[level-membership-for-anesthesiology-category]
Drugs Acting on the Cardiovascular System
THE AUTONOMIC NERVOUS SYSTEM
The term autonomic nervous system (ANS) refers to the nervous and humoral mechanisms which modify the function of the autonomous or automatic organs. These include heart rate and force of contraction, calibre of blood vessels, contraction and relaxation of smooth muscle in gut, bladder and bronchi, visual accommodation and pupillary size. Other functions include regulation of secretion from exocrine and other glands and aspects of metabolism (e.g. glycogenolysis and lipolysis) (Table 8.1). There is constant activity of both the sympathetic and parasympathetic nervous systems even at rest. This is termed sympathetic or parasympathetic tone and allows alterations in autonomic activity to produce rapid two-way regulation of physiological effect. The ANS is controlled by centres in the spinal cord, brainstem and hypothalamus, which are in turn influenced by higher centres in the cerebral and particularly the limbic cortex. The ANS is also influenced by visceral reflexes whereby afferent signals enter the autonomic ganglia, spinal cord, hypothalamus or brainstem and directly elicit appropriate reflex responses via the visceral organs. The efferent autonomic signals are transmitted through the body to two major subdivisions (separated by anatomical, physiological and pharmacological criteria), the sympathetic and the parasympathetic nervous systems.
TABLE 8.1
Effects of the Sympathetic and Parasympathetic Nervous Systems on Peripheral Effector Organs, and Receptor Subtypes Mediating these Functions (Where Known)
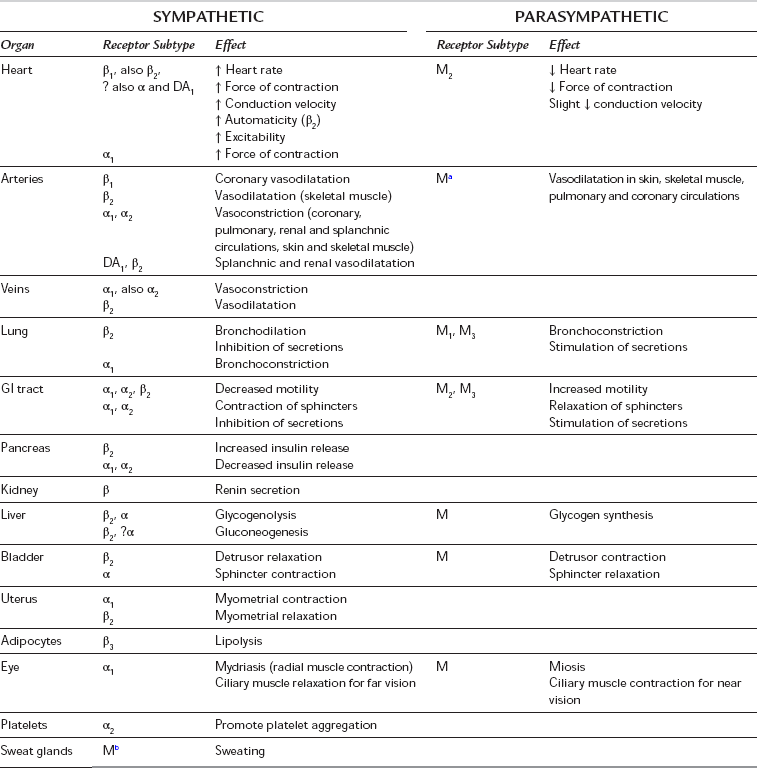
aMuscarinic receptors are present on vascular smooth muscle, but they are independent of parasympathetic innervation and have little or no physiological role in the control of vasomotor tone.
bSympathetic cholinergic fibres supply sweat glands and arterioles in some sites.
All postganglionic parasympathetic fibres are muscarinic (M), but in many sites the subtype has not been identified.
The Sympathetic Nervous System
The sympathetic nervous system includes nerves which originate in the spinal cord between the first thoracic and second lumbar segments (T1 to L2). Fibres leave the spinal cord with the anterior nerve roots and then branch off as white rami communicantes to synapse in the bilateral paravertebral sympathetic ganglionic chains, although some preganglionic fibres synapse instead in the paravertebral ganglia (e.g. coeliac, mesenteric and hypogastric) in the abdomen before travelling to their effector organ with the relevant arteries. Postganglionic fibres travel from paravertebral ganglia in sympathetic nerves (to supply the internal viscera, including the heart) and spinal nerves (which innervate the peripheral vasculature and sweat glands). Sympathetic nerves throughout the circulation contain vasoconstrictor fibres, particularly in the kidneys, spleen, gut and skin; however, sympathetic vasodilator fibres predominate in skeletal muscle, and coronary and cerebral vessels. Sympathetic stimulation therefore causes predominantly vasoconstriction but also a redistribution of blood flow to skeletal muscle; constriction of venous capacitance vessels may decrease their volume and thereby increase venous return. The effects of sympathetic stimulation at different receptors and effector organs are summarized in Table 8.1. The distribution of sympathetic nerve fibres to an organ or region may differ from the sensory or motor supply, according to its embryonic origin. For example, sympathetic fibres to the heart arise from T1 to T5 (but predominantly from T1 to T4), the neck is supplied by fibres from T2, the chest by fibres from T3 to T6 and the abdomen by fibres from T7 to T11.
Sympathetic Neurotransmitters
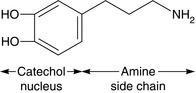
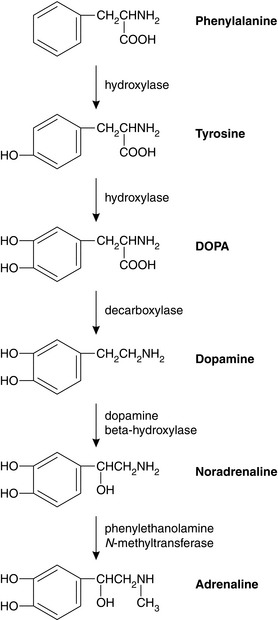
Activation of preganglionic nicotinic fibres to the adrenal medulla causes the release of adrenaline (adrenaline), which is released primarily as a circulating hormone and is only found in insignificant amounts in the nerve endings. Endogenous catecholamines (adrenaline, noradrenaline (norepinephrine) and dopamine) are synthesized from the essential amino acid phenylalanine. Their structure is based on a catechol ring (i.e. a benzene ring with -OH groups in the 3 and 4 positions), and an ethylamine side chain (Figs 8.1 and 8.2); substitutions in the side chain produce the different compounds. Dopamine may act as a precursor for both adrenaline and noradrenaline when administered exogenously (see below).



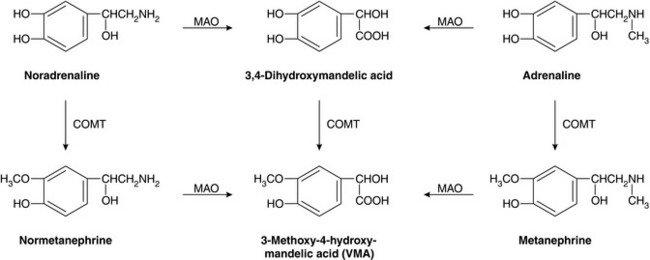
Most noradrenaline released from sympathetic nerves is taken back into the presynaptic nerve ending for storage and subsequent reuse. Re-uptake is by active transport back into the nerve terminal cytoplasm and then into cytoplasmic vesicles. This mechanism of presynaptic re-uptake, termed uptake1, is dependent on adenosine triphosphate (ATP) and Mg2 +, is enhanced by Li+ and may be blocked by cocaine and tricyclic antidepressants. Endogenous catecholamines entering the circulation by diffusion from sympathetic nerve endings or by release from the adrenal gland are metabolized rapidly by the enzymes monamine oxidase (MAO) and catechol O-methyltransferase (COMT) in the liver, kidneys, gut and many other tissues. The metabolites are conjugated before being excreted in urine as 3-methoxy-4-hydroxymandelic acid, metanephrine (from adrenaline) and normetanephrine (from noradrenaline) (Fig. 8.3). Noradrenaline taken up into the nerve terminal may also be deaminated by cytoplasmic MAO.
Adrenergic Receptor Pharmacology
These subdivisions and the functions of the autonomic nervous system are summarized in Table 8.1.
The Parasympathetic Nervous System
The parasympathetic nervous system controls vegetative functions, e.g. the digestion and absorption of nutrients, excretion of waste products and the conservation and restoration of energy. Parasympathetic neurones arise from cell bodies of the motor nuclei of cranial nerves III, VII, IX and X in the brainstem, and from the sacral segments of the spinal cord (‘the craniosacral outflow’). Preganglionic fibres run almost to the organ innervated and synapse in ganglia within the organ, giving rise to postganglionic fibres which then supply the relevant tissues. The ganglion cells may be well organized (e.g. the myenteric plexus of the intestine) or diffuse (e.g. in the bladder or vasculature). As the majority of all parasympathetic nerves are contained in branches of the vagus nerve, which innervates the viscera of the thorax and abdomen, increased parasympathetic activity is characterized by signs of vagal overactivity. Parasympathetic fibres also pass to the eye via the oculomotor (third cranial) nerve, and to the lacrimal, nasal and salivary glands via the facial (fifth) and glossopharyngeal (ninth) nerves. Fibres originating in the sacral portion of the spinal cord pass to the distal GI tract, bladder and reproductive organs. The effects of parasympathetic stimulation at different receptors and effector organs are summarized in Table 8.1
Parasympathetic Receptor Pharmacology
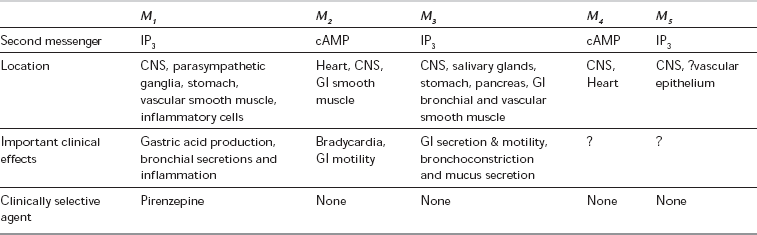
Parasympathetic receptors have been classified according to the actions of the alkaloids muscarine and nicotine. The actions of ACh at the postganglionic membrane are mimicked by muscarine and are termed muscarinic, whereas preganglionic transmission is termed nicotinic. ACh is also the neurotransmitter at the neuromuscular junction, via nicotinic receptor sites. Five subtypes of muscarinic receptors (M1–M5) have been characterized; all five subtypes exist in the CNS, but there are differences in their peripheral distribution and function (Table 8.2). M1-receptors are found in the stomach, where they mediate acid secretion, and in inflammatory cells in the lung (including mast cells and eosinophils) where they may have a role in airway inflammation. M2-receptors predominate in the myocardium, where they modulate heart rate and impulse conduction. Prejunctional M2-receptors are also involved in the regulation of synaptic noradrenaline and postganglionic ACh release. M3-receptors are present in classic postsynaptic sites in glandular tissue (of the GI and respiratory tract) and bronchial smooth muscle, where they mediate most of the post-junctional effects of ACh. M4-receptors have been isolated in cardiac and lung tissue in animal models and may have inhibitory effects, but the distribution and functions of M5-receptors are not yet defined. In common with adrenergic receptors, muscarinic receptors are coupled to membrane-bound G-proteins although the subtypes differ in the second messenger system with which they interact. Currently available anticholinergics probably act at all muscarinic receptor subtypes but their clinical spectra differ, which suggests that they may have differential effects at different subtypes.
