[level-membership-for-critical-care-medicine-category]
110 Disorders of Water Balance
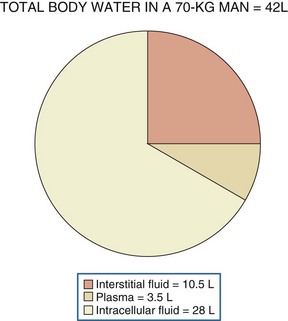
Water, the body’s most abundant constituent, accounts for approximately 50% of lean body mass in females and 60% of lean body mass in males. As shown in Figure 110-1, total body water is distributed between the intracellular compartment (two-thirds of total body water) and the extracellular compartment (one-third of total body water). The extracellular compartment is subdivided into the interstitial compartment (three-fourths of extracellular body water) and the plasma compartment (one-fourth of extracellular body water).1
The concentration of solutes in body fluids, as reflected in extracellular fluid by the serum sodium ion concentration, is tightly regulated between 138 and 142 mmol/L. This precise control is achieved by the maintenance of water balance; intake and losses are matched in a steady-state situation, despite marked fluctuations in daily solute and water intake. Water intake is determined primarily by thirst. Water excretion is controlled by the hypothalamic secretion of vasopressin (antidiuretic hormone [ADH]) and its target tissue, the renal collecting tubule. This allows for enormous flexibility because the kidney is able to dilute or concentrate urine (osmolality as low as 50 mOsm/kg H2O or as high as 1200 mOsm/kg H2O), depending on the body’s need to excrete or retain water, respectively. Under water-loading conditions, the kidney can excrete up to 20 to 25 L of urine a day. Likewise, the kidney has the ability to excrete as little as 0.5 L of urine per day (under conditions of water deprivation).1
 Control of Serum Sodium Concentration
Control of Serum Sodium Concentration
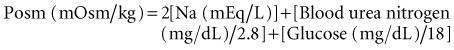
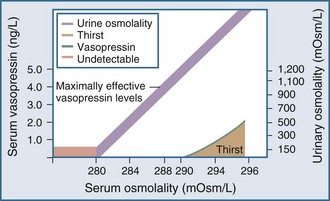
Under normal physiologic conditions, plasma osmolality is maintained between 280 and 290 mOsm/kg. Fluctuations in plasma osmolality outside this range are sensed by osmoreceptors in the hypothalamus, which is normally the primary determinant of the secretion of vasopressin, a cyclic octapeptide synthesized and secreted by supraoptic and paraventricular nuclei within the hypothalamus. The threshold for the osmotic release of vasopressin is 280 to 290 mOsm/kg, and the receptors are sensitive to changes in plasma osmolality of as little as 1% (Figure 110-2). The stimulus for vasopressin release is not limited to changes in osmolality. The primary nonosmotic stimulus for vasopressin secretion is decreased effective arterial blood volume, which can achieve a far greater rise in vasopressin levels than hyperosmolality can. Additional nonosmotic stimuli for vasopressin secretion include nausea, hypotension, and pain.
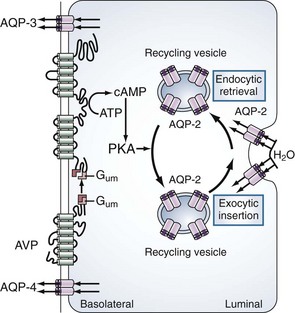
The primary site of action for vasopressin is within the principal cells of the renal collecting ducts. As illustrated in Figure 110-3, vasopressin binds to the V2 receptors on the basolateral membrane of these cells. Through a G protein–activated cascade, this results in increased insertion of a specific water (aquaporin 2) in the luminal membrane2 and renders the collecting tubule permeable to water.
Thirst also plays an important role in water balance. The most potent stimulus for thirst is hypertonicity; a change of 2% to 3% in plasma osmolality produces a strong desire to consume water. The threshold that triggers the sensation of thirst is higher than that for the release of vasopressin and usually occurs at a plasma osmolality of 290 to 295 mOsm/kg (see Figure 110-2). A decrease in effective arterial blood volume also stimulates thirst.
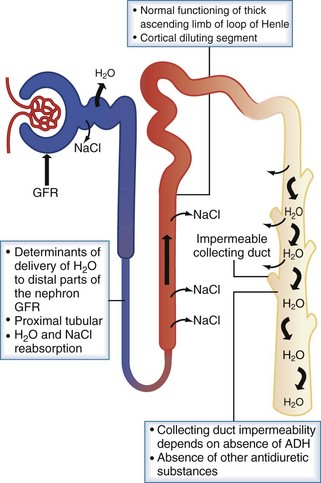
Protection against states of water excess is provided by the normally functioning renal diluting system. The three essential components of the diluting mechanism are depicted in Figure 110-4. First, because the major site of urine dilution is the water-impermeable ascending limb of the loop of Henle and the distal convoluted tubule, it is necessary to have normal delivery of tubular fluid to the distal nephron. Therefore, either a decreased glomerular filtration rate or increased proximal tubule fluid reabsorption limits the volume of dilute urine available for excretion. Second, the diluting segment of the nephron must be functioning normally. Thiazide diuretics, for example, impair the distal convoluted tubule’s ability to maximally dilute tubular fluid by blocking the thiazide-sensitive Na+/Cl− channel. Third, in order to excrete a dilute urine, vasopressin must be absent so that the collecting duct remains impermeable to water. With this diluting system intact, the kidney can handle a large load of free water (up to 1 L/h) without changes in serum sodium and thus serum osmolality.
