[level-membership-for-pediatrics-category]
Duodenal and Intestinal Atresia and Stenosis
Congenital intestinal obstruction occurs in approximately 1 : 2000 live births and is a common cause of admission to a neonatal surgical unit, accounting for up to one-third of all admissions.1 Morphologically, congenital defects related to continuity of the intestine can be divided into either atresia or stenosis. Together, they constitute one of the most common etiologies of neonatal intestinal obstruction.2–4 See Chapter 29 for information about pyloric atresia.
Duodenal Atresia and Stenosis
Congenital duodenal atresia and stenosis is a frequent cause of intestinal obstruction and occurs in 1 per 5000 to 10,000 live births, affecting boys more commonly than girls.5 More than 50% of affected patients have associated congenital anomalies, with trisomy 21 occurring in approximately 30% of patients.6,7 Operative correction is accomplished via a duodenoduodenostomy, with or without tapering duodenoplasty. This can be performed either laparoscopically or open. Early postoperative survival rates of greater than 90% should be expected.7–11
Etiology
Congenital duodenal obstruction can occur due to an intrinsic or extrinsic lesion.12 The most common cause of duodenal obstruction is atresia.7 This intrinsic lesion is most commonly believed to be caused by a failure of recanalization of the fetal duodenum resulting in complete obstruction. Early in the fourth week of gestation, the duodenum begins to develop from the distal foregut and the proximal midgut. During the fifth and sixth weeks of gestation, the duodenal lumen temporarily obliterates due to proliferation of its epithelial cells. Vacuolation due to degeneration of the epithelial cells during the 11th week of gestation then leads to duodenal recanalization.13 An embryologic insult during this period can lead to an intrinsic web, atresia, or stenosis. The extrinsic form of duodenal obstruction is due to defects in the development of neighboring structures such as the pancreas, a preduodenal portal vein, or malrotation and Ladd’s bands.14,15
Annular pancreas as an etiology for duodenal obstruction warrants special mention as this form of obstruction is likely due to failure of duodenal development rather than a true constricting lesion. Thus, the presence of an annular pancreas is simply a visible indication of an underlying atresia or stenosis.16 Between the fourth and eighth week of gestation, the pancreatic buds merge. In annular pancreas, the tip of the ventral pancreas becomes fixed to the duodenal wall forming a nondistensible, ring-like or annular portion of pancreatic tissue surrounding the descending part of the duodenum.13 In annular pancreas associated with duodenal obstruction, the distal biliary tree is often abnormal and may open proximal or distal to the atresia or stenosis.17,18 Other reported biliary abnormalities associated with duodenal obstruction include biliary atresia, gallbladder agenesis, stenosis of the common bile duct, choledochal cyst, and immune deficiency.19–24
Classification
Anatomically, duodenal obstructions are classified as either atresias or stenoses. An incomplete obstruction, due to a fenestrated web or diaphragm, is considered a stenosis. Most stenoses involve the third and/or fourth part of the duodenum. Atresias, or complete obstruction, are further classified into three morphologic types (Fig. 30-1). Type I atresias account for more than 90% of all duodenal obstructions and contain a lumenal diaphragm that includes mucosal and submucosal layers. A diaphragm that has ballooned distally (windsock) is a type I atresia.25,26 It is important to understand that the anatomy of the windsock may lead to a portion of the dilated duodenum actually being distal to the actual obstruction (Fig. 30-2). Type II atresias are characterized by a dilated proximal and collapsed distal segment connected by a fibrous cord. Type III atresias have an obvious gap separating the proximal and distal duodenal segments.27
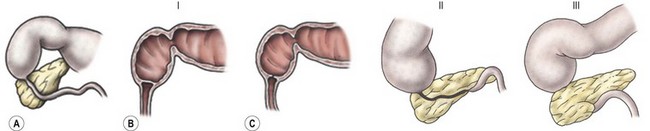
FIGURE 30-1 Duodenal atresia (and stenosis) is depicted. In type I (A), either a membrane (B) or web (C) causes the intrinsic duodenal obstruction. There is no fibrous cord and the duodenum remains in continuity. Type II is characterized by complete obliteration of a segment of the duodenum with the proximal and distal portions attached via a fibrous cord. Type III is associated with complete separation of the dilated proximal duodenum from the collapsed distal duodenum.
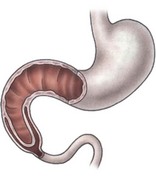
FIGURE 30-2 Illustration of the ‘windsock’ deformity, a variant of type I duodenal atresia. Note the actual position of the origin of the web in relation to the extent of proximal duodenal dilation and the distal collapsed duodenum.
More than 50% of affected patients with duodenal atresia have associated congenital anomalies.28 Approximately 30% are associated with trisomy 21, 30% with isolated cardiac defects, and 25% with other gastrointestinal (GI) anomalies.29,30 Approximately 45% of patients are premature, and about one-third exhibit growth retardation.6,7
Pathology
The obstruction can be classified as either preampullary or postampullary, with approximately 85% of obstructions located distal to the ampulla.30 With complete or almost complete obstruction, the stomach and proximal duodenum become significantly dilated. The pylorus is usually distended and hypertrophic. The bowel distal to the obstruction is collapsed, except in the case of a windsock deformity in which the distal bowel is dilated to a variable length depending on the length of the windsock (see Fig. 30-2). In most cases of duodenal obstruction, the gastrointestinal tract can be decompressed proximally. With complete obstruction of the duodenum, the incidence of polyhydramnios ranges from 32–81%.1,31–34 Growth retardation is also common, presumably from nutritional deprivation from the swallowed amniotic fluid.
Diagnosis
There are multiple benefits to the antenatal diagnosis of duodenal obstruction, including parental counseling. The diagnosis can often be suggested by prenatal ultrasound (US). Sonographic evaluation in fetuses of mothers with a history of polyhydramnios can detect two fluid-filled structures consistent with a double bubble in up to 44% of cases.35–37 Despite duodenal obstruction usually occurring by week 12, the reason for failure of early prenatal detection is not entirely clear. Most cases of duodenal atresia are detected between 7 and 8 months gestation.38 It is currently believed that immature gastric emptying in-utero may contribute to low gastric pressures, failing to dilate the proximal duodenum until later in gestation. While both circular and longitudinal muscle layers are present in the stomach by week 8 of gestation, pressure amplitudes at 25 weeks are only 60% of term gastric pressures.39,40
The presentation of the neonate with duodenal obstruction varies depending on whether the obstruction is complete or incomplete, and the location of the ampulla of Vater in relation to the obstruction. The classic presentation is that of bilious emesis within the first hours of life in an otherwise stable neonate. In about 10% of cases, however, the atresia is pre-ampullary and the emesis is nonbilious.15 Abdominal distention may or may not be present. In neonates with duodenal atresia, the abdomen is scaphoid. Aspiration via a nasogastric (NG) tube of more than 20 mL of gastric contents in a newborn suggests intestinal obstruction, as normal aspirate is less than 5 mL.41 For patients with stenosis, the diagnosis is often delayed until the neonate has started on enteral feeds and feeding intolerance develops with emesis and gastric distention.
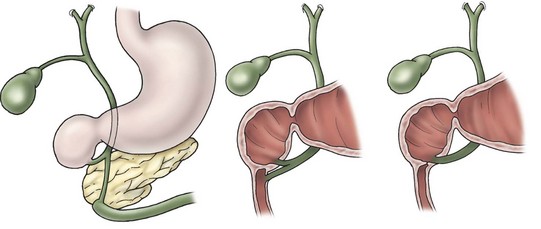
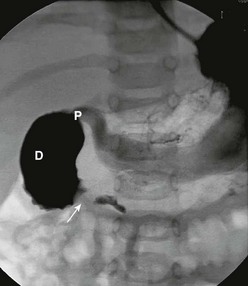
In antenatally suspected cases of duodenal obstruction, as well as in neonates with a clinical presentation consistent with a proximal bowel obstruction, an upright abdominal radiograph is usually sufficient to confirm the diagnosis of duodenal atresia. The diagnostic radiographic presentation of duodenal atresia is that of a double bubble sign with no distal bowel gas (Fig. 30-3). The proximal left-sided bubble represents the air and fluid-filled stomach while the dilated proximal duodenum represents the second bubble to the right of midline.42 In almost all cases of duodenal atresia, the distal bowel is gasless. However, the presence of distal gas does not necessarily exclude the diagnosis of atresia as there are reports of bifed common bile ducts with insertion of one of the ducts proximal and the other distal to the atretic segment which allows the air to bypass the atresia.43 In neonates whose stomach has been decompressed by either NG aspiration or vomiting, 40–60 mL of instilled air into the stomach will reproduce the double bubble.27 Rarely, the biliary tree is air filled, and a variety of pancreatic and biliary anomalies have been demonstrated (Fig. 30-4).43 At our institution, neonates who present with bilious emesis and a decompressed stomach on plain abdominal films receive a limited upper GI contrast study to exclude malrotation and volvulus. With duodenal stenosis, a double bubble sign is often not present and the diagnosis is usually made with a contrast study (Fig. 30-5).
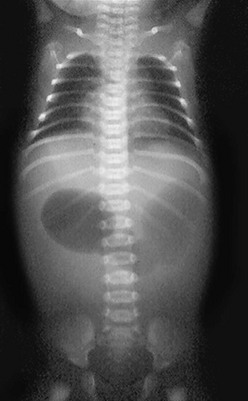
FIGURE 30-3 Classic ‘double bubble’ sign. This abdominal radiograph in a newborn shows a markedly distended stomach and duodenal bulb without evidence of distal intestinal air.
Management
After the diagnosis is made, appropriate resuscitation is required with correction of fluid balance and electrolyte abnormalities, in addition to gastric decompression. At our institution, all neonates diagnosed with duodenal obstruction receive a complete metabolic profile, complete blood count, coagulation studies, an abdominal and spinal ultrasound, and two-dimensional echocardiography prior to any operation. An emergency operation is only performed in cases where malrotation with concurrent volvulus cannot be excluded.
Prior to the mid-1970s, duodenojejunostomy was the preferred technique for correcting duodenal atresia or stenosis.7,44,45 Since then, the various techniques utilized include side-to-side duodenoduodenostomy, diamond-shaped duodenoduodenostomy, partial web resection with Heineke–Mickulicz-type duodenoplasty, and tapering duodenoplasty.44–46 The long side-to-side duodenoduodenostomy, although effective, is associated with a high incidence of anastomotic dysfunction and prolonged obstruction.30 Blind-loop syndrome appears to be more common in patients treated with duodenojejunostomy.47 Gastrojejunostomy should not be performed as it is associated with a high incidence of marginal ulceration and bleeding.27
Currently, the preferred technique is either laparoscopic or open duodenoduodenostomy.9–11,30 Originally, a side-to-side anastomosis was performed. A proximal transverse to distal longitudinal (diamond-shaped) anastomosis is now preferred.7,44,45,48–50 For the open approach, either a right upper quadrant supraumbilical transverse incision or an umbilical crease incision is utilized.49 After mobilizing the ascending and transverse colon to the left, the duodenal obstruction is readily exposed. Malrotation should be evaluated at this point as it can occur in association with congenital duodenal obstruction in up to 30% of patients.1 A sufficient length of duodenum distal to the atresia is mobilized to allow for a tension-free anastomosis. A transverse duodenotomy is made in the anterior wall of the distal portion of the dilated proximal duodenum and a similar length duodenotomy is made in a vertical orientation on the antimesenteric border of the distal duodenum. The anastomosis is then fashioned by approximating the end of each incision to the appropriate mid-portion of the other incision (Fig. 30-6). Tapering duodenoplasty is generally not necessary as the proximal duodenal dilation usually resolves after relief of the obstruction. Muscular continuity of the duodenal wall suggests a windsock deformity or diaphragm. This finding should precipitate extra vigilance in the operative correction because the dilated and collapsed bowel are both distal to the windsock, and have been anastomosed in error.26,51
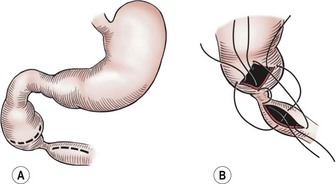
FIGURE 30-6 The technique of duodenoduodenostomy. A diamond-shaped anastomosis is created via the proximal transversely oriented and distal vertically oriented duodenotomies.
