[level-membership-for-internal-medicine-category]
FIGURE 411-1 Pubertal events in males. Sexual maturity ratings for genitalia and pubic hair and divided into five stages. (From WA Marshall, JM Tanner: Variations in the pattern of pubertal changes in boys. Arch Dis Child 45:13, 1970.)
The early stages of puberty are characterized by nocturnal surges of LH and FSH. Growth of the testes is usually the first sign of puberty, reflecting an increase in seminiferous tubule volume. Increasing levels of testosterone deepen the voice and increase muscle growth. Conversion of testosterone to DHT leads to growth of the external genitalia and pubic hair. DHT also stimulates prostate and facial hair growth and initiates recession of the temporal hairline. The growth spurt occurs at a testicular volume of about 10–12 mL. GH increases early in puberty and is stimulated in part by the rise in gonadal steroids. GH increases the level of insulin-like growth factor I (IGF-I), which enhances linear bone growth. The prolonged pubertal exposure to gonadal steroids (mainly estradiol) ultimately causes epiphyseal closure and limits further bone growth.
REGULATION OF TESTICULAR FUNCTION
REGULATION OF THE HYPOTHALAMIC-PITUITARY-TESTIS AXIS IN ADULT MAN
Hypothalamic GnRH regulates the production of the pituitary gonadotropins LH and FSH (Fig. 411-2). GnRH is released in discrete pulses approximately every 2 h, resulting in corresponding pulses of LH and FSH. These dynamic hormone pulses account in part for the wide variations in LH and testosterone, even within the same individual. LH acts primarily on the Leydig cell to stimulate testosterone synthesis. The regulatory control of androgen synthesis is mediated by testosterone and estrogen feedback on both the hypothalamus and the pituitary. FSH acts on the Sertoli cell to regulate spermatogenesis and the production of Sertoli products such as inhibin B, which acts to selectively suppress pituitary FSH. Despite these somewhat distinct Leydig and Sertoli cell–regulated pathways, testis function is integrated at several levels: GnRH regulates both gonadotropins; spermatogenesis requires high levels of testosterone; and numerous paracrine interactions between Leydig and Sertoli cells are necessary for normal testis function.
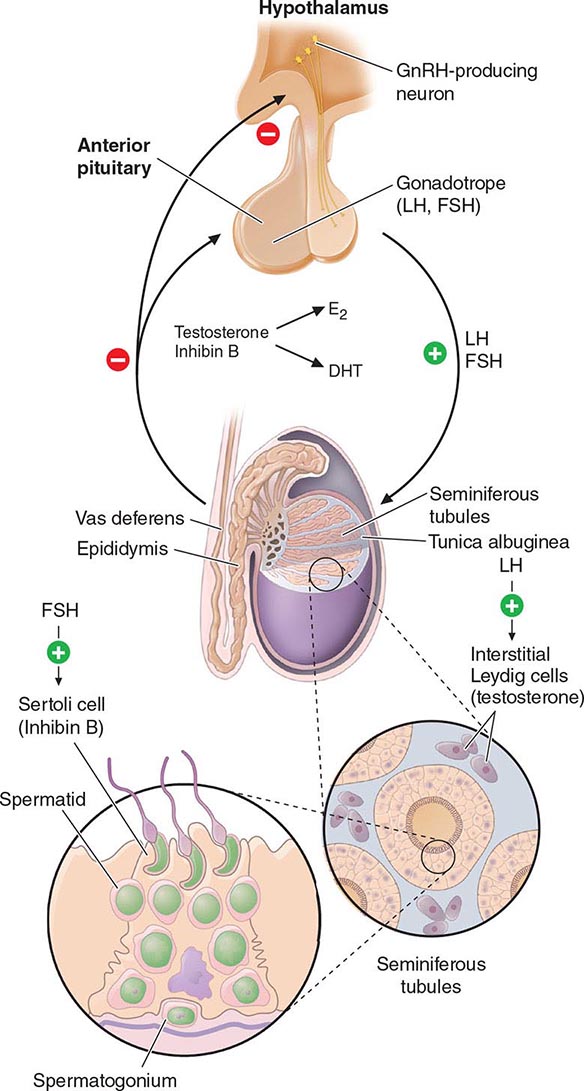
FIGURE 411-2 Human pituitary gonadotropin axis, structure of testis, and seminiferous tubule. E2, 17β-estradiol; DHT, dihydrotestosterone; FSH, follicle-stimulating hormones; GnRH, gonadotropin-releasing; LH, luteinizing hormone.
THE LEYDIG CELL: ANDROGEN SYNTHESIS
LH binds to its seven-transmembrane, G protein–coupled receptor to activate the cyclic AMP pathway. Stimulation of the LH receptor induces steroid acute regulatory (StAR) protein, along with several steroidogenic enzymes involved in androgen synthesis. LH receptor mutations cause Leydig cell hypoplasia or agenesis, underscoring the importance of this pathway for Leydig cell development and function. The rate-limiting process in testosterone synthesis is the delivery of cholesterol by the StAR protein to the inner mitochondrial membrane. Peripheral benzodiazepine receptor, a mitochondrial cholesterol-binding protein, is also an acute regulator of Leydig cell steroidogenesis. The five major enzymatic steps involved in testosterone synthesis are summarized in Fig. 411-3. After cholesterol transport into the mitochondrion, the formation of pregnenolone by CYP11A1 (side chain cleavage enzyme) is a limiting enzymatic step. The 17α-hydroxylase and the 17,20-lyase reactions are catalyzed by a single enzyme, CYP17; posttranslational modification (phosphorylation) of this enzyme and the presence of specific enzyme cofactors confer 17,20-lyase activity selectively in the testis and zona reticularis of the adrenal gland. Testosterone can be converted to the more potent DHT by 5α-reductase, or it can be aromatized to estradiol by CYP19 (aromatase). Two isoforms of steroid 5α-reductase, SRD5A1 and SRD5A2, have been described; all known kindreds with 5α-reductase deficiency have had mutations in SRD5A2, the predominant form in the prostate and the skin.
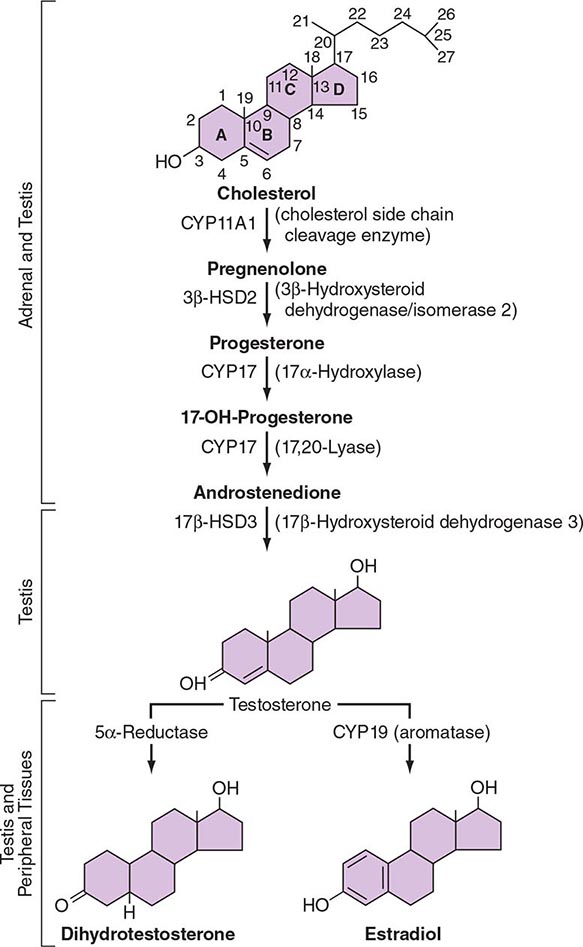
FIGURE 411-3 The biochemical pathway in the conversion of 27-carbon sterol cholesterol to androgens and estrogens.
Testosterone Transport and Metabolism In males, 95% of circulating testosterone is derived from testicular production (3–10 mg/d). Direct secretion of testosterone by the adrenal and the peripheral conversion of androstenedione to testosterone collectively account for another 0.5 mg/d of testosterone. Only a small amount of DHT (70 μg/d) is secreted directly by the testis; most circulating DHT is derived from peripheral conversion of testosterone. Most of the daily production of estradiol (~45 μg/d) in men is derived from aromatase-mediated peripheral conversion of testosterone and androstenedione.
Circulating testosterone is bound to two plasma proteins: sex hormone–binding globulin (SHBG) and albumin (Fig. 411-4). SHBG binds testosterone with much greater affinity than albumin. Only 0.5–3% of testosterone is unbound. According to the “free hormone” hypothesis, only the unbound fraction is biologically active; however, albumin-bound hormone dissociates readily in the capillaries and may be bioavailable. SHBG-bound testosterone also may be internalized through endocytic pits by binding to a protein called megalin. SHBG concentrations are decreased by androgens, obesity, diabetes mellitus, insulin, and nephrotic syndrome. Conversely, estrogen administration, hyperthyroidism, many chronic inflammatory illnesses, infections such as HIV or hepatitis B and C, and aging are associated with high SHBG concentrations.
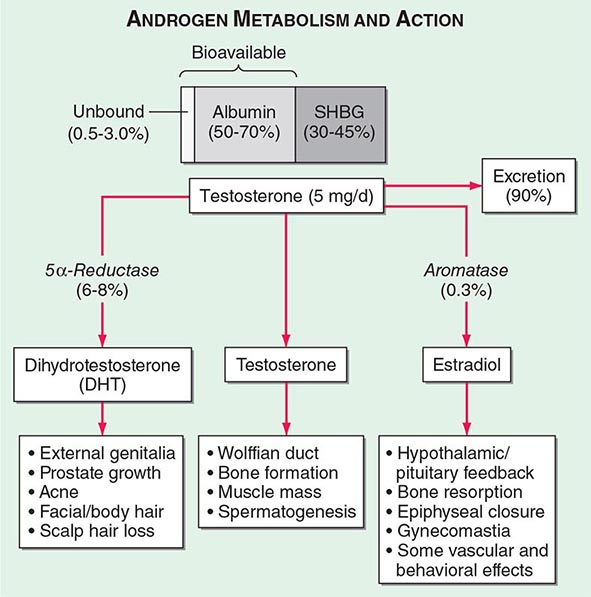
FIGURE 411-4 Androgen metabolism and actions. SHBG, sex hormone–binding globulin.
Testosterone is metabolized predominantly in the liver, although some degradation occurs in peripheral tissues, particularly the prostate and the skin. In the liver, testosterone is converted by a series of enzymatic steps that involve 5α- and 5β-reductases, 3α- and 3β-hydroxysteroid dehydrogenases, and 17β-hydroxysteroid dehydrogenase into androsterone, etiocholanolone, DHT, and 3-α-androstanediol. These compounds undergo glucuronidation or sulfation before being excreted by the kidneys.
Mechanism of Androgen Action Testosterone exerts some of its biologic effects by binding to androgen receptor, either directly or after its conversion to DHT by the steroid 5-α reductase. Testosterone’s effects on the skeletal muscle, erythropoiesis, and bone in men do not require its obligatory conversion to DHT. However, the conversion of testosterone to DHT is necessary for the masculinization of the urogenital sinus and genital tubercle. Aromatization of testosterone to estradiol mediates additional effects of testosterone on the bone resorption, epiphyseal closure, sexual desire, vascular endothelium, and fat. DHT can also be converted in some tissues by 3-keto reductase/3β-hydroxysteroid dehydrogenase enzymes to 5α-androstane-3β,17β-diol, which is a high-affinity ligand and agonist of estrogen receptor β.
The androgen receptor (AR) is structurally related to the nuclear receptors for estrogen, glucocorticoids, and progesterone (Chap. 400e). The AR is encoded by a gene on the long arm of the × chromosome and has a molecular mass of about 110 kDa. A polymorphic region in the amino terminus of the receptor, which contains a variable number of glutamine repeats, modifies the transcriptional activity of the receptor. The AR protein is distributed in both the cytoplasm and the nucleus. The ligand binding to the AR induces conformational changes that allow the recruitment and assembly of tissue-specific cofactors and causes it to translocate into the nucleus, where it binds to DNA or other transcription factors already bound to DNA. Thus, the AR is a ligand-regulated transcription factor that regulates the expression of androgen-dependent genes in a tissue-specific manner. Some androgen effects may be mediated by nongenomic AR signal transduction pathways. Testosterone binds to AR with half the affinity of DHT. The DHT-AR complex also has greater thermostability and a slower dissociation rate than the testosterone-AR complex. However, the molecular basis for selective testosterone versus DHT actions remains incompletely explained.
THE SEMINIFEROUS TUBULES: SPERMATOGENESIS
The seminiferous tubules are convoluted, closed loops with both ends emptying into the rete testis, a network of progressively larger efferent ducts that ultimately form the epididymis (Fig. 411-2). The seminiferous tubules total about 600 m in length and comprise about two-thirds of testis volume. The walls of the tubules are formed by polarized Sertoli cells that are apposed to peritubular myoid cells. Tight junctions between Sertoli cells create a blood-testis barrier. Germ cells compose the majority of the seminiferous epithelium (~60%) and are intimately embedded within the cytoplasmic extensions of the Sertoli cells, which function as “nurse cells.” Germ cells progress through characteristic stages of mitotic and meiotic divisions. A pool of type A spermatogonia serve as stem cells capable of self-renewal. Primary spermatocytes are derived from type B spermatogonia and undergo meiosis before progressing to spermatids that undergo spermiogenesis (a differentiation process involving chromatin condensation, acquisition of an acrosome, elongation of cytoplasm, and formation of a tail) and are released from Sertoli cells as mature spermatozoa. The complete differentiation process into mature sperm requires 74 days. Peristaltic-type action by peritubular myoid cells transports sperm into the efferent ducts. The spermatozoa spend an additional 21 days in the epididymis, where they undergo further maturation and capacitation. The normal adult testes produce >100 million sperm per day.
Naturally occurring mutations in the FSHβ gene and in the FSH receptor confirm an important, but not essential, role for this pathway in spermatogenesis. Females with these mutations are hypogonadal and infertile because ovarian follicles do not mature; males exhibit variable degrees of reduced spermatogenesis, presumably because of impaired Sertoli cell function. Because Sertoli cells produce inhibin B, an inhibitor of FSH, seminiferous tubule damage (e.g., by radiation) causes a selective increase of FSH. Testosterone reaches very high concentrations locally in the testis and is essential for spermatogenesis. The cooperative actions of FSH and testosterone are important in the progression of meiosis and spermiation. FSH and testosterone regulate germ cell survival via the intrinsic and the extrinsic apoptotic mechanisms. FSH may also play an important role in supporting spermatogonia. Gonadotropin-regulated testicular RNA helicase (GRTH/DDX25), a testis-specific gonadotropin/androgen-regulated RNA helicase, is present in germ cells and Leydig cells and may be an important factor in the paracrine regulation of germ cell development. Several cytokines and growth factors are also involved in the regulation of spermatogenesis by paracrine and autocrine mechanisms. A number of knockout mouse models exhibit impaired germ cell development or spermatogenesis, presaging possible mutations associated with male infertility. The human Y chromosome contains a small pseudoautosomal region that can recombine with homologous regions of the × chromosome. Most of the Y chromosome does not recombine with the × chromosome and is referred to as the male-specific region of the Y (MSY). The MSY contains 156 transcription units that encode for 26 proteins, including nine families of Y-specific multicopy genes; many of these Y-specific genes are also testis-specific and necessary for spermatogenesis. Microdeletions of several Y chromosome azoospermia factor (AZF) genes (e.g., RNA-binding motif, RBM; deleted in azoospermia, DAZ) are associated with oligospermia or azoospermia.
|
TREATMENT |
MALE FACTOR INFERTILITY |
Treatment options for male factor infertility have expanded greatly in recent years. Secondary hypogonadism is highly amenable to treatment with pulsatile GnRH or gonadotropins (see below). Assisted reproductive technologies such as the in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI) have provided new opportunities for patients with primary testicular failure and disorders of sperm transport. Choice of initial treatment options depends on sperm concentration and motility. Expectant management should be attempted initially in men with mild male factor infertility (sperm count of 15–20 × 106/mL and normal motility). Moderate male factor infertility (10–15 × 106/mL and 20–40% motility) should begin with intrauterine insemination alone or in combination with treatment of the female partner with clomiphene or gonadotropins, but it may require IVF with or without ICSI. For men with a severe defect (sperm count of <10 × 106/mL, 10% motility), IVF with ICSI or donor sperm should be used.
CLINICAL AND LABORATORY EVALUATION OF MALE REPRODUCTIVE FUNCTION
HISTORY AND PHYSICAL EXAMINATION
The history should focus on developmental stages such as puberty and growth spurts, as well as androgen-dependent events such as early morning erections, frequency and intensity of sexual thoughts, and frequency of masturbation or intercourse. Although libido and the overall frequency of sexual acts are decreased in androgen-deficient men, young hypogonadal men may achieve erections in response to visual erotic stimuli. Men with acquired androgen deficiency often report decreased energy and increased irritability.
The physical examination should focus on secondary sex characteristics such as hair growth, gynecomastia, testicular volume, prostate, and height and body proportions. Eunuchoid proportions are defined as an arm span >2 cm greater than height and suggest that androgen deficiency occurred before epiphyseal fusion. Hair growth in the face, axilla, chest, and pubic regions is androgen-dependent; however, changes may not be noticeable unless androgen deficiency is severe and prolonged. Ethnicity also influences the intensity of hair growth (Chap. 68). Testicular volume is best assessed by using a Prader orchidometer. Testes range from 3.5 to 5.5 cm in length, which corresponds to a volume of 12–25 mL. Advanced age does not influence testicular size, although the consistency becomes less firm. Asian men generally have smaller testes than Western Europeans, independent of differences in body size. Because of its possible role in infertility, the presence of varicocele should be sought by palpation while the patient is standing; it is more common on the left side. Patients with Klinefelter’s syndrome have markedly reduced testicular volumes (1–2 mL). In congenital hypogonadotropic hypogonadism, testicular volumes provide a good index for the degree of gonadotropin deficiency and the likelihood of response to therapy.
GONADOTROPIN AND INHIBIN MEASUREMENTS
LH and FSH are measured using two-site immunoradiometric, immunofluorometric, or chemiluminescent assays, which have very low cross-reactivity with other pituitary glycoprotein hormones and human chorionic gonadotropin (hCG) and have sufficient sensitivity to measure the low levels present in patients with hypogonadotropic hypogonadism. In men with a low testosterone level, an LH level can distinguish primary (high LH) versus secondary (low or inappropriately normal LH) hypogonadism. An elevated LH level indicates a primary defect at the testicular level, whereas a low or inappropriately normal LH level suggests a defect at the hypothalamic-pituitary level. LH pulses occur about every 1–3 h in normal men. Thus, gonadotropin levels fluctuate, and samples should be pooled or repeated when results are equivocal. FSH is less pulsatile than LH because it has a longer half-life. Selective increase in FSH suggests damage to the seminiferous tubules. Inhibin B, a Sertoli cell product that suppresses FSH, is reduced with seminiferous tubule damage. Inhibin B is a dimer with α-βB subunits and is measured by two-site immunoassays.
GnRH Stimulation Testing The GnRH test is performed by measuring LH and FSH concentrations at baseline and at 30 and 60 min after intravenous administration of 100 μg of GnRH. A minimally acceptable response is a twofold LH increase and a 50% FSH increase. In the prepubertal period or with severe GnRH deficiency, the gonadotrope may not respond to a single bolus of GnRH because it has not been primed by endogenous hypothalamic GnRH; in these patients, GnRH responsiveness may be restored by chronic, pulsatile GnRH administration. With the availability of sensitive and specific LH assays, GnRH stimulation testing is used rarely except to evaluate gonadotrope function in patients who have undergone pituitary surgery or have a space-occupying lesion in the hypothalamic-pituitary region.
TESTOSTERONE ASSAYS
Total Testosterone Total testosterone includes both unbound and protein-bound testosterone and is measured by radioimmunoassays, immunometric assays, or liquid chromatography tandem mass spectrometry (LC-MS/MS). LC-MS/MS involves extraction of serum by organic solvents, separation of testosterone from other steroids by high-performance liquid chromatography and mass spectrometry, and quantitation of unique testosterone fragments by mass spectrometry. LC-MS/MS provides accurate and sensitive measurements of testosterone levels even in the low range and is emerging as the method of choice for testosterone measurement. Laboratories that have been certified by the Centers for Disease Control and Prevention (CDC) Hormone Standardization Program for Testosterone (HoST) can ensure that testosterone measurements are accurate and calibrated to an international standard. A single fasting morning sample provides a good approximation of the average testosterone concentration with the realization that testosterone levels fluctuate in response to pulsatile LH. Testosterone is generally lower in the late afternoon and is reduced by acute illness. The testosterone concentration in healthy young men ranges from 300 to 1000 ng/dL in most laboratories, and efforts are under way to generate harmonized population-based reference ranges that can be applied to all CDC-certified laboratories. Alterations in SHBG levels due to aging, obesity, diabetes mellitus, hyperthyroidism, some types of medications, or chronic illness or on a congenital basis can affect total testosterone levels. Heritable factors contribute substantially to the population-level variation in testosterone levels, and genome-wide association studies have revealed polymorphisms in the SHBG gene as important contributors to variation in testosterone levels.
Measurement of Unbound Testosterone Levels Most circulating testosterone is bound to SHBG and to albumin; only 0.5–3% of circulating testosterone is unbound, or “free.” The unbound testosterone concentration can be measured by equilibrium dialysis or calculated from total testosterone, SHBG, and albumin concentrations. Recent research has shown that testosterone binding to SHBG is a multistep process that involves complex homoallostery within the SHBG dimer; a novel allosteric model of testosterone binding to SHBG dimers provides good estimates of free testosterone concentrations. The previous law of mass action equations based on linear models of testosterone binding to SHBG have been shown to be erroneous. Tracer analogue methods are relatively inexpensive and convenient, but they are inaccurate. Bioavailable testosterone refers to unbound testosterone plus testosterone that is loosely bound to albumin; it can be determined by the ammonium sulfate precipitation method.
hCG Stimulation Test The hCG stimulation test is performed by administering a single injection of 1500–4000 IU of hCG intramuscularly and measuring testosterone levels at baseline and 24, 48, 72, and 120 h after hCG injection. An alternative regimen involves three injections of 1500 units of hCG on successive days and measuring testosterone levels 24 h after the last dose. An acceptable response to hCG is a doubling of the testosterone concentration in adult men. In prepubertal boys, an increase in testosterone to >150 ng/dL indicates the presence of testicular tissue. No response may indicate an absence of testicular tissue or marked impairment of Leydig cell function. Measurement of MIS, a Sertoli cell product, is also used to detect the presence of testes in prepubertal boys with cryptorchidism.
SEMEN ANALYSIS
Semen analysis is the most important step in the evaluation of male infertility. Samples are collected by masturbation following a period of abstinence for 2–3 days. Semen volumes and sperm concentrations vary considerably among fertile men, and several samples may be needed before concluding that the results are abnormal. Analysis should be performed within an hour of collection. Using semen samples from over 4500 men in 14 countries, whose partners had a time-to-pregnancy of less than 12 months, the World Health Organization (WHO) has generated the following one-sided reference limits for semen parameters: semen volume, 1.5 mL; total sperm number, 39 million per ejaculate; sperm concentration, 15 million/mL; vitality, 58% live; progressive motility, 32%; total (progressive + nonprogressive) motility, 40%; morphologically normal forms, 4.0%. Some men with low sperm counts are nevertheless fertile. A variety of tests for sperm function can be performed in specialized laboratories, but these add relatively little to the treatment options.
TESTICULAR BIOPSY
Testicular biopsy is useful in some patients with oligospermia or azoospermia as an aid in diagnosis and indication for the feasibility of treatment. Using local anesthesia, fine-needle aspiration biopsy is performed to aspirate tissue for histology. Alternatively, open biopsies can be performed under local or general anesthesia when more tissue is required. A normal biopsy in an azoospermic man with a normal FSH level suggests obstruction of the vas deferens, which may be correctable surgically. Biopsies are also used to harvest sperm for ICSI and to classify disorders such as hypospermatogenesis (all stages present but in reduced numbers), germ cell arrest (usually at primary spermatocyte stage), and Sertoli cell–only syndrome (absent germ cells) or hyalinization (sclerosis with absent cellular elements).
DISORDERS OF SEXUAL DIFFERENTIATION
See Chap. 410.
DISORDERS OF PUBERTY
The onset and tempo of puberty varies greatly in the general population and is affected by genetic and environmental factors. Although some of the variance in the timing of puberty is explained by heritable factors, the genes involved remain unknown.
PRECOCIOUS PUBERTY
Puberty in boys before age 9 is considered precocious. Isosexual precocity refers to premature sexual development consistent with phenotypic sex and includes features such as the development of facial hair and phallic growth. Isosexual precocity is divided into gonadotropin-dependent and gonadotropin-independent causes of androgen excess (Table 411-1). Heterosexual precocity refers to the premature development of estrogenic features in boys, such as breast development.
|
CAUSES OF PRECOCIOUS OR DELAYED PUBERTY IN BOYS |
Abbreviations: CNS, central nervous system; GnRH, gonadotropin-releasing hormone; hCG, human chronic gonadotropin; LH, luteinizing hormone.
Gonadotropin-Dependent Precocious Puberty This disorder, called central precocious puberty (CPP), is less common in boys than in girls. It is caused by premature activation of the GnRH pulse generator, sometimes because of central nervous system (CNS) lesions such as hypothalamic hamartomas, but it is often idiopathic. CPP is characterized by gonadotropin levels that are inappropriately elevated for age. Because pituitary priming has occurred, GnRH elicits LH and FSH responses typical of those seen in puberty or in adults. Magnetic resonance imaging (MRI) should be performed to exclude a mass, structural defect, infection, or inflammatory process. Mutations in MKRN3, an imprinted gene encoding makorin ring-finger protein 3, which is expressed only from the paternally inherited allele, have been associated with CPP.
Gonadotropin-Independent Precocious Puberty In gonadotropin-independent precocious puberty, androgens from the testis or the adrenal are increased, but gonadotropins are low. This group of disorders includes hCG-secreting tumors; congenital adrenal hyperplasia; sex steroid–producing tumors of the testis, adrenal, and ovary; accidental or deliberate exogenous sex steroid administration; hypothyroidism; and activating mutations of the LH receptor or Gsα subunit.
FAMILIAL MALE-LIMITED PRECOCIOUS PUBERTY Also called testotoxicosis, familial male-limited precocious puberty is an autosomal dominant disorder caused by activating mutations in the LH receptor, leading to constitutive stimulation of the cyclic AMP pathway and testosterone production. Clinical features include premature androgenization in boys, growth acceleration in early childhood, and advanced bone age followed by premature epiphyseal fusion. Testosterone is elevated, and LH is suppressed. Treatment options include inhibitors of testosterone synthesis (e.g., ketoconazole), AR antagonists (e.g., flutamide and bicalutamide), and aromatase inhibitors (e.g., anastrazole).
MCCUNE-ALBRIGHT SYNDROME This is a sporadic disorder caused by somatic (postzygotic) activating mutations in the Gsα subunit that links G protein–coupled receptors to intracellular signaling pathways (Chap. 426e). The mutations impair the guanosine triphosphatase activity of the Gsα protein, leading to constitutive activation of adenylyl cyclase. Like activating LH receptor mutations, this stimulates testosterone production and causes gonadotropin-independent precocious puberty. In addition to sexual precocity, affected individuals may have autonomy in the adrenals, pituitary, and thyroid glands. Café au lait spots are characteristic skin lesions that reflect the onset of the somatic mutations in melanocytes during embryonic development. Polyostotic fibrous dysplasia is caused by activation of the parathyroid hormone receptor pathway in bone. Treatment is similar to that in patients with activating LH receptor mutations. Bisphosphonates have been used to treat bone lesions.
CONGENITAL ADRENAL HYPERPLASIA Boys with congenital adrenal hyperplasia (CAH) who are not well controlled with glucocorticoid suppression of adrenocorticotropic hormone (ACTH) can develop premature virilization because of excessive androgen production by the adrenal gland (Chaps. 406 and 410). LH is low, and the testes are small. Adrenal rests may develop within the testis of poorly controlled patients with CAH because of chronic ACTH stimulation; adrenal rests do not require surgical removal and regress with effective glucocorticoid therapy. Some children with CAH may develop gonadotropin-dependent precocious puberty with early maturation of the hypothalamic-pituitary-gonadal axis, elevated gonadotropins, and testicular growth.
Heterosexual Sexual Precocity Breast enlargement in prepubertal boys can result from familial aromatase excess, estrogen-producing tumors in the adrenal gland, Sertoli cell tumors in the testis, marijuana smoking, or exogenous estrogens or androgens. Occasionally, germ cell tumors that secrete hCG can be associated with breast enlargement due to excessive stimulation of estrogen production (see “Gynecomastia,” below).
|
TREATMENT |
PRECOCIOUS PUBERTY |
In patients with a known cause (e.g., a CNS lesion or a testicular tumor), therapy should be directed toward the underlying disorder. In patients with idiopathic CPP, long-acting GnRH analogues can be used to suppress gonadotropins and decrease testosterone, halt early pubertal development, delay accelerated bone maturation, prevent early epiphyseal closure, promote final height gain, and mitigate the psychosocial consequences of early pubertal development without causing osteoporosis. The treatment is most effective for increasing final adult height if it is initiated before age 6. Puberty resumes after discontinuation of the GnRH analogue. Counseling is an important aspect of the overall treatment strategy.
In children with gonadotropin-independent precocious puberty, inhibitors of steroidogenesis, such as ketoconazole, and AR antagonists have been used empirically. Long-term treatment with spironolactone (a weak androgen antagonist) and ketoconazole has been reported to normalize growth rate and bone maturation and to improve predicted height in small, nonrandomized trials in boys with familial male-limited precocious puberty. Aromatase inhibitors, such as testolactone and letrozole, have been used as an adjunct to antiandrogen and GnRH analogue therapy for children with familial male-limited precocious puberty, CAH, and McCune-Albright syndrome.
DELAYED PUBERTY
Puberty is delayed in boys if it has not ensued by age 14, an age that is 2–2.5 standard deviations above the mean for healthy children. Delayed puberty is more common in boys than in girls. There are four main categories of delayed puberty: (1) constitutional delay of growth and puberty (~60% of cases); (2) functional hypogonadotropic hypogonadism caused by systemic illness or malnutrition (~20% of cases); (3) hypogonadotropic hypogonadism caused by genetic or acquired defects in the hypothalamic-pituitary region (~10% of cases); and (4) hypergonadotropic hypogonadism secondary to primary gonadal failure (~15% of cases) (Table 411-1). Functional hypogonadotropic hypogonadism is more common in girls than in boys. Permanent causes of hypogonadotropic or hypergonadotropic hypogonadism are identified in >25% of boys with delayed puberty.
|
TREATMENT |
DELAYED PUBERTY |
If therapy is considered appropriate, it can begin with 25–50 mg testosterone enanthate or testosterone cypionate every 2 weeks, or by using a 2.5-mg testosterone patch or 25-mg testosterone gel. Because aromatization of testosterone to estrogen is obligatory for mediating androgen effects on epiphyseal fusion, concomitant treatment with aromatase inhibitors may allow attainment of greater final adult height. Testosterone treatment should be interrupted after 6 months to determine if endogenous LH and FSH secretion have ensued. Other causes of delayed puberty should be considered when there are associated clinical features or when boys do not enter puberty spontaneously after a year of observation or treatment.
