[level-membership-for-otolaryngology-category]
CHAPTER 123 Disorders of the Thyroid Gland
Physiology of the Thyroid Gland
The thyroid gland produces two hormones, 3,5,3′-triiodothyronine (T3) and 3,5,3′,5′-tetraiodothyronine or thyroxine (T4). Both are iodinated derivatives of tyrosine. Hormone production depends on an external iodine supply and on intrathyroidal mechanisms for concentrating ingested iodide and then incorporating it into the tissue-specific protein, thyroglobulin. The thyroid gland is unique within the endocrine system in having a large extracellular space, the follicular lumen, which is used for storage of the hormones and their precursors. As hormone is needed by the organism, thyroglobulin is retrieved by the cell, where the biologically active hormones are released from thyroglobulin before being passed into circulation (Fig. 123-1).
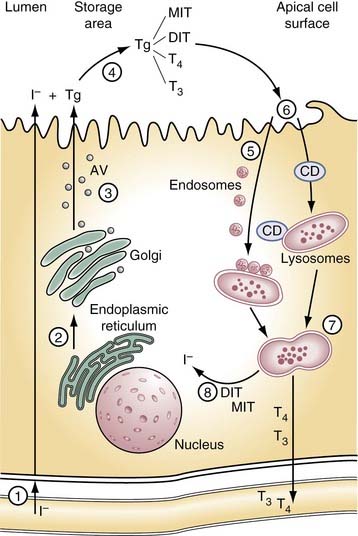
Iodide Transport
A daily dietary intake of at least 100 µg of iodine per day is required in humans to ensure adequate production of thyroid hormone. In North America, the average daily intake is higher than this, largely because of the use of iodine as a food additive.1 In many parts of the world, however, consumption is significantly below the minimum level, and iodine deficiency is the leading cause of thyroid-related disorders.
The thyroid normally concentrates iodide 20-fold to 40-fold over the extracellular space and against an electrical gradient of approximately 40 mV. Key to this trapping action is a protein located in the basal membrane of the thyroid cell known as the sodium/iodide symporter (NIS).2 NIS couples with the influx of Na+ down its electrochemical gradient with the simultaneous influx of I− up its electrochemical gradient. An Na+,K+-ATPase acts to maintain the Na+ gradient. Iodide travels down its electrochemical gradient to the apical surface of the thyrocyte, where it is incorporated into thyroglobulin. More recent evidence suggests an apical membrane protein, pendrin, aids in releasing iodide into the follicular lumen.3 Mutations in the gene and coding for this protein are responsible for the common hereditary disorder Pendred’s syndrome, which is associated with mild hypothyroidism, goiter, and hearing loss.3 Mutations in the gene coding for NIS have been identified in patients with iodide trapping defects, a rare cause of congenital hypothyroidism.2
Thyroglobulin
Thyroglobulin is essential to thyroid physiology. It is a tissue-specific protein that serves as a matrix for the synthesis of hormone and as a vehicle for its storage.4 The human thyroglobulin gene has been cloned and is located on the long arm of chromosome 8Q24. Thyroglobulin is a large dimeric glycoprotein of approximately 660 kD, consisting in humans of two identical polypeptide chains each of 2750 amino acids. About 10% of its weight is carbohydrate, and about 0.1% to 1% is iodine. Synthesis and maturation of thyroglobulin follow a pathway typical of proteins destined for secretion. The polypeptide chain is synthesized on the surface of the rough endoplasmic reticulum. As that passes through a series of intracellular compartments, it undergoes important post-translational modifications before reaching the follicular lumen.5 Carbohydrate units are added to the polypeptide chain as it is translocated into the lumen of the rough endoplasmic reticulum. Folding and dimerization of the polypeptide chain occurs within this compartment, aided by folding enzymes and a group of proteins known as molecular chaperones. Perturbations of this process result in block of protein transport beyond this point and cause congenital hypothyroidism.5 Under normal circumstances, the properly folded thyroglobulin dimers migrate to the Golgi complex, where processing of the carbohydrate units are completed. Mature but as of yet uniodinated thyroglobulin is transferred from the Golgi complex to the apical cell surface in small vesicles.
Iodination and Thyroperoxidase
Newly formed thyroglobulin and iodide meet at the apical cell surface, where hormone synthesis occurs. This process includes (1) the oxidation of iodide; (2) its subsequent transfer to thyrosyl residues on thyroglobulin, producing monoiodotyrosine (MIT) and di-iodotyrosine (DIT); and (3) coupling of two iodotyrosine molecules, either one each of MIT and DIT to form T3 or two of DIT to form T4. Thyroperoxidase (TPO), an enzyme present in the apical cell membrane, is responsible for each of these steps.6 Hydrogen peroxide (H2O2), required in the iodinating and coupling reactions, is generated at the apical membrane by a reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.7 Mutations in the TPO gene have been found in patients with congenital hypothyroidism caused by defective organification. Abnormalities in H2O2 generation seem to be more rare.
Under normal circumstances, iodide, when trapped, is rapidly incorporated into thyroglobulin so that little free iodide exists within the thyroid gland at any given time. The extent to which thyroglobulin is iodinated depends on the thyroid’s iodide supply. At a level of 0.5% iodine, the thyroglobulin dimer in humans contains on average 5 residues of MIT, 5 of DIT, 2.5 of T4, and 0.7 of T3 of a total of 132 residues of tyrosine.4
Hormone formation involves the coupling of two residues of iodotyrosine within the thyroglobulin polypeptide chain. At the hormonogenic site, the “acceptor” DIT receives the iodinated phenol ring of the “donor” iodothyrosyl (either MIT or DIT) located at some distal site on the polypeptide chain. In the process, the alanine side chain of the donor remains behind, now presumably in the form of hydroalanine. Iodination in vitro of low iodine human thyroglobulin indicates that certain thyrocele sites are favored for early iodination, and that three or four major sites exist for hormone formation. The most important hormonogenic site is located five residues from the amino terminal thyroglobulin, whereas a second major site is located three residues from the carboxy terminal. The locations of donor thyrosyls are incomplete. To date, only one has been identified in human thyroglobulin, and this resides in the amino terminal region of the molecule.4
Storage and Release of Hormone
Hormone release is initiated by the retrieval of thyroglobulin from the follicular lumen. Under stimulatory conditions in some species, this process may occur by macropinocytosis. Pseudopods form at the thyrocytes’ apical surface and engulf thyroglobulin as large colloid droplets. Under physiologic conditions in most species, however, including humans, thyroglobulin is retrieved by micropinocytosis into small vesicles. It is then passed through the endosome-lysosomal system, where the combined action of several acid proteases, including cathepsins B, D, and L, and lysosomal dipeptidase 1, releases the hormones and their iodotyrosine precursors from the polypeptide backbone.4 Evidence suggests the iodo-amino acids may be preferentially cleaved first, but ultimately thyroglobulin is broken down into amino acids or small peptides within the lysosomes.
When released from thyroglobulin, the thyroid hormones and their precursors enter the cytosol; there MIT and DIT are deiodinated by an iodotyrosine-specific deiodinase, and the released iodide re-enters the iodine pool. Some T4 is deiodinated to T3 before it is released into the circulation by 5′-iodothyronine deiodinase similar to that found in peripheral tissue.8 The mechanism by which T4 and T3 are released from the thyrocyte is unknown, but more recent evidence suggests a carrier protein may be involved.9
Circulating Thyroid Hormones
Less than 1% of circulating thyroid hormones exist as free iodo-amino acids. The remainder are bound in reversible, noncovalent linkage to one of several plasma proteins.10 In humans, the most important of these is thyroxine-binding protein (TBG), accounting for approximately 70% of circulating hormone. The TBG molecule has one hormone-binding site with a very high affinity for T4 and lower affinity for T3. A second plasma protein transthyretin accounts for approximately 10% of circulating T4 and T3. Each transthyretin molecule has two hormone-binding sites, but the affinity of the first is lower than that of TBG, and that of the second site is very low for both hormones. Albumin also serves as a thyroid hormone transport protein. Although it has low affinity, its abundance allows it to account for 10% to 20% of bound circulating hormone.
The bound hormones are in equilibrium with the minute fraction of free circulating hormone that is available for use in peripheral tissue. Under euthyroid conditions, approximately 0.2% of T4 and about 0.3% of T3 in circulation is unbound. The larger free-to-bound ratio of T3 to T4 is caused by the lower affinity of TBG for T3.11 To date, no change in thyroid state has been attributed to abnormalities in these hormone-binding proteins, despite their apparent role in thyroid function homeostasis.
Metabolism of Thyroid Hormones
T4 must first be deiodinated to T3 to exert most of its biologic actions. Because little T3 is directly synthesized on thyroglobulin, this transformation becomes an important step in hormonogenesis. Three iodothyronine deiodinases are present in mammals.8 These are membrane-bound enzymes that are closely related structurally and are distinguished by the presence of selenocysteine at their active sites. Each has distinctive substrate preferences, activity characteristics, inhibitor sensitivities, and relative tissue specificity. Type I and II deiodinases, through their combined action, are responsible for generating approximately 80% of the total T3 production.
Type I deiodinase is the primary source of circulating T3 and is found in liver, kidney, and thyroid (where it is activated by TSH) tissues, and to a lesser extent in other tissues. Type I deiodinase is positively regulated by thyroid hormones and is greatly reduced under pathophysiologic states, such as starvation and nonthyroidal illnesses. It is inhibited by the antithyroid drug propylthiouracil. Type II deiodinase is present primarily in the central nervous system, the pituitary, the placenta, and the skin, and has more recently been found in the thyroid.12 Its major role is thought to be in the local production of T3, but it may also contribute to circulating T3. In contrast to type I deiodinase, the type II enzyme is negatively regulated by thyroid hormone and is unaffected by propylthiouracil. Type III deiodinase inactivates T4 and T3 by inner ring deiodination in the five position, forming reversed T3. The enzyme is present in the adult brain, skin, and placenta, and is present in high levels in fetal tissues, where it is thought to be important in protecting developing tissue from excess levels of thyroid hormone.13
Control of Thyroid Function
The anterior pituitary is the primary internal regulator of thyroid function, influencing virtually all phases of thyroid metabolism.14 It secretes TSH, also known as thyrotropin, which is a 28- to 30-kD lipoprotein consisting of two subunits, α and luteinizing hormone. The α subunit is common to the pituitary hormones follicle-stimulating hormone and luteinizing hormone and to chorionic gonadotropin. The α subunit is unique to TSH, however, and is responsible for the binding of the hormone to its receptor in the basal membrane of the thyroid cell. On interaction with TSH, the receptor, a member of a family of G protein–coupled receptors, undergoes conformational changes that activate one or two regulatory pathways. Most TSH effects are mediated by the activation of the cyclic adenosine monophosphate (cAMP) pathway; others involve the Ca2+/phosphatidylinositol cascade. The pathway used to elicit a given effect may vary among species. TSH stimulates the efflux of iodide into the follicle and the resorption of colloid into the cell within minutes. Later effects include increased expression of the NIS, thyroglobulin, and TPO genes; stimulation of H2O2 production; promotion of glycosylation; and increased production of T3 relative to T4.
Circulating levels of TSH are controlled by the opposing influences of thyroid hormone and of thyrotropin-releasing hormone (TRH) from the hypothalamus.15 The latter is a modified tripeptide secreted to the anterior pituitary by way of the hypothalamohypophyseal portal system. TRH binds to the plasma membrane of the thyrotrope and stimulates the release of TSH and the expression of its gene. Levels of circulating TSH are under strict control by the thyroid in a classic negative feedback system. As levels of thyroid hormone increase in response to TSH stimulation, T4 and T3 block the TRH-stimulated release of TSH in the thyrotrope. The thyroid hormones also act indirectly by inhibiting TRH gene expression in the hypothalamus.
Withdrawal of iodide from the diet leads to a rapid decrease in serum T4 and an increase in serum TSH. Serum T3 levels initially are unaffected, but eventually decline with prolonged withdrawal. In response to TSH stimulation, the thyroid increases iodide uptake and organification, alters the distribution of iodo-amino acids within thyroglobulin by increasing the ratios of MIT to DIT and T3 to T4, and increases the intrathyroidal conversion of T4 to T3 by type I and II deiodinases.8 With prolonged iodine deficiency, TSH-stimulated cell proliferation eventually leads to goiter.
Antithyroid Agents
Antithyroid drugs can inhibit thyroid hormone synthesis secretion or metabolism.16 Common agents and their major actions are summarized in Table 123-1. Numerous agents used in the treatment of nonthyroidal illnesses may have profound effects on thyroid hormone production. Notable among these are the iodinated radiocontrast agents that are potent inhibitors of thyroid hormone deiodination, and can interfere with hepatic uptake of T4 and binding of T3 to nuclear receptors.17 The antiarrhythmic agent amiodarone, which is also heavily iodinated, elicits similar alterations in thyroid hormone metabolism and action. Lithium, used in the treatment of bipolar illness, is a potent inhibitor of thyroid hormone release and acts by blocking thyroglobulin endocytosis.18
| Drug | Usual Starting Dose |
|---|---|
| Propylthiouracil | 200 mg PO tid |
| Methimazole | 20 mg PO bid |
| Propranolol | 10-40 mg PO qid |
| Saturated solution of potassium iodide (SSKI) | 1-2 drops PO qd-tid |
| Compound solution of iodine (Lugol’s solution) | 2-5 drops PO qd-tid |
| Dexamethasone | 2 mg PO qid |
| Prednisone | 40-60 mg po qd |
| Ipodate | 1 g PO qd |
| Lithium | 300-450 mg PO tid |
| Perchlorate | 1 g PO qd |
| Cholestyramine | 2-4 g PO bid-qid |
| Colestipol | 5 g PO one to five times a day |
| Octreotide | 50-100 µg SC bid-tid |
| Diltiazem | 120 mg PO tid |
Thyroid Hormone Mechanism of Action
The thyroid has multiple effects on development, growth, and metabolism. The effects on development are widespread phylogenetically and can be dramatically observed during the course of amphibian metamorphosis. The appropriate levels of thyroid hormone during fetal and neonatal stages in humans are crucial for the normal maturation of the central nervous system, muscle, bone, and lung. In severe cases of thyroid hormone deficiency during this period, the syndrome of cretinism results with its associated mental retardation, deafness, mutism, and stunted growth.19 Similarly, an excess of thyroid hormone during these critical developmental periods can result in neurologic abnormalities. The metabolic effects of thyroid hormone seem to be confined to birds and mammals, presumably evolving in response to the increased metabolic pressures of thermogenesis. Oxygen consumption and the metabolism of proteins, carbohydrates, and fats all are under thyroid hormone control.