The laparoscopic approach was first described by Rothenberg in 2002.9 The standard laparoscopic approach begins with the patient supine and the abdomen is insufflated through the umbilicus. Two other instruments are inserted, one in the baby’s right lower quadrant and one in the right mid-epigastric region, respectively. A liver retractor can be placed in the right or left upper quadrant if necessary. Alternatively, the liver can be elevated by placing a transabdominal wall suture around the falciform ligament and tying it outside the abdomen (Fig. 30-7). The duodenum is mobilized and the location of obstruction is identified. Using the same principles that have been described for the open approach, a standard diamond-shaped anastomosis is created. Although some surgeons will perform the laparoscopic anastomosis with interrupted sutures, this can be technically demanding because of the significant number of sutures required and the thin nature of the distal duodenum. We reported our results using Nitinol U-clips (Medtronic, Minneapolis, MN) to create the duodenoduodenostomy with no leaks and more rapid initiation of feeds when compared to the traditional open approach (Fig. 30-8).10,11 Although the U-clips are no longer commercially available, this study highlighted the advantages of early feeding. The historical approach to enteral feeding following duodenal atresia repair involved a period of waiting for the gastric output to become less bilious and the volume of gastric drainage to decrease, indicating return of intestinal function. This report showed that the time spent waiting for the gastric output to decrease is likely not necessary as all of the patients undergong the laparoscopic duodenoplasty had initiation of feeds without adverse events after an upper gastrointestinal contrast study on day 5 revealed no leak. When compared to infants undergoing an open operation with the historical postoperative management mentioned previously, there was a marked reduction in hospitalization for the laparoscopically corrected infants, primarily due to the early feeding.
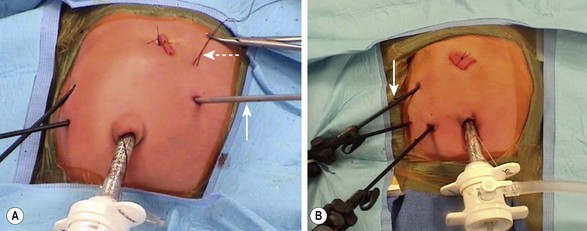
FIGURE 30-7 Two approaches to placement of the instruments for a laparoscopic duodenal atresia repair. (A) The two right-sided instruments are the primary working sites for the surgeon. The liver retractor (arrow) has been placed in the left midepigastric region. The falciform ligament has been elevated by a suture placed under it and tied over the red rubber catheter, which is used as a bolster. The suture (dotted arrow) exteriorized in the infant’s left upper abdomen was placed in the dilated proximal duodenum so that it could be easily manipulated. (B) This is a similar configuration except the instrument elevating the liver (arrow) is placed in the infant’s right upper abdomen rather than the left upper abdomen. The suture that was placed through the proximal dilated duodenum in A was not needed in this particular case.
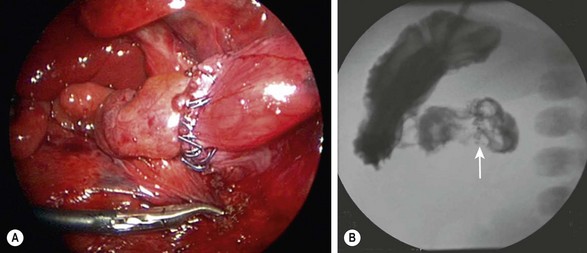
FIGURE 30-8 (A) Laparoscopic view of a completed duodenoduodenostomy using the Nitinol U-clips. (B) A postoperative contrast study at five days showed no evidence of obstruction or leak at the anastomosis. The U-clips (arrow) can be seen marking the anastomosis.
Historically, during repair of duodenal atresia, it has been emphasized that inspecting the entire small bowel to identify a second atresia is important. Given that duodenal atresia and jejunoileal atresia do not share common embryologic etiologies, a multi-institutional review of duodenal atresia patients was undertaken to quantify the incidence of jejunoileal atresia in this population.52 In the largest series to date, the rate of concomitant jejunoileal atresia in patients with duodenal atresia was less than 1%. With the low incidence of a concomitant distal atresia, extensive inspection of the entire bowel does not appear necessary.
Early postoperative mortality for duodenal atresia repair has been reported to be as low as 3–5% with the majority of deaths occurring secondary to complications related to associated congenital abnormalities.53,54 Long-term survival approaches 90%.7,53–55 Long-term complications have been noted following repair and include delayed gastric emptying, severe gastroesophageal reflux, bleeding peptic ulcer, megaduodenum, duodenogastric reflux, gastritis, blind-loop syndrome and intestinal obstruction related to adhesions.30
Jejunoileal Atresia and Stenosis
Etiology
Jejunoileal atresia occurs in approximately 1 in 5,000 live births. It occurs equally in males and females, and about one in three infants is premature.56 Although the majority of cases are thought to occur sporadically, familial cases of intestinal atresias have been described.57 It is generally accepted that jejunoileal atresia occurs as a result of an intrauterine ischemic insult to the midgut, affecting single or multiple segments of the already developed intestine.13,58–61 Intrauterine vascular disruption can lead to ischemic necrosis of the bowel with subsequent resorption of the affected segment or segments (Fig. 30-9).
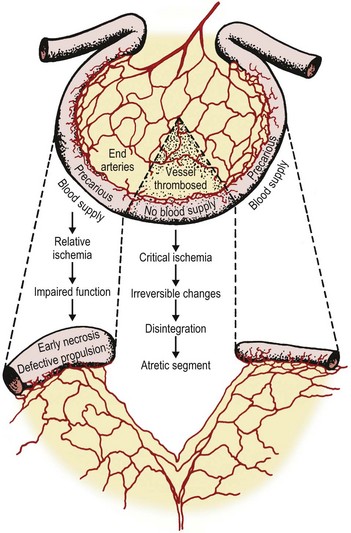
FIGURE 30-9 The proposed mechanism of vascular compromise and subsequent development of jejunoileal atresias is depicted.
The hypothesis that most cases of jejunoileal atresia occur secondary to vascular disruption during fetal life is derived from experimental as well as clinical evidence. Isolated mesenteric vascular insults and interference with the segmental blood supply to the small intestine were created in fetal dogs, and resulted in different degrees and patterns of intraluminal obstruction, reproducing the spectrum of stenosis and atresia found in humans.62–64 Moreover, the presence of bile, lanugo hair, and squamous epithelial cells from swallowed amniotic fluid distal to an atresia suggests that the atresia occurs subsequent to some event, but that at some time in gestation the intestinal lumen was patent, thus allowing passage of these contents. Additionally, atresias seen in association with other intrauterine vascular insults such as fetal intussusception, midgut volvulus, thromboembolic occlusions, transmesenteric internal hernias, and incarceration or snaring of bowel in an omphalocele or gastroschisis have contributed to wide acceptance of this hypothesis.58,64–68
The presence of associated extra-abdominal organ abnormalities in jejunoileal atresia is low (<10%) due to its occurrence later in fetal life and the localized nature of the vascular insult.69 Rarely, jejunoileal atresia has been found in patients with Hirschsprung disease, cystic fibrosis, malrotation, Down syndrome, anorectal and vertebral anomalies, neural tube defects, congenital heart disease, and other GI atresias.56,69,70 Methylene blue, previously used for amniocentesis in twin pregnancies, has been implicated in causing small bowel atresia.71
Although jejunoileal atresias are usually not hereditary, there is a well-documented autosomal recessive pattern of inheritance of multiple atresias.72 In these cases, intestinal rotation was normal, mesenteric defects were never observed, and lanugo hairs and squamous cells were not identified distal to the most proximal atresia. All these findings suggest an early intrauterine event. Survival is poor in these infants, even with successful bowel resection.
No correlations have been found between jejunoileal atresia and parental or maternal disease. However, the use of maternal vasoconstrictive medications, as well as maternal cigarette smoking in the first trimester of pregnancy, has been shown to increase the risk of small bowel atresia.73 Chromosomal abnormalities are seen in less than 1% of the patients with jejunoileal atresia.74
Pathology
The Grosfeld classification system separates these defects into four groups, with an additional consideration for type III(b) (Fig. 30-10).4 This classification has significant prognostic and therapeutic value as it emphasizes the importance of associated loss of intestinal length, abnormal collateral intestinal blood supply, and concomitant atresia or stenosis.75 Regarding classification, the most proximal atresia determines whether the atresia is classified as jejunal or ileal atresia. Multiple atresias are found in up to 30% of patients.56,76
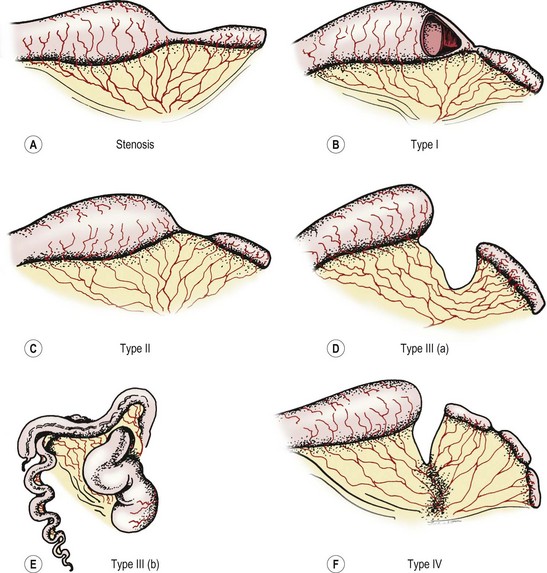
Stenosis
Stenosis is defined as a localized narrowing of the intestinal lumen without disruption in the intestinal wall or a defect in the mesentery (see Fig. 30-10A). At the stenotic site, a short, narrow, somewhat rigid segment of intestine with a small lumen is found. Often the muscularis is irregular and the submucosa is thickened. Stenosis may also take the form of a type I atresia with a fenestrated web. Patients with jejunoileal stenosis usually have a normal length of small intestine.
Type I Atresia
In type I jejunoileal atresia, the intestinal obstruction occurs secondary to a membrane or web formed by both mucosa and submucosa, while the muscularis and serosa remain intact (see Figs 30-10B and 30-11). On gross inspection, the bowel and its mesentery appear to be in continuity. However, the proximal bowel is dilated while the distal bowel is collapsed. With the increased intraluminal pressure in the proximal bowel, bulging of the web into the distal intestine can create a windsock effect. As with stenosis, there is no foreshortening of the bowel in type I atresias.
Type II Atresia
The clinical findings of a type II atresia are a dilated, blind-ending proximal bowel loop connected by a fibrous cord to the collapsed distal bowel with an intact mesentery (see Fig. 30-10C). Increased intraluminal pressure in the dilated and hypertrophied proximal bowel may lead to focal proximal small bowel ischemia. The distal collapsed bowel commences as a blind end, which sometimes assumes a bulbous appearance owing to the remains of an intussusception. Again, the total small bowel length is usually normal.