DRUGS ACTING ON THE SYMPATHETIC NERVOUS SYSTEM
 directly on the adrenergic receptor, e.g. the catecholamines, phenylephrine, methoxamine
directly on the adrenergic receptor, e.g. the catecholamines, phenylephrine, methoxamine
 indirectly causing release of noradrenaline from the adrenergic nerve ending, e.g. amphetamine
indirectly causing release of noradrenaline from the adrenergic nerve ending, e.g. amphetamine

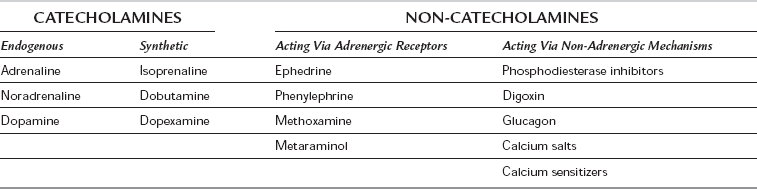
The drugs may be classified according to their structure (catecholamine/non-catecholamine), their origin (endogenous/synthetic) and their mechanism of action (via adrenergic receptors or via a non-adrenergic mechanism) (Table 8.3). Drugs which affect myocardial contractility are termed inotropes, although this term is usually applied to those drugs that increase cardiac contractility (strictly ‘positive inotropes’). Myocardial contractility may be increased by:
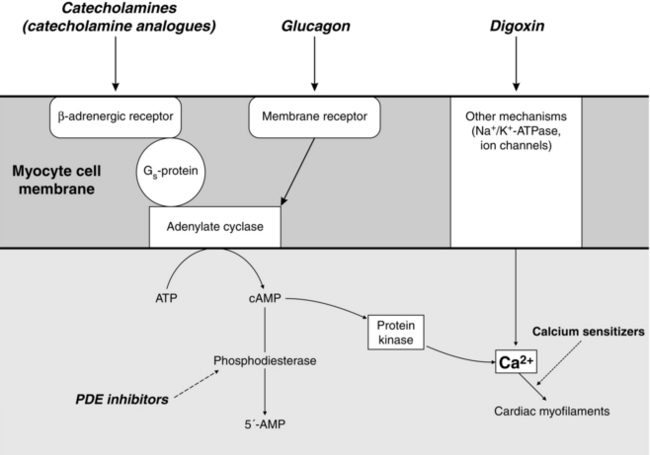
 increasing intracellular cAMP by activation of the adenylate cyclase system (e.g. catecholamines and other drugs acting via the adrenergic receptor)
increasing intracellular cAMP by activation of the adenylate cyclase system (e.g. catecholamines and other drugs acting via the adrenergic receptor)
 decreasing breakdown of cAMP (e.g. phosphodiesterase inhibitors)
decreasing breakdown of cAMP (e.g. phosphodiesterase inhibitors)
 increasing intracellular calcium availability (e.g. digoxin, calcium salts, glucagon)
increasing intracellular calcium availability (e.g. digoxin, calcium salts, glucagon)
 increasing the response of contractile proteins to calcium (e.g. levosimendan) (Fig. 8.4).
increasing the response of contractile proteins to calcium (e.g. levosimendan) (Fig. 8.4).
Catecholamines
Endogenous Catecholamines
Adrenaline: Adrenaline comprises 80–90% of adrenal medullary catecholamine content and is also an important CNS neurotransmitter. It is a powerful agonist at both α- and β-adrenergic receptors, being slightly less potent than noradrenaline at α1-receptors but more potent at β-receptors. It is the treatment of choice in acute allergic (anaphylactic) reactions and is used in the management of cardiac arrest and shock, and occasionally as a bronchodilator. Except in emergency situations, i.v. injection is avoided because of the risk of inducing cardiac arrhythmias. Subcutaneous administration produces local vasoconstriction and so smoothes out its own effect by slowing absorption.
Noradrenaline: Noradrenaline acts as a potent arteriolar and venous vasoconstrictor, acting predominantly at α-receptors, with a slightly greater potency there than adrenaline. It is also an agonist at β-receptors, but β2 effects are not apparent in clinical use. Infusions of noradrenaline increase venous return, systolic and diastolic systemic and pulmonary arterial pressures, and central venous pressure. Cardiac output increases but heart rate decreases because of baroreflex activity. At higher doses, the α-mediated effects of widespread intense vasoconstriction overcome β1 effects on cardiac contractility, leading to a decrease in cardiac output at the cost of increased myocardial oxygen demand in conjunction with reductions in renal blood flow and glomerular filtration rate. Its principal use is in the management of septic shock when systemic vascular resistance is low.
Dopamine: Dopamine is the natural precursor of adrenaline and noradrenaline. It stimulates both α- and β-adrenergic receptors in addition to specific dopamine DA1-receptors in renal and mesenteric arteries. Dopamine has a direct positive inotropic action on the myocardium via β-receptors and also by release of noradrenaline from adrenergic nerve terminals. The overall effects of dopamine are highly dose-dependent. In low dosage (< 3 μg kg–1 min–1), renal and mesenteric vascular resistances are reduced by an action on DA1-receptors, resulting in increased splanchnic and renal blood flows, glomerular filtration rate and sodium excretion. At doses of 5–10 μg kg–1 min–1, the increasing direct β-mediated inotropic action predominates, increasing cardiac output and systolic pressure with little effect on diastolic pressure; peripheral resistance is usually unchanged. At doses > 15 μg kg–1 min–1, α-receptor activity predominates, with direct vasoconstriction and increased cardiac stimulation (similar to noradrenaline). Renal and splanchnic blood flows decrease, and arrhythmias may occur. Dopamine receptors are widely present in the CNS, particularly in the basal ganglia, pituitary (where they mediate prolactin secretion) and the chemoreceptor trigger zone on the floor of the fourth ventricle (where they mediate nausea and vomiting). Recently, dopamine infusions have been associated with decreased prolactin secretion, and the use of ‘prophylactic’ dopamine infusions in an attempt to preserve renal function in perioperative or critically ill patients has declined.
Synthetic Catecholamines
Isoprenaline: Isoprenaline is a potent β1– and β2-agonist, with virtually no activity at α-receptors. It acts via cardiac β1-receptors, and at β2-receptors in the smooth muscle of bronchi, the vasculature of skeletal muscle and the gut. After intravenous infusion, heart rate increases and peripheral resistance is reduced. Cardiac output may increase because of increased heart rate and contractility but effects on arterial pressure are variable. Isoprenaline also reduces coronary perfusion pressure, increases myocardial oxygen consumption and causes arrhythmias. Other β2-mediated effects include relaxation of bronchial smooth muscle and stabilization of mast cells. It has been superseded for use in the treatment of severe asthma by newer specific β2-agonists with fewer cardiac effects. Its current indication is as an infusion in the treatment of bradyarrhythmias or atrioventricular heart block associated with low cardiac output (e.g. following acute myocardial infarction) because it increases heart rate and conduction by a direct action on the subsidiary pacemaker. This indication is usually an interim measure before insertion of a temporary pacing wire.
Dobutamine: Dobutamine is primarily a β1-agonist, with moderate β2– and mild α1-agonist activity, and no action at DA-receptors. Its primary effect is an increase in cardiac output via increased contractility (β1 effect) augmented by a reduction in afterload. Heart rate also increases (β2 effect). Systolic arterial pressure may increase but peripheral resistance is reduced or unchanged. There is no direct effect on venous tone or renal blood flow but preload may decrease and urine output and sodium excretion increase as a consequence of the increased cardiac output. Dobutamine increases SA node automaticity and conduction velocity in the atria, ventricles and AV node, but to a lesser extent than isoprenaline. Dobutamine infusion produces a progressive increase in cardiac output which is greater than with comparable doses of dopamine, although arterial pressure may remain unchanged. At higher doses, tachycardia and arrhythmias may occur, but dobutamine has less effect on myocardial oxygen consumption compared with other catecholamines. Dobutamine is widely used to optimize cardiac output in septic shock, often in combination with noradrenaline. It is also used alone or in combination with vasodilator drugs in heart failure when peripheral resistance is high, and to increase heart rate and cardiac output in myocardial stress testing.
Dopexamine: Dopexamine is a synthetic dopamine analogue which is an agonist at β2– and DA1-receptors. It is also a weak DA2-agonist and it inhibits the neuronal re-uptake of noradrenaline (uptake1), but has no direct effects at β1– or α-receptors. Its principal effect is β2-agonism, producing vasodilatation in skeletal muscle; it is less potent at DA1-receptors, but a more potent β2-agonist than dopamine. It produces mild increases in heart rate, contractility and cardiac output (effects on β2-receptors and noradrenaline uptake), renal and mesenteric vasodilatation (β2 and DA1 effects), and natriuresis (DA1 effect). Coronary and cerebral blood flows are also increased. Systemic vascular resistance decreases and arterial pressure may decrease if intravascular volume is not maintained. Dopexamine has theoretical advantages in maintaining cardiac output and splanchnic blood flow in patients with systemic sepsis or heart failure. It is also used for this purpose in patients undergoing major abdominal surgery. Dopexamine also has anti-inflammatory effects (in common with other β-agonists) which are independent of its effects on gut mucosal perfusion. It is metabolized by hepatic methylation and conjugation and is eliminated mostly via the kidneys.
Non-Catecholamine Sympathomimetics
Ephedrine: Ephedrine is a naturally occurring sympathomimetic amine which is now produced synthetically. It acts directly and indirectly as an agonist at α-, β1– and β2-receptors. The indirect actions are increased endogenous noradrenaline release and inhibition of MAO. Its cardiovascular effects are similar to those of adrenaline, but the duration of action is up to 10 times longer. It causes increases in heart rate, contractility, cardiac output and arterial pressure (systolic > diastolic). It may predispose to arrhythmias. Systemic vascular resistance is usually unchanged because α-mediated vasoconstriction in some vascular beds is balanced by β-mediated vasodilatation in others, but renal and splanchnic blood flows decrease. It relaxes bronchial and other smooth muscle, and is occasionally used as a bronchodilator. It is active orally because it is not metabolized by MAO in the gut, and is useful by intramuscular injection because muscle blood flow is preserved. Ephedrine undergoes hepatic deamination and conjugation but significant amounts are excreted unchanged in urine. This accounts for its long duration of action and elimination half-life (3–6 h). Tachyphylaxis (a decreased response to repeated doses of the drug) occurs because of persistent occupation of adrenergic receptors and depletion of noradrenaline stores.
Phenylephrine: Phenylephrine is a potent synthetic direct-acting α1-agonist, which has minimal agonist effects at α2-and β-receptors. It has effects similar to those of noradrenaline, causing widespread vasoconstriction, increased arterial pressure, bradycardia (as a result of baroreflex activation) and a decrease in cardiac output. Venoconstriction predominates and diastolic pressure increases more than systolic pressure, so that coronary blood flow may increase. It is used as intermittent boluses (50-100 μg by slow injection) or an infusion (50–150 μg min− 1) to maintain arterial pressure during general or regional anaesthesia, and also topically as a nasal decongestant and mydriatic. Absorption of phenylephrine from mucous membranes may occasionally produce systemic side-effects.
Methoxamine: Methoxamine is a direct-acting α1-agonist which also has a weak β-antagonist action. It produces vasoconstriction, increased diastolic arterial pressure and decreases in cardiac output and heart rate from baroreflex activation and the mild β-blocking effect. It is no longer available in the UK.
Metaraminol: Metaraminol is a direct- and indirect-acting α- and β-agonist, which acts partly by being taken up into sympathetic nerve terminals and acting as a false transmitter for noradrenaline. Its α effects predominate, causing pronounced vasoconstriction; arterial pressure increases and a reflex bradycardia may occur.
