[level-membership-for-emergency-medicine-category]Chapter 79
Dysrhythmias
Cardiac Cellular Electrophysiology
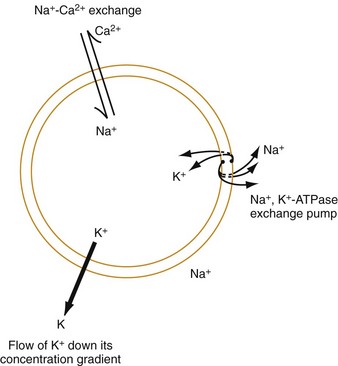
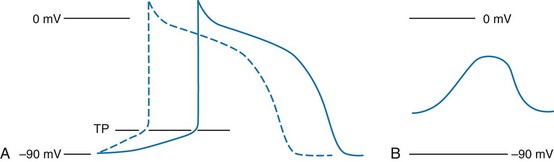
The function of individual cells in the conductive and contractile tissues of the heart depends on an intact resting membrane potential. Na+, K+, and Ca2+ ions create the membrane potential and regulate conduction and contractility. The membrane potential is the result of a differential concentration of Na+ and K+ on either side of the cell membrane, measuring approximately −90 mV in normal resting, nonpacemaker cells. This electrical gradient exists mainly because of the Na+-K+ exchange pump and the natural concentration-dependent flow of K+ out of the cell. Adenosine triphosphate (ATP) fuels Na+ transport out to the extracellular fluid, with Mg2+ used as a cofactor (Fig. 79-1). This process creates an osmotic gradient, allowing Ca2+ to be passively exchanged for Na2+ and promoting conduction as well as myofibril contraction. The resting membrane potential is generated from the flow of K+ down a concentration gradient toward the extracellular fluid. The cell membrane is far more permeable to potassium than sodium ions, resulting in a net loss of intracellular positive charge.
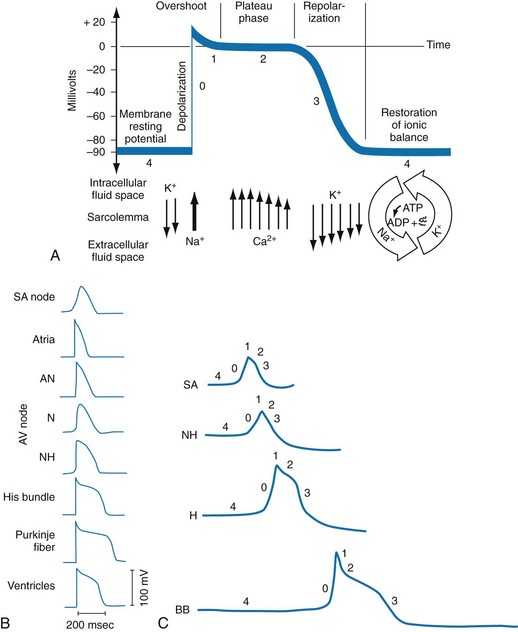
In normal nonpacemaker cells, the application of an electrical stimulus causes the membrane potential to become less negative, termed depolarization. When the membrane potential reaches −70 mV, specialized Na2+ channels open, causing a rapid influx of positive charge into the cell. This “fast” channel activity further decreases the membrane potential and is augmented at 30 to 40 mV by a second “slow” channel that allows Ca2+ influx. When these channels close, resting potential is restored by the sodium-potassium pump, an event known as repolarization (Fig. 79-2).
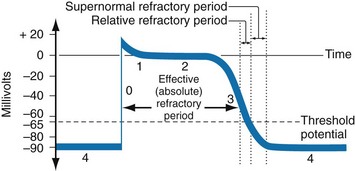
In nonpacemaker cells, additional depolarization from a second electrical stimulus is not possible when the membrane potential is more positive than −60 mV. This period is termed the effective refractory period (Fig. 79-3). At a membrane potential of −60 to −70 mV, a strong impulse can cause a response that is likely to be propagated, although abnormally; this response represents the relative refractory period. At a membrane potential of −70 mV or less, virtually all fast channels are ready for activity if properly stimulated (see Fig. 79-3).
Anatomy and Conduction
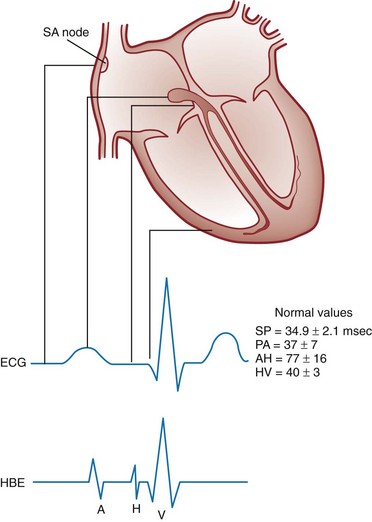
Figure 79-4 correlates the surface ECG tracings with tissue electrical events. Impulses generated from within the SA node itself, imperceptible on the surface ECG, are propagated through the atrial tissue to the AV node. Atrial depolarization is characterized by the P wave on the surface ECG.
The AV node is an area of specialized tissue between the atria and the ventricles of the heart, located in the posterior-inferior region of the interatrial septum. Its blood supply is from a branch of the RCA in 90% of patients (right dominant) and from the LCA in the remaining 10% (left dominant). Transmission of impulses within the AV node is slower than in parts of the conducting system (Table 79-1) because of a dependence on slow-channel ion influx for membrane depolarization. In some patients, pathologic “accessory pathways” connect atrial and ventricular tissues. These accessory pathways do not share the normal conduction delay of the AV node and may allow for rapid ventricular rates. Preexcitation refers to the early depolarization of ventricular myocardium when accessory paths are used instead of the normal conduction system.

Mechanisms of Dysrhythmia Formation
There are three electrophysiologic mechanisms: enhanced automaticity, triggered activity, and reentry. Enhanced automaticity includes spontaneous depolarization of nonpacemaker cells or a lower threshold for depolarization in normal pacemaker cells (Fig. 79-5). Both types of enhanced automaticity can occur in the setting of ischemia, electrolyte disturbances, or drugs. Examples of enhanced automaticity include idioventricular rhythms in the setting of acute myocardial infarction, and atrial tachycardias or junctional tachycardias (JTs) seen with digitalis toxicity.
Reentry dysrhythmias occur as a consequence of abnormal conduction (Fig. 79-6). For a reentry mechanism, two alternate pathways for conduction must be present and one path must have a longer refractory period. The unequal responsiveness of the limbs creates a functional unidirectional block such that when the impulse exits one limb, it may then reenter the other in retrograde fashion. The cycle is then repeated, creating a self-sustaining or “circus movement” tachycardia that can appear orderly or disorderly (i.e., fibrillatory).
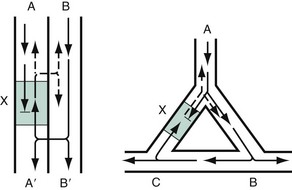
Figure 79-6 Mechanism of reentry.
Classification of Antidysrhythmic Drugs
Medications used to treat dysrhythmias are classified into four major categories on the basis of their electrophysiologic effects (Box 79-1). Other agents fall outside this classification system and are discussed separately.
All antidysrhythmics can cause “prodysrhythmic effects.” This occurs most often in patients with existing structural heart disease and in patients receiving new or higher doses of antidysrhythmic agents. The class I and III agents are associated with prodysrhythmic effects in up to 15% of patients.1,2
Class IC Agents
The class IC agents profoundly slow depolarization and conduction and have pronounced antidysrhythmic properties.1,2 Up to 15% of patients treated with class IC agents experience new or increased ventricular dysrhythmias. Class IC agents are approved only for oral use in the United States.
Class II Agents
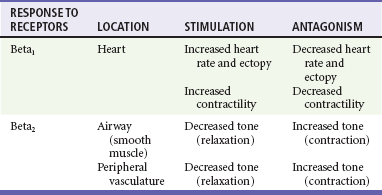
Side effects of beta-blockers include bronchospasm, bradycardia, hypotension, and heart failure. All beta-blockers are active at both beta1 and beta2 receptors (Table 79-2) to varying degrees. Those with more prominent beta1 effects are said to be cardioselective. Relative contraindications to the use of beta-blockers include asthma or chronic obstructive lung disease, advanced congestive heart failure, and third-trimester pregnancy. Beta-blockers should not be used in patients with preexisting bradycardia or heart block beyond first-degree. Intravenous beta-blockers should be used cautiously in conjunction with calcium channel blockers because of the risk of additive side effects. Acute side effects of beta-blockers include bronchospasm, heart failure, excessive bradycardia, and hypotension.
Table 79-2
Cardiac and Respiratory Beta-Adrenergic Receptors and Responses to Pharmacologic Manipulation
Class III Agents
All class III agents prolong the refractory period by blocking K+ channels, and they have variable effects on the QT interval.1–4 In general, class III agents are alternatives to the class I agents for the treatment of many ventricular and atrial dysrhythmias.