Most effects of thyroid hormone are now believed to be exerted by interactions with specific nuclear thyroid hormone receptors, resulting in the altered expression of specific genes.20 T4 has little affinity for the nuclear receptors and first must be converted to T3 to be effective. The receptors themselves belong to a large superfamily of nuclear receptors, which includes the steroid hormones, retinoic acid, and vitamin D. The thyroid hormone receptors are closely related isoforms, despite being encoded by two different genes (α and β).
Thyroid Function Studies
Circulating Thyroid Hormone Measurement
Elevated total T4 levels may also occur when there is production of endogenous antibodies to T4, especially in patients with Hashimoto’s thyroiditis or other autoimmune disorders, and occasionally in patients with Waldenström’s macroglobulinemia associated with a benign monoclonal gammopathy.21 Another condition of elevated total T4 level is peripheral resistance to thyroid hormone. Individuals with this condition may have goiter, and they may be hyperactive.22 Patients with this disorder are euthyroid. Although rarely found, this disorder has led to inappropriate treatment for hyperthyroidism.
The most widely used measurement of thyrometabolic status is measurement of serum free T4 by equilibrium dialysis.23 When measured by the dialysis method, free T4 is not affected by changes in binding protein concentrations or by nonthyroidal illness. This method is cumbersome and expensive, and it is not routinely performed. Commercial free T4 levels are most commonly measured by immunoassay techniques, but their reliability is suboptimal because they may be affected by illness or significant changes in binding proteins.24 The clinical usefulness of free T4 measurements by any method may be limited.25
Although the thyrometabolic status is best reflected by the free T4 level, from a clinical standpoint, an index or estimate of free T4 is generally adequate. The free T4 index is obtained by multiplying the serum total T4 and an indirect assessment of thyroglobulin. Serum thyroglobulin is generally estimated by one of two methods—the thyroid uptake test and the T3 uptake test.25 The thyroid uptake test is directly proportional to thyroglobulin levels in serum, whereas the T3 uptake test is inversely proportional to thyroglobulin levels.26 The result, by use of either method, is that variances in serum thyroglobulin levels are largely eliminated, and the calculated free T4 index accurately reflects actual free T4 status. Extreme changes in thyroglobulin levels, or the presence of the severe nonthyroidal illness, may result in poor correlation between calculated and measured free T4 levels.
The principles used for obtaining the serum T3 are to determine the severity of hyperthyroidism and to confirm the diagnosis of suspected thyrotoxicosis in cases in which serum T4 levels are normal or equivocal. In addition, the serum T3 may be indicated in evaluating patients with autonomously functioning thyroid adenomas, in whom so-called T3 toxicosis may be present. Such patients may have normal or borderline elevated serum T4 levels along with suppressed serum TSH levels.27
Serum Thyrotropin Measurement
Until approximately 10 years ago, virtually all clinical TSH assays were performed by radioimmunoassay. By the mid-1980s, many commercial laboratories began using more sensitive immunometric TSH methods with either monoclonal or polyclonal antibodies. Functional sensitivity of these assays represented a 10-fold improvement in sensitivity over radioimmunoassay methods. More recently, nonisotopic immunometric TSH assays have been developed with a chemiluminescent label. These newer assays have a 10-fold greater sensitivity than the early immunometric TSH assays and are 100 times more sensitive than radioimmunoassay methods. These latest TSH assays, with a sensitivity of 0.01 mU/L, are currently termed third-generation TSH assays and represent the most sensitive method for detecting TSH level.28
The clinical application of TSH detection may be summarized as follows:
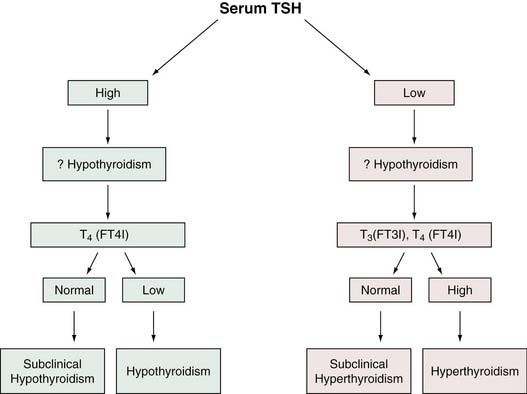
The fact that serum TSH is abnormal in hypothyroidism and hyperthyroidism would seem to make it ideally suited as a screen of thyroid status because, with rare exceptions, a normal TSH level would suggest normal thyroid hormone homeostasis. Experience in ambulatory individuals suggests that a normal TSH virtually excludes the possibility of thyroid dysfunction.34 In addition, the serum TSH level is more sensitive than the serum T4 level as a test for thyroid dysfunction because TSH can detect subclinical thyroid disorders in which serum total T4 (and T3) is usually normal. As a result of advances in TSH methodology, measurement of circulating thyroid hormones may become assigned to a second line of assessment of suspected thyroid dysfunction. Many investigators believe that the serum TSH is preferable as a screening method for thyrometabolic status.11,29 Figure 123-2 is an algorithm for the use of TSH level in the evaluation of thyroid function.
Serum Thyroglobulin Measurement
Thyroglobulin is elevated in the serum of patients with nearly all types of thyroid disorders, limiting its usefulness as a diagnostic test. Its greatest clinical value is in managing patients with well-differentiated thyroid carcinoma. An elevated or increasing thyroglobulin level (after initial surgical and ablation therapy) suggests persistence or recurrence of tumor.35 Thyroglobulin is measured by either radioimmunoassay or immunometric technique. Although antithyroid antibodies may cause interference with accurate thyroglobulin measurement in 10% of individuals, in these patients measurements of thyroglobulin and antithyroglobulin antibodies may be used concurrently to provide information regarding the tumor status.35
Thyroid Antibody Status
Circulating antithyroid antibodies, specifically antimicrosomal (AMA) and antithyroglobulin (ATA) antibodies, are usually present in patients with autoimmune thyroid disease.36 Since the introduction of immunoassay techniques, the term antithyroperoxidase (anti-TPO) has become interchangeable with AMA. AMAs are detectable in more than 90% of patients with chronic autoimmune thyroid disease; nearly 100% of patients with Hashimoto’s thyroiditis and more than 80% of patients with Graves’ disease have positive titers.37 Although ATAs are more specific than AMAs, they are less sensitive, and they are not as useful in the detection of autoimmune thyroid disease.38 Elevated levels of AMA are also frequently positive in various other organ-specific autoimmune diseases, such as lupus, rheumatoid arthritis, autoimmune anemia, Sjögren’s syndrome, type 1 diabetes mellitus, and Addison’s disease.39
Approximately 15% of adults (especially women) in the United States have elevated AMA titers.31 Prevalence of positive AMA titers increases with age, as does the incidence of primary hypothyroidism. The presence of a positive AMA titer should alert one to the possibility of hypothyroidism. Individuals with positive AMA and elevated TSH levels, even with normal serum total T4 levels (subclinical hypothyroidism), had a 3% to 5% per year likelihood of clinical hypothyroidism developing.40 In this manner, determination of AMA levels may be useful in the diagnosis of individuals with suspected autoimmune thyroid disease and in providing prognostic information when used in conjunction with TSH levels.
Thyroid-Stimulating Antibody Measurement
The immunopathogenesis of Graves’ disease was first suspected in the mid-1950s, when it was observed that injecting sera of patients with Graves’ disease into rats produced a prolonged uptake of radioactive iodine in the rat thyroid glands. The term long-acting thyroid stimulator (LATS) was coined.41 Later, LATS was characterized as a 7S immunoglobulin, and in recent years several assays have been developed for the detection of LATS, or thyroid-stimulating antibodies. Two methods are commonly used; one depends on generation of cAMP, and the other is a radioreceptor method that relies on the TSH binding inhibitory properties of the immunoglobulin. The cAMP-generating assay is termed thyroid-stimulating immunoglobulin, and it is detectable in 90% to 95% of hyperthyroid patients with Graves’ disease. The other assay detects stimulating and blocking antibodies termed TBII; it is detected in 85% of patients with hyperthyroid Graves’ disease.42 Thyroid-stimulating antibody measurements are not indicated for the routine diagnostic evaluation of suspected Graves’ disease, but they may be useful when the diagnosis of Graves’ disease is not evident.
Thyrotoxicosis
Thyrotoxicosis is a common disorder. The overall prevalence rates of overt and subclinical thyrotoxicosis gleaned from clinical surveys in the United States and Europe show an incidence of approximately 0.5 to 1 (United States) and 10 to 40 (Europe) per 1000 people.43–46 Within these ranges, the rates are generally higher in older individuals, especially women, and seem to be lower in community-based screening programs.
Thyrotoxicosis is about 10 times more common in women than in men, especially with regard to overt thyrotoxicosis. The etiology is Graves’ disease in 60% to 85% of patients, toxic nodular goiter in 10% to 30%, and toxic thyroid adenoma in 2% to 20%, with the remainder being represented by some type of thyroiditis.47,48 The frequency of toxic multinodular goiter and toxic adenoma varies the most, being higher in areas of lower iodine intake.48 Most individuals with Graves’ disease are 30 to 60 years old, whereas individuals with toxic multinodular goiter or toxic thyroid adenoma are 40 to 70 years old.49
Pathophysiology
Table 123-2 lists the causes of thyrotoxicosis. The cause of thyrotoxicosis can usually be identified with reasonable certainty by history and physical examination. The most important findings to elicit concern are the duration of symptoms, the degree and pattern of thyroid enlargement, and the presence or absence of thyroid pain and tenderness.
Graves’ Disease
Well-differentiated thyroid cancer is approximately two times more prevalent in patients with Graves’ disease than the general population.50 Well-differentiated thyroid cancers may contain TSH receptors that can be stimulated by the thyroid-stimulating immunoglobulins. These tumors associated with Graves’ disease tend to be larger and more aggressive and have more local invasion with more regional lymph node metastases than cancers occurring without Graves’ disease.51 When a palpable, hypofunctional thyroid nodule is found in a patient with Graves’ disease, it has about 45% probability of being a thyroid malignancy.51 A palpable hypofunctional nodule in a diffuse toxic goiter of Graves’ disease should be regarded with great suspicion and, if proven to be malignant, should be managed aggressively.50
An antithyroid drug and 131I are the two best treatments for patients with Graves’ thyrotoxicosis. Both methods are effective, safe, and relatively inexpensive. They represent treatments for hyperthyroidism, rather than for the autoimmune process itself, although some antithyroid drugs may also have an immunosuppressive effect. The antithyroid drugs used in the United States are methimazole and propylthiouracil (PTU). These drugs inhibit thyroid hormone biosynthesis by inhibiting the oxidation and organification of iodine and the coupling of iodotyrosines, reactions that are catalyzed by thyroid peroxidase.52 PTU also inhibits the thyroidal and extrathyroidal conversion of T4 to T3.53 Both medications are concentrated in the thyroid, and intrathyroidal concentrations, especially of methimazole, remain high for considerably longer than do serum concentrations.54
Methimazole and PTU have immunosuppressive actions that may contribute to the occurrence of remissions of Graves’ disease. Both drugs reduce the number of intrathyroidal T cells and inhibit lymphocyte function, including thyroid autoantibody production in vitro, although the latter actions require very high concentrations.55
Adverse reactions to antithyroid drugs are uncommon and probably occur with equal frequency with either methimazole or PTU. Pruritus, urticaria or other rashes, arthralgia or myalgia, and fever occur in approximately 5% of patients taking either drug.16 Both drugs may result in dysgeusia. The most dangerous adverse effect is agranulocytosis, which occurs in 0.2% or less of patients taking these medications.46 Rare adverse effects include aplastic anemia, thrombocytopenia, hepatocellular hepatitis (with PTU), cholestatic hepatitis (with methimazole), and a lupus-like vasculitis (with PTU).56,57
Inorganic iodine inhibits thyroid hormone secretion, primarily by inhibiting thyroglobulin proteolysis, and inhibits thyroidal iodine transport, oxidation, and organification.52 These actions require only a few milligrams of iodine daily, which may be administered in dosages of 5 to 10 drops of a saturated solution of potassium iodide (Lugol’s solution) several times daily. This compound is often given in preparation for thyroidectomy, for its antithyroid action and because it reduces thyroid blood flow, theoretically reducing hemorrhage at the time of surgery. Lithium carbonate shows antithyroid action similar to that of inorganic iodine and has proved effective in doses of 300 mg three or four times daily.58 Cholestyramine, when added to thionamides and β-blockers, leads to a more rapid decrease in thyroid hormone levels, especially in the first few weeks.53
β-Blockers are valuable in the management of hyperthyroidism. Independent of alteration of thyroid function, these drugs minimize many of the sympathetic overdrive symptoms found in hyperthyroidism, such as tachycardia, excessive sweating, nervousness, tremors, and hyperdynamic cardiac activity. They are contraindicated in patients with severe thyrotoxic cardiomyopathy and heart failure, but may benefit patients with atrial fibrillation and heart failure.59 β-Blockers are useful for reducing symptoms of thyrotoxicosis before and for several weeks after 131I therapy, before subtotal thyroidectomy, in thyroiditis, and in thyroid storm.