Type III(a) Atresia
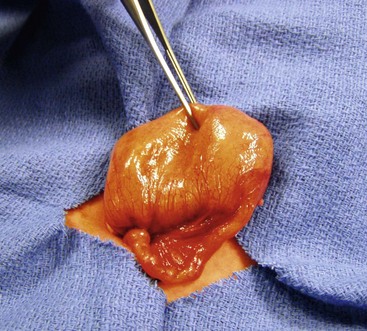
In type III(a) atresia, the proximal bowel ends blindly with no fibrous connecting cord to the distal intestine. A V-shaped mesenteric defect of varying size is present between the two ends of intestine (see Figs 30-10D and 30-12). The dilated, blind-ending proximal bowel is often aperistaltic and frequently undergoes torsion or becomes overdistended, with subsequent necrosis and perforation occurring as a secondary event.77 In this scenario, the total length of the small bowel is variable (but usually less than normal), owing to intrauterine resorption of the affected bowel.
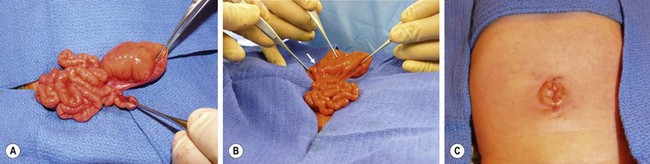
FIGURE 30-12 This baby was suspected of having a proximal jejunal atresia based on symptoms of bilious emesis and the abdominal film. (A, B) At operation, this type III(a) jejunal atresia was found. Note the V-shaped mesenteric defect between the dilated proximal atretic bowel and the distal decompressed bowel. Due to the size discrepancy between the two ends of the intestine, the proximal dilated bowel was resected at the arrow and an end-to-side anastomosis was performed. (C) The operation was performed through a slightly extended umbilical incision. The baby recovered nicely and no complications developed.
Type III(b) Atresia
Type III(b) atresia (apple-peel, Christmas tree, or Maypole deformity) consists of a proximal jejunal atresia, absence of the superior mesenteric artery beyond the origin of the middle colic branch, agenesis of the dorsal mesentery, a significant loss of intestinal length, and a large mesenteric defect (see Fig. 30-10E). The decompressed distal small bowel lies free in the abdomen and assumes a helical configuration around a single perfusing vessel arising from the ileocolic or right colic arcades (Fig. 30-13). Occasionally, additional type I or type II atresias are found distal to the initial atresia. Also, the vascularity of the distal bowel is often impaired. This type of atresia has been found in families with a pattern suggestive of an autosomal recessive mode of inheritance. It also has been encountered in siblings with identical lesions and in twins.77–80
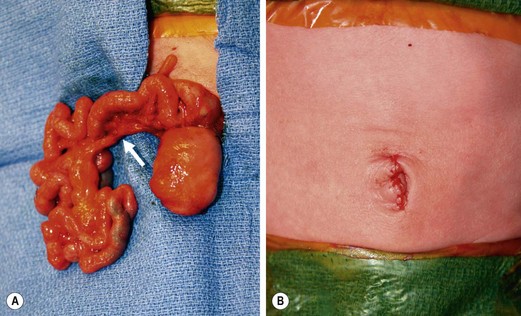
FIGURE 30-13 (A) The operative findings in neonate with a type III(b) intestinal atresia are seen. Note the classic ‘apple-core’ or ‘Christmas tree’ deformity as well as the wide mesenteric gap between the proximal dilated bowel and distal decompressed ileum. Also, the colon and distal small bowel are perfused through a single artery (arrow) running through the mesentery of the distal bowel. (B) This operation was performed through a small umbilical incision. This baby recovered nicely and has not developed any complications.
The occurrence of intestinal atresia in other siblings, the association of multiple atresias (15%), and the discordance in a set of apparently monozygotic twins may point to more complex genetic transmission with an overall recurrence rate of 18%.78,81,82 Infants with this anomaly are often premature, and up to 50% may have malrotation. Short bowel syndrome is present in nearly 75% of cases. Accordingly, there is increased morbidity (63%) and mortality (54%) in this population.78,83 Type III(b) atresias are most likely the result of a proximal superior mesenteric arterial occlusion with extensive infarction of the proximal segment of the midgut. Also, it can develop from a midgut volvulus.58,84 Primary failure of development of the distal superior mesenteric artery has also been suggested as an etiologic factor. However, this is unlikely because meconium is usually found in the bowel distal to the atresia. This finding indicates that the atresia develops after bile secretion begins, which occurs around week 12 of intrauterine life. The superior mesenteric artery develops much earlier than 12 weeks.85
Type IV atresia
Multiple-segment atresias or a combination of types I to III are classified as type IV (see Fig. 30-10F). Twenty to 35% of infants affected with jejunoileal atresia present with multiple atresias.56,76 The majority of cases of multiple-segment atresias are sporadic with no other family history of intestinal abnormalities. They are likely a result of multiple vascular insults to the mesentery, intrauterine inflammatory processes, or a malformation of the GI tract occurring during embryonic development.76,86 Embolic material from a nonviable fetus to a living monochorionic twin through placental vascular connections could also account for single or multiple intestinal atresias.87 Associated defects, particularly abnormalities of the central nervous system, have been noted in approximately 25% of nonfamilial multiple intestinal atresia patients.76 Multiple atresias have also been seen in association with severe immunodeficiency.23
A familial form of multiple intestinal atresia (FMIA) involving the stomach, duodenum, and both the small and large bowel has been described.72,88 It is associated with prematurity and shortened bowel length. To date, it has been uniformly fatal. It is associated with type I and II atresias, with type II predominating. An autosomal recessive mode of transmission has been suggested for this familial condition because it is unlikely that an isolated prenatal vascular accident is responsible for such extensive involvement of the GI tract. In addition, infants affected with this familial form are found to have long segments of completely occluded small or large intestine without a recognizable lumen.88–90 Another pathognomonic feature seen in FMIA is the sieve-like appearance of the intestine on histologic examination where multiple lumina are surrounded by epithelial cells and muscularis mucosa.88
Pathophysiology
The vascular and subsequent ischemic insult not only causes morphologic abnormalities but also adversely influences the structure and subsequent function of the remaining proximal and distal bowel.58,59,91 The blind-ended proximal bowel is dilated and hypertrophied with histologically normal villi, but without effective peristaltic activity. A deficiency of mucosal enzymes and muscular adenosine triphosphatase has also been found.92 At the level of the atresia, the ganglia of the enteric nervous system are atrophic with minimal acetylcholinesterase activity. These changes are most likely secondary to local ischemia. Obstruction alone can elicit similar, but less severe, morphologic and functional abnormalities.92
Experimental studies showing that the intestinal atresia results from ischemic necrosis of the intestine also imply that there is a precarious blood supply to the proximally dilated bowel. This has been confirmed with postmortem injection of barium sulfate into the mesenteric vessels.58,59,85 However, it has also been postulated that the intestine is not ischemic at birth, but rather becomes so only with swallowing air. Distention and increased intraluminal pressure or torsion can then occur. The good results obtained with tapering procedures without resection of the bulbous portion would support the contention that the blood and nerve supply to the bowel adjacent to the atresia is normal.58 However, this ischemic insult may interfere with mucosal and neural function. Defective peristalsis is commonly noticed in the atretic area, thus supporting resection of the dilated bulbous proximal end for better function.93 Because the proximal end of the distal atretic bowel has been subjected to a similar insult, a small portion of it should be resected at the time of operative correction as well.
Diagnosis
The diagnosis of jejunoileal atresia can usually be made by radiographic examination of the abdomen using swallowed air as contrast. Swallowed air reaches the proximal bowel by one hour and the distal small bowel by three hours in a normal vigorous infant in whom its passage is blocked, but this pattern may be delayed in premature or sick infants with poor sucking.94,95 Jejunal atresia patients can have a few gas-filled and fluid-filled loops of small bowel, but the remainder of the abdomen is gasless (Fig. 30-14). When the atresia is associated with cystic fibrosis, fewer air-fluid levels are evident, and the typical ground-glass appearance of inspissated meconium is seen. A limited-contrast study may be useful if intestinal stenosis is suspected.
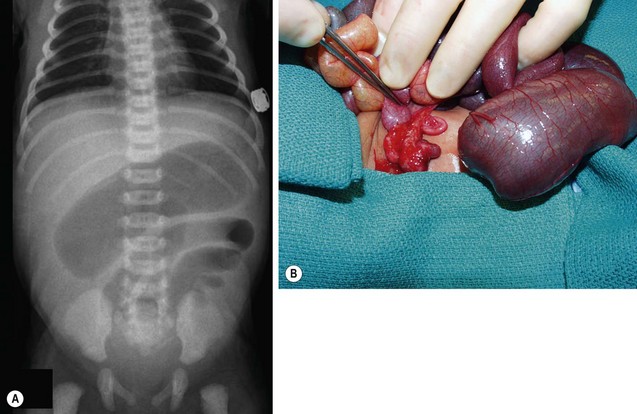
FIGURE 30-14 (A) The abdominal radiograph in this neonate shows several proximally dilated intestinal loops consistent with jejunal atresia. (B) A type III(a) distal atresia was found at operation.
As haustral markings are rarely seen in neonates, distal ileal atresia may be difficult to differentiate from colonic atresia (Fig. 30-15). A contrast enema will reveal an unused appearance to the colon. Reliance on intraoperative injection of saline into the large bowel to confirm distal bowel patency may fail to identify an associated colonic or rectal atresia.96,97 If the small bowel atresia occurred late in gestation, the bowel distal to the atresia may have a more normal caliber. Occasionally, air and meconium can accumulate proximal to an atresia, mimicking the radiologic appearance of meconium ileus. Additionally, total colonic aganglionosis may be difficult to differentiate from small bowel atresia.
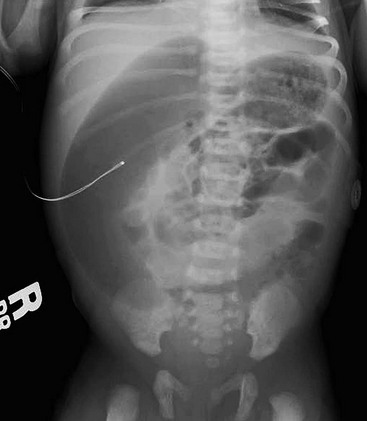
FIGURE 30-15 The diagnosis of colonic atresia can be difficult on the plain abdominal radiograph. This radiograph shows multiple dilated intestinal loops and appears similar to the findings shown in Figure 30-14. At operation, the infant was found to have atresia of the transverse colon (see Fig. 30-16).