Amiodarone
The serum half-life of amiodarone is 25 hours after a single intravenous dose and up to 50 days during long-term oral use. Because of the unusual pharmacokinetics, oral regimens vary widely. The acute side effects of amiodarone are primarily limited to hypotension, bradycardia, and heart failure (Box 79-2). There is an additive risk of bradycardia and hypotension when amiodarone is used in conjunction with calcium channel or beta-adrenergic blockers. Rates of prodysrhythmia are relatively low (1-3%). Long-term amiodarone use, however, is associated with significant extracardiac side effects, including irreversible lung and thyroid disease. Amiodarone alters the pharmacokinetics of numerous other drugs, including digoxin and warfarin.
Ibutilide
Ibutilide has a unique mechanism of action characterized by induction of a slow inward Na2+ current, thereby prolonging the refractory period. Intravenous ibutilide is approved for cardioversion of atrial fibrillation and atrial flutter.5 Ibutilide is associated with QT interval prolongation, and polymorphic VT is more common than with amiodarone although still relatively rare (0.9-2.5%).
Dofetilide
Dofetilide is a pure class III agent that blocks K+ efflux, increasing the refractory period. It is approved for chemical cardioversion and maintenance of sinus rhythm in patients with atrial fibrillation or flutter,6 but because of its prodysrhythmic effects, it has limited usefulness.
Dronedarone
Structurally related to amiodarone, dronedarone displays class III properties plus those of other antidysrhythmic classes. The incidence of polymorphic VT associated with dronedarone is very low. Dronedarone is approved for oral use to maintain sinus rhythm in patients with atrial fibrillation or flutter but is contraindicated in patients with severe or recent heart failure decompensation.7 Long-term effects and the ideal populations for the use of this drug remain unclear.8
Class IV Agents
Miscellaneous Agents
Digoxin (0.25-0.5 mg IV) can control the ventricular rate in patients with SVTs, including atrial fibrillation and atrial flutter. Because of its delayed onset of action and narrow therapeutic window, however, digitalis is not a first-line agent for emergency therapy. It is not true that digoxin promotes conversion to a sinus rhythm any more than other rate-controlling agents.9
Side effects of digoxin are listed in Box 79-3 and are aggravated by hypokalemia, hypercalcemia, hypomagnesemia, increased catecholamines, and acid-base disturbances. Digoxin overdose therapy is covered elsewhere.
Adenosine
Adenosine is a naturally occurring purine nucleoside used for the termination of narrow-complex tachydysrhythmias. Administered as an intravenous bolus, adenosine causes an abrupt slowing of AV conduction in both anterograde and retrograde pathways. Adenosine usually has an onset of action of 5 to 20 seconds and a duration of effect of 30 to 40 seconds. Except in rare cases of catecholamine-induced ventricular dysrhythmias, adenosine has little or no effect on infranodal conduction. For this reason, adenosine is often used as a diagnostic agent in patients with wide-complex tachydysrhythmias when the cause is unclear.10–12 An initial dose of 6 mg as a rapid bolus for adults weighing 50 kg or greater is recommended, with flush through a large peripheral vein. If no response is seen within 1 to 2 minutes, the second dose is doubled (12 mg) and administered. If no effect is seen after a final 12-mg dose, the rhythm should be reassessed and another agent used. Pediatric doses are 0.05 mg/kg initially with doubling at similar intervals up to a total dose of 0.25 mg/kg. Side effects coincide with the onset of clinical effects and occur in up to a third of patients but are usually minor. These include flushing, dyspnea, chest pressure, nausea, headache, dizziness, transient bradycardia or heart block, and hypotension (seen rarely, from the vasodilatory properties). These side effects usually resolve rapidly without treatment, although many patients are intensely uncomfortable for a short period. Asystole can occur but is usually transient though unsettling to those observing.
Approach to Dysrhythmia Recognition and Management
• Narrow-complex (QRS less than 0.12 second) tachycardias (regular and irregular)
• Wide-complex (QRS 0.12 second or greater) tachycardias (regular and irregular)
Initial Assessment of Stable Patients
The 12-lead ECG is essential for evaluation of any patient with a suspected dysrhythmia. Use of a single ECG lead is often adequate for diagnosis, but multiple leads are often required to detect such things as the presence or absence of P waves (often best seen in inferior leads or V1-2; Fig. 79-7), the relationship between P waves and QRS complexes, prolongation of the QRS and QT interval, and evidence of ischemia or prior myocardial infarction (Box 79-4). For certain conditions, such as Brugada’s syndrome, the 12-lead ECG together with a history of syncope is diagnostic.
Pseudodysrhythmias
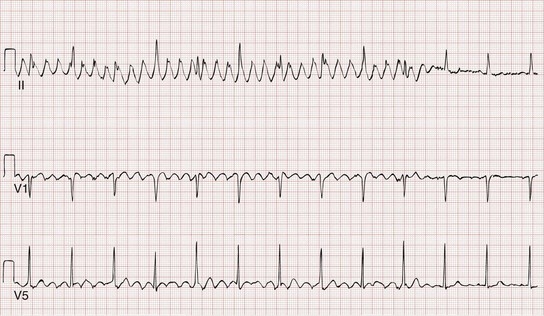
Loose leads, muscle contraction, shivering, tremors, and other patient movement can produce artifactual findings on a monitor, rhythm strip, or 12-lead ECG (Fig. 79-8). Such pseudodysrhythmias can mimic and are often mistaken for serious dysrhythmias, including ventricular fibrillation, illustrating the need to avoid making decisions on the basis of ECG tracings without incorporating the clinical context.
Specific Dysrhythmias
Sinus Bradycardia, Sinoatrial and Atrioventricular Block
Sinus Bradycardia
Sinus bradycardia is characterized by a P wave with normal morphology and a fixed P-P interval that is equal to the R-R interval, with a ventricular rate below 60 beats/min (Fig. 79-9). This pattern may be found in healthy adults, particularly among well-conditioned athletes. It can also be seen in virtually any condition associated with vagal stimulation, ranging from a simple fainting event to hemoperitoneum from a ruptured ectopic pregnancy. Sinus bradycardia is described in the early stages of an acute inferior wall myocardial infarction as a consequence of parasympathetic stimulation. Other pathologic causes of sinus bradycardia include hypothermia, hypoxia, drug effects (especially of beta-blockers and calcium channel blockers), and sick sinus syndrome (SSS; see later). In cases of profound sinus bradycardia (i.e., rates below 40-50), it is not uncommon for a junctional escape rhythm to emerge.
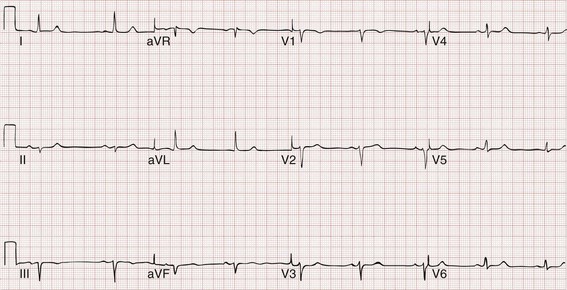
Figure 79-9 Sinus bradycardia.
Sinus Dysrhythmia
Sinus dysrhythmia is similar to sinus bradycardia and is managed in the same manner; it is diagnosed when a variable P-P interval is seen (Fig. 79-10). It is considered a normal variant and is seen frequently in children and young adults.
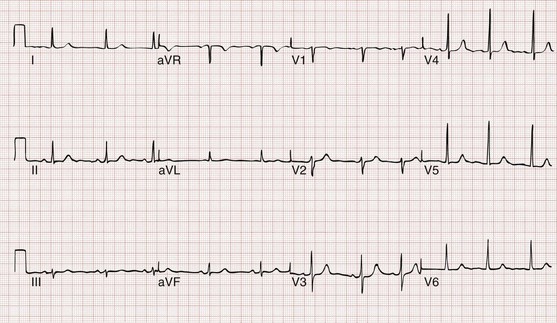
Figure 79-10 Sinus dysrhythmia (note slight irregularity).
Sinus Arrest and Sinoatrial Exit Block
Aside from sinus bradycardia and sinus dysrhythmia, disturbances in sinus rhythm are characterized by an absence of atrial depolarization, diagnosed by missing P waves on the ECG (Fig. 79-11). A lack of atrial depolarization can occur because of either failure of the sinus node to generate an impulse (sinus arrest) or failure of impulse conduction out of the SA node (SA exit block). When incomplete block (i.e., one or more missing P waves) exists, a compensatory pause equal to the P-P interval implies the presence of SA exit block, whereas a shorter pause more likely represents sinus arrest. Also, with SA exit block, it is not uncommon to see dropped P waves in regularly occurring patterns, representing 2 : 1, 3 : 1, or 4 : 1 block. Both sinus arrest and SA exit block are often manifestations of intrinsic SA node disease or looming SSS, but they can also be seen under conditions of increased vagal tone, both benign and pathologic. When the patient is symptomatic, the approach to treatment is similar to that for sinus bradycardia.