An individual’s average daily solute load is approximately 600 mOsm. In states of low water intake, the kidney can concentrate the urine to 1200 mOsm/kg, therefore allowing for the excretion of as little as 0.5 L of urine per day. For this to occur, the renal concentrating mechanism must operate normally. The determinants of the renal concentrating mechanism are depicted in Figure 110-5. The water-impermeable thick ascending loop of Henle actively reabsorbs sodium chloride into the medullary interstitium while leaving water behind in the tubular fluid. The reabsorbed sodium increases the osmolality of the interstitium, which reaches its maximum at the papillary tip of the medulla. In the presence of vasopressin, water in the collecting duct is able to travel down its osmotic gradient and is reabsorbed. Once vasopressin is secreted, the collecting duct must be able to respond to it. Any disorder or pharmacologic agent that impairs the ability of vasopressin to act on the collecting ducts will incapacitate the renal concentrating mechanism and lead to dilute urine excretion.
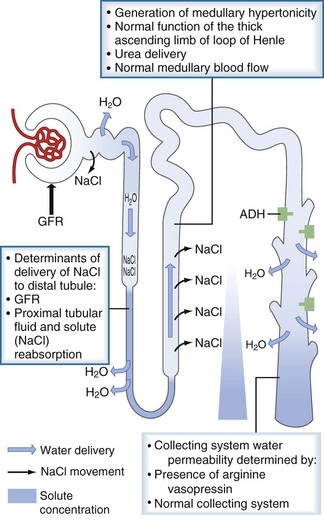
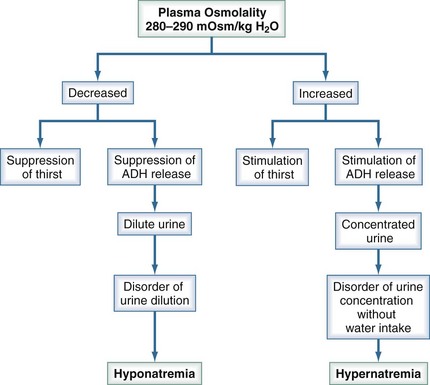
Figure 110-6 summarizes the mechanisms that maintain plasma tonicity and culminate in altered serum sodium values when impaired. These disorders arise whenever there is a disturbance in the body’s regulation of the relative amount of water to sodium. Hypernatremia results from a decrease in water relative to sodium (a water deficit state), and hyponatremia is caused by an increase in water relative to sodium (a water excess state).
 Hypernatremia
Hypernatremia
Hypernatremia is defined as a serum sodium concentration greater than 145 mEq/L. Its incidence in hospitalized patients ranges from 0.63% to 2.23%, with the elderly being more susceptible.3 Hypernatremia results in significant morbidity and mortality, ranging from 42% to 70% in adult patients. Acute elevations of serum sodium above 160 mEq/L are associated with a mortality rate of 75%, whereas mortality in chronic hypernatremia is 10%.
Hypernatremia develops whenever intake is less than the sum of extrarenal and renal water losses or, less commonly, when too much salt is introduced without adequate water intake. The primary defense mechanism against water depletion and hyperosmolarity is the renal concentrating capacity. However, even maximally concentrated urine does not prevent all water losses. Thirst also plays an important role in preventing water depletion. So long as water losses can be replaced, normal serum sodium concentration can be maintained. Most hypernatremic patients therefore have either an inability to obtain free water or an impaired thirst sensation. Both hypernatremia and hyponatremia can be assessed by the extracellular volume state: hypovolemic, isovolemic, or hypervolemic (see Figure 110-6).
Isovolemic Hypernatremia
Specific Isovolemic Hypernatremic Disorders
Central Diabetes Insipidus
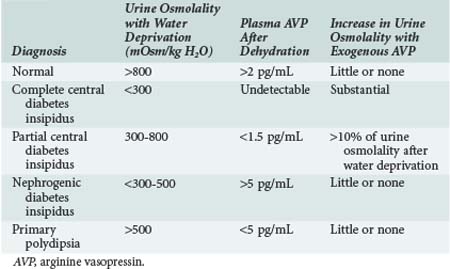
Central diabetes insipidus results from impaired secretion of vasopressin from the supraoptic and paraventricular nuclei of the hypothalamus. Known causes include congenital defects and infection, tumor, or trauma affecting the central nervous system (CNS); however, approximately 50% of cases are idiopathic (Box 110-1). Differentiating central diabetes insipidus, nephrogenic diabetes insipidus, and primary polydipsia can be a diagnostic challenge because all three present with polyuria and polydipsia. Several clinical features may assist in this effort. Central diabetes insipidus is often abrupt in onset with patients experiencing a constant need for water, whereas a compulsive water drinker often provides a more vague history of onset. Similarly, nocturia is common in patients with central diabetes insipidus but is unusual in compulsive water drinkers. The plasma osmolality is also a helpful measurement, with values above 295 mOsm/kg suggestive of central diabetes insipidus and values below 270 mOsm/kg favoring a diagnosis of compulsive water drinking. Distinguishing among the three entities is best accomplished by measuring vasopressin levels and monitoring the response to a water deprivation test followed by vasopressin administration (Table 110-1). Pituitary magnetic resonance imaging (MRI) can also be used to make the diagnosis of central diabetes insipidus. The T1-weighted images of a healthy posterior pituitary gland demonstrate a hyperintense signal, whereas this signal is absent in most patients with central diabetes insipidus (although it may be present in rare inherited forms of the condition).4
The treatment of central diabetes insipidus relies primarily on pharmacologic agents (Table 110-2). In the acute setting, aqueous vasopressin (Pitressin) is advantageous; its short duration of action makes complications such as water intoxication less likely. For a patient with chronic central diabetes insipidus, desmopressin acetate (DDAVP) is the agent of choice; it has a long half-life and can be administered intranasally (10-20 µg) every 12 to 24 hours. DDAVP does not have the strong vasoconstrictive properties of aqueous vasopressin, which must be used with caution in patients with coronary and peripheral vascular disease. In patients with partial diabetes insipidus, additional agents that increase the release of vasopressin (e.g., carbamazepine, chlorpropamide, clofibrate) can be used.