Ten per cent of babies with jejunoileal atresia present with meconium peritonitis.4 The intestinal perforation usually occurs proximal to the obstruction, near the bulbous blind end. The radiologic appearance of a meconium pseudocyst containing a large air/fluid level is related to the late intrauterine bowel perforation. Intraluminal calcification of meconium or intramural calcification in the form of diffuse punctate or rounded aggregations have been reported with intestinal stenosis or atresia.98 Meconium calcification in patients with hereditary FMIA produces a ‘string of pearls,’ which is pathognomonic of this condition.72,88
Differential Diagnosis
Diseases that mimic jejunoileal atresia include colonic atresia, midgut volvulus, meconium ileus, duplication cysts, internal hernias, ileus due to sepsis, birth trauma, maternal medications, prematurity, and hypothyroidism.4,99,100 Special investigations including an upper GI contrast study, contrast enema, rectal biopsy, and a delta F508 gene deletion assay or sweat test to exclude associated cystic fibrosis may be needed.99,101
Surgical Considerations
The operative management of intestinal atresias is based on the location of the lesion, anatomic findings, associated conditions noted at operation, and the length of the remaining instestine.56 Resection of the dilated and hypertrophied proximal bowel (see Fig. 30-12B), with primary end-to-end anastomosis with or without tapering of the proximal bowel, is the most common technique.4, 77,100
As recently as the 1950s, the surgical mortality for newborns with intestinal atresia was 80% to 90%.56,99 This high mortality rate was mostly related to late presentation and dysmotility of the proximal dilated bowel which led to complications related to chronic obstruction and inanition. Fortunately, the current survival rate is greater than 90%.56 The understanding that the proximal bowel is dysfunctional, improvements in the anastomotic technique and suture material, and the development of total parenteral nutrition (TPN) are the primary reasons for this significantly improved survival in recent years. Currently, only infants with severe associated congenital abnormalities or short bowel syndrome should not have a good prognosis.
Operative Considerations
The repair of small intestinal atresia can be undertaken via several approaches. One option is to evaluate using a laparoscopic approach, with subsequent resection and anastomosis performed in an extra-corporeal fashion. Although this approach seems attractive, it can be difficult to identify the atresia due to the markedly dilated small intestine and the small working space of the neonate’s abdominal cavity. To overcome these limitations, we explore the abdomen through the umbilicus. With this technique, the umbilical skin is incised and the fascia is opened vertically in the midline to the extent allowed by the umbilical skin incision. The small intestine can be exteriorized relatively easy through the umbilical incision (see Figs 30-12C and 30-13B). In a retrospective report, a circumumbilical incision for neonatal surgery was found to as effective as the transverse abdominal incision with less morbidity and better cosmetic results.102 The traditional transverse supra- or infraumbilical incision is also appropriate. Regardless of the approach, access to the entire intestine and peritoneal cavity is necessary. Careful inspection of the entire bowel is performed and the site and type of obstruction should be noted as well as any other abnormalities. In addition, the length of bowel should be assessed. The most distal limb of the atretic bowel can then be cannulated with a red rubber catheter and irrigated with warm saline to evaluate for distal obstruction. Continuity of the colon can be established preoperatively by a contrast enema or with a prepositioned transrectal catheter placed prior to prepping.103 Failure to adequately evaluate for distal obstruction or stenosis can lead to postoperative complications, including an anastomotic leak. If present, malrotation should be corrected with a Ladd procedure. Because the length of functional bowel has important prognostic significance, and determines the most appropriate method of repair, the length of functional bowel should be carefully measured along the antimesenteric border and documented in the operative report.
Delayed intestinal function in the blind proximal atretic segment as well as functional obstruction after performance of a side-to-side anastomosis without resection of the dilated proximal atretic bowel have been described.100 Therefore, if the length of functional bowel is adequate, the bulbous hypertrophied proximal bowel should be resected to approximately normal caliber bowel. Ultimately, the goal is to restore bowel continuity while maintaining both intestinal function and length. Intestinal imbrication has also been shown to be an effective method to reduce the caliber of the dilated bowel while maintaining mucosal absorptive surface.104 Regarding the distal segment, a short length (4–5 cm) of bowel is obliquely resected, leaving the mesenteric side longer than the antimesenteric aspect. An incision along the antimesenteric border to create a ‘fish mouth’ may be needed to create an adequate distal enterotomy for the anastomosis.
Although there are multiple techniques for the anastomosis, we generally perform a one-layer modification of the end-to-back technique using 5-0 or 6-0 sutures. Once the anastomosis is completed, the suture line is tested for leaks, and reinforcing sutures are placed as needed. The mesenteric defect is repaired with careful attention to avoid rotation or kinking of the anastomosis, or injury to the blood supply. A temporary enterostomy should be performed if there is a question of bowel viability.56 However, neither decompressive gastrostomy nor transanastomotic stents are usually needed.105,106
Prognostic Factors
If proximal resection will lead to significant, or unacceptable bowel loss, tapering or plication of the dilated bowel is a useful technique.104,107 Tapering enteroplasty as far proximal as the second portion of the duodenum can be accomplished by resecting an antimesenteric strip of the dilated proximal bowel.108 During tapering duodenojejunoplasty, particularly with type III atresias, the duodenum is de-rotated, thus allowing a direct caudal descent from the stomach which decreases the risk for obstruction. Additionally, the mesentery should be maximally opened, while meticulously protecting the small bowel blood supply. During this process, the cecum can be mobilized to the left which results in a broader mesentery and also allows the anastomosis to lie in manner that will help avoid kinking.109 The tapering can be safely performed up to 35 cm.107 The tapered bowel may then be anastomosed to the distal bowel or exteriorized as a stoma.
A primary anastomosis may be contraindicated in cases of peritonitis, volvulus with vascular compromise, meconium ileus, or type III(b) atresia.110,111 Under these circumstances, exteriorization of both ends of the atresia may be needed.
Intestinal atresia encountered in a baby with gastroschisis may be single or multiple, and may be located in either the small or large bowel. In a series from our institution, 12.6% of 199 patients with gastroschisis had an associated atresia.112 The most common location for the atresia was jejunoileal and most were type III(a). Our current management algorithm for patients with gastroschisis and atresia is to first assess the extent of reactive change (peel) on the intestine. If there is minimal peel, primary anastomosis may be an option. This is rare and should be considered only in the most optimal situations. In nearly all instances, the atresia should be left undisturbed at the initial operation. After fascial closure is accomplished, management should include gastric decompression and TPN support with subsequent atresia repair four to six weeks later.
With type III(b) atresia, restricting bands along the free edge of the distal coiled and narrow mesentery should be divided to optimize the blood supply. The bowel should be returned to the abdomen with careful inspection of the mesentery to prevent torsing the single marginal artery and vein. In cases of questionable intestinal viability, improved long-term results have been achieved with resection and tapering of the dilated proximal bowel with limited resection of the distal bowel.113,114
Bowel-length conservation methods, such as multiple anastomoses for multiple atresias, may result in increased morbidity. A silicone (Silastic) catheter stent can be used with multiple primary anastomoses and serves as a conduit for radiologic evidence of anastomotic integrity, luminal patency, and enteral feeding.115 If multiple atresias are grouped closely together and there is adequate bowel length, a single resection and anastomosis can be performed.
No attempt at any bowel lengthening procedures should be entertained at the initial operation. However, such procedures may ultimately obviate the need for prolonged TPN in patients with short gut syndrome.
Postoperative Care
Transient GI dysfunction is frequently observed in infants with jejunal and ileal atresia, and its etiology is multifactorial.4,116 Lactose intolerance, malabsorption (owing to stasis with bacterial overgrowth), and diarrhea may be significant in infants who have undergone repair of type III(b) atresia, or in those with short bowel syndrome after surgery for multiple atresias. Regular monitoring for clinical signs of intestinal overload or intolerance is required. Water-loss stools, increasing frequency of stooling, hematochezia, fecal-reducing substances, or a decreased stool pH warrant biochemical evaluation of the stool for disaccharide or monosaccharide intolerance.117 Unintentional injury to the mucosa can be caused by sugars, high-osmolarity feeds, oral medications, and bacterial or viral infections. Pharmacologic control of altered GI function may hasten adaptation. Loperamide hydrochloride decreases intestinal peristaltic activity and cholestyramine is effective in binding bile salts.117,118 Cholestyramine should not be given unless water-loss stools are evident. Vitamin B12 and folic acid should be given regularly to the patient without a terminal ileum to prevent megaloblastic anemia.
Functional outcome ultimately depends on the following factors: (1) the location of the atresia (the ileum adapts to a greater degree than the jejunum); (2) the maturity of the intestine (the small intestine in a premature infant still has time for maturation and growth); and (3) the length of the small intestine, which can be difficult to determine accurately after birth.119 The ileocecal valve is critically important as it allows for more rapid intestinal adaptation when the residual small bowel length is short.
Colonic Atresia
Colonic atresia (CA) is a rare cause of intestinal obstruction and comprises 1.8–15% of all GI atresias.120,121 The reported incidence of CA varies greatly from 1 : 5000 to 1 : 60,000 live births.122–125 The accepted incidence is approximately 1 in 20,000 live births. Although it is most commonly reported as an isolated anomaly, approximately one-third of babies have associated congenital lesions.123,124,126 There are various classifications of CA, but the one most commonly used divides CA into three types. Type I consists of mucosal atresia with an intact bowel wall and mesentery. In type II, the atretic ends are separated by a fibrous cord. In type III, the atretic ends are separated by a V-shaped mesenteric gap (Fig. 30-16). Type III lesions are the most commonly occurring lesions overall, while types I and II are seen more commonly distal to the splenic flexure.122,127
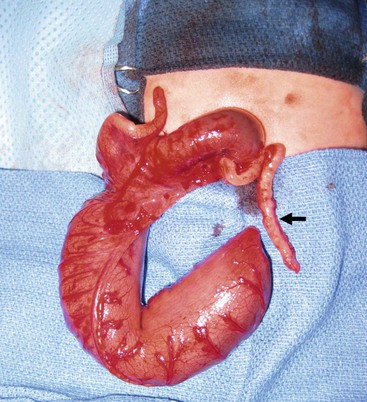
FIGURE 30-16 A type III colonic atresia was found at operation in this infant with intestinal obstruction. Note the cecum and appendix and the very dilated right colon. Also, note the extremely small distal colon (arrow). A colostomy was performed as the initial procedure in this infant.
The rate of associated anomalies with CA is much smaller when compared to other atresias. CAs have been found in approximately 2.5% of neonates with gastroschisis.112 There are less than 25 published cases of CA and Hirschsprung disease (HD).126 Complex urologic abnormalities, multiple small intestinal atresias, an unfixed mesentery, and skeletal anomalies have also been reported with CA.122,127–129 Similar to small bowel atresias, a vascular insult to the colon continues to be the accepted etiology for all types of CA.130,131
The characteristic clinical features of CA are abdominal distention, bilious emesis, and failure to pass meconium. On plain radiographs, air-fluid levels are usually appreciated as well as dilated intestinal loops of large bowel often associated with a ‘ground-glass’ appearance of meconium mixed with air. Occasionally, the dilation can be so massive that it mimics pneumoperitoneum (Fig. 30-17). The diagnosis is made with a contrast enema showing a small diameter distal colon that comes to an abrupt halt at the level of the obstruction (Fig. 30-18).
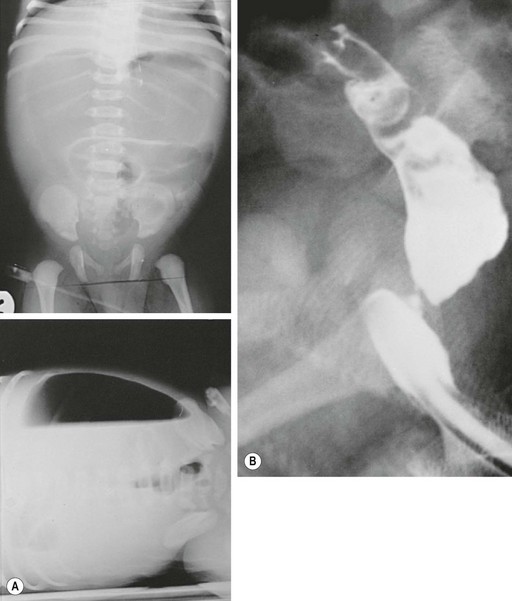
FIGURE 30-17 (A) Abdominal radiograph of colonic atresia showing huge air-filled proximal colon mimicking a pneumoperitoneum. (B) Right colonic atresia with rectal stenosis.