Reassurance without hormonal treatment is appropriate for many individuals with presumed constitutional delay of puberty. However, the impact of delayed growth and pubertal progression on a child’s social relationships and school performance should be weighed. Also, boys with constitutional delay of puberty are less likely to achieve their full genetic height potential and have reduced total-body bone mass as adults, mainly due to narrow limb bones and vertebrae as a result of impaired periosteal expansion during puberty. Administration of androgen therapy to boys with constitutional delay does not affect final height, and when administered with an aromatase inhibitor, it may improve final height.
DISORDERS OF THE MALE REPRODUCTIVE AXIS DURING ADULTHOOD
HYPOGONADOTROPIC HYPOGONADISM
Because LH and FSH are trophic hormones for the testes, impaired secretion of these pituitary gonadotropins results in secondary hypogonadism, which is characterized by low testosterone in the setting of low LH and FSH. Those with the most severe deficiency have complete absence of pubertal development, sexual infantilism, and, in some cases, hypospadias and undescended testes. Patients with partial gonadotropin deficiency have delayed or arrested sex development. The 24-h LH secretory profiles are heterogeneous in patients with hypogonadotropic hypogonadism, reflecting variable abnormalities of LH pulse frequency or amplitude. In severe cases, basal LH is low and there are no LH pulses. A smaller subset of patients has low-amplitude LH pulses or markedly reduced pulse frequency. Occasionally, only sleep-entrained LH pulses occur, reminiscent of the pattern seen in the early stages of puberty. Hypogonadotropic hypogonadism can be classified into congenital and acquired disorders. Congenital disorders most commonly involve GnRH deficiency, which leads to gonadotropin deficiency. Acquired disorders are much more common than congenital disorders and may result from a variety of sellar mass lesions or infiltrative diseases of the hypothalamus or pituitary.
Congenital Disorders Associated with Gonadotropin Deficiency Congenital hypogonadotropic hypogonadism is a heterogeneous group of disorders characterized by decreased gonadotropin secretion and testicular dysfunction either due to impaired function of the GnRH pulse generator or the gonadotrope. The disorders characterized by GnRH deficiency represent a family of oligogenic disorders whose phenotype spans a wide spectrum. Some individuals with GnRH deficiency may suffer from complete absence of pubertal development, while others may manifest varying degrees of gonadotropin deficiency and pubertal delay, and a subset that carries the same mutations as their affected family members may even have normal reproductive function. In approximately 10% of men with idiopathic hypogonadotropic hypogonadism, reversal of gonadotropin deficiency may occur in adult life after sex steroid therapy. Also, a small fraction of men with idiopathic hypogonadotropic hypogonadism may present with androgen deficiency and infertility in adult life after having gone through apparently normal pubertal development. Nutritional, emotional, or metabolic stress may unmask gonadotropin deficiency and reproductive dysfunction (analogous to hypothalamic amenorrhea) in some patients who harbor mutations in the candidate genes but who previously had normal reproductive function. The clinical phenotype may include isolated anosmia or hyposmia. These striking variations in phenotypic presentation of GnRH deficiency have highlighted the important role of oligogenicity and gene-gene and gene-environment interactions in shaping the clinical phenotype.
Mutations in a number of genes involved in the development and migration of GnRH neurons or in the regulation of GnRH secretion have been linked to GnRH deficiency, although the genetic defect remains elusive in nearly two-thirds of cases. Familial hypogonadotropic hypogonadism can be transmitted as an X-linked (20%), autosomal recessive (30%), or autosomal dominant (50%) trait. Some individuals with idiopathic hypogonadotropic hypogonadism (IHH) have sporadic mutations in the same genes that cause inherited forms of the disorder. The genetic defects associated with GnRH deficiency can be conveniently classified as anosmic (Kallmann’s syndrome) or normosmic (Table 411-2), although the occurrence of both anosmic and normosmic forms of GnRH deficiency in the same families suggests commonality of pathophysiologic mechanisms. Kallmann’s syndrome, the anosmic form of GnRH deficiency, can result from mutations in one or more genes associated with olfactory bulb morphogenesis and the migration of GnRH neurons from their origin in the region of the olfactory placode, along the scaffold established by the olfactory nerves, through the cribriform plate into their final location into the preoptic region of the hypothalamus. Thus, mutations in KAL1, FGF8, FGFR1, NELF, PROK2, PROK2R, and CHD7 have been described in patients with Kallmann’s syndrome. An X-linked form of IHH is caused by mutations in the KAL1 gene, which encodes anosmin, a protein that mediates the migration of neural progenitors of the olfactory bulb and GnRH-producing neurons. These individuals have GnRH deficiency and variable combinations of anosmia or hyposmia, renal defects, and neurologic abnormalities including mirror movements. Mutations in the FGFR1 gene cause an autosomal dominant form of hypogonadotropic hypogonadism that clinically resembles Kallmann’s syndrome; mutations in its putative ligand, FGF8 gene product, have also been associated with IHH. Prokineticin 2 (PROK2) also encodes a protein involved in migration and development of olfactory and GnRH neurons. Recessive mutations in PROK2 or in its receptor, PROKR2, have been associated with both anosmic and normosmic forms of hypogonadotropic hypogonadism.
|
CAUSES OF CONGENITAL HYPOGONADOTROPIC HYPOGONADISM |
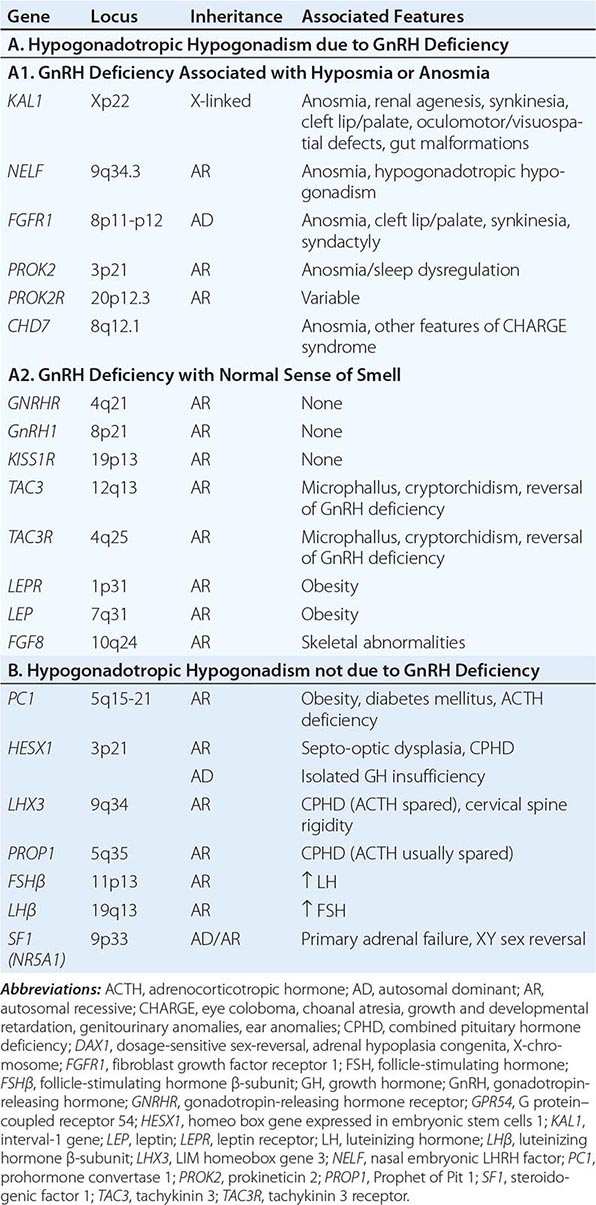
Normosmic GnRH deficiency results from defects in pulsatile GnRH secretion, its regulation, or its action on the gonadotrope and has been associated with mutations in GnRHR, GNRH1, KISS1R, TAC3, TACR3, and NROB1 (DAX1). Some mutations, such as those in PROK2, PROKR2, and CHD7, have been associated with both the anosmic and normosmic forms of IHH. GnRHR mutations, the most frequent identifiable cause of normosmic IHH, account for ~40% of autosomal recessive and 10% of sporadic cases of hypogonadotropic hypogonadism. These patients have decreased LH response to exogenous GnRH. Some receptor mutations alter GnRH binding affinity, allowing apparently normal responses to pharmacologic doses of exogenous GnRH, whereas other mutations may alter signal transduction downstream of hormone binding. Mutations of the GnRH1 gene have also been reported in patients with hypogonadotropic hypogonadism, although they are rare. G protein–coupled receptor KISS1R (GPR54) and its cognate ligand, kisspeptin (KISS1), are important regulators of sexual maturation in primates. Recessive mutations in GPR54 cause gonadotropin deficiency without anosmia. Patients retain responsiveness to exogenous GnRH, suggesting an abnormality in the neural pathways controlling GnRH release. The genes encoding neurokinin B (TAC3), which is involved in preferential activation of GnRH release in early development, and its receptor (TAC3R) have been implicated in some families with normosmic IHH. Mutations in more than one gene (digenicity or oligogenicity) may contribute to clinical heterogeneity in IHH patients. X-linked hypogonadotropic hypogonadism also occurs in adrenal hypoplasia congenita, a disorder caused by mutations in the DAX1 gene, which encodes a nuclear receptor in the adrenal gland and reproductive axis. Adrenal hypoplasia congenita is characterized by absent development of the adult zone of the adrenal cortex, leading to neonatal adrenal insufficiency. Puberty usually does not occur or is arrested, reflecting variable degrees of gonadotropin deficiency. Although sexual differentiation is normal, most patients have testicular dysgenesis and impaired spermatogenesis despite gonadotropin replacement. Less commonly, adrenal hypoplasia congenita, sex reversal, and hypogonadotropic hypogonadism can be caused by mutations of steroidogenic factor 1 (SF1). Rarely, recessive mutations in the LHβ or FSHβ gene have been described in patients with selective deficiencies of these gonadotropins. In approximately 10% of men with IHH, reversal of gonadotropin deficiency may occur in adult life. Also, a small fraction of men with IHH may present with androgen deficiency and infertility in adult life after having gone through apparently normal pubertal development.
A number of homeodomain transcription factors are involved in the development and differentiation of the specialized hormone-producing cells within the pituitary gland (Table 411-2). Patients with mutations of PROP1 have combined pituitary hormone deficiency that includes GH, prolactin (PRL), thyroid-stimulating hormone (TSH), LH, and FSH, but not ACTH. LHX3 mutations cause combined pituitary hormone deficiency in association with cervical spine rigidity. HESX1 mutations cause septo-optic dysplasia and combined pituitary hormone deficiency.
Prader-Willi syndrome is characterized by obesity, hypotonic musculature, mental retardation, hypogonadism, short stature, and small hands and feet. Prader-Willi syndrome is a genomic imprinting disorder caused by deletions of the proximal portion of the paternally derived chromosome 15q11-15q13 region, which contains a bipartite imprinting center, uniparental disomy of the maternal alleles, or mutations of the genes/loci involved in imprinting (Chap. 83e). Laurence-Moon syndrome is an autosomal recessive disorder characterized by obesity, hypogonadism, mental retardation, polydactyly, and retinitis pigmentosa. Recessive mutations of leptin, or its receptor, cause severe obesity and pubertal arrest, apparently because of hypothalamic GnRH deficiency (Chap. 415e).
Acquired Hypogonadotropic Disorders • SEVERE ILLNESS, STRESS, MALNUTRITION, AND EXERCISE These factors may cause reversible gonadotropin deficiency. Although gonadotropin deficiency and reproductive dysfunction are well documented in these conditions in women, men exhibit similar but less pronounced responses. Unlike women, most male runners and other endurance athletes have normal gonadotropin and sex steroid levels, despite low body fat and frequent intensive exercise. Testosterone levels fall at the onset of illness and recover during recuperation. The magnitude of gonadotropin suppression generally correlates with the severity of illness. Although hypogonadotropic hypogonadism is the most common cause of androgen deficiency in patients with acute illness, some have elevated levels of LH and FSH, which suggest primary gonadal dysfunction. The pathophysiology of reproductive dysfunction during acute illness is unknown but likely involves a combination of cytokine and/or glucocorticoid effects. There is a high frequency of low testosterone levels in patients with chronic illnesses such as HIV infection, end-stage renal disease, chronic obstructive lung disease, and many types of cancer and in patients receiving glucocorticoids. About 20% of HIV-infected men with low testosterone levels have elevated LH and FSH levels; these patients presumably have primary testicular dysfunction. The remaining 80% have either normal or low LH and FSH levels; these men have a central hypothalamic-pituitary defect or a dual defect involving both the testis and the hypothalamic-pituitary centers. Muscle wasting is common in chronic diseases associated with hypogonadism, which also leads to debility, poor quality of life, and adverse outcome of disease. There is great interest in exploring strategies that can reverse androgen deficiency or attenuate the sarcopenia associated with chronic illness.
Men using opioids for relief of cancer or noncancerous pain or because of addiction often have suppressed testosterone and LH levels and high prevalence of sexual dysfunction and osteoporosis; the degree of suppression is dose-related and particularly severe with long-acting opioids such as methadone. Opioids suppress GnRH secretion and alter the sensitivity to feedback inhibition by gonadal steroids. Men who are heavy users of marijuana have decreased testosterone secretion and sperm production. The mechanism of marijuana-induced hypogonadism is decreased GnRH secretion. Gynecomastia observed in marijuana users can also be caused by plant estrogens in crude preparations. Androgen deprivation therapy in men with prostate cancer has been associated with increased risk of bone fractures, diabetes mellitus, cardiovascular events, fatigue, sexual dysfunction, and poor quality of life.
OBESITY In men with mild to moderate obesity, SHBG levels decrease in proportion to the degree of obesity, resulting in lower total testosterone levels. However, free testosterone levels usually remain within the normal range. The decrease in SHBG levels is caused by increased circulating insulin, which inhibits SHBG production. Estradiol levels are higher in obese men compared to healthy, nonobese controls, because of aromatization of testosterone to estradiol in adipose tissue. Weight loss is associated with reversal of these abnormalities including an increase in total and free testosterone levels and a decrease in estradiol levels. A subset of obese men with moderate to severe obesity may have a defect in the hypothalamic-pituitary axis as suggested by low free testosterone in the absence of elevated gonadotropins. Weight gain in adult men can accelerate the rate of age-related decline in testosterone levels.
HYPERPROLACTINEMIA (See also Chap. 403) Elevated PRL levels are associated with hypogonadotropic hypogonadism. PRL inhibits hypothalamic GnRH secretion either directly or through modulation of tuberoinfundibular dopaminergic pathways. A PRL-secreting tumor may also destroy the surrounding gonadotropes by invasion or compression of the pituitary stalk. Treatment with dopamine agonists reverses gonadotropin deficiency, although there may be a delay relative to PRL suppression.
SELLAR MASS LESIONS Neoplastic and nonneoplastic lesions in the hypothalamus or pituitary can directly or indirectly affect gonadotrope function. In adults, pituitary adenomas constitute the largest category of space-occupying lesions affecting gonadotropin and other pituitary hormone production. Pituitary adenomas that extend into the suprasellar region can impair GnRH secretion and mildly increase PRL secretion (usually <50 μg/L) because of impaired tonic inhibition by dopaminergic pathways. These tumors should be distinguished from prolactinomas, which typically secrete higher PRL levels. The presence of diabetes insipidus suggests the possibility of a craniopharyngioma, infiltrative disorder, or other hypothalamic lesions (Chap. 404).
HEMOCHROMATOSIS (See also Chap. 428) Both the pituitary and testis can be affected by excessive iron deposition. However, the pituitary defect is the predominant lesion in most patients with hemochromatosis and hypogonadism. The diagnosis of hemochromatosis is suggested by the association of characteristic skin discoloration, hepatic enlargement or dysfunction, diabetes mellitus, arthritis, cardiac conduction defects, and hypogonadism.
PRIMARY TESTICULAR CAUSES OF HYPOGONADISM
Common causes of primary testicular dysfunction include Klinefelter’s syndrome, uncorrected cryptorchidism, cancer chemotherapy, radiation to the testes, trauma, torsion, infectious orchitis, HIV infection, anorchia syndrome, and myotonic dystrophy. Primary testicular disorders may be associated with impaired spermatogenesis, decreased androgen production, or both. See Chap. 410 for disorders of testis development, androgen synthesis, and androgen action.
Klinefelter’s Syndrome (See also Chap. 410) Klinefelter’s syndrome is the most common chromosomal disorder associated with testicular dysfunction and male infertility. It occurs in about 1 in 600 live-born males. Azoospermia is the rule in men with Klinefelter’s syndrome who have the 47,XXY karyotype; however, men with mosaicism may have germ cells, especially at a younger age. The clinical phenotype of Klinefelter’s syndrome can be heterogeneous possibly because of mosaicism, polymorphisms in AR gene, variable testosterone levels, or other genetic factors. Testicular histology shows hyalinization of seminiferous tubules and absence of spermatogenesis. Although their function is impaired, the number of Leydig cells appears to increase. Testosterone is decreased and estradiol is increased, leading to clinical features of undervirilization and gynecomastia. Men with Klinefelter’s syndrome are at increased risk of systemic lupus erythematosus, Sjögren’s syndrome, breast cancer, diabetes mellitus, osteoporosis, non-Hodgkin’s lymphoma, and lung cancer, and reduced risk of prostate cancer. Periodic mammography for breast cancer surveillance is recommended for men with Klinefelter’s syndrome. Fertility has been achieved by intracytoplasmic injection of sperm retrieved surgically from testicular biopsies of men with Klinefelter’s syndrome, including some men with the nonmosaic form of Klinefelter’s syndrome. The karyotypes 48,XXXY and 49,XXXXY are associated with a more severe phenotype, increased risk of congenital malformations, and lower intelligence than 47,XXY individuals.
Cryptorchidism Cryptorchidism occurs when there is incomplete descent of the testis from the abdominal cavity into the scrotum. About 3% of full-term and 30% of premature male infants have at least one undescended testis at birth, but descent is usually complete by the first few weeks of life. The incidence of cryptorchidism is <1% by 9 months of age. Androgens regulate predominantly the inguinoscrotal descent of the testes through degeneration of the craniosuspensory ligament and a shortening of the gubernaculums, respectively. Mutations in INSL3 and leucine-rich repeat family of G protein–coupled receptor 8 (LGR8), which regulate the transabdominal portion of testicular descent, have been found in some patients with cryptorchidism.
Cryptorchidism is associated with increased risk of malignancy, infertility, inguinal hernia, and torsion. Unilateral cryptorchidism, even when corrected before puberty, is associated with decreased sperm count, possibly reflecting unrecognized damage to the fully descended testis or other genetic factors. Epidemiologic, clinical, and molecular evidence supports the idea that cryptorchidism, hypospadias, impaired spermatogenesis, and testicular cancer may be causally related to common genetic and environment perturbations and are components of the testicular dysgenesis syndrome.
Acquired Testicular Defects Viral orchitis may be caused by the mumps virus, echovirus, lymphocytic choriomeningitis virus, and group B arboviruses. Orchitis occurs in as many as one-fourth of adult men with mumps; the orchitis is unilateral in about two-thirds and bilateral in the remainder. Orchitis usually develops a few days after the onset of parotitis but may precede it. The testis may return to normal size and function or undergo atrophy. Semen analysis returns to normal for three-fourths of men with unilateral involvement but for only one-third of men with bilateral orchitis. Trauma, including testicular torsion, can also cause secondary atrophy of the testes. The exposed position of the testes in the scrotum renders them susceptible to both thermal and physical trauma, particularly in men with hazardous occupations.
The testes are sensitive to radiation damage. Doses >200 mGy (20 rad) are associated with increased FSH and LH levels and damage to the spermatogonia. After ~800 mGy (80 rad), oligospermia or azoospermia develops, and higher doses may obliterate the germinal epithelium. Permanent androgen deficiency in adult men is uncommon after therapeutic radiation; however, most boys given direct testicular radiation therapy for acute lymphoblastic leukemia have permanently low testosterone levels. Sperm banking should be considered before patients undergo radiation treatment or chemotherapy.
Drugs interfere with testicular function by several mechanisms, including inhibition of testosterone synthesis (e.g., ketoconazole), blockade of androgen action (e.g., spironolactone), increased estrogen (e.g., marijuana), or direct inhibition of spermatogenesis (e.g., chemotherapy).
Combination chemotherapy for acute leukemia, Hodgkin’s disease, and testicular and other cancers may impair Leydig cell function and cause infertility. The degree of gonadal dysfunction depends on the type of chemotherapeutic agent and the dose and duration of therapy. Because of high response rates and the young age of these men, infertility and androgen deficiency have emerged as important long-term complications of cancer chemotherapy. Cyclophosphamide and combination regimens containing procarbazine are particularly toxic to germ cells. Thus, 90% of men with Hodgkin’s lymphoma receiving MOPP (mechlorethamine, vincristine, procarbazine, prednisone) therapy develop azoospermia or extreme oligozoospermia; newer regimens that do not include procarbazine, such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine), are less toxic to germ cells.
Alcohol, when consumed in excess for prolonged periods, decreases testosterone, independent of liver disease or malnutrition. Elevated estradiol and decreased testosterone levels may occur in men taking digitalis.
The occupational and recreational history should be carefully evaluated in all men with infertility because of the toxic effects of many chemical agents on spermatogenesis. Known environmental hazards include pesticides (e.g., vinclozolin, dicofol, atrazine), sewage contaminants (e.g., ethinyl estradiol in birth control pills, surfactants such as octylphenol, nonyphenol), plasticizers (e.g., pthalates), flame retardants (e.g., polychlorinated biphenyls, polybrominated diphenol ethers), industrial pollutants (e.g., heavy metals cadmium and lead, dioxins, polycyclic aromatic hydrocarbons), microwaves, and ultrasound. In some populations, sperm density is said to have declined by as much as 40% in the past 50 years. Environmental estrogens or antiandrogens may be partly responsible.
Testicular failure also occurs as a part of polyglandular autoimmune insufficiency (Chap. 408). Sperm antibodies can cause isolated male infertility. In some instances, these antibodies are secondary phenomena resulting from duct obstruction or vasectomy. Granulomatous diseases can affect the testes, and testicular atrophy occurs in 10–20% of men with lepromatous leprosy because of direct tissue invasion by the mycobacteria. The tubules are involved initially, followed by endarteritis and destruction of Leydig cells.
Systemic disease can cause primary testis dysfunction in addition to suppressing gonadotropin production. In cirrhosis, a combined testicular and pituitary abnormality leads to decreased testosterone production independent of the direct toxic effects of ethanol. Impaired hepatic extraction of adrenal androstenedione leads to extraglandular conversion to estrone and estradiol, which partially suppresses LH. Testicular atrophy and gynecomastia are present in approximately one-half of men with cirrhosis. In chronic renal failure, androgen synthesis and sperm production decrease despite elevated gonadotropins. The elevated LH level is due to reduced clearance, but it does not restore normal testosterone production. About one-fourth of men with renal failure have hyperprolactinemia. Improvement in testosterone production with hemodialysis is incomplete, but successful renal transplantation may return testicular function to normal. Testicular atrophy is present in one-third of men with sickle cell anemia. The defect may be at either the testicular or the hypothalamic-pituitary level. Sperm density can decrease temporarily after acute febrile illness in the absence of a change in testosterone production. Infertility in men with celiac disease is associated with a hormonal pattern typical of androgen resistance, namely elevated testosterone and LH levels.
Neurologic diseases associated with altered testicular function include myotonic dystrophy, spinobulbar muscular atrophy, and paraplegia. In myotonic dystrophy, small testes may be associated with impairment of both spermatogenesis and Leydig cell function. Spinobulbar muscular atrophy is caused by an expansion of the glutamine repeat sequences in the amino-terminal region of the AR; this expansion impairs function of the AR, but it is unclear how the alteration is related to the neurologic manifestations. Men with spinobulbar muscular atrophy often have undervirilization and infertility as a late manifestation. Spinal cord lesions that cause paraplegia can lead to a temporary decrease in testosterone levels and may cause persistent defects in spermatogenesis; some patients retain the capacity for penile erection and ejaculation.
ANDROGEN INSENSITIVITY SYNDROMES
Mutations in the AR cause resistance to the action of testosterone and DHT. These X-linked mutations are associated with variable degrees of defective male phenotypic development and undervirilization (Chap. 410). Although not technically hormone-insensitivity syndromes, two genetic disorders impair testosterone conversion to active sex steroids. Mutations in the SRD5A2 gene, which encodes 5α-reductase type 2, prevent the conversion of testosterone to DHT, which is necessary for the normal development of the male external genitalia. Mutations in the CYP19 gene, which encodes aromatase, prevent testosterone conversion to estradiol. Males with CYP19 mutations have delayed epiphyseal fusion, tall stature, eunuchoid proportions, and osteoporosis, consistent with evidence from an estrogen receptor–deficient individual that these testosterone actions are mediated indirectly via estrogen.
GYNECOMASTIA
Gynecomastia refers to enlargement of the male breast. It is caused by excess estrogen action and is usually the result of an increased estrogen-to-androgen ratio. True gynecomastia is associated with glandular breast tissue that is >4 cm in diameter and often tender. Glandular tissue enlargement should be distinguished from excess adipose tissue: glandular tissue is firmer and contains fibrous-like cords. Gynecomastia occurs as a normal physiologic phenomenon in the newborn (due to transplacental transfer of maternal and placental estrogens), during puberty (high estrogen-to-androgen ratio in early stages of puberty), and with aging (increased fat tissue and increased aromatase activity), but it can also result from pathologic conditions associated with androgen deficiency or estrogen excess. The prevalence of gynecomastia increases with age and body mass index (BMI), likely because of increased aromatase activity in adipose tissue. Medications that alter androgen metabolism or action may also cause gynecomastia. The relative risk of breast cancer is increased in men with gynecomastia, although the absolute risk is relatively small.
PATHOLOGIC GYNECOMASTIA
Any cause of androgen deficiency can lead to gynecomastia, reflecting an increased estrogen-to-androgen ratio, because estrogen synthesis still occurs by aromatization of residual adrenal and gonadal androgens. Gynecomastia is a characteristic feature of Klinefelter’s syndrome (Chap. 410). Androgen insensitivity disorders also cause gynecomastia. Excess estrogen production may be caused by tumors, including Sertoli cell tumors in isolation or in association with Peutz-Jeghers syndrome or Carney complex. Tumors that produce hCG, including some testicular tumors, stimulate Leydig cell estrogen synthesis. Increased conversion of androgens to estrogens can be a result of increased availability of substrate (androstenedione) for extraglandular estrogen formation (CAH, hyperthyroidism, and most feminizing adrenal tumors) or of diminished catabolism of androstenedione (liver disease) so that estrogen precursors are shunted to aromatase in peripheral sites. Obesity is associated with increased aromatization of androgen precursors to estrogens. Extraglandular aromatase activity can also be increased in tumors of the liver or adrenal gland or rarely as an inherited disorder. Several families with increased peripheral aromatase activity inherited as an autosomal dominant or as an X-linked disorder have been described. In some families with this disorder, an inversion in chromosome 15q21.2-3 causes the CYP19 gene to be activated by the regulatory elements of contiguous genes, resulting in excessive estrogen production in the fat and other extragonadal tissues. Drugs can cause gynecomastia by acting directly as estrogenic substances (e.g., oral contraceptives, phytoestrogens, digitalis) or by inhibiting androgen synthesis (e.g., ketoconazole) or action (e.g., spironolactone).
Because up to two-thirds of pubertal boys and half of hospitalized men have palpable glandular tissue that is benign, detailed investigation or intervention is not indicated in all men presenting with gynecomastia (Fig. 411-5). In addition to the extent of gynecomastia, recent onset, rapid growth, tender tissue, and occurrence in a lean subject should prompt more extensive evaluation. This should include a careful drug history, measurement and examination of the testes, assessment of virilization, evaluation of liver function, and hormonal measurements including testosterone, estradiol, and androstenedione, LH, and hCG. A karyotype should be obtained in men with very small testes to exclude Klinefelter’s syndrome. Despite extensive evaluation, the etiology is established in fewer than one-half of patients.
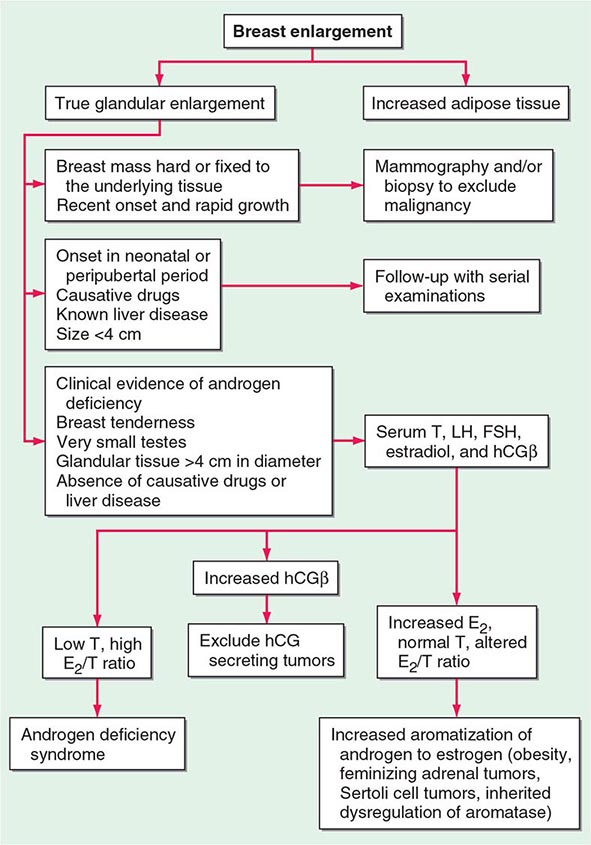
FIGURE 411-5 Evaluation of gynecomastia. E2, 17β-estradiol; hCGβ, human chorionic gonadotropin β; T, testosterone.
|
TREATMENT |
GYNECOMASTIA |
When the primary cause can be identified and corrected, breast enlargement usually subsides over several months. However, if gynecomastia is of long duration, surgery is the most effective therapy. Indications for surgery include severe psychological and/or cosmetic problems, continued growth or tenderness, or suspected malignancy. In patients who have painful gynecomastia and in whom surgery cannot be performed, treatment with antiestrogens such as tamoxifen (20 mg/d) can reduce pain and breast tissue size in over half the patients. Estrogen receptor antagonists, tamoxifen and raloxifene, have been reported in small trials to reduce breast size in men with pubertal gynecomastia, although complete regression of breast enlargement is unusual with the use of estrogen receptor antagonists. Aromatase inhibitors can be effective in the early proliferative phase of the disorder. However, in a randomized trial in men with established gynecomastia, anastrozole proved no more effective than placebo in reducing breast size. Tamoxifen is effective in the prevention and treatment of breast enlargement and breast pain in men with prostate cancer who are receiving antiandrogen therapy.
AGING-RELATED CHANGES IN MALE REPRODUCTIVE FUNCTION
A number of cross-sectional and longitudinal studies (e.g., The Baltimore Longitudinal Study of Aging, the Framingham Heart Study, the Massachusetts Male Aging Study, and the European Male Aging Study) have established that testosterone concentrations decrease with advancing age. This age-related decline starts in the third decade of life and progresses slowly; the rate of decline in testosterone concentrations is greater in obese men, men with chronic illness, and those taking medications than in healthy older men. Because SHBG concentrations are higher in older men than in younger men, free or bioavailable testosterone concentrations decline with aging to a greater extent than total testosterone concentrations. The age-related decline in testosterone is due to defects at all levels of the hypothalamic-pituitary-testicular axis: pulsatile GnRH secretion is attenuated, LH response to GnRH is reduced, and testicular response to LH is impaired. However, the gradual rise of LH with aging suggests that testis dysfunction is the main cause of declining androgen levels. The term andropause has been used to denote age-related decline in testosterone concentrations; this term is a misnomer because there is no discrete time when testosterone concentrations decline abruptly. The approach to evaluating hypogonadism is summarized in Fig. 411-6.
