper studies, slit-lamp examination |
| Brain MRI | |
| DYT1 gene analysis | |
| Trial of L-dopa | |
| Late onset (>28 years) | Copper studies, slit-lamp examination if younger than 50 years |
| Brain MRI | |
| Spine MRI if dystonia fixed or painful | |
| EMG if painful axial muscle spasm | |
| Secondary dystonia | Brain/spine MRI |
| Nerve conduction studies | |
| Copper studies, slit-lamp examination, liver biopsy | |
| Genetic test for neurodegenerative disorders (e.g., Huntington’s disease) | |
| White blood cell enzymes | |
| α-Fetoprotein, immunoglobulins | |
| Lactate, pyruvate, mtDNA analysis; muscle biopsy | |
| Blood film for acanthocytes | |
| Urine amino acid, organic acid, oligosaccharide measurements | |
| Bone marrow biopsy | |
| Phenylalanine loading test, CSF pterin measurement | |
| ERG, retinal examination | |
CSF, cerebrospinal fluid; EMG, electromyography; ERG, electroretinogram; MRI, magnetic resonance imaging; mtDNA, mitochondrial deoxyribonucleic acid.
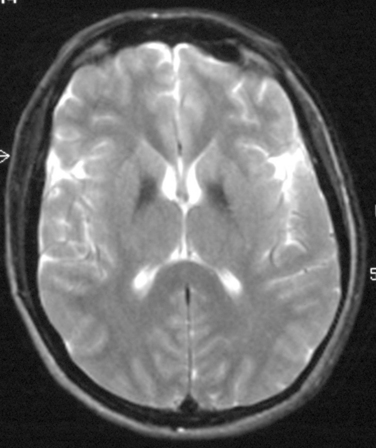
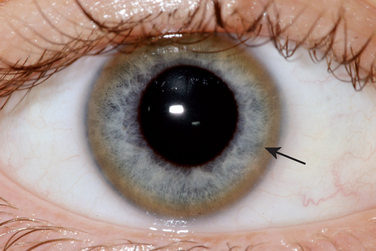
Hemidystonia is highly suggestive of a contralateral lesion of the basal ganglia, and cranial imaging with MRI is mandatory. For young-onset generalized dystonia, imaging may also aid in diagnosis, such as revealing the “eye of the tiger” sign in brain iron accumulation type 1 (Fig. 35-1), typical changes in the midbrain of a patient with Wilson’s disease, or basal ganglia calcification in Fahr’s disease. If MRI appearance is normal, further tests should be performed to exclude Wilson’s disease as it is potentially treatable. Slit-lamp examination for Kayser-Fleischer rings (Fig. 35-2), serum ceruloplasmin measurement, and 24-hour urinary copper excretion measurement are required.
PATHOPHYSIOLOGY OF DYSTONIA
Neurophysiological Studies
The hallmark of dystonia is involuntary sustained muscle contractions, which are characterized by an abnormal pattern of EMG activity: excessive co-contraction of antagonist muscles during an action and overflow into extraneous muscles. Other findings include prolongation of EMG bursts. These findings have been described in patients with primary focal hand dystonia, and it was also noted that there was loss of selectivity to perform independent finger movements, occasional failure of willed activity, and tremor. These features emphasize that in dystonia, there is excessiveness of movements and lack of fine control.
However, it has become clear that dystonia is not a pure motor disorder and that individuals with dystonia have sensory abnormalities that play an important role in causing motor dysfunction. Studies have demonstrated evidence of abnormalities in both somatosensory spatial discrimination and temporal discrimination (the shortest time two successive stimuli are perceived as separate). The importance of the sensory system in dystonia is also evident from study of the sensory tricks (geste antagoniste), which are various maneuvers used by patients with focal dystonia to temporarily relieve the dystonic spasms; for example, a finger placed on the face of an individual with cervical dystonia can eliminate neck muscle spasm. There is also evidence to suggest that abnormal sensory input can trigger dystonia, such as trauma to a body part before the dystonia. Another line of evidence comes from study of processing of muscle spindle input. In patients with hand cramps, vibration can induce the patient’s dystonia, and cutaneous input similar to that which produces the sensory trick can reverse this vibration-induced dystonia.
Summary
Evidence therefore suggests that dystonia is characterized pathophysiologically by abnormal sensory processing and deficient cortical inhibition.4 The current model implicates abnormal surround inhibition as the substrate that leads to generation of uncontrolled dystonic movements. Dystonia could result from lesions in the basal ganglia, which disrupt intricate pathways and push normal movements to abnormal. Genetic defects (such as those found in DYT1 dystonia) may also affect these pathways, possibly through abnormal dopaminergic neurotransmission. Repetitive movements or use of a body part may also lead to dystonia. Thus, a combination of factors may lead to the cortical abnormality that results in the production of dystonic movements.