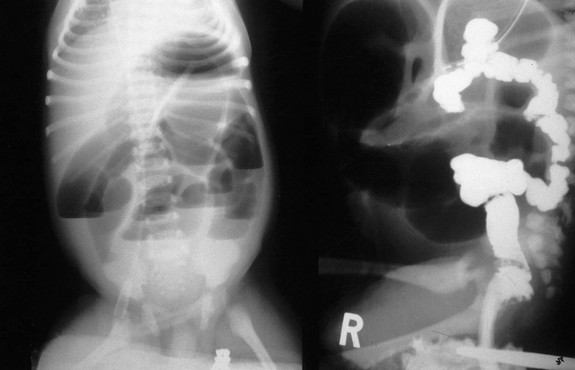
FIGURE 30-18 The contrast enema on the right in a patient with a distal intestinal obstruction (left) shows a small colon and failure of the contrast to move proximally past the mid-transverse colon.
The diagnosis of CA is an indication for urgent operative management as the risk for perforation is higher than is seen in jejunoileal atresias. The operative approach depends on the clinical status of the patient, the level of the atresia, any associated small intestinal atresias, and the patency of the bowel distal to the atresia. It is important to exclude other intestinal atresias and stenoses at the time of operation as they occur with some frequency.132 A diagnosis of associated HD, although rare, must be made by frozen section analysis of rectal biopsies during the initial surgery as unrecognized HD can lead to an anastomotic leak or functional obstruction.
A staged approach consisting of colostomy with mucous fistula is generally preferred for CA. Because the proximal and distal ends adjacent to the atresia are abnormal in both innervation and vascularity, resection of the bulbous proximal colon as well as a portion of the distal microcolon is suggested.133,134 Primary resection with anastomosis has a higher incidence of complications, usually due to undiagnosed distal pathology.124,135
A recent report of a small case series of rectal and sigmoid atresias described a transanal approach for the repair of the atresias.136 An initial colostomy at birth was performed followed by a transanal approach three to six months later. Closure of the colostomy was then carried out one to two months later.
In the absence of other serious co-morbidities, the prognosis in CA is excellent. If diagnosed early, the overall mortality is less than 10%.126 A delay in diagnosis beyond 72 hours, however, may result in a mortality of greater than 60%.129 This high mortality is due, in part, to the formation of a closed loop obstruction between an intact ileocecal valve and the atresia, leading to massive colonic distention and perforation.
References
1. Irving, IM. Duodenal atresia and stenosis: Annular pancreas. In: Lister J, Irving IM, eds. Neonatal Surgery. 3rd ed. London: Buttersworths; 1990:424.
2. Adeyemi, D. Neonatal intestinal obstruction in a developing tropical country: Patterns, problems, and prognosis. J Trop Pediatr. 1989; 35:66–70.
3. Cywes, S, Davies, MRQ, Rode, H. Congenital jejuno-ileal atresia and stenosis. S Afr Med J. 1980; 57:630–639.
4. Grosfeld, JL. Jejunoileal atresia and stenosis, section 3: The small intestine. In: Ravitch MM, Welch KJ, Benson CD, et al, eds. Pediatric Surgery. Chicago: Year Book Medical; 1986:838.
5. Kimura, K, Loening-Baucke, V. Bilious vomiting in the newborn: Rapid diagnosis of intestinal obstruction. Am Fam Physician. 2000; 61:2791–2798.
6. Chhabra, R, Suresh, BR, Weinberg, G, et al. Duodenal atresia presenting as hematemesis in a premature infant with Down syndrome. Case report and review of the literature. J Perinatol. 1992; 12:25–27.
7. Grosfeld, JL, Rescorla, FJ. Duodenal atresia and stenosis: Reassessment of treatment and outcome based on antenatal diagnosis, pathologic variance, and long-term follow-up. World J Surg. 1993; 17:301309.
8. Adzick, NS, Harrison, MR, de Lorimier, AA. Tapering duodenoplasty for megaduodenum associated with duodenal atresia. J Pediatr Surg. 1986; 21:311–312.
9. Rothenberg, SS. Laparoscopic duodenoduodenostomy for duodenal obstruction in infants and children. J Pediatr Surg. 2002; 37:1088–1089.
10. Valusek, PA, Spilde, TL, Tsao, K, et al. Laparoscopic duodenal atresia repair using surgical U-clips: A novel technique. Surg Endosc. 2007; 21:1023–1024.
11. Spilde, TL, St Peter, SD, Keckler, SJ, et al. Open vs. laparoscopic repair of congenital duodenal obstructions: A concurrent series. J Pediatr Surg. 2008; 43:1002–1005.
12. Ladd, WE. Congenital obstruction of the duodenum in children. N Engl J Med. 1931; 206:277–283.
13. Moore, KL, Persaud, TVN. The digestive system. In The Developing Human, 8th ed, Philadelphia: WB Saunders; 2007:218.
14. Schnaufer, L. Duodenal atresia, stenosis and annular pancreas. In: Welch RJ, Randolph JG, et al, eds. Pediatric Surgery. Chicago: Year Book Medical; 1986:929.
15. Shawis, R, Antao, B. Prenatal bowel dilatation and the subsequent postnatal management. Early Hum Dev. 2006; 82:297–303.
16. Elliot, GB, Kliman, R, Elliot, KA. Pancreatic annulus: A sign or a cause of duodenal obstruction? Can J Surg. 1968; 11:357.
17. Gourevitch, A. Duodenal atresia in the newborn. Ann R Coll Surg Engl. 1971; 48:141–158.
18. Jona, JZ, Belin, RP. Duodenal anomalies and the ampulla of Vater. Surg Gynecol Obstet. 1976; 143:565–569.
19. Irving, IM, Rickham, PP. Duodenal atresia and stenosis: Annular pancreas. In: Rickham PP, Lister J, Irving IM, eds. Neonatal Surgery. 2nd ed. Boston: Butterworths; 1978:355.
20. Brereton, RJ, Cudmore, RE, Bouton, JM. Double atresia of the duodenum. Z Kinderchir. 1980; 31:60–65.
21. Coughlin, JP, Rector, FE, Klein, MD. Agenesis of the gallbladder in duodenal atresia: Two case reports. J Pediatr Surg. 1992; 27:1304.
22. Davenport, M, Saxena, R, Howard, E. Acquired biliary atresia. J Pediatr Surg. 1996; 31:1721–1723.
23. Moore, SW, de Jongh, G, Bouic, P, et al. Immune deficiency in familial duodenal atresia. J Pediatr Surg. 1996; 31:1733–1735.
24. Mali, V, Wagener, S, Sharif, K, et al. Foregut atresias and bile duct anomalies: Rare, infrequent or common?!. Pediatr Surg Int. 2007; 23:889–895.
25. Bill, AH, Jr., Pope, WM. Congenital duodenal diaphragm. Surgery. 1954; 35:482–486.
26. Rowe, M, Buckner, D, Clatworthy, HW, Jr. Wind sock web of the duodenum. Am J Surg. 1968; 116:444–449.
27. Magnuson, DK, Schwartz, MZ. Stomach and duodenum. In: Oldham KT, Colombani PM, Foglia RP, et al, eds. Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott Williams & Wilkins, 2004.
28. Kimble, RM, Harding, J, Kolbe, A. Additional congenital anomalies in babies with gut atresia or stenosis: When to investigate, and which investigation. Pediatr Surg Int. 1997; 12:565–570.
29. Mustafawi, AR, Hassan, ME. Congenital duodenal obstruction in children: A decade’s experience. Eur J Pediatr Surg. 2008; 18:93–97.
30. Escobar, MA, Ladd, AP, Grosfeld, JL, et al. Duodenal atresia and stenosis: Long-term follow-up over 30 years. J Pediatr Surg. 2004; 39:867–871.
31. Fonkalsrud, EW, DeLorimier, AA, Hays, DM. Congenital atresia and stenosis of the duodenum: A review compiled from the members of the Surgical Section of the American Academy of Pediatrics. Pediatrics. 1969; 43:79–83.
32. al-Salem, AH, Khwaja, S, Grant, C, et al. Congenital intrinsic duodenal obstruction: Problems in the diagnosis and management. J Pediatr Surg. 1989; 24:1247–1249.
33. Longo, MF, Lynn, HB. Congenital duodenal obstruction: Review of 29 cases encountered in a 30-year period. Mayo Clin Proc. 1967; 42:423–430.
34. Kimble, RM, Harding, JE, Kolbe, A. Does gut atresia cause polyhydramnios? Pediatr Surg Int. 1998; 13:115–117.
35. Stubbs, TM, Horger, EO. Sonographic detection of fetal duodenal atresia [Letter]. Obstet Gynecol. 1989; 73:146.
36. Akhtar, J, Guiney, EJ. Congenital duodenal obstruction. Br J Surg. 1992; 79:133–135.
37. Bittnecourt, DG, Barini, R, Marba, S, et al. Congenital duodenal obstruction: Does prenatal diagnosis improve the outcome? Pediatr Surg Int. 2004; 20:582–585.
38. Lawrence, MJ, Ford, WD, Furness, ME, et al. Congenital duodenal obstruction: Early antenatal ultrasound diagnosis. Pediatr Surg Int. 2000; 16:342–345.
39. Dumont, RC, Rudolph, CD. Development of gastrointestinal motility in the infant and child. Gastroenterol Clin North Am. 1994; 23:655–671.
40. Berseth, CL. Gestational evolution of small intestinal motility in preterm and term infants. J Pediatr. 1989; 115:646–651.
41. Britton, JR, Britton, HL. Gastric aspirate volume at birth as an indicator of congenital intestinal obstruction. Acta Paediatr. 1995; 84:945–946.
42. Traubici, J. The double bubble sign. Radiology. 2001; 220:463–464.
43. Kassner, EG, Sutton, A, De Groot, TJ. Bile duct anomalies associated with duodenal atresia: Paradoxical presence of small bowel gas. Am J Roentgenol Radium Ther Nucl Med. 1972; 116:577–583.
44. Kimura, K, Mukohara, N, Nishijima, E, et al. Diamond-shaped anastomosis for duodenal atresia: An experience with 44 patients over 15 years. J Pediatr Surg. 1990; 25:977–979.
45. Weber, TR, Lewis, JE, Mooney, D, et al. Duodenal atresia: A comparison of techniques of repair. J Pediatr Surg. 1986; 21:1133–1136.
46. Singh, SJ, Dickson, R, Baskaranathan, S, et al. Excision duodenoplasty: A new technique for congenital duodenal obstruction. Pediatr Surg Int. 2002; 18:75–78.
47. Rescorla, FJ, Grosfeld, JL. Duodenal atresia in infancy and childhood: Improved survival and long-term follow-up. Contemp Surg. 1988; 33:22–27.
48. Takayashi, Y, Tajiri, T, Masumoto, K, et al. Umbilical crease incision for duodenal atresia achieves excellent cosmetic results. Pediatr Surg Int. 2010; 26:963–966.
49. Ein, SH, Kim, PC, Miller, HA. The late nonfunctioning duodenal atresia repair-A second look. J Pediatr Surg. 2000; 35:690–691.