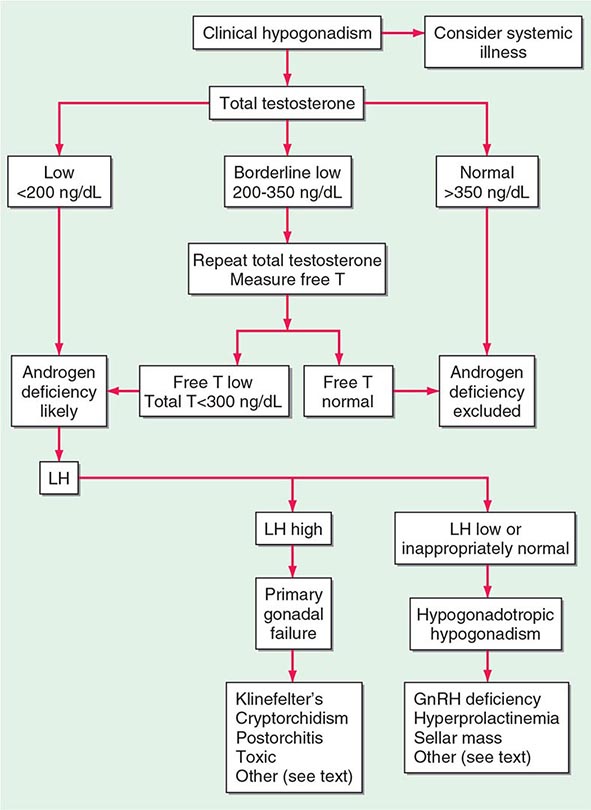
FIGURE 411-6 Evaluation of hypogonadism. GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; T, testosterone.
In epidemiologic surveys, low total and bioavailable testosterone concentrations have been associated with decreased appendicular skeletal muscle mass and strength, decreased self-reported physical function, higher visceral fat mass, insulin resistance, and increased risk of coronary artery disease and mortality, although the associations are weak. An analysis of signs and symptoms in older men in the European Male Aging Study revealed a syndromic association of sexual symptoms with total testosterone levels below 320 ng/dL and free testosterone levels below 64 pg/mL in community-dwelling older men. In systematic reviews of randomized controlled trials, testosterone therapy of healthy older men with low or low-normal testosterone levels was associated with greater increments in lean body mass, grip strength, and self-reported physical function compared with placebo. Testosterone therapy also induced greater improvement in vertebral but not femoral bone mineral density. Testosterone therapy of older men with sexual dysfunction and unequivocally low testosterone levels improves libido, but testosterone effects on erectile function and response to selective phosphodiesterase inhibitors have been inconsistent. Testosterone therapy has not been shown to improve depression scores, fracture risk, cognitive function, response to phosphodiesterase inhibitors, or clinical outcomes in older men. Furthermore, neither the long-term risks nor clinical benefits of testosterone therapy in older men have been demonstrated in adequately powered trials. Although there is no evidence that testosterone causes prostate cancer, there is concern that testosterone therapy might cause subclinical prostate cancers to grow. Testosterone therapy is associated with increased risk of detection of prostate events (Fig. 411-7).
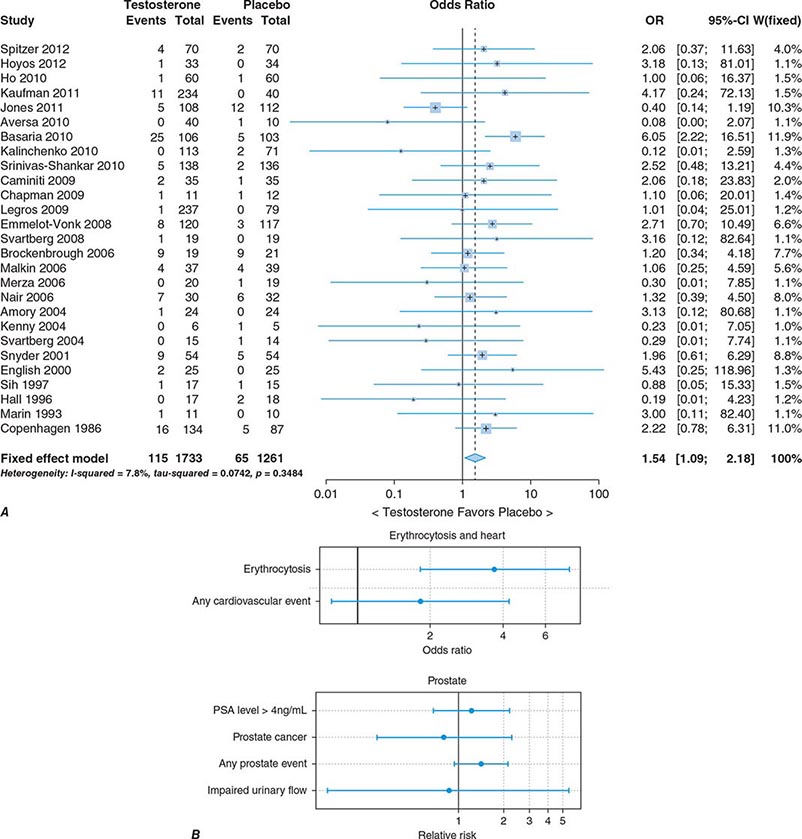
FIGURE 411-7 Meta-analyses of cardiovascular and prostate adverse events associated with testosterone therapy. A. A meta-analysis of cardiovascular-related events in randomized testosterone trials of 12 weeks or longer in duration. Randomization to testosterone was associated with a significantly increased risk of cardiovascular-related event (odds ratio [OR] 1.54). (Modified with permission from L Xu et al: Testosterone therapy and cardiovascular events among men: a systematic review and meta-analysis of placebo-controlled randomized trials BMC Med 11:108, 2013.) B. The relative risk of prostate events and the associated 95% confidence intervals (CIs) in a meta-analysis of randomized testosterone trials. PSA, prostate-specific antigen. (Data were derived from a meta-analysis by MM Fernández-Balsells et al: J Clin Endocrinol Metab 95:2560, 2010, and the figure was reproduced with permission from M Spitzer et al: Nat Rev Endocrinol 9:414, 2013.)
One randomized testosterone trial in older men with mobility limitation and high burden of chronic conditions, such as diabetes, heart disease, hypertension, and hyperlipidemia, reported a greater number of cardiovascular events in men randomized to the testosterone arm of the study than in those randomized to the placebo arm. Since then, two large retrospective analyses of patient databases have reported higher frequency of cardiovascular events, including myocardial infarction, in older men with preexisting heart disease (Fig. 411-7).
Population screening of all older men for low testosterone levels is not recommended, and testing should be restricted to men who have symptoms or physical features attributable to androgen deficiency. Testosterone therapy is not recommended for all older men with low testosterone levels. In older men with significant symptoms of androgen deficiency who have testosterone levels below 200 ng/dL, testosterone therapy may be considered on an individualized basis and should be instituted after careful discussion of the risks and benefits (see “Testosterone Replacement,” below).
Testicular morphology, semen production, and fertility are maintained up to a very old age in men. Although concern has been expressed about age-related increases in germ cell mutations and impairment of DNA repair mechanisms, there is no clear evidence that the frequency of chromosomal aneuploidy is increased in the sperm of older men. However, the incidence of autosomal dominant diseases, such as achondroplasia, polyposis coli, Marfan’s syndrome, and Apert’s syndrome, increases in the offspring of men who are advanced in age, consistent with transmission of sporadic missense mutations. Advanced paternal age may be associated with increased rates of de novo mutations, which may contribute to an increased risk of neurodevelopmental diseases such as schizophrenia and autism. The somatic mutations in male germ cells that enhance the proliferation of germ cells could lead to within-testis expansion of mutant clonal lines, thus favoring the propagation of germ cells carrying these pathogenic mutations and increasing the risk of mutations in the offspring of older fathers (the “selfish spermatogonial selection” hypothesis).
|
TREATMENT |
ANDROGEN DEFICIENCY |
GONADOTROPINS
Gonadotropin therapy is used to establish or restore fertility in patients with gonadotropin deficiency of any cause. Several gonadotropin preparations are available. Human menopausal gonadotropin (hMG; purified from the urine of postmenopausal women) contains 75 IU FSH and 75 IU LH per vial. hCG (purified from the urine of pregnant women) has little FSH activity and resembles LH in its ability to stimulate testosterone production by Leydig cells. Recombinant LH is now available. Because of the expense of hMG, treatment is usually begun with hCG alone, and hMG is added later to promote the FSH-dependent stages of spermatid development. Recombinant human FSH (hFSH) is now available and is indistinguishable from purified urinary hFSH in its biologic activity and pharmacokinetics in vitro and in vivo, although the mature β subunit of recombinant hFSH has seven fewer amino acids. Recombinant hFSH is available in ampoules containing 75 IU (~7.5 μg FSH), which accounts for >99% of protein content. Once spermatogenesis is restored using combined FSH and LH therapy, hCG alone is often sufficient to maintain spermatogenesis.
Although a variety of treatment regimens are used, 1000–2000 IU of hCG or recombinant human LH (rhLH) administered intramuscularly three times weekly is a reasonable starting dose. Testosterone levels should be measured 6–8 weeks later and 48–72 h after the hCG or rhLH injection; the hCG/rhLH dose should be adjusted to achieve testosterone levels in the mid-normal range. Sperm counts should be monitored on a monthly basis. It may take several months for spermatogenesis to be restored; therefore, it is important to forewarn patients about the potential length and expense of the treatment and to provide conservative estimates of success rates. If testosterone levels are in the mid-normal range but the sperm concentrations are low after 6 months of therapy with hCG alone, FSH should be added. This can be done by using hMG, highly purified urinary hFSH, or recombinant hFSH. The selection of FSH dose is empirical. A common practice is to start with the addition of 75 IU FSH three times each week in conjunction with the hCG/rhLH injections. If sperm densities are still low after 3 months of combined treatment, the FSH dose should be increased to 150 IU. Occasionally, it may take ≥18–24 months for spermatogenesis to be restored.
The two best predictors of success using gonadotropin therapy in hypogonadotropic men are testicular volume at presentation and time of onset. In general, men with testicular volumes >8 mL have better response rates than those who have testicular volumes >4 mL. Patients who became hypogonadotropic after puberty experience higher success rates than those who have never undergone pubertal changes. Spermatogenesis can usually be reinitiated by hCG alone, with high rates of success for men with postpubertal onset of hypogonadotropism. The presence of a primary testicular abnormality, such as cryptorchidism, will attenuate testicular response to gonadotropin therapy. Prior androgen therapy does not preclude subsequent response to gonadotropin therapy, although some studies suggest that it may attenuate response to subsequent gonadotropin therapy.
TESTOSTERONE REPLACEMENT
Androgen therapy is indicated to restore testosterone levels to normal to correct features of androgen deficiency. Testosterone replacement improves libido and overall sexual activity; increases energy, lean muscle mass, and bone density; and decreases fat mass. The benefits of testosterone replacement therapy have only been proven in men who have documented androgen deficiency, as demonstrated by testosterone levels that are well below the lower limit of normal.
Testosterone is available in a variety of formulations with distinct pharmacokinetics (Table 411-3). Testosterone serves as a prohormone and is converted to 17β-estradiol by aromatase and to 5α-dihydrotestosterone by steroid 5α-reductase. Therefore, when evaluating testosterone formulations, it is important to consider whether the formulation being used can achieve physiologic estradiol and DHT concentrations, in addition to normal testosterone concentrations. Although testosterone concentrations at the lower end of the normal male range can restore sexual function, it is not clear whether low-normal testosterone levels can maintain bone mineral density and muscle mass. The current recommendation is to restore testosterone levels to the mid-normal range.
|
CLINICAL PHARMACOLOGY OF SOME TESTOSTERONE FORMULATIONS |
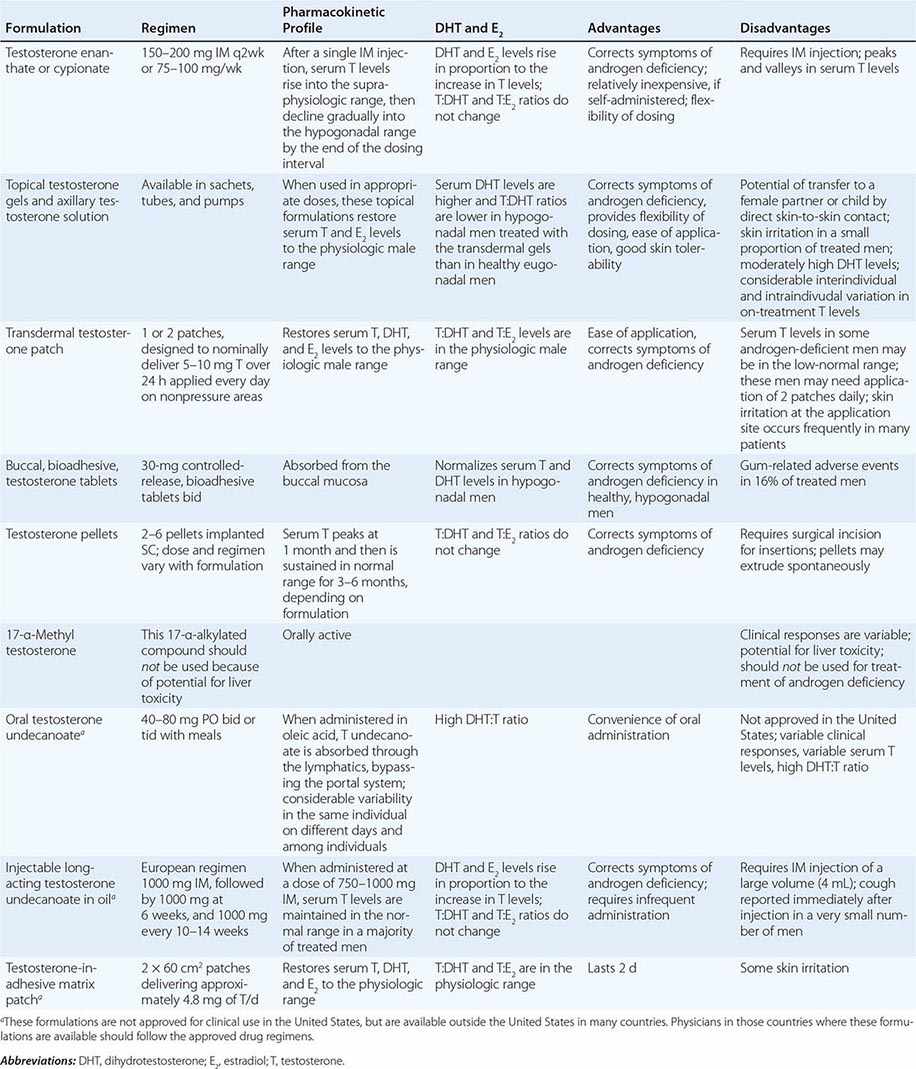
Oral Derivatives of Testosterone Testosterone is well-absorbed after oral administration but is quickly degraded during the first pass through the liver. Therefore, it is difficult to achieve sustained blood levels of testosterone after oral administration of crystalline testosterone. 17α-Alkylated derivatives of testosterone (e.g., 17α-methyl testosterone, oxandrolone, fluoxymesterone) are relatively resistant to hepatic degradation and can be administered orally; however, because of the potential for hepatotoxicity, including cholestatic jaundice, peliosis, and hepatoma, these formulations should not be used for testosterone replacement. Hereditary angioedema due to C1 esterase deficiency is the only exception to this general recommendation; in this condition, oral 17α-alkylated androgens are useful because they stimulate hepatic synthesis of the C1 esterase inhibitor.
Injectable Forms of Testosterone The esterification of testosterone at the 17β-hydroxy position makes the molecule hydrophobic and extends its duration of action. The slow release of testosterone ester from an oily depot in the muscle accounts for its extended duration of action. The longer the side chain, the greater is the hydrophobicity of the ester and the longer is the duration of action. Thus, testosterone enanthate, cypionate, and undecanoate with longer side chains have longer duration of action than testosterone propionate. Within 24 h after intramuscular administration of 200 mg testosterone enanthate or cypionate, testosterone levels rise into the high-normal or supraphysiologic range and then gradually decline into the hypogonadal range over the next 2 weeks. A bimonthly regimen of testosterone enanthate or cypionate therefore results in peaks and troughs in testosterone levels that are accompanied by changes in a patient’s mood, sexual desire, and energy level. The kinetics of testosterone enanthate and cypionate are similar. Estradiol and DHT levels are normal if testosterone replacement is physiologic.
Transdermal Testosterone Patch The nongenital testosterone patch, when applied in an appropriate dose, can normalize testosterone, DHT, and estradiol levels 4–12 h after application. Sexual function and well-being are restored in androgen-deficient men treated with the nongenital patch. One 5-mg patch may not be sufficient to increase testosterone into the mid-normal male range in all hypogonadal men; some patients may need two 5-mg patches daily to achieve the targeted testosterone concentrations. The use of testosterone patches may be associated with skin irritation in some individuals.
Testosterone Gel Several transdermal testosterone gels (e.g., Androgel, Testim, Fortesta, and Axiron), when applied topically to the skin in appropriate doses (Table 411-3), can maintain total and free testosterone concentrations in the normal range in hypogonadal men. The current recommendations are to begin with an initial U.S. Food and Drug Administration–approved dose and adjust the dose based on testosterone levels. The advantages of the testosterone gel include the ease of application and its flexibility of dosing. A major concern is the potential for inadvertent transfer of the gel to a sexual partner or to children who may come in close contact with the patient. The ratio of DHT to testosterone concentrations is higher in men treated with the testosterone gel than in healthy men. Also, there is considerable intra- and interindividual variation in serum testosterone levels in men treated with the transdermal gel due to variations in transdermal absorption and plasma clearance of testosterone. Therefore, monitoring of serum testosterone levels and multiple dose adjustments may be required to achieve and maintain testosterone levels in the target range.
Buccal Adhesive Testosterone A buccal testosterone tablet, which adheres to the buccal mucosa and releases testosterone as it is slowly dissolved, has been approved. After twice-daily application of 30-mg tablets, serum testosterone levels are maintained within the normal male range in a majority of treated hypogonadal men. The adverse effects include buccal ulceration and gum problems in a few subjects. The effects of food and brushing on absorption have not been studied in detail.
Implants of crystalline testosterone can be inserted in the subcutaneous tissue by means of a trocar through a small skin incision. Testosterone is released by surface erosion of the implant and absorbed into the systemic circulation. Two to six 200-mg implants can maintain testosterone in the mid- to high-normal range for up to 6 months. Potential drawbacks include incising the skin for insertion and removal and spontaneous extrusions and fibrosis at the site of the implant.
Testosterone Formulations Not Available in the United States Testosterone undecanoate, when administered orally in oleic acid, is absorbed preferentially through the lymphatics into the systemic circulation and is spared the first-pass degradation in the liver. Doses of 40–80 mg orally, two or three times daily, are typically used. However, the clinical responses are variable and suboptimal. DHT-to-testosterone ratios are higher in hypogonadal men treated with oral testosterone undecanoate, as compared to eugonadal men.
After initial priming, long-acting testosterone undecanoate in oil, when administered intramuscularly every 12 weeks, maintains serum testosterone, estradiol, and DHT in the normal male range and corrects symptoms of androgen deficiency in a majority of treated men. However, large injection volume (4 mL) is its relative drawback.
Novel Androgen Formulations A number of androgen formulations with better pharmacokinetics or more selective activity profiles are under development. A long-acting ester, testosterone undecanoate, when injected intramuscularly, can maintain circulating testosterone concentrations in the male range for 7–12 weeks. Initial clinical trials have demonstrated the feasibility of administering testosterone by the sublingual or buccal routes. 7α-Methyl-19-nortestosterone is an androgen that cannot be 5α-reduced; therefore, compared to testosterone, it has relatively greater agonist activity in muscle and gonadotropin suppression but lesser activity on the prostate.
Selective AR modulators (SARMs) are a class of AR ligands that bind the AR and display tissue-selective actions. A number of nonsteroidal SARMs that act as full agonists on the muscle and bone and that spare the prostate to varying degrees have advanced to phase 3 human trials. Nonsteroidal SARMs do not serve as substrates for either the steroid 5α-reductase or the CYP19 aromatase. SARM binding to AR induces specific conformational changes in the AR protein, which then modulates protein-protein interactions between AR and its coregulators, resulting in tissue-specific regulation of gene expression.
Pharmacologic Uses of Androgens Androgens and SARMs are being evaluated as anabolic therapies for functional limitations associated with aging and chronic illness. Testosterone supplementation increases skeletal muscle mass, maximal voluntary strength, and muscle power in healthy men, hypogonadal men, older men with low testosterone levels, HIV-infected men with weight loss, and men receiving glucocorticoids. These anabolic effects of testosterone are related to testosterone dose and circulating concentrations. Systematic reviews have confirmed that testosterone therapy of HIV-infected men with weight loss promotes improvements in body weight, lean body mass, muscle strength, and depression indices, leading to the recommendation that testosterone be considered as an adjunctive therapy in HIV-infected men who are experiencing unexplained weight loss and who have low testosterone levels. Similarly, in glucocorticoid-treated men, testosterone therapy should be considered to maintain muscle mass and strength and vertebral bone mineral density. It is unknown whether testosterone therapy of older men with functional limitations is safe and effective in improving physical function, vitality, and health-related quality of life and reducing disability. Concerns about potential adverse effects of testosterone on prostate and cardiovascular event rates have encouraged the development of SARMs that are preferentially anabolic and spare the prostate.
Testosterone administration induces hypertrophy of both type 1 and 2 fibers and increases satellite cell (muscle progenitor cells) and myonuclear number. Androgens promote the differentiation of mesenchymal, multipotent progenitor cells into the myogenic lineage and inhibit their differentiation into the adipogenic lineage. Testosterone may have additional effects on satellite cell replication and muscle protein synthesis, which may contribute to an increase in skeletal muscle mass.
Other indications for androgen therapy are in selected patients with anemia due to bone marrow failure (an indication largely supplanted by erythropoietin) or for hereditary angioedema.
Male Hormonal Contraception Based on Combined Administration of Testosterone and Gonadotropin Inhibitors Supraphysiologic doses of testosterone (200 mg testosterone enanthate weekly) suppress LH and FSH secretion and induce azoospermia in 50% of Caucasian men and >95% of Chinese men. The WHO-supported multicenter efficacy trials have demonstrated that suppression of spermatogenesis to azoospermia or severe oligozoospermia (<3 million/mL) by administration of testosterone enanthate to men results in highly effective contraception. Because of concern about long-term adverse effects of supraphysiologic testosterone doses, regimens that combine other gonadotropin inhibitors, such as GnRH antagonists and progestins with replacement doses of testosterone, are being investigated. Oral etonogestrel daily in combination with intramuscular testosterone decanoate every 4–6 weeks induced azoospermia or severe oligozoospermia (sperm density <1 million/mL) in 99% of treated men over a 1-year period. This regimen was associated with weight gain, deceased testicular volume, and decreased plasma high-density lipoprotein (HDL) cholesterol, and its long-term safety has not been demonstrated. SARMs that are more potent inhibitors of gonadotropins than testosterone and spare the prostate hold promise for their contraceptive potential.
Recommended Regimens for Androgen Replacement Testosterone esters are administered typically at doses of 75–100 mg intramuscularly every week, or 150–200 mg every 2 weeks. One or two 5-mg nongenital testosterone patches can be applied daily over the skin of the back, thigh, or upper arm away from pressure areas. Testosterone gels are typically applied over a covered area of skin at initial doses that vary with the formulation; patients should wash their hands after gel application. Bioadhesive buccal testosterone tablets at a dose of 30 mg are typically applied twice daily on the buccal mucosa.
Establishing Efficacy of Testosterone Replacement Therapy Because a clinically useful marker of androgen action is not available, restoration of testosterone levels to the mid-normal range remains the goal of therapy. Measurements of LH and FSH are not useful in assessing the adequacy of testosterone replacement. Testosterone should be measured 3 months after initiating therapy to assess adequacy of therapy. There is substantial interindividual variability in serum testosterone levels, especially with transdermal gels, presumably due to genetic differences in testosterone clearance and transdermal absorption. In patients who are treated with testosterone enanthate or cypionate, testosterone levels should be 350–600 ng/dL 1 week after the injection. If testosterone levels are outside this range, adjustments should be made either in the dose or in the interval between injections. In men on transdermal patch, gel, or buccal testosterone therapy, testosterone levels should be in the mid-normal range (500–700 ng/dL) 4–12 h after application. If testosterone levels are outside this range, the dose should be adjusted. Multiple dose adjustments are often necessary to achieve testosterone levels in the desired therapeutic range.
Restoration of sexual function, secondary sex characteristics, energy, and well-being and maintenance of muscle and bone health are important objectives of testosterone replacement therapy. The patient should be asked about sexual desire and activity, the presence of early morning erections, and the ability to achieve and maintain erections adequate for sexual intercourse. Some hypogonadal men continue to complain about sexual dysfunction even after testosterone replacement has been instituted; these patients may benefit from counseling. The hair growth in response to androgen replacement is variable and depends on ethnicity. Hypogonadal men with prepubertal onset of androgen deficiency who begin testosterone therapy in their late twenties or thirties may find it difficult to adjust to their newly found sexuality and may benefit from counseling. If the patient has a sexual partner, the partner should be included in counseling because of the dramatic physical and sexual changes that occur with androgen treatment.
Contraindications for Androgen Administration Testosterone administration is contraindicated in men with a history of prostate or breast cancer (Table 411-4). Testosterone therapy should not be administered without further urologic evaluation to men with a palpable prostate nodule or induration; to men with prostate-specific antigen levels >4 ng/mL or >3 ng/mL in men at high risk for prostate cancer such as African Americans or men with first-degree relatives with prostate cancer; or to men with severe lower urinary tract symptoms (American Urological Association lower urinary tract symptom score >19). Testosterone replacement should not be administered to men with baseline hematocrit ≥50%, severe untreated obstructive sleep apnea, uncontrolled or poorly controlled congestive heart failure, or myocardial infarction, stroke, or acute coronary syndrome in the preceding 6 months.
|
CONDITIONS IN WHICH TESTOSTERONE ADMINISTRATION IS ASSOCIATED WITH A RISK OF ADVERSE OUTCOME |
Abbreviation: PSA, prostate-specific antigen.
Source: Reproduced from the Endocrine Society Guideline for Testosterone Therapy of Androgen Deficiency Syndromes in Men (S Bhasin et al: J Clin Endocrinol Metab 95:2536, 2010).
Monitoring Potential Adverse Experiences The clinical effectiveness and safety of testosterone replacement therapy should be assessed 3 to 6 months after initiating testosterone therapy and annually thereafter (Table 411-5). Potential adverse effects include acne, oiliness of skin, erythrocytosis, breast tenderness and enlargement, leg edema, induction and exacerbation of obstructive sleep apnea, and increased risk of detection of prostate events. In addition, there may be formulation-specific adverse effects such as skin irritation with transdermal patch, risk of gel transfer to a sexual partner with testosterone gels, buccal ulceration and gum problems with buccal testosterone, and pain and mood fluctuation with injectable testosterone esters. Older men with preexisting heart disease may be at increased risk of cardiovascular events after initiation of testosterone therapy.
|
MONITORING MEN RECEIVING TESTOSTERONE THERAPY |
aNot approved for clinical use in the United States.
Abbreviations: AUA/IPSS, American Urological Association/International Prostate Symptom Score; PSA, prostate-specific antigen.
Source: Reproduced with permission from the Endocrine Society Guideline for Testosterone Therapy of Androgen Deficiency Syndromes in Adult Men (S Bhasin et al: J Clin Endocrinol Metab 95:2536, 2010).
HEMOGLOBIN LEVELS Administration of testosterone to androgen-deficient men is typically associated with a ~3% increase in hemoglobin levels, due to increased erythropoiesis, suppression of hepcidin, and increased iron availability for erythropoiesis. The magnitude of hemoglobin increase during testosterone therapy is greater in older men than younger men and in men who have sleep apnea, a significant smoking history, or chronic obstructive lung disease. The frequency of erythrocytosis is higher in hypogonadal men treated with injectable testosterone esters than in those treated with transdermal formulations, presumably due to the higher testosterone dose delivered by the typical regimens of testosterone esters. Erythrocytosis is the most frequent adverse event reported in testosterone trials in middle-aged and older men and is also the most frequent cause of treatment discontinuation in these trials. If hematocrit rises above 54%, testosterone therapy should be stopped until hematocrit has fallen to <50%. After evaluation of the patient for hypoxia and sleep apnea, testosterone therapy may be reinitiated at a lower dose.
PROSTATE AND SERUM PROSTATE-SPECIFIC ANTIGEN LEVELS Testosterone replacement therapy increases prostate volume to the size seen in age-matched controls but does not increase prostate volume beyond that expected for age. There is no evidence that testosterone therapy causes prostate cancer. However, androgen administration can exacerbate preexisting metastatic prostate cancer. Many older men harbor microscopic foci of cancer in their prostates. It is not known whether long-term testosterone administration will induce these microscopic foci to grow into clinically significant cancers.
Prostate-specific antigen (PSA) levels are lower in testosterone-deficient men and are restored to normal after testosterone replacement. There is considerable test-retest variability in PSA measurements. Increments in PSA levels after testosterone supplementation in androgen-deficient men are generally <0.5 ng/mL, and increments >1.0 ng/mL over a 3- to 6-month period are unusual. The 90% confidence interval for the change in PSA values in men with benign prostatic hypertrophy, measured 3–6 months apart, is 1.4 ng/mL. Therefore, the Endocrine Society expert panel suggested that an increase in PSA >1.4 ng/mL in any 1 year after starting testosterone therapy, if confirmed, should lead to urologic evaluation. PSA velocity criterion can be used for patients who have sequential PSA measurements for >2 years; a change of >0.40 ng/mL per year merits closer urologic follow-up.
CARDIOVASCULAR RISK In epidemiologic studies, testosterone concentrations are negatively related to the risk of diabetes mellitus, heart disease, and all-cause and cardiovascular mortality. A recent testosterone trial in older men with mobility limitation was stopped early because of the higher rates of cardiovascular events in the testosterone arm than in the placebo arm of this trial. Meta-analyses of testosterone trials have found a significant increase in cardiovascular event rates in older men receiving testosterone therapy. Inferences about adverse events from previous trials included in these meta-analyses are limited by poor ascertainment, small numbers of events, heterogeneity of study populations, and small numbers of participants. Two retrospective analyses also found a higher frequency of cardiovascular events in association with testosterone therapy in older men with preexisting heart disease. Retrospective database analyses are limited by their inherent inability to verify the indication for treatment, diagnoses, or other relevant quantitative information and are susceptible to confounding by many other factors. Adequately powered prospective studies are needed to determine the effect on testosterone replacement on cardiovascular risk.
Androgen Abuse by Athletes and Recreational Bodybuilders The illicit use of androgenic-anabolic steroids (AAS) to enhance athletic performance first surfaced in the 1950s among power lifters and spread rapidly to other sports, professional as well as high school athletes, and recreational bodybuilders. In the early 1980s, the use of AAS spread beyond the athletic community into the general population, and now as many as 3 million Americans, most of them men, have likely used these compounds. Most AAS users are not athletes, but rather recreational weightlifters, who use these drugs to look lean and more muscular. The most commonly used AAS include testosterone esters, nandrolone, stanozolol, methandienone, and methenolol. AAS users generally use increasing doses of multiple steroids in a practice known as stacking.