TREATMENT OF DYSTONIA
Treatment options for dystonia have increased dramatically since the mid-1980s. This has been the result of the use of botulinum toxin and a renewed interest in functional neurosurgery for dystonia. Drug treatment has some use, particularly for DRD and MDS and the more severe childhood-onset primary dystonias. Table 35-7 lists the treatment options for various forms of dystonia.
| Therapy | Agent/Procedure | Uses |
|---|---|---|
| Drugs | Dopaminergic agents | DRD, sometimes primary and secondary dystonia |
| Anticholinergics | Primary and secondary dystonia and DRD | |
| Baclofen (oral or intrathecal) | Childhood primary dystonia | |
| Benzodiazepines | Primary and secondary dystonia, MDS | |
| Antidopaminergic agents | Occasional primary/secondary dystonia | |
| Botulinum toxin | Type A and B | Main treatment of focal and segmental dystonia |
| Destructive neurosurgery | Selective peripheral denervation | Cervical dystonia |
| Myectomy/myotomy | Cervical dystonia/blepharospasm | |
| Intradural rhizotomy/nerve sectioning | Cervical dystonia (rarely used) | |
| Functional stereotactic neurosurgery | Pallidotomy, thalamotomy | Primary and secondary generalized or hemidystonia |
| Deep brain stimulation | Primary generalized and segmental dystonia |
DRD, dopa-responsive dystonia; MDS, myoclonus-dystonia syndrome.
Drug Therapy
Anticholinergic Drugs
Trihexyphenidyl can be useful for childhood-onset primary dystonia. One prospective placebo-controlled trial revealed that 67% of treated patients showed improvement and 68% continued to benefit after 2.4 years at a mean daily dose of 40 mg. It appears to be less useful for adult-onset primary focal dystonia. In both children and adults, the dose of anticholinergic must be titrated up gradually to minimize side effects, particularly of dry mouth, gastrointestinal upset, and confusion.
Botulinum Toxin
Botulinum toxin injections are the first line of treatment for focal and segmental dystonias.5 Botulinum toxin consists of a number of serotypes of a potent neurotoxin that acts by inhibiting neurotransmitter release at the neuromuscular junction, which leads to temporary weakness of the muscle. Botulinum toxin types A and B are most commonly used in clinical practice. Its principal mode of action in dystonia is to cause denervation of motor end plates, although there is evidence that its effect on sensory symptoms may also be important, possibly by modulating muscle spindle input to the central nervous system. Local injection of botulinum toxin type A into overactive dystonic muscles can provide very effective relief of symptoms. Double-blind placebo-controlled trials have shown botulinum toxin type A to be efficacious in treating cervical dystonia and blepharospasm (60% to 70% patients showed improvement), and retrospective and open-label studies have also demonstrated its efficacy for laryngeal dystonia, writer’s cramp, and limb dystonias and for selected cases of oromandibular dystonia. Botulinum toxin has a temporary effect; patients require repeat injections at intervals, usually every 12 to 16 weeks. For uncomplicated cases of blepharospasm and cervical dystonia, muscles to receive injection are usually selected clinically. For more complex cases and other types of dystonia, EMG guidance is often required.
Surgery
Functional Stereotactic Surgery
Current functional stereotactic options include lesion induction and deep-brain stimulation (DBS) of the globus pallidus internus and the thalamus. Because of the relatively small numbers of patient series, variations in surgical technique, and inconsistent use of outcome measures, no definite recommendations about ideal surgical targets or optimal methods can be made. In general, however, the globus pallidus internus appears to be the preferred target in primary dystonias. The pallidal target is located in the posteroventral lateral globus pallidus internus, which is the same site as that used in Parkinson’s disease. Pallidotomy has been reported to be effective in various forms of dystonia, including primary generalized and segmental dystonia and hemidystonia, with a number of studies reporting 50 to 80% improvement in symptoms. In general, patients with primary dystonia (especially DYT1) had a better response than did those with secondary dystonia.6
PAROXYSMAL DYSKINESIAS
Paroxysmal Nonkinesigenic Dyskinesia (PKND)
PKND is characterized by attacks of dyskinesia that are frequently precipitated by alcohol ingestion, caffeine ingestion, stress, or fatigue. The episodes are often dystonic or choreic and have longer duration (minutes to hours) and are less frequent (on to three per day) than those in PKD. There may be longer attack-free intervals, and, again, boys are more often affected than girls. It is often familial, showing autosomal dominant inheritance, with onset in childhood and attacks diminishing in adulthood. A locus on chromosome 2q31-36 has been mapped in a number of families. Most patients with PKND do not benefit from antiepileptic drugs, but some response to clonazepam or clobazam has been reported. In general, it is more difficult to treat than PKD, and patient often learn to avoid precipitants.
CONCLUSIONS
Albanese A. The clinical expression of primary dystonia. J Neurol. 2003;250:1145-1151.
Comella CL, Pullman SL. Botulinum toxin in neurological disease. Muscle Nerve. 2004;29:628-644.
Fahn S, Bressman S, Marsden CD. Classification of dystonia. Adv Neurol. 1998;78:1-10.
Hallett M. Abnormal movements result from loss of inhibition. Adv Neurol. 2004;94:1-11.
Vidailhet M, Vercueil L, Houeto JL, et al. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med. 2005;352:459-500.
1 Fahn S, Bressman S, Marsden CD. Classification of dystonia. Adv Neurol. 1998;78:1-10.
2 Muller J, Kiechl S, Wenning GK, et al. The prevalence of primary dystonia in the general community. Neurology. 2002;59:941-943.
3 Albanese A. The clinical expression of primary dystonia. J Neurol. 2003;250:1145-1151.
4 Hallett M. Abnormal movements result from loss of inhibition. Adv Neurol. 2004;94:1-11.
5 Comella CL, Pullman SL. Botulinum toxin in neurological disease. Muscle Nerve. 2004;29:628-644.
6 Vidailhet M, Vercueil L, Houeto JL, et al. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med. 2005;352:459-500.