50. Kimura, K, Tsugawa, C, Ogawa, K, et al. Diamond-shaped anastomosis for congenital duodenal obstruction. Arch Surg. 1977; 112:1262–1263.
51. Richardson, WR, Martin, LW. Pitfalls in the surgical management of the incomplete duodenal diaphragm. J Pediatr Surg. 1969; 4:303–312.
52. St. Peter, SD, Little, DC, Barsness, KS, et al. Should we be concerned about jejunoileal atresia during repair of duodenal atresia? J Laparoendoscopic Adv Surg Tech. 2010; 20:773–775.
53. Feggetter, S. A review of the long-term results of operations for duodenal atresia. Br J Surg. 1969; 56:68–72.
54. Stauffer, UG, Irving, I. Duodenal atresia and stenosis–long-term results. Prog Pediatr Surg. 1977; 10:49–60.
55. Kokkonen, ML, Kalima, T, Jaaskelainen, J, et al. Duodenal atresia: Late follow-up. J Pediatr Surg. 1988; 23:216–220.
56. Dalla Vecchia, LK, Grosfeld, JL, West, KW, et al. Intestinal atresia and stenosis: A 25-year experience with 277 cases. Arch Surg. 1998; 133:490–496.
57. Kumaran, N, Shankar, KR, Lloyd, DA, et al. Trends in the management and outcome of jejuno-ileal atresia. Eur J Pediatr Surg. 2002; 12:163–167.
58. Louw, JH. Congenital intestinal atresia and stenosis in the newborn. Observations on its pathogenesis and treatment. Ann R Coll Surg Engl. 1959; 25:209–234.
59. Louw, JH, Barnard, CN. Congenital intestinal atresia: Observations on its origin. Lancet. 1955; 269:1065–1067.
60. Abrams, JS. Experimental intestinal atresia. Surgery. 1968; 64:185–191.
61. Puri, P, Fujimoto, T. New observations on the pathogenesis of multiple intestinal atresias. J Pediatr Surg. 1988; 23:221–225.
62. Koga, Y, Hayashida, Y, Ikeda, K, et al. Intestinal atresia in fetal dogs produced by localized ligation of mesenteric vessels. J Pediatr Surg. 1975; 10:949–953.
63. Tibboel, D, van der Kamp, AW, Molenaar, JC. An experimental study of the effect of an intestinal perforation at various developmental stages. Z Kinderchir. 1982; 37:62–66.
64. Khen, N, Jaubert, F, Sauvat, F, et al. Fetal intestinal obstruction induces alteration of enteric nervous system development in human intestinal atresia. Pediatr Res. 2004; 56:975–980.
65. Amoury, RA, Ashcraft, KW, Holder, TM. Gastroschisis complicated by intestinal atresia. Surgery. 1977; 82:373–381.
66. Grosfeld, JL, Clatworthy, HW, Jr. The nature of ileal atresia due to intrauterine intussusception. Arch Surg. 1970; 100:714–717.
67. Murphy, DA. Internal hernias in infancy and childhood. Surgery. 1964; 55:311–316.
68. Vassy, LE, Boles, ET, Jr. Iatrogenic ileal atresia secondary to clamping of an occult omphalocele. J Pediatr Surg. 1975; 10:797–800.
69. Sweeney, B, Surana, R, Puri, P. Jejunoileal atresia and associated malformations: Correlation with timing of in-utero insult. J Pediatr Surg. 2001; 36:774–776.
70. Moore, SW, Rode, H, Millar, AJW, et al. Intestinal atresia and Hirschsprung’s disease. Pediatr Surg Int. 1990; 5:182–184.
71. Nicolini, U, Monni, G. Intestinal obstruction in babies exposed in-utero to methylene blue. Lancet. 1990; 336:1258–1259.
72. Guttman, FM, Braun, P, Garance, PH, et al. Multiple atresias and a new syndrome of hereditary multiple atresias involving the gastrointestinal tract from stomach to rectum. J Pediatr Surg. 1973; 8:633–640.
73. Werler, MM, Sheehan, JE, Mitchell, AA. Association of vasoconstrictive exposures with risks of gastroschisis and small intestinal atresia. Epidemiology. 2003; 14:349–354.
74. Cywes, S, Davies, MR, Rode, H. Congenital jejuno-ileal atresia and stenosis. S Afr Med J. 1980; 57:630–639.
75. Davies, MR, Louw, JH, Cywes, S, et al. The classification of congenital intestinal atresias [letter]. J Pediatr Surg. 1982; 17:224.
76. Baglaj, M, Carachi, R, Lawther, S. Multiple atresia of the small intestine: A 20-year review. Eur J Pediatr Surg. 2008; 18:13–18.
77. Louw, JH. Congenital intestinal atresia and severe stenosis in the newborn. S Afr J Clin Sci. 1952; 3:109–129.
78. Seashore, JH, Collins, FS, Markowitz, RI, et al. Familial apple peel jejunal atresia: Surgical, genetic, and radiographic aspects. Pediatrics. 1987; 80:540–544.
79. Mishalany, HG, Der Kaloustian, VM. Familial multiple-level intestinal atresias: Report of two siblings. J Pediatr. 1971; 79:124–125.
80. Weitzman, JJ, Vanderhoof, RS. Jejunal atresia with agenesis of the dorsal mesentery with “Christmas tree” deformity of the small intestine. Am J Surg. 1966; 111:443–449.
81. Zerella, JT, Martin, LW. Jejunal atresia with absent mesentery and a helical ileum. Surgery. 1976; 80:550–553.
82. Smith, MB, Smith, L, Wells, JW, et al. Concurrent jejunal atresia with “apple peel” deformity in premature twins. Pediatr Surg Int. 1991; 6:425–428.
83. DeLorimier, AA, Fonkalsrud, EW, Hays, DM. Congenital atresia and stenosis of the jejunum and ileum. Surgery. 1969; 65:819–827.
84. Dickson, JA. Apple peel small bowel: An uncommon variant of duodenal and jejunal atresia. J Pediatr Surg. 1970; 5:595–600.
85. Jimenez, FA, Reiner, L. Arteriographic findings in congenital abnormalities of the mesentery and intestines. Surg Gynecol Obstet. 1961; 113:346–352.
86. Tsujimoto, K, Sherman, FE, Ravitch, MM. Experimental intestinal atresia in the rabbit fetus. Sequential pathological studies. Johns Hopkins Med J. 1972; 131:287–297.
87. Komuro, H, Amagai, T, Hori, T, et al. Placental vascular compromise in jejunoileal atresia. J Pediatr Surg. 2004; 39:1701–1705.
88. Bilodeau, A, Prasil, P, Cloutier, R, et al. Hereditary multiple intestinal atresia: Thirty years later. J Pediatr Surg. 2004; 39:726–730.
89. Hasegawa, T, Sumimura, J, Nose, K, et al. Congenital multiple intestinal atresia successfully treated with multiple anastomoses in a premature neonate: Report of a case. Surg Today. 1996; 26:849–851.
90. Puri, P, Guiney, E, Carroll, R. Multiple gastrointestinal atresias in three consecutive siblings: Observations on pathogenesis. J Pediatr Surg. 1985; 20:22–24.
91. Baglaj, SM, Czernik, J, Koryszko, J, et al. Natural history of experimental intestinal atresia: Morphologic and ultrastructural study. J Pediatr Surg. 2001; 36:1428–1434.
92. Pickard, LR, Santoro, S, Wyllie, RG, et al. Histochemical studies of experimental fetal intestinal obstruction. J Pediatr Surg. 1981; 16:256–260.
93. Doolin, EJ, Ormsbee, HS, Hill, JL. Motility abnormalities in intestinal atresia. J Pediatr Surg. 1987; 22:320–324.
94. Cremin, BJ, Cywes, S, Louw, JH. Small intestine. In: Cremin BJ, Cywes S, Louw JH, eds. Radiological Diagnosis of Digestive Tract Disorders in the Newborn: A Guide to Radiologists, Surgeons, and Paediatricians. London: Butterworths; 1973:62.
95. Wasch, MG, Marck, A. The radiographic appearance of the gastrointestinal tract during the first day of life. J Pediatr. 1948; 32:479–489.
96. Benson, CD, Lofti, MW, Brogh, AJ. Congenital atresia and stenosis of the colon. J Pediatr Surg. 1968; 3:253–257.
97. Jackman, S, Brereton, RJ. A lesson in intestinal atresias. J Pediatr Surg. 1988; 23:852–853.
98. Aharon, M, Kleinhaus, U, Lichtig, C. Neonatal intramural intestinal calcifications associated with bowel atresia. AJR Am J Roentgenol. 1986; 130:999–1000.
99. Hays, DM. Intestinal atresia and stenosis. In: Ravitch M, ed. Current Problems in Surgery. Chicago: Year Book Medical; 1969:3.
100. Louw, JH. Resection and end-to-end anastomosis in the management of atresia and stenosis of the small bowel. Surgery. 1967; 62:940–950.
101. Blanck, C, Okmian, L, Robbe, H. Mucoviscidosis and intestinal atresia. A study of four cases in the same family. Acta Paediatr Scand. 1965; 54:557–565.
102. Suri, M, Langer, JC. A comparison of circumbilical and transverse abdominal incisions for neonatal abdominal surgery. J Pediatr Surg. 2011; 46:1076–1080.
103. McKee, MA, Jejunoileal Atresia edsitorsOldham KT, Colombani PM, Foglia RP, et al, eds. Principles and Practice of Pediatric Surgery. Lippincott Williams & Wilkins: Philadelphia, 2004; 1149.
104. de Lorimier, AA, Harrison, MR. Intestinal plication in the treatment of atresia. J Pediatr Surg. 1983; 18:734–737.
105. Holder, TM, Gross, RE. Temporary gastrostomy in pediatric surgery. Experience with 187 cases. Pediatrics. 1960; 26:36–41.
106. Howard, ER, Othersen, HB. Proximal jejunoplasty in the treatment of jejunoileal atresia. J Pediatr Surg. 1973; 8:685–690.
107. Ramanujan, TM. Functional capability of blind small bowel loops after intestinal remodeling techniques. Aust N Z J Surg. 1984; 54:145–150.
108. Kimura, K, Perdzynski, W, Soper, RT. Elliptical seromuscular resection for tapering the proximal dilated bowel in duodenal or jejunal atresia. J Pediatr Surg. 1996; 31:1405–1406.
109. Kling, K, Applebaum, H, Dunn, J, et al. A novel technique for correction of intestinal atresia at the ligament of Treitz. J Pediatr Surg. 2000; 35:353–355.
110. Grosfeld, JL, Ballantine, TV, Shoemaker, R. Operative management of intestinal atresia and stenosis based on pathologic findings. J Pediatr Surg. 1979; 14:368–375.
111. Touloukian, RJ. Intestinal atresia. Clin Perinatol. 1978; 5:3–18.
112. Snyder, CL, Miller, KA, Sharp, RJ, et al. Management of intestinal atresia in patients with gastroschisis. J Pediatr Surg. 2001; 36:1542–1545.
113. Festen, S, Brevoord, JC, Goldhoorn, GA, et al. Excellent long-term outcome for survivors of apple peel atresia. J Pediatr Surg. 2002; 37:61–65.
114. Waldhausen, JH, Sawin, RS. Improved long-term outcome for patients with jejunoileal apple peel atresia. J Pediatr Surg. 1997; 32:1307–1309.
115. Chaet, MS, Warner, BW, Sheldon, CA. Management of multiple jejunoileal atresias with an intraluminal Silastic stent. J Pediatr Surg. 1994; 29:1604–1606.