| Type of Diabetes Insipidus | Drug | Dose |
|---|---|---|
| Complete central | DDAVP | 10-20 g intranasally every 12-24 h |
| Partial central | Aqueous vasopressin | 5-10 U subcutaneously every 4-6 h |
| Chlorpropamide | 250-500 mg/d | |
| Clofibrate | 500 mg 3-4 times daily | |
| Carbamazepine | 400-600 mg/d | |
| Nephrogenic | Thiazide diuretics | |
| NSAIDs | ||
| Amiloride (for lithium-related disease) | 5 mg/d | |
| Gestational | DDAVP | As for complete central |
DDAVP, desmopressin; NSAIDs, nonsteroidal antiinflammatory drugs.
Adapted from Lanese D, Teitelbaum I. Hypernatremia. In: Jacobson HR, Striker GE, Klahr S, editors. The principles and practice of nephrology. Philadelphia: CV Mosby; 1998.
Nephrogenic Diabetes Insipidus
One form of congenital nephrogenic diabetes insipidus follows an X-linked inheritance pattern, with only males exhibiting the complete disease phenotype. Females can have a subclinical form, which suggests the presence of variable penetrance. Affected males with X-linked congenital nephrogenic diabetes insipidus have an inability to concentrate urine in the presence of vasopressin. The defect has been located on the X chromosome where the V2 receptor protein is encoded. There appear to be multiple disease-causing mutations in this area of the X chromosome; 87 such mutations in the V2 receptor were found in 106 presumably unrelated affected families.5 The autosomal recessive form of congenital nephrogenic diabetes insipidus is the result of mutations in the gene encoding for aquaporin-2 (AQP2). This form is much less common than the X-linked variety, but multiple disease-causing mutations have been described.6
Hypernatremia Management
Hypernatremia requires prompt treatment tailored to the patient’s volume status, with the end goal being restoration of serum tonicity (Figure 110-7). The rate of correction of hypernatremia depends on its rate of development and on the presence or absence of neurologic symptoms. If corrected too rapidly, water moves into the brain cells, resulting in cerebral edema. If symptoms are present and the hypernatremia is thought to be acute in onset, rapid correction over the first several hours is appropriate, with the maximum correction rate not exceeding 2 mEq/L/h. An accepted goal is to correct half the water deficit over the first 24 hours, with the remaining deficit being corrected over the next 48 hours. The serum sodium should be closely monitored during the course of treatment, with careful assessment of ongoing fluid losses.
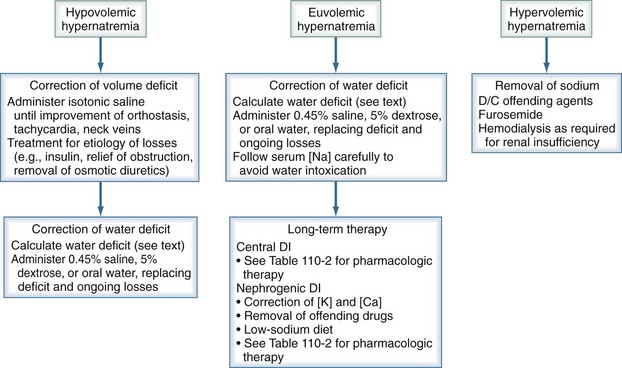
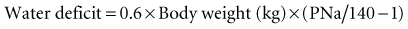
To take into account any ongoing urinary water losses, it is necessary to calculate an electrolyte-free water clearance:
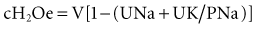
The sum total of the water deficit and ongoing urinary water losses serves as a guide to the amount and duration of water replacement, with the understanding that these calculations are not static and may need frequent adjustments. In the case of acute severe central diabetes insipidus, in addition to the preceding water replacement therapy, it may be necessary to use short-acting aqueous vasopressin (Pitressin, 5 U subcutaneously every 6 hours), depending on the response to therapy. In the chronic setting, DDAVP should be used as previously described. In patients with chronic nephrogenic diabetes insipidus, the primary intervention is treatment or removal of the underlying cause. A rare form of diabetes insipidus can occur with pregnancy when the placenta produces vasopressinase. These patients respond to treatment with DDAVP, which is not degraded by this enzyme.7
 Hyponatremia
Hyponatremia
Hyponatremia is defined as a serum sodium less than 135 mEq/L and is among the most common electrolyte disorders encountered in clinical practice.8 It is generally associated with hypo-osmolality. There are, however, clinical settings in which plasma osmolality is normal or even high (Figure 110-8).