The adverse effects of long-term AAS abuse remain poorly understood. Most of the information about the adverse effects of AAS has emerged from case reports, uncontrolled studies, or clinical trials that used replacement doses of testosterone. The adverse event data from clinical trials using physiologic replacement doses of testosterone have been extrapolated unjustifiably to AAS users who may administer 10–100 times the replacement doses of testosterone over many years and to support the claim that AAS use is safe. A substantial fraction of androgenic steroid users also use other drugs that are perceived to be muscle building or performance enhancing, such as GH; erythropoiesis-stimulating agents; insulin; and stimulants such as amphetamine, clenbuterol, cocaine, ephedrine, and thyroxine; and drugs perceived to reduce adverse effects such as hCG, aromatase inhibitors, or estrogen antagonists. The men who abuse androgenic steroids are more likely to engage in other high-risk behaviors than nonusers. The adverse events associated with AAS use may be due to AAS themselves, concomitant use of other drugs, high-risk behaviors, and host characteristics that may render these individuals more susceptible to AAS use or to other high-risk behaviors.
The high rates of mortality and morbidities observed in AAS users are alarming. One Finnish study reported 4.6 times the risk of death among elite power lifters than in age-matched men from the general population. The causes of death among power lifters included suicides, myocardial infarction, hepatic coma, and non-Hodgkin’s lymphoma. A retrospective review of patient records in Sweden also reported higher standardized mortality ratios for AAS users than for nonusers. Thiblin and colleagues found that 32% of deaths among AAS users were suicidal, 26% homicidal, and 35% accidental. The median age of death among AAS users (24 years) is even lower than that for heroin or amphetamine users.
Numerous reports of cardiac death among young AAS users raise concerns about the adverse cardiovascular effects of AAS. High doses of AAS may induce proatherogenic dyslipidemia, increase thrombosis risk via effects on clotting factors and platelets, and induce vasospasm through their effects on vascular nitric oxide.
Replacement doses of testosterone, when administered parenterally, are associated with only a small decrease in HDL cholesterol and little or no effect on total cholesterol, low-density lipoprotein (LDL) cholesterol, and triglyceride levels. In contrast, supraphysiologic doses of testosterone and orally administered, 17α-alkylated, nonaromatizable AAS are associated with marked reductions in HDL cholesterol and increases in LDL cholesterol.
Recent studies of AAS users using tissue Doppler and strain imaging and MRI have reported diastolic and systolic dysfunction, including significantly lower early and late diastolic tissue velocities, reduced E/A ratio, and reduced peak systolic strain in AAS users than in nonusers. Power athletes using AAS often have short QT intervals but increased QT dispersion, which may predispose them to ventricular arrhythmias. Long-term AAS use may be associated with myocardial hypertrophy and fibrosis. Myocardial tissue of power lifters using AAS has been shown to be infiltrated with fibrous tissue and fat droplets. The finding of ARs on myocardial cells suggests that AAS might be directly toxic to myocardial cells.
Long-term AAS use suppresses LH and FSH secretion and inhibits endogenous testosterone production and spermatogenesis. Men who have used AAS for more than a few months experience marked suppression of the hypothalamic-pituitary-testicular (HPT) axis after stopping AAS that may be associated with sexual dysfunction, fatigue, infertility, and depression; in some AAS users, HPT suppression may last more than a year, and in a few individuals, complete recovery may never occur. The symptoms of androgen deficiency caused by androgen withdrawal may cause some men to revert back to using AAS, leading to continued use and AAS dependence. As many as 30% of AAS users develop a syndrome of AAS dependence, characterized by long-term AAS use despite adverse medical and psychiatric effects.
Supraphysiologic doses of testosterone may also impair insulin sensitivity. Orally administered androgens also have been associated with insulin resistance and diabetes.
Unsafe injection practices, high-risk behaviors, and increased rates of incarceration render AAS users at increased risk of HIV and hepatitis B and C. In one survey, nearly 1 in 10 gay men had injected AAS or other substances, and AAS users were more likely to report high-risk unprotected anal sex than other men.
Some AAS users develop hypomanic or manic symptoms during AAS exposure (irritability, aggressiveness, reckless behavior, and occasional psychotic symptoms, sometimes associated with violence) and major depression (sometimes associated with suicidality) during AAS withdrawal. Users may also develop other forms of illicit drug use, which may be potentiated or exacerbated by AAS.
Elevated liver enzymes, cholestatic jaundice, hepatic neoplasms, and peliosis hepatis have been reported with oral, 17α-alkylated AAS. AAS use may cause muscle hypertrophy without compensatory adaptations in tendons, ligaments, and joints, thus increasing the risk of tendon and joint injuries. AAS use is associated with acne, baldness, and increased body hair.
The suspicion of AAS use may be raised by the increased hemoglobin and hematocrit, suppressed LH and FSH and testosterone levels, low high-density lipoproteins cholesterol, and low testicular volume and sperm density in a person who looks highly muscular. Accredited laboratories use gas chromatography–mass spectrometry or liquid chromatography–mass spectrometry to detect anabolic steroid abuse. In recent years, the availability of high-resolution mass spectrometry and tandem mass spectrometry has further improved the sensitivity of detecting androgen abuse. Illicit testosterone use is detected generally by the application of the measurement of the urinary testosterone-to-epitestosterone ratio and further confirmed by the use of the 13C:12C ratio in testosterone by the use of isotope ratio combustion mass spectrometry. Exogenous testosterone administration increases urinary testosterone glucuronide excretion and consequently the testosterone-to-epitestosterone ratio. Ratios >4 suggest exogenous testosterone use but can also reflect genetic variation. Genetic variations in the uridine diphosphoglucuronyl transferase 2B17 (UGT2B17), the major enzyme for testosterone glucuronidation, affect the testosterone-to-epitestosterone ratio. Synthetic testosterone has a lower 13C:12C ratio than endogenously produced testosterone, and these differences in 13C:12C ratio can be detected by isotope ratio combustion mass spectrometry, which is used to confirm exogenous testosterone use in individuals with a high testosterone-to-epitestosterone ratio.
412 |
Disorders of the Female Reproductive System |
The female reproductive system regulates the hormonal changes responsible for puberty and adult reproductive function. Normal reproductive function in women requires the dynamic integration of hormonal signals from the hypothalamus, pituitary, and ovary, resulting in repetitive cycles of follicle development, ovulation, and preparation of the endometrial lining of the uterus for implantation should conception occur. It is critical to understand pubertal development in normal girls (and boys) as a yardstick for identifying precocious and delayed puberty.
For further discussion of related topics, see the following chapters: amenorrhea and pelvic pain (Chap. 69), infertility and contraception (Chap. 414), menopause (Chap. 413), disorders of sex development (Chap. 410), and disorders of the male reproductive system (Chap. 411).
DEVELOPMENT OF THE OVARY AND EARLY FOLLICULAR GROWTH
The ovary orchestrates the development and release of a mature oocyte and also elaborates hormones (e.g., estrogen, progesterone, inhibin, relaxin) that are critical for pubertal development and preparation of the uterus for conception, implantation, and the early stages of pregnancy. To achieve these functions in repeated monthly cycles, the ovary undergoes some of the most dynamic changes of any organ in the body. Primordial germ cells can be identified by the third week of gestation, and their migration to the genital ridge is complete by 6 weeks of gestation. Germ cells persist within the genital ridge, are then referred to as oogonia, and are essential for induction of ovarian development. Although one × chromosome undergoes × inactivation in somatic cells, it is reactivated in oogonia and genes on both × chromosomes are required for normal ovarian development. A streak ovary containing only stromal cells is found in patients with 45,X Turner’s syndrome (Chap. 410).
The germ cell population expands, and starting at ~8 weeks of gestation, oogonia begin to enter prophase of the first meiotic division and become primary oocytes. This allows the oocyte to be surrounded by a single layer of flattened granulosa cells to form a primordial follicle (Fig. 412-1). Granulosa cells are derived from mesonephric cells that invade the ovary early in its development, pushing the germ cells to the periphery. The weight of evidence supports the concept that for the most part, the ovary contains a nonrenewable pool of germ cells. Through the combined processes of mitosis, meiosis, and atresia, the population of oogonia reaches its maximum of 6–7 million by 20 weeks of gestation, after which there is a progressive loss of both oogonia and primordial follicles through the process of atresia. At birth, oogonia are no longer present in the ovary, and only 1–2 million germ cells remain in the form of primordial follicles (Fig. 412-2). The oocyte persists in prophase of the first meiotic division until just before ovulation, when meiosis resumes.
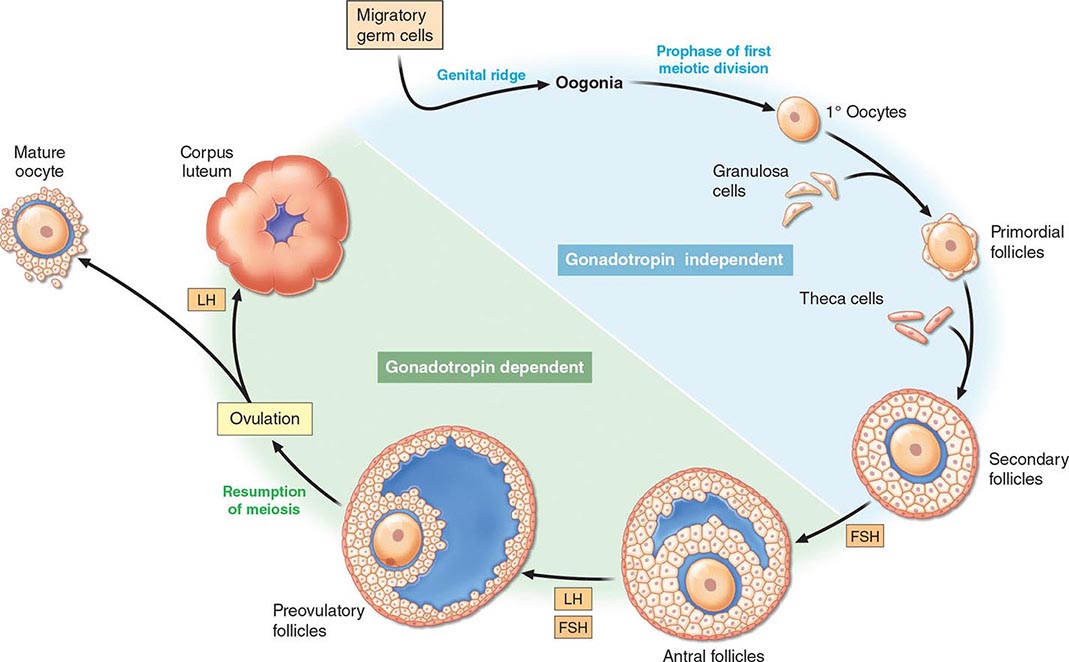
FIGURE 412-1 Stages of ovarian development from the arrival of the migratory germ cells at the genital ridge through gonadotropin-independent and gonadotropin-dependent phases that ultimately result in ovulation of a mature oocyte. FSH, follicle-stimulating hormone; LH, luteinizing hormone.
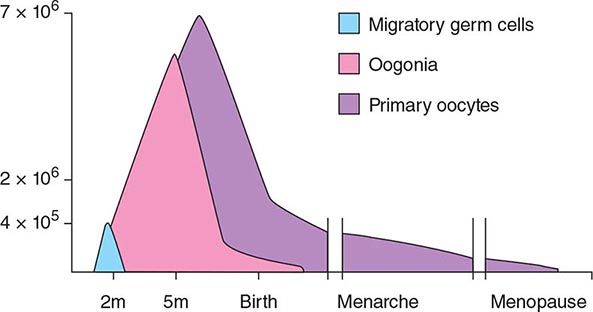
FIGURE 412-2 Ovarian germ cell number is maximal at mid-gestation and then decreases precipitously.
The quiescent primordial follicles are recruited to further growth and differentiation through a highly regulated process that limits the size of the developing cohort to ensure that folliculogenesis can continue throughout the reproductive life span. This initial recruitment of primordial follicles to form primary follicles (Fig. 412-1) is characterized by growth of the oocyte and the transition from squamous to cuboidal granulosa cells. The theca interna cells that surround the developing follicle begin to form as the primary follicle grows. Acquisition of a zona pellucida by the oocyte and the presence of several layers of surrounding cuboidal granulosa cells mark the development of secondary follicles. It is at this stage that granulosa cells develop follicle-stimulating hormone (FSH), estradiol, and androgen receptors and communicate with one another through the development of gap junctions.
Bidirectional signaling between the germ cells and the somatic cells in the ovary is a necessary component underlying the maturation of the oocyte and the capacity for hormone secretion. For example, oocyte-derived growth differentiation factor 9 (GDF-9) and bone morphogenic protein-15 (BMP-15), also known as GDF-9b, are required for migration of pregranulosa and pretheca cells to the outer surface of the developing follicle and, hence, initial follicle formation. GDF-9 is also required for formation of secondary follicles, as are granulosa cell–derived KIT ligand (KITL) and the forkhead transcription factor (FOXL2). All of these genes are potential candidates for premature ovarian failure in women, and mutations in the human FOXL2 gene have been shown to cause the syndrome of blepharophimosis/ptosis/epicanthus inversus, which is associated with ovarian failure.
DEVELOPMENT OF A MATURE FOLLICLE
The early stages of follicle growth are primarily driven by intraovarian factors and may take up to a year from development of the primary follicle to the dominant follicle stage. Further maturation to the preovulatory state, including the resumption of meiosis in the oocyte, requires the combined stimulus of FSH and luteinizing hormone (LH) (Fig. 412-1) and can be accomplished within weeks. Recruitment of secondary follicles from the resting follicle pool requires the direct action of FSH, whereas anti-müllerian hormone (AMH) produced from small growing follicles, restrains this effect of FSH. Accumulation of follicular fluid between the layers of granulosa cells creates an antrum that divides the granulosa cells into two functionally distinct groups: mural cells that line the follicle wall and cumulus cells that surround the oocyte (Fig. 412-3). Recent evidence suggests that, in addition to its role in normal development of the müllerian system, the WNT signaling pathway is required for normal antral follicle development and may also play a role in ovarian steroidogenesis. A single dominant follicle emerges from the growing follicle pool within the first 5–7 days after the onset of menses, and the majority of follicles fall off their growth trajectory and become atretic. Autocrine actions of activin and BMP-6, derived from the granulosa cells, and paracrine actions of GDF-9, BMP-15, BMP-6, and Gpr149, derived from the oocyte, are involved in granulosa cell proliferation and modulation of FSH responsiveness. Differential exposure to these factors may explain the mechanism whereby a given follicle is selected for continued growth to the preovulatory stage. The dominant follicle can be distinguished by its size, evidence of granulosa cell proliferation, large number of FSH receptors, high aromatase activity, and elevated concentrations of estradiol and inhibin A in follicular fluid.
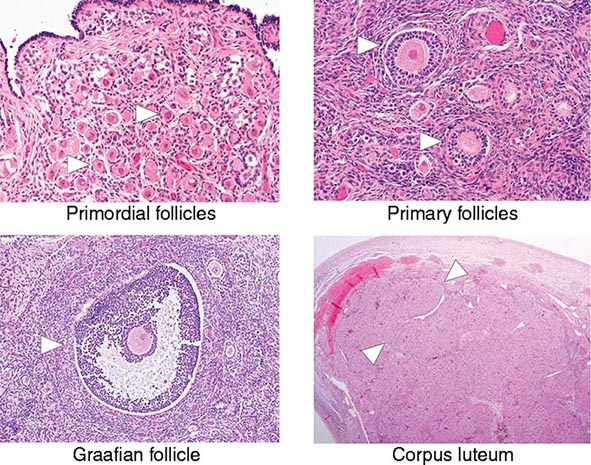
FIGURE 412-3 Development of ovarian follicles. The Graafian follicle is also known as a tertiary or preovulatory follicle. (Courtesy of JH Eichhorn and D. Roberts, Massachusetts General Hospital; with permission.)
The dominant follicle undergoes rapid expansion during the 5–6 days prior to ovulation, reflecting granulosa cell proliferation and accumulation of follicular fluid. FSH induces LH receptors on the granulosa cells, and the preovulatory, or Graafian, follicle moves to the outer ovarian surface in preparation for ovulation. The LH surge triggers the resumption of meiosis, the suppression of granulosa cell proliferation, and the induction of cyclooxygenase 2 (COX-2), prostaglandins, the progesterone receptor, and the epidermal growth factor (EGF)-like growth factors amphiregulin, epiregulin, betacellulin, and neuroregulin 1, all of which are required for ovulation. EGF-like factors are thought to mediate these follicular responses to LH. Ovulation also involves production of extracellular matrix leading to expansion of the cumulus cell population that surrounds the oocyte and the controlled expulsion of the egg and follicular fluid. Both progesterone and prostaglandins (induced by the ovulatory stimulus) are essential for this process. After ovulation, luteinization is induced by LH in conjunction with the acquisition of a rich vascular network in response to vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (FGF). Traditional regulators of central reproductive control, gonadotropin-releasing hormone (GnRH) and its receptor (GnRHR), are also produced in the ovary and may be involved in corpus luteum function.
REGULATION OF OVARIAN FUNCTION
HYPOTHALAMIC AND PITUITARY SECRETION
GnRH neurons develop from epithelial cells outside the central nervous system and migrate, initially alongside the olfactory neurons, to the medial basal hypothalamus. Studies in GnRH-deficient patients who fail to undergo puberty have provided insights into genes that control the ontogeny and function of GnRH neurons (Fig. 412-4). KAL1, FGF8/FGFR1, PROK2/PROKR2, NSMF, and CDH7, among others (Chap. 411), have been implicated in the migration of GnRH neurons to the hypothalamus. Approximately 7000 GnRH neurons, scattered throughout the medial basal hypothalamus, establish contacts with capillaries of the pituitary portal system in the median eminence. GnRH is secreted into the pituitary portal system in discrete pulses to stimulate synthesis and secretion of LH and FSH from pituitary gonadotropes, which comprise ~10% of cells in the pituitary (Chap. 401e). Functional connections of GnRH neurons with the portal system are established by the end of the first trimester, coinciding with the production of pituitary gonadotropins. Thus, like the ovary, the hypothalamic and pituitary components of the reproductive system are present before birth. However, the high levels of estradiol and progesterone produced by the placenta suppress hypothalamic-pituitary stimulation of ovarian hormonal secretion in the fetus.
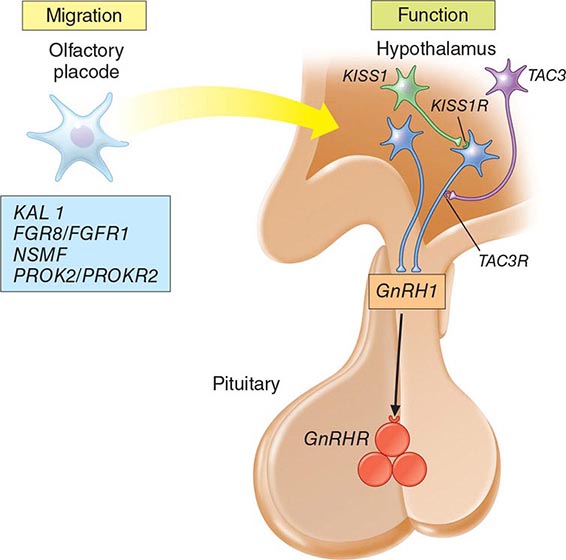
FIGURE 412-4 Establishment of a functional gonadotropin-releasing hormone (GnRH) system requires the participation of a number of genes that are essential for development and migration of GnRH neurons from the olfactory placode to the hypothalamus in addition to genes involved in the functional control of GnRH secretion and action.
After birth and the loss of placenta-derived steroids, gonadotropin levels rise. FSH levels are much higher in girls than in boys. This rise in FSH results in ovarian activation (evident on ultrasound) and increased inhibin B and estradiol levels. Studies that have identified mutations in TAC3, which encodes neurokinin B, and its receptor, TAC3R, in patients with GnRH deficiency indicate that both are involved in control of GnRH secretion and may be particularly important at this early stage of development. By 12–20 months of age, the reproductive axis is again suppressed, and a period of relative quiescence persists until puberty (Fig. 412-5). At the onset of puberty, pulsatile GnRH secretion induces pituitary gonadotropin production. In the early stages of puberty, LH and FSH secretion are apparent only during sleep, but as puberty develops, pulsatile gonadotropin secretion occurs throughout the day and night.
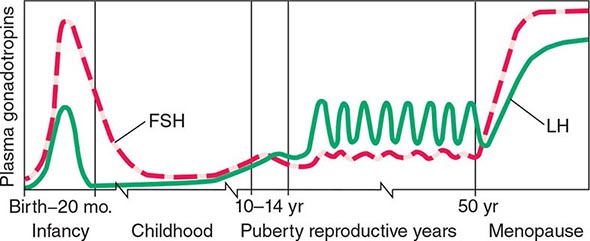
FIGURE 412-5 Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) are increased during the neonatal years but go through a period of childhood quiescence before increasing again during puberty. Gonadotropin levels are cyclic during the reproductive years and increase dramatically with the loss of negative feedback that accompanies menopause.
The mechanisms responsible for the childhood quiescence and pubertal reactivation of the reproductive axis remain incompletely understood. GnRH neurons in the hypothalamus respond to both excitatory and inhibitory factors. Increased sensitivity to the inhibitory influence of gonadal steroids has long been implicated in the inhibition of GnRH secretion during childhood but has not been definitively established in the human. Metabolic signals, such as adipocyte-derived leptin, play a permissive role in reproductive function (Chap. 415e). Studies of patients with isolated GnRH deficiency reveal that mutations in the G protein–coupled receptor 54 (GPR54) gene (now known as KISS1R) preclude the onset of puberty. The ligand for this receptor, metastin, is derived from the parent peptide, kisspeptin-1 (KISS1), and is a powerful stimulant for GnRH release. A potential role for kisspeptin in the onset of puberty has been suggested by upregulation of KISS1 and KISS1R transcripts in the hypothalamus at the time of puberty. TAC3 and dynorphin (Dyn), which appear to play an inhibitory rather than stimulatory role in GnRH control, are co-expressed with KISS1 in KNDy neurons that project to GnRH neurons. This system is intimately involved with estrogen negative feedback regulation of GnRH secretion.
OVARIAN STEROIDS
Ovarian steroid-producing cells do not store hormones but produce them in response to LH and FSH during the normal menstrual cycle. The sequence of steps and the enzymes involved in the synthesis of steroid hormones are similar in the ovary, adrenal, and testis. However, the enzymes required to catalyze specific steps are compartmentalized and may not be abundant or even present in all cell types. Within the developing ovarian follicle, estrogen synthesis from cholesterol requires close integration between theca and granulosa cells—sometimes called the two-cell model for steroidogenesis (Fig. 412-6). FSH receptors are confined to the granulosa cells, whereas LH receptors are restricted to the theca cells until the late stages of follicular development, when they are also found on granulosa cells. The theca cells surrounding the follicle are highly vascularized and use cholesterol, derived primarily from circulating lipoproteins, as the starting point for the synthesis of androstenedione and testosterone under the control of LH. Androstenedione and testosterone are transferred across the basal lamina to the granulosa cells, which receive no direct blood supply. The mural granulosa cells are particularly rich in aromatase and, under the control of FSH, produce estradiol, the primary steroid secreted from the follicular phase ovary and the most potent estrogen. Theca cell–produced androstenedione and, to a lesser extent, testosterone are also secreted into peripheral blood, where they can be converted to dihydrotestosterone in skin and to estrogens in adipose tissue. The hilar interstitial cells of the ovary are functionally similar to Leydig cells and are also capable of secreting androgens. Although stromal cells proliferate in response to androgens (as in polycystic ovarian syndrome [PCOS]), they do not secrete androgens.
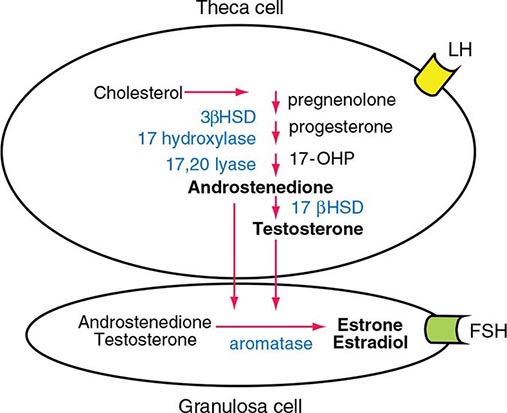
FIGURE 412-6 Estrogen production in the ovary requires the cooperative function of the theca and granulosa cells under the control of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). HSD, hydroxysteroid dehydrogenase; OHP, hydroxyprogesterone.
Development of the rich capillary network following rupture of the follicle at the time of ovulation makes it possible for large molecules such as low-density lipoprotein (LDL) to reach the luteinized granulosa and theca lutein cells. As in the follicle, both cell types are required for steroidogenesis in the corpus luteum. The large luteinized granulosa cells are the main source of progesterone production, whereas the smaller theca lutein cells produce 17-hydroxyprogesterone, a substrate for aromatization to estradiol by the luteinized granulosa cells. LH is critical for normal structure and function of the corpus luteum. Because LH and human chorionic gonadotropin (hCG) bind to a common receptor, the role of LH in support of the corpus luteum can be replaced by hCG in the first 10 weeks after conception, and hCG is commonly used for luteal phase support in the treatment of infertility.
Steroid Hormone Actions Both estrogen and progesterone play critical roles in the expression of secondary sexual characteristics in women (Chap. 400e). Estrogen promotes development of the ductule system in the breast, whereas progesterone is responsible for glandular development. In the reproductive tract, estrogens create a receptive environment for fertilization and support pregnancy and parturition through carefully coordinated changes in the endometrium, thickening of the vaginal mucosa, thinning of the cervical mucus, and uterine growth and contractions. Progesterone induces secretory activity in the estrogen-primed endometrium, increases the viscosity of cervical mucus, and inhibits uterine contractions. Both gonadal steroids play critical roles in the negative and positive feedback controls of gonadotropin secretion. Progesterone also increases basal body temperature and has therefore been used clinically as a marker of ovulation.
The vast majority of circulating estrogens and androgens are carried in the blood bound to carrier proteins, which restrain their free diffusion into cells and prolong their clearance, serving as a reservoir. High-affinity binding proteins include sex hormone–binding globulin (SHBG), which binds androgens with somewhat greater affinity than estrogens, and corticosteroid-binding globulin (CBG), which also binds progesterone. Modulations in binding protein levels by insulin, androgens, and estrogens contribute to high bioavailable testosterone levels in PCOS and to high circulating estrogen and progesterone levels during pregnancy.
Estrogens act primarily through binding to the nuclear receptors, estrogen receptor (ER) α and β. Transcriptional coactivators and co-repressors modulate ER action (Chap. 400e). Both ER subtypes are present in the hypothalamus, pituitary, ovary, and reproductive tract. Although ERα and -β exhibit some functional redundancy, there is also a high degree of specificity, particularly in expression within cell types. For example, ERα functions in the ovarian theca cells, whereas ERβ is critical for granulosa cell function. There is also evidence for membrane-initiated signaling by estrogen. Similar signaling mechanisms pertain for progesterone with evidence of transcriptional regulation through progesterone receptor (PR) A and B protein isoforms, as well as rapid membrane signaling.
OVARIAN PEPTIDES
Inhibin was initially isolated from gonadal fluids based on its ability to selectively inhibit FSH secretion from pituitary cells. Inhibin is a heterodimer composed of an α subunit and a βA or βB subunit to form inhibin A or inhibin B, both of which are secreted from the ovary. Activin is a homodimer of inhibin β subunits with the capacity to stimulate the synthesis and secretion of FSH. Inhibins and activins are members of the transforming growth factor β (TGF-β) superfamily of growth and differentiation factors. During the purification of inhibin, follistatin, an unrelated monomeric protein that inhibits FSH secretion, was discovered. Within the pituitary, follistatin inhibits FSH secretion indirectly through binding and neutralizing activin.
Inhibin B is secreted from the granulosa cells of small antral follicles, whereas inhibin A is present in both granulosa and theca cells and is secreted by dominant follicles. Inhibin A is also present in luteinized granulosa cells and is a major secretory product of the corpus luteum. Inhibin B is constitutively secreted by granulosa cells and increases in serum in conjunction with recruitment of secondary follicles to the pool of actively growing follicles under the control of FSH. Inhibin B has been used clinically as a marker of ovarian reserve. Inhibin B is an important inhibitor of FSH, independent of estradiol, during the menstrual cycle. Although activin is also secreted from the ovary, the excess of follistatin in serum, combined with its nearly irreversible binding of activin, make it unlikely that ovarian activin plays an endocrine role in FSH regulation. However, there is evidence that activin plays an autocrine/paracrine role in the ovary, in addition to its intrapituitary role in modulation of FSH production.
AMH (also known as müllerian-inhibiting substance) is important in ovarian biology in addition to the function from which it derived its name (i.e., promotion of the degeneration of the müllerian system during embryogenesis in the male). AMH is produced by granulosa cells from small follicles and, like inhibin B, is a marker of ovarian reserve. AMH inhibits the recruitment of primordial follicles into the follicle pool and counters FSH stimulation of aromatase expression.
Relaxin, which is produced by the theca lutein cells of the corpus luteum, is thought to play a role in decidualization of the endometrium and suppression of myometrial contractile activity, both of which are essential for the early establishment of pregnancy.
HORMONAL INTEGRATION OF THE NORMAL MENSTRUAL CYCLE
The sequence of changes responsible for mature reproductive function is coordinated through a series of negative and positive feedback loops that alter pulsatile GnRH secretion, the pituitary response to GnRH, and the relative secretion of LH and FSH from the gonadotrope. The frequency and amplitude of pulsatile GnRH secretion differentially modulate the synthesis and secretion of LH and FSH, with slow frequencies favoring FSH synthesis and increased amplitudes favoring LH synthesis. Activin is produced in both pituitary gonadotropes and folliculostellate cells and stimulates the synthesis and secretion of FSH. Inhibins function as potent antagonists of activins through sequestration of the activin receptors. Although inhibin is expressed in the pituitary, gonadal inhibin is the principal source of feedback inhibition of FSH.
For the majority of the cycle, the reproductive system functions in a classic endocrine negative feedback mode. Estradiol and progesterone inhibit GnRH secretion, and the inhibins act at the pituitary to selectively inhibit FSH synthesis and secretion (Fig. 412-7). This negative feedback control of FSH is critical for development of the single mature oocyte that characterizes normal reproductive function in women. In addition to these negative feedback controls, the menstrual cycle is uniquely dependent on estrogen-induced positive feedback to produce an LH surge that is essential for ovulation of a mature follicle. Estrogen negative feedback in women occurs primarily at the hypothalamus with a small pituitary contribution, whereas estrogen positive feedback occurs at the pituitary with hypothalamic GnRH secretion playing a permissive role.
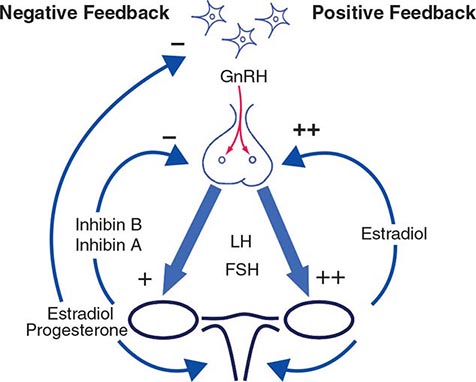
FIGURE 412-7 The reproductive system in women is critically dependent on both negative feedback of gonadal steroids and inhibin to modulate follicle-stimulating hormone (FSH) secretion and on estrogen positive feedback to generate the preovulatory luteinizing hormone (LH) surge. GnRH, gonadotropin-releasing hormone.