Atrioventricular Block
First-Degree Atrioventricular Block.: First-degree AV block is characterized by prolonged conduction of atrial impulses without the loss of any single impulse. This can occur at the level of the atria, the AV node (most common), or the His-Purkinje system. On the ECG, first-degree AV block is defined by a prolonged PR interval (>0.20 second), typically with a narrow QRS complex (Fig. 79-12). First-degree AV block is a normal variant in up to 2% of healthy young adults but is also seen in pathologic conditions associated with depression of AV node conduction. First-degree AV block requires no specific treatment, but nodal depressing agents should be given with caution in this setting.
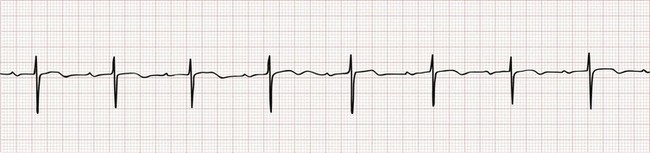
Figure 79-12 First-degree atrioventricular block.
Second-Degree Atrioventricular Block.: Second-degree AV block is characterized by one or more (but not all) sinus impulses failing to reach the ventricles. The conduction ratio is defined as the ratio of the number of P waves to the number of QRS complexes over a period of time (e.g., 3 : 2, 2 : 1). A 2 : 1 conduction ratio does not always mean worse conduction system disease than a 4 : 2 ratio, and not all 2 : 1 conduction is pathologic. For example, an atrial impulse rate of 300 complexes per minute (common in atrial flutter) presented to the AV node usually results in conduction of half the impulses, producing a ventricular rate of 150 beats/min. This conduction ratio does not represent AV dysfunction or important block because the AV node is responding normally and preventing excessive ventricular stimulation. Conversely, a sinus rhythm at a rate of 70 P waves per minute with a 2 : 1 ratio (a ventricular rate of 35 beats/min) represents profound AV block and dysfunction. The term high-grade second-degree block is best applied to conduction disturbances that prevent physiologic ventricular response rates and not solely to higher conduction ratios. Second-degree AV block can be further classified into two types on the basis of the underlying pathophysiology and ECG appearance (Table 79-3).
Table 79-3
Features of Type I and Type II Second-Degree Atrioventricular Block
| FEATURE | TYPE I | TYPE II |
| Clinical | Usually acute | Often chronic |
| Inferior myocardial infarction | Anteroseptal | |
| Rheumatic fever | Lenègre’s disease | |
| Lev disease | ||
| Digitalis or beta-blockers | Cardiomyopathy | |
| Anatomic | Usually AV node | Infranodal |
| Electrophysiology | Increased relative refractory period | No relative refractory period |
| Decremental conduction | All-or-none conduction | |
| ECG features | RP/PR reciprocity | PR interval stable |
| Prolonged PR interval | PR interval usually normal | |
| QRS duration normal | QRS duration prolonged | |
| Response to atropine and exercise | Improves | Worsens |
| Response to carotid massage | Worsens | Improves* |
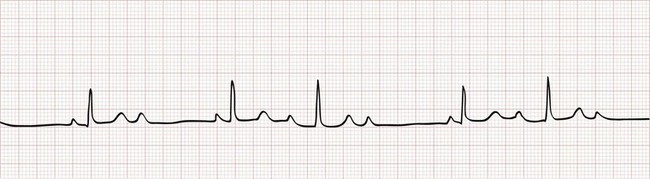
Type I Second-Degree Atrioventricular Block.: Type I second-degree AV block, also called Wenckebach or Mobitz I AV block, is associated with a conduction disturbance within the AV node. The surface ECG is characterized by a progressive lengthening of the PR interval until an impulse is not conducted (“dropped beat”). The progressive lengthening of the PR interval gives the appearance of successive P waves retreating into the preceding QRS complexes (Fig. 79-13). By definition, the longest R-R interval (i.e., following the dropped beat) is less than twice the length of the shortest. On a rhythm strip, type I second-degree AV block gives the appearance of “grouped beating,” especially pairs or trios (i.e., 3 : 2 or 4 : 3 block), but occasionally in larger multiples. Grouped beating is not unique to type I second-degree AV block; it occurs in a variety of other conditions (Box 79-5), including SA exit block, type II second-degree AV block, and extrasystoles with or without block.
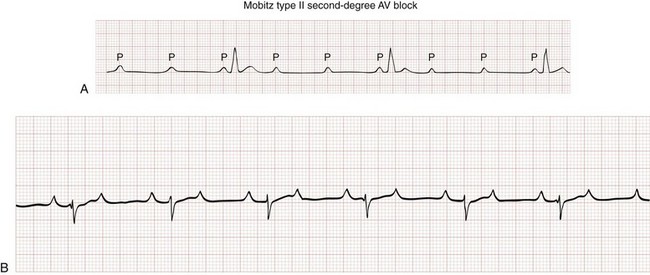
Type II Second-Degree Atrioventricular Block.: Type II second-degree AV block, or Mobitz II block, represents a conduction block just below the level of the AV node and is never a normal variant. On the surface ECG, conduction of atrial impulses is sporadic and typically periodic, but the PR interval does not widen (Fig. 79-14). The QRS complex is intrinsically narrow, but concomitant infranodal conduction disturbances (i.e., bundle branch blocks) are not uncommon with type II second-degree AV block.
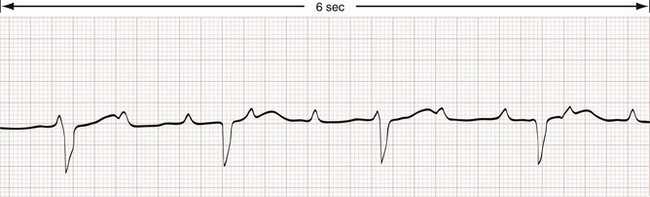
Third-Degree Atrioventricular Block.: Third-degree AV block (also known as complete AV block or complete heart block) is characterized by absent conduction of all atrial impulses (Fig. 79-15) accompanied by a slow escape rhythm. The width and frequency of QRS complexes depends on the site of the escape-rhythm pacemaker. Pacemakers above the His bundle are associated with a narrow-complex QRS at a rate of 45 to 60 beats/min, whereas pacemakers at or below the His bundle produce a wide-complex QRS at a rate of 30 to 45 beats/min.
Extrasystoles
An extrasystole is an electrical impulse originating from an ectopic atrial or ventricular focus. Depending on the site of origin and the timing of the impulse, there may not be an associated mechanical contraction. The terms “premature atrial contraction” and “premature ventricular contraction” are misleading but widespread and used here for ease. The extrasystole and its preceding impulse are referred to as a couplet, and the coupling interval refers to the period between these two beats. Bigeminy (Fig. 79-16) occurs when there is an extrasystole after every native beat, so that every other impulse is extrasystolic; trigeminy (every third beat) and quadrigeminy (every fourth beat) are similar. Most extrasystoles are the result of enhanced automaticity, whether in the atria, AV node, His-Purkinje system, or ventricles.
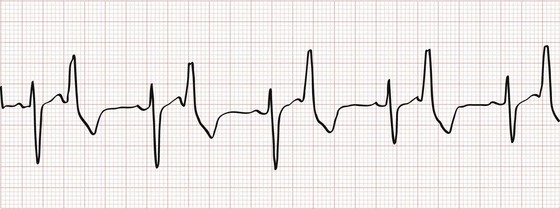
Figure 79-16 Ventricular bigeminy.
Premature Atrial Contractions
Premature atrial contractions (PACs; Fig. 79-17) are common and usually have little clinical significance. On occasion, a PAC can be the precipitant of a more important dysrhythmia, such as atrial fibrillation, atrial flutter, or SVT. PACs can be distinguished on the ECG by the presence of an abnormal P wave early within a cardiac cycle, although sometimes the P wave may be difficult to detect if it is buried within the preceding T wave.
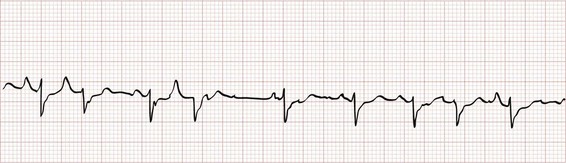
Figure 79-17 Premature atrial contractions.
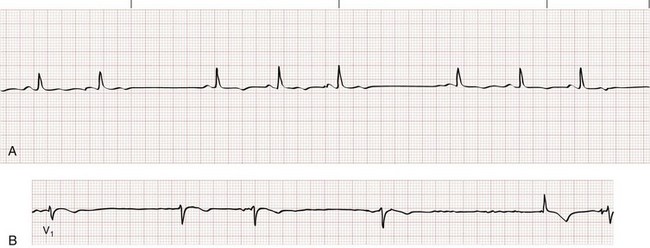
Most PACs will depolarize the sinus node, resetting its refractory period. Because of this, the P-P interval between two sinus beats surrounding a PAC will be less than twice the intrinsic P-P cycle length (see Fig. 79-17). If a PAC reaches the AV node or the infranodal conducting system during its absolute refractory period, there will be no ventricular depolarization. A nonconducted (or blocked) PAC typically results in a noncompensatory pause (i.e., R-R interval less than twice the intrinsic R-R cycle; Fig. 79-18) because the sinus node is reset. Blocked PACs are among the most common causes of pauses seen on an ECG but can be easily overlooked if one is not careful to inspect for ectopic P waves.