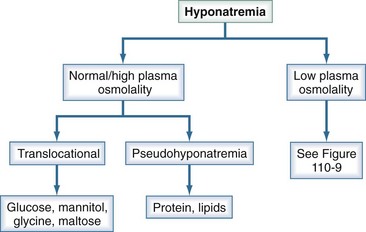
Translocational hyponatremia occurs when water moves from the intracellular space to the extracellular space in response to an osmotically active solute; as such, this does not reflect a change in total body water. In clinical practice, translocational hyponatremia is most frequently associated with hyperglycemia, which accounts for 15% of hyponatremia in hospitalized patients.9 The decrease in plasma sodium can be approximated as 1.6 mEq/L for every 100 mg/dL increase in plasma glucose concentration. However, this correction factor has been challenged in that it leads to a serious underestimation of serum sodium values in association with serum glucose concentrations over 500 mg/dL. It has been recommended that a correction factor of 2.4 be used in patients with severe hyperglycemia.10 This hyponatremia resolves with correction of the serum glucose.
Once it is established that a patient has true hypotonic hyponatremia, it is helpful to determine the patient’s volume status as previously stated. A thorough history and physical examination, supported by measurements of urinary sodium concentration, are essential in making this categorization (Figure 110-9).
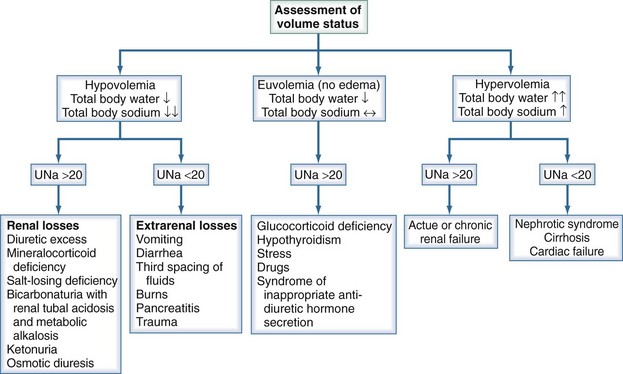
Figure 110-9 Diagnostic algorithm for hyponatremia. UNa, urinary sodium concentration.
(Adapted from Parix G, Kumar S, Beil T. Disorders of water metabolism. In: Johnson R, Feehally J, editors. Comprehensive clinical nephrology. St Louis: Mosby; 2003, p. 93.)
Hypovolemic Hyponatremia
Diuretic use is one of the more common causes of hypovolemic hyponatremia, particularly thiazide diuretics. Loop diuretics inhibit the sodium-potassium-chloride pump in the thick ascending loop of Henle, resulting in urine sodium levels above 20 mmol/L (see Figure 110-5). However, because this inhibition also interferes with generation of the hypertonic medullary interstitium, the responsiveness to vasopressin is decreased, and adequate urine dilution is still possible. In contrast, thiazide diuretics block the sodium-chloride co-transporter in the distal tubule, directly impairing the urinary diluting capacity (see Figure 110-4). Underweight women and elderly patients appear to be especially at risk for developing hyponatremia with thiazide use. Several proposed mechanisms for diuretic-induced hyponatremia, which usually occurs within 2 weeks after starting the drug, have been put forth. One is that hypovolemia causes increased vasopressin secretion, decreased delivery of fluid to the diluting segment of the nephron, and potassium depletion, resulting in increased thirst by alterations in osmoreceptor sensitivity.
Isovolemic Hyponatremia
Isovolemic hyponatremia is the most commonly encountered dysnatremia in hospitalized patients. These patients have increased total body water but no clinical signs of increased total body sodium. There are many causes of isovolemic hyponatremia (see Figure 110-9), including many pharmacologic agents (Box 110-2), hypothyroidism, and glucocorticoid deficiencies. The most common cause, however, is the syndrome of inappropriate antidiuretic hormone (SIADH) secretion. This syndrome is characterized by an impaired suppression of vasopressin secretion relative to the degree of hypotonicity. CNS disturbances, certain solid organ tumors, and human immunodeficiency virus (HIV) are some of the more notable causes, although many others exist. SIADH remains a diagnosis of exclusion, and certain criteria need be met. Essential diagnostic criteria are a plasma osmolality less than 270 mOsm/kg H2O, inappropriately concentrated urine osmolality greater than 100 mOsm/kg H2O, clinical isovolemia, elevated urine sodium concentration under conditions of normal salt and water intake, and absence of adrenal, thyroid, pituitary, or renal insufficiency or diuretic use.11
Hypervolemic Hyponatremia
Congestive heart failure, cirrhosis, nephrotic syndrome, and renal failure can all result in hypervolemic states with increased total body sodium and water. Hyponatremia occurs when the increase in total body water exceeds that of sodium. All these conditions are associated with impaired water and salt excretion (Figure 110-10).