Vasopressin: Arginine vasopressin (AVP) (formerly termed antidiuretic hormone) is a peptide hormone secreted by the hypothalamus. Its primary role is the regulation of body fluid balance. It is secreted in response to hypotension and promotes retention of water by action on specific cAMP-coupled V2-receptors. It causes vasoconstriction by stimulating V1-receptors in vascular smooth muscle and is particularly potent in hypotensive patients. It is increasingly used in the treatment of refractory vasodilatory shock which is resistant to catecholamines, although it can cause peripheral or splanchnic ischaemia. The vasopressin analogue desmopressin is used to treat diabetes insipidus. Another analogue, terlipressin, is used to limit bleeding from oesophageal varices in patients with portal hypertension as an adjunct to definitive treatment.
Phosphodiesterase Inhibitors: Phosphodiesterase inhibitors increase intracellular cAMP concentrations by inhibition of the enzyme responsible for cAMP breakdown (Fig. 8.4). Increased intracellular cAMP concentrations promote the activation of protein kinases, which lead to an increase in intracellular Ca2 +. In cardiac muscle cells, this causes a positive inotropic effect and also facilitates diastolic relaxation and cardiac filling (termed ‘positive lusitropy’). In vascular smooth muscle, increased cAMP decreases intracellular Ca2 + and causes marked vasodilatation. Several subtypes of phosphodiesterase (PDE) isoenzyme exist in different tissues. Theophylline is a non-specific PDE inhibitor, but the newer drugs (e.g. enoximone and milrinone) are selective for the PDE type III isoenzyme present in the myocardium, vascular smooth muscle and platelets. PDE III inhibitors are positive inotropes and potent arterial, coronary and venodilators. They decrease preload, afterload, pulmonary vascular resistance and pulmonary capillary wedge pressure (PCWP), and increase cardiac index. Heart rate may increase or remain unchanged. In contrast to sympathomimetics, they improve myocardial function without increasing oxygen demand or causing tachyphylaxis. Their effects are augmented by the co-administration of β1-agonists (i.e. increases in cAMP production are synergistic with decreased cAMP breakdown). They have particular advantages in patients with chronic heart failure, in whom downregulation of myocardial β-adrenergic receptors occurs, so that there is a decreased inotropic response to β-sympathomimetic drugs. A similar phenomenon occurs with advanced age, prolonged (> 72 h) catecholamine therapy and possibly with surgical stress.
Glucagon: Glucagon is a polypeptide secreted by the α cells of the pancreatic islets. Its physiological actions include stimulation of hepatic gluconeogenesis in response to hypoglycaemia, amino acids and as part of the stress response. These effects are mediated by increasing adenylate cyclase activity and intracellular cAMP, by a mechanism independent of the β-adrenergic receptor (Fig. 8.4). It increases cAMP in myocardial cells and so increases cardiac contractility. Glucagon causes nausea and vomiting, hyperglycaemia and hyperkalaemia and is not used as an inotrope except in the management of β-blocker poisoning.
Calcium: Calcium ions are involved in cellular excitation, excitation-contraction coupling and muscle contraction in cardiac, skeletal and smooth muscle cells. Increased extracellular Ca2 + increases intracellular Ca2 + concentrations and consequently the force of contraction of cardiac myocytes and vascular smooth muscle cells. Massive blood loss and replacement with large volumes of calcium-free fluids or citrated blood (which chelates Ca2 +) may cause a decrease in serum Ca2 + concentration, especially in the critically ill. Therefore, Ca2 + salts (e.g. calcium chloride or gluconate) may be administered, particularly during and after cardiopulmonary bypass. Intravenous calcium 5 mg kg–1 may increase mean arterial pressure, but the effects on cardiac output and systemic vascular resistance are variable and there is little good evidence for the efficacy of Ca2 + salts. Moreover, high Ca2 + concentrations may cause cardiac arrhythmias and vasoconstriction, may be cytotoxic and may worsen the cellular effects of ischaemia. Calcium salts may be indicated for the treatment of hypocalcaemia (ionized Ca2 + < 0.8 mmol L–1), hyperkalaemia and calcium channel blocker toxicity.
Calcium Sensitizers: Levosimendan and pimobendan are positive inotropic drugs which act by stabilizing the troponin molecule in cardiac myocytes (by a cAMP- independent mechanism) and so increase myocyte Ca2 + sensitivity without increasing Ca2 + influx. Contractility is increased without an increase in oxygen consumption or a tendency to arrhythmias. Levosimendan also causes vasodilatation by opening K+ channels via an ATP-dependent mechanism and may be used as a second line agent in acute heart failure. It is available in Europe and South America. Pimobendan also inhibits phosphodiesterase-III and has been investigated for use in chronic heart failure. It is available in Japan.
Selective β2-Agonists
Selective β2-receptor agonists (e.g. salbutamol, terbutaline, formoterol and salmeterol) relax bronchial, uterine and vascular smooth muscle while having much less effect on the heart than isoprenaline. These drugs are partial agonists (their maximal effect at β2-receptors is less than that of isoprenaline) and are only partially selective for β2-receptors. They are used widely in the treatment of bronchospasm (see Ch 9). Although less cardiotoxic than isoprenaline, dose-related tremor, tachyarrhythmias, hyperglycaemia, hypokalaemia and hypomagnesaemia may occur. β2-Agonists are resistant to metabolism by COMT and therefore have a prolonged duration of action (mostly 3–5 h). Salmeterol is highly lipophilic, has a strong affinity for the β2-adrenergic receptor, is longer acting than the other β2-agonists and so is used for maintenance therapy in chronic asthma in combination with inhaled steroids. β2-Agonists are usually administered by the inhaled (metered dose inhaler or nebulizer) or intravenous routes because of unpredictable oral absorption and a high hepatic extraction ratio. When inhaled, only 10–20% of the administered dose reaches the lower airways; this proportion is reduced further when administered via a tracheal tube. Nevertheless, systemic absorption does occur, although adverse effects are less common during long-term therapy.
Salbutamol: Salbutamol is the β2-agonist used most commonly for the prevention and treatment of bronchospasm. When administered by metered dose inhaler (1 or 2 puffs, each delivering 100 μg), it acts within a few minutes, with a peak action at 30–60 min. In severe cases, it may be given by nebulizer (2.5–5.0 mg), repeated if required, or intravenously (either 250 μg by slow i.v. injection or as an infusion starting at 5 μg min–1 and titrated to response). It is metabolized in the liver and excreted in urine both as metabolites and as unchanged drug; the proportions are dependent on the route of administration.
Sympatholytic Drugs
Peripherally Acting Sympatholytic Drugs
Ganglion Blocking Drugs: Nicotinic receptor antagonists (e.g. hexamethonium, pentolinium, trimetaphan) competitively inhibit the effects of ACh at autonomic ganglia and block both parasympathetic and sympathetic transmission. Sympathetic blockade produces venodilatation, decreased myocardial contractility and hypotension, but the effects vary depending on pre-existing sympathetic tone. Tachyphylaxis develops rapidly and these drugs have now been superseded.
Adrenergic Neurone Blocking Drugs: Guanethidine decreases peripheral sympathetic nervous system activity by competitively binding to noradrenaline binding sites in storage vesicles in the cytoplasm of postganglionic sympathetic nerve terminals. Further uptake of noradrenaline into the vesicles is inhibited and it is metabolized by cytoplasmic MAO, so the nerve terminals become depleted of noradrenaline. Guanethidine has local anaesthetic properties and does not cross the blood–brain barrier. It is sometimes used to produce intravenous regional sympathetic blockade in the treatment of chronic limb pain associated with excessive autonomic activity (reflex sympathetic dystrophy or complex regional pain syndromes). Bretylium has a similar mode of action; it is used in the treatment of resistant ventricular arrhythmias (see below).
α-Adrenergic Receptor Antagonists: α-Adrenergic antagonists (α-blockers) selectively inhibit the action of catecholamines at α-adrenergic receptors. They are used mainly as vasodilators for the second-line treatment of hypertension or as urinary tract smooth muscle relaxants in patients with benign prostatic hyperplasia. They also have an important role in the preoperative management of phaeochromocytoma (see Ch 37).
α1-Selective Antagonists: Selective α1-blockers include prazosin, doxazosin, indoramin, and urapidil. Doxazosin has largely succeeded prazosin as it has a more prolonged duration of action. Reflex tachycardia and postural hypotension are less common than with direct-acting vasodilators (e.g. hydralazine) and the non-selective α-blockers, but may still occur on initiating therapy. Nasal congestion, sedation and inhibition of ejaculation may occur.
α2-Selective Antagonists: Drugs of this type, e.g. yohimbine, are not used because of the unacceptable incidence of adverse effects.
Non-Selective α-Antagonists: Non-selective α-blockers, e.g. phentolamine and phenoxybenzamine, produce more postural hypotension, reflex tachycardia and adverse gastrointestinal effects (e.g. abdominal cramps, diarrhoea) than α1-selective drugs. Phentolamine 2–5 mg i.v. produces a rapid decrease in arterial pressure lasting 10–15 min and is used for the treatment of hypertensive crises.
Phenoxybenzamine binds covalently (i.e. irreversibly and non-competitively) to α-receptors so that its effects last up to several days, and may be cumulative on repeated dosing. It also reduces central sympathetic activity, which enhances vasodilatation, and has antagonist effects at 5-HT receptors. It is used for the preoperative preparation of patients with phaeochromocytoma (see Ch 37).
β-Adrenergic Receptor Antagonists




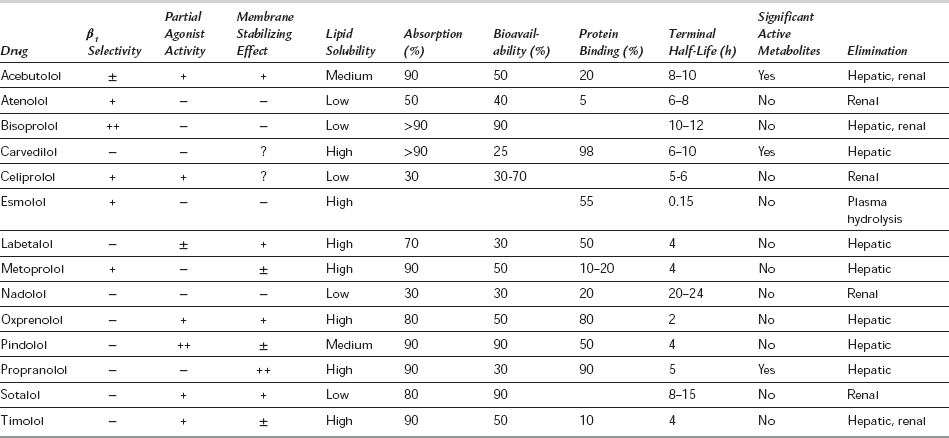
β1– or β2-Adrenergic Receptor Affinity: The relative potency of β-blockers is less important than their relative effects on the different β-receptor subtypes. Compounds are available which block preferentially either β1– or β2-receptors, although in clinical practice the β1-selective drugs are more important. The first generation of β-blockers (e.g. propranolol, timolol) were non-selective; second-generation drugs (e.g. atenolol, metoprolol, bisoprolol) are selective for β1-receptors but have no ancillary effects. The third generation of β-blockers are β1-selective, but also have effects on other receptors (e.g. labetalol and carvedilol are antagonists at α1-adrenergic receptors, and celiprolol produces vasodilatation by a mechanism involving endothelial nitric oxide). β1-Selective (or ‘cardioselective’) drugs have theoretical advantages because some of the adverse effects of β-blockers are related to β2-antagonism, but the selectivity of both drugs and tissues is only relative: all β1-selective drugs antagonize β2-receptors at higher doses, and 25% of cardiac β-receptors are of the β2 subtype. However β1-selective drugs appear to have fewer adverse effects on blood glucose control in diabetics, less effect on serum lipids and less effect on bronchial tone in patients with chronic obstructive pulmonary disease.