Premature Ventricular Contractions
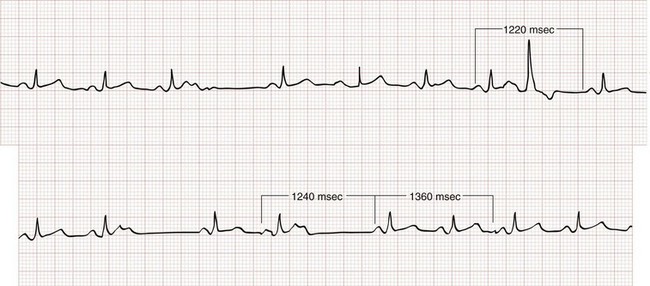
A PVC appears as a wide–QRS complex extrasystole without a preceding P wave (Fig. 79-19). Most PVCs capture the AV node, making it refractory to the next arriving atrial impulse. Because retrograde conduction of a PVC rarely extends far enough to capture and reset the SA node, however, atrial impulses continue to arrive at the AV node at the intrinsic sinus rate. As a result, the R-R interval surrounding a PVC ends up being equal to twice the intrinsic R-R interval length (see Fig. 79-19), a phenomenon known as a compensatory pause. Rarely, a PVC will capture the SA node, resulting in a noncompensatory pause, or will fail to capture the AV node, leaving the underlying rhythm completely unaffected (a so-called interpolated PVC; Fig. 79-20).
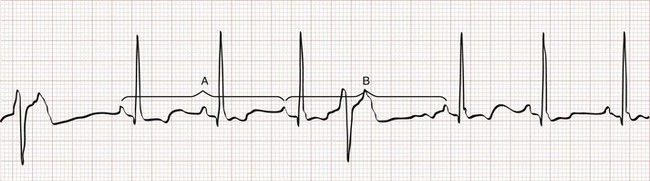
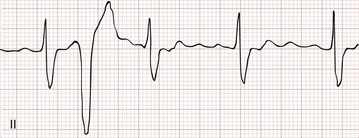
Figure 79-20 Interpolated premature ventricular contraction.
The morphology of a PVC depends on the origin of the impulse, with a left bundle branch block (LBBB) appearance resulting from an extrasystolic focus in the right ventricle and vice versa. Multiform (or “multifocal”) PVCs come from more than one source and have variable morphologies. When a PVC occurs at or around the time that a supraventricular impulse is set to depolarize the ventricle, the result is a fusion QRS complex (Fig. 79-21). Table 79-4 notes common features of PACs and PVCs.
Table 79-4
| PREMATURE ATRIAL CONTRACTIONS | PREMATURE VENTRICULAR CONTRACTIONS |
| No compensatory pause | Fully compensatory pause (unless interpolated) |
| Preceding P wave (different from sinus P wave; occasionally buried in T wave) | No preceding P waves (although retrograde atrial conduction can cause inverted P wave after QRS) |
| Usually classic right bundle branch block pattern (especially if long-short cycle sequence appears) identical to sinus QRS | Left bundle branch block, right bundle branch block, or hybrid pattern |
| QRS axis normal or near normal | Frequently bizarre QRS axis |
| QRS rarely >0.14 s | QRS often >0.14 s |
Therapy for PVCs is directed toward correcting any precipitating condition, be it a catecholamine excess, a drug effect, an electrolyte imbalance, or cardiac ischemia (Box 79-6). When occurring in isolation, symptomatic PVCs can be treated with low-dose beta-blocker therapy, but this is rarely an emergency need. Although lidocaine will suppress PVCs, it is not recommended in the absence of VT owing to limited clinical benefit and the risk of asystole.
Narrow-Complex Tachycardia
Sinus Tachycardia
Sinus tachycardia is characterized electrocardiographically by a regular, narrow-complex tachycardia with normal P waves preceding each QRS complex (Fig. 79-22). In adults, sinus tachycardia rarely exceeds a rate of 170 beats/min, but in infants and young children, it is not unusual to see rates above 200 to 225 beats/min. As opposed to other forms of narrow-complex tachycardia, sinus tachycardia tends to speed up or slow down in a graded and continuous manner over time.
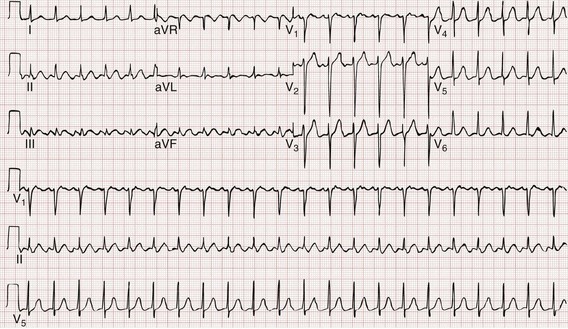
Figure 79-22 Sinus tachycardia.
Atrial Tachycardia
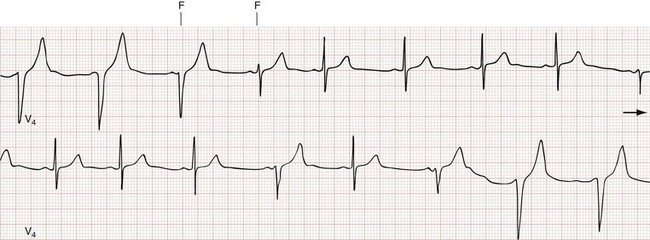
Atrial tachycardia (AT) is an atrial rhythm occurring at a rate of more than 100 beats/min arising from a non–sinus node site (or sites) within the left or right atrium. The hallmark of AT is the presence morphologically abnormal P waves on the surface ECG (Fig. 79-23). If the site of origin is close to the sinus node, atrial depolarization waves may look like a normal P wave. Depending on the atrial rate, the AV conduction ratio may be 1 : 1, 2 : 1 or higher.
Multifocal atrial tachycardia (MAT) is a form of AT with three or more distinct P wave morphologies, and varying PR and P-P intervals from the multiple ectopic atrial foci (Fig. 79-24). MAT is associated with pulmonary disease (chronic obstructive pulmonary disease [COPD] in particular) in up to 60% of cases, but it can also be seen in the presence of primary cardiac pathology. On the surface ECG, MAT can be mistaken for course atrial fibrillation because of the nonuniform atrial activity and irregular R-R intervals.
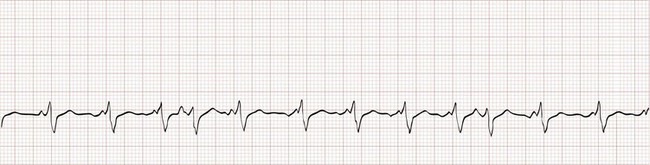
Atrial Fibrillation
Electrocardiographically, the hallmark of atrial fibrillation is a “irregularly irregular” QRS pattern (Fig. 79-25). Although atrial fibrillation is not the sole cause of an irregular ventricular rhythm, it is by far the most common (Box 79-7). Atrial fibrillatory waves appear “coarse” or “fine” on the basis of their amplitude and are often best appreciated in the inferior leads or lead V1.
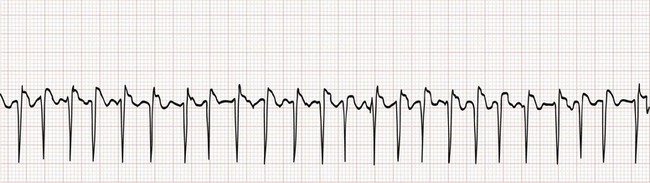
Figure 79-25 Atrial fibrillation with rapid ventricular response. Note that the irregularity could be easily overlooked.
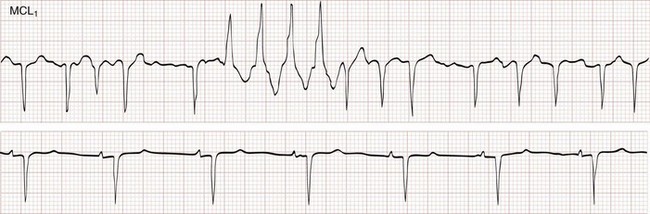
The Ashman phenomenon refers to aberrant ventricular conduction of an early-arriving atrial impulse following a relatively long R-R interval, as a result of a partially refractory His bundle. Such aberrantly conducted impulses are often seen in atrial fibrillation but can occur in any irregular rhythm in which long-short cycle sequences occur, and they typically assume an RBB block pattern (Fig. 79-26). Ashman beats can be mistaken for PVCs or paroxysmal VT.
Atrial fibrillation is most commonly associated with underlying heart disease or hypertension (Box 79-8), but it can also occur in isolation (so-called lone atrial fibrillation) or as a manifestation of hyperthyroidism. As many as one third of patients with congestive heart failure also have atrial fibrillation.