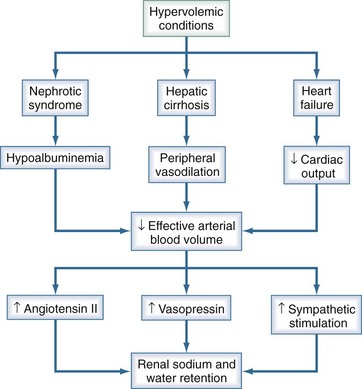
In contrast to those with congestive heart failure and cirrhosis, most patients with nephrotic syndrome have intravascular volume contraction resulting from an alteration in Starling forces from hypoalbuminemia and lowered plasma oncotic pressure. Volume contraction has been shown to stimulate vasopressin release in nephrotic subjects.12 In advanced renal failure, the water-excreting capacity of the kidney is greatly reduced. Just as edema occurs when sodium intake exceeds the excretory capacity of the diseased kidney, hyponatremia occurs when the free water intake is greater than the ability to excrete solute-free water. Even with maximum suppression of vasopressin, a patient with a glomerular filtration rate of 5 mL/min may be able to excrete only a little more than 2 liters of solute-free urine daily.9
Hyponatremia Clinical Presentation
Patients with serum sodium concentrations above 125 mmol/L are usually asymptomatic, although some patients may have nausea and vomiting. Once serum sodium concentrations go below 125 mmol/L, neuropsychiatric symptoms predominate, mostly as a result of increasing cerebral edema. These include headaches, lethargy, ataxia, psychosis, seizures, coma, and death. Severe cerebral edema resulting in tentorial herniation can also occur, more commonly with the rapid development of hyponatremia. The mortality of severe hyponatremia approaches 50% if left untreated; therefore, the presence of any signs and symptoms warrants prompt intervention.13,14
Hyponatremia Management
Certain patient populations are at increased risk of developing cerebral edema during hyponatremia (Table 110-3). Postoperative premenopausal women with hyponatremia are more likely to develop neurologic complications than are either postmenopausal women or men; thus, hypotonic fluids should not be used perioperatively in these patients. Patients on thiazide diuretics, particularly elderly women, are more susceptible to severe hyponatremia and its complications. Children, psychiatric polydipsic patients, and patients with hypoxia also seem to be at higher risk.
TABLE 110-3 Hyponatremic Patients at Risk for Neurologic Complications
| Acute Cerebral Edema | Osmotic Demyelination Syndrome |
|---|---|
| Postoperative menstruant females | Alcoholics |
| Elderly women taking thiazides | Malnourished patients |
| Children | Hypokalemic patients |
| Psychiatric polydipsic patients | Burn patients |
| Hypoxemic patients | Elderly women taking thiazides |
Certain subpopulations of patients are at greater risk of developing osmotic demyelination syndromes during treatment for hyponatremia (see Table 110-3). Susceptibility to osmotic demyelination is related to the severity and chronicity of the hyponatremia. Osmotic demyelination is rarely seen with serum sodium greater than 120 mmol/L or if the duration of hyponatremia is less than 24 to 48 hours. Severely hyponatremic patients with alcoholism, malnutrition, hypokalemia, or severe burns, as well as elderly women prescribed thiazide diuretics, appear to be at increased risk.15 Osmotic demyelination initially presents as a generalized encephalopathy associated with the rapid correction of serum sodium. The classic symptoms follow 2 to 3 days after the serum sodium is corrected; these include behavioral changes, cranial nerve palsies, and quadriplegia with a “locked-in” syndrome. MRI is diagnostic, but the typical lesions may not appear for up to 2 weeks after symptoms begin.16
The optimal treatment strategy for hyponatremia should focus on four factors: (1) presence or absence of symptoms, (2) duration of hyponatremia if known, (3) patient’s volume status, and (4) degree of hyponatremia (Figure 110-11).
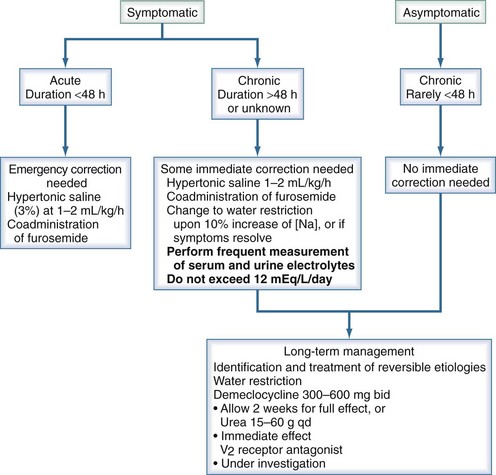
Figure 110-11 Treatment of severe (<125 mM/L) euvolemic hyponatremia.
(Adapted from Thurman JM, Halterman RK, Berl T. Theory of dysnatremic disorders. In: Brady H, Wilcox C, editors. Therapy in nephrology and hypertension. 2nd ed. Philadelphia: Saunders; 2003, p. 335-48.)
Symptomatic hyponatremia for more than 48 hours must be approached with extreme caution; these patients have the greatest risk of complications. Partial correction of serum sodium in patients with chronic symptomatic hyponatremia should proceed without delay, because failure to correct is associated with poor outcome.17 Cerebral water increases by about 10% during severe hyponatremia. With this in mind, it is safe to increase the serum sodium by 10%, followed by water restriction. This aggressive treatment should continue until either the symptoms resolve or this 10% increase is reached. Thereafter, the correction rate should be less than 0.5 mmol/L/h and should certainly not exceed 1 to 1.5 mmol/L/h or 12 mmol/L/d. To prevent overcorrection, it is important to monitor the rate and electrolyte content of infused fluids and urine output.