Partial Agonist Activity: Some β-blockers have intrinsic sympathomimetic activity (ISA), i.e. they are partial agonists. This stimulant effect is apparent at low levels of sympathetic activity, but at high levels of sympathetic discharge, blockade of endogenously released catecholamines is the major clinical effect. Partial agonists may be advantageous in patients with a low resting heart rate because they reduce the risk of AV conduction disturbance, and have theoretical advantages in patients with peripheral vascular disease or hyperlipidaemia. However, only β-blockers without partial agonist activity have been shown to be beneficial after myocardial infarction.
Membrane-Stabilizing Effect: Some β-blockers have a quinidine-like action, inhibiting Na+ transport in nerve and cardiac conducting tissue (‘membrane-stabilizing effect’). This may be demonstrated in vivo as a stabilizing effect on the cardiac action potential, reducing the slope of phase 4 noticeably, thus decreasing excitability and automaticity of the myocardium (see below). However, the membrane-stabilizing activity probably has little clinical significance because it occurs only at plasma concentrations above the therapeutic range, and the antiarrhythmic effect of β-blockade occurs via inhibition of the effects of catecholamines.
Ancillary Effects: Some of the newer β-blockers drugs have additional effects, e.g. action at α-receptors, vasodilatation, antioxidant effects and other actions. The relevance of these properties is discussed below.
Pharmacological Properties of β-Blockers: The pharmacological properties of β-blockers are summarized in Table 8.4. All are weak bases and most are well absorbed to produce peak plasma concentrations 1–3 h after oral administration. The more lipid-soluble drugs are almost completely absorbed, but are metabolized to a greater extent and tend to have a marked first-pass effect through the liver. This reduces their bioavailability, but is offset by the fact that the 4-hydroxylated metabolites so formed are also active. These active metabolites are excreted via the kidneys and may accumulate in patients with renal failure. Propranolol decreases both the clearance of amide local anaesthetics (by decreasing hepatic blood flow and inhibiting metabolism) and the pulmonary first-pass uptake of fentanyl. The first-pass metabolic pathways may also become saturated, so that proportionately higher plasma concentrations of the parent drug occur at higher oral doses. The first-pass effect is also a source of wide interindividual variation in plasma concentrations achieved from the same dose of primarily metabolized drugs, although β-blockers have a flat dose-response curve and large changes in plasma concentration may give rise to only a small change in degree of β-blockade. However, differences in individual plasma concentration–response relationships may occur, possibly as a result of variations in sympathetic tone or the formation of active metabolites. All β-blockers are distributed widely throughout the body and significant concentrations occur in the CNS, particularly for the more lipid-soluble drugs (e.g. propranolol).
Indications for β-Blockade: See Tables 8.5 and 8.6 for indications for β-blockade.
TABLE 8.5
Clinical Indications for β-Blockade
Hypertension
Ischaemic heart disease
Secondary prevention of myocardial infarction
Obstructive cardiomyopathy
Congestive heart failure
Arrhythmias
Miscellaneous
TABLE 8.6
Specific Perioperative Indications for β-Blockers
Prevention or treatment of intraoperative hypertension, tachycardia and supraventricular tachyarrhythmias associated with excessive sympathetic activity
Treatment of postoperative hypertension
Controlled hypotension
Prophylaxis or treatment of perioperative myocardial ischaemia
Pre- and perioperative management of phaeochromocytoma
Pre- and perioperative management of thyrotoxic patients




Adverse Reactions to β-Blockers: All available β-blockers have similar adverse effects, although their magnitude depends on β1-selectivity and the presence or absence of partial agonist activity. The reactions may be classified as follows.
Reactions Resulting from β-Blockade:
Cardiovascular effects: β-Blockers may precipitate heart failure in patients with poor LV function, and accentuate AV block. Their negative inotropic and chronotropic effects may be additive with other drugs affecting cardiac conduction or drugs used during anaesthesia. They prevent the compensatory tachycardia which accompanies hypovolaemia so that severe hypotension may occur if intravascular replacement is delayed. Bradycardia caused by excessive β-blockade may be treated by atropine, β-agonists (e.g. dobutamine or isoprenaline), glucagon or calcium chloride. Occasionally cardiac pacing may be required.
Induction of bronchospasm: This occurs in patients with asthma or chronic bronchitis who rely on sympathetically (β2) mediated bronchodilation. Theoretically, β1-selective drugs are less likely to aggravate bronchospasm in asthmatics, but because their selectivity is only relative, they should not be considered completely safe.
Other: Increased muscle fatigue can occur, possibly resulting from blockade of β2-mediated vasodilatation in muscles during exercise. A withdrawal phenomenon may occur after abrupt cessation of long-term therapy for angina, causing rebound tachycardia, worsening angina or precipitation of myocardial infarction. Impotence occurs commonly during chronic β-blocker therapy.
Idiosyncratic Reactions: Central nervous system effects occur with some β-blockers, including nightmares, hallucinations, insomnia and depression. These effects are more common with the lipophilic drugs (e.g. propranolol, acebutolol, oxprenolol and metoprolol). Gastrointestinal reactions include nausea, vomiting and diarrhoea.
Newer β-Blockers: Recently introduced third- generation β-blockers (e.g. labetalol, celiprolol, nevibolol and carvedilol) are mostly non-selective (β1 > β2) antagonists which also produce vasodilatation by several mechanisms. Labetalol and carvedilol are also α1-antagonists; bucindolol produces direct vasodilatation by a cAMP-dependent mechanism. The severity of some of the adverse effects of β-blockade may be less with those drugs with vasodilating properties.
Labetalol is a competitive α1-, β1– and β2-antagonist, which is a partial agonist at β2-receptors. It is four to seven times more potent at β- than at α-receptors and is useful for the prevention and treatment of perioperative hypertension, or to produce controlled hypotension (see Ch 21). It is also available as an oral preparation for the treatment of chronic hypertension or the preoperative management of phaeochromocytoma (see Ch 37). Intravenous labetalol in small increments (e.g. 5–10 mg) produces a controlled decrease in arterial pressure over 5–10 min with no change in cardiac output or reflex tachycardia, suggesting that at this dose the vasodilating action predominates. At higher doses, the β-effect becomes more prominent, with negative inotropic and chronotropic effects.
DRUGS ACTING ON THE PARASYMPATHETIC NERVOUS SYSTEM
The major drugs in use which act on the parasympathetic nervous system are muscarinic antagonists (e.g. atropine, hyoscine and propantheline), and parasympathetic agonists (e.g. the anticholinesterases neostigmine and pyridostigmine). Neuromuscular blocking drugs act at nicotinic receptors, and are described in Chapter 6.
Parasympathetic Antagonists
Parasympathetic Agonists
Anticholinesterase Drugs
Neostigmine and pyridostigmine antagonize acetylcholinesterase, thereby decreasing the breakdown of released ACh. They exert both nicotinic and muscarinic effects and are used in anaesthesia to reverse non-depolarizing neuromuscular blockade (see Ch 6), accompanied by an antimuscarinic drug to minimize the adverse vagal effects. Other anticholinesterases include edrophonium (short-acting) and pyridostigmine (long-acting), used for the diagnosis and symptomatic management of myasthenia gravis, respectively.
VASODILATORS
Vasodilators dilate arteries or veins and may reduce afterload, preload or both. Acute and chronic heart failure are both associated with a reflex increase in sympathetic tone and an increase in systemic vascular resistance. By lowering this resistance (afterload), myocardial work and oxygen requirements are reduced. Vasodilators acting on the venous side of the circulation (e.g. nitrates) increase venous capacitance, reduce venous return to the heart and so decrease left ventricular filling pressure (preload), myocardial fibre length and myocardial oxygen consumption for the same degree of cardiac work performed. They have several clinical indications (Table 8.7).
TABLE 8.7
Acute and chronic left ventricular failure
Prophylaxis and treatment of unstable and stable angina
Treatment of acute myocardial ischaemia and infarction
Chronic hypertension
Acute hypertensive episodes
Elective controlled hypotensive anaesthesia
CALCIUM CHANNEL BLOCKERS
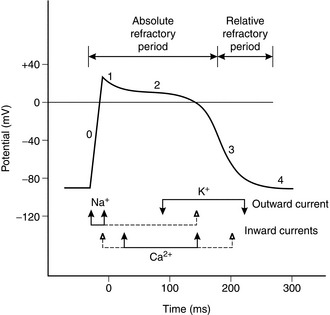
Cardiac cells in the SA and AV nodes depend on the slow inward calcium current for depolarization. CCBs which act here decrease calcium entry during phase 0 of the action potential of SA node and AV node cells, decreasing heart rate and AV node conduction. Calcium entry during phase 2 of the action potential of ventricular myocytes may be decreased (Fig. 8.5) and excitation–contraction coupling inhibited, causing decreased myocardial contractility. Some CCBs may also have favourable effects on endothelial function.
Classification
CCBs are a diverse group of compounds which have been classified in several ways, according to their structure, mechanism of action and specificity for slow calcium channels. They are classified here by their chemical structure, tissue selectivity and pharmacokinetic properties (Table 8.8).
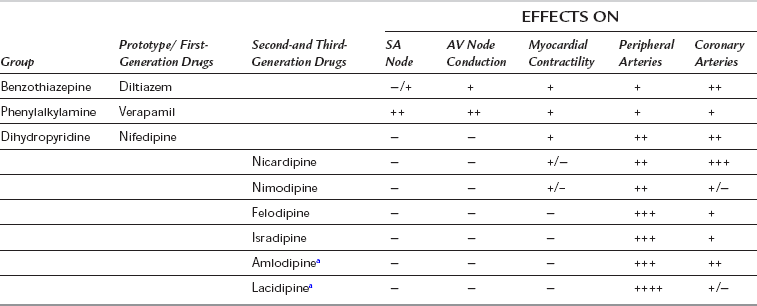
First-Generation Calcium Channel Blockers
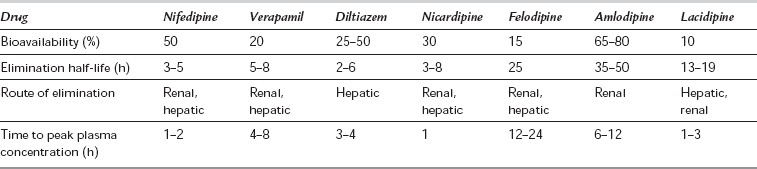
The first-generation CCBs (verapamil, diltiazem and nifedipine) have a rapid onset of action which may reduce arterial pressure acutely and produce reflex sympathetic activation. They have marked negative dromotropic and inotropic effects (especially verapamil and diltiazem). Their intrinsic duration of action is short, but slow-release formulations have been developed (see below). All are well absorbed but undergo a significant first-pass effect, leading to low bioavailability. They are highly protein-bound and metabolized extensively by hepatic demethylation and dealkylation, with wide individual pharmacokinetic variability (Table 8.9). Most CCBs possess one or more chiral centres, and the different enantiomeric forms have different pharmacokinetic and pharmacodynamic properties. For example, L-verapamil undergoes higher first-pass metabolism than the D-form, so that plasma concentrations of L-verapamil are relatively higher after intravenous administration, producing more pronounced negative inotropic and chronotropic effects.