In the United States, radioactive iodine is the preferred management for most adults, and for children in cases where thionamides fail or where the patient does not respond well to these medications. The goal of 131I therapy is to reduce the amount of functioning thyroid tissue, and its efficacy is independent of whether a remission of Graves’ disease occurs. A major advantage of 131I therapy for patients with Graves’ thyrotoxicosis are that usually only a single dose is necessary, it reduces thyroid size to normal in most patients, and it is safe.60 Thionamides should be stopped for about 3 days before and after radioactive iodine therapy, which is usually effective in 2 to 4 months.
Radioactive iodine usually causes a transient exacerbation of thyrotoxicosis and rarely precipitates thyroid storm. This occurs within 1 to 2 weeks and is caused by radiation-induced thyroiditis; it is a major problem in seriously thyrotoxic or elderly patients. Thyrotoxicosis may also be exacerbated by stopping thionamides before radioactive iodine therapy. Hypothyroidism is not so much a complication of 131I treatment as an almost inevitable consequence of it. Early hypothyroidism, defined as occurring within 1 year after treatment, is caused by the acute destructive effects of 131I. Its frequency ranges from 40% to 80% in patients treated with higher doses of radioactive iodine.61 Lower doses result in early hypothyroidism less often and persistent thyrotoxicosis more often.
Surgical therapy for thyrotoxic Graves’ disease is effective and expeditious. The classic operation for surgical treatment of Graves’ disease is subtotal thyroidectomy. Performing this operation either unilaterally or bilaterally, a narrow margin of thyroid tissue in the superolateral aspect of the thyroid where the recurrent laryngeal nerve enters the larynx is preserved by dividing across thyroid tissue at that location.62 This operation provides the additional benefit of preserving the blood supply to the superior parathyroid gland on one or both sides. The intended goal of subtotal thyroidectomy is to leave 3 to 6 g of thyroid tissue, providing the benefit that patients become euthyroid without hormone replacement therapy.32 The amount of thyroid tissue remnant preserved directly affects the recurrence rate of hyperthyroidism and the development of long-term hypothyroidism.63 For patients who have larger remnants preserved, an increased incidence of recurrent hyperthyroidism is noted that can often be treated efficiently with radioactive iodine ablation because of the preserved small amount of residual thyroid tissue.64 Patients in whom more complete thyroid removal is performed are generally guaranteed to have resolution of their hyperthyroidism; however, long-term hypothyroidism would be the outcome of therapy.65
Authors favoring more complete surgical resection of thyroid tissue in the form of near-total thyroidectomy for Graves’ disease point out that long-term hypothyroidism may be easily remedied by appropriate hormone replacement therapy, whereas recurrent hyperthyroidism, as a result of leaving behind a larger than intended thyroid remnant, carries with it the need for further treatment.66 Arguments have been raised in favor of subtotal thyroidectomy as a means by which complications such as permanent hypoparathyroidism and recurrent laryngeal nerve injury may be avoided.66 The rate of permanent recurrent laryngeal nerve injury approaches zero, however, as does the rate of long-term hypocalcemia, in patients operated by experienced surgeons.67 The general recommendation for the surgical approach in patients with indications for thyroidectomy is a near-total thyroidectomy, completely eliminating the potential for recurrent or persistent hyperthyroidism.68,69
The absolute indications for surgical treatment for Graves’ disease are in patients who have significant adverse reactions to thionamide drugs and cannot be appropriately blocked before radioactive iodine administration.70 Included in this category are patients with very severe skin reactions, hepatic damage, or agranulocytosis. Other indications for surgery in patients with Graves’ disease include very large thyroid glands in excess of 75 g and neoplasia either suspected or proven within the setting of diffuse toxic nodular goiter.
Surgical therapy is relatively indicated in young women of childbearing age, who wish to attempt achieving pregnancy or who are in the process of lactating and want to continue to do so. An additional relative indication for surgery as opposed to radioactive iodine is moderate or severe ocular symptoms related to Graves’ ophthalmopathy. Ophthalmopathy may be worsened with radioactive iodine administration secondary to the development of tissue edema and worsening of the ocular symptoms.71
Patients undergoing surgery for Graves’ thyrotoxicosis require preoperative preparation so as not to induce thyrotoxicosis on induction of general anesthesia and subsequent manipulation of the thyroid intraoperatively. This is accomplished by administering antithyroid drug treatment 4 to 6 weeks and inorganic iodine treatment for 7 to 10 days preoperatively. Treatment with a β-adrenergic antagonist drug for several weeks, with or without concomitant inorganic iodide for 10 to 14 days, also has proved to be safe and effective preoperative therapy.72
Postoperative problems after thyroidectomy include wound hematoma, transient or permanent hypocalcemia, vocal paresis or paralysis, recurrent thyrotoxicosis, and transient or permanent hypothyroidism. The frequency of nonthyroid complications is low, especially in experienced surgical hands. Transient hypocalcemia may occur secondary to temporary hypoparathyroidism or the healing of thyrotoxic osteopenia. The incidence of wound hematoma is less than 1%, as is the incidence of permanent hypoparathyroidism. In addition, the risk of permanent injury to the recurrent laryngeal nerves in the setting of initial surgery is a fraction of 1%.74
Thyroiditis
The thyrotoxicosis that occurs in all forms of thyroiditis is caused by T4 and T3 release from thyroglobulin as a result of thyroid inflammation and disruption of thyroid follicles. Because the stores of thyroglobulin are limited, and the new T4 and T3 synthesis ceases, thyrotoxicosis is generally transient. Approximately half of patients with subacute or granulomatous thyroiditis have clinical manifestations of thyrotoxicosis, and a significant proportion of the remainder have high serum T4 and T3 concentrations.75 This illness is dominated by nonspecific systemic manifestations of inflammation, including fever, malaise, and myalgias. In addition, local symptoms of thyroid pain and tenderness are also noted that may be severe. Approximately 50% of the patients have a history of a recent upper respiratory tract infection preceding the illness. Any manifestations of thyrotoxicosis together with thyroid pain and tenderness are usually short-lived, lasting approximately 4 to 6 weeks or less. This inflammatory and thyrotoxic phase may be followed by transient hypothyroidism, but permanent hypothyroidism is rare. The thyroid gland is usually firm in consistency and may be quite hard. Cervical lymphadenopathy is uncommon. If pursued, thyroid radionuclide uptake scanning is generally low, and ultrasonography reveals thyroid hypoechogenicity.76
Indirect evidence suggests that subacute thyroiditis may be a viral illness, but conclusive proof is lacking. The disorder has been associated with mumps, influenza, adenovirus, and other viral infections, and small epidemics of subacute thyroiditis have been reported.77
Acute suppurative thyroiditis is most commonly caused by Staphylococcus aureus, hemolytic streptococcus, or Streptococcus pneumoniae, but occasionally is caused by other organisms, such as Fusobacterium and Haemophilus.78 This bacterial infection of the thyroid gland may be the result of trauma, hematologic seeding from a distant infected site, or direct extension from a deep cervical infection. The infection is usually localized to a single lobe, and most commonly develops an abscess cavity that may rupture through the glans capsule, extending into the mediastinum or the deep neck spaces along fascial planes. This disorder is especially common in children, in whom a prodrome of malaise is followed by the acute onset of fever, neck pain and tenderness, severe systemic symptoms, and marked leukocytosis. Referred pain to the homolateral mandible and ear may be present, and typically the child fixes the head and neck in a single position similar to torticollis. Localized tenderness over the gland and pain on head movement is commonly noted. This disorder may be difficult to distinguish from subacute nonsuppurative thyroiditis, but the pain is generally more severe, the thyroid hormone levels are generally normal, the erythrocyte sedimentation rate is normal, and the leukocyte count is high. Although the diagnosis is generally made on clinical grounds, needle aspiration of the abscess cavity establishes the bacterial organism causing the infection.
Exogenous Thyrotoxicosis
Thyrotoxicosis may occur as a result of either intentional or accidental administration of inappropriately high doses of thyroid hormone initiated by caregivers or patients themselves. Important clues to the presence of exogenous thyrotoxicosis are the absence of thyroid enlargement or the failure of thyroid enlargement to regress much if the treatment was given for this purpose. This occurs together with normal or low serum T4 concentrations if the patient is taking T3 or preparations containing T3. These patients also show low thyroid radioactive iodine uptake values and low serum thyroglobulin concentrations. Despite the ability of iodine supplementation to decrease the size of goiter and to improve thyroid function in patients living in regions of endemic goiter, it has the potential to induce thyrotoxicosis in these same patients. This usually occurs as a result of a preexisting thyroid abnormality that results in autonomous thyroid secretion—Graves’ disease or, more commonly, a nodular goiter—but insufficient iodine intake to permit excessive production of T4 and T3. Iodine-induced thyrotoxicosis may also occur in nonendemic goiter regions.79 Most of these patients have autonomously functioning thyroid tissue, such as a multinodular goiter or a thyroid adenoma, that transports iodide poorly.
Amiodarone, because it contains iodine, can cause iodine-induced thyrotoxicosis in patients with nodular goiter.80 It may also cause painless thyroiditis that is sufficiently severe to cause thyrotoxicosis, apparently because of a direct toxic effect of the drug or one of its metabolites.
Approximately 2% of patients who are treated with interferon-α have thyrotoxicosis develop, caused mostly by painless thyroiditis, but sometimes by overt Graves’ disease.81 This thyrotoxicosis is generally more subclinical than overt.
Toxic Thyroid Adenoma
Toxic uninodular goiters or toxic thyroid adenoma are autonomously functioning thyroid neoplasms.82 Among patients with these adenomas, about 20% have overt and 20% have subclinical thyrotoxicosis at the time of diagnosis.83 Although these neoplasms occur in adults of all ages and occasionally in children, most patients having thyrotoxicosis are in older age groups. Hemorrhagic infarction of a nontoxic thyroid adenoma may result in a transient thyrotoxicosis.84
Management of the toxic adenoma is required unless spontaneous infarction occurs because the resultant thyrotoxicosis is usually permanent. Definitive treatment may be obtained by either surgical resection of the adenoma through thyroid lobectomy or 131I ablative therapy. Definitive 131I therapy carries a slight to moderate risk of either transient or permanent hypothyroidism after completion of therapy, although with appropriate dosing of 131I, this risk may be minimized. Surgical resection may be carried out after 4 to 6 weeks of antithyroid drug administration and a 7- to 10-day course of inorganic iodine therapy in the form of Lugol’s solution. Complications after lobectomy are generally exceedingly rare, and the surgical resection is usually definitive, resulting in no evidence of recrudescent thyrotoxicosis or evidence of hypothyroidism.85
Toxic Multinodular Goiter
131I is generally the treatment of choice for patients with thyrotoxicosis caused by a multinodular goiter, primarily because spontaneous remission does not occur, and because surgical resection generally requires removal of most of the thyroid gland. Patients who are not candidates for radioactive iodine therapy or who refuse this modality may undergo surgery after preparation with an antithyroid medication and inorganic iodine therapy, similar to patients being treated for Graves’ disease surgically. The usual operation is either bilateral subtotal thyroidectomy or near-total thyroidectomy with preservation of a 3- to 6-g remnant of thyroid tissue. Surgery is more effective in rapidly reducing the effects of thyrotoxicosis than is 131I, and is attractive in terms of volume reduction of goiter size. Surgery may be more likely to result in long-term or permanent hypothyroidism.86
Ectopic Thyrotoxicosis
The only recognized etiologies of thyrotoxicosis secondary to excessive ectopic thyroid hormone secretion are dermoid tumors and teratomas of the ovary. Most of the uncommon patients with substantial amounts of thyroid tissue in their tumors (struma ovarii) who have thyrotoxicosis also have Graves’ disease or a multinodular goiter.87 They have one of the common causes of thyrotoxicosis, affecting the thyroid gland and the ectopic thyroid tissue within the ovarian tumor. In the absence of a functional thyroid gland because of surgical removal or radioactive iodine ablation, the ovarian tumor may be the only source of excessive thyroid hormone in these patients when it contains a toxic thyroid adenoma.88
Special Situations in Thyrotoxicosis
Subclinical Thyrotoxicosis
Subclinical thyrotoxicosis is characterized chemically by a normal serum T4 and T3 with low TSH concentrations. Most patients with subclinical thyrotoxicosis are asymptomatic, but a few may have nonspecific symptoms or physical signs compatible with overt thyrotoxicosis. The course of this disorder generally varies, with some patients showing resolution within weeks to years, others maintaining a state of subclinical thyrotoxicosis, and a smaller percentage (approximately 10%) showing the development of overt thyrotoxicosis.89 There seems to be some increased risk of progression to overt thyrotoxicosis in patients who also have thyroid adenomas, multinodular goiters, or history of Graves’ disease.90,91
Thyroid Storm
The index clinical findings in patients with thyroid storm are fever greater than 38.5°C, tachycardia, and generally some type of central nervous system dysfunction. Central nervous system abnormalities include anxiety; agitation and delirium; possibly acute psychosis or seizures; and, as a terminal event, coma. Severe cardiovascular effects, such as congestive heart failure or atrial fibrillation, may also be present.92
Determinative laboratory abnormalities in patients with thyroid storm are generally not found. Serum T4 and T3 concentrations may be high, but no more so than in ordinary thyrotoxicosis. Serum free T4 and T3 concentrations may be more elevated than in less ill patients with thyrotoxicosis.93
Hypothyroidism
Prevalence
Hypothyroidism affects women fourfold to sixfold more often than men and increases with advancing age. The National Health and Nutrition Examination Survey (NHANES III), a sample of 17,353 individuals 12 years old and older representing the geographic and ethnic distribution of the U.S. population from 1988 to 1994, reported a prevalence of clinical hypothyroidism at 0.3% and subclinical hypothyroidism at 4.3%.94 Thyroid peroxidase antibodies were elevated in 11.3%, and thyroglobulin antibodies were elevated in 10.4%. Thyroid peroxidase antibody positivity was associated with hypothyroidism (and hyperthyroidism), but thyroglobulin antibodies were not.