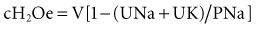
where cH2Oe is electrolyte-free water clearance, V is urine volume, UNa is urinary sodium concentration, UK is urinary potassium concentration, and PNa is serum sodium concentration. To increase serum sodium, the amount of water intake has to be less than the sum of the insensible losses and the free water excretion. This formula can be used to guide therapy as follows15:
Secondary to its pivotal role in body water regulation, vasopressin has long been considered a potential target for the treatment of hyponatremia. Several oral nonpeptide vasopressin antagonists have been introduced in recent years.18 A 2010 systematic review and meta-analysis evaluated the short-term efficacy and safety of vasopressin receptor antagonists.19 The authors concluded that vasopressin antagonists are effective and safe for the treatment of isovolemic and hypervolemic hypernatremia. Further studies are needed to elucidate their exact role in this electrolyte disorder.
Hypervolemic hyponatremia can be very difficult to treat because it is often a sign of severe underlying cardiac, hepatic, or renal disease. Water restriction is important; however, these patients often experience extreme thirst, making compliance difficult. Loop diuretics increase free water excretion and can therefore be beneficial in raising serum sodium values as well as treating edema. Thiazide diuretics should generally be avoided because they impair urinary dilution and may worsen the hyponatremia. Vasopressin receptor antagonists are also under investigation in these disorders but are not yet available for clinical use.20 A study by Wong and colleagues investigated the efficacy of the vasopressin V2 antagonist, VPA-985, in correcting hyponatremia in a group of patients including 33 with cirrhosis and 6 with congestive heart failure.21 VPA-985 produced a significant aquaresis, with significant increases in free water clearance and serum sodium levels. Unless the underlying disease process can somehow be improved, treating hyponatremia in these cases represents a significant clinical challenge.
Key Points
Rozen-Zvi B, Yahav D, Gheorghiade M, Korzets A, Leibovici L, Gafter U. Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta-analysis. Am J Kidney Dis. 2010;56:325-337. Epub 2010 Jun 9
Decaux G. Long-term treatment of patients with inappropriate secretion of antidiuretic hormone by the vasopressin receptor antagonist conivaptan, urea or furosemide. Am J Med. 2002;110:582-584.
Furst H, Hallows KR, Post J, Chen S, Kotzker W, Goldfarb S, et al. The urine/plasma electrolyte ratio: a predictive guide to water restriction. Am J Med Sci. 2000;319:240-244.
Knepper MA. Molecular physiology of urinary concentrating mechanisms: regulation of aquaporin water channels by vasopressin. Am J Physiol. 1997;272:F3-12.
Palevsky PM, Bhagrath R, Greenberg A. Hypernatremia in hospitalized patients. Ann Intern Med. 1996;124:197-203.
1 Berl T, Verbalis J. Pathophysiology of water metabolism. In Brenner B, editor: The Kidney, 7th ed, Philadelphia: WB Saunders, 2003.
2 Knepper MA. Molecular physiology of urinary concentrating mechanisms: Regulation of aquaporin water channels by vasopressin. Am J Physiol. 1997;272:F3-12.
3 Palevsky PM, Bhagrath R, Greenberg A. Hypernatremia in hospitalized patients. Ann Intern Med. 1996;124:197-203.
4 Tien R, Kucharczyk W. MR imaging of the brain in patients with diabetes insipidus. AJNR Am J Neuroradiol. 1991;12:533-542.
5 Bichet DG, Oksche A, Rosenthal W. Congenital nephrogenic diabetes insipidus. J Am Soc Nephrol. 1997;8:1951-1958.
6 Canfield MC, Tamarappoo BK, Moses AM, et al. Identification and characterization of aquaporin-2 water channel mutations causing nephrogenic diabetes insipidus with partial vasopressin response. Hum Mol Genet. 1997;6:1865-1871.
7 Durr HA, Hoggard JG, Hunt JM, Schrier RW. Diabetes insipidus in pregnancy associated with abnormally high vasopressinase activity. N Engl J Med. 1987;316:1070.
8 Anderson RJ, Chung HM, Kluge R, Schrier RW. Hyponatremia: a prospective analysis of its epidemiology and the pathogenic role of vasopressin. Ann Intern Med. 1985;102:164-168.
9 Berl T, Schrier RW. Disorders of water metabolism. In: Schrier RW, editor. Renal and Electrolyte Disorders. 6th ed. New York: Lippincott-Raven; 2000:1-71.
10 Hillier TA, Abbott RD, Borrett EJ. Hyponatremia: evaluating correction factors for hyperglycemia. Am J Med. 1999;106:399-403.
11 Verbalis J. The syndrome of inappropriate antidiuretic hormone secretion and other hypoosmolar disorders. 7th ed. Schrier RW, editor. Diseases of the Kidney and Urinary Tract. vol 3. New York: Lippincott Williams & Wilkins; 2001:2511-2548.
12 Krishna GG, Danovitch GM. Effects of water immersion on renal function in the nephritic syndrome. Kidney Int. 1982;21:385.