116. Haller, JA, Jr., Tepas, JJ, Pickard, LR, et al. Intestinal atresia. Current concepts of pathogenesis, pathophysiology, and operative management. Am Surg. 1983; 49:385–391.
117. Dowling, RH. Small bowel adaptation and its regulation. Scand J Gastroenterol Suppl. 1982; 17:53–74.
118. Remmington, M, Malagelada, JR, Zinsmeister, A, et al. Abnormalities in gastrointestinal motor activity in patients with short bowels: Effect of a synthetic opiate. Gastroenterology. 1983; 85:629–636.
119. Rode, H, Millar, AJW. Jejuno-ileal atresia and stenosis. In: Puri P, ed. Newborn Surgery. 2nd ed. London: Hodder Arnold; 2003:445.
120. Boles, ET, Jr., Vassy, LE, Ralston, M. Atresia of the colon. J Pediatr Surg. 1976; 11:69–75.
121. Benson, CD, Lotfi, MW, Brogh, AJ. Congenital atresia and stenosis of the colon. J Pediatr Surg. 1968; 3:253–257.
122. Powell, RW, Raffensperger, JG. Congenital colonic atresia. J Pediatr Surg. 1982; 17:166–170.
123. Croaker, GD, Harvey, JG, Cass, DT. Hirschsprung’s disease, colonic atresia, and absent hand: A new triad. J Pediatr Surg. 1997; 32:1368–1370.
124. Kim, PC, Superina, RA, Ein, S. Colonic atresia combined with Hirschsprung’s disease: A diagnostic and therapeutic challenge. J Pediatr Surg. 1995; 30:1216–1217.
125. Davenport, M, Bianchi, A, Doig, CM, et al. Colonic atresia: Current results of treatment. J R Coll Surg Edinb. 1990; 35:25–28.
126. Williams, MD, Burrington, JD. Hirschsprung’s disease complicating colon atresia. J Pediatr Surg. 1993; 28:637–639.
127. Karnak, I, Ciftci, AO, Senocak, ME, et al. Colonic atresia: Surgical management and outcome. Pediatr Surg Int. 2001; 17:631–635.
128. Sui, KL, Kwok, WK, Lee, WY, et al. A male newborn with colonic atresia and total colonic aganglionosis. Pediatr Surg Int. 1999; 15:141–142.
129. Cox, SG, Numanoglu, A, Millar, AJ, et al. Colonic atresia: Spectrum of presentation and pitfalls in management. A review of 14 cases. Pediatr Surg Int. 2005; 21:813–818.
130. Louw, JH. Investigations into the etiology of congenital atresia of the colon. Dis Colon Rectum. 1964; 7:471–478.
131. Santulli, TV, Blanc, WA. Congenital atresia of the intestine: Pathogenesis and treatment. Ann Surg. 1961; 154:939–948.
132. Rescorla, FJ, Grosfeld, JL. Intestinal atresia and stenosis: Analysis of survival in 120 cases. Surgery. 1985; 98:668–676.
133. Watts, AC, Sabharwal, AJ, Mackinlay, GA, et al. Congenital colonic atresia: Should primary anastomosis always be the goal? Pediatr Surg Int. 2003; 19:14–17.
134. DeFore, WW, Garcia-Rinaldi, R, Mattox, KL, et al. Surgical management of colon atresia. Surg Gynecol Obstet. 1976; 143:767–769.
135. Pohlson, EC, Hatch, EI, Jr., Glick, PL, et al. Individualized management of colonic atresia. Am J Surg. 1988; 155:690–692.
136. Hamzaoui, M, Ghribi, A, Makni, W, et al. Rectal and sigmoid atresia: Transanal approach. J Pediatr Surg. 2012; 47:E41–E44.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Duodenal and Intestinal Atresia and Stenosis
Congenital intestinal obstruction occurs in approximately 1 : 2000 live births and is a common cause of admission to a neonatal surgical unit, accounting for up to one-third of all admissions.1 Morphologically, congenital defects related to continuity of the intestine can be divided into either atresia or stenosis. Together, they constitute one of the most common etiologies of neonatal intestinal obstruction.2–4 See Chapter 29 for information about pyloric atresia.
Duodenal Atresia and Stenosis
Congenital duodenal atresia and stenosis is a frequent cause of intestinal obstruction and occurs in 1 per 5000 to 10,000 live births, affecting boys more commonly than girls.5 More than 50% of affected patients have associated congenital anomalies, with trisomy 21 occurring in approximately 30% of patients.6,7 Operative correction is accomplished via a duodenoduodenostomy, with or without tapering duodenoplasty. This can be performed either laparoscopically or open. Early postoperative survival rates of greater than 90% should be expected.7–11
Etiology
Congenital duodenal obstruction can occur due to an intrinsic or extrinsic lesion.12 The most common cause of duodenal obstruction is atresia.7 This intrinsic lesion is most commonly believed to be caused by a failure of recanalization of the fetal duodenum resulting in complete obstruction. Early in the fourth week of gestation, the duodenum begins to develop from the distal foregut and the proximal midgut. During the fifth and sixth weeks of gestation, the duodenal lumen temporarily obliterates due to proliferation of its epithelial cells. Vacuolation due to degeneration of the epithelial cells during the 11th week of gestation then leads to duodenal recanalization.13 An embryologic insult during this period can lead to an intrinsic web, atresia, or stenosis. The extrinsic form of duodenal obstruction is due to defects in the development of neighboring structures such as the pancreas, a preduodenal portal vein, or malrotation and Ladd’s bands.14,15
Annular pancreas as an etiology for duodenal obstruction warrants special mention as this form of obstruction is likely due to failure of duodenal development rather than a true constricting lesion. Thus, the presence of an annular pancreas is simply a visible indication of an underlying atresia or stenosis.16 Between the fourth and eighth week of gestation, the pancreatic buds merge. In annular pancreas, the tip of the ventral pancreas becomes fixed to the duodenal wall forming a nondistensible, ring-like or annular portion of pancreatic tissue surrounding the descending part of the duodenum.13 In annular pancreas associated with duodenal obstruction, the distal biliary tree is often abnormal and may open proximal or distal to the atresia or stenosis.17,18 Other reported biliary abnormalities associated with duodenal obstruction include biliary atresia, gallbladder agenesis, stenosis of the common bile duct, choledochal cyst, and immune deficiency.19–24
Classification
Anatomically, duodenal obstructions are classified as either atresias or stenoses. An incomplete obstruction, due to a fenestrated web or diaphragm, is considered a stenosis. Most stenoses involve the third and/or fourth part of the duodenum. Atresias, or complete obstruction, are further classified into three morphologic types (Fig. 30-1). Type I atresias account for more than 90% of all duodenal obstructions and contain a lumenal diaphragm that includes mucosal and submucosal layers. A diaphragm that has ballooned distally (windsock) is a type I atresia.25,26 It is important to understand that the anatomy of the windsock may lead to a portion of the dilated duodenum actually being distal to the actual obstruction (Fig. 30-2). Type II atresias are characterized by a dilated proximal and collapsed distal segment connected by a fibrous cord. Type III atresias have an obvious gap separating the proximal and distal duodenal segments.27
FIGURE 30-1 Duodenal atresia (and stenosis) is depicted. In type I (A), either a membrane (B) or web (C) causes the intrinsic duodenal obstruction. There is no fibrous cord and the duodenum remains in continuity. Type II is characterized by complete obliteration of a segment of the duodenum with the proximal and distal portions attached via a fibrous cord. Type III is associated with complete separation of the dilated proximal duodenum from the collapsed distal duodenum.
FIGURE 30-2 Illustration of the ‘windsock’ deformity, a variant of type I duodenal atresia. Note the actual position of the origin of the web in relation to the extent of proximal duodenal dilation and the distal collapsed duodenum.
More than 50% of affected patients with duodenal atresia have associated congenital anomalies.28 Approximately 30% are associated with trisomy 21, 30% with isolated cardiac defects, and 25% with other gastrointestinal (GI) anomalies.29,30 Approximately 45% of patients are premature, and about one-third exhibit growth retardation.6,7
Pathology
The obstruction can be classified as either preampullary or postampullary, with approximately 85% of obstructions located distal to the ampulla.30 With complete or almost complete obstruction, the stomach and proximal duodenum become significantly dilated. The pylorus is usually distended and hypertrophic. The bowel distal to the obstruction is collapsed, except in the case of a windsock deformity in which the distal bowel is dilated to a variable length depending on the length of the windsock (see Fig. 30-2). In most cases of duodenal obstruction, the gastrointestinal tract can be decompressed proximally. With complete obstruction of the duodenum, the incidence of polyhydramnios ranges from 32–81%.1,31–34 Growth retardation is also common, presumably from nutritional deprivation from the swallowed amniotic fluid.
Diagnosis
There are multiple benefits to the antenatal diagnosis of duodenal obstruction, including parental counseling. The diagnosis can often be suggested by prenatal ultrasound (US). Sonographic evaluation in fetuses of mothers with a history of polyhydramnios can detect two fluid-filled structures consistent with a double bubble in up to 44% of cases.35–37 Despite duodenal obstruction usually occurring by week 12, the reason for failure of early prenatal detection is not entirely clear. Most cases of duodenal atresia are detected between 7 and 8 months gestation.38 It is currently believed that immature gastric emptying in-utero may contribute to low gastric pressures, failing to dilate the proximal duodenum until later in gestation. While both circular and longitudinal muscle layers are present in the stomach by week 8 of gestation, pressure amplitudes at 25 weeks are only 60% of term gastric pressures.39,40
The presentation of the neonate with duodenal obstruction varies depending on whether the obstruction is complete or incomplete, and the location of the ampulla of Vater in relation to the obstruction. The classic presentation is that of bilious emesis within the first hours of life in an otherwise stable neonate. In about 10% of cases, however, the atresia is pre-ampullary and the emesis is nonbilious.15 Abdominal distention may or may not be present. In neonates with duodenal atresia, the abdomen is scaphoid. Aspiration via a nasogastric (NG) tube of more than 20 mL of gastric contents in a newborn suggests intestinal obstruction, as normal aspirate is less than 5 mL.41 For patients with stenosis, the diagnosis is often delayed until the neonate has started on enteral feeds and feeding intolerance develops with emesis and gastric distention.
In antenatally suspected cases of duodenal obstruction, as well as in neonates with a clinical presentation consistent with a proximal bowel obstruction, an upright abdominal radiograph is usually sufficient to confirm the diagnosis of duodenal atresia. The diagnostic radiographic presentation of duodenal atresia is that of a double bubble sign with no distal bowel gas (Fig. 30-3). The proximal left-sided bubble represents the air and fluid-filled stomach while the dilated proximal duodenum represents the second bubble to the right of midline.42 In almost all cases of duodenal atresia, the distal bowel is gasless. However, the presence of distal gas does not necessarily exclude the diagnosis of atresia as there are reports of bifed common bile ducts with insertion of one of the ducts proximal and the other distal to the atretic segment which allows the air to bypass the atresia.43 In neonates whose stomach has been decompressed by either NG aspiration or vomiting, 40–60 mL of instilled air into the stomach will reproduce the double bubble.27 Rarely, the biliary tree is air filled, and a variety of pancreatic and biliary anomalies have been demonstrated (Fig. 30-4).43 At our institution, neonates who present with bilious emesis and a decompressed stomach on plain abdominal films receive a limited upper GI contrast study to exclude malrotation and volvulus. With duodenal stenosis, a double bubble sign is often not present and the diagnosis is usually made with a contrast study (Fig. 30-5).