For stable patients with new or recurrent rapid atrial fibrillation, administration of a nodal blocking agent with a goal of achieving a target ventricular rate of 120 beats/min or less is a first step.13 Intravenous calcium channel blockers (diltiazem and verapamil) or beta-blockers (metoprolol) offer the advantage of being easily titrated and can be followed by an oral agent before discharge. Nodal agents should not be used for rate control in the setting of accessory pathway conduction because AV conduction (with retrograde conduction into the accessory pathway) may be the only thing preventing the ventricular rate from accelerating even further and degenerating into ventricular fibrillation. The debate as to whether asymptomatic patients with persistent atrial fibrillation benefit additionally from rhythm control, either via cardioversion or by other means (e.g., ablation), is ongoing without a definitive answer. In choosing rate control for the long term, the ideal target rate is debated, with some advocating less than 80 beats/min at rest instead of the common goal of 110 beats/min or less; no clear evidence of either target being superior exists yet.
For stable patients with new-onset or newly recurrent atrial fibrillation—defined as having a duration of 72 hours or less—ED cardioversion is an option. If atrial fibrillation has been present longer or for an uncertain interval in the absence of ongoing anticoagulation, however, cardioversion is not recommended to avoid the risk (1-4% at 30 days) of systemic embolization. The choice of electrical versus pharmacologic cardioversion is dependent on institutional factors and patient preference, although published success rates are higher (80% or greater) with electrical conversion.13,14 Among patients with new or recurrent atrial fibrillation of less than 48 to 72 hours’ duration, up to 50% will convert spontaneously to sinus rhythm within 24 hours. Prehospital cardioversion is not recommended.
Various agents are available for pharmacologic cardioversion of patients with stable atrial fibrillation in the ED, including the class IA, IC, and III antidysrhythmics (Box 79-9). In practice, intravenous procainamide, amiodarone, and ibutilide are the agents most commonly used in ED setting. Although differences in success rates exist among agents, the overall response is 40 to 65% for drug-based ED cardioversion. The class IC antidysrhythmics are contraindicated in patients with structural or ischemic heart disease. For atrial fibrillation with accessory pathway conduction, procainamide is recommended because it has no effect on AV conduction.
Atrial Flutter
Atrial flutter is defined by atrial depolarization occurring at a regular rate of 250 to 350 beats/min (300 beats/min is classic) caused by an atrial reentry mechanism (Fig. 79-27). So-called flutter waves are distinguished by their broad, sawtooth appearance on the surface ECG. The ventricular rate in atrial flutter is often rapid, but in the absence of a bypass tract (in which 1 : 1 conduction is possible), the conduction ratio is limited by the refractory period of the AV node. With 2 : 1 conduction, the ventricular rate is approximately 150 beats/min, often making flutter waves themselves difficult to appreciate and allowing the rhythm to be mistaken for sinus tachycardia. Often the conduction ratio will change from beat to beat, called atrial flutter with variable conduction, with a resultant irregular ventricular rate; this is hard to distinguish from atrial fibrillation.
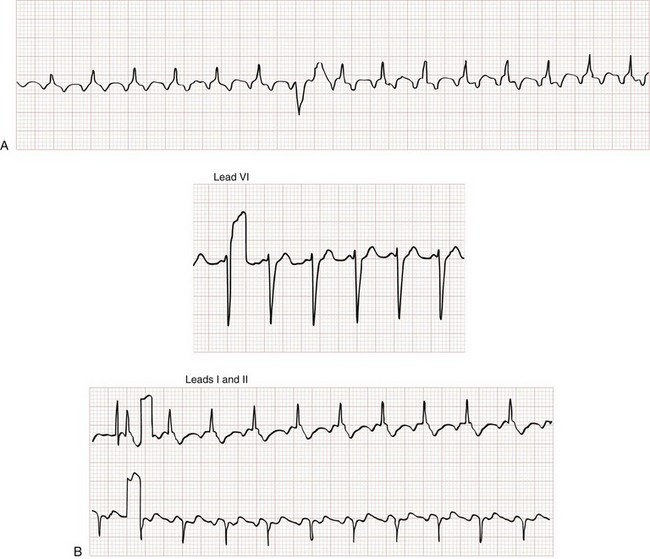
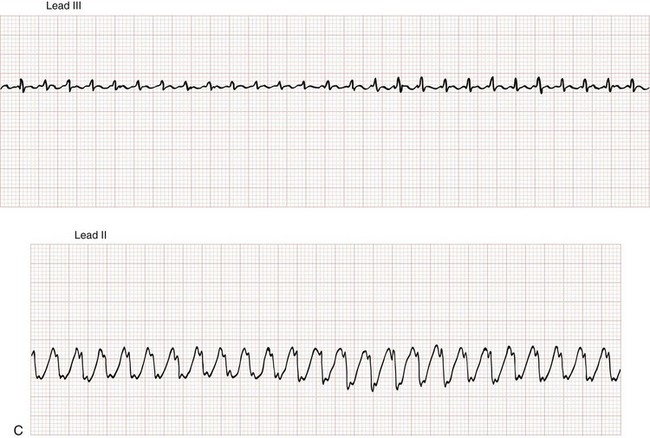
Atrial flutter can be associated with structural heart disease, particularly valvular heart disease and cardiomyopathies. The acute management of atrial flutter is similar to that of atrial fibrillation with a few special considerations. Because AV conduction occurs at fixed ratios in atrial flutter, the administration of beta-blocker or calcium channel blocker therapy can result in an abrupt rate change, making it more challenging to titrate therapy to a desired target rate. Atrial flutter is more sensitive to DC cardioversion (up to 90% conversion rate) than atrial fibrillation, and usually requires lower energy (20-50 J) for conversion to sinus rhythm.15 Conversely, atrial flutter is more resistant to chemical cardioversion (less than 50%) than new-onset nonvalvular atrial fibrillation.
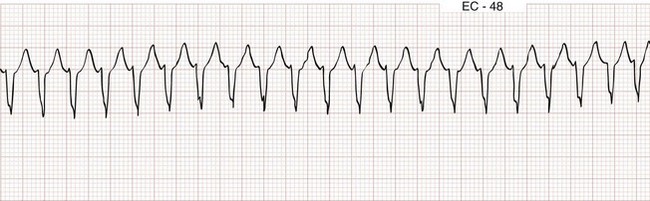
Atrioventricular Nodal Reentrant Tachycardia
Also known by the less precise term paroxysmal supraventricular tachycardia (PSVT), AVNRT is characterized by a regular narrow-complex rhythm and a ventricular rate of 160 beats/min or greater (Fig. 79-28). It is the most common nonsinus tachydysrhythmia in young adults. As its name suggests, AVNRT occurs as the result of a reentry circuit within the AV node, with normal conduction (narrow QRS) down the bundles of His, and with retrograde conduction (inverted P waves typically buried within the QRS) up into the atria (Fig. 79-29).
Preexcitation and Accessory Pathway Syndromes
Preexcitation refers to depolarization of the ventricular myocardium by circumvention of the AV node via an accessory pathway or bypass tract linking the atria to the ventricles. Accessory pathways lend themselves to reentry tachycardias and very rapid ventricular rates. Wolff-Parkinson-White (WPW) syndrome is the classic accessory pathway syndrome, characterized by paroxysmal tachycardia and the following three resting ECG features (Fig. 79-30):
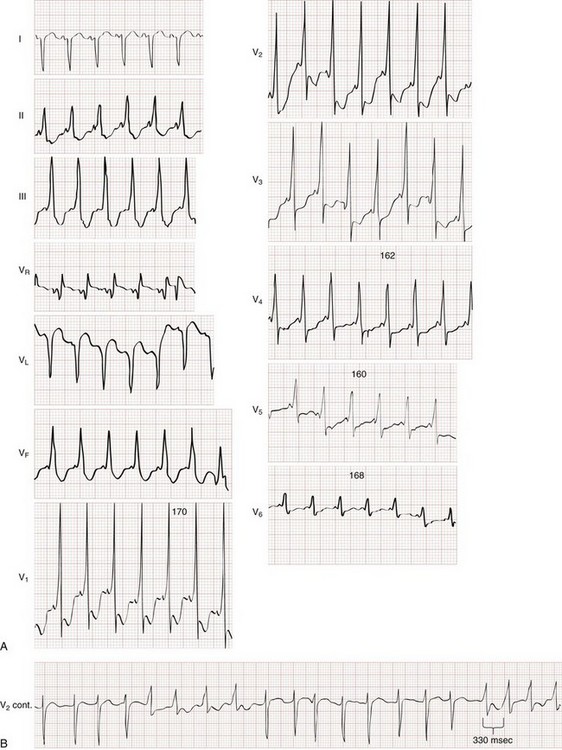
• A short PR interval (<0.12 second)
• QRS duration greater than 0.10 second
• A slurred upstroke to the QRS complex, referred to as a delta wave
Together with the AV node, an accessory pathway can form a reentry circuit to produce and sustain a tachycardia (Fig. 79-31). When the AV node is being used for anterograde conduction to the ventricles and the accessory path is used for retrograde conduction, it is called an orthodromic AV-reentrant tachycardia; the QRS complex will be narrow and the ventricular rate is constrained. Conversely, when the accessory pathway is being used as the anterograde limb and the AV node as the retrograde limb of the reentry circuit, it is called an antidromic AV-reentrant tachycardia with a wide QRS complex. The ventricular rate can be normal or rapid and more than 200 beats/min. Fortunately, in patients with WPW and most other preexcitation syndromes, orthodromic tachycardia is much more common than antidromic tachycardia. WPW syndrome is associated with a variety of conditions, although the majority of patients have no underlying structural heart disease (Box 79-10).