13 Sterns RH. The management of symptomatic hyponatremia. Semin Nephrol. 1990;10:503-514.
14 Berl T. Treating hyponatremia: Damned if we do and damned if we don’t (nephrology forum). Kidney Int. 1990;37:1006.
15 Sterns RH. Severe symptomatic hyponatremia: Treatment and outcome. A study of 64 cases. Ann Intern Med. 1987;107:656-664.
16 Laureno R, Karp BI. Myelinolysis after correction of hyponatremia. Ann Intern Med. 1997;126:57-62.
17 Ayus JC, Arieff AI. Chronic hyponatremic encephalopathy in postmenopausal women: Association of therapies with morbidity and mortality. JAMA. 1999;281:2299-2304.
18 Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet. 2008;371:162401632.
19 Rozen-Zvi B, Yahav D, Gheorghiade M, Korzets A, Leibovici L, Gafter U. Vasopressin receptor antagonists for the treatment of hyponatremia: systematic review and meta-analysis. Am J Kidney Dis. 2010;56:325-337. Epub 2010 Jun 9
20 Inoue, Ohnishi, Matsuo A, et al. Therapeutic and diagnostic potential of a vasopressin 2 antagonist for impaired water handling in cirrhosis. Clin Pharmacol Ther. 1998;63:561-570.
21 Wong F, Blei AT, Blendis LM, Thuluvath PJ. A vasopressin receptor antagonist (VPA-985) improves serum sodium concentration in patients with hyponatremia: A multi-center, randomized, placebo-controlled trial. Hepatology. 2003;37:182-191.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
110 Disorders of Water Balance
Water, the body’s most abundant constituent, accounts for approximately 50% of lean body mass in females and 60% of lean body mass in males. As shown in Figure 110-1, total body water is distributed between the intracellular compartment (two-thirds of total body water) and the extracellular compartment (one-third of total body water). The extracellular compartment is subdivided into the interstitial compartment (three-fourths of extracellular body water) and the plasma compartment (one-fourth of extracellular body water).1
The concentration of solutes in body fluids, as reflected in extracellular fluid by the serum sodium ion concentration, is tightly regulated between 138 and 142 mmol/L. This precise control is achieved by the maintenance of water balance; intake and losses are matched in a steady-state situation, despite marked fluctuations in daily solute and water intake. Water intake is determined primarily by thirst. Water excretion is controlled by the hypothalamic secretion of vasopressin (antidiuretic hormone [ADH]) and its target tissue, the renal collecting tubule. This allows for enormous flexibility because the kidney is able to dilute or concentrate urine (osmolality as low as 50 mOsm/kg H2O or as high as 1200 mOsm/kg H2O), depending on the body’s need to excrete or retain water, respectively. Under water-loading conditions, the kidney can excrete up to 20 to 25 L of urine a day. Likewise, the kidney has the ability to excrete as little as 0.5 L of urine per day (under conditions of water deprivation).1
Control of Serum Sodium Concentration
Under normal physiologic conditions, plasma osmolality is maintained between 280 and 290 mOsm/kg. Fluctuations in plasma osmolality outside this range are sensed by osmoreceptors in the hypothalamus, which is normally the primary determinant of the secretion of vasopressin, a cyclic octapeptide synthesized and secreted by supraoptic and paraventricular nuclei within the hypothalamus. The threshold for the osmotic release of vasopressin is 280 to 290 mOsm/kg, and the receptors are sensitive to changes in plasma osmolality of as little as 1% (Figure 110-2). The stimulus for vasopressin release is not limited to changes in osmolality. The primary nonosmotic stimulus for vasopressin secretion is decreased effective arterial blood volume, which can achieve a far greater rise in vasopressin levels than hyperosmolality can. Additional nonosmotic stimuli for vasopressin secretion include nausea, hypotension, and pain.
The primary site of action for vasopressin is within the principal cells of the renal collecting ducts. As illustrated in Figure 110-3, vasopressin binds to the V2 receptors on the basolateral membrane of these cells. Through a G protein–activated cascade, this results in increased insertion of a specific water (aquaporin 2) in the luminal membrane2 and renders the collecting tubule permeable to water.
Thirst also plays an important role in water balance. The most potent stimulus for thirst is hypertonicity; a change of 2% to 3% in plasma osmolality produces a strong desire to consume water. The threshold that triggers the sensation of thirst is higher than that for the release of vasopressin and usually occurs at a plasma osmolality of 290 to 295 mOsm/kg (see Figure 110-2). A decrease in effective arterial blood volume also stimulates thirst.
Protection against states of water excess is provided by the normally functioning renal diluting system. The three essential components of the diluting mechanism are depicted in Figure 110-4. First, because the major site of urine dilution is the water-impermeable ascending limb of the loop of Henle and the distal convoluted tubule, it is necessary to have normal delivery of tubular fluid to the distal nephron. Therefore, either a decreased glomerular filtration rate or increased proximal tubule fluid reabsorption limits the volume of dilute urine available for excretion. Second, the diluting segment of the nephron must be functioning normally. Thiazide diuretics, for example, impair the distal convoluted tubule’s ability to maximally dilute tubular fluid by blocking the thiazide-sensitive Na+/Cl− channel. Third, in order to excrete a dilute urine, vasopressin must be absent so that the collecting duct remains impermeable to water. With this diluting system intact, the kidney can handle a large load of free water (up to 1 L/h) without changes in serum sodium and thus serum osmolality.