Etiology
Hypothyroidism can be classified (in order of decreasing frequency) as thyroid (primary), pituitary (secondary), or hypothalamic (tertiary) failure and thyroid hormone receptor resistance. Causes of primary hypothyroidism are listed in Box 123-1. Worldwide, the most common cause of hypothyroidism is iodine deficiency. In iodine-sufficient areas, such as the United States, the most common cause is chronic autoimmune (Hashimoto’s) thyroiditis. With current administered doses of 131I for patients with Graves’ disease, approximately 90% become hypothyroid by the first year.95 External neck irradiation in a cohort of 1677 patients with Hodgkin’s disease followed for a mean of 9.9 years was associated with a cumulative incidence of hypothyroidism of 30.6%, highlighting the importance of continued clinical and biochemical evaluation.96
Box 123-1 Causes of Primary (Thyroidal) Hypothyroidism
Modified from Braverman LE, Utiger RD. Introduction to hypothyroidism. In: Braverman LE, Utiger RD, eds. Werner and Ingbar’s the Thyroid: A Fundamental and Clinical Text. 7th ed. Philadelphia: Lippincott-Raven; 1996:736.
Clinical Features
The severity of clinical features depends on the severity of thyroid hormone deficiency, rather than etiology. Individuals with mild hypothyroidism with elevated TSH but normal free T4 (subclinical hypothyroidism) may have few or no symptoms. At the opposite extreme, individuals with severe hypothyroidism may have myxedema coma. Even in individuals with overt biochemical hypothyroidism, severity of symptoms varies. Generally, patients are more symptomatic if hypothyroidism develops rapidly. Elderly patients have fewer symptoms than younger patients.4 Common symptoms of hypothyroidism, such as fatigue, constipation, dry skin, and cold intolerance, may be mistakenly misinterpreted as part of the normal aging process.
Hypothyroidism should be suspected in individuals with goiter and risk factors (Box 123-2). With the widespread use of the serum TSH assay, hypothyroidism is frequently detected at an earlier stage. The classic symptoms and signs of hypothyroidism are now less frequently found (Box 123-3).
Otolaryngologic Manifestations
Hearing Loss
Hearing loss may be conductive, mixed, or sensorineural in origin. It occurs more frequently and with greater severity in congenital than in adult hypothyroidism.97 Progressive mixed hearing loss is reported in one half to nearly all children with endemic cretinism,98 but only approximately 30% to 40% of adults with myxedema have bilateral sensorineural hearing loss. Substantial deafness persists after T4 therapy in 10% of children with congenital hypothyroidism.99 Although it mainly occurs in primary hypothyroidism, deafness has been reported with panhypopituitarism.100
Children with cretinism may have anomalous ossicles involving any bone in the middle ear, and may have atrophy of the organ of Corti.101 The tectorial membrane is the first structure to change, followed by degeneration of hair cells at the basal turn of the cochlea, with prolongation of wave I; outer hair cells remain intact.102 Patients with acquired hypothyroidism who have hearing loss may display similar abnormalities. Only a few adults and almost no children with a well-established hearing loss improve with thyroid hormone therapy.
Vertigo
Vertigo is experienced in two thirds of patients with hypothyroidism. Attacks are usually mild and brief, and are not associated with electronystagmography changes or concurrent hearing loss.103
Hoarseness
Gradual and progressive hoarseness occurs in hypothyroidism as a result of mucopolysaccharide infiltration of the vocal cords and possibly tissue edema in the ambiguous nucleus or the cricothyroid muscles.104 Finding bilaterally edematous, mobile vocal cords should raise the suspicion of hypothyroidism. Hoarseness almost invariably dissipates with thyroid hormone replacement alone.
Goitrous Hypothyroidism
The most common cause of goitrous hypothyroidism in adults in the United States is autoimmune thyroiditis (Hashimoto’s disease).105 Other less common causes are drugs (lithium, amiodarone, sulfisoxazole, large doses of iodides, aminosalicylic acid, interferon, and antithyroid drugs), infiltration of the gland with tumor or inflammatory processes, and familial defects in thyroid hormonogenesis.
Transient Hypothyroidism
Hypothyroidism resulting from Hashimoto’s thyroiditis is transient in approximately 10% of cases. Spontaneous remission is associated with the presence of a larger goiter, a high initial TSH level, and a family history of thyroid disease.106 Autoimmune thyroid dysfunction may become apparent after surgery for Cushing’s disease.107 Smoking increases the metabolic effects of overt and subclinical hypothyroidism in a dose-dependent way.108
Excessive Iodine Intake
In iodine-sufficient areas of the world such as the United States, excess iodine intake can cause hypothyroidism in individuals with autoimmune thyroiditis, 131I-treated or surgically treated Graves’ disease patients, and patients treated with hemithyroidectomy for thyroid nodules.46 Hypothyroidism may develop in individuals taking amiodarone, especially individuals with an underlying thyroid abnormality. Thyroid autoantibodies are risk factors for the development of hypothyroidism.109
Endemic Goiter
Endemic goiter is uncommon in the United States, but TSH levels are elevated in more than 50% of patients with this disorder, many of whom have no clinical features of thyroid failure.110
Familial Hypothyroidism
Kindreds with hypothyroidism usually have inherited defects in hormonogenesis, but rarely may have generalized thyroid hormone resistance.111
Nongoitrous Hypothyroidism
Nongoitrous hypothyroidism is most often caused by thyroid disease—most commonly autoimmune diffuse thyroid atrophy and management of Graves’ disease with 131I, thionamides, or thyroidectomy—but may be caused by pituitary and hypothalamic disorders.112,113
Hypothyroidism after Laryngectomy and Radiotherapy
Hypothyroidism may start within 4 months of surgery, but may not become clinically apparent for 1 year.114 In a multivariate analysis of 221 patients, risk factors for hypothyroidism were high radiation dose, combination of radiotherapy and cervical surgery, time from therapy, and no shielding of the midline neck. Patients receiving irradiation to the neck—particularly patients undergoing neck dissections or total laryngectomy—should have routine thyroid function studies performed every 3 to 6 months the first year after management and annually thereafter.
Subclinical Hypothyroidism
Prevalence
In population-based studies, the prevalence of subclinical hypothyroidism is approximately 8% in women and 3% in men, higher in whites (versus blacks), and in individuals older than 75 years (versus 55 to 64 years).115,116 NHANES III reported that in the 16,533 participants who reported no known thyroid disease, goiter, or thyroid hormone use, 4.3% had subclinical hypothyroidism.94
Natural History
Progression from subclinical to overt hypothyroidism is not inevitable in all individuals. In a large population study in Great Britain followed for more than 20 years, women with an elevated TSH and elevated antithyroid antibody titers progressed to overt hypothyroidism at a rate of 4.3% per year, greater than women with elevated TSH alone (2.6% per year) or antithyroid antibodies alone (2.1% per year).46 In this study, there was no increase in all-cause or cardiac mortality in participants with subclinical hypothyroidism at baseline. In a natural history study of 26 elderly subjects with subclinical hypothyroidism, one third had overt biochemical hypothyroidism develop within 4 years of follow-up. Progression to overt hypothyroidism occurred in subjects with an initial TSH greater than 20 µIU/mL and in 80% with high-titer AMAs greater than 1 : 1600.78 In a more recent prospective study of 82 women with subclinical hypothyroidism, the cumulative incidence of overt hypothyroidism was 43% of women with TSH 6 to 12 µIU/mL and 77% with TSH greater than 12 µIU/mL, and no women with TSH less than 6 µIU/mL followed for 10 years. TPO antibody positivity was associated with development to overt hypothyroidism.117
Effects on Lipids, Hypothyroid Symptoms, and Mood
The relationship between subclinical hypothyroidism and effects of lipid profile are inconsistent. Some studies show that individuals with subclinical hypothyroidism have an atherogenic lipid profile (higher total cholesterol, low-density lipoprotein [LDL] cholesterol, lipoprotein (a), and apolipoprotein B, and lower high-density lipoprotein [HDL] cholesterol) than euthyroid individuals,118–120 but other studies show no difference.121–123 In the largest cross-sectional study of 25,862 subjects in the United States, subjects with subclinical hypothyroidism had a higher total cholesterol than the euthyroid group (223 mg/dL versus 216 mg/dL; P < .003), and had higher LDL cholesterol than the euthyroid group (144 mg/dL versus 140 mg/dL; P < .003).43 In small studies, T4 therapy of patients with subclinical hypothyroidism leads to an increase in HDL cholesterol124 and decrease in total and LDL cholesterol.119,121,125 Meta-analysis of T4 therapy in subclinical hypothyroidism shows a 10 mg/dL decrease in LDL cholesterol and 7.9 mg/dL decrease in total cholesterol concentration.126 Greater improvement was seen in subjects with baseline total cholesterol levels 240 mg/dL or greater versus subjects with total cholesterol less than 240 mg/dL.
T4 therapy leads to significantly increased cardiac output, increased mean arterial pressure, and decreased systemic vascular resistance.127 In a survey of postmenopausal women, subclinical hypothyroidism is associated with an increased risk of myocardial infarction (odds ratio 2.3) and aortic atherosclerosis (odds ratio 1.7), but no subsequent risk of myocardial infarction at 4.6 years of follow-up.128 It is unclear whether thyroid hormone therapy in subclinical hypothyroidism improves cardiac mortality.
Subclinical hypothyroidism is associated with depression in some,129 but not in all, studies.130 Similarly, some randomized placebo-controlled trials in subjects with subclinical hypothyroidism show improvement in symptoms of hypothyroidism,131,132 but one reported no difference.133 Depressed patients with subclinical hypothyroidism have a poorer response to antidepressant therapy than depressed patients who are euthyroid.134 Individuals with subclinical hypothyroidism show impairment in neurobehavioral scores, such as memory, which improve with T4 therapy. Treatment of subclinical hypothyroidism is reasonable in pregnant women to avoid impairment of intellectual potential of the fetus,135 and in women who have ovulatory dysfunction with infertility.131
Treatment
Patients with subclinical hypothyroidism and positive TPO antibodies, with TSH greater than 10 µIU/mL, are prone to have overt hypothyroidism develop and should receive thyroid hormone replacement. Risk of overt disease may depend on the etiology of subclinical hypothyroidism. Individuals receiving radioactive iodine therapy or high-dose external radiation are likely to progress to overt hypothyroidism and should probably be treated with thyroid hormone. Others who may benefit include patients with goiter, individuals with elevated total or LDL cholesterol, pregnant women, and women with ovulatory dysfunction with infertility.131 Small doses are usually needed (e.g., 50 to 75 µg daily), with monitoring of TSH and dose titration in 4 to 6 weeks until TSH is normalized. Patients with coronary artery disease should begin at a lower dose of 25 µg daily.
Nonthyroidal Illness
TSH elevation may occur in conditions other than hypothyroidism, including recovery from nonthyroidal illness, also known as sick euthyroid syndrome. Hospitalized and critically ill patients may have a decreased free T4 index or free T4 concentration by radioimmunoassay. When measured by equilibrium dialysis, however, free T4 is normal or elevated. In one report, serum total T4 levels less than 3 µg/dL were associated with mortality in 84% of critically ill patients.136 In a randomized prospective study, T4 treatment in an intensive care unit did not alter mortality.137
Laboratory Diagnosis
There is a set-point for optimal serum free T4 concentration in a given individual. Because of the log-linear relationship between serum TSH and T4 concentrations, small changes in free T4 from this set-point lead to relatively large changes in TSH by negative feedback. The most sensitive test for hypothyroidism is an elevated serum TSH. In subclinical hypothyroidism, TSH is elevated, whereas free T4 remains normal. If the disorder progresses to overt hypothyroidism, free T4 is decreased (see Table 123-3 for thyroid function tests in hypothyroidism and other low T4 syndromes). Radioactive iodine uptake is not indicated for the diagnosis of hypothyroidism because low, normal, or high values can occur, depending on the cause.