THE FOLLICULAR PHASE
The follicular phase is characterized by recruitment of a cohort of secondary follicles and the ultimate selection of a dominant preovulatory follicle (Fig. 412-8). The follicular phase begins, by convention, on the first day of menses. However, follicle recruitment is initiated by the rise in FSH that begins in the late luteal phase of the previous cycle in conjunction with the loss of negative feedback of gonadal steroids and likely inhibin A. The fact that a 20–30% increase in FSH is adequate for follicular recruitment speaks to the marked sensitivity of the resting follicle pool to FSH. The resultant granulosa cell proliferation is responsible for increasing early follicular phase levels of inhibin B. Inhibin B in conjunction with rising levels of estradiol, and probably inhibin A, restrain FSH secretion during this critical period such that only a single follicle matures in the vast majority of cycles. The increased risk of multiple gestation associated with the increased levels of FSH characteristic of advanced maternal age, or with exogenous gonadotropin administration in the treatment of infertility, attests to the importance of negative feedback regulation of FSH. With further growth of the dominant follicle, estradiol and inhibin A increase exponentially and the follicle acquires LH receptors. Increasing levels of estradiol are responsible for proliferative changes in the endometrium. The exponential rise in estradiol results in positive feedback on the pituitary, leading to the generation of an LH surge (and a smaller FSH surge), thereby triggering ovulation and luteinization of the granulosa cells.
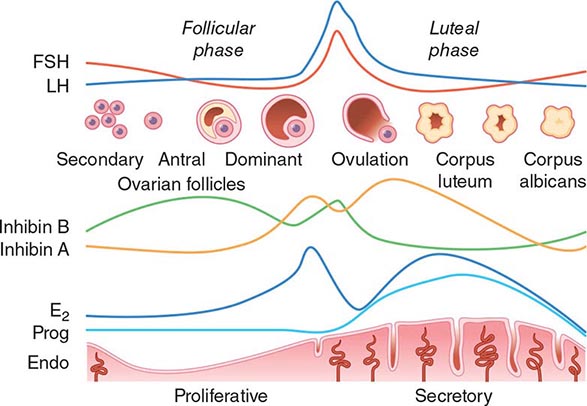
FIGURE 412-8 Relationship between gonadotropins, follicle development, gonadal secretion, and endometrial changes during the normal menstrual cycle. E2, estradiol; Endo, endometrium; FSH, follicle-stimulating hormone; LH, luteinizing hormone; Prog, progesterone.
THE LUTEAL PHASE
The luteal phase begins with the formation of the corpus luteum from the ruptured follicle (Fig. 412-8). Progesterone and inhibin A are produced from the luteinized granulosa cells, which continue to aromatize theca-derived androgen precursors, producing estradiol. The combined actions of estrogen and progesterone are responsible for the secretory changes in the endometrium that are necessary for implantation. The corpus luteum is supported by LH but has a finite life span because of diminished sensitivity to LH. The demise of the corpus luteum results in a progressive decline in hormonal support of the endometrium. Inflammation or local hypoxia and ischemia result in vascular changes in the endometrium, leading to the release of cytokines, cell death, and shedding of the endometrium.
If conception occurs, hCG produced by the trophoblast binds to LH receptors on the corpus luteum, maintaining steroid hormone production and preventing involution of the corpus luteum. The corpus luteum is essential for the hormonal maintenance of the endometrium during the first 6–10 weeks of pregnancy until this function is taken over by the placenta.
CLINICAL ASSESSMENT OF OVARIAN FUNCTION
Menstrual bleeding should become regular within 2–4 years of menarche, although anovulatory and irregular cycles are common before that. For the remainder of adult reproductive life, the cycle length counted from the first day of menses to the first day of subsequent menses is ~28 days, with a range of 25–35 days. However, cycle-to-cycle variability for an individual woman is ±2 days. Luteal phase length is relatively constant between 12 and 14 days in normal cycles; thus, the major variability in cycle length is due to variations in the follicular phase. The duration of menstrual bleeding in ovulatory cycles varies between 4 and 6 days. There is a gradual shortening of cycle length with age such that women over the age of 35 have cycles that are shorter than during their younger reproductive years. Anovulatory cycles increase as women approach menopause, and bleeding patterns may be erratic.
Women who report regular monthly bleeding with cycles that do not vary by >4 days generally have ovulatory cycles, but several other clinical signs can be used to assess the likelihood of ovulation. Some women experience mittelschmerz, described as midcycle pelvic discomfort that is thought to be caused by the rapid expansion of the dominant follicle at the time of ovulation. A constellation of premenstrual moliminal symptoms such as bloating, breast tenderness, and food cravings often occur several days before menses in ovulatory cycles, but their absence cannot be used as evidence of anovulation. Methods that can be used to determine whether ovulation is likely include a serum progesterone level >5 ng/mL ~7 days before expected menses, an increase in basal body temperature of 0.24°C (>0.5°F) in the second half of the cycle due to the thermoregulatory effect of progesterone, or the detection of the urinary LH surge using ovulation predictor kits. Because ovulation occurs ~36 h after the LH surge, urinary LH can be helpful in timing intercourse to coincide with ovulation.
Ultrasound can be used to detect the growth of the fluid-filled antrum of the developing follicle and to assess endometrial proliferation in response to increasing estradiol levels in the follicular phase, as well as the characteristic echogenicity of the secretory endometrium of the luteal phase.
PUBERTY
NORMAL PUBERTAL DEVELOPMENT IN GIRLS
The first menstrual period (menarche) occurs relatively late in the series of developmental milestones that characterize normal pubertal development (Table 412-1). Menarche is preceded by the appearance of pubic and then axillary hair (adrenarche) as a result of maturation of the zona reticularis in the adrenal gland and increased adrenal androgen secretion, particularly dehydroepiandrosterone (DHEA). The triggers for adrenarche remain unknown but may involve increases in body mass index, as well as in utero and neonatal factors. Menarche is also preceded by breast development (thelarche). The breast is exquisitely sensitive to the very low levels of estrogen that result from peripheral conversion of adrenal androgens and the low levels of estrogen secreted from the ovary early in pubertal maturation. Breast development precedes the appearance of pubic and axillary hair in ~60% of girls. The interval between the onset of breast development and menarche is ~2 years. There has been a gradual decline in the age of menarche over the past century, attributed in large part to improvement in nutrition, and there is a relationship between adiposity and earlier sexual maturation in girls. In the United States, menarche occurs at an average age of 12.5 years (Table 412-1). Much of the variation in the timing of puberty is due to genetic factors, with heritability estimates of 50–80%. Both adrenarche and thelarche occur ~1 year earlier in black compared with white girls, although the timing of menarche differs by only 6 months between these ethnic groups.
|
MEAN AGE (YEARS) OF PUBERTAL MILESTONES IN GIRLS |
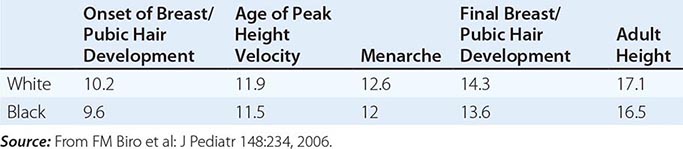
Other important hormonal changes also occur in conjunction with puberty. Growth hormone (GH) levels increase early in puberty, stimulated in part by the pubertal increases in estrogen secretion. GH increases insulin-like growth factor-I (IGF-I), which enhances linear growth. The growth spurt is generally less pronounced in girls than in boys, with a peak growth velocity of ~7 cm/year. Linear growth is ultimately limited by closure of epiphyses in the long bones as a result of prolonged exposure to estrogen. Puberty is also associated with mild insulin resistance.
DISORDERS OF PUBERTY
The differential diagnosis of precocious and delayed puberty is similar in boys (Chap. 411) and girls. However, there are differences in the timing of normal puberty and differences in the relative frequency of specific disorders in girls compared with boys.
Precocious Puberty Traditionally, precocious puberty has been defined as the development of secondary sexual characteristics before the age of 8 in girls based on data from Marshall and Tanner in British girls studied in the 1960s. More recent studies led to recommendations that girls be evaluated for precocious puberty if breast development or pubic hair is present at <7 years of age for white girls or <6 years for black girls.
Precocious puberty in girls is most often centrally mediated (Table 412-2), resulting from early activation of the hypothalamic-pituitary-ovarian axis. It is characterized by pulsatile LH secretion (which is initially associated with deep sleep) and an enhanced LH and FSH response to exogenous GnRH (two- to threefold stimulation) (Table 412-3). True precocity is marked by advancement in bone age of >2 standard deviations, a recent history of growth acceleration, and progression of secondary sexual characteristics. In girls, centrally mediated precocious puberty is idiopathic in ~85% of cases; however, neurogenic causes must be considered. Mutations in genes associated with GnRH deficiency have been reported in small numbers of patients with idiopathic precocious puberty (KISS, KISS1R, TAC3, TAC3R, and DAX-1), but their frequency is insufficient to warrant their use in clinical testing. GnRH agonists that induce pituitary desensitization are the mainstay of treatment to prevent premature epiphyseal closure and preserve adult height, as well as to manage psychosocial repercussions of precocious puberty.
|
DIFFERENTIAL DIAGNOSIS OF PRECOCIOUS PUBERTY |
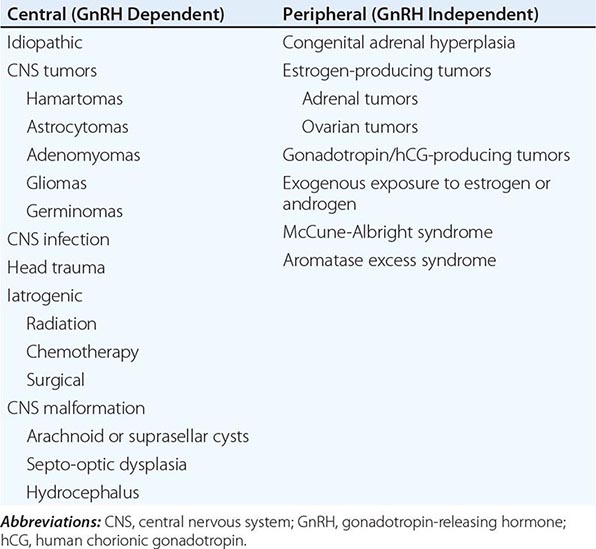
|
EVALUATION OF PRECOCIOUS AND DELAYED PUBERTY |
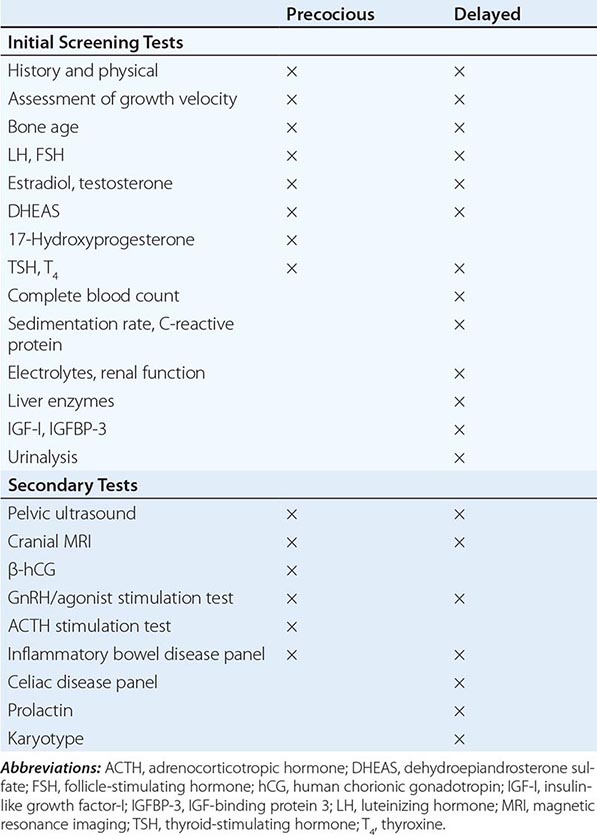
Peripherally mediated precocious puberty does not involve activation of the hypothalamic-pituitary-ovarian axis and is characterized by suppressed gonadotropins in the presence of elevated estradiol. Management of peripheral precocious puberty involves treating the underlying disorder (Table 412-2) and limiting the effects of gonadal steroids using aromatase inhibitors, inhibitors of steroidogenesis, and ER blockers. It is important to be aware that central precocious puberty can also develop in girls whose precocity was initially peripherally mediated, as in McCune-Albright syndrome and congenital adrenal hyperplasia.
Incomplete and intermittent forms of precocious puberty may also occur. For example, premature breast development may occur in girls before the age of 2 years, with no further progression and without significant advancement in bone age, estrogen production, or compromised height. Premature adrenarche can also occur in the absence of progressive pubertal development, but it must be distinguished from late-onset congenital adrenal hyperplasia and androgen-secreting tumors, in which case it may be termed heterosexual precocity. Premature adrenarche may be associated with obesity, hyperinsulinemia, and the subsequent predisposition to PCOS.
Delayed Puberty Delayed puberty (Table 412-4) is defined as the absence of secondary sexual characteristics by age 13 in girls. The diagnostic considerations are very similar to those for primary amenorrhea (Chap. 69). Between 25 and 40% of delayed puberty in girls is of ovarian origin, with Turner’s syndrome accounting for the majority of such patients. Functional hypogonadotropic hypogonadism encompasses diverse etiologies such as systemic illnesses, including celiac disease and chronic renal disease, and endocrinopathies such as diabetes and hypothyroidism. In addition, girls appear to be particularly susceptible to the adverse effects of decreased energy balance resulting from exercise, dieting, and/or eating disorders. Together these reversible conditions account for ~25% of delayed puberty in girls. Congenital hypogonadotropic hypogonadism in girls or boys can be caused by mutations in several different genes or combinations of genes (Fig. 412-4, Chap. 411, Table 411-2). Approximately 50% of girls with congenital hypogonadotropic hypogonadism, with or without anosmia, have a history of some degree of breast development, and 10% report one to two episodes of vaginal bleeding. Family studies suggest that genes identified in association with absent puberty may also cause delayed puberty, and recent reports have further suggested that a genetic susceptibility to environmental stresses such as diet and exercise may account for at least some cases of functional hypothalamic amenorrhea. Although neuroanatomic causes of delayed puberty are considerably less common in girls than in boys, it is always important to rule these out in the setting of hypogonadotropic hypogonadism.
|
DIFFERENTIAL DIAGNOSIS OF DELAYED PUBERTY |
Abbreviations: CHD7, chromodomain-helicase-DNA-binding protein 7; CNS, central nervous system; FGF8, fibroblast growth factor 8; FGFR1, fibroblast growth factor 1 receptor; FSHβ, follicle-stimulating hormone β chain; FSHR, FSH receptor; GNRHR, gonadotropin-releasing hormone receptor; HESX1, homeobox, embryonic stem cell expressed 1; HS6ST1, heparin sulfate 6-O sulfotransferase 1; IHH, idiopathic hypogonadotropic hypogonadism; KAL, Kallmann; KISS1, kisspeptin 1; KISSR1, KISS1 receptor; LHR, luteinizing hormone receptor; NSMF, NMDA receptor synaptonuclear signaling and neuronal migration factor; PROK2, prokineticin 2; PROKR2 prokineticin receptor 2; PROP1, prophet of Pit1, paired-like homeodomain transcription factor SEMA3A, semaphorin-3A; WDR11, WD repeat-containing protein 11.
413 |
Menopause and Postmenopausal Hormone Therapy |
Menopause is the permanent cessation of menstruation due to loss of ovarian follicular function. It is diagnosed retrospectively after 12 months of amenorrhea. The average age at menopause is 51 years among U.S. women. Perimenopause refers to the time period preceding menopause, when fertility wanes and menstrual cycle irregularity increases, until the first year after cessation of menses. The onset of perimenopause precedes the final menses by 2–8 years, with a mean duration of 4 years. Smoking accelerates the menopausal transition by 2 years.
Although the peri- and postmenopausal transitions share many symptoms, the physiology and clinical management of the two differ. Low-dose oral contraceptives have become a therapeutic mainstay in perimenopause, whereas postmenopausal hormone therapy (HT) has been a common method of symptom alleviation after menstruation ceases.
PERIMENOPAUSE
PHYSIOLOGY
Ovarian mass and fertility decline sharply after age 35 and even more precipitously during perimenopause; depletion of primary follicles, a process that begins before birth, occurs steadily until menopause (Chap. 412). In perimenopause, intermenstrual intervals shorten significantly (typically by 3 days) as a result of an accelerated follicular phase. Follicle-stimulating hormone (FSH) levels rise because of altered folliculogenesis and reduced inhibin secretion. In contrast to the consistently high FSH and low estradiol levels seen in menopause, perimenopause is characterized by “irregularly irregular” hormone levels. The propensity for anovulatory cycles can produce a hyperestrogenic, hypoprogestagenic environment that may account for the increased incidence of endometrial hyperplasia or carcinoma, uterine polyps, and leiomyoma observed among women of perimenopausal age. Mean serum levels of selected ovarian and pituitary hormones during the menopausal transition are shown in Fig. 413-1. With transition into menopause, estradiol levels fall markedly, whereas estrone levels are relatively preserved, a pattern reflecting peripheral aromatization of adrenal and ovarian androgens. Levels of FSH increase more than those of luteinizing hormone, presumably because of the loss of inhibin as well as estrogen feedback.
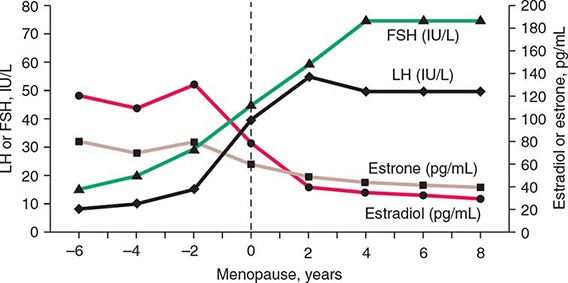
FIGURE 413-1 Mean serum levels of ovarian and pituitary hormones during the menopausal transition. FSH, follicle-stimulating hormone; LH, luteinizing hormone. (From JL Shifren, I Schiff: J Womens Health Gend Based Med 9 Suppl 1:S3, 2000.)
DIAGNOSTIC TESTS
The Stages of Reproductive Aging Workshop +10 (STRAW+10) classification provides a comprehensive framework for the clinical assessment of ovarian aging. As shown in Fig. 413-2, menstrual cycle characteristics are the principal criteria for characterizing the menopausal transition, with biomarker measures as supportive criteria. Because of their extreme intraindividual variability, FSH and estradiol levels are imperfect diagnostic indicators of perimenopause in menstruating women. However, a consistently low FSH level in the early follicular phase (days 2–5) of the menstrual cycle does not support a diagnosis of perimenopause, while levels >25 IU/L in a random blood sample are characteristic of the late menopause transition. FSH measurement can also aid in assessing fertility; levels of <20 IU/L, 20 to <30 IU/L, and ≥30 IU/L measured on day 3 of the cycle indicate a good, fair, and poor likelihood of achieving pregnancy, respectively. Antimüllerian hormone and inhibin B may also be useful for assessing reproductive aging.
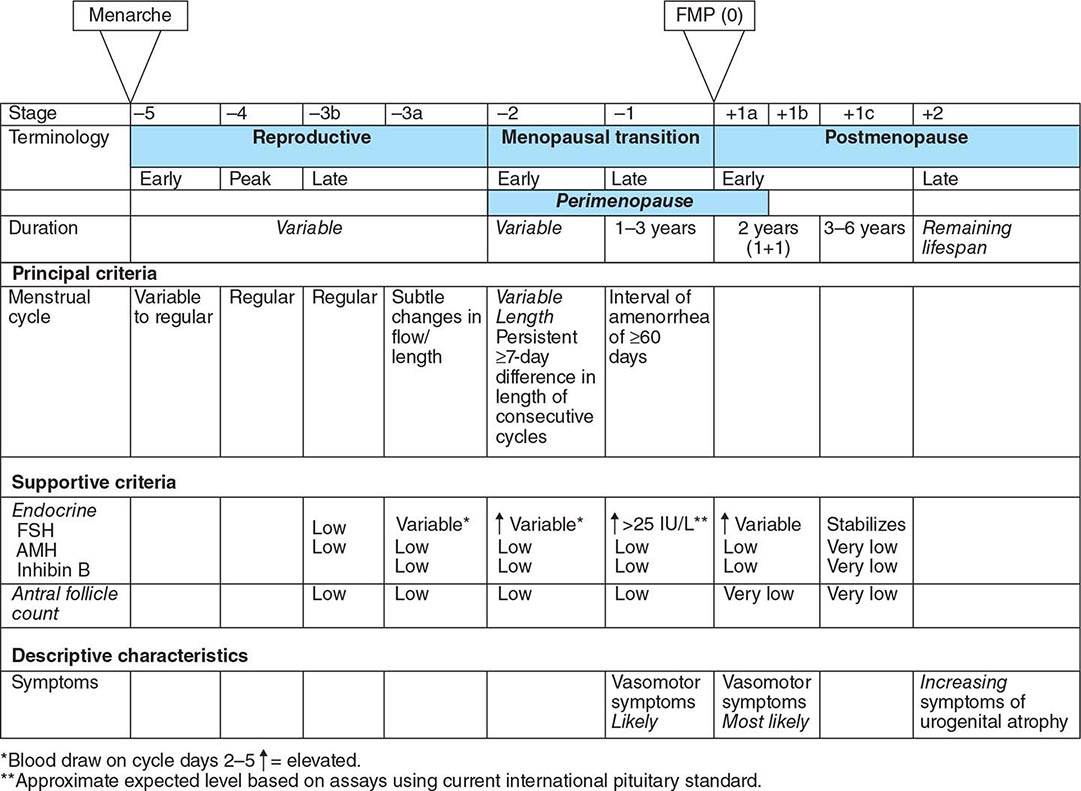
FIGURE 413-2 The Stages of Reproductive Aging Workshop +10 (STRAW +10) staging system for reproductive aging in women. AMH, antimüllerian hormone; FSH, follicle-stimulating hormone. (From SD Harlow et al: Menopause 14:387, 2012. Reproduced with permission.)
SYMPTOMS
Determining whether symptoms that develop in midlife are due to ovarian senescence or to other age-related changes is difficult. There is strong evidence that the menopausal transition can cause hot flashes, night sweats, irregular bleeding, and vaginal dryness, and there is moderate evidence that it can cause sleep disturbances in some women. There is inconclusive or insufficient evidence that ovarian aging is a major cause of mood swings, depression, impaired memory or concentration, somatic symptoms, urinary incontinence, or sexual dysfunction. In one U.S. study, nearly 60% of women reported hot flashes in the 2 years before their final menses. Symptom intensity, duration, frequency, and effects on quality of life are highly variable.
|
TREATMENT |
PERIMENOPAUSE |
PERIMENOPAUSAL THERAPY
For women with irregular or heavy menses or hormone-related symptoms that impair quality of life, low-dose combined oral contraceptives are a staple of therapy. Static doses of estrogen and progestin (e.g., 20 μg of ethinyl estradiol and 1 mg of norethindrone acetate daily for 21 days each month) can eliminate vasomotor symptoms and restore regular cyclicity. Oral contraceptives provide other benefits, including protection against ovarian and endometrial cancers and increased bone density, although it is not clear whether use during perimenopause decreases fracture risk later in life. Moreover, the contraceptive benefit is important, given that the unintentional pregnancy rate among women in their forties rivals that of adolescents. Contraindications to oral contraceptive use include cigarette smoking, liver disease, a history of thromboembolism or cardiovascular disease, breast cancer, or unexplained vaginal bleeding. Progestin-only formulations (e.g., 0.35 mg of norethindrone daily) or medroxyprogesterone (Depo-Provera) injections (e.g., 150 mg IM every 3 months) may provide an alternative for the treatment of perimenopausal menorrhagia in women who smoke or have cardiovascular risk factors. Although progestins neither regularize cycles nor reduce the number of bleeding days, they reduce the volume of menstrual flow.
Nonhormonal strategies to reduce menstrual flow include the use of nonsteroidal anti-inflammatory agents such as mefenamic acid (an initial dose of 500 mg at the start of menses, then 250 mg qid for 2–3 days) or, when medical approaches fail, endometrial ablation. It should be noted that menorrhagia requires an evaluation to rule out uterine disorders. Transvaginal ultrasound with saline enhancement is useful for detecting leiomyomata or polyps, and endometrial aspiration can identify hyperplastic changes.
TRANSITION TO MENOPAUSE
For sexually active women using contraceptive hormones to alleviate perimenopausal symptoms, the question of when and if to switch to HT must be individualized. Doses of estrogen and progestogen (either synthetic progestins or natural forms of progesterone) in HT are lower than those in oral contraceptives and have not been documented to prevent pregnancy. Although a 1-year absence of spontaneous menses reliably indicates ovulation cessation, it is not possible to assess the natural menstrual pattern while a woman is taking an oral contraceptive. Women willing to switch to a barrier method of contraception should do so; if menses occur spontaneously, oral contraceptive use can be resumed. The average age of final menses among relatives can serve as a guide for when to initiate this process, which can be repeated yearly until menopause has occurred.
MENOPAUSE AND POSTMENOPAUSAL HORMONE THERAPY
One of the most complex health care decisions facing women is whether to use postmenopausal HT. Once prescribed primarily to relieve vasomotor symptoms, HT has been promoted as a strategy to forestall various disorders that accelerate after menopause, including osteoporosis and cardiovascular disease. In 2000, nearly 40% of postmenopausal women age 50–74 in the United States had used HT. This widespread use occurred despite the paucity of conclusive data, until recently, on the health consequences of such therapy. Although many women rely on their health care providers for a definitive answer to the question of whether to use postmenopausal hormones, balancing the benefits and risks for an individual patient is challenging.
Although observational studies suggest that HT prevents cardiovascular and other chronic diseases, the apparent benefits may result at least in part from differences between women who opt to take postmenopausal hormones and women who do not. Those choosing HT tend to be healthier, have greater access to medical care, are more compliant with prescribed treatments, and maintain a more health-promoting lifestyle. Randomized trials, which eliminate these confounding factors, have not consistently confirmed the benefits found in observational studies. Indeed, the largest HT trial to date, the Women’s Health Initiative (WHI), which examined more than 27, 000 postmenopausal women age 50–79 (mean age, 63) for an average of 5–7 years, was stopped early because of an overall unfavorable benefit-risk ratio in the estrogen-progestin arm and an excess risk of stroke that was not offset by a reduced risk of coronary heart disease (CHD) in the estrogen-only arm.
The following summary offers a decision-making guide based on a synthesis of currently available evidence. Prevention of cardiovascular disease is eliminated from the equation due to lack of evidence for such benefits in recent randomized clinical trials.
BENEFITS AND RISKS OF POSTMENOPAUSAL HORMONE THERAPY
See Table 413-1.
|
BENEFITS AND RISKS OF POSTMENOPAUSAL HORMONE THERAPY IN THE OVERALL STUDY POPULATION OF WOMEN 50–79 YEARS OF AGE IN THE INTERVENTION PHASE OF THE WOMEN’S HEALTH INITIATIVE (WHI) ESTROGEN-PROGESTIN AND ESTROGEN-ALONE TRIALSa |
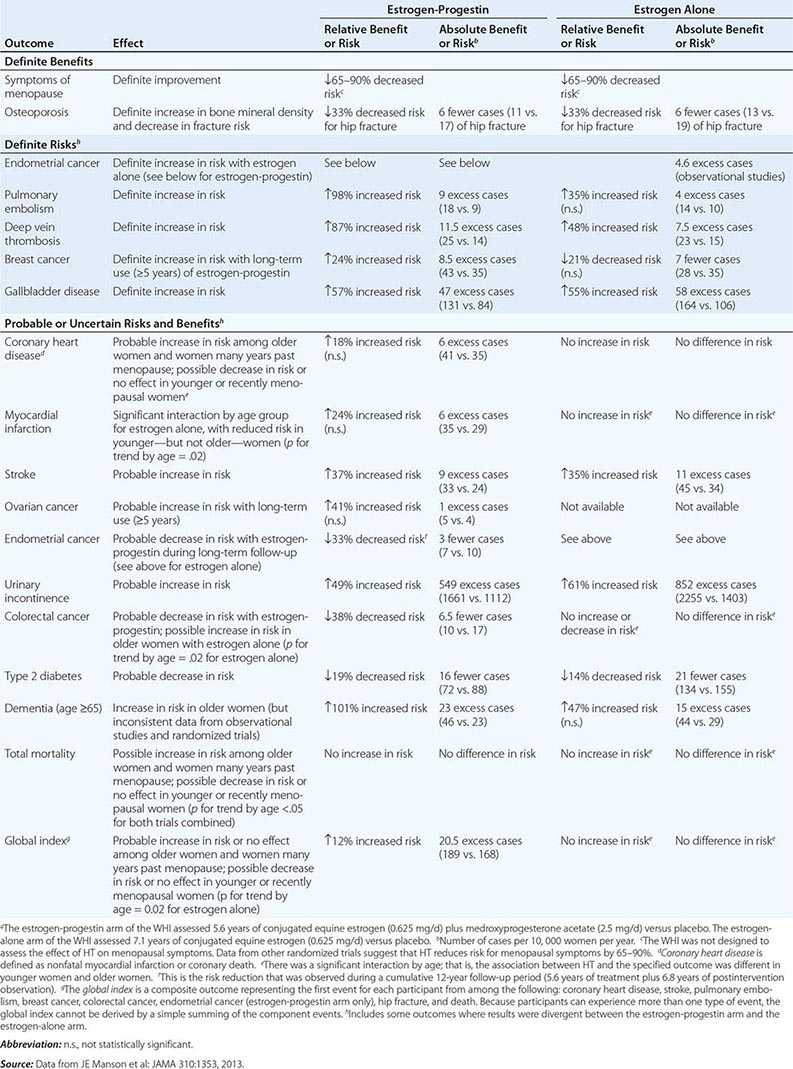
Definite Benefits • SYMPTOMS OF MENOPAUSE Compelling evidence, including data from randomized clinical trials, indicates that estrogen therapy is highly effective for controlling vasomotor and genitourinary symptoms. Alternative approaches, including the use of antidepressants (such as paroxetine, 7.5 mg/d; or venlafaxine, 75–150 mg/d), gabapentin (300–900 mg/d), clonidine (0.1–0.2 mg/d), or vitamin E (400–800 IU/d), or the consumption of soy-based products or other phytoestrogens, may also alleviate vasomotor symptoms, although they are less effective than HT. Paroxetine is the only nonhormonal drug approved by the U.S. Food and Drug Administration for treatment of vasomotor symptoms. Bazedoxifene, an estrogen agonist/antagonist, in combination with conjugated estrogens has also received approval for vasomotor symptom management. For genitourinary symptoms, the efficacy of vaginal estrogen is similar to that of oral or transdermal estrogen; oral ospemifene is an additional option.
OSTEOPOROSIS (See also Chap. 425)
Bone density By reducing bone turnover and resorption rates, estrogen slows the aging-related bone loss experienced by most postmenopausal women. More than 50 randomized trials have demonstrated that postmenopausal estrogen therapy, with or without a progestogen, rapidly increases bone mineral density at the spine by 4–6% and at the hip by 2–3% and that those increases are maintained during treatment.
Fractures Data from observational studies indicate a 50–80% lower risk of vertebral fracture and a 25–30% lower risk of hip, wrist, and other peripheral fractures among current estrogen users; addition of a progestogen does not appear to modify this benefit. In the WHI, 5–7 years of either combined estrogen-progestin or estrogen-only therapy was associated with a 33% reduction in hip fractures and 25–30% fewer total fractures among a population unselected for osteoporosis. Bisphosphonates (such as alendronate, 10 mg/d or 70 mg once per week; risedronate, 5 mg/d or 35 mg once per week; or ibandronate, 2.5 mg/d or 150 mg once per month or 3 mg every 3 months IV) and raloxifene (60 mg/d), a selective estrogen receptor modulator (SERM), have been shown in randomized trials to increase bone mass density and decrease fracture rates. Other options for treatment of osteoporosis are bazedoxifene in combination with conjugated estrogens and parathyroid hormone (teriparatide, 20 μg/d SC). These agents, unlike estrogen, do not appear to have adverse effects on the endometrium or breast. Increased physical activity, adequate calcium intake (1000–1200 mg/d through diet or supplements in two or three divided doses), and adequate vitamin D intake (600–1000 IU/d) may also reduce the risk of osteoporosis-related fractures. According to the Institute of Medicine’s 2011 report, 25-hydroxyvitamin D blood levels of ≥50 nmol/L are sufficient for bone-density maintenance and fracture prevention. The Fracture Risk Assessment (FRAX®) score, an algorithm that combines an individual’s bone-density score with age and other risk factors to predict her 10-year risk of hip and major osteoporotic fracture, may be of use in guiding decisions about pharmacologic treatment (see www.shef.ac.uk/FRAX/).