DRUGS ACTING VIA THE RENIN–ANGIOTENSIN–ALDOSTERONE SYSTEM
The production of AT-II from angiotensinogen occurs in the walls of small blood vessels in the lungs, kidneys and other organs, and in the plasma. The rate-limiting step for this cascade is the plasma concentration of renin (Fig. 8.6). AT-II is metabolized by several peptidases to several breakdown products including angiotensin III (AT-III), which has some activity at angiotensin receptors. Four subtypes of angiotensin receptor have been defined (AT1–4). AT1-receptors are found principally in vascular smooth muscle, adrenal cortex, kidney, liver and some areas of the brain, and mediate all the known physiological functions of AT-II. AT2-receptors are present in the kidney, adrenal medulla, uterus, ovary and the brain; they may play a role in cell growth and differentiation. The roles of AT3– and AT4-receptors are unclear.
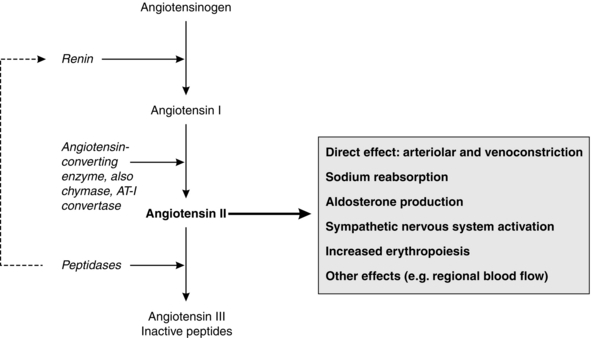
AT-II (see Fig. 8.6) is a potent vasoconstrictor (by direct action on vascular smooth muscle of arterioles and veins) and it promotes sodium reabsorption both by direct action at the proximal tubules and by stimulating aldosterone secretion. AT-II produces preglomerular vasoconstriction and efferent arteriolar vasoconstriction, and so maintains glomerular filtration rate in response to a decrease in renal blood flow. It also affects the local regulation of blood flow in other vascular beds, e.g. the splanchnic circulation, and stimulates the sympathetic nervous system via direct and indirect methods to increase noradrenaline and adrenaline release; it may also inhibit cardiac vagal activity. AT-II stimulates erythropoiesis and has direct trophic effects on vascular smooth muscle and cardiac muscle, promoting cellular proliferation, migration and hypertrophy. There is some evidence for relative downregulation of AT1-receptors and upregulation of AT2-receptors in heart failure, with AT2-receptors being responsible for some cardioprotective effects.
Drugs Acting on the Renin–Angiotensin System
The activity of the RAS may be inhibited by several mechanisms:
 Suppression of renin release or inhibition of renin activity. Sympatholytic drugs (e.g. β-blockers or central α-antagonists) directly inhibit renin secretion. Renin inhibitors competitively inhibit the reaction between renin and angiotensinogen, preventing the production of AT-II. Because renin release still occurs, the consequent reduction of plasma AT-II concentrations leads to a secondary increase in renin secretion, limiting the effect of such drugs.
Suppression of renin release or inhibition of renin activity. Sympatholytic drugs (e.g. β-blockers or central α-antagonists) directly inhibit renin secretion. Renin inhibitors competitively inhibit the reaction between renin and angiotensinogen, preventing the production of AT-II. Because renin release still occurs, the consequent reduction of plasma AT-II concentrations leads to a secondary increase in renin secretion, limiting the effect of such drugs.



ACE Inhibitors
Clinical Applications of ACE Inhibitors: ACE inhibitors are established in the treatment of hypertension and congestive heart failure. They improve left ventricular dysfunction after myocardial infarction, delay the progression of diabetic nephropathy and have a protective effect in non-diabetic chronic renal failure. ACE inhibitors improve vascular endothelial function by their effects on AT-II and bradykinin, and improve long-term cardiovascular outcome in patients with established vascular disease. ACE inhibitors have a common mechanism of action, differing in the chemical structure of their active moieties in potency, bioavailability, plasma half-life, route of elimination, distribution and affinity for tissue-bound ACE (Table 8.10). Most of the newer compounds are prodrugs, converted to an active metabolite by the liver, and have a prolonged duration of action. Most are excreted via the kidneys, and dosage should be reduced in the elderly and those with impaired renal or cardiac function. Enalapril is also available as the active drug, enalaprilat, and may be administered i.v. for the treatment of hypertensive emergencies.
TABLE 8.10
Pharmacology of ACE Inhibitors
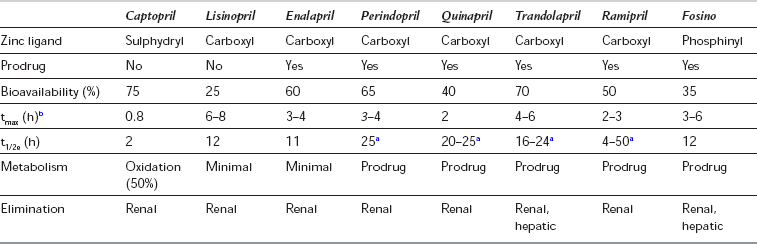
aThese drugs have polyphasic pharmacokinetics, with a dose-dependent prolonged terminal elimination phase from plasma of over 24 h.
bThe prodrugs are metabolized in the gut mucosa and liver to active compounds. Pharmacokinetic data refer to the active compounds.
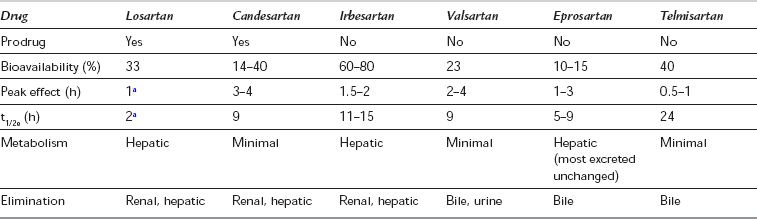
Angiotensin-II Receptor Blockers
Angiotensin-II receptor blockers (ARBs) specifically and non-competitively block the AT1-receptor, inhibiting the RAS independently and blocking any effects of AT-II resulting from compensatory stimulation of renin. Hence, they reduce afterload and increase cardiac output without causing tachycardia and are used in the treatment of hypertension, diabetic nephropathy and heart failure. As they have no effect on bradykinin metabolism or prostaglandin synthesis, ARBs do not produce the cough or rash associated with ACE inhibitors, though angio-oedema has been reported. However, plasma renin, AT-I and AT-II concentrations increase, and aldosterone concentrations decrease, during long-term therapy: hyperkalaemia may occur if potassium-sparing diuretics are also administered. Losartan is also uricosuric. All available ARBs are non-peptide imidazole compounds and are highly protein bound, with a prolonged duration of action exceeding their plasma half-life, and a maximum antihypertensive effect 2–4 weeks after starting therapy. In common with ACE inhibitors, they are contraindicated in pregnancy and are likely to have an adverse effect in patients with renal artery stenosis or those taking NSAIDs. The pharmacological properties of some ARBs are shown in Table 8.11. There are few data describing the effects of ARBs in the perioperative period, but caution would be appropriate when large fluid or blood losses are expected.
ANTIARRHYTHMIC DRUGS
Cardiac arrhythmias are irregular or abnormal heart rhythms and include bradycardias or tachycardias outside the physiological range. Patients may present for surgery with a pre-existing arrhythmia; alternatively, arrhythmias may be precipitated or accentuated during anaesthesia by several surgical, pharmacological or physiological factors (Table 8.12). Although several drugs (including anaesthetic drugs) have effects on heart rate and rhythm, the term antiarrhythmic is applied to drugs which primarily affect ionic currents within myocardial conducting tissue. Therapy for long-term arrhythmias has changed during the last two decades with the development of non- pharmacological techniques, e.g. DC cardioversion, implantable cardioverter-defibrillator (ICD) devices or radiofrequency ablation of ectopic foci. Most forms of supraventricular tachycardia may be controlled by radiofrequency ablation and ICDs are increasingly used in patients who have suffered an episode of ventricular tachycardia. Long-term drug therapy has therefore declined and is now largely confined to patients with atrial fibrillation, or as an adjunct in patients with ICDs or benign arrhythmias resistant to catheter ablation. However, owing to the frequency of arrhythmias during anaesthesia, knowledge of the available drugs and their interactions is important for the anaesthetist. Antiarrhythmic drugs are classified according to their effects on the action potential (see below).
TABLE 8.12
Precipitants of Arrhythmias During Anaesthesia
Myocardial ischaemia
Hypoxia
Hypercapnia
Halogenated hydrocarbons (volatile anaesthetic agents, e.g. trichloroethylene, cyclopropane, halothane)
Catecholamines (endogenous or exogenous)
Electrolyte abnormalities (hypo- or hyperkalaemia, hypocalcaemia, hypomagnesaemia)
Hypotension
Autonomic effects (e.g. reflex vagal stimulation, brain tumours or trauma)
Acid–base abnormalities
Mechanical stimuli (e.g. during CVP or PAFC catheter insertion)
Drugs (toxicity or adverse reactions)
Medical conditions (e.g. sepsis, myocarditis, pneumonia, alcohol abuse, thyrotoxicosis)
The Cardiac Action Potential
The cardiac action potential (AP) is generated by movement of charged ions across the cell membrane and comprises five phases (see Fig. 8.5). At rest, the cells are polarized and the resting membrane potential is negative (− 50 to − 60 mV in sinus node pacemaker cells and − 80 to − 90 mV in Purkinje, atrial and ventricular muscle fibres). The AP is triggered by a low intra-cellular leak of Na+ ions (and Ca2 + ions at the AV node) until a threshold point is reached, when sudden rapid influx of Na+ ions causes an increase in positive charge within the cell and generates an impulse (phase 0, depolarization). The AP starts to reverse (phase 1), but is sustained because of slower inward movement of Ca2 + ions (phase 2). Efflux of K+ ions brings about repolarization (phase 3) and the gradual termination of the AP. Thereafter, re-equilibration of Na+ and K+ takes place and the resting membrane potential is restored (phase 4). The AP spreads between adjacent cells and is transmitted through the specialized conducting system from the AV node to the bundle of His and ventricular muscle fibres via the Purkinje fibres. The SA node pacemaker cells have the fastest spontaneous discharge rate and usually initiate the coordinated action potential. However, action potentials may also be generated by the AV node and other cells in the conducting system.
Mechanisms of Arrhythmias
 Altered automaticity. Increased pacemaker activity in the SA node (e.g. caused by increased sympathetic tone) may cause sinus tachycardia, or atrial or ventricular tachyarrhythmias. Decreased SA node automaticity (e.g. as a result of enhanced vagal activity) may allow the emergence of latent pacemaker activity in distal conducting tissues, e.g. AV node or the bundle of His–Purkinje system, causing sinus bradycardia, AV nodal or idioventricular escape rhythms. These rhythms are common during halothane anaesthesia.
Altered automaticity. Increased pacemaker activity in the SA node (e.g. caused by increased sympathetic tone) may cause sinus tachycardia, or atrial or ventricular tachyarrhythmias. Decreased SA node automaticity (e.g. as a result of enhanced vagal activity) may allow the emergence of latent pacemaker activity in distal conducting tissues, e.g. AV node or the bundle of His–Purkinje system, causing sinus bradycardia, AV nodal or idioventricular escape rhythms. These rhythms are common during halothane anaesthesia.





Arrhythmias and Anaesthesia
Arrhythmias are common during anaesthesia and in intensive care, especially in patients with preoperative arrhythmia, cardiomyopathy, ischaemia or valvular or pericardial disease. They may be precipitated by several factors (see Table 8.13). Some arrhythmias are immediately life-threatening but all warrant attention because they usually imply the presence of other disturbances, and the effects of specific therapy (e.g. drugs, electrical cardioversion or cardiac pacing) are enhanced by prior corrective measures. Specific antiarrhythmic treatment is usually reserved for those arrhythmias affecting cardiac output or those which may progress to dangerous tachyarrhythmias.