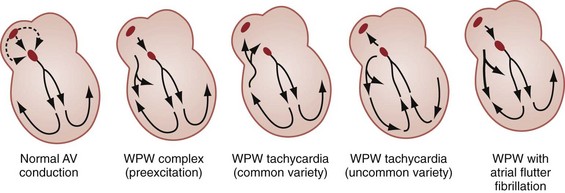
In contrast, antidromic AV-reentrant tachycardia often causes a very rapid rate (200 beats/min or greater) and a wide-complex QRS. In this situation, AV nodal blocking agents are contraindicated because it is the presence of anterograde AV conduction that provides a brake on even more rapid retrograde conduction. Unopposed retrograde conduction through the AV node can result in ventricular fibrillation.16 Likewise, with a very rapid irregular tachycardia (i.e., atrial fibrillation with a ventricular rate exceeding 200) accompanied by a wide-complex QRS, the likelihood of accessory pathway conduction should be considered and nodal blocking agents again withheld. Procainamide is recommended any time an accessory pathway with wide QRS complexes or very rapid rates exists, with amiodarone as a secondary choice despite theoretic concerns about its nodal blocking properties. Electrical cardioversion (100-200 J) is indicated for ventricular rates greater than 250 beats/min, clinical deterioration, or failure of pharmacologic therapy.
Wide-Complex Tachycardias
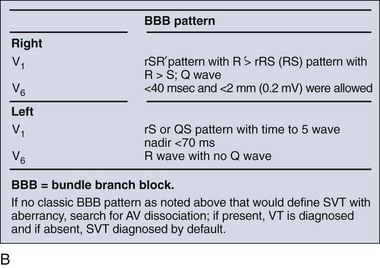
Wide-complex tachycardia refers to any tachydysrhythmia accompanied by a QRS duration of 0.12 second or more. Wide-complex tachycardias can derive from the ventricles themselves (i.e., VT) or can result from a tachycardia originating at or above the AV node accompanied by aberrant AV conduction, caused by either an accessory pathway or a preexisting or rate-related conduction delay, such as a bundle-branch block. The presence of AV dissociation or fusion beats on the 12-lead ECG clearly points to VT; all else being equal, older age and a prior history of myocardial infarction make VT much more likely than a supraventricular tachycardia with aberrancy. On the other hand, an irregular tachycardia with a wide QRS approximating a bundle-branch morphology is most likely atrial fibrillation with aberrant conduction. For stable patients, there are decision tools that can be used to distinguish VT with relatively good accuracy (Table 79-5 and Fig. 79-32).17–21
Table 79-5
| VENTRICULAR TACHYCARDIA | SVT PLUS ABERRANCY | |
| Clinical features | Age 50 or older | Age 35 or younger |
| History of myocardial infarction, congestive heart failure, CABG, or ASHD | None | |
| Previous history of ventricular tachycardia | Previous history of SVT | |
| Physical examination | Cannon A waves | Absent |
| Variation in arterial pulse | Absence of variability | |
| Variable first heart sound | Absence of variability | |
| Electrocardiogram | Fusion beats | None |
| AV dissociation | Preceding P waves with QRS complexes | |
| QRS >0.14 s | QRS usually <0.14 s | |
| Extreme LAD artery (30 degrees) | Axis normal or near normal | |
| No response to vagal maneuvers | Slow or terminate with vagal maneuvers | |
| Specific QRS patterns | V1: R, qR, or RS | V1: rSR′ |
| V6: S, rS, or qR | V6: qRs | |
| Identical to previous ventricular tachycardia tracing* | Identical to previous SVT tracing* | |
| Concordance of positivity or negativity† |
*If proven by electrophysiologic studies or by a preponderance of evidence.
†Main deflection of QRS complex either positive or negative in every precordial lead.
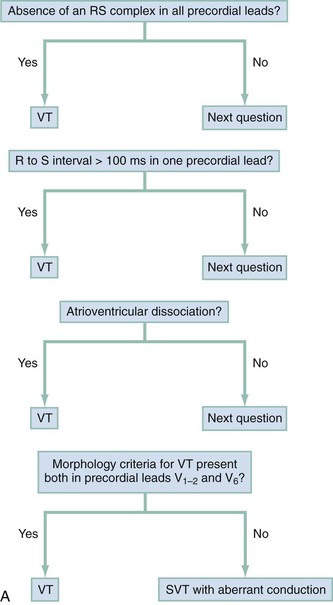
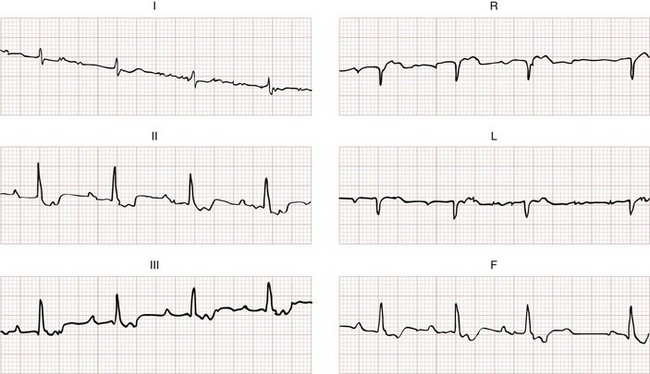
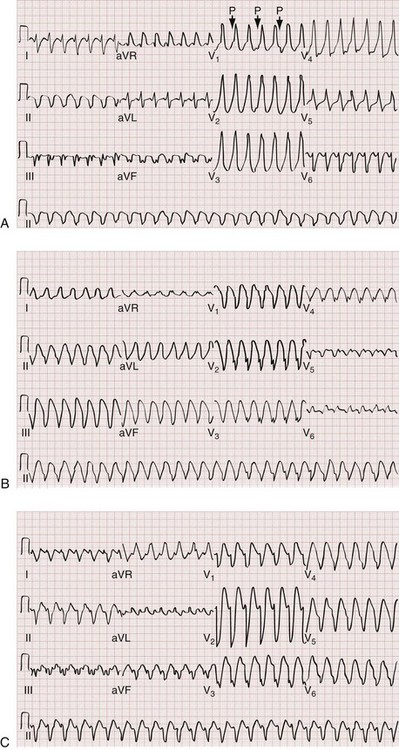
The Brugada ECG criteria search for four pieces of evidence of VT from among those listed previously; as soon as one is found,18,20,21 the diagnosis of VT is made. The rhythm needs to be regular for these to be used (chaos suggests atrial fibrillation with altered conduction). The sequential criteria are as follows (see Figs. 79-32 and 79-33):
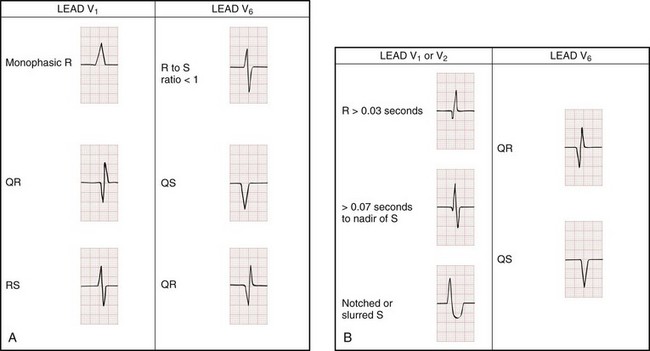
1. Absence of any RS complexes in the chest leads
2. RS duration (measured from beginning of R to deepest part of S wave) greater than 100 msec
3. AV dissociation (often present but overlooked; may be best appreciated in inferior limb leads and V1-2; Fig. 79-34)
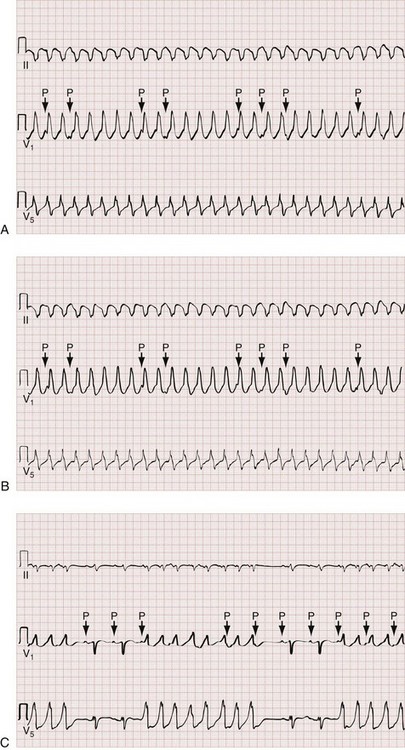
The Griffith criteria use a three-step approach to identify aberrancy,19,20 first through classic RBBB or LBBB morphologies in V1 and V6 to identify SVT, then seeking AV dissociation in the remainder to identify VT (see Figs. 79-32 and 79-34).This approach has a reported sensitivity of 92%, although specificity may be less than seen with the Brugada approach (though head-to-head in-practice comparative data do not exist).