Definite Risks • ENDOMETRIAL CANCER (WITH ESTROGEN ALONE) A combined analysis of 30 observational studies found a tripling of endometrial cancer risk among short-term users (1–5 years) of unopposed estrogen and a nearly tenfold increased risk among long-term users (≥10 years). These findings are supported by results from the randomized Postmenopausal Estrogen/Progestin Interventions (PEPI) trial, in which 24% of women assigned to unopposed estrogen for 3 years developed atypical endometrial hyperplasia—a premalignant lesion—as opposed to only 1% of women assigned to placebo. Use of a progestogen, which opposes the effects of estrogen on the endometrium, eliminates these risks and may even reduce risk (see later).
VENOUS THROMBOEMBOLISM A meta-analysis of observational studies found that current oral estrogen use was associated with a 2.5-fold increase in risk of venous thromboembolism in postmenopausal women. A meta-analysis of randomized trials, including the WHI, found a 2.1-fold increase in risk. Results from the WHI indicate a nearly twofold increase in risk of pulmonary embolism and deep vein thrombosis with estrogen-progestin and a 35–50% increase in these risks with estrogen-only therapy. Transdermal estrogen, taken alone or with certain progestogens (micronized progesterone or pregnane derivatives), appears to be a safer alternative with respect to thrombotic risk.
BREAST CANCER (WITH ESTROGEN-PROGESTIN) An increased risk of breast cancer has been found among current or recent estrogen users in observational studies; this risk is directly related to duration of use. In a meta-analysis of 51 case-control and cohort studies, short-term use (<5 years) of postmenopausal HT did not appreciably elevate breast cancer incidence, whereas long-term use (≥5 years) was associated with a 35% increase in risk. In contrast to findings for endometrial cancer, combined estrogen-progestin regimens appear to increase breast cancer risk more than estrogen alone. Data from randomized trials also indicate that estrogen-progestin raises breast cancer risk. In the WHI, women assigned to receive combination hormones for an average of 5.6 years were 24% more likely to develop breast cancer than women assigned to placebo, but 7.1 years of estrogen-only therapy did not increase risk. Indeed, the WHI showed a trend toward a reduction in breast cancer risk with estrogen alone, although it is unclear whether this finding would pertain to formulations of estrogen other than conjugated equine estrogens or to treatment durations of >7 years. In the Heart and Estrogen/Progestin Replacement Study (HERS), 4 years of combination therapy was associated with a 27% increase in breast cancer risk. Although the latter finding was not statistically significant, the totality of evidence strongly implicates estrogen-progestin therapy in breast carcinogenesis.
Some observational data suggest that the length of the interval between menopause onset and HT initiation may influence the association between such therapy and breast cancer risk, with a “gap time” of <3–5 years conferring a higher HT-associated breast cancer risk. (This pattern of findings contrasts with that for CHD, as discussed later in this Chapter.) However, this association remains inconclusive and may be a spurious finding attributable to higher rates of screening mammography and thus earlier cancer detection in HT users than in nonusers, especially in early menopause. Indeed, in the WHI trial, hazard ratios for HT and breast cancer risk did not differ among women 50–59, those 60–69, and those 70–79 years of age at trial entry. (There was insufficient power to examine finer age categories.) Additional research is needed to clarify the issue.
GALLBLADDER DISEASE Large observational studies report a two- to threefold increased risk of gallstones or cholecystectomy among postmenopausal women taking oral estrogen. In the WHI, women randomized to estrogen-progestin or estrogen alone were ∼55% more likely to develop gallbladder disease than those assigned to placebo. Risks were also increased in HERS. Transdermal HT might be a safer alternative, but further research is needed.
Probable or Uncertain Risks and Benefits • CORONARY HEART DISEASE/STROKE Until recently, HT had been enthusiastically recommended as a possible cardioprotective agent. In the past three decades, multiple observational studies suggested, in the aggregate, that estrogen use leads to a 35–50% reduction in CHD incidence among postmenopausal women. The biologic plausibility of such an association is supported by data from randomized trials demonstrating that exogenous estrogen lowers plasma low-density lipoprotein (LDL) cholesterol levels and raises high-density lipoprotein (HDL) cholesterol levels by 10–15%. Administration of estrogen also favorably affects lipoprotein(a) levels, LDL oxidation, endothelial vascular function, fibrinogen, and plasminogen activator inhibitor 1. However, estrogen therapy has unfavorable effects on other biomarkers of cardiovascular risk: it boosts triglyceride levels; promotes coagulation via factor VII, prothrombin fragments 1 and 2, and fibrinopeptide A elevations; and raises levels of the inflammatory marker C-reactive protein.
Randomized trials of estrogen or combined estrogen-progestin in women with preexisting cardiovascular disease have not confirmed the benefits reported in observational studies. In HERS (a secondary-prevention trial designed to test the efficacy and safety of estrogen-progestin therapy with regard to clinical cardiovascular outcomes), the 4-year incidence of coronary death and nonfatal myocardial infarction was similar in the active-treatment and placebo groups, and a 50% increase in risk of coronary events was noted during the first year among participants assigned to the active-treatment group. Although it is possible that progestin may mitigate estrogen’s benefits, the Estrogen Replacement and Atherosclerosis (ERA) trial indicated that angiographically determined progression of coronary atherosclerosis was unaffected by either opposed or unopposed estrogen treatment. Moreover, no cardiovascular benefit was found in the Papworth Hormone Replacement Therapy Atherosclerosis Study, a trial of transdermal estradiol with and without norethindrone; the Women’s Estrogen for Stroke Trial (WEST), a trial of oral 17β-estradiol; or the Estrogen in the Prevention of Reinfarction Trial (ESPRIT), a trial of oral estradiol valerate. Thus, in clinical trials, HT has not proved effective for the secondary prevention of cardiovascular disease in postmenopausal women.
Primary-prevention trials also suggest an early increase in cardiovascular risk and an absence of cardioprotection with postmenopausal HT. In the WHI, women assigned to 5.6 years of estrogen-progestin therapy were 18% more likely to develop CHD (defined in primary analyses as nonfatal myocardial infarction or coronary death) than those assigned to placebo, although this risk elevation was not statistically significant. However, during the trial’s first year, there was a significant 80% increase in risk, which diminished in subsequent years (p for trend by time = .03). In the estrogen-only arm of the WHI, no overall effect on CHD was observed during the 7.1 years of the trial or in any specific year of follow-up. This pattern of results was similar to that for the outcome of total myocardial infarction.
However, a closer look at available data suggests that timing of initiation of HT may critically influence the association between such therapy and CHD. Estrogen may slow early stages of atherosclerosis but have adverse effects on advanced atherosclerotic lesions. It has been hypothesized that the prothrombotic and proinflammatory effects of estrogen manifest themselves predominantly among women with subclinical lesions who initiate HT well after the menopausal transition, whereas women with less arterial damage who start HT early in menopause may derive cardiovascular benefit because they have not yet developed advanced lesions. Nonhuman primate data support this concept. Conjugated estrogens had no effect on the extent of coronary artery plaque in cynomolgus monkeys assigned to receive estrogen alone or combined with progestin starting 2 years (∼6 years in human terms) after oophorectomy and well after the establishment of atherosclerosis. However, administration of exogenous hormones immediately after oophorectomy, during the early stages of atherosclerosis, reduced the extent of plaque by 70%.
Lending further credence to this hypothesis are results of subgroup analyses of observational and clinical trial data. For example, among women who entered the WHI trial with a relatively favorable cholesterol profile, estrogen with or without progestin led to a 40% lower risk of incident CHD. Among women who entered with a worse cholesterol profile, therapy resulted in a 73% higher risk (p for interaction = .02). The presence or absence of the metabolic syndrome (Chap. 422) also strongly influenced the relation between HT and incident CHD. Among women with the metabolic syndrome, HT more than doubled CHD risk, whereas no association was observed among women without the syndrome. Moreover, although there was no association between estrogen-only therapy and CHD in the WHI trial cohort as a whole, such therapy was associated with a CHD risk reduction of 40% among participants age 50–59; in contrast, a risk reduction of only 5% was observed among those age 60–69, and a risk increase of 9% was found among those age 70–79 (p for trend by age = .08). For the outcome of total myocardial infarction, estrogen alone was associated with a borderline-significant 45% reduction and a nonsignificant 24% increase in risk among the youngest and oldest women, respectively (p for trend by age = .02). Estrogen was also associated with lower levels of coronary artery calcified plaque in the younger age group. Although age did not have a similar effect in the estrogen-progestin arm of the WHI, CHD risks increased with years since menopause (p for trend = .08), with a significantly elevated risk among women who were ≥20 years past menopause. For the outcome of total myocardial infarction, estrogen-progestin was associated with a 9% risk reduction among women <10 years past menopause as opposed to a 16% increase in risk among women 10–19 years past menopause and a twofold increase in risk among women >20 years past menopause (p for trend = .01). In the large observational Nurses’ Health Study, women who chose to start HT within 4 years of menopause experienced a lower risk of CHD than did nonusers, whereas those who began therapy ≥10 years after menopause appeared to receive little coronary benefit. Observational studies include a high proportion of women who begin HT within 3–4 years of menopause, whereas clinical trials include a high proportion of women ≥12 years past menopause; this difference helps to reconcile some of the apparent discrepancies between the two types of studies.
For the outcome of stroke, WHI participants assigned to estrogen-progestin or estrogen alone were ∼35% more likely to suffer a stroke than those assigned to placebo. Whether or not age at initiation of HT influences stroke risk is not well understood. In the WHI and the Nurses’ Health Study, HT was associated with an excess risk of stroke in all age groups. Further research is needed on age, time since menopause, and other individual characteristics (including biomarkers) that predict increases or decreases in cardiovascular risk associated with exogenous HT. Furthermore, it remains uncertain whether different doses, formulations, or routes of administration of HT will produce different cardiovascular effects.
COLORECTAL CANCER Observational studies have suggested that HT reduces risks of colon and rectal cancer, although the estimated magnitudes of the relative benefits have ranged from 8% to 34% in various meta-analyses. In the WHI (the sole trial to examine the issue), estrogen-progestin was associated with a significant 38% reduction in colorectal cancer over a 5.6-year period, although no benefit was seen with 7 years of estrogen-only therapy. However, a modifying effect of age was observed, with a doubling of risk with HT in women age 70–79 but no risk elevation in younger women (p for trend by age = .02).
COGNITIVE DECLINE AND DEMENTIA A meta-analysis of 10 case-control and two cohort studies suggested that postmenopausal HT is associated with a 34% decreased risk of dementia. Subsequent randomized trials (including the WHI), however, have failed to demonstrate any benefit of estrogen or estrogen-progestin therapy on the progression of mild to moderate Alzheimer’s disease and/or have indicated a potential adverse effect of HT on the incidence of dementia, at least in women ≥65 years of age. Among women randomized to HT (as opposed to placebo) at age 50–55 in the WHI, no effect on cognition was observed during the postintervention phase. Determining whether timing of initiation of HT influences cognitive outcomes will require further study.
OVARIAN CANCER AND OTHER DISORDERS On the basis of limited observational and randomized data, it has been hypothesized that HT increases the risk of ovarian cancer and reduces the risk of type 2 diabetes mellitus. Results from the WHI support these hypotheses. The WHI also found that HT use was associated with an increased risk of urinary incontinence and that estrogen-progestin was associated with increased rates of lung cancer mortality.
ENDOMETRIAL CANCER (WITH ESTROGEN-PROGESTIN) In the WHI, use of estrogen-progestin was associated with a nonsignificant 17% reduction in risk of endometrial cancer. A significant reduction in risk emerged during the postintervention period (see later).
ALL-CAUSE MORTALITY In the overall WHI cohort, estrogen with or without progestin was not associated with all-cause mortality. However, there was a trend toward reduced mortality in younger women, particularly with estrogen alone. For women 50–59, 60–69, and 70–79 years of age, relative risks (RRs) associated with estrogen-only therapy were 0.70, 1.01, and 1.21, respectively (p for trend = .04).
OVERALL BENEFIT-RISK PROFILE Estrogen-progestin was associated with an unfavorable benefit-risk profile (excluding relief from menopausal symptoms) as measured by a “global index”—a composite outcome including CHD, stroke, pulmonary embolism, breast cancer, colorectal cancer, endometrial cancer, hip fracture, and death (Table 413-1)—in the WHI cohort as a whole, and this association did not vary by 10-year age group. Estrogen-only therapy was associated with a neutral benefit-risk profile in the WHI cohort as a whole. However, there was a significant trend toward a more favorable benefit-risk profile among younger women and a less favorable profile among older women, with RRs of 0.84, 0.99, and 1.17 for women 50–59, 60–69, and 70–79 years of age, respectively (p for trend by age = .02).
CHANGES IN HEALTH STATUS AFTER DISCONTINUATION OF HORMONE THERAPY In the WHI, many but not all risks and benefits associated with active use of HT dissipated within 5–7 years after discontinuation of therapy. For estrogen-progestin, an elevated risk of breast cancer persisted (RR = 1.28 [95% confidence interval, 1.11–1.48]) during a cumulative 12-year follow-up period (5.6 years of treatment plus 6.8 years of postintervention observation), but most cardiovascular disease risks became neutral. A reduction in hip fracture risk persisted (RR = 0.81 [0.68–0.97]), and a significant reduction in endometrial cancer risk emerged (RR = 0.67 [0.49–0.91]). For estrogen alone, the reduction in breast cancer risk became statistically significant (RR = 0.79 [0.65–0.97]) during a cumulative 12-year follow-up period (6.8 years of treatment plus 5.1 years of postintervention observation), and significant differences by age group persisted for total myocardial infarction and the global index, with more favorable results for younger women.
APPROACH TO THE PATIENT:
Postmenopausal Hormone Therapy
The rational use of postmenopausal HT requires balancing the potential benefits and risks. Figure 413-3 provides one approach to decision making. The clinician should first determine whether the patient has moderate to severe menopausal symptoms—the primary indication for initiation of systemic HT. Systemic HT may also be used to prevent osteoporosis in women at high risk of fracture who cannot tolerate alternative osteoporosis therapies. (Vaginal estrogen or other medications may be used to treat urogenital symptoms in the absence of vasomotor symptoms.) The benefits and risks of such therapy should be reviewed with the patient, giving more emphasis to absolute than to relative measures of effect and pointing out uncertainties in clinical knowledge where relevant. Because chronic disease rates generally increase with age, absolute risks tend to be greater in older women, even when relative risks remain similar. Potential side effects—especially vaginal bleeding that may result from use of the combined estrogen-progestogen formulations recommended for women with an intact uterus—should be noted. The patient’s own preference regarding therapy should be elicited and factored into the decision. Contraindications to HT should be assessed routinely and include unexplained vaginal bleeding, active liver disease, venous thromboembolism, history of endometrial cancer (except stage 1 without deep invasion) or breast cancer, and history of CHD, stroke, transient ischemic attack, or diabetes. Relative contraindications include hypertriglyceridemia (>400 mg/dL) and active gallbladder disease; in such cases, transdermal estrogen may be an option. Primary prevention of heart disease should not be viewed as an expected benefit of HT, and an increase in the risk of stroke as well as a small early increase in the risk of coronary artery disease should be considered. Nevertheless, such therapy may be appropriate if the noncoronary benefits of treatment clearly outweigh the risks. A woman who suffers an acute coronary event or stroke while taking HT should discontinue therapy immediately.
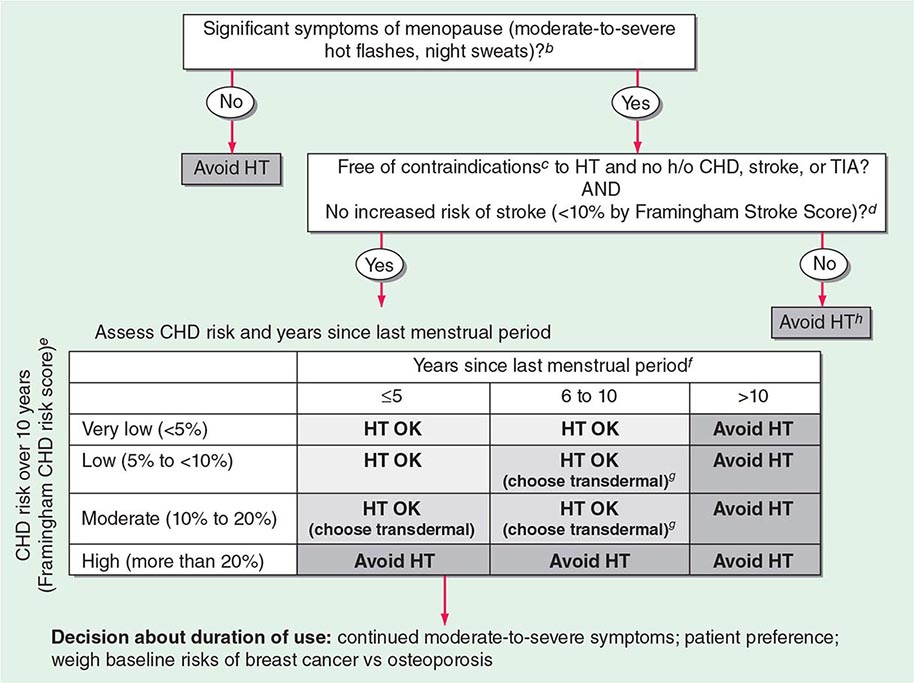
FIGURE 413-3 Chart for identifying appropriate candidates for postmenopausal hormone therapy (HT).a
aReassess each step at least once every 6–12 months (assuming the patient’s continued preference for HT). bWomen who are at high risk of osteoporotic fracture but are unable to tolerate alternative preventive medications may also be reasonable candidates for systemic HT even if they do not have moderate to severe vasomotor symptoms. Women who have vaginal dryness without moderate to severe vasomotor symptoms may be candidates for vaginal estrogen. cTraditional contraindications are unexplained vaginal bleeding; active liver disease; history of venous thromboembolism due to pregnancy, oral contraceptive use, or an unknown etiology; blood-clotting disorder; history of breast or endometrial cancer; and diabetes. Oral HT should be avoided but transdermal HT may be an option (see g below) for other contraindications, including high triglyceride levels (>400 mg/dL); active gallbladder disease; and history of venous thromboembolism due to past immobility, surgery, or bone fracture. dTen-year risk of stroke, based on Framingham Stroke Risk Score (RB D’Agostino et al: Stroke risk profile: Adjustment for antihypertensive medication. The Framingham Study. Stroke 25:40, 1994), as modified by JE Manson, SS Bassuk: Hot Flashes, Hormones & Your Health. New York, McGraw-Hill, 2007. eTen-year risk of CHD, based on Framingham Coronary Heart Disease Risk Score (Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults: JAMA 285:2486, 2001), as modified by JE Manson, SS Bassuk: Hot Flashes, Hormones & Your Health. New York, McGraw-Hill, 2007. f Women >10 years past menopause are not good candidates for initiation (first use) of HT. gAvoid oral HT. Transdermal HT may be an option because it has a less adverse effect on clotting factors, triglyceride levels, and inflammation factors than oral HT. hConsider selective serotonin or serotonin–norepinephrine reuptake inhibitor, gabapentin, clonidine, soy, or another alternative.
Abbreviations: CHD, coronary heart disease; h/o, history of; TIA, transient ischemic attack. (Adapted from JE Manson, SS Bassuk: Hot Flashes, Hormones & Your Health. New York, McGraw-Hill, 2007. Copyright © 2007 by the President and Fellows of Harvard College. All rights reserved.)
Short-term use (<5 years for estrogen-progestogen and <7 years for estrogen alone) is appropriate for relief of menopausal symptoms among women without contraindications to such use. However, such therapy should be avoided by women with an elevated baseline risk of future cardiovascular events. Women who have contraindications for or are opposed to HT may derive benefit from the use of certain antidepressants (including venlafaxine, fluoxetine, or paroxetine), gabapentin, clonidine, soy, or black cohosh and, for genitourinary symptoms, intravaginal estrogen creams or devices, or ospemifene.
Long-term use (≥5 years for estrogen-progestogen and ≥7 years for estrogen alone) is more problematic because a heightened risk of breast cancer must be factored into the decision, especially for estrogen-progestogen. Reasonable candidates for such use include the small percentage of postmenopausal women who have persistent severe vasomotor symptoms along with an increased risk of osteoporosis (e.g., those with osteopenia, a personal or family history of nontraumatic fracture, or a weight below 125 lbs), who also have no personal or family history of breast cancer in a first-degree relative or other contraindications, and who have a strong personal preference for therapy. Poor candidates are women with elevated cardiovascular risk, those at increased risk of breast cancer (e.g., women who have a first-degree relative with breast cancer, susceptibility genes such as BRCA1 or BRCA2, or a personal history of cellular atypia detected by breast biopsy), and those at low risk of osteoporosis. Even for reasonable candidates, strategies to minimize dose and duration of use should be employed. For example, women using HT to relieve intense vasomotor symptoms in early postmenopause should consider discontinuing therapy within 5 years, resuming it only if such symptoms persist. Because of the role of progestogens in increasing breast cancer risk, regimens that employ cyclic rather than continuous progestogen exposure as well as formulations other than medroxyprogesterone acetate should be considered if treatment is extended. For prevention of osteoporosis, alternative therapies such as bisphosphonates or SERMs should be considered. Research on alternative progestogens and androgen-containing preparations has been limited, particularly with respect to long-term safety. Additional research on the effects of these agents on cardiovascular disease, glucose tolerance, and breast cancer will be of particular interest.
In addition to HT, lifestyle choices such as smoking abstention, adequate physical activity, and a healthy diet can play a role in controlling symptoms and preventing chronic disease. An expanding array of pharmacologic options (e.g., bisphosphonates, SERMs, and other agents for osteoporosis; cholesterol-lowering or antihypertensive agents for cardiovascular disease) should also reduce the widespread reliance on hormone use. However, short-term HT may still benefit some women.
414 |
Infertility and Contraception |
INFERTILITY
DEFINITION AND PREVALENCE
Infertility has traditionally been defined as the inability to conceive after 12 months of unprotected sexual intercourse. In women who ultimately conceived, pregnancy occurred in ~50% within 3 months, 75–82% within 6 months, and 85–92% within 12 months. The World Health Organization (WHO) considers infertility as a disability (an impairment of function) and thus access to health care falls under the Convention on the Rights of Persons with Disability. Thirty-four million women, predominantly from developing countries, have infertility resulting from maternal sepsis and unsafe abortion. In populations <60 years old, infertility is ranked the fifth highest serious global disability. In the United States, the rate of infertility in married women age 15–44 is 6% based on the National Survey of Family Growth, although prospective studies suggest that it may be as high as 12–15%. The infertility rate has remained relatively stable over the past 30 years in most countries. However, the proportion of couples without children has risen, reflecting both higher numbers of couples in childbearing years and a trend to delay childbearing. This trend has important implications because of an age-related decrease in fecundability: the incidence of primary infertility increases from ~8% between the ages of 18 and 38 to 25% and 30% between the ages of 35 and 39 and 40 and 44, respectively. It is estimated that 14% of couples in the United States have received medical assistance for infertility; of these, two-thirds received counseling, ~12% underwent infertility testing of the female and/or male partner, and 17% received drugs to induce ovulation.
CAUSES OF INFERTILITY
The spectrum of infertility ranges from reduced conception rates or the need for medical intervention to irreversible causes of infertility. Infertility can be attributed primarily to male factors in 25% of couples and female factors in 58% of couples and is unexplained in about 17% of couples (Fig. 414-1). Not uncommonly, both male and female factors contribute to infertility. Decreases in the ability to conceive as a function of age in women has led to recommendations that women >34 years old who are not at increased risk of infertility seek attention after 6 months, rather than 12 months as suggested for younger women, and receive an expedited work-up and approach to treatment.
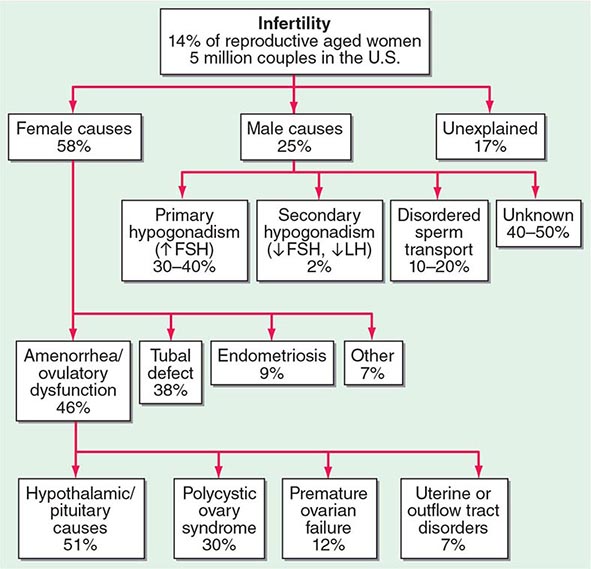
FIGURE 414-1 Causes of infertility. FSH, follicle-stimulating hormone; LH, luteinizing hormone.
|
TREATMENT |
INFERTILITY |
In addition to addressing the negative impact of smoking on fertility and pregnancy outcome, counseling about nutrition and weight is a fundamental component of infertility and pregnancy management. Both low and increased body mass index (BMI) are associated with infertility in women and with increased morbidity during pregnancy. Obesity has also been associated with infertility in men. The treatment of infertility should be tailored to the problems unique to each couple. In many situations, including unexplained infertility, mild-to-moderate endometriosis, and/or borderline semen parameters, a stepwise approach to infertility is optimal, beginning with low-risk interventions and moving to more invasive, higher risk interventions only if necessary. After determination of all infertility factors and their correction, if possible, this approach might include, in increasing order of complexity: (1) expectant management, (2) clomiphene citrate or an aromatase inhibitor (see below) with or without intrauterine insemination (IUI), (3) gonadotropins with or without IUI, and (4) in vitro fertilization (IVF). The time used for evaluation, correction of problems identified, and expectant management can be longer in women age <30 years, but this process should be advanced rapidly in women age >35 years. In some situations, expectant management will not be appropriate.
OVULATORY DYSFUNCTION
Treatment of ovulatory dysfunction should first be directed at identification of the etiology of the disorder to allow specific management when possible. Dopamine agonists, for example, may be indicated in patients with hyperprolactinemia (Chap. 403); lifestyle modification may be successful in women with obesity, low body weight, or a history of intensive exercise.
Medications used for ovulation induction include agents that increase FSH through alteration of negative feedback, gonadotropins, and pulsatile GnRH. Clomiphene citrate is a nonsteroidal estrogen antagonist that increases FSH and LH levels by blocking estrogen negative feedback at the hypothalamus. The efficacy of clomiphene for ovulation induction is highly dependent on patient selection. In appropriate patients, it induces ovulation in ~60% of women with PCOS and has traditionally been the initial treatment of choice. Combination with agents that modify insulin levels such as metformin does not appear to improve outcome. Clomiphene citrate is less successful in patients with hypogonadotropic hypogonadism. Aromatase inhibitors have also been investigated for the treatment of infertility. Studies suggest they may have advantages over clomiphene, but these medications have not been approved for this indication.
Gonadotropins are highly effective for ovulation induction in women with hypogonadotropic hypogonadism and PCOS and are used to induce the development of multiple follicles in unexplained infertility and in older reproductive-age women. Disadvantages include a significant risk of multiple gestation and the risk of ovarian hyperstimulation, particularly in women with polycystic ovaries, with or without other features of PCOS. Careful monitoring and a conservative approach to ovarian stimulation reduce these risks. Currently available gonadotropins include urinary preparations of LH and FSH, highly purified FSH, and recombinant FSH. Although FSH is the key component, LH is essential for steroidogenesis in hypogonadotropic patients, and LH or human chorionic gonadotropin (hCG) may improve results through effects on terminal differentiation of the oocyte. These methods are commonly combined with IUI.
None of these methods are effective in women with premature ovarian failure, in whom donor oocyte or adoption is the method of choice.
TUBAL DISEASE
If hysterosalpingography suggests a tubal or uterine cavity abnormality or if a patient is age ≥35 at the time of initial evaluation, laparoscopy with tubal lavage is recommended, often with a hysteroscopy. Although tubal reconstruction may be attempted if tubal disease is identified, it is generally being replaced by the use of IVF. These patients are at increased risk of developing an ectopic pregnancy.
ENDOMETRIOSIS
Although 60% of women with minimal or mild endometriosis may conceive within 1 year without treatment, laparoscopic resection or ablation appears to improve conception rates. Medical management of advanced stages of endometriosis is widely used for symptom control but has not been shown to enhance fertility. In moderate and severe endometriosis, conservative surgery is associated with pregnancy rates of 50 and 39%, respectively, compared with rates of 25 and 5% with expectant management alone. In some patients, IVF may be the treatment of choice.
MALE FACTOR INFERTILITY
The treatment options for male factor infertility have expanded greatly in recent years (Chap. 411). Secondary hypogonadism is highly amenable to treatment with gonadotropins or pulsatile gonadotropin-releasing hormone (GnRH) where available. In vitro techniques have provided new opportunities for patients with primary testicular failure and disorders of sperm transport. Choice of initial treatment options depends on sperm concentration and motility. Expectant management should be attempted initially in men with mild male factor infertility (sperm count of 15 to 20 × 106/mL and normal motility). Moderate male factor infertility (10 to 15 × 106/mL and 20–40% motility) should begin with IUI alone or in combination with treatment of the female partner with ovulation induction, but it may require IVF with or without intracytoplasmic sperm injection (ICSI). For men with a severe defect (sperm count of <10 × 106/mL, 10% motility), IVF with ICSI or donor sperm should be used. If ICSI is performed because of azoospermia due to congenital bilateral absence of the vas deferens, genetic testing and counseling should be provided because of the risk of cystic fibrosis.
ASSISTED REPRODUCTIVE TECHNOLOGIES
The development of assisted reproductive technologies (ARTs) has dramatically altered the treatment of male and female infertility. IVF is indicated for patients with many causes of infertility that have not been successfully managed with more conservative approaches. IVF or ICSI is often the treatment of choice in couples with a significant male factor or tubal disease, whereas IVF using donor oocytes is used in patients with premature ovarian failure and in women of advanced reproductive age. Success rates are influenced by cause of infertility and age, varying between 15 and 40%. Success rates are highest in anovulatory women and lowest in women with decreased ovarian reserve. In the United States, success rates are higher in white than in black, Asian, or Hispanic women. Although often effective, IVF is expensive and requires careful monitoring of ovulation induction and invasive techniques, including the aspiration of multiple follicles. IVF is associated with a significant risk of multiple gestation, particularly in women age <35, in whom the rate can be as high as 30%, which has led to specific recommendations for numbers of embryos or blastocysts to transfer based on age and specific prognostic factors.
CONTRACEPTION
Although use of contraception worldwide has increased in the last two decades, as of 2010, 146 million women worldwide age 15–49 years who were married or in a union had an unmet need for family planning. The absolute number of married women who use contraception or have an unmet need for family planning is projected to grow from 900 million (876–922 million) in 2010 to 962 million (927–992 million) in 2015.