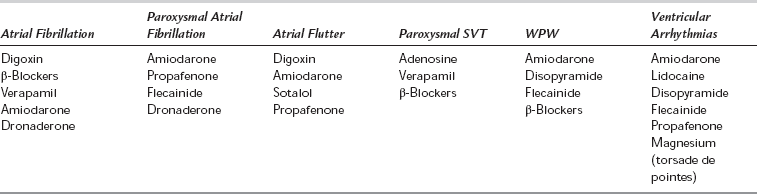
Mechanisms of Action of Antiarrhythmic Drugs
 The automaticity (tendency to spontaneous discharge) of cells may be reduced. This result can be achieved by reducing the rate of leakage of sodium (reducing the slope of phase 4), by increasing the electronegativity of the resulting membrane potential or by decreasing the electronegativity of the threshold potential.
The automaticity (tendency to spontaneous discharge) of cells may be reduced. This result can be achieved by reducing the rate of leakage of sodium (reducing the slope of phase 4), by increasing the electronegativity of the resulting membrane potential or by decreasing the electronegativity of the threshold potential.


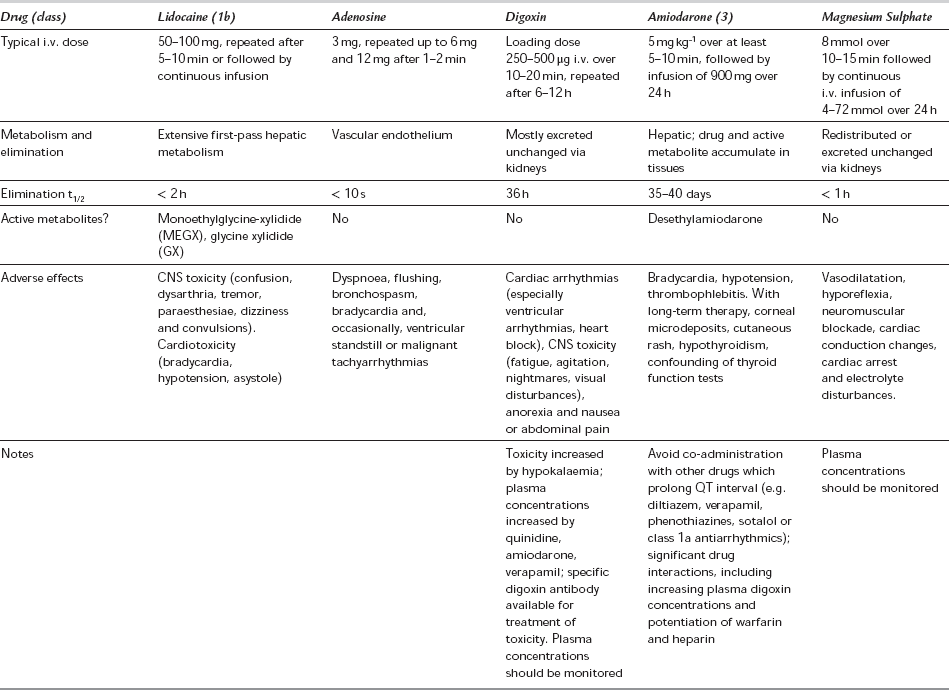
All antiarrhythmic drugs may themselves induce arrhythmias. Many (particularly class 1 antiarrhythmics) have a narrow therapeutic index and some have been associated with an increase in mortality in large-scale studies. Antiarrhythmic agents may be classified empirically on the basis of their effectiveness in supraventricular tachycardias (e.g. digoxin, β-blockers and verapamil) or in ventricular arrhythmias (lidocaine, mexiletine, magnesium, phenytoin). Many drugs (disopyramide, amiodarone, quinidine and procainamide) are effective in both supraventricular and ventricular arrhythmias (Table 8.13). The Vaughan Williams classification (Table 8.14) is based on electrophysiological mechanisms. This classification has limitations (some drugs belong to more than one class, some arrhythmias may be caused by several mechanisms, some drugs, e.g. digoxin and adenosine, do not fit into the classification) but it remains in use and is therefore described below.
TABLE 8.14
Vaughan Williams Classification of Antiarrhythmic Drugs
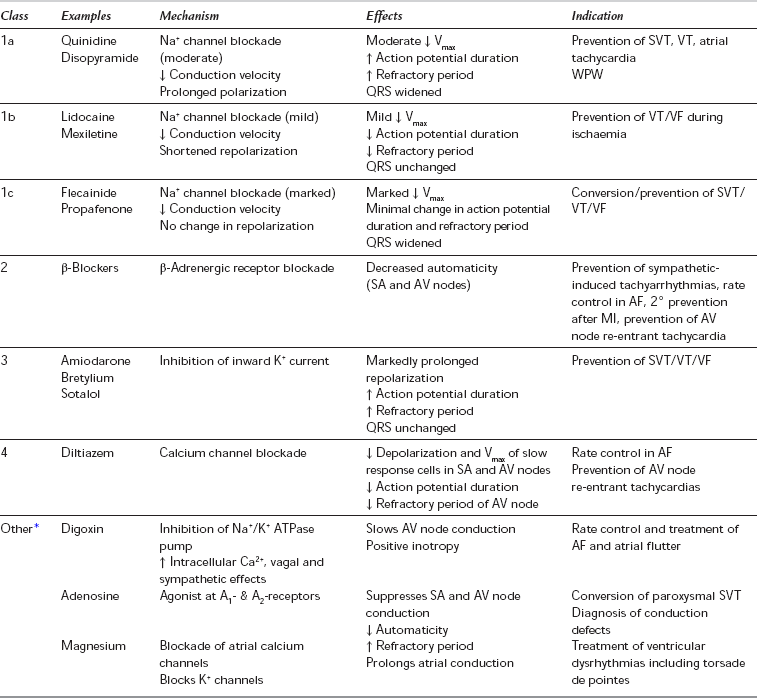
*The original Vaughan Williams classification included Classes 1-4 only. Digoxin, adenosine and other drugs which do not fit into this classification are sometimes termed ‘Class 5’ but are included here as ‘Other’.
Class 1 antiarrhythmic drugs (Table 8.15) inhibit the fast Na+ influx during depolarization; they inhibit arrhythmias caused by abnormal automaticity or re-entry. All class 1 drugs decrease the maximum rate of rise of phase 0, and decrease conduction velocity, excitability and automaticity to varying degrees. In addition to these local anaesthetic properties, some have membrane-stabilizing effects. Class 1a drugs antagonize primarily the fast influx of Na+ ions and so reduce conduction velocity through the AV node and His–Purkinje system, whilst prolonging the duration of the action potential and the refractory period. They also have varying antimuscarinic and sympathomimetic effects. Class 1b drugs have much less effect on conduction velocity in usual therapeutic doses and they shorten the refractory period. Agents in class 1c affect conduction profoundly without altering the refractory period.
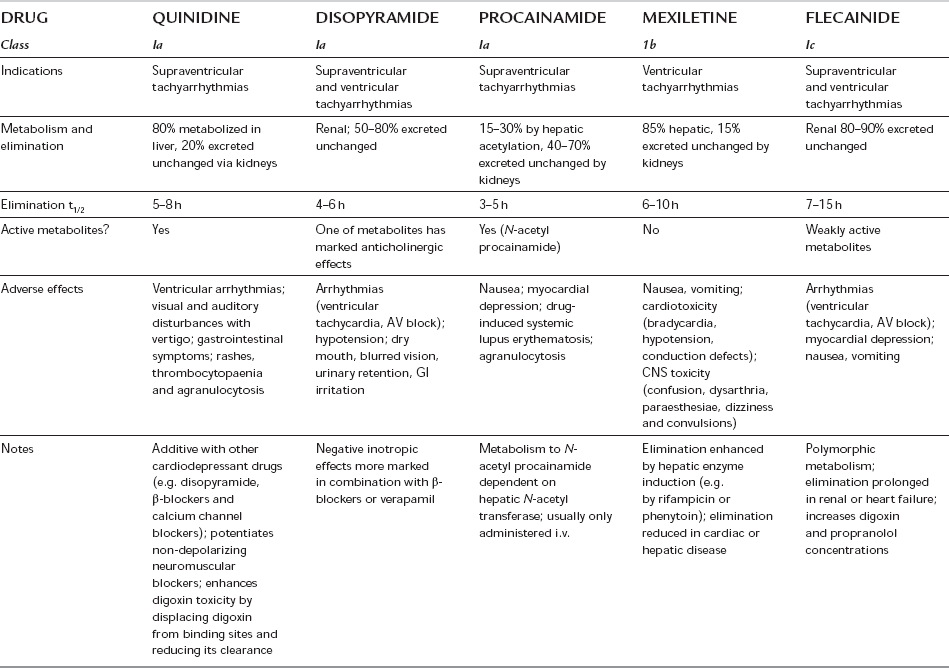
Class 1 Antiarrhythmics
Class 1a: These drugs (see Tables 8.14 and 8.15) are used for the treatment and prevention of ventricular and supraventricular arrhythmias. Their use in the prevention of atrial fibrillation has declined because of proarrhythmic effects causing increased mortality, especially in patients with ischaemic heart disease or poor LV function. They may induce torsades de pointes (a form of polymorphic ventricular tachycardia) even in patients without structural heart disease.
Class 1b: Class 1b drugs are useful for the prevention and treatment of premature ventricular contractions, ventricular tachycardia and ventricular fibrillation, particularly associated with ischaemia.
Class 1c: Class 1c drugs are used for the prevention and treatment of supraventricular and ventricular tachyarrhythmias and junctional tachycardias with or without an accessory pathway. They are proarrhythmogenic, particularly in patients with myocardial ischaemia, poor left ventricular function or after myocardial infarction, and although effective in chronic atrial fibrillation, they are reserved for life-threatening arrhythmias.
Class 3 Antiarrhythmic Drugs
Amiodarone is primarily a class 3 drug; it acts by inhibition of inward K+ current. It also blocks sodium and calcium channels, and has competitive inhibitory actions at α- and β-adrenoceptors, and may therefore be considered to have class 1, 2 and 4 antiarrhythmic activity. It prolongs AP duration, repolarization and refractory periods in the atria and ventricles. In addition, AV node conduction is markedly slowed and refractory period increased. Ventricular conduction velocity is slowed. Amiodarone is effective against a wide variety of supraventricular and ventricular arrhythmias, including WPW syndrome, and is preferred to other drugs in the presence of left ventricular dysfunction. Intravenous amiodarone is contraindicated in the presence of bradycardia or AV block but is less likely than other agents to cause arrhythmias. Bradycardia unresponsive to atropine, and hypotension, have been reported during general anaesthesia in patients receiving amiodarone therapy. Long-term oral therapy may produce a number of adverse effects. Amiodarone is an iodinated compound, which explains its effects on the thyroid (Table 8.16).
Calvey, T.N., Williams, N.E. Principles and practice of pharmacology for anaesthetists, fifth ed. Oxford: Blackwell Science; 2008.
Stoelting, R.K. Pharmacology and physiology in anesthetic practice, fourth ed. Philadelphia: Lippincott, Williams & Wilkins; 2005.