Ventricular Tachycardia
Monomorphic ventricular tachycardia is the most common form of VT and is characterized by morphologically consistent QRS complexes, usually in a regular pattern and at a rate of 150 to 200 beats/min (Fig. 79-35). Polymorphic ventricular tachycardia is seen with varying QRS morphologies and suggests more severe underlying disease (Figs. 79-36 and 79-37). VT is prevalent in patients with both ischemic and nonischemic cardiomyopathy.
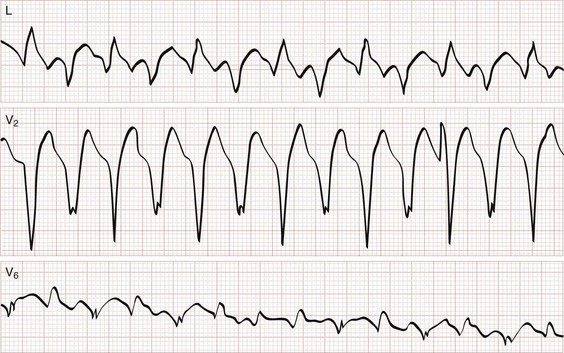
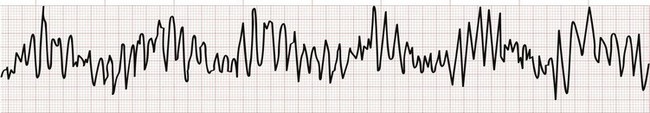
All patients with new or symptomatic VT should be admitted, the exception being those who have implanted defibrillators that are functioning appropriately (see Chapter 80).
Torsades de Pointes
Torsades de pointes is literally translated as “twisting of the points” and is a paroxysmal form of polymorphic VT that meets the following clinical criteria (see Fig. 79-37):
1. Ventricular rate greater than 200 beats/min
2. Undulating QRS axis, with the polarity of the complexes appearing to shift about the baseline
Acquired QT prolongation is often multifactorial (Box 79-11). Common triggers include electrolyte disturbances (hypokalemia and hypomagnesemia) and many different drugs (notably class IA and IC agents but also many others; see Box 79-11), especially when used in combination. Treatment of torsades de pointes in stable patients involves correcting any underlying metabolic or electrolyte abnormalities and increasing the heart rate to shorten ventricular repolarization. For obvious reasons, classes IA and IC antidysrhythmics are contraindicated. Intravenous magnesium sulfate should be given empirically and is effective in treating torsades de pointes even in the absence of hypomagnesemia.
Brugada’s Syndrome
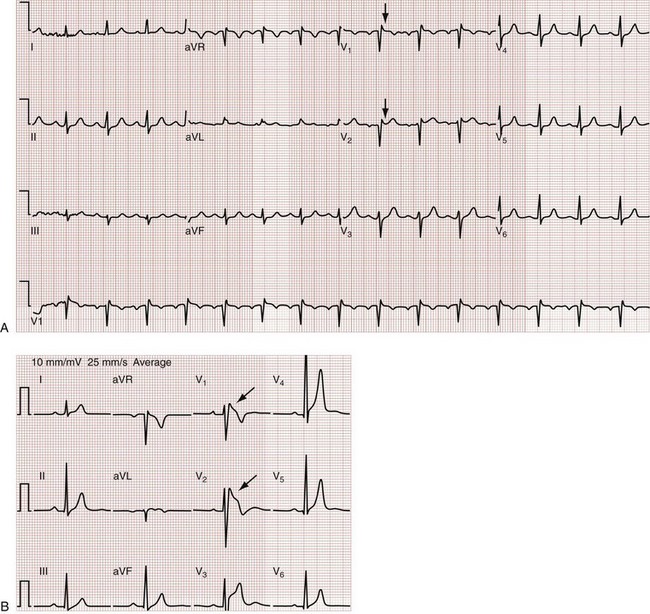
Brugada’s syndrome is also associated with ventricular dysrhythmias and syncope or sudden cardiac death in the absence of structural heart disease.22 Brugada’s syndrome results from an inherited disorder of sodium channels and is most commonly present in males during young adulthood. The Brugada syndrome ECG pattern is characterized by a downward coved or humped (saddleback) ST segment elevation in leads V1 to V3 (Fig. 79-38), sometimes simulating an RBBB appearance. The ST segment findings may be transient or elicited only with pharmacologic stimulation.





References
1. Roden, DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013.
2. Nygaard, TW, Sellers, TD, Cook, TS, DiMarco, JP. Adverse reactions to antiarrhythmic drugs during therapy for ventricular arrhythmias. JAMA. 1986;256:55.
3. Schrickel, JW, et al. Pro-arrhythmic effects of amiodarone and concomitant rate-control medication. Europace. 2006;8:403–407.
4. Goldschlager, N, et al. A practical guide for clinicians who treat patients with amiodarone. Heart Rhythm. 2007;4:1250–1259.
5. Naccarelli, GV, Lee, KS, Gibson, JK, VanderLugt, J. Electrophysiology and pharmacology of ibutilide. Am J Cardiol. 1996;78:12–16.
6. Banchs, JE, et al. Efficacy and safety of dofetilide in patients with atrial fibrillation and atrial flutter. J Interv Card Electrophysiol. 2008;23:111–115.
7. Singh, BN, et al. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N Engl J Med. 2007;357:987–999.
8. Hohnloser, SH, et al. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med. 2009;360:668–678.
9. Falk, RH, Knowlton, AA, Bernard, SA, Gotlieb, NE, Battinelli, NJ. Digoxin for converting recent-onset atrial fibrillation to sinus rhythm: A randomized double-blinded trial. Ann Intern Med. 1987;106:503–508.
10. Tan, HL, Spekhorst, HHM, Peters, RJG, Wilde, AAM. Adenosine induced ventricular dysrhythmias in the emergency room. Pacing Clin Electrophysiol. 2011;24:450–455.
11. Sharma, AD, Klein, GJ, Yee, R. Intravenous adenosine triphosphate during wide QRS complex tachycardia: Safety, therapeutic efficacy, and diagnostic utility. Am J Med. 1990;88:337.
12. Marill, KA, et al. Adenosine for wide-complex tachycardia: Efficacy and safety. Crit Care Med. 2009;37:2512–2518.
13. Stiell, IG, Macle, L, CCS Atrial Fibrillation Guidelines Committee. Canadian Cardiovascular Society Atrial Fibrillation Guidelines 2010: Management of recent-onset atrial fibrillation and flutter in the emergency department. Can J Cardiol. 2011;27:38–46.
14. Burton, JH, et al. Electrical cardioversion of emergency department patients with atrial fibrillation. Ann Emerg Med. 2004;44:20–30.
15. Scheuermeyer, FX, Grafstein, E, Heilbron, B, Innes, G. Emergency department management and 1-year outcomes of patients with atrial flutter. Ann Emerg Med. 2011;57:564–571.
16. Montoya, PT, et al. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. Eur Heart J. 1991;12:144–150.
17. Wellens, HJ, Bar, FW, Lie, KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med. 1977;64:27–33.
18. Brugada, P, Brugada, J, Mont, L, Smeets, J, Andries, EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991;83:1649–1659.
19. Griffith, MJ, Garratt, CJ, Mounsey, P, Camm, AJ. Ventricular tachycardia as default diagnosis in broad complex tachycardia. Lancet. 1994;343:386–388.
20. Lau, EW, Ng, GA. Comparison of the performance of three diagnostic algorithms for regular broad complex tachycardia in practical application. Pacing Clin Electrophysiol. 2002;25:822–827.
21. Herbert, ME, Votey, SR, Morgan, MT, Cameron, P, Dziukas, L. Failure to agree on the electrocardiographic diagnosis of ventricular tachycardia. Ann Emerg Med. 1996;27:35–38.
22. Wilde, AA, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation. 2002;106:2514–2519.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]Chapter 79
Dysrhythmias
Cardiac Cellular Electrophysiology
The function of individual cells in the conductive and contractile tissues of the heart depends on an intact resting membrane potential. Na+, K+, and Ca2+ ions create the membrane potential and regulate conduction and contractility. The membrane potential is the result of a differential concentration of Na+ and K+ on either side of the cell membrane, measuring approximately −90 mV in normal resting, nonpacemaker cells. This electrical gradient exists mainly because of the Na+-K+ exchange pump and the natural concentration-dependent flow of K+ out of the cell. Adenosine triphosphate (ATP) fuels Na+ transport out to the extracellular fluid, with Mg2+ used as a cofactor (Fig. 79-1). This process creates an osmotic gradient, allowing Ca2+ to be passively exchanged for Na2+ and promoting conduction as well as myofibril contraction. The resting membrane potential is generated from the flow of K+ down a concentration gradient toward the extracellular fluid. The cell membrane is far more permeable to potassium than sodium ions, resulting in a net loss of intracellular positive charge.