Only 15% of couples in the United States report having unprotected sexual intercourse in the past 3 months. However, despite the wide availability and widespread use of a variety of effective methods of contraception, approximately one-half of all births in the United States are the result of unintended pregnancy. Teenage pregnancies continue to represent a serious public health problem in the United States, with >1 million unintended pregnancies each year—a significantly greater incidence than in other industrialized nations.
Of the contraceptive methods available (Table 414-1), a reversible form of contraception is used by >50% of couples, whereas sterilization (male or female) has been used as a permanent form of contraception by over one-third of couples. Pregnancy termination is relatively safe when directed by health care professionals but is rarely the option of choice.
|
EFFECTIVENESS OF DIFFERENT FORMS OF CONTRACEPTION |
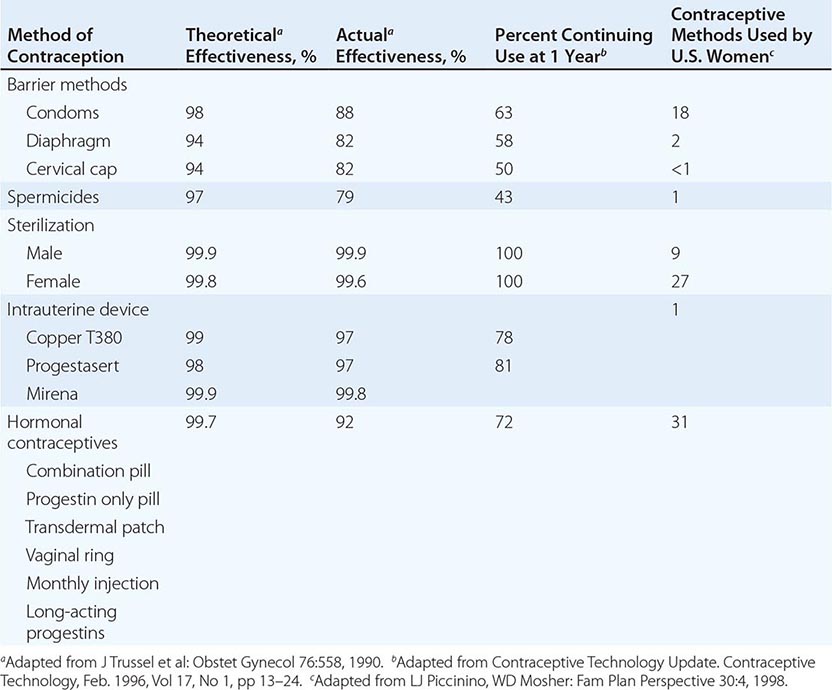
No single contraceptive method is ideal, although all are safer than carrying a pregnancy to term. The effectiveness of a given method of contraception does not just depend on the efficacy of the method itself. Discrepancies between theoretical and actual effectiveness emphasize the importance of patient education and compliance when considering various forms of contraception (Table 414-1). Knowledge of the advantages and disadvantages of each contraceptive is essential for counseling an individual about the methods that are safest and most consistent with his or her lifestyle. The WHO has extensive family planning resources for the physician and patient that can be accessed online. Similar resources for determining medical eligibility are available through the Centers for Disease Control and Prevention (CDC). Considerations for contraceptive use in obese patients and after bariatric surgery are discussed below.
BARRIER METHODS
Barrier contraceptives (such as condoms, diaphragms, and cervical caps) and spermicides are easily available, reversible, and have fewer side effects than hormonal methods. However, their effectiveness is highly dependent on adherence and proper use (Table 414-1). A major advantage of barrier contraceptives is the protection provided against sexually transmitted infections (STIs) (Chap. 163). Consistent use is associated with a decreased risk of HIV, gonorrhea, nongonococcal urethritis, and genital herpes, probably due in part to the concomitant use of spermicides. Natural membrane condoms may be less effective than latex condoms, and petroleum-based lubricants can degrade condoms and decrease their efficacy for preventing HIV infection. Barrier methods used by women include the diaphragm, cervical cap, and contraceptive sponge. The cervical cap and sponge are less effective than the diaphragm, and there have been rare reports of toxic shock syndrome with the diaphragm and contraceptive sponge.
STERILIZATION
Sterilization is the method of birth control most frequently chosen by fertile men and multiparous women >30 years old (Table 414-1). Sterilization refers to a procedure that prevents fertilization by surgical interruption of the fallopian tubes in women or the vas deferens in men. Although tubal ligation and vasectomy are potentially reversible, these procedures should be considered permanent and should not be undertaken without patient counseling.
Several methods of tubal ligation have been developed, all of which are highly effective with a 10-year cumulative pregnancy rate of 1.85 per 100 women. However, when pregnancy does occur, the risk of ectopic pregnancy may be as high as 30%. The success rate of tubal reanastomosis depends on the method of ligation used, but even after successful reversal, the risk of ectopic pregnancy remains high. In addition to prevention of pregnancy, tubal ligation reduces the risk of ovarian cancer, possibly by limiting the upward migration of potential carcinogens.
Vasectomy is a highly effective outpatient surgical procedure that has little risk. The development of azoospermia may be delayed for 2–6 months, and other forms of contraception must be used until two sperm-free ejaculations provide proof of sterility. Reanastomosis may restore fertility in 30–50% of men, but the success rate declines with time after vasectomy and may be influenced by factors such as the development of antisperm antibodies.
INTRAUTERINE DEVICES
IUDs inhibit pregnancy through several mechanisms, primarily via a spermicidal effect caused by a sterile inflammatory reaction induced by the presence of a foreign body in the uterine cavity (copper IUDs) or by the release of progestins (Progestasert, Mirena). IUDs provide a high level of efficacy in the absence of systemic metabolic effects, and ongoing motivation is not required to ensure efficacy once the device has been placed. However, only 1% of women in the United States use this method compared to a utilization rate of 15–30% in much of Europe and Canada, despite evidence that the newer devices are not associated with increased rates of pelvic infection and infertility, as occurred with earlier devices. An IUD should not be used in women at high risk for development of STI or in women at high risk for bacterial endocarditis. The IUD may not be effective in women with uterine leiomyomas because they alter the size or shape of the uterine cavity. IUD use is associated with increased menstrual blood flow, although this is less pronounced with the progestin-releasing IUD, which is associated with a more frequent occurrence of spotting or amenorrhea.
HORMONAL METHODS
Oral Contraceptive Pills Because of their ease of use and efficacy, oral contraceptive pills are the most widely used form of hormonal contraception. They act by suppressing ovulation, changing cervical mucus, and altering the endometrium. The current formulations are made from synthetic estrogens and progestins. The estrogen component of the pill consists of ethinyl estradiol or mestranol, which is metabolized to ethinyl estradiol. Multiple synthetic progestins are used. Norethindrone and its derivatives are used in many formulations. Low-dose norgestimate and the more recently developed (third-generation) progestins (desogestrel, gestodene, drospirenone) have a less androgenic profile; levonorgestrel appears to be the most androgenic of the progestins and should be avoided in patients with hyperandrogenism. The three major formulations of oral contraceptives are (1) fixed-dose estrogen-progestin combination, (2) phasic estrogen-progestin combination, and (3) progestin only. Each of these formulations is administered daily for 3 weeks followed by a week of no medication during which menstrual bleeding generally occurs. Two extended oral contraceptives are approved for use in the United States; Seasonale is a 3-month preparation with 84 days of active drug and 7 days of placebo, whereas Lybrel is a continuous preparation. Current doses of ethinyl estradiol range from 10 to 50 μg. However, indications for the 50-μg dose are rare, and the majority of formulations contain 30–35 μg of ethinyl estradiol. The reduced estrogen and progestin content in the second- and third-generation pills has decreased both side effects and risks associated with oral contraceptive use (Table 414-2). At the currently used doses, patients must be cautioned not to miss pills due to the potential for ovulation. Side effects, including breakthrough bleeding, amenorrhea, breast tenderness, and weight gain, often respond to a change in formulation. Even the lower dose oral contraceptives have been associated with an increased risk of cardiovascular disease (myocardial infarction, stroke, venous thromboembolism [VTE]), but the absolute excess risk is extremely low. VTE risk is higher with the third-generation than the second-generation progestins, and the risk of stroke and VTE is also higher with drospirenone (although not cyproterone), but the absolute excess risk is small and may be outweighed by contraceptive benefits and reduction in ovarian and endometrial cancer risk.
|
ORAL CONTRACEPTIVES: CONTRAINDICATIONS AND DISEASE RISK |
Abbreviation: OCP, oral contraceptive pill.
The microdose progestin-only minipill is less effective as a contraceptive, having a pregnancy rate of 2–7 per 100 women-years. However, it may be appropriate for women at increased risk for cardiovascular disease or for women who cannot tolerate synthetic estrogens.
Alternative Methods A weekly contraceptive patch (Ortho Evra) is available and has similar efficacy to oral contraceptives. Approximately 2% of patches fail to adhere, and a similar percentage of women have skin reactions. Efficacy is lower in women weighing >90 kg. The amount of estrogen delivered may be comparable to that of a 40-μg ethinyl estradiol oral contraceptive, raising the possibility of increased risk of VTE, which must be balanced against potential benefits for women not able to successfully use other methods. A monthly contraceptive estrogen/progestin injection (Lunelle) is highly effective, with a first-year failure rate of <0.2%, but it may be less effective in obese women. Its use is associated with bleeding irregularities that diminish over time. Fertility returns rapidly after discontinuation. A monthly vaginal ring (NuvaRing) that is intended to be left in place during intercourse is also available for contraceptive use. It is highly effective, with a 12-month failure rate of 0.7%. Ovulation returns within the first recovery cycle after discontinuation.
Long-Term Contraceptives Long-term progestin administration acts primarily by inhibiting ovulation and causing changes in the endometrium and cervical mucus that result in decreased implantation and sperm transport. Depot medroxyprogesterone acetate (Depo-Provera, DMPA), the only injectable form available in the United States, is effective for 3 months, but return of fertility after discontinuation may be delayed for up to 12–18 months. DMPA is now available for both SC and IM injection. Irregular bleeding, amenorrhea, and weight gain are the most common side effects. This form of contraception may be particularly good for women in whom an estrogen-containing contraceptive is contraindicated (e.g., migraine exacerbation, sickle cell anemia, fibroids).
POSTCOITAL CONTRACEPTION
The probability of pregnancy without relation to time of the month is 8%, but the probability varies significantly in relation to proximity to ovulation and may be as high has 30%. In order of efficacy, methods of postcoital contraception include the following:
1. Copper IUD insertion within a maximum of 5 days has a reported efficacy of 99–100% and prevents pregnancy by its spermicidal effect; insertion is frequently available through family planning clinics.
2. Oral antiprogestins (ulipristal acetate, 30 mg single dose, available worldwide, or mifepristone, 600 mg single dose, not available for this indication in the United States) prevent pregnancy by delaying or preventing ovulation; when administered, ideally within 72 h but up to 120 h after intercourse, they have an efficacy of 98–99%; require a prescription.
3. Levonorgestrel (1.5 mg as a single dose) delays or prevents ovulation and is not effective after ovulation; should be taken within 72 h of unprotected intercourse, and has an efficacy that varies between 60 and 94%; it is available over the counter.
Combined estrogen and progestin regimens have lower efficacy and are no longer recommended. A pregnancy test is not necessary before the use of oral methods, but pregnancy should be excluded before IUD insertion. Risk factors for failure of oral regimens include close proximity to ovulation and unprotected intercourse after use. In addition, there is an increased risk of pregnancy in obese and overweight women using levonorgestrel for postcoital contraception and an increased risk in obese women using an antiprogestin.
IMPACT OF OBESITY ON CONTRACEPTIVE CHOICE
Approximately one-third of adults in the United States are obese. Although obesity is associated with some reduction in fertility, the vast majority of obese women can conceive. The risk of pregnancy-associated complications is higher in obese women. Intrauterine contraception may be more effective than oral or transdermal methods for obese women. The WHO guidelines provide no restrictions (class 1) for the use of intrauterine contraception, DMPA, and progestin-only pills for obese women (BMI ≥30) in the absence of coexistent medical problems, whereas methods that include estrogen (pill, patch, ring) are considered class 2 (advantages generally outweigh theoretical or proven risks) due to the increased risk of thromboembolic disease. There are no restrictions to the use of any contraceptive methods following restrictive bariatric surgery procedures, but both combined and progestin-only pills are relatively less effective following procedures associated with malabsorption.
SECTION 3 |
OBESITY, DIABETES MELLITUS, AND METABOLIC SYNDROME |
415e |
Biology of Obesity |
In a world where food supplies are intermittent, the ability to store energy in excess of what is required for immediate use is essential for survival. Fat cells, residing within widely distributed adipose tissue depots, are adapted to store excess energy efficiently as triglyceride and, when needed, to release stored energy as free fatty acids for use at other sites. This physiologic system, orchestrated through endocrine and neural pathways, permits humans to survive starvation for as long as several months. However, in the presence of nutritional abundance and a sedentary lifestyle, and influenced importantly by genetic endowment, this system increases adipose energy stores and produces adverse health consequences.
DEFINITION AND MEASUREMENT
Obesity is a state of excess adipose tissue mass. Although often viewed as equivalent to increased body weight, this need not be the case—lean but very muscular individuals may be overweight by numerical standards without having increased adiposity. Body weights are distributed continuously in populations, so that choice of a medically meaningful distinction between lean and obese is somewhat arbitrary. Obesity is therefore defined by assessing its linkage to morbidity or mortality.
Although not a direct measure of adiposity, the most widely used method to gauge obesity is the body mass index (BMI), which is equal to weight/height2 (in kg/m2) (Fig. 415e-1). Other approaches to quantifying obesity include anthropometry (skinfold thickness), densitometry (underwater weighing), computed tomography (CT) or magnetic resonance imaging (MRI), and electrical impedance. Using data from the Metropolitan Life Tables, BMIs for the midpoint of all heights and frames among both men and women range from 19 to 26 kg/m2; at a similar BMI, women have more body fat than men. Based on data of substantial morbidity, a BMI of 30 is most commonly used as a threshold for obesity in both men and women. Most but not all large-scale epidemiologic studies suggest that all-cause, metabolic, cancer, and cardiovascular morbidity begin to rise (albeit at a slow rate) when BMIs are ≥25. Most authorities use the term overweight (rather than obese) to describe individuals with BMIs between 25 and 30. A BMI between 25 and 30 should be viewed as medically significant and worthy of therapeutic intervention in the presence of risk factors that are influenced by adiposity, such as hypertension and glucose intolerance.

FIGURE 415e-1 Nomogram for determining body mass index. To use this nomogram, place a ruler or other straight edge between the body weight (without clothes) in kilograms or pounds located on the left-hand line and the height (without shoes) in centimeters or inches located on the right-hand line. The body mass index is read from the middle of the scale and is in metric units. (Copyright 1979, George A. Bray, MD; used with permission.)
The distribution of adipose tissue in different anatomic depots also has substantial implications for morbidity. Specifically, intraabdominal and abdominal subcutaneous fat have more significance than subcutaneous fat present in the buttocks and lower extremities. This distinction is most easily made clinically by determining the waist-to-hip ratio, with a ratio >0.9 in women and >1.0 in men being abnormal. Many of the most important complications of obesity, such as insulin resistance, diabetes, hypertension, hyperlipidemia, and hyperandrogenism in women, are linked more strongly to intraabdominal and/or upper body fat than to overall adiposity (Chap. 422). The mechanism underlying this association is unknown but may relate to the fact that intraabdominal adipocytes are more lipolytically active than those from other depots. Release of free fatty acids into the portal circulation has adverse metabolic actions, especially on the liver. Adipokines and cytokines that are differentially secreted by adipocyte depots may play a role in the systemic complications of obesity.
PREVALENCE
Data from the National Health and Nutrition Examination Surveys (NHANES) show that the percentage of the American adult population with obesity (BMI >30) has increased from 14.5% (between 1976 and 1980) to 35.7% (between 2009 and 2010). As many as 68% of U.S. adults aged ≥20 years were overweight (defined as BMI >25) between the years of 2007 and 2008. Extreme obesity (BMI ≥40) has also increased and affects 5.7% of the population. The increasing prevalence of medically significant obesity raises great concern. Overall, the prevalence of obesity is comparable in men and women. In women, poverty is associated with increased prevalence. Obesity is more common among blacks and Hispanics. The prevalence in children and adolescents has been rising at a worrisome rate, reaching 15.9% in 2009/2010, but may be leveling off.
PHYSIOLOGIC REGULATION OF ENERGY BALANCE
Substantial evidence suggests that body weight is regulated by both endocrine and neural components that ultimately influence the effector arms of energy intake and expenditure. This complex regulatory system is necessary because even small imbalances between energy intake and expenditure will ultimately have large effects on body weight. For example, a 0.3% positive imbalance over 30 years would result in a 9-kg (20-lb) weight gain. This exquisite regulation of energy balance cannot be monitored easily by calorie-counting in relation to physical activity. Rather, body weight regulation or dysregulation depends on a complex interplay of hormonal and neural signals. Alterations in stable weight by forced overfeeding or food deprivation induce physiologic changes that resist these perturbations: with weight loss, appetite increases and energy expenditure falls; with overfeeding, appetite falls and energy expenditure increases. This latter compensatory mechanism frequently fails, however, permitting obesity to develop when food is abundant and physical activity is limited. A major regulator of these adaptive responses is the adipocyte-derived hormone leptin, which acts through brain circuits (predominantly in the hypothalamus) to influence appetite, energy expenditure, and neuroendocrine function (see below).
Appetite is influenced by many factors that are integrated by the brain, most importantly within the hypothalamus (Fig. 415e-2). Signals that impinge on the hypothalamic center include neural afferents, hormones, and metabolites. Vagal inputs are particularly important, bringing information from viscera, such as gut distention. Hormonal signals include leptin, insulin, cortisol, and gut peptides. Among the latter is ghrelin, which is made in the stomach and stimulates feeding, and peptide YY (PYY) and cholecystokinin, which is made in the small intestine and signals to the brain through direct action on hypothalamic control centers and/or via the vagus nerve. Metabolites, including glucose, can influence appetite, as seen by the effect of hypoglycemia to induce hunger; however, glucose is not normally a major regulator of appetite. These diverse hormonal, metabolic, and neural signals act by influencing the expression and release of various hypothalamic peptides (e.g., neuropeptide Y [NPY], Agouti-related peptide [AgRP], α-melanocyte-stimulating hormone [α-MSH], and melanin-concentrating hormone [MCH]) that are integrated with serotonergic, catecholaminergic, endocannabinoid, and opioid signaling pathways (see below). Psychological and cultural factors also play a role in the final expression of appetite. Apart from rare genetic syndromes involving leptin, its receptor, and the melanocortin system, specific defects in this complex appetite control network that influence common cases of obesity are not well defined.
FIGURE 415e-2 The factors that regulate appetite through effects on central neural circuits. Some factors that increase or decrease appetite are listed. AgRP, Agouti-related peptide; CART, cocaine- and amphetamine-related transcript; CCK, cholecystokinin; GLP-1, glucagon-related peptide-1; MCH, melanin-concentrating hormone; α-MSH, α-melanocyte-stimulating hormone; NPY, neuropeptide Y.
Energy expenditure includes the following components: (1) resting or basal metabolic rate; (2) the energy cost of metabolizing and storing food; (3) the thermic effect of exercise; and (4) adaptive thermogenesis, which varies in response to long-term caloric intake (rising with increased intake). Basal metabolic rate accounts for ~70% of daily energy expenditure, whereas active physical activity contributes 5–10%. Thus, a significant component of daily energy consumption is fixed.
Genetic models in mice indicate that mutations in certain genes (e.g., targeted deletion of the insulin receptor in adipose tissue) protect against obesity, apparently by increasing energy expenditure. Adaptive thermogenesis occurs in brown adipose tissue (BAT), which plays an important role in energy metabolism in many mammals. In contrast to white adipose tissue, which is used to store energy in the form of lipids, BAT expends stored energy as heat. A mitochondrial uncoupling protein (UCP-1) in BAT dissipates the hydrogen ion gradient in the oxidative respiration chain and releases energy as heat. The metabolic activity of BAT is increased by a central action of leptin, acting through the sympathetic nervous system that heavily innervates this tissue. In rodents, BAT deficiency causes obesity and diabetes; stimulation of BAT with a specific adrenergic agonist (β3 agonist) protects against diabetes and obesity. BAT exists in humans (especially neonates), and although its physiologic role is not yet established, identification of functional BAT in many adults using positron emission tomography (PET) imaging has increased interest in the implications of the tissue for pathogenesis and therapy of obesity. Beige fat cells, recently described, resemble BAT cells in expressing UCP-1. They are scattered through white adipose tissue, and their thermogenic potential is uncertain.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
FIGURE 411-1 Pubertal events in males. Sexual maturity ratings for genitalia and pubic hair and divided into five stages. (From WA Marshall, JM Tanner: Variations in the pattern of pubertal changes in boys. Arch Dis Child 45:13, 1970.)
The early stages of puberty are characterized by nocturnal surges of LH and FSH. Growth of the testes is usually the first sign of puberty, reflecting an increase in seminiferous tubule volume. Increasing levels of testosterone deepen the voice and increase muscle growth. Conversion of testosterone to DHT leads to growth of the external genitalia and pubic hair. DHT also stimulates prostate and facial hair growth and initiates recession of the temporal hairline. The growth spurt occurs at a testicular volume of about 10–12 mL. GH increases early in puberty and is stimulated in part by the rise in gonadal steroids. GH increases the level of insulin-like growth factor I (IGF-I), which enhances linear bone growth. The prolonged pubertal exposure to gonadal steroids (mainly estradiol) ultimately causes epiphyseal closure and limits further bone growth.
REGULATION OF TESTICULAR FUNCTION
REGULATION OF THE HYPOTHALAMIC-PITUITARY-TESTIS AXIS IN ADULT MAN
Hypothalamic GnRH regulates the production of the pituitary gonadotropins LH and FSH (Fig. 411-2). GnRH is released in discrete pulses approximately every 2 h, resulting in corresponding pulses of LH and FSH. These dynamic hormone pulses account in part for the wide variations in LH and testosterone, even within the same individual. LH acts primarily on the Leydig cell to stimulate testosterone synthesis. The regulatory control of androgen synthesis is mediated by testosterone and estrogen feedback on both the hypothalamus and the pituitary. FSH acts on the Sertoli cell to regulate spermatogenesis and the production of Sertoli products such as inhibin B, which acts to selectively suppress pituitary FSH. Despite these somewhat distinct Leydig and Sertoli cell–regulated pathways, testis function is integrated at several levels: GnRH regulates both gonadotropins; spermatogenesis requires high levels of testosterone; and numerous paracrine interactions between Leydig and Sertoli cells are necessary for normal testis function.
FIGURE 411-2 Human pituitary gonadotropin axis, structure of testis, and seminiferous tubule. E2, 17β-estradiol; DHT, dihydrotestosterone; FSH, follicle-stimulating hormones; GnRH, gonadotropin-releasing; LH, luteinizing hormone.
THE LEYDIG CELL: ANDROGEN SYNTHESIS
LH binds to its seven-transmembrane, G protein–coupled receptor to activate the cyclic AMP pathway. Stimulation of the LH receptor induces steroid acute regulatory (StAR) protein, along with several steroidogenic enzymes involved in androgen synthesis. LH receptor mutations cause Leydig cell hypoplasia or agenesis, underscoring the importance of this pathway for Leydig cell development and function. The rate-limiting process in testosterone synthesis is the delivery of cholesterol by the StAR protein to the inner mitochondrial membrane. Peripheral benzodiazepine receptor, a mitochondrial cholesterol-binding protein, is also an acute regulator of Leydig cell steroidogenesis. The five major enzymatic steps involved in testosterone synthesis are summarized in Fig. 411-3. After cholesterol transport into the mitochondrion, the formation of pregnenolone by CYP11A1 (side chain cleavage enzyme) is a limiting enzymatic step. The 17α-hydroxylase and the 17,20-lyase reactions are catalyzed by a single enzyme, CYP17; posttranslational modification (phosphorylation) of this enzyme and the presence of specific enzyme cofactors confer 17,20-lyase activity selectively in the testis and zona reticularis of the adrenal gland. Testosterone can be converted to the more potent DHT by 5α-reductase, or it can be aromatized to estradiol by CYP19 (aromatase). Two isoforms of steroid 5α-reductase, SRD5A1 and SRD5A2, have been described; all known kindreds with 5α-reductase deficiency have had mutations in SRD5A2, the predominant form in the prostate and the skin.
FIGURE 411-3 The biochemical pathway in the conversion of 27-carbon sterol cholesterol to androgens and estrogens.
Testosterone Transport and Metabolism In males, 95% of circulating testosterone is derived from testicular production (3–10 mg/d). Direct secretion of testosterone by the adrenal and the peripheral conversion of androstenedione to testosterone collectively account for another 0.5 mg/d of testosterone. Only a small amount of DHT (70 μg/d) is secreted directly by the testis; most circulating DHT is derived from peripheral conversion of testosterone. Most of the daily production of estradiol (~45 μg/d) in men is derived from aromatase-mediated peripheral conversion of testosterone and androstenedione.
Circulating testosterone is bound to two plasma proteins: sex hormone–binding globulin (SHBG) and albumin (Fig. 411-4). SHBG binds testosterone with much greater affinity than albumin. Only 0.5–3% of testosterone is unbound. According to the “free hormone” hypothesis, only the unbound fraction is biologically active; however, albumin-bound hormone dissociates readily in the capillaries and may be bioavailable. SHBG-bound testosterone also may be internalized through endocytic pits by binding to a protein called megalin. SHBG concentrations are decreased by androgens, obesity, diabetes mellitus, insulin, and nephrotic syndrome. Conversely, estrogen administration, hyperthyroidism, many chronic inflammatory illnesses, infections such as HIV or hepatitis B and C, and aging are associated with high SHBG concentrations.
FIGURE 411-4 Androgen metabolism and actions. SHBG, sex hormone–binding globulin.
Testosterone is metabolized predominantly in the liver, although some degradation occurs in peripheral tissues, particularly the prostate and the skin. In the liver, testosterone is converted by a series of enzymatic steps that involve 5α- and 5β-reductases, 3α- and 3β-hydroxysteroid dehydrogenases, and 17β-hydroxysteroid dehydrogenase into androsterone, etiocholanolone, DHT, and 3-α-androstanediol. These compounds undergo glucuronidation or sulfation before being excreted by the kidneys.
Mechanism of Androgen Action Testosterone exerts some of its biologic effects by binding to androgen receptor, either directly or after its conversion to DHT by the steroid 5-α reductase. Testosterone’s effects on the skeletal muscle, erythropoiesis, and bone in men do not require its obligatory conversion to DHT. However, the conversion of testosterone to DHT is necessary for the masculinization of the urogenital sinus and genital tubercle. Aromatization of testosterone to estradiol mediates additional effects of testosterone on the bone resorption, epiphyseal closure, sexual desire, vascular endothelium, and fat. DHT can also be converted in some tissues by 3-keto reductase/3β-hydroxysteroid dehydrogenase enzymes to 5α-androstane-3β,17β-diol, which is a high-affinity ligand and agonist of estrogen receptor β.
The androgen receptor (AR) is structurally related to the nuclear receptors for estrogen, glucocorticoids, and progesterone (Chap. 400e). The AR is encoded by a gene on the long arm of the × chromosome and has a molecular mass of about 110 kDa. A polymorphic region in the amino terminus of the receptor, which contains a variable number of glutamine repeats, modifies the transcriptional activity of the receptor. The AR protein is distributed in both the cytoplasm and the nucleus. The ligand binding to the AR induces conformational changes that allow the recruitment and assembly of tissue-specific cofactors and causes it to translocate into the nucleus, where it binds to DNA or other transcription factors already bound to DNA. Thus, the AR is a ligand-regulated transcription factor that regulates the expression of androgen-dependent genes in a tissue-specific manner. Some androgen effects may be mediated by nongenomic AR signal transduction pathways. Testosterone binds to AR with half the affinity of DHT. The DHT-AR complex also has greater thermostability and a slower dissociation rate than the testosterone-AR complex. However, the molecular basis for selective testosterone versus DHT actions remains incompletely explained.
THE SEMINIFEROUS TUBULES: SPERMATOGENESIS
The seminiferous tubules are convoluted, closed loops with both ends emptying into the rete testis, a network of progressively larger efferent ducts that ultimately form the epididymis (Fig. 411-2). The seminiferous tubules total about 600 m in length and comprise about two-thirds of testis volume. The walls of the tubules are formed by polarized Sertoli cells that are apposed to peritubular myoid cells. Tight junctions between Sertoli cells create a blood-testis barrier. Germ cells compose the majority of the seminiferous epithelium (~60%) and are intimately embedded within the cytoplasmic extensions of the Sertoli cells, which function as “nurse cells.” Germ cells progress through characteristic stages of mitotic and meiotic divisions. A pool of type A spermatogonia serve as stem cells capable of self-renewal. Primary spermatocytes are derived from type B spermatogonia and undergo meiosis before progressing to spermatids that undergo spermiogenesis (a differentiation process involving chromatin condensation, acquisition of an acrosome, elongation of cytoplasm, and formation of a tail) and are released from Sertoli cells as mature spermatozoa. The complete differentiation process into mature sperm requires 74 days. Peristaltic-type action by peritubular myoid cells transports sperm into the efferent ducts. The spermatozoa spend an additional 21 days in the epididymis, where they undergo further maturation and capacitation. The normal adult testes produce >100 million sperm per day.
Naturally occurring mutations in the FSHβ gene and in the FSH receptor confirm an important, but not essential, role for this pathway in spermatogenesis. Females with these mutations are hypogonadal and infertile because ovarian follicles do not mature; males exhibit variable degrees of reduced spermatogenesis, presumably because of impaired Sertoli cell function. Because Sertoli cells produce inhibin B, an inhibitor of FSH, seminiferous tubule damage (e.g., by radiation) causes a selective increase of FSH. Testosterone reaches very high concentrations locally in the testis and is essential for spermatogenesis. The cooperative actions of FSH and testosterone are important in the progression of meiosis and spermiation. FSH and testosterone regulate germ cell survival via the intrinsic and the extrinsic apoptotic mechanisms. FSH may also play an important role in supporting spermatogonia. Gonadotropin-regulated testicular RNA helicase (GRTH/DDX25), a testis-specific gonadotropin/androgen-regulated RNA helicase, is present in germ cells and Leydig cells and may be an important factor in the paracrine regulation of germ cell development. Several cytokines and growth factors are also involved in the regulation of spermatogenesis by paracrine and autocrine mechanisms. A number of knockout mouse models exhibit impaired germ cell development or spermatogenesis, presaging possible mutations associated with male infertility. The human Y chromosome contains a small pseudoautosomal region that can recombine with homologous regions of the × chromosome. Most of the Y chromosome does not recombine with the × chromosome and is referred to as the male-specific region of the Y (MSY). The MSY contains 156 transcription units that encode for 26 proteins, including nine families of Y-specific multicopy genes; many of these Y-specific genes are also testis-specific and necessary for spermatogenesis. Microdeletions of several Y chromosome azoospermia factor (AZF) genes (e.g., RNA-binding motif, RBM; deleted in azoospermia, DAZ) are associated with oligospermia or azoospermia.
|
TREATMENT |
MALE FACTOR INFERTILITY |
Treatment options for male factor infertility have expanded greatly in recent years. Secondary hypogonadism is highly amenable to treatment with pulsatile GnRH or gonadotropins (see below). Assisted reproductive technologies such as the in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI) have provided new opportunities for patients with primary testicular failure and disorders of sperm transport. Choice of initial treatment options depends on sperm concentration and motility. Expectant management should be attempted initially in men with mild male factor infertility (sperm count of 15–20 × 106/mL and normal motility). Moderate male factor infertility (10–15 × 106/mL and 20–40% motility) should begin with intrauterine insemination alone or in combination with treatment of the female partner with clomiphene or gonadotropins, but it may require IVF with or without ICSI. For men with a severe defect (sperm count of <10 × 106/mL, 10% motility), IVF with ICSI or donor sperm should be used.