Sweetman S.C., ed. Martindale: the complete drug reference, thirty-seventh ed., London: Pharmaceutical Press, 2011.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Drugs Acting on the Cardiovascular System
THE AUTONOMIC NERVOUS SYSTEM
The term autonomic nervous system (ANS) refers to the nervous and humoral mechanisms which modify the function of the autonomous or automatic organs. These include heart rate and force of contraction, calibre of blood vessels, contraction and relaxation of smooth muscle in gut, bladder and bronchi, visual accommodation and pupillary size. Other functions include regulation of secretion from exocrine and other glands and aspects of metabolism (e.g. glycogenolysis and lipolysis) (Table 8.1). There is constant activity of both the sympathetic and parasympathetic nervous systems even at rest. This is termed sympathetic or parasympathetic tone and allows alterations in autonomic activity to produce rapid two-way regulation of physiological effect. The ANS is controlled by centres in the spinal cord, brainstem and hypothalamus, which are in turn influenced by higher centres in the cerebral and particularly the limbic cortex. The ANS is also influenced by visceral reflexes whereby afferent signals enter the autonomic ganglia, spinal cord, hypothalamus or brainstem and directly elicit appropriate reflex responses via the visceral organs. The efferent autonomic signals are transmitted through the body to two major subdivisions (separated by anatomical, physiological and pharmacological criteria), the sympathetic and the parasympathetic nervous systems.
TABLE 8.1
Effects of the Sympathetic and Parasympathetic Nervous Systems on Peripheral Effector Organs, and Receptor Subtypes Mediating these Functions (Where Known)
aMuscarinic receptors are present on vascular smooth muscle, but they are independent of parasympathetic innervation and have little or no physiological role in the control of vasomotor tone.
bSympathetic cholinergic fibres supply sweat glands and arterioles in some sites.
All postganglionic parasympathetic fibres are muscarinic (M), but in many sites the subtype has not been identified.
The Sympathetic Nervous System
The sympathetic nervous system includes nerves which originate in the spinal cord between the first thoracic and second lumbar segments (T1 to L2). Fibres leave the spinal cord with the anterior nerve roots and then branch off as white rami communicantes to synapse in the bilateral paravertebral sympathetic ganglionic chains, although some preganglionic fibres synapse instead in the paravertebral ganglia (e.g. coeliac, mesenteric and hypogastric) in the abdomen before travelling to their effector organ with the relevant arteries. Postganglionic fibres travel from paravertebral ganglia in sympathetic nerves (to supply the internal viscera, including the heart) and spinal nerves (which innervate the peripheral vasculature and sweat glands). Sympathetic nerves throughout the circulation contain vasoconstrictor fibres, particularly in the kidneys, spleen, gut and skin; however, sympathetic vasodilator fibres predominate in skeletal muscle, and coronary and cerebral vessels. Sympathetic stimulation therefore causes predominantly vasoconstriction but also a redistribution of blood flow to skeletal muscle; constriction of venous capacitance vessels may decrease their volume and thereby increase venous return. The effects of sympathetic stimulation at different receptors and effector organs are summarized in Table 8.1. The distribution of sympathetic nerve fibres to an organ or region may differ from the sensory or motor supply, according to its embryonic origin. For example, sympathetic fibres to the heart arise from T1 to T5 (but predominantly from T1 to T4), the neck is supplied by fibres from T2, the chest by fibres from T3 to T6 and the abdomen by fibres from T7 to T11.
Sympathetic Neurotransmitters
Activation of preganglionic nicotinic fibres to the adrenal medulla causes the release of adrenaline (adrenaline), which is released primarily as a circulating hormone and is only found in insignificant amounts in the nerve endings. Endogenous catecholamines (adrenaline, noradrenaline (norepinephrine) and dopamine) are synthesized from the essential amino acid phenylalanine. Their structure is based on a catechol ring (i.e. a benzene ring with -OH groups in the 3 and 4 positions), and an ethylamine side chain (Figs 8.1 and 8.2); substitutions in the side chain produce the different compounds. Dopamine may act as a precursor for both adrenaline and noradrenaline when administered exogenously (see below).
Most noradrenaline released from sympathetic nerves is taken back into the presynaptic nerve ending for storage and subsequent reuse. Re-uptake is by active transport back into the nerve terminal cytoplasm and then into cytoplasmic vesicles. This mechanism of presynaptic re-uptake, termed uptake1, is dependent on adenosine triphosphate (ATP) and Mg2 +, is enhanced by Li+ and may be blocked by cocaine and tricyclic antidepressants. Endogenous catecholamines entering the circulation by diffusion from sympathetic nerve endings or by release from the adrenal gland are metabolized rapidly by the enzymes monamine oxidase (MAO) and catechol O-methyltransferase (COMT) in the liver, kidneys, gut and many other tissues. The metabolites are conjugated before being excreted in urine as 3-methoxy-4-hydroxymandelic acid, metanephrine (from adrenaline) and normetanephrine (from noradrenaline) (Fig. 8.3). Noradrenaline taken up into the nerve terminal may also be deaminated by cytoplasmic MAO.
Adrenergic Receptor Pharmacology
These subdivisions and the functions of the autonomic nervous system are summarized in Table 8.1.
The Parasympathetic Nervous System
The parasympathetic nervous system controls vegetative functions, e.g. the digestion and absorption of nutrients, excretion of waste products and the conservation and restoration of energy. Parasympathetic neurones arise from cell bodies of the motor nuclei of cranial nerves III, VII, IX and X in the brainstem, and from the sacral segments of the spinal cord (‘the craniosacral outflow’). Preganglionic fibres run almost to the organ innervated and synapse in ganglia within the organ, giving rise to postganglionic fibres which then supply the relevant tissues. The ganglion cells may be well organized (e.g. the myenteric plexus of the intestine) or diffuse (e.g. in the bladder or vasculature). As the majority of all parasympathetic nerves are contained in branches of the vagus nerve, which innervates the viscera of the thorax and abdomen, increased parasympathetic activity is characterized by signs of vagal overactivity. Parasympathetic fibres also pass to the eye via the oculomotor (third cranial) nerve, and to the lacrimal, nasal and salivary glands via the facial (fifth) and glossopharyngeal (ninth) nerves. Fibres originating in the sacral portion of the spinal cord pass to the distal GI tract, bladder and reproductive organs. The effects of parasympathetic stimulation at different receptors and effector organs are summarized in Table 8.1
Parasympathetic Receptor Pharmacology
Parasympathetic receptors have been classified according to the actions of the alkaloids muscarine and nicotine. The actions of ACh at the postganglionic membrane are mimicked by muscarine and are termed muscarinic, whereas preganglionic transmission is termed nicotinic. ACh is also the neurotransmitter at the neuromuscular junction, via nicotinic receptor sites. Five subtypes of muscarinic receptors (M1–M5) have been characterized; all five subtypes exist in the CNS, but there are differences in their peripheral distribution and function (Table 8.2). M1-receptors are found in the stomach, where they mediate acid secretion, and in inflammatory cells in the lung (including mast cells and eosinophils) where they may have a role in airway inflammation. M2-receptors predominate in the myocardium, where they modulate heart rate and impulse conduction. Prejunctional M2-receptors are also involved in the regulation of synaptic noradrenaline and postganglionic ACh release. M3-receptors are present in classic postsynaptic sites in glandular tissue (of the GI and respiratory tract) and bronchial smooth muscle, where they mediate most of the post-junctional effects of ACh. M4-receptors have been isolated in cardiac and lung tissue in animal models and may have inhibitory effects, but the distribution and functions of M5-receptors are not yet defined. In common with adrenergic receptors, muscarinic receptors are coupled to membrane-bound G-proteins although the subtypes differ in the second messenger system with which they interact. Currently available anticholinergics probably act at all muscarinic receptor subtypes but their clinical spectra differ, which suggests that they may have differential effects at different subtypes.
DRUGS ACTING ON THE SYMPATHETIC NERVOUS SYSTEM
directly on the adrenergic receptor, e.g. the catecholamines, phenylephrine, methoxamine
indirectly causing release of noradrenaline from the adrenergic nerve ending, e.g. amphetamine
The drugs may be classified according to their structure (catecholamine/non-catecholamine), their origin (endogenous/synthetic) and their mechanism of action (via adrenergic receptors or via a non-adrenergic mechanism) (Table 8.3). Drugs which affect myocardial contractility are termed inotropes, although this term is usually applied to those drugs that increase cardiac contractility (strictly ‘positive inotropes’). Myocardial contractility may be increased by:
increasing intracellular cAMP by activation of the adenylate cyclase system (e.g. catecholamines and other drugs acting via the adrenergic receptor)
decreasing breakdown of cAMP (e.g. phosphodiesterase inhibitors)
increasing intracellular calcium availability (e.g. digoxin, calcium salts, glucagon)
increasing the response of contractile proteins to calcium (e.g. levosimendan) (Fig. 8.4).
Catecholamines
Endogenous Catecholamines
Adrenaline: Adrenaline comprises 80–90% of adrenal medullary catecholamine content and is also an important CNS neurotransmitter. It is a powerful agonist at both α- and β-adrenergic receptors, being slightly less potent than noradrenaline at α1-receptors but more potent at β-receptors. It is the treatment of choice in acute allergic (anaphylactic) reactions and is used in the management of cardiac arrest and shock, and occasionally as a bronchodilator. Except in emergency situations, i.v. injection is avoided because of the risk of inducing cardiac arrhythmias. Subcutaneous administration produces local vasoconstriction and so smoothes out its own effect by slowing absorption.
Noradrenaline: Noradrenaline acts as a potent arteriolar and venous vasoconstrictor, acting predominantly at α-receptors, with a slightly greater potency there than adrenaline. It is also an agonist at β-receptors, but β2 effects are not apparent in clinical use. Infusions of noradrenaline increase venous return, systolic and diastolic systemic and pulmonary arterial pressures, and central venous pressure. Cardiac output increases but heart rate decreases because of baroreflex activity. At higher doses, the α-mediated effects of widespread intense vasoconstriction overcome β1 effects on cardiac contractility, leading to a decrease in cardiac output at the cost of increased myocardial oxygen demand in conjunction with reductions in renal blood flow and glomerular filtration rate. Its principal use is in the management of septic shock when systemic vascular resistance is low.
Dopamine: Dopamine is the natural precursor of adrenaline and noradrenaline. It stimulates both α- and β-adrenergic receptors in addition to specific dopamine DA1-receptors in renal and mesenteric arteries. Dopamine has a direct positive inotropic action on the myocardium via β-receptors and also by release of noradrenaline from adrenergic nerve terminals. The overall effects of dopamine are highly dose-dependent. In low dosage (< 3 μg kg–1 min–1), renal and mesenteric vascular resistances are reduced by an action on DA1-receptors, resulting in increased splanchnic and renal blood flows, glomerular filtration rate and sodium excretion. At doses of 5–10 μg kg–1 min–1, the increasing direct β-mediated inotropic action predominates, increasing cardiac output and systolic pressure with little effect on diastolic pressure; peripheral resistance is usually unchanged. At doses > 15 μg kg–1 min–1, α-receptor activity predominates, with direct vasoconstriction and increased cardiac stimulation (similar to noradrenaline). Renal and splanchnic blood flows decrease, and arrhythmias may occur. Dopamine receptors are widely present in the CNS, particularly in the basal ganglia, pituitary (where they mediate prolactin secretion) and the chemoreceptor trigger zone on the floor of the fourth ventricle (where they mediate nausea and vomiting). Recently, dopamine infusions have been associated with decreased prolactin secretion, and the use of ‘prophylactic’ dopamine infusions in an attempt to preserve renal function in perioperative or critically ill patients has declined.
Synthetic Catecholamines
Isoprenaline: Isoprenaline is a potent β1– and β2-agonist, with virtually no activity at α-receptors. It acts via cardiac β1-receptors, and at β2-receptors in the smooth muscle of bronchi, the vasculature of skeletal muscle and the gut. After intravenous infusion, heart rate increases and peripheral resistance is reduced. Cardiac output may increase because of increased heart rate and contractility but effects on arterial pressure are variable. Isoprenaline also reduces coronary perfusion pressure, increases myocardial oxygen consumption and causes arrhythmias. Other β2-mediated effects include relaxation of bronchial smooth muscle and stabilization of mast cells. It has been superseded for use in the treatment of severe asthma by newer specific β2-agonists with fewer cardiac effects. Its current indication is as an infusion in the treatment of bradyarrhythmias or atrioventricular heart block associated with low cardiac output (e.g. following acute myocardial infarction) because it increases heart rate and conduction by a direct action on the subsidiary pacemaker. This indication is usually an interim measure before insertion of a temporary pacing wire.