In normal nonpacemaker cells, the application of an electrical stimulus causes the membrane potential to become less negative, termed depolarization. When the membrane potential reaches −70 mV, specialized Na2+ channels open, causing a rapid influx of positive charge into the cell. This “fast” channel activity further decreases the membrane potential and is augmented at 30 to 40 mV by a second “slow” channel that allows Ca2+ influx. When these channels close, resting potential is restored by the sodium-potassium pump, an event known as repolarization (Fig. 79-2).
In nonpacemaker cells, additional depolarization from a second electrical stimulus is not possible when the membrane potential is more positive than −60 mV. This period is termed the effective refractory period (Fig. 79-3). At a membrane potential of −60 to −70 mV, a strong impulse can cause a response that is likely to be propagated, although abnormally; this response represents the relative refractory period. At a membrane potential of −70 mV or less, virtually all fast channels are ready for activity if properly stimulated (see Fig. 79-3).
Anatomy and Conduction
Figure 79-4 correlates the surface ECG tracings with tissue electrical events. Impulses generated from within the SA node itself, imperceptible on the surface ECG, are propagated through the atrial tissue to the AV node. Atrial depolarization is characterized by the P wave on the surface ECG.
The AV node is an area of specialized tissue between the atria and the ventricles of the heart, located in the posterior-inferior region of the interatrial septum. Its blood supply is from a branch of the RCA in 90% of patients (right dominant) and from the LCA in the remaining 10% (left dominant). Transmission of impulses within the AV node is slower than in parts of the conducting system (Table 79-1) because of a dependence on slow-channel ion influx for membrane depolarization. In some patients, pathologic “accessory pathways” connect atrial and ventricular tissues. These accessory pathways do not share the normal conduction delay of the AV node and may allow for rapid ventricular rates. Preexcitation refers to the early depolarization of ventricular myocardium when accessory paths are used instead of the normal conduction system.
Mechanisms of Dysrhythmia Formation
There are three electrophysiologic mechanisms: enhanced automaticity, triggered activity, and reentry. Enhanced automaticity includes spontaneous depolarization of nonpacemaker cells or a lower threshold for depolarization in normal pacemaker cells (Fig. 79-5). Both types of enhanced automaticity can occur in the setting of ischemia, electrolyte disturbances, or drugs. Examples of enhanced automaticity include idioventricular rhythms in the setting of acute myocardial infarction, and atrial tachycardias or junctional tachycardias (JTs) seen with digitalis toxicity.
Reentry dysrhythmias occur as a consequence of abnormal conduction (Fig. 79-6). For a reentry mechanism, two alternate pathways for conduction must be present and one path must have a longer refractory period. The unequal responsiveness of the limbs creates a functional unidirectional block such that when the impulse exits one limb, it may then reenter the other in retrograde fashion. The cycle is then repeated, creating a self-sustaining or “circus movement” tachycardia that can appear orderly or disorderly (i.e., fibrillatory).
Figure 79-6 Mechanism of reentry.
Classification of Antidysrhythmic Drugs
Medications used to treat dysrhythmias are classified into four major categories on the basis of their electrophysiologic effects (Box 79-1). Other agents fall outside this classification system and are discussed separately.
All antidysrhythmics can cause “prodysrhythmic effects.” This occurs most often in patients with existing structural heart disease and in patients receiving new or higher doses of antidysrhythmic agents. The class I and III agents are associated with prodysrhythmic effects in up to 15% of patients.1,2
Class IC Agents
The class IC agents profoundly slow depolarization and conduction and have pronounced antidysrhythmic properties.1,2 Up to 15% of patients treated with class IC agents experience new or increased ventricular dysrhythmias. Class IC agents are approved only for oral use in the United States.
Class II Agents
Side effects of beta-blockers include bronchospasm, bradycardia, hypotension, and heart failure. All beta-blockers are active at both beta1 and beta2 receptors (Table 79-2) to varying degrees. Those with more prominent beta1 effects are said to be cardioselective. Relative contraindications to the use of beta-blockers include asthma or chronic obstructive lung disease, advanced congestive heart failure, and third-trimester pregnancy. Beta-blockers should not be used in patients with preexisting bradycardia or heart block beyond first-degree. Intravenous beta-blockers should be used cautiously in conjunction with calcium channel blockers because of the risk of additive side effects. Acute side effects of beta-blockers include bronchospasm, heart failure, excessive bradycardia, and hypotension.
Table 79-2
Cardiac and Respiratory Beta-Adrenergic Receptors and Responses to Pharmacologic Manipulation
Class III Agents
All class III agents prolong the refractory period by blocking K+ channels, and they have variable effects on the QT interval.1–4 In general, class III agents are alternatives to the class I agents for the treatment of many ventricular and atrial dysrhythmias.
Amiodarone
The serum half-life of amiodarone is 25 hours after a single intravenous dose and up to 50 days during long-term oral use. Because of the unusual pharmacokinetics, oral regimens vary widely. The acute side effects of amiodarone are primarily limited to hypotension, bradycardia, and heart failure (Box 79-2). There is an additive risk of bradycardia and hypotension when amiodarone is used in conjunction with calcium channel or beta-adrenergic blockers. Rates of prodysrhythmia are relatively low (1-3%). Long-term amiodarone use, however, is associated with significant extracardiac side effects, including irreversible lung and thyroid disease. Amiodarone alters the pharmacokinetics of numerous other drugs, including digoxin and warfarin.
Ibutilide
Ibutilide has a unique mechanism of action characterized by induction of a slow inward Na2+ current, thereby prolonging the refractory period. Intravenous ibutilide is approved for cardioversion of atrial fibrillation and atrial flutter.5 Ibutilide is associated with QT interval prolongation, and polymorphic VT is more common than with amiodarone although still relatively rare (0.9-2.5%).
Dofetilide
Dofetilide is a pure class III agent that blocks K+ efflux, increasing the refractory period. It is approved for chemical cardioversion and maintenance of sinus rhythm in patients with atrial fibrillation or flutter,6 but because of its prodysrhythmic effects, it has limited usefulness.
Dronedarone
Structurally related to amiodarone, dronedarone displays class III properties plus those of other antidysrhythmic classes. The incidence of polymorphic VT associated with dronedarone is very low. Dronedarone is approved for oral use to maintain sinus rhythm in patients with atrial fibrillation or flutter but is contraindicated in patients with severe or recent heart failure decompensation.7 Long-term effects and the ideal populations for the use of this drug remain unclear.8
Class IV Agents
Miscellaneous Agents
Digoxin (0.25-0.5 mg IV) can control the ventricular rate in patients with SVTs, including atrial fibrillation and atrial flutter. Because of its delayed onset of action and narrow therapeutic window, however, digitalis is not a first-line agent for emergency therapy. It is not true that digoxin promotes conversion to a sinus rhythm any more than other rate-controlling agents.9
Side effects of digoxin are listed in Box 79-3 and are aggravated by hypokalemia, hypercalcemia, hypomagnesemia, increased catecholamines, and acid-base disturbances. Digoxin overdose therapy is covered elsewhere.
Adenosine
Adenosine is a naturally occurring purine nucleoside used for the termination of narrow-complex tachydysrhythmias. Administered as an intravenous bolus, adenosine causes an abrupt slowing of AV conduction in both anterograde and retrograde pathways. Adenosine usually has an onset of action of 5 to 20 seconds and a duration of effect of 30 to 40 seconds. Except in rare cases of catecholamine-induced ventricular dysrhythmias, adenosine has little or no effect on infranodal conduction. For this reason, adenosine is often used as a diagnostic agent in patients with wide-complex tachydysrhythmias when the cause is unclear.10–12 An initial dose of 6 mg as a rapid bolus for adults weighing 50 kg or greater is recommended, with flush through a large peripheral vein. If no response is seen within 1 to 2 minutes, the second dose is doubled (12 mg) and administered. If no effect is seen after a final 12-mg dose, the rhythm should be reassessed and another agent used. Pediatric doses are 0.05 mg/kg initially with doubling at similar intervals up to a total dose of 0.25 mg/kg. Side effects coincide with the onset of clinical effects and occur in up to a third of patients but are usually minor. These include flushing, dyspnea, chest pressure, nausea, headache, dizziness, transient bradycardia or heart block, and hypotension (seen rarely, from the vasodilatory properties). These side effects usually resolve rapidly without treatment, although many patients are intensely uncomfortable for a short period. Asystole can occur but is usually transient though unsettling to those observing.
Approach to Dysrhythmia Recognition and Management
• Narrow-complex (QRS less than 0.12 second) tachycardias (regular and irregular)
• Wide-complex (QRS 0.12 second or greater) tachycardias (regular and irregular)
Initial Assessment of Stable Patients
The 12-lead ECG is essential for evaluation of any patient with a suspected dysrhythmia. Use of a single ECG lead is often adequate for diagnosis, but multiple leads are often required to detect such things as the presence or absence of P waves (often best seen in inferior leads or V1-2; Fig. 79-7), the relationship between P waves and QRS complexes, prolongation of the QRS and QT interval, and evidence of ischemia or prior myocardial infarction (Box 79-4). For certain conditions, such as Brugada’s syndrome, the 12-lead ECG together with a history of syncope is diagnostic.