An individual’s average daily solute load is approximately 600 mOsm. In states of low water intake, the kidney can concentrate the urine to 1200 mOsm/kg, therefore allowing for the excretion of as little as 0.5 L of urine per day. For this to occur, the renal concentrating mechanism must operate normally. The determinants of the renal concentrating mechanism are depicted in Figure 110-5. The water-impermeable thick ascending loop of Henle actively reabsorbs sodium chloride into the medullary interstitium while leaving water behind in the tubular fluid. The reabsorbed sodium increases the osmolality of the interstitium, which reaches its maximum at the papillary tip of the medulla. In the presence of vasopressin, water in the collecting duct is able to travel down its osmotic gradient and is reabsorbed. Once vasopressin is secreted, the collecting duct must be able to respond to it. Any disorder or pharmacologic agent that impairs the ability of vasopressin to act on the collecting ducts will incapacitate the renal concentrating mechanism and lead to dilute urine excretion.
Figure 110-6 summarizes the mechanisms that maintain plasma tonicity and culminate in altered serum sodium values when impaired. These disorders arise whenever there is a disturbance in the body’s regulation of the relative amount of water to sodium. Hypernatremia results from a decrease in water relative to sodium (a water deficit state), and hyponatremia is caused by an increase in water relative to sodium (a water excess state).
Hypernatremia
Hypernatremia is defined as a serum sodium concentration greater than 145 mEq/L. Its incidence in hospitalized patients ranges from 0.63% to 2.23%, with the elderly being more susceptible.3 Hypernatremia results in significant morbidity and mortality, ranging from 42% to 70% in adult patients. Acute elevations of serum sodium above 160 mEq/L are associated with a mortality rate of 75%, whereas mortality in chronic hypernatremia is 10%.
Hypernatremia develops whenever intake is less than the sum of extrarenal and renal water losses or, less commonly, when too much salt is introduced without adequate water intake. The primary defense mechanism against water depletion and hyperosmolarity is the renal concentrating capacity. However, even maximally concentrated urine does not prevent all water losses. Thirst also plays an important role in preventing water depletion. So long as water losses can be replaced, normal serum sodium concentration can be maintained. Most hypernatremic patients therefore have either an inability to obtain free water or an impaired thirst sensation. Both hypernatremia and hyponatremia can be assessed by the extracellular volume state: hypovolemic, isovolemic, or hypervolemic (see Figure 110-6).
Isovolemic Hypernatremia
Specific Isovolemic Hypernatremic Disorders
Central Diabetes Insipidus
Central diabetes insipidus results from impaired secretion of vasopressin from the supraoptic and paraventricular nuclei of the hypothalamus. Known causes include congenital defects and infection, tumor, or trauma affecting the central nervous system (CNS); however, approximately 50% of cases are idiopathic (Box 110-1). Differentiating central diabetes insipidus, nephrogenic diabetes insipidus, and primary polydipsia can be a diagnostic challenge because all three present with polyuria and polydipsia. Several clinical features may assist in this effort. Central diabetes insipidus is often abrupt in onset with patients experiencing a constant need for water, whereas a compulsive water drinker often provides a more vague history of onset. Similarly, nocturia is common in patients with central diabetes insipidus but is unusual in compulsive water drinkers. The plasma osmolality is also a helpful measurement, with values above 295 mOsm/kg suggestive of central diabetes insipidus and values below 270 mOsm/kg favoring a diagnosis of compulsive water drinking. Distinguishing among the three entities is best accomplished by measuring vasopressin levels and monitoring the response to a water deprivation test followed by vasopressin administration (Table 110-1). Pituitary magnetic resonance imaging (MRI) can also be used to make the diagnosis of central diabetes insipidus. The T1-weighted images of a healthy posterior pituitary gland demonstrate a hyperintense signal, whereas this signal is absent in most patients with central diabetes insipidus (although it may be present in rare inherited forms of the condition).4
The treatment of central diabetes insipidus relies primarily on pharmacologic agents (Table 110-2). In the acute setting, aqueous vasopressin (Pitressin) is advantageous; its short duration of action makes complications such as water intoxication less likely. For a patient with chronic central diabetes insipidus, desmopressin acetate (DDAVP) is the agent of choice; it has a long half-life and can be administered intranasally (10-20 µg) every 12 to 24 hours. DDAVP does not have the strong vasoconstrictive properties of aqueous vasopressin, which must be used with caution in patients with coronary and peripheral vascular disease. In patients with partial diabetes insipidus, additional agents that increase the release of vasopressin (e.g., carbamazepine, chlorpropamide, clofibrate) can be used.
| Type of Diabetes Insipidus | Drug | Dose |
|---|---|---|
| Complete central | DDAVP | 10-20 g intranasally every 12-24 h |
| Partial central | Aqueous vasopressin | 5-10 U subcutaneously every 4-6 h |
| Chlorpropamide | 250-500 mg/d | |
| Clofibrate | 500 mg 3-4 times daily | |
| Carbamazepine | 400-600 mg/d | |
| Nephrogenic | Thiazide diuretics | |
| NSAIDs | ||
| Amiloride (for lithium-related disease) | 5 mg/d | |
| Gestational | DDAVP | As for complete central |
DDAVP, desmopressin; NSAIDs, nonsteroidal antiinflammatory drugs.
[/not-level-membership-for-critical-care-medicine-category]