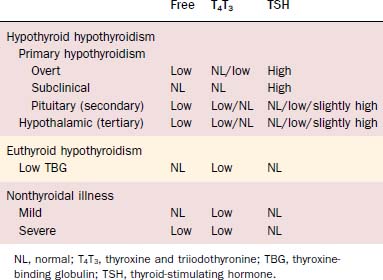
Central hypothyroidism caused by pituitary or hypothalamic disorder shows a low free T4 and TSH that is low, inappropriately normal, or mildly elevated. TRH stimulation testing of TSH has traditionally been used to distinguish between these two entities, but is unreliable.138,139
Management
Oral synthetic levothyroxine (T4) is the therapy of choice to correct hypothyroidism. Gastrointestinal absorption is 81%.140 Because the plasma half-life of T4 is long (6.7 days),141 once-daily administration leads to stable T4 and T3 concentrations. Numerous brand name (Euthyrox, Levothroid, Levoxyl, Synthroid, Unithroid) and generic preparations of T4 are available, each in varying doses with different color-coded tablets to allow dose titration at precise increments. In one study, comparison of two brand-name and two generic preparations in the United States showed bioequivalency.142 Equivalent doses of different formulations of T4 are generally interchangeable. One should repeat the TSH level 4 to 6 weeks after switching, however.140,143
In young, otherwise healthy adults, a full replacement dose can be prescribed at 1.6 µg/kg/day for nonmalignant conditions. In patients with known coronary disease, patients with multiple coronary risk factors, and elderly patients who may have previously silent coronary disease, conservative therapy with an initial dose of 25 µg/day is advisable. One should repeat TSH measurements with dose adjustment every 4 to 6 weeks (four to six half-lives of T4) until serum TSH normalizes or cardiac symptoms arise that may limit therapy to less than a full replacement dose. In individuals without residual thyroid tissue, such as a patient with thyroid cancer who has undergone thyroidectomy, the mean T4 dose required to achieve euthyroidism is generally higher (2.1 µg/kg/day).144
In patients with primary hypothyroidism, the goal of therapy is to normalize the serum TSH level. After initiation or change in dose of T4, TSH should be repeated in 4 to 6 weeks. Ultimately, TSH measurements are needed annually, or sooner depending on clinical status. In individuals with central hypothyroidism, free T4 alone should be normalized, also with repeat measurements free in 4 to 6 weeks. The use of the patient’s symptoms to judge the adequacy of T4 dosing is frequently inaccurate. When subjective symptoms were used to determine T4 dosing, patients chose a dose that produced mild hyperthyroidism.145
Potential adverse effects of overtreatment with an excessive dose of T4 include bone loss in postmenopausal, but not premenopausal, women,146,147 and in elderly patients, cardiac complications, including cardiac arrhythmias, heart failure, angina, and myocardial infarction.88 Occasionally, patients have manic behavior develop with T4 replacement. Severe behavioral manifestations of T4 therapy for juvenile hypothyroidism are uncommon, but mild behavioral symptoms and poorer school achievement may occur in approximately 25% of patients, who represent the most severe cases at the time of diagnosis.148
Poor patient compliance with taking thyroid hormone leads to therapeutic failure. Alternatives to a daily regimen include twice-weekly149 or once-weekly regimens.150 These probably should not be used in patients with coronary artery disease. Numerous medications may bind to and interfere with intestinal absorption of T4, including aluminum hydroxide,151 ferrous sulfate,152 sucralfate,153 cholestyramine,94 and calcium carbonate.139 Thyroid hormone administration should be separated in time from these medications by a few hours.
Thyroid hormone preparations that contain T3 alone (e.g., Cytomel), combinations of T4 and T3 (e.g., Thyrolar), and desiccated thyroid extract (Armour Thyroid) should not be used for the treatment of hypothyroidism. Serum T3 levels fluctuate widely because of the short half-life of T3. Temporary T3 therapy is indicated in patients with thyroid cancer who have undergone thyroidectomy and await thyroid remnant ablation to shorten the period of hypothyroidism. T3 can be discontinued 2 weeks before 131I.154 In addition, temporary switching from T4 to T3 therapy in individuals undergoing thyroid hormone withdrawal whole-body scanning also reduces the period of hypothyroidism.
Thyroid hormone requirements are increased during pregnancy by an average of 45%151 because of an estrogen-mediated increase in TBG, fetal T4 transfer, and increased T4 clearance. Serum TSH should be obtained at each trimester of pregnancy. If the T4 dose requires adjustment, TSH should be remeasured in 4 weeks with further dose adjustment as necessary. After delivery, the prepregnancy T4 dose should be resumed.155 A hypothyroid woman starting oral estrogen therapy such as with hormone replacement therapy may also require a higher thyroid hormone dose,156 and TSH should be obtained 3 months after initiation of estrogen to determine whether a dose increase is needed. Increases in dose may be necessary in patients who start medications that increase T4 catabolism (e.g., phenytoin, carbamazepine, phenobarbital, rifampin), have gastrointestinal malabsorption, or have nephrotic syndrome develop.157 A decreased thyroid hormone dose requirement may be seen in elderly patients158 and in women with breast cancer treated with androgens.159
Surgery
With mild to moderate hypothyroidism, postoperative complications are frequent, but are rarely serious or lasting, and necessary surgery should not be postponed simply to replete thyroid hormone.160 This is not true for patients with severe myxedema, who should be given preoperative thyroid hormone except in the most urgent surgical emergencies or uncontrolled ischemic heart disease.
In euthyroid patients, total T4 tends to decrease in the first postoperative day, then spontaneously normalizes in 7 days; the same occurs in hypothyroid patients, but T4 levels do not normalize until thyroid supplementation is given.161 It is usually unnecessary to increase the postoperative dose of T4, however, and almost never necessary to use parenteral T4, unless the patient cannot take medication by mouth for several weeks. If parenteral T4 therapy is necessary, half of the patient’s usual daily T4 dose is ordinarily given, and attention is given to the patient’s cardiac status because this therapy may precipitate cardiac arrhythmias, angina, and heart failure.
Cardiac and pulmonary problems are prevalent in elderly patients with hypothyroidism. The prevalence of coronary artery disease is high, but the diagnosis is easily overlooked because patients often have few symptoms because of their low metabolic activity or because they fail to communicate their symptoms clearly.103 Pericardial effusions are often apparent and rarely cause tamponade. Patients with severe hypothyroidism also respond poorly to stress by having hypothermia and hypotension develop, and they do not have tachycardia develop in response to infection or hypotension. Shock responds poorly to vasoconstrictors.
Patients with severe hypothyroidism often display upper airway obstruction caused by oropharyngeal muscle dysfunction and tissue infiltration with mucopolysaccharide.162 They may have central sleep apnea, insensitivity to hypoxia and hypercarbia,163 and respiratory muscle weakness,164 changes that often lead to severe postoperative hypoxia and difficulty in weaning from a ventilator. These defects are reversible with T4 replacement therapy, but obstructive sleep apnea may be more closely related to obesity and male gender than hypothyroidism.165
A von Willebrand’s disease–like defect is common in hypothyroidism, which may lead to bleeding.166,167 It resolves promptly with infusion of desmopressin, suggesting that it acts through the β-adrenergic receptor.89 This can be helpful in the acute management of bleeding. It resolves permanently with T4 therapy.168,169
Bennedaek FN, Hegedus L. The value of ultrasonography in the diagnosis and follow-up of subacute thyroiditis. Thyroid. 1997;7:45.
Brent GA. The molecular basis of thyroid hormone action. N Engl J Med. 1994;331:847.
Burch HB, Wartofsky L. Life-threatening thyrotoxicosis: thyroid storm. Endocrinol Metab Clin North Am. 1993;22:263.
Erickson D, Gharib H, Li H, van Heerden JA. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8:277.
Klee GG, Hay ID. Biochemical thyroid function testing. Mayo Clin Proc. 1994;69:469.
Mazzaferri EL. Thyroid cancer and Graves’ disease. J Clin Endocrinol Metab. 1990;70:826.
McConahey WM. Hashimoto’s thyroiditis. Med Clin North Am. 1972;56:885.
Miccoli P, Vitti P, Rago T, et al. Surgical treatment of Graves’ disease: subtotal or total thyroidectomy? Surgery. 1996;120:1020.
Razack MS, Lore JM, Lippes HA, et al. Total thyroidectomy for Graves’ disease. Head Neck. 1997;19:278.
Ruf J, Feldt-Rasmussen U, Hegedus L, et al. Bispecific thyroglobulin and thyroperoxidase autoantibodies in patients with various thyroid and autoimmune diseases. J Clin Endocrinol Metab. 1994;79:1404.
Singer PA, Cooper DS, Levy EG, et al. Treatment guidelines for patients with hyperthyroidism and hypothyroidism. JAMA. 1995;273:808.
Spencer CA, Wang CC. Thyroid globulin measurement: techniques, clinical benefits and pitfalls. Endocrinol Metab Clin North Am. 1995;24:841.
1. Dunn JT. Sources of dietary iodine in industrialized countries. In: Delange F, Dunn JT, Glinoer D, editors. Iodine Deficiency in Europe: A Continuing Concern. New York: Plenum Press; 1993:17.
2. Levy O, De la Vieja A, Carrasco N. The Na+/I− symporter (NIS): recent advances. J Bioenerg Biomembr. 1998;30:195.
3. Royaux IE, et al. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology. 2000;141:839.
4. Doucet J, et al. Does age play a role in the clinical presentation of hypothyroidism? J Am Geriatr Soc. 1994;42:984.
5. Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev. 1998;19:173.
6. Ohtaki S, et al. Thyroid peroxidase: experimental and clinical integration. Endocr J. 1996;43:1.
7. Michot JL, et al. Relationship between thyroid peroxidase, H2O2 generating system and NADPH-dependent reductase activities in thyroid particulate fractions. Mol Cell Endocrinol. 1985;41:211.
8. Kohrle J. Local activation and inactivation of thyroid hormones: the deiodinase family. Mol Cell Endocrinol. 1999;151:103.
9. Cavalieri RR, et al. Thyroid hormone export in rat FRTL-5 thyroid cells and mouse NIH-3T3 cells is carrier-mediated, verapamil-sensitive, and stereospecific. Endocrinology. 1999;140:4948.
10. Schussler GC. The thyroxine-binding proteins. Thyroid. 2000;10:141.
11. Singer PA, et al. Treatment guidelines for patients with hyperthyroidism and hypothyroidism. JAMA. 1995;273:808.
12. Salvatore D, et al. Type 2 iodothyronine deiodinase is highly expressed in human thyroid. J Clin Invest. 1996;98:962.
13. Bates JM, St. Germain DL, Galton VA. Expression profiles of the three iodothyronine deiodinase, D1, D2, and D3, in the developing rat. Endocrinology. 1999;140:844.
14. Vassart G, Dumont JE. The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocr Rev. 1992;13:596.
15. Scanlon MF, Toft AD. Regulation of thyrotropin secretion. In: Braverman LE, Utiger RD, editors. The Thyroid: A Fundamental and Clinical Text. 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:234.
16. Cooper DS. Antithyroid drugs for the treatment of hyperthyroidism caused by Graves’ disease. Endocrinol Metab Clin North Am. 1998;27:225.
17. DeGroot LJ, Rue PA. Roentgenographic contrast agents inhibit triiodothyronine binding to nuclear receptors in vitro. J Clin Endocrinol Metab. 1979;49:538.
18. Lazarus JH. The effects of lithium therapy on thyroid and thyrotropin-releasing hormone. Thyroid. 1998;8:909.
19. Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid. 1993;3:59.
20. Brent GA. The molecular basis of thyroid hormone action. N Engl J Med. 1994;331:847.
21. Sakata S, Nakamura S, Miura K. Auto-antibodies against thyroid hormone or iodothyronines: implications in diagnosis, thyroid function, treatment and pathogenesis. Ann Intern Med. 1985;103:579.
22. Rich EJ, Menelman PM. Acute suppurative thyroiditis in pediatric patients. Pediatr Infect Dis J. 1987;6:936.
23. Nelson JC, Tomel RT. Direct determination of free thyroxine in undiluted serum by equilibrium dialysis. Clin Chem. 1988;34:1737.
24. Spencer CA. Clinical evaluation of free T4 techniques. J Endocrinol Invest. 1986;9:57.
25. Kaptein EM. Clinical applications of free thyroxine determinations. Clin Lab Med. 1993;13:653.
26. Larsen PR, et al. Revised nomenclature for tests of thyroid hormones and thyroid related proteins in serum. J Clin Endocrinol Metab. 1987;64:1089.
27. Bitton RN, Wexler C. Free triiodothyronine toxicosis: a distinct entity. Am J Med. 1990;88:531-533.
28. Spencer CA, Nicoloff JT. Serum TSH measurement: a 1990 status report. Thyroid Today. 1990;13:1.
29. Klee GG, Hay ID. Biochemical thyroid function testing. Mayo Clin Proc. 1994;69:469.
30. Ross DS. Hyperthyroidism, thyroid hormone therapy and bone. Thyroid. 1994;4:319.
31. Sawin CT, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in order persons. N Engl J Med. 1994;331:1249.
32. Sugino K, et al. Follow-up evaluation of patients with Graves’ disease treated by subtotal thyroidectomy and risk factor analysis for post-operative thyroid dysfunction. J Endocrinol Invest. 1993;16:195.
33. Ross DS. Subclinical hyperthyroidism. In: Braverman LE, Utiger RD, editors. Werner and Ingbar’s the Thyroid: A Fundamental and Clinical Text. 6th ed. Philadelphia: Lippincott; 1991:1249.
34. Chopra IJ, et al. Thyroid function in nonthyroidal illnesses. Ann Intern Med. 1983;98:946.
35. Spencer CA, Wang CC. Thyroglobulin measurement: techniques, clinical benefits, and pitfalls. Endocrinol Metab Clin North Am. 1995;24:841.
36. Brown J, et al. Autoimmune thyroid disease—Graves’ and Hashimoto’s. Ann Intern Med. 1978;88:379.
37. Kaufman KD, et al. Recombinant human thyroid peroxidase generated in eukaryotic cells: a source of specific antigen for the immunological assay of antimicrosomal antibodies in the sera of patients with autoimmune thyroid disease. J Clin Endocrinol Metab. 1990;70:724.
38. Beever K, et al. Highly sensitive assays of autoantibodies to thyroglobulin and to thyroid peroxidase. Clin Chem. 1989;35:1949.
39. Ruf J, et al. Bispecific thyroglobulin and thyroperoxidase autoantibodies in patients with various thyroid and autoimmune diseases. J Clin Endocrinol Metab. 1994;79:1404.
40. Gordin A, Lamberg BA. Spontaneous hypothyroidism in symptomless autoimmune thyroiditis: a long term followup study. Clin Endocrinol. 1981;15:537.
41. Adams DD, Purves HD. Abnormal responses in the assay of thyrotropin. Proc Univ Otago Med Sch. 1956;34:11.
42. Oppenheim DS. TSH and other glycoprotein producing pituitary adenomas: alpha-subunit as a tumor marker. Thyroid Today. 1991;14:1.