FIGURE 30-3 Classic ‘double bubble’ sign. This abdominal radiograph in a newborn shows a markedly distended stomach and duodenal bulb without evidence of distal intestinal air.
Management
After the diagnosis is made, appropriate resuscitation is required with correction of fluid balance and electrolyte abnormalities, in addition to gastric decompression. At our institution, all neonates diagnosed with duodenal obstruction receive a complete metabolic profile, complete blood count, coagulation studies, an abdominal and spinal ultrasound, and two-dimensional echocardiography prior to any operation. An emergency operation is only performed in cases where malrotation with concurrent volvulus cannot be excluded.
Prior to the mid-1970s, duodenojejunostomy was the preferred technique for correcting duodenal atresia or stenosis.7,44,45 Since then, the various techniques utilized include side-to-side duodenoduodenostomy, diamond-shaped duodenoduodenostomy, partial web resection with Heineke–Mickulicz-type duodenoplasty, and tapering duodenoplasty.44–46 The long side-to-side duodenoduodenostomy, although effective, is associated with a high incidence of anastomotic dysfunction and prolonged obstruction.30 Blind-loop syndrome appears to be more common in patients treated with duodenojejunostomy.47 Gastrojejunostomy should not be performed as it is associated with a high incidence of marginal ulceration and bleeding.27
Currently, the preferred technique is either laparoscopic or open duodenoduodenostomy.9–11,30 Originally, a side-to-side anastomosis was performed. A proximal transverse to distal longitudinal (diamond-shaped) anastomosis is now preferred.7,44,45,48–50 For the open approach, either a right upper quadrant supraumbilical transverse incision or an umbilical crease incision is utilized.49 After mobilizing the ascending and transverse colon to the left, the duodenal obstruction is readily exposed. Malrotation should be evaluated at this point as it can occur in association with congenital duodenal obstruction in up to 30% of patients.1 A sufficient length of duodenum distal to the atresia is mobilized to allow for a tension-free anastomosis. A transverse duodenotomy is made in the anterior wall of the distal portion of the dilated proximal duodenum and a similar length duodenotomy is made in a vertical orientation on the antimesenteric border of the distal duodenum. The anastomosis is then fashioned by approximating the end of each incision to the appropriate mid-portion of the other incision (Fig. 30-6). Tapering duodenoplasty is generally not necessary as the proximal duodenal dilation usually resolves after relief of the obstruction. Muscular continuity of the duodenal wall suggests a windsock deformity or diaphragm. This finding should precipitate extra vigilance in the operative correction because the dilated and collapsed bowel are both distal to the windsock, and have been anastomosed in error.26,51
FIGURE 30-6 The technique of duodenoduodenostomy. A diamond-shaped anastomosis is created via the proximal transversely oriented and distal vertically oriented duodenotomies.
The laparoscopic approach was first described by Rothenberg in 2002.9 The standard laparoscopic approach begins with the patient supine and the abdomen is insufflated through the umbilicus. Two other instruments are inserted, one in the baby’s right lower quadrant and one in the right mid-epigastric region, respectively. A liver retractor can be placed in the right or left upper quadrant if necessary. Alternatively, the liver can be elevated by placing a transabdominal wall suture around the falciform ligament and tying it outside the abdomen (Fig. 30-7). The duodenum is mobilized and the location of obstruction is identified. Using the same principles that have been described for the open approach, a standard diamond-shaped anastomosis is created. Although some surgeons will perform the laparoscopic anastomosis with interrupted sutures, this can be technically demanding because of the significant number of sutures required and the thin nature of the distal duodenum. We reported our results using Nitinol U-clips (Medtronic, Minneapolis, MN) to create the duodenoduodenostomy with no leaks and more rapid initiation of feeds when compared to the traditional open approach (Fig. 30-8).10,11 Although the U-clips are no longer commercially available, this study highlighted the advantages of early feeding. The historical approach to enteral feeding following duodenal atresia repair involved a period of waiting for the gastric output to become less bilious and the volume of gastric drainage to decrease, indicating return of intestinal function. This report showed that the time spent waiting for the gastric output to decrease is likely not necessary as all of the patients undergong the laparoscopic duodenoplasty had initiation of feeds without adverse events after an upper gastrointestinal contrast study on day 5 revealed no leak. When compared to infants undergoing an open operation with the historical postoperative management mentioned previously, there was a marked reduction in hospitalization for the laparoscopically corrected infants, primarily due to the early feeding.
FIGURE 30-7 Two approaches to placement of the instruments for a laparoscopic duodenal atresia repair. (A) The two right-sided instruments are the primary working sites for the surgeon. The liver retractor (arrow) has been placed in the left midepigastric region. The falciform ligament has been elevated by a suture placed under it and tied over the red rubber catheter, which is used as a bolster. The suture (dotted arrow) exteriorized in the infant’s left upper abdomen was placed in the dilated proximal duodenum so that it could be easily manipulated. (B) This is a similar configuration except the instrument elevating the liver (arrow) is placed in the infant’s right upper abdomen rather than the left upper abdomen. The suture that was placed through the proximal dilated duodenum in A was not needed in this particular case.
FIGURE 30-8 (A) Laparoscopic view of a completed duodenoduodenostomy using the Nitinol U-clips. (B) A postoperative contrast study at five days showed no evidence of obstruction or leak at the anastomosis. The U-clips (arrow) can be seen marking the anastomosis.
Historically, during repair of duodenal atresia, it has been emphasized that inspecting the entire small bowel to identify a second atresia is important. Given that duodenal atresia and jejunoileal atresia do not share common embryologic etiologies, a multi-institutional review of duodenal atresia patients was undertaken to quantify the incidence of jejunoileal atresia in this population.52 In the largest series to date, the rate of concomitant jejunoileal atresia in patients with duodenal atresia was less than 1%. With the low incidence of a concomitant distal atresia, extensive inspection of the entire bowel does not appear necessary.
Early postoperative mortality for duodenal atresia repair has been reported to be as low as 3–5% with the majority of deaths occurring secondary to complications related to associated congenital abnormalities.53,54 Long-term survival approaches 90%.7,53–55 Long-term complications have been noted following repair and include delayed gastric emptying, severe gastroesophageal reflux, bleeding peptic ulcer, megaduodenum, duodenogastric reflux, gastritis, blind-loop syndrome and intestinal obstruction related to adhesions.30
Jejunoileal Atresia and Stenosis
Etiology
Jejunoileal atresia occurs in approximately 1 in 5,000 live births. It occurs equally in males and females, and about one in three infants is premature.56 Although the majority of cases are thought to occur sporadically, familial cases of intestinal atresias have been described.57 It is generally accepted that jejunoileal atresia occurs as a result of an intrauterine ischemic insult to the midgut, affecting single or multiple segments of the already developed intestine.13,58–61 Intrauterine vascular disruption can lead to ischemic necrosis of the bowel with subsequent resorption of the affected segment or segments (Fig. 30-9).
FIGURE 30-9 The proposed mechanism of vascular compromise and subsequent development of jejunoileal atresias is depicted.
The hypothesis that most cases of jejunoileal atresia occur secondary to vascular disruption during fetal life is derived from experimental as well as clinical evidence. Isolated mesenteric vascular insults and interference with the segmental blood supply to the small intestine were created in fetal dogs, and resulted in different degrees and patterns of intraluminal obstruction, reproducing the spectrum of stenosis and atresia found in humans.62–64 Moreover, the presence of bile, lanugo hair, and squamous epithelial cells from swallowed amniotic fluid distal to an atresia suggests that the atresia occurs subsequent to some event, but that at some time in gestation the intestinal lumen was patent, thus allowing passage of these contents. Additionally, atresias seen in association with other intrauterine vascular insults such as fetal intussusception, midgut volvulus, thromboembolic occlusions, transmesenteric internal hernias, and incarceration or snaring of bowel in an omphalocele or gastroschisis have contributed to wide acceptance of this hypothesis.58,64–68
The presence of associated extra-abdominal organ abnormalities in jejunoileal atresia is low (<10%) due to its occurrence later in fetal life and the localized nature of the vascular insult.69 Rarely, jejunoileal atresia has been found in patients with Hirschsprung disease, cystic fibrosis, malrotation, Down syndrome, anorectal and vertebral anomalies, neural tube defects, congenital heart disease, and other GI atresias.56,69,70 Methylene blue, previously used for amniocentesis in twin pregnancies, has been implicated in causing small bowel atresia.71
Although jejunoileal atresias are usually not hereditary, there is a well-documented autosomal recessive pattern of inheritance of multiple atresias.72 In these cases, intestinal rotation was normal, mesenteric defects were never observed, and lanugo hairs and squamous cells were not identified distal to the most proximal atresia. All these findings suggest an early intrauterine event. Survival is poor in these infants, even with successful bowel resection.
No correlations have been found between jejunoileal atresia and parental or maternal disease. However, the use of maternal vasoconstrictive medications, as well as maternal cigarette smoking in the first trimester of pregnancy, has been shown to increase the risk of small bowel atresia.73 Chromosomal abnormalities are seen in less than 1% of the patients with jejunoileal atresia.74
Pathology
The Grosfeld classification system separates these defects into four groups, with an additional consideration for type III(b) (Fig. 30-10).4 This classification has significant prognostic and therapeutic value as it emphasizes the importance of associated loss of intestinal length, abnormal collateral intestinal blood supply, and concomitant atresia or stenosis.75 Regarding classification, the most proximal atresia determines whether the atresia is classified as jejunal or ileal atresia. Multiple atresias are found in up to 30% of patients.56,76
Stenosis
Stenosis is defined as a localized narrowing of the intestinal lumen without disruption in the intestinal wall or a defect in the mesentery (see Fig. 30-10A). At the stenotic site, a short, narrow, somewhat rigid segment of intestine with a small lumen is found. Often the muscularis is irregular and the submucosa is thickened. Stenosis may also take the form of a type I atresia with a fenestrated web. Patients with jejunoileal stenosis usually have a normal length of small intestine.
Type I Atresia
In type I jejunoileal atresia, the intestinal obstruction occurs secondary to a membrane or web formed by both mucosa and submucosa, while the muscularis and serosa remain intact (see Figs 30-10B and 30-11). On gross inspection, the bowel and its mesentery appear to be in continuity. However, the proximal bowel is dilated while the distal bowel is collapsed. With the increased intraluminal pressure in the proximal bowel, bulging of the web into the distal intestine can create a windsock effect. As with stenosis, there is no foreshortening of the bowel in type I atresias.
Type II Atresia
The clinical findings of a type II atresia are a dilated, blind-ending proximal bowel loop connected by a fibrous cord to the collapsed distal bowel with an intact mesentery (see Fig. 30-10C). Increased intraluminal pressure in the dilated and hypertrophied proximal bowel may lead to focal proximal small bowel ischemia. The distal collapsed bowel commences as a blind end, which sometimes assumes a bulbous appearance owing to the remains of an intussusception. Again, the total small bowel length is usually normal.
Type III(a) Atresia
In type III(a) atresia, the proximal bowel ends blindly with no fibrous connecting cord to the distal intestine. A V-shaped mesenteric defect of varying size is present between the two ends of intestine (see Figs 30-10D and 30-12). The dilated, blind-ending proximal bowel is often aperistaltic and frequently undergoes torsion or becomes overdistended, with subsequent necrosis and perforation occurring as a secondary event.77 In this scenario, the total length of the small bowel is variable (but usually less than normal), owing to intrauterine resorption of the affected bowel.
[/not-level-membership-for-pediatrics-category]