Dobutamine: Dobutamine is primarily a β1-agonist, with moderate β2– and mild α1-agonist activity, and no action at DA-receptors. Its primary effect is an increase in cardiac output via increased contractility (β1 effect) augmented by a reduction in afterload. Heart rate also increases (β2 effect). Systolic arterial pressure may increase but peripheral resistance is reduced or unchanged. There is no direct effect on venous tone or renal blood flow but preload may decrease and urine output and sodium excretion increase as a consequence of the increased cardiac output. Dobutamine increases SA node automaticity and conduction velocity in the atria, ventricles and AV node, but to a lesser extent than isoprenaline. Dobutamine infusion produces a progressive increase in cardiac output which is greater than with comparable doses of dopamine, although arterial pressure may remain unchanged. At higher doses, tachycardia and arrhythmias may occur, but dobutamine has less effect on myocardial oxygen consumption compared with other catecholamines. Dobutamine is widely used to optimize cardiac output in septic shock, often in combination with noradrenaline. It is also used alone or in combination with vasodilator drugs in heart failure when peripheral resistance is high, and to increase heart rate and cardiac output in myocardial stress testing.
Dopexamine: Dopexamine is a synthetic dopamine analogue which is an agonist at β2– and DA1-receptors. It is also a weak DA2-agonist and it inhibits the neuronal re-uptake of noradrenaline (uptake1), but has no direct effects at β1– or α-receptors. Its principal effect is β2-agonism, producing vasodilatation in skeletal muscle; it is less potent at DA1-receptors, but a more potent β2-agonist than dopamine. It produces mild increases in heart rate, contractility and cardiac output (effects on β2-receptors and noradrenaline uptake), renal and mesenteric vasodilatation (β2 and DA1 effects), and natriuresis (DA1 effect). Coronary and cerebral blood flows are also increased. Systemic vascular resistance decreases and arterial pressure may decrease if intravascular volume is not maintained. Dopexamine has theoretical advantages in maintaining cardiac output and splanchnic blood flow in patients with systemic sepsis or heart failure. It is also used for this purpose in patients undergoing major abdominal surgery. Dopexamine also has anti-inflammatory effects (in common with other β-agonists) which are independent of its effects on gut mucosal perfusion. It is metabolized by hepatic methylation and conjugation and is eliminated mostly via the kidneys.
Non-Catecholamine Sympathomimetics
Ephedrine: Ephedrine is a naturally occurring sympathomimetic amine which is now produced synthetically. It acts directly and indirectly as an agonist at α-, β1– and β2-receptors. The indirect actions are increased endogenous noradrenaline release and inhibition of MAO. Its cardiovascular effects are similar to those of adrenaline, but the duration of action is up to 10 times longer. It causes increases in heart rate, contractility, cardiac output and arterial pressure (systolic > diastolic). It may predispose to arrhythmias. Systemic vascular resistance is usually unchanged because α-mediated vasoconstriction in some vascular beds is balanced by β-mediated vasodilatation in others, but renal and splanchnic blood flows decrease. It relaxes bronchial and other smooth muscle, and is occasionally used as a bronchodilator. It is active orally because it is not metabolized by MAO in the gut, and is useful by intramuscular injection because muscle blood flow is preserved. Ephedrine undergoes hepatic deamination and conjugation but significant amounts are excreted unchanged in urine. This accounts for its long duration of action and elimination half-life (3–6 h). Tachyphylaxis (a decreased response to repeated doses of the drug) occurs because of persistent occupation of adrenergic receptors and depletion of noradrenaline stores.
Phenylephrine: Phenylephrine is a potent synthetic direct-acting α1-agonist, which has minimal agonist effects at α2-and β-receptors. It has effects similar to those of noradrenaline, causing widespread vasoconstriction, increased arterial pressure, bradycardia (as a result of baroreflex activation) and a decrease in cardiac output. Venoconstriction predominates and diastolic pressure increases more than systolic pressure, so that coronary blood flow may increase. It is used as intermittent boluses (50-100 μg by slow injection) or an infusion (50–150 μg min− 1) to maintain arterial pressure during general or regional anaesthesia, and also topically as a nasal decongestant and mydriatic. Absorption of phenylephrine from mucous membranes may occasionally produce systemic side-effects.
Methoxamine: Methoxamine is a direct-acting α1-agonist which also has a weak β-antagonist action. It produces vasoconstriction, increased diastolic arterial pressure and decreases in cardiac output and heart rate from baroreflex activation and the mild β-blocking effect. It is no longer available in the UK.
Metaraminol: Metaraminol is a direct- and indirect-acting α- and β-agonist, which acts partly by being taken up into sympathetic nerve terminals and acting as a false transmitter for noradrenaline. Its α effects predominate, causing pronounced vasoconstriction; arterial pressure increases and a reflex bradycardia may occur.
Vasopressin: Arginine vasopressin (AVP) (formerly termed antidiuretic hormone) is a peptide hormone secreted by the hypothalamus. Its primary role is the regulation of body fluid balance. It is secreted in response to hypotension and promotes retention of water by action on specific cAMP-coupled V2-receptors. It causes vasoconstriction by stimulating V1-receptors in vascular smooth muscle and is particularly potent in hypotensive patients. It is increasingly used in the treatment of refractory vasodilatory shock which is resistant to catecholamines, although it can cause peripheral or splanchnic ischaemia. The vasopressin analogue desmopressin is used to treat diabetes insipidus. Another analogue, terlipressin, is used to limit bleeding from oesophageal varices in patients with portal hypertension as an adjunct to definitive treatment.
Phosphodiesterase Inhibitors: Phosphodiesterase inhibitors increase intracellular cAMP concentrations by inhibition of the enzyme responsible for cAMP breakdown (Fig. 8.4). Increased intracellular cAMP concentrations promote the activation of protein kinases, which lead to an increase in intracellular Ca2 +. In cardiac muscle cells, this causes a positive inotropic effect and also facilitates diastolic relaxation and cardiac filling (termed ‘positive lusitropy’). In vascular smooth muscle, increased cAMP decreases intracellular Ca2 + and causes marked vasodilatation. Several subtypes of phosphodiesterase (PDE) isoenzyme exist in different tissues. Theophylline is a non-specific PDE inhibitor, but the newer drugs (e.g. enoximone and milrinone) are selective for the PDE type III isoenzyme present in the myocardium, vascular smooth muscle and platelets. PDE III inhibitors are positive inotropes and potent arterial, coronary and venodilators. They decrease preload, afterload, pulmonary vascular resistance and pulmonary capillary wedge pressure (PCWP), and increase cardiac index. Heart rate may increase or remain unchanged. In contrast to sympathomimetics, they improve myocardial function without increasing oxygen demand or causing tachyphylaxis. Their effects are augmented by the co-administration of β1-agonists (i.e. increases in cAMP production are synergistic with decreased cAMP breakdown). They have particular advantages in patients with chronic heart failure, in whom downregulation of myocardial β-adrenergic receptors occurs, so that there is a decreased inotropic response to β-sympathomimetic drugs. A similar phenomenon occurs with advanced age, prolonged (> 72 h) catecholamine therapy and possibly with surgical stress.
Glucagon: Glucagon is a polypeptide secreted by the α cells of the pancreatic islets. Its physiological actions include stimulation of hepatic gluconeogenesis in response to hypoglycaemia, amino acids and as part of the stress response. These effects are mediated by increasing adenylate cyclase activity and intracellular cAMP, by a mechanism independent of the β-adrenergic receptor (Fig. 8.4). It increases cAMP in myocardial cells and so increases cardiac contractility. Glucagon causes nausea and vomiting, hyperglycaemia and hyperkalaemia and is not used as an inotrope except in the management of β-blocker poisoning.
Calcium: Calcium ions are involved in cellular excitation, excitation-contraction coupling and muscle contraction in cardiac, skeletal and smooth muscle cells. Increased extracellular Ca2 + increases intracellular Ca2 + concentrations and consequently the force of contraction of cardiac myocytes and vascular smooth muscle cells. Massive blood loss and replacement with large volumes of calcium-free fluids or citrated blood (which chelates Ca2 +) may cause a decrease in serum Ca2 + concentration, especially in the critically ill. Therefore, Ca2 + salts (e.g. calcium chloride or gluconate) may be administered, particularly during and after cardiopulmonary bypass. Intravenous calcium 5 mg kg–1 may increase mean arterial pressure, but the effects on cardiac output and systemic vascular resistance are variable and there is little good evidence for the efficacy of Ca2 + salts. Moreover, high Ca2 + concentrations may cause cardiac arrhythmias and vasoconstriction, may be cytotoxic and may worsen the cellular effects of ischaemia. Calcium salts may be indicated for the treatment of hypocalcaemia (ionized Ca2 + < 0.8 mmol L–1), hyperkalaemia and calcium channel blocker toxicity.
Calcium Sensitizers: Levosimendan and pimobendan are positive inotropic drugs which act by stabilizing the troponin molecule in cardiac myocytes (by a cAMP- independent mechanism) and so increase myocyte Ca2 + sensitivity without increasing Ca2 + influx. Contractility is increased without an increase in oxygen consumption or a tendency to arrhythmias. Levosimendan also causes vasodilatation by opening K+ channels via an ATP-dependent mechanism and may be used as a second line agent in acute heart failure. It is available in Europe and South America. Pimobendan also inhibits phosphodiesterase-III and has been investigated for use in chronic heart failure. It is available in Japan.
Selective β2-Agonists
Selective β2-receptor agonists (e.g. salbutamol, terbutaline, formoterol and salmeterol) relax bronchial, uterine and vascular smooth muscle while having much less effect on the heart than isoprenaline. These drugs are partial agonists (their maximal effect at β2-receptors is less than that of isoprenaline) and are only partially selective for β2-receptors. They are used widely in the treatment of bronchospasm (see Ch 9). Although less cardiotoxic than isoprenaline, dose-related tremor, tachyarrhythmias, hyperglycaemia, hypokalaemia and hypomagnesaemia may occur. β2-Agonists are resistant to metabolism by COMT and therefore have a prolonged duration of action (mostly 3–5 h). Salmeterol is highly lipophilic, has a strong affinity for the β2-adrenergic receptor, is longer acting than the other β2-agonists and so is used for maintenance therapy in chronic asthma in combination with inhaled steroids. β2-Agonists are usually administered by the inhaled (metered dose inhaler or nebulizer) or intravenous routes because of unpredictable oral absorption and a high hepatic extraction ratio. When inhaled, only 10–20% of the administered dose reaches the lower airways; this proportion is reduced further when administered via a tracheal tube. Nevertheless, systemic absorption does occur, although adverse effects are less common during long-term therapy.
Salbutamol: Salbutamol is the β2-agonist used most commonly for the prevention and treatment of bronchospasm. When administered by metered dose inhaler (1 or 2 puffs, each delivering 100 μg), it acts within a few minutes, with a peak action at 30–60 min. In severe cases, it may be given by nebulizer (2.5–5.0 mg), repeated if required, or intravenously (either 250 μg by slow i.v. injection or as an infusion starting at 5 μg min–1 and titrated to response). It is metabolized in the liver and excreted in urine both as metabolites and as unchanged drug; the proportions are dependent on the route of administration.