43. Canaris GJ, et al. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160:526.
44. Helfand M, Redfern CC. Screening for thyroid disease: an update. Ann Intern Med. 1998;129:144.
45. Okamura K, et al. Thyroid disorders in the general population of Hisayama, Japan, with special reference to prevalence and sex differences. Int J Epidemiol. 1987;16:545.
46. Vanderpump MPJ, et al. The incidence of thyroid disorders in the community: a twenty-year follow-up of the Whickham survey. Clin Endocrinol (Oxf). 1995;43:55.
47. Brownlie BEW, Wells JE. The epidemiology of thyrotoxicosis in New Zealand: incidence and geographical distribution in North Canterbury, 1983-1985. Clin Endocrinol (Oxf). 1990;33:249.
48. Williams I, et al. Aetiology of hyperthyroidism in Canada and Wales. J Epidemiol Community Health. 1983;37:245.
49. Reinwein D, et al. The different types of hyperthyroidism in Europe: results of a prospective survey of 924 patients. J Endocrinol Invest. 1988;11:193.
50. Mazzaferri EL. Thyroid cancer and Graves’ disease. J Clin Endocrinol Metab. 1990;70:826.
51. Belfiore A, et al. Increased aggressiveness of thyroid cancer in patients with Graves’ disease. J Clin Endocrinol Metab. 1990;70:830.
52. Taurog A. Hormone synthesis: thyroid iodine metabolism. In: Braverman LE, Utiger RD, editors. The Thyroid: A Fundamental and Clinical Text. 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2000:61.
53. Mercardo M, et al. Treatment of hyperthyroidism with a combination of methimazole and cholestyramine. J Clin Endocrinol Metab. 1996;81:3191.
54. Jansson R, et al. Intrathyroidal concentrations of methimazole in patients with Graves’ disease. J Clin Endocrinol Metab. 1983;57:129.
55. Weetman AP. The immunomodulatory effects of antithyroid drugs. Thyroid. 1994;4:145.
56. Escobar-Morreale HF, et al. Methimazole-induced severe aplastic anemia: unsuccessful treatment with recombinant granulocyte-monocyte colony-stimulating factor. Thyroid. 1997;7:67.
57. Liaw Y-F, et al. Hepatic injury during propylthiouracil therapy in patients with hyperthyroidism: a cohort study. Ann Intern Med. 1993;118:424.
58. Kristensen O, Andersen HH, Pallisgaard G. Lithium carbonate in the treatment of thyrotoxicosis: a controlled trial. Lancet. 1976;1:603.
59. Klein I, et al. Symptom rating scale for assessing hyperthyroidism. Arch Intern Med. 1988;148:387.
60. Chiovato L, et al. Outcome of thyroid function in Graves’ patients treated with radioiodine: role of thyroid-stimulating and thyrotropin-blocking antibodies and of radioiodine-induced damage. J Clin Endocrinol Metab. 1998;83:40.
61. Beckers C. Regulations and policies on radioiodine 131I therapy in Europe. Thyroid. 1997;7:221.
62. Menegaux F, Reprecht T, Chigot JP. The surgical treatment of Graves’ disease. Surg Gynecol Obstet. 1993;176:277.
63. Sugino K, et al. Management of recurrent hyperthyroidism in patients with Graves’ disease treated by subtotal thyroidectomy. J Endocrinol Invest. 1995;18:415.
64. Sugino K, et al. Early recurrence of hyperthyroidism in patients with Graves’ disease treated by subtotal thyroidectomy. World J Surg. 1995;19:648.
65. Razack MS, et al. Total thyroidectomy for Graves’ disease. Head Neck. 1997;19:378.
66. Kuma K, et al. Natural course of Graves’ disease after subtotal thyroidectomy and management of patients with postoperative thyroid dysfunction. Am J Med Sci. 1991;302:8.
67. Yamashita H, et al. Postoperative tetany in patients with Graves’ disease: a risk factor. Clin Endocrinol. 1997;47:71.
68. Miccoli P, et al. Surgical treatment of Graves’ disease: subtotal or total thyroidectomy? Surgery. 1996;120:1020.
69. Winsa B, et al. Total thyroidectomy in therapy-resistant Graves’ disease. Surgery. 1994;116:1068.
70. Patwardhan NA, et al. Surgery still has a role in Graves’ hyperthyroidism. Surgery. 1993;114:1108.
71. Winsa B, et al. Retrospective evaluation of subtotal and total thyroidectomy in Graves’ disease with and without endocrine ophthalmology. Eur J Endocrinol. 1995;132:406.
72. Peek CM, et al. Combination of potassium iodide and propranolol in preparation of patients with Graves’ disease for thyroid surgery. N Engl J Med. 1980;302:883.
73. Soreide JA, van Heerden JA, Lo CY, et al. Surgical management of Graves disease in patients younger than 18 years. World J Surg.. 1996;20:794-799.
74. Andaker L, et al. Surgery for hyperthyroidism: hemithyroidectomy plus contralateral resection or bilateral resection? A prospective randomized study of postoperative complications and long-term results. World J Surg. 1992;16:765.
75. Christiansen NJ, et al. Serum thyroxine in the early phase of subacute thyroiditis. Acta Endocrinol (Copenh). 1970;64:359.
76. Bennedaek FN, Hegedus L. The value of ultrasonography in the diagnosis and followup of subacute thyroiditis. Thyroid. 1997;7:45.
77. Tomer Y, Davies TF. Infection, thyroid disease, and autoimmunity. Endocr Rev. 1993;14:107.
78. Rosenthal MJ, et al. Thyroid failure in the elderly: microsomal antibodies as discriminant for therapy. JAMA. 1987;258:209.
79. Stanbury JB, et al. Iodine-induced hyperthyroidism: occurrence and epidemiology. Thyroid. 1998;8:83.
80. Bartalena L, et al. Treatment of amiodarone-induced thyrotoxicosis, a difficult challenge: results of a prospective study. J Clin Endocrinol Metab. 1996;81:2930.
81. Fernandez-Soto L, et al. Increased risk of autoimmune thyroid disease in hepatitis C vs hepatitis B before, during and after discontinuing interferon therapy. Arch Intern Med. 1998;158:1445.
82. Burch HB, et al. Diagnosis and management of the autonomously functioning thyroid nodule: the Walter Reed Army Medical Center experience, 1975-1996. Thyroid. 1998;8:871.
83. Hamburger JI, Taylor CL. Transient thyrotoxicosis associated with acute hemorrhagic infarction of autonomously functioning thyroid nodules. Ann Intern Med. 1979;91:406.
84. Ferrari C, Reschini E, Paracchi A. Treatment of autonomous thyroid nodule: a review. Eur J Endocrinol. 1996;135:383.
85. Erickson D, et al. Treatment of patients with toxic multinodular goiter. Thyroid. 1998;8:277.
86. Bayot MR, Chopra IJ. Coexistence of struma ovarii and Graves’ disease. Thyroid. 1995;5:469.
87. Brown WW, Shetty KR, Rosenfeld PS. Hyperthyroidism due to struma ovarii: demonstration by radioiodine scan. Acta Endocrinol (Copenh). 1973;73:266.
88. Mazzaferi EL. Adult hypothyroidism. Postgrad Med. 1986;79:75.
89. Marqusse E, Haden ST, Utiger RD. Subclinical thyrotoxicosis. Endocrinol Metab Clin North Am. 1998;27:37.
90. Elte JWF, Bussemaker JK, Haak A. The natural history of euthyroid multinodular goiter. Postgrad Med J. 1990;66:186.
91. Sandrock D, et al. Long-term follow-up in patients with autonomous thyroid adenoma. Acta Endocrinol (Copenh). 1993;128:51.
92. Burch HB, Wartofsky L. Life-threatening thyrotoxicosis: thyroid storm. Endocrinol Metab Clin North Am. 1993;22:263.
93. Brooks MH, Waldstein SS. Free thyroxine concentrations in thyroid storm. Ann Intern Med. 1980;93:694.
94. Hollowell JG, et al. Serum TSH, T4, and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87:489.
95. Cunnien AJ, et al. Radioiodine-induced hypothyroidism in Graves’ disease: factors associated. J Nucl Med. 1982;23:978.
96. Hancock SL, et al. Thyroid diseases after treatment of Hodgkin’s disease. N Engl J Med. 1991;325:599.
97. Nilsson LR, et al. Nonendemic goiter and deafness. Acta Paediatr. 1964;53:117.
98. Meyerhoff WL. Hypothyroidism and the ear: electrophysical, morphological and chemical considerations. Laryngoscope. 1979;89:1.
99. Debruyne F, Vanderschueren-Lodeweyckx M, Bastinjns P. Hearing in congenital hypothyroidism. Audiology. 1983;22:404.
100. de Luca F, et al. Sensorineural deafness in congenital hypopituitarism with severe hypothyroidism. Acta Paediatr Scand. 1985;74:148.
101. Meyerhoff WL. The thyroid and audition. Laryngoscope. 1976;86:483.
102. Francois M, et al. Audiological assessment of eleven congenital hypothyroid infants before and after treatment. Acta Otolaryngol (Stockh). 1993;113:39.
103. Bhatia PF, et al. Audiological and vestibular function tests in hypothyroidism. Laryngoscope. 1997;87:2082.
104. Rapp MF, et al. Laryngeal involvement in scleromyxedema: a case report. Otolaryngol Head Neck Surg. 1991;104:362.
105. McConahey WM. Hashimoto’s thyroiditis. Med Clin North Am. 1972;56:885.
106. Comtois R, Faucher L, Lafleche L. Outcome of hypothyroidism caused by Hashimoto’s thyroiditis. Arch Intern Med. 1995;155:1404.
107. Takasu N, et al. Simple and reliable method for predicting the remission of Graves’ disease: revised triiodothyronine-suppression test, indexed by serum thyroxine. J Endocrinol Invest. 1995;18:288.
108. Muller B, et al. Impaired action of thyroid hormone associated with smoking in women with hypothyroidism. N Engl J Med. 1995;333:964.
109. Martino E, et al. Amiodarone iodine-induced hypothyroidism: risk factors and follow-up in 28 cases. Clin Endocrinol (Oxf). 1987;26:227.
110. Biel MA, Maisel RA. Indications for performing hemithyroidectomy for tumors requiring total laryngectomy. Am J Surg. 1985;150:435.
111. Usala SJ, Weintraub BD. Familial thyroid hormone resistance: clinical and molecular studies. In: Mazzaferri EL, editor. Advances in Endocrinology Metabolism, No. 2. St Louis: Mosby, 1991.
112. Cevallos JL, et al. Low-dosage 131I therapy of thyrotoxicosis (diffuse goiters): a five-year follow-up study. N Engl J Med. 1974;290:141.
113. Tamai H, et al. Development of spontaneous hypothyroidism in patients with Graves’ disease treated with antithyroidal drugs: clinical, immunological, and histological findings in 26 patients. J Clin Endocrinol Metab. 1989;69:49.
114. de Jong JM, et al. Primary hypothyroidism as a complication after treatment of tumors of the head and neck. Acta Radiol. 1982;21:299.
115. Bagchi N, et al. Thyroid dysfunction in adults over age 55 years: a study in an urban US community. Arch Intern Med. 1990;150:785.
116. Tunbridge WM, et al. The spectrum of thyroid disease in a community: the Wickham survey. Clin Endocrinol. 1977;7:481.
117. Huber G, et al. Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve, and thyroid antibodies. J Clin Endocrinol Metab. 2002;87:3221.
118. Althaus BU, et al. LDL/HDL-changes in subclinical hypothyroidism: possible risk factors of coronary heart disease. Clin Endocrinol (Oxf). 1988:28-157.
119. Caraccio N, et al. Lipoprotein profile in subclinical hypothyroidism: response to levothyroxine replacement, a randomized placebo-controlled study. J Clin Endocrinol Metab. 2002;87:1533.
120. Kung AW, et al. Elevated serum lipoprotein (a) in subclinical hypothyroidism. Clin Endocrinol (Oxf). 1995;43:445.
121. Bogner U, et al. Subclinical hypothyroidism and hyperlipiproteinaemia: indiscriminant L-thyroxine treatment not justified. Acta Endocrinol (Copenh). 1993;128:202.
122. Kutty KM, et al. Serum lipids in hypothyroidism—a re-evaluation. J Clin Endocrinol Metab. 1978;46:55.
123. Parle JV, et al. Circulating lipids and minor abnormalities of thyroid function. Clin Endocrinol (Oxf). 1992;37:411.
124. Caron P, et al. Decreased HDL cholesterol in subclinical hypothyroidism: the effect of L-thyroxine therapy. Clin Endocrinol (Oxf). 1990;33:519.
125. Arem R, et al. Effect of L-thyroxine therapy on lipoprotein fractions in overt and subclinical hypothyroidism, with special reference to lipoprotein (a). Metabolism. 1995;44:1559.
126. Danese MD, et al. Clinical review 115: effect of thyroxine therapy on lipoproteins in patients with mild thyroid failure: a quantitative review of the literature. J Clin Endocrinol Metab. 2000;85:2993.
127. Faber J, et al. Hemodynamic changes after levothyroxine treatment in subclinical hypothyroidism. Thyroid. 2002;12:319.
128. Hak AE, et al. Subclinical hypothyroidism is an independent risk factor for atherosclerosis and myocardial infarction in elderly women: the Rotterdam Study. Ann Intern Med. 2000;132:270.
129. Haggerty JJJr, et al. Subclinical hypothyroidism: a modifiable risk factor for depression? Am J Psychiatry. 1993;150:508.
130. Pop VJ, et al. Are autoimmune thyroid dysfunction and depression related? J Clin Endocrinol Metab. 1998;83:3194.
131. Cooper DS, et al. L-thyroxine therapy in subclinical hypothyroidism: a double-blind, placebo-controlled trial. Ann Intern Med. 1984;101:18.
132. Nystrom E, et al. A double-blind cross-over 12-month study of L-thyroxine treatment of women with “subclinical” hypothyroidism. Clin Endocrinol (Oxf). 1988;29:63.
133. Jaeschke R, et al. Does treatment with L-thyroxine influence health status in middle-aged and older adults with subclinical hypothyroidism? J Gen Intern Med. 1996;11:744.
134. Joffe RT, et al. Major depression and subclinical (grade 2) hypothyroidism. Psychoneuroendocrinology. 1992;17:215.
135. Haddow JE, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med. 1999;341:549.
136. Slag MF, et al. Hypothyroxinemia in critically ill patients as a predictor of high mortality. JAMA. 1981;245:43.
137. Brent GA, et al. Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration. J Clin Endocrinol Metab. 1986;63:1.
138. Faglia G, et al. Plasma thyrotropin response to thyrotropin releasing hormone in patients with pituitary and hypothalamic disorders. J Clin Endocrinol Metab. 1973;37:5951.
139. Snyder PJ, et al. Diagnostic value of thyrotropin-releasing hormone in pituitary and hypothalamic disorders. Ann Intern Med. 1974;81:751.
140. Fish LH, et al. Replacement dose, metabolism, and bioavailability of levothyroxine in the treatment of hypothyroidism: role of triiodothyronine in pituitary feedback in humans. N Engl J Med. 1987;316:764.
141. Gregerman RI, et al. Thyroxine turnover in euthyroid man with special reference to changes with age. J Clin Invest. 1962;41:2065.
142. Dong BJ, et al. Bioequivalence of generic and brand-name levothyroxine products in the treatment of hypothyroidism. JAMA. 1997;277:1205.
143. Copeland PM. Two cases of therapeutic failure associated with levothyroxine brand interchange. Ann Pharmacother. 1995;29:482.
144. Gordon MB, et al. Variations in adequate levothyroxine therapy in patients with different causes of hypothyroidism. Endocr Pract. 1999;5:233.
145. Carr D, et al. Fine adjustment of thyroxine replacement dosage: comparison of the thyrotrophin releasing hormone test using a sensitive thyrotrophin assay with measurement of free thyroid hormones and clinical assessment. Clin Endocrinol (Oxf). 1988;28:325.
146. Banovac K, et al. Evidence of hyperthyroidism in apparently euthyroid patients treated with thyroxine. Arch Intern Med. 1998;149:809.
147. Uzzan B, et al. Effects on bone mass of long term treatment with thyroid hormones: a meta-analysis. J Clin Endocrinol Metab. 1996;81:4278.
148. Rovet JF, et al. Psychologic and psychoeducational consequences of thyroxine therapy for juvenile acquired hypothyroidism. J Pediatr. 1993;122:543.
149. Taylor J, et al. Twice-weekly dosing for thyroxine replacement in elderly patients with primary hypothyroidism. J Int Med Res. 1994;22:273.
150. Grebe SK, et al. Treatment with hypothyroidism with once weekly thyroxine. J Clin Endocrinol Metab. 1997;82:870.
151. Mandel SJ, et al. Increased need for thyroxine during pregnancy in women with primary hypothyroidism. N Engl J Med. 1990;323:91.
152. Campbell NR, et al. Ferrous sulfate reduces thyroxine efficacy in patients with hypothyroidism. Ann Intern Med. 1992;117:1010.
153. Campbell JA, et al. Sucralfate and the absorption of L-thyroxine. Ann Intern Med. 1994;121:152.
154. Goldman JM, et al. Influence of triiodothyronine withdrawal time on 131I uptake post thyroidectomy for thyroid cancer. J Clin Endocrinol Metab. 1980;50:734.
155. Kaplan MM. Management of thyroxine therapy during pregnancy. Endocr Pract. 1996;2:281.
156. Arafah BM. Increased need for thyroxine in women with hypothyroidism during estrogen therapy. N Engl J Med. 2001;344:1743.
157. Afrasiabi MA, et al. Thyroid function studies in the nephritic syndrome. Ann Intern Med. 1979;90:335.
158. Sawin CT, et al. Aging and the thyroid: decreased requirement for thyroid hormone in older hypothyroid patients. Am J Med. 1983;75:206.
159. Arafah BM. Decreased levothyroxine requirement in women with hypothyroidism during androgen therapy for breast cancer. Ann Intern Med. 1994;121:247.
160. Ladenson PW, et al. Complications of surgery in hypothyroid patients. Am J Med. 1984;77:261.
161. Kawasuji M, et al. Coronary artery bypass surgery in patients with angina pectoris and hypothyroidism. Eur J Cardiothorac Surg. 1991;5:230.
162. Orr WC, Males JL, Imes NK. Myxedema and obstruction sleep apnea. Am J Med. 1981;70:1061.
163. Duranti R, et al. Control of breathing in patients with severe hypothyroidism. Am J Med. 1993;95:29.
164. Siafakas NM, et al. Respiratory muscle strength in hypothyroidism. Chest. 1992;102:189.
165. Pelttari L, et al. Upper airway obstruction in hypothyroidism. J Intern Med. 1994;236:177.
166. Levesque H, et al. Acquired von Willebrand’s syndrome associated with decrease of plasminogen activator and its inhibitor during hypothyroidism. Eur J Med. 1993;2:284.
167. Myrup B, Bregenfrd C, Faber J. Primary haemostasis in thyroid disease. J Intern Med. 1995;238:59.
168. Bruggers CS, McElligott K, Rallison ML. Acquired von Willebrand disease in twins with autoimmune hypothyroidism: response to desmopressin and L-thyroxine therapy. J Pediatr. 1994;125:911.
169. Erfurth EMT, et al. Effect of acute desmopressin and of long-term thyroxine replacement on hemostasis in hypothyroidism. Clin Endocrinol (Oxf). 1995;42:373.
[/level-membership-for-otolaryngology-category][not-level-membership-for-otolaryngology-category]
CHAPTER 123 Disorders of the Thyroid Gland
Physiology of the Thyroid Gland
The thyroid gland produces two hormones, 3,5,3′-triiodothyronine (T3) and 3,5,3′,5′-tetraiodothyronine or thyroxine (T4). Both are iodinated derivatives of tyrosine. Hormone production depends on an external iodine supply and on intrathyroidal mechanisms for concentrating ingested iodide and then incorporating it into the tissue-specific protein, thyroglobulin. The thyroid gland is unique within the endocrine system in having a large extracellular space, the follicular lumen, which is used for storage of the hormones and their precursors. As hormone is needed by the organism, thyroglobulin is retrieved by the cell, where the biologically active hormones are released from thyroglobulin before being passed into circulation (Fig. 123-1).
Iodide Transport
A daily dietary intake of at least 100 µg of iodine per day is required in humans to ensure adequate production of thyroid hormone. In North America, the average daily intake is higher than this, largely because of the use of iodine as a food additive.1 In many parts of the world, however, consumption is significantly below the minimum level, and iodine deficiency is the leading cause of thyroid-related disorders.
The thyroid normally concentrates iodide 20-fold to 40-fold over the extracellular space and against an electrical gradient of approximately 40 mV. Key to this trapping action is a protein located in the basal membrane of the thyroid cell known as the sodium/iodide symporter (NIS).2 NIS couples with the influx of Na+ down its electrochemical gradient with the simultaneous influx of I− up its electrochemical gradient. An Na+,K+-ATPase acts to maintain the Na+ gradient. Iodide travels down its electrochemical gradient to the apical surface of the thyrocyte, where it is incorporated into thyroglobulin. More recent evidence suggests an apical membrane protein, pendrin, aids in releasing iodide into the follicular lumen.3 Mutations in the gene and coding for this protein are responsible for the common hereditary disorder Pendred’s syndrome, which is associated with mild hypothyroidism, goiter, and hearing loss.3 Mutations in the gene coding for NIS have been identified in patients with iodide trapping defects, a rare cause of congenital hypothyroidism.2
Thyroglobulin
Thyroglobulin is essential to thyroid physiology. It is a tissue-specific protein that serves as a matrix for the synthesis of hormone and as a vehicle for its storage.4 The human thyroglobulin gene has been cloned and is located on the long arm of chromosome 8Q24. Thyroglobulin is a large dimeric glycoprotein of approximately 660 kD, consisting in humans of two identical polypeptide chains each of 2750 amino acids. About 10% of its weight is carbohydrate, and about 0.1% to 1% is iodine. Synthesis and maturation of thyroglobulin follow a pathway typical of proteins destined for secretion. The polypeptide chain is synthesized on the surface of the rough endoplasmic reticulum. As that passes through a series of intracellular compartments, it undergoes important post-translational modifications before reaching the follicular lumen.5 Carbohydrate units are added to the polypeptide chain as it is translocated into the lumen of the rough endoplasmic reticulum. Folding and dimerization of the polypeptide chain occurs within this compartment, aided by folding enzymes and a group of proteins known as molecular chaperones. Perturbations of this process result in block of protein transport beyond this point and cause congenital hypothyroidism.5 Under normal circumstances, the properly folded thyroglobulin dimers migrate to the Golgi complex, where processing of the carbohydrate units are completed. Mature but as of yet uniodinated thyroglobulin is transferred from the Golgi complex to the apical cell surface in small vesicles.
Iodination and Thyroperoxidase
Newly formed thyroglobulin and iodide meet at the apical cell surface, where hormone synthesis occurs. This process includes (1) the oxidation of iodide; (2) its subsequent transfer to thyrosyl residues on thyroglobulin, producing monoiodotyrosine (MIT) and di-iodotyrosine (DIT); and (3) coupling of two iodotyrosine molecules, either one each of MIT and DIT to form T3 or two of DIT to form T4. Thyroperoxidase (TPO), an enzyme present in the apical cell membrane, is responsible for each of these steps.6 Hydrogen peroxide (H2O2), required in the iodinating and coupling reactions, is generated at the apical membrane by a reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.7 Mutations in the TPO gene have been found in patients with congenital hypothyroidism caused by defective organification. Abnormalities in H2O2 generation seem to be more rare.
Under normal circumstances, iodide, when trapped, is rapidly incorporated into thyroglobulin so that little free iodide exists within the thyroid gland at any given time. The extent to which thyroglobulin is iodinated depends on the thyroid’s iodide supply. At a level of 0.5% iodine, the thyroglobulin dimer in humans contains on average 5 residues of MIT, 5 of DIT, 2.5 of T4, and 0.7 of T3 of a total of 132 residues of tyrosine.4
Hormone formation involves the coupling of two residues of iodotyrosine within the thyroglobulin polypeptide chain. At the hormonogenic site, the “acceptor” DIT receives the iodinated phenol ring of the “donor” iodothyrosyl (either MIT or DIT) located at some distal site on the polypeptide chain. In the process, the alanine side chain of the donor remains behind, now presumably in the form of hydroalanine. Iodination in vitro of low iodine human thyroglobulin indicates that certain thyrocele sites are favored for early iodination, and that three or four major sites exist for hormone formation. The most important hormonogenic site is located five residues from the amino terminal thyroglobulin, whereas a second major site is located three residues from the carboxy terminal. The locations of donor thyrosyls are incomplete. To date, only one has been identified in human thyroglobulin, and this resides in the amino terminal region of the molecule.4
Storage and Release of Hormone
Hormone release is initiated by the retrieval of thyroglobulin from the follicular lumen. Under stimulatory conditions in some species, this process may occur by macropinocytosis. Pseudopods form at the thyrocytes’ apical surface and engulf thyroglobulin as large colloid droplets. Under physiologic conditions in most species, however, including humans, thyroglobulin is retrieved by micropinocytosis into small vesicles. It is then passed through the endosome-lysosomal system, where the combined action of several acid proteases, including cathepsins B, D, and L, and lysosomal dipeptidase 1, releases the hormones and their iodotyrosine precursors from the polypeptide backbone.4 Evidence suggests the iodo-amino acids may be preferentially cleaved first, but ultimately thyroglobulin is broken down into amino acids or small peptides within the lysosomes.
When released from thyroglobulin, the thyroid hormones and their precursors enter the cytosol; there MIT and DIT are deiodinated by an iodotyrosine-specific deiodinase, and the released iodide re-enters the iodine pool. Some T4 is deiodinated to T3 before it is released into the circulation by 5′-iodothyronine deiodinase similar to that found in peripheral tissue.8 The mechanism by which T4 and T3 are released from the thyrocyte is unknown, but more recent evidence suggests a carrier protein may be involved.9
Circulating Thyroid Hormones
Less than 1% of circulating thyroid hormones exist as free iodo-amino acids. The remainder are bound in reversible, noncovalent linkage to one of several plasma proteins.10 In humans, the most important of these is thyroxine-binding protein (TBG), accounting for approximately 70% of circulating hormone. The TBG molecule has one hormone-binding site with a very high affinity for T4 and lower affinity for T3. A second plasma protein transthyretin accounts for approximately 10% of circulating T4 and T3. Each transthyretin molecule has two hormone-binding sites, but the affinity of the first is lower than that of TBG, and that of the second site is very low for both hormones. Albumin also serves as a thyroid hormone transport protein. Although it has low affinity, its abundance allows it to account for 10% to 20% of bound circulating hormone.
The bound hormones are in equilibrium with the minute fraction of free circulating hormone that is available for use in peripheral tissue. Under euthyroid conditions, approximately 0.2% of T4 and about 0.3% of T3 in circulation is unbound. The larger free-to-bound ratio of T3 to T4 is caused by the lower affinity of TBG for T3.11 To date, no change in thyroid state has been attributed to abnormalities in these hormone-binding proteins, despite their apparent role in thyroid function homeostasis.
Metabolism of Thyroid Hormones
T4 must first be deiodinated to T3 to exert most of its biologic actions. Because little T3 is directly synthesized on thyroglobulin, this transformation becomes an important step in hormonogenesis. Three iodothyronine deiodinases are present in mammals.8 These are membrane-bound enzymes that are closely related structurally and are distinguished by the presence of selenocysteine at their active sites. Each has distinctive substrate preferences, activity characteristics, inhibitor sensitivities, and relative tissue specificity. Type I and II deiodinases, through their combined action, are responsible for generating approximately 80% of the total T3 production.
Type I deiodinase is the primary source of circulating T3 and is found in liver, kidney, and thyroid (where it is activated by TSH) tissues, and to a lesser extent in other tissues. Type I deiodinase is positively regulated by thyroid hormones and is greatly reduced under pathophysiologic states, such as starvation and nonthyroidal illnesses. It is inhibited by the antithyroid drug propylthiouracil. Type II deiodinase is present primarily in the central nervous system, the pituitary, the placenta, and the skin, and has more recently been found in the thyroid.12 Its major role is thought to be in the local production of T3, but it may also contribute to circulating T3. In contrast to type I deiodinase, the type II enzyme is negatively regulated by thyroid hormone and is unaffected by propylthiouracil. Type III deiodinase inactivates T4 and T3 by inner ring deiodination in the five position, forming reversed T3. The enzyme is present in the adult brain, skin, and placenta, and is present in high levels in fetal tissues, where it is thought to be important in protecting developing tissue from excess levels of thyroid hormone.13
Control of Thyroid Function
The anterior pituitary is the primary internal regulator of thyroid function, influencing virtually all phases of thyroid metabolism.14 It secretes TSH, also known as thyrotropin, which is a 28- to 30-kD lipoprotein consisting of two subunits, α and luteinizing hormone. The α subunit is common to the pituitary hormones follicle-stimulating hormone and luteinizing hormone and to chorionic gonadotropin. The α subunit is unique to TSH, however, and is responsible for the binding of the hormone to its receptor in the basal membrane of the thyroid cell. On interaction with TSH, the receptor, a member of a family of G protein–coupled receptors, undergoes conformational changes that activate one or two regulatory pathways. Most TSH effects are mediated by the activation of the cyclic adenosine monophosphate (cAMP) pathway; others involve the Ca2+/phosphatidylinositol cascade. The pathway used to elicit a given effect may vary among species. TSH stimulates the efflux of iodide into the follicle and the resorption of colloid into the cell within minutes. Later effects include increased expression of the NIS, thyroglobulin, and TPO genes; stimulation of H2O2 production; promotion of glycosylation; and increased production of T3 relative to T4.
Circulating levels of TSH are controlled by the opposing influences of thyroid hormone and of thyrotropin-releasing hormone (TRH) from the hypothalamus.15 The latter is a modified tripeptide secreted to the anterior pituitary by way of the hypothalamohypophyseal portal system. TRH binds to the plasma membrane of the thyrotrope and stimulates the release of TSH and the expression of its gene. Levels of circulating TSH are under strict control by the thyroid in a classic negative feedback system. As levels of thyroid hormone increase in response to TSH stimulation, T4 and T3 block the TRH-stimulated release of TSH in the thyrotrope. The thyroid hormones also act indirectly by inhibiting TRH gene expression in the hypothalamus.
Withdrawal of iodide from the diet leads to a rapid decrease in serum T4 and an increase in serum TSH. Serum T3 levels initially are unaffected, but eventually decline with prolonged withdrawal. In response to TSH stimulation, the thyroid increases iodide uptake and organification, alters the distribution of iodo-amino acids within thyroglobulin by increasing the ratios of MIT to DIT and T3 to T4, and increases the intrathyroidal conversion of T4 to T3 by type I and II deiodinases.8 With prolonged iodine deficiency, TSH-stimulated cell proliferation eventually leads to goiter.
Antithyroid Agents
Antithyroid drugs can inhibit thyroid hormone synthesis secretion or metabolism.16 Common agents and their major actions are summarized in Table 123-1. Numerous agents used in the treatment of nonthyroidal illnesses may have profound effects on thyroid hormone production. Notable among these are the iodinated radiocontrast agents that are potent inhibitors of thyroid hormone deiodination, and can interfere with hepatic uptake of T4 and binding of T3 to nuclear receptors.17 The antiarrhythmic agent amiodarone, which is also heavily iodinated, elicits similar alterations in thyroid hormone metabolism and action. Lithium, used in the treatment of bipolar illness, is a potent inhibitor of thyroid hormone release and acts by blocking thyroglobulin endocytosis.18
| Drug | Usual Starting Dose |
|---|---|
| Propylthiouracil | 200 mg PO tid |
| Methimazole | 20 mg PO bid |
| Propranolol | 10-40 mg PO qid |
| Saturated solution of potassium iodide (SSKI) | 1-2 drops PO qd-tid |
| Compound solution of iodine (Lugol’s solution) | 2-5 drops PO qd-tid |
| Dexamethasone | 2 mg PO qid |
| Prednisone | 40-60 mg po qd |
| Ipodate | 1 g PO qd |
| Lithium | 300-450 mg PO tid |
| Perchlorate | 1 g PO qd |
| Cholestyramine | 2-4 g PO bid-qid |
| Colestipol | 5 g PO one to five times a day |
| Octreotide | 50-100 µg SC bid-tid |
| Diltiazem | 120 mg PO tid |
Thyroid Hormone Mechanism of Action
The thyroid has multiple effects on development, growth, and metabolism. The effects on development are widespread phylogenetically and can be dramatically observed during the course of amphibian metamorphosis. The appropriate levels of thyroid hormone during fetal and neonatal stages in humans are crucial for the normal maturation of the central nervous system, muscle, bone, and lung. In severe cases of thyroid hormone deficiency during this period, the syndrome of cretinism results with its associated mental retardation, deafness, mutism, and stunted growth.19 Similarly, an excess of thyroid hormone during these critical developmental periods can result in neurologic abnormalities. The metabolic effects of thyroid hormone seem to be confined to birds and mammals, presumably evolving in response to the increased metabolic pressures of thermogenesis. Oxygen consumption and the metabolism of proteins, carbohydrates, and fats all are under thyroid hormone control.
Most effects of thyroid hormone are now believed to be exerted by interactions with specific nuclear thyroid hormone receptors, resulting in the altered expression of specific genes.20 T4 has little affinity for the nuclear receptors and first must be converted to T3 to be effective. The receptors themselves belong to a large superfamily of nuclear receptors, which includes the steroid hormones, retinoic acid, and vitamin D. The thyroid hormone receptors are closely related isoforms, despite being encoded by two different genes (α and β).
Thyroid Function Studies
Circulating Thyroid Hormone Measurement
Elevated total T4 levels may also occur when there is production of endogenous antibodies to T4, especially in patients with Hashimoto’s thyroiditis or other autoimmune disorders, and occasionally in patients with Waldenström’s macroglobulinemia associated with a benign monoclonal gammopathy.21 Another condition of elevated total T4 level is peripheral resistance to thyroid hormone. Individuals with this condition may have goiter, and they may be hyperactive.22 Patients with this disorder are euthyroid. Although rarely found, this disorder has led to inappropriate treatment for hyperthyroidism.
The most widely used measurement of thyrometabolic status is measurement of serum free T4 by equilibrium dialysis.23 When measured by the dialysis method, free T4 is not affected by changes in binding protein concentrations or by nonthyroidal illness. This method is cumbersome and expensive, and it is not routinely performed. Commercial free T4 levels are most commonly measured by immunoassay techniques, but their reliability is suboptimal because they may be affected by illness or significant changes in binding proteins.24 The clinical usefulness of free T4 measurements by any method may be limited.25
Although the thyrometabolic status is best reflected by the free T4 level, from a clinical standpoint, an index or estimate of free T4 is generally adequate. The free T4 index is obtained by multiplying the serum total T4 and an indirect assessment of thyroglobulin. Serum thyroglobulin is generally estimated by one of two methods—the thyroid uptake test and the T3 uptake test.25 The thyroid uptake test is directly proportional to thyroglobulin levels in serum, whereas the T3 uptake test is inversely proportional to thyroglobulin levels.26 The result, by use of either method, is that variances in serum thyroglobulin levels are largely eliminated, and the calculated free T4 index accurately reflects actual free T4 status. Extreme changes in thyroglobulin levels, or the presence of the severe nonthyroidal illness, may result in poor correlation between calculated and measured free T4 levels.
The principles used for obtaining the serum T3 are to determine the severity of hyperthyroidism and to confirm the diagnosis of suspected thyrotoxicosis in cases in which serum T4 levels are normal or equivocal. In addition, the serum T3 may be indicated in evaluating patients with autonomously functioning thyroid adenomas, in whom so-called T3 toxicosis may be present. Such patients may have normal or borderline elevated serum T4 levels along with suppressed serum TSH levels.27
Serum Thyrotropin Measurement
Until approximately 10 years ago, virtually all clinical TSH assays were performed by radioimmunoassay. By the mid-1980s, many commercial laboratories began using more sensitive immunometric TSH methods with either monoclonal or polyclonal antibodies. Functional sensitivity of these assays represented a 10-fold improvement in sensitivity over radioimmunoassay methods. More recently, nonisotopic immunometric TSH assays have been developed with a chemiluminescent label. These newer assays have a 10-fold greater sensitivity than the early immunometric TSH assays and are 100 times more sensitive than radioimmunoassay methods. These latest TSH assays, with a sensitivity of 0.01 mU/L, are currently termed third-generation TSH assays and represent the most sensitive method for detecting TSH level.28
The clinical application of TSH detection may be summarized as follows:
The fact that serum TSH is abnormal in hypothyroidism and hyperthyroidism would seem to make it ideally suited as a screen of thyroid status because, with rare exceptions, a normal TSH level would suggest normal thyroid hormone homeostasis. Experience in ambulatory individuals suggests that a normal TSH virtually excludes the possibility of thyroid dysfunction.34 In addition, the serum TSH level is more sensitive than the serum T4 level as a test for thyroid dysfunction because TSH can detect subclinical thyroid disorders in which serum total T4 (and T3) is usually normal. As a result of advances in TSH methodology, measurement of circulating thyroid hormones may become assigned to a second line of assessment of suspected thyroid dysfunction. Many investigators believe that the serum TSH is preferable as a screening method for thyrometabolic status.11,29 Figure 123-2 is an algorithm for the use of TSH level in the evaluation of thyroid function.
Serum Thyroglobulin Measurement
Thyroglobulin is elevated in the serum of patients with nearly all types of thyroid disorders, limiting its usefulness as a diagnostic test. Its greatest clinical value is in managing patients with well-differentiated thyroid carcinoma. An elevated or increasing thyroglobulin level (after initial surgical and ablation therapy) suggests persistence or recurrence of tumor.35 Thyroglobulin is measured by either radioimmunoassay or immunometric technique. Although antithyroid antibodies may cause interference with accurate thyroglobulin measurement in 10% of individuals, in these patients measurements of thyroglobulin and antithyroglobulin antibodies may be used concurrently to provide information regarding the tumor status.35
Thyroid Antibody Status
Circulating antithyroid antibodies, specifically antimicrosomal (AMA) and antithyroglobulin (ATA) antibodies, are usually present in patients with autoimmune thyroid disease.36 Since the introduction of immunoassay techniques, the term antithyroperoxidase (anti-TPO) has become interchangeable with AMA. AMAs are detectable in more than 90% of patients with chronic autoimmune thyroid disease; nearly 100% of patients with Hashimoto’s thyroiditis and more than 80% of patients with Graves’ disease have positive titers.37 Although ATAs are more specific than AMAs, they are less sensitive, and they are not as useful in the detection of autoimmune thyroid disease.38 Elevated levels of AMA are also frequently positive in various other organ-specific autoimmune diseases, such as lupus, rheumatoid arthritis, autoimmune anemia, Sjögren’s syndrome, type 1 diabetes mellitus, and Addison’s disease.39
Approximately 15% of adults (especially women) in the United States have elevated AMA titers.31 Prevalence of positive AMA titers increases with age, as does the incidence of primary hypothyroidism. The presence of a positive AMA titer should alert one to the possibility of hypothyroidism. Individuals with positive AMA and elevated TSH levels, even with normal serum total T4 levels (subclinical hypothyroidism), had a 3% to 5% per year likelihood of clinical hypothyroidism developing.40 In this manner, determination of AMA levels may be useful in the diagnosis of individuals with suspected autoimmune thyroid disease and in providing prognostic information when used in conjunction with TSH levels.
Thyroid-Stimulating Antibody Measurement
The immunopathogenesis of Graves’ disease was first suspected in the mid-1950s, when it was observed that injecting sera of patients with Graves’ disease into rats produced a prolonged uptake of radioactive iodine in the rat thyroid glands. The term long-acting thyroid stimulator (LATS) was coined.41 Later, LATS was characterized as a 7S immunoglobulin, and in recent years several assays have been developed for the detection of LATS, or thyroid-stimulating antibodies. Two methods are commonly used; one depends on generation of cAMP, and the other is a radioreceptor method that relies on the TSH binding inhibitory properties of the immunoglobulin. The cAMP-generating assay is termed thyroid-stimulating immunoglobulin, and it is detectable in 90% to 95% of hyperthyroid patients with Graves’ disease. The other assay detects stimulating and blocking antibodies termed TBII; it is detected in 85% of patients with hyperthyroid Graves’ disease.42 Thyroid-stimulating antibody measurements are not indicated for the routine diagnostic evaluation of suspected Graves’ disease, but they may be useful when the diagnosis of Graves’ disease is not evident.