CLINICAL AND LABORATORY EVALUATION OF MALE REPRODUCTIVE FUNCTION
HISTORY AND PHYSICAL EXAMINATION
The history should focus on developmental stages such as puberty and growth spurts, as well as androgen-dependent events such as early morning erections, frequency and intensity of sexual thoughts, and frequency of masturbation or intercourse. Although libido and the overall frequency of sexual acts are decreased in androgen-deficient men, young hypogonadal men may achieve erections in response to visual erotic stimuli. Men with acquired androgen deficiency often report decreased energy and increased irritability.
The physical examination should focus on secondary sex characteristics such as hair growth, gynecomastia, testicular volume, prostate, and height and body proportions. Eunuchoid proportions are defined as an arm span >2 cm greater than height and suggest that androgen deficiency occurred before epiphyseal fusion. Hair growth in the face, axilla, chest, and pubic regions is androgen-dependent; however, changes may not be noticeable unless androgen deficiency is severe and prolonged. Ethnicity also influences the intensity of hair growth (Chap. 68). Testicular volume is best assessed by using a Prader orchidometer. Testes range from 3.5 to 5.5 cm in length, which corresponds to a volume of 12–25 mL. Advanced age does not influence testicular size, although the consistency becomes less firm. Asian men generally have smaller testes than Western Europeans, independent of differences in body size. Because of its possible role in infertility, the presence of varicocele should be sought by palpation while the patient is standing; it is more common on the left side. Patients with Klinefelter’s syndrome have markedly reduced testicular volumes (1–2 mL). In congenital hypogonadotropic hypogonadism, testicular volumes provide a good index for the degree of gonadotropin deficiency and the likelihood of response to therapy.
GONADOTROPIN AND INHIBIN MEASUREMENTS
LH and FSH are measured using two-site immunoradiometric, immunofluorometric, or chemiluminescent assays, which have very low cross-reactivity with other pituitary glycoprotein hormones and human chorionic gonadotropin (hCG) and have sufficient sensitivity to measure the low levels present in patients with hypogonadotropic hypogonadism. In men with a low testosterone level, an LH level can distinguish primary (high LH) versus secondary (low or inappropriately normal LH) hypogonadism. An elevated LH level indicates a primary defect at the testicular level, whereas a low or inappropriately normal LH level suggests a defect at the hypothalamic-pituitary level. LH pulses occur about every 1–3 h in normal men. Thus, gonadotropin levels fluctuate, and samples should be pooled or repeated when results are equivocal. FSH is less pulsatile than LH because it has a longer half-life. Selective increase in FSH suggests damage to the seminiferous tubules. Inhibin B, a Sertoli cell product that suppresses FSH, is reduced with seminiferous tubule damage. Inhibin B is a dimer with α-βB subunits and is measured by two-site immunoassays.
GnRH Stimulation Testing The GnRH test is performed by measuring LH and FSH concentrations at baseline and at 30 and 60 min after intravenous administration of 100 μg of GnRH. A minimally acceptable response is a twofold LH increase and a 50% FSH increase. In the prepubertal period or with severe GnRH deficiency, the gonadotrope may not respond to a single bolus of GnRH because it has not been primed by endogenous hypothalamic GnRH; in these patients, GnRH responsiveness may be restored by chronic, pulsatile GnRH administration. With the availability of sensitive and specific LH assays, GnRH stimulation testing is used rarely except to evaluate gonadotrope function in patients who have undergone pituitary surgery or have a space-occupying lesion in the hypothalamic-pituitary region.
TESTOSTERONE ASSAYS
Total Testosterone Total testosterone includes both unbound and protein-bound testosterone and is measured by radioimmunoassays, immunometric assays, or liquid chromatography tandem mass spectrometry (LC-MS/MS). LC-MS/MS involves extraction of serum by organic solvents, separation of testosterone from other steroids by high-performance liquid chromatography and mass spectrometry, and quantitation of unique testosterone fragments by mass spectrometry. LC-MS/MS provides accurate and sensitive measurements of testosterone levels even in the low range and is emerging as the method of choice for testosterone measurement. Laboratories that have been certified by the Centers for Disease Control and Prevention (CDC) Hormone Standardization Program for Testosterone (HoST) can ensure that testosterone measurements are accurate and calibrated to an international standard. A single fasting morning sample provides a good approximation of the average testosterone concentration with the realization that testosterone levels fluctuate in response to pulsatile LH. Testosterone is generally lower in the late afternoon and is reduced by acute illness. The testosterone concentration in healthy young men ranges from 300 to 1000 ng/dL in most laboratories, and efforts are under way to generate harmonized population-based reference ranges that can be applied to all CDC-certified laboratories. Alterations in SHBG levels due to aging, obesity, diabetes mellitus, hyperthyroidism, some types of medications, or chronic illness or on a congenital basis can affect total testosterone levels. Heritable factors contribute substantially to the population-level variation in testosterone levels, and genome-wide association studies have revealed polymorphisms in the SHBG gene as important contributors to variation in testosterone levels.
Measurement of Unbound Testosterone Levels Most circulating testosterone is bound to SHBG and to albumin; only 0.5–3% of circulating testosterone is unbound, or “free.” The unbound testosterone concentration can be measured by equilibrium dialysis or calculated from total testosterone, SHBG, and albumin concentrations. Recent research has shown that testosterone binding to SHBG is a multistep process that involves complex homoallostery within the SHBG dimer; a novel allosteric model of testosterone binding to SHBG dimers provides good estimates of free testosterone concentrations. The previous law of mass action equations based on linear models of testosterone binding to SHBG have been shown to be erroneous. Tracer analogue methods are relatively inexpensive and convenient, but they are inaccurate. Bioavailable testosterone refers to unbound testosterone plus testosterone that is loosely bound to albumin; it can be determined by the ammonium sulfate precipitation method.
hCG Stimulation Test The hCG stimulation test is performed by administering a single injection of 1500–4000 IU of hCG intramuscularly and measuring testosterone levels at baseline and 24, 48, 72, and 120 h after hCG injection. An alternative regimen involves three injections of 1500 units of hCG on successive days and measuring testosterone levels 24 h after the last dose. An acceptable response to hCG is a doubling of the testosterone concentration in adult men. In prepubertal boys, an increase in testosterone to >150 ng/dL indicates the presence of testicular tissue. No response may indicate an absence of testicular tissue or marked impairment of Leydig cell function. Measurement of MIS, a Sertoli cell product, is also used to detect the presence of testes in prepubertal boys with cryptorchidism.
SEMEN ANALYSIS
Semen analysis is the most important step in the evaluation of male infertility. Samples are collected by masturbation following a period of abstinence for 2–3 days. Semen volumes and sperm concentrations vary considerably among fertile men, and several samples may be needed before concluding that the results are abnormal. Analysis should be performed within an hour of collection. Using semen samples from over 4500 men in 14 countries, whose partners had a time-to-pregnancy of less than 12 months, the World Health Organization (WHO) has generated the following one-sided reference limits for semen parameters: semen volume, 1.5 mL; total sperm number, 39 million per ejaculate; sperm concentration, 15 million/mL; vitality, 58% live; progressive motility, 32%; total (progressive + nonprogressive) motility, 40%; morphologically normal forms, 4.0%. Some men with low sperm counts are nevertheless fertile. A variety of tests for sperm function can be performed in specialized laboratories, but these add relatively little to the treatment options.
TESTICULAR BIOPSY
Testicular biopsy is useful in some patients with oligospermia or azoospermia as an aid in diagnosis and indication for the feasibility of treatment. Using local anesthesia, fine-needle aspiration biopsy is performed to aspirate tissue for histology. Alternatively, open biopsies can be performed under local or general anesthesia when more tissue is required. A normal biopsy in an azoospermic man with a normal FSH level suggests obstruction of the vas deferens, which may be correctable surgically. Biopsies are also used to harvest sperm for ICSI and to classify disorders such as hypospermatogenesis (all stages present but in reduced numbers), germ cell arrest (usually at primary spermatocyte stage), and Sertoli cell–only syndrome (absent germ cells) or hyalinization (sclerosis with absent cellular elements).
DISORDERS OF SEXUAL DIFFERENTIATION
See Chap. 410.
DISORDERS OF PUBERTY
The onset and tempo of puberty varies greatly in the general population and is affected by genetic and environmental factors. Although some of the variance in the timing of puberty is explained by heritable factors, the genes involved remain unknown.
PRECOCIOUS PUBERTY
Puberty in boys before age 9 is considered precocious. Isosexual precocity refers to premature sexual development consistent with phenotypic sex and includes features such as the development of facial hair and phallic growth. Isosexual precocity is divided into gonadotropin-dependent and gonadotropin-independent causes of androgen excess (Table 411-1). Heterosexual precocity refers to the premature development of estrogenic features in boys, such as breast development.
|
CAUSES OF PRECOCIOUS OR DELAYED PUBERTY IN BOYS |
Abbreviations: CNS, central nervous system; GnRH, gonadotropin-releasing hormone; hCG, human chronic gonadotropin; LH, luteinizing hormone.
Gonadotropin-Dependent Precocious Puberty This disorder, called central precocious puberty (CPP), is less common in boys than in girls. It is caused by premature activation of the GnRH pulse generator, sometimes because of central nervous system (CNS) lesions such as hypothalamic hamartomas, but it is often idiopathic. CPP is characterized by gonadotropin levels that are inappropriately elevated for age. Because pituitary priming has occurred, GnRH elicits LH and FSH responses typical of those seen in puberty or in adults. Magnetic resonance imaging (MRI) should be performed to exclude a mass, structural defect, infection, or inflammatory process. Mutations in MKRN3, an imprinted gene encoding makorin ring-finger protein 3, which is expressed only from the paternally inherited allele, have been associated with CPP.
Gonadotropin-Independent Precocious Puberty In gonadotropin-independent precocious puberty, androgens from the testis or the adrenal are increased, but gonadotropins are low. This group of disorders includes hCG-secreting tumors; congenital adrenal hyperplasia; sex steroid–producing tumors of the testis, adrenal, and ovary; accidental or deliberate exogenous sex steroid administration; hypothyroidism; and activating mutations of the LH receptor or Gsα subunit.
FAMILIAL MALE-LIMITED PRECOCIOUS PUBERTY Also called testotoxicosis, familial male-limited precocious puberty is an autosomal dominant disorder caused by activating mutations in the LH receptor, leading to constitutive stimulation of the cyclic AMP pathway and testosterone production. Clinical features include premature androgenization in boys, growth acceleration in early childhood, and advanced bone age followed by premature epiphyseal fusion. Testosterone is elevated, and LH is suppressed. Treatment options include inhibitors of testosterone synthesis (e.g., ketoconazole), AR antagonists (e.g., flutamide and bicalutamide), and aromatase inhibitors (e.g., anastrazole).
MCCUNE-ALBRIGHT SYNDROME This is a sporadic disorder caused by somatic (postzygotic) activating mutations in the Gsα subunit that links G protein–coupled receptors to intracellular signaling pathways (Chap. 426e). The mutations impair the guanosine triphosphatase activity of the Gsα protein, leading to constitutive activation of adenylyl cyclase. Like activating LH receptor mutations, this stimulates testosterone production and causes gonadotropin-independent precocious puberty. In addition to sexual precocity, affected individuals may have autonomy in the adrenals, pituitary, and thyroid glands. Café au lait spots are characteristic skin lesions that reflect the onset of the somatic mutations in melanocytes during embryonic development. Polyostotic fibrous dysplasia is caused by activation of the parathyroid hormone receptor pathway in bone. Treatment is similar to that in patients with activating LH receptor mutations. Bisphosphonates have been used to treat bone lesions.
CONGENITAL ADRENAL HYPERPLASIA Boys with congenital adrenal hyperplasia (CAH) who are not well controlled with glucocorticoid suppression of adrenocorticotropic hormone (ACTH) can develop premature virilization because of excessive androgen production by the adrenal gland (Chaps. 406 and 410). LH is low, and the testes are small. Adrenal rests may develop within the testis of poorly controlled patients with CAH because of chronic ACTH stimulation; adrenal rests do not require surgical removal and regress with effective glucocorticoid therapy. Some children with CAH may develop gonadotropin-dependent precocious puberty with early maturation of the hypothalamic-pituitary-gonadal axis, elevated gonadotropins, and testicular growth.
Heterosexual Sexual Precocity Breast enlargement in prepubertal boys can result from familial aromatase excess, estrogen-producing tumors in the adrenal gland, Sertoli cell tumors in the testis, marijuana smoking, or exogenous estrogens or androgens. Occasionally, germ cell tumors that secrete hCG can be associated with breast enlargement due to excessive stimulation of estrogen production (see “Gynecomastia,” below).
|
TREATMENT |
PRECOCIOUS PUBERTY |
In patients with a known cause (e.g., a CNS lesion or a testicular tumor), therapy should be directed toward the underlying disorder. In patients with idiopathic CPP, long-acting GnRH analogues can be used to suppress gonadotropins and decrease testosterone, halt early pubertal development, delay accelerated bone maturation, prevent early epiphyseal closure, promote final height gain, and mitigate the psychosocial consequences of early pubertal development without causing osteoporosis. The treatment is most effective for increasing final adult height if it is initiated before age 6. Puberty resumes after discontinuation of the GnRH analogue. Counseling is an important aspect of the overall treatment strategy.
In children with gonadotropin-independent precocious puberty, inhibitors of steroidogenesis, such as ketoconazole, and AR antagonists have been used empirically. Long-term treatment with spironolactone (a weak androgen antagonist) and ketoconazole has been reported to normalize growth rate and bone maturation and to improve predicted height in small, nonrandomized trials in boys with familial male-limited precocious puberty. Aromatase inhibitors, such as testolactone and letrozole, have been used as an adjunct to antiandrogen and GnRH analogue therapy for children with familial male-limited precocious puberty, CAH, and McCune-Albright syndrome.
DELAYED PUBERTY
Puberty is delayed in boys if it has not ensued by age 14, an age that is 2–2.5 standard deviations above the mean for healthy children. Delayed puberty is more common in boys than in girls. There are four main categories of delayed puberty: (1) constitutional delay of growth and puberty (~60% of cases); (2) functional hypogonadotropic hypogonadism caused by systemic illness or malnutrition (~20% of cases); (3) hypogonadotropic hypogonadism caused by genetic or acquired defects in the hypothalamic-pituitary region (~10% of cases); and (4) hypergonadotropic hypogonadism secondary to primary gonadal failure (~15% of cases) (Table 411-1). Functional hypogonadotropic hypogonadism is more common in girls than in boys. Permanent causes of hypogonadotropic or hypergonadotropic hypogonadism are identified in >25% of boys with delayed puberty.
|
TREATMENT |
DELAYED PUBERTY |
If therapy is considered appropriate, it can begin with 25–50 mg testosterone enanthate or testosterone cypionate every 2 weeks, or by using a 2.5-mg testosterone patch or 25-mg testosterone gel. Because aromatization of testosterone to estrogen is obligatory for mediating androgen effects on epiphyseal fusion, concomitant treatment with aromatase inhibitors may allow attainment of greater final adult height. Testosterone treatment should be interrupted after 6 months to determine if endogenous LH and FSH secretion have ensued. Other causes of delayed puberty should be considered when there are associated clinical features or when boys do not enter puberty spontaneously after a year of observation or treatment.
Reassurance without hormonal treatment is appropriate for many individuals with presumed constitutional delay of puberty. However, the impact of delayed growth and pubertal progression on a child’s social relationships and school performance should be weighed. Also, boys with constitutional delay of puberty are less likely to achieve their full genetic height potential and have reduced total-body bone mass as adults, mainly due to narrow limb bones and vertebrae as a result of impaired periosteal expansion during puberty. Administration of androgen therapy to boys with constitutional delay does not affect final height, and when administered with an aromatase inhibitor, it may improve final height.
DISORDERS OF THE MALE REPRODUCTIVE AXIS DURING ADULTHOOD
HYPOGONADOTROPIC HYPOGONADISM
Because LH and FSH are trophic hormones for the testes, impaired secretion of these pituitary gonadotropins results in secondary hypogonadism, which is characterized by low testosterone in the setting of low LH and FSH. Those with the most severe deficiency have complete absence of pubertal development, sexual infantilism, and, in some cases, hypospadias and undescended testes. Patients with partial gonadotropin deficiency have delayed or arrested sex development. The 24-h LH secretory profiles are heterogeneous in patients with hypogonadotropic hypogonadism, reflecting variable abnormalities of LH pulse frequency or amplitude. In severe cases, basal LH is low and there are no LH pulses. A smaller subset of patients has low-amplitude LH pulses or markedly reduced pulse frequency. Occasionally, only sleep-entrained LH pulses occur, reminiscent of the pattern seen in the early stages of puberty. Hypogonadotropic hypogonadism can be classified into congenital and acquired disorders. Congenital disorders most commonly involve GnRH deficiency, which leads to gonadotropin deficiency. Acquired disorders are much more common than congenital disorders and may result from a variety of sellar mass lesions or infiltrative diseases of the hypothalamus or pituitary.
Congenital Disorders Associated with Gonadotropin Deficiency Congenital hypogonadotropic hypogonadism is a heterogeneous group of disorders characterized by decreased gonadotropin secretion and testicular dysfunction either due to impaired function of the GnRH pulse generator or the gonadotrope. The disorders characterized by GnRH deficiency represent a family of oligogenic disorders whose phenotype spans a wide spectrum. Some individuals with GnRH deficiency may suffer from complete absence of pubertal development, while others may manifest varying degrees of gonadotropin deficiency and pubertal delay, and a subset that carries the same mutations as their affected family members may even have normal reproductive function. In approximately 10% of men with idiopathic hypogonadotropic hypogonadism, reversal of gonadotropin deficiency may occur in adult life after sex steroid therapy. Also, a small fraction of men with idiopathic hypogonadotropic hypogonadism may present with androgen deficiency and infertility in adult life after having gone through apparently normal pubertal development. Nutritional, emotional, or metabolic stress may unmask gonadotropin deficiency and reproductive dysfunction (analogous to hypothalamic amenorrhea) in some patients who harbor mutations in the candidate genes but who previously had normal reproductive function. The clinical phenotype may include isolated anosmia or hyposmia. These striking variations in phenotypic presentation of GnRH deficiency have highlighted the important role of oligogenicity and gene-gene and gene-environment interactions in shaping the clinical phenotype.
Mutations in a number of genes involved in the development and migration of GnRH neurons or in the regulation of GnRH secretion have been linked to GnRH deficiency, although the genetic defect remains elusive in nearly two-thirds of cases. Familial hypogonadotropic hypogonadism can be transmitted as an X-linked (20%), autosomal recessive (30%), or autosomal dominant (50%) trait. Some individuals with idiopathic hypogonadotropic hypogonadism (IHH) have sporadic mutations in the same genes that cause inherited forms of the disorder. The genetic defects associated with GnRH deficiency can be conveniently classified as anosmic (Kallmann’s syndrome) or normosmic (Table 411-2), although the occurrence of both anosmic and normosmic forms of GnRH deficiency in the same families suggests commonality of pathophysiologic mechanisms. Kallmann’s syndrome, the anosmic form of GnRH deficiency, can result from mutations in one or more genes associated with olfactory bulb morphogenesis and the migration of GnRH neurons from their origin in the region of the olfactory placode, along the scaffold established by the olfactory nerves, through the cribriform plate into their final location into the preoptic region of the hypothalamus. Thus, mutations in KAL1, FGF8, FGFR1, NELF, PROK2, PROK2R, and CHD7 have been described in patients with Kallmann’s syndrome. An X-linked form of IHH is caused by mutations in the KAL1 gene, which encodes anosmin, a protein that mediates the migration of neural progenitors of the olfactory bulb and GnRH-producing neurons. These individuals have GnRH deficiency and variable combinations of anosmia or hyposmia, renal defects, and neurologic abnormalities including mirror movements. Mutations in the FGFR1 gene cause an autosomal dominant form of hypogonadotropic hypogonadism that clinically resembles Kallmann’s syndrome; mutations in its putative ligand, FGF8 gene product, have also been associated with IHH. Prokineticin 2 (PROK2) also encodes a protein involved in migration and development of olfactory and GnRH neurons. Recessive mutations in PROK2 or in its receptor, PROKR2, have been associated with both anosmic and normosmic forms of hypogonadotropic hypogonadism.
|
CAUSES OF CONGENITAL HYPOGONADOTROPIC HYPOGONADISM |
Normosmic GnRH deficiency results from defects in pulsatile GnRH secretion, its regulation, or its action on the gonadotrope and has been associated with mutations in GnRHR, GNRH1, KISS1R, TAC3, TACR3, and NROB1 (DAX1). Some mutations, such as those in PROK2, PROKR2, and CHD7, have been associated with both the anosmic and normosmic forms of IHH. GnRHR mutations, the most frequent identifiable cause of normosmic IHH, account for ~40% of autosomal recessive and 10% of sporadic cases of hypogonadotropic hypogonadism. These patients have decreased LH response to exogenous GnRH. Some receptor mutations alter GnRH binding affinity, allowing apparently normal responses to pharmacologic doses of exogenous GnRH, whereas other mutations may alter signal transduction downstream of hormone binding. Mutations of the GnRH1 gene have also been reported in patients with hypogonadotropic hypogonadism, although they are rare. G protein–coupled receptor KISS1R (GPR54) and its cognate ligand, kisspeptin (KISS1), are important regulators of sexual maturation in primates. Recessive mutations in GPR54 cause gonadotropin deficiency without anosmia. Patients retain responsiveness to exogenous GnRH, suggesting an abnormality in the neural pathways controlling GnRH release. The genes encoding neurokinin B (TAC3), which is involved in preferential activation of GnRH release in early development, and its receptor (TAC3R) have been implicated in some families with normosmic IHH. Mutations in more than one gene (digenicity or oligogenicity) may contribute to clinical heterogeneity in IHH patients. X-linked hypogonadotropic hypogonadism also occurs in adrenal hypoplasia congenita, a disorder caused by mutations in the DAX1 gene, which encodes a nuclear receptor in the adrenal gland and reproductive axis. Adrenal hypoplasia congenita is characterized by absent development of the adult zone of the adrenal cortex, leading to neonatal adrenal insufficiency. Puberty usually does not occur or is arrested, reflecting variable degrees of gonadotropin deficiency. Although sexual differentiation is normal, most patients have testicular dysgenesis and impaired spermatogenesis despite gonadotropin replacement. Less commonly, adrenal hypoplasia congenita, sex reversal, and hypogonadotropic hypogonadism can be caused by mutations of steroidogenic factor 1 (SF1). Rarely, recessive mutations in the LHβ or FSHβ gene have been described in patients with selective deficiencies of these gonadotropins. In approximately 10% of men with IHH, reversal of gonadotropin deficiency may occur in adult life. Also, a small fraction of men with IHH may present with androgen deficiency and infertility in adult life after having gone through apparently normal pubertal development.
A number of homeodomain transcription factors are involved in the development and differentiation of the specialized hormone-producing cells within the pituitary gland (Table 411-2). Patients with mutations of PROP1 have combined pituitary hormone deficiency that includes GH, prolactin (PRL), thyroid-stimulating hormone (TSH), LH, and FSH, but not ACTH. LHX3 mutations cause combined pituitary hormone deficiency in association with cervical spine rigidity. HESX1 mutations cause septo-optic dysplasia and combined pituitary hormone deficiency.
Prader-Willi syndrome is characterized by obesity, hypotonic musculature, mental retardation, hypogonadism, short stature, and small hands and feet. Prader-Willi syndrome is a genomic imprinting disorder caused by deletions of the proximal portion of the paternally derived chromosome 15q11-15q13 region, which contains a bipartite imprinting center, uniparental disomy of the maternal alleles, or mutations of the genes/loci involved in imprinting (Chap. 83e). Laurence-Moon syndrome is an autosomal recessive disorder characterized by obesity, hypogonadism, mental retardation, polydactyly, and retinitis pigmentosa. Recessive mutations of leptin, or its receptor, cause severe obesity and pubertal arrest, apparently because of hypothalamic GnRH deficiency (Chap. 415e).
Acquired Hypogonadotropic Disorders • SEVERE ILLNESS, STRESS, MALNUTRITION, AND EXERCISE These factors may cause reversible gonadotropin deficiency. Although gonadotropin deficiency and reproductive dysfunction are well documented in these conditions in women, men exhibit similar but less pronounced responses. Unlike women, most male runners and other endurance athletes have normal gonadotropin and sex steroid levels, despite low body fat and frequent intensive exercise. Testosterone levels fall at the onset of illness and recover during recuperation. The magnitude of gonadotropin suppression generally correlates with the severity of illness. Although hypogonadotropic hypogonadism is the most common cause of androgen deficiency in patients with acute illness, some have elevated levels of LH and FSH, which suggest primary gonadal dysfunction. The pathophysiology of reproductive dysfunction during acute illness is unknown but likely involves a combination of cytokine and/or glucocorticoid effects. There is a high frequency of low testosterone levels in patients with chronic illnesses such as HIV infection, end-stage renal disease, chronic obstructive lung disease, and many types of cancer and in patients receiving glucocorticoids. About 20% of HIV-infected men with low testosterone levels have elevated LH and FSH levels; these patients presumably have primary testicular dysfunction. The remaining 80% have either normal or low LH and FSH levels; these men have a central hypothalamic-pituitary defect or a dual defect involving both the testis and the hypothalamic-pituitary centers. Muscle wasting is common in chronic diseases associated with hypogonadism, which also leads to debility, poor quality of life, and adverse outcome of disease. There is great interest in exploring strategies that can reverse androgen deficiency or attenuate the sarcopenia associated with chronic illness.
Men using opioids for relief of cancer or noncancerous pain or because of addiction often have suppressed testosterone and LH levels and high prevalence of sexual dysfunction and osteoporosis; the degree of suppression is dose-related and particularly severe with long-acting opioids such as methadone. Opioids suppress GnRH secretion and alter the sensitivity to feedback inhibition by gonadal steroids. Men who are heavy users of marijuana have decreased testosterone secretion and sperm production. The mechanism of marijuana-induced hypogonadism is decreased GnRH secretion. Gynecomastia observed in marijuana users can also be caused by plant estrogens in crude preparations. Androgen deprivation therapy in men with prostate cancer has been associated with increased risk of bone fractures, diabetes mellitus, cardiovascular events, fatigue, sexual dysfunction, and poor quality of life.
OBESITY In men with mild to moderate obesity, SHBG levels decrease in proportion to the degree of obesity, resulting in lower total testosterone levels. However, free testosterone levels usually remain within the normal range. The decrease in SHBG levels is caused by increased circulating insulin, which inhibits SHBG production. Estradiol levels are higher in obese men compared to healthy, nonobese controls, because of aromatization of testosterone to estradiol in adipose tissue. Weight loss is associated with reversal of these abnormalities including an increase in total and free testosterone levels and a decrease in estradiol levels. A subset of obese men with moderate to severe obesity may have a defect in the hypothalamic-pituitary axis as suggested by low free testosterone in the absence of elevated gonadotropins. Weight gain in adult men can accelerate the rate of age-related decline in testosterone levels.
HYPERPROLACTINEMIA (See also Chap. 403) Elevated PRL levels are associated with hypogonadotropic hypogonadism. PRL inhibits hypothalamic GnRH secretion either directly or through modulation of tuberoinfundibular dopaminergic pathways. A PRL-secreting tumor may also destroy the surrounding gonadotropes by invasion or compression of the pituitary stalk. Treatment with dopamine agonists reverses gonadotropin deficiency, although there may be a delay relative to PRL suppression.
SELLAR MASS LESIONS Neoplastic and nonneoplastic lesions in the hypothalamus or pituitary can directly or indirectly affect gonadotrope function. In adults, pituitary adenomas constitute the largest category of space-occupying lesions affecting gonadotropin and other pituitary hormone production. Pituitary adenomas that extend into the suprasellar region can impair GnRH secretion and mildly increase PRL secretion (usually <50 μg/L) because of impaired tonic inhibition by dopaminergic pathways. These tumors should be distinguished from prolactinomas, which typically secrete higher PRL levels. The presence of diabetes insipidus suggests the possibility of a craniopharyngioma, infiltrative disorder, or other hypothalamic lesions (Chap. 404).
HEMOCHROMATOSIS (See also Chap. 428) Both the pituitary and testis can be affected by excessive iron deposition. However, the pituitary defect is the predominant lesion in most patients with hemochromatosis and hypogonadism. The diagnosis of hemochromatosis is suggested by the association of characteristic skin discoloration, hepatic enlargement or dysfunction, diabetes mellitus, arthritis, cardiac conduction defects, and hypogonadism.
PRIMARY TESTICULAR CAUSES OF HYPOGONADISM
Common causes of primary testicular dysfunction include Klinefelter’s syndrome, uncorrected cryptorchidism, cancer chemotherapy, radiation to the testes, trauma, torsion, infectious orchitis, HIV infection, anorchia syndrome, and myotonic dystrophy. Primary testicular disorders may be associated with impaired spermatogenesis, decreased androgen production, or both. See Chap. 410 for disorders of testis development, androgen synthesis, and androgen action.
Klinefelter’s Syndrome (See also Chap. 410) Klinefelter’s syndrome is the most common chromosomal disorder associated with testicular dysfunction and male infertility. It occurs in about 1 in 600 live-born males. Azoospermia is the rule in men with Klinefelter’s syndrome who have the 47,XXY karyotype; however, men with mosaicism may have germ cells, especially at a younger age. The clinical phenotype of Klinefelter’s syndrome can be heterogeneous possibly because of mosaicism, polymorphisms in AR gene, variable testosterone levels, or other genetic factors. Testicular histology shows hyalinization of seminiferous tubules and absence of spermatogenesis. Although their function is impaired, the number of Leydig cells appears to increase. Testosterone is decreased and estradiol is increased, leading to clinical features of undervirilization and gynecomastia. Men with Klinefelter’s syndrome are at increased risk of systemic lupus erythematosus, Sjögren’s syndrome, breast cancer, diabetes mellitus, osteoporosis, non-Hodgkin’s lymphoma, and lung cancer, and reduced risk of prostate cancer. Periodic mammography for breast cancer surveillance is recommended for men with Klinefelter’s syndrome. Fertility has been achieved by intracytoplasmic injection of sperm retrieved surgically from testicular biopsies of men with Klinefelter’s syndrome, including some men with the nonmosaic form of Klinefelter’s syndrome. The karyotypes 48,XXXY and 49,XXXXY are associated with a more severe phenotype, increased risk of congenital malformations, and lower intelligence than 47,XXY individuals.
Cryptorchidism Cryptorchidism occurs when there is incomplete descent of the testis from the abdominal cavity into the scrotum. About 3% of full-term and 30% of premature male infants have at least one undescended testis at birth, but descent is usually complete by the first few weeks of life. The incidence of cryptorchidism is <1% by 9 months of age. Androgens regulate predominantly the inguinoscrotal descent of the testes through degeneration of the craniosuspensory ligament and a shortening of the gubernaculums, respectively. Mutations in INSL3 and leucine-rich repeat family of G protein–coupled receptor 8 (LGR8), which regulate the transabdominal portion of testicular descent, have been found in some patients with cryptorchidism.
Cryptorchidism is associated with increased risk of malignancy, infertility, inguinal hernia, and torsion. Unilateral cryptorchidism, even when corrected before puberty, is associated with decreased sperm count, possibly reflecting unrecognized damage to the fully descended testis or other genetic factors. Epidemiologic, clinical, and molecular evidence supports the idea that cryptorchidism, hypospadias, impaired spermatogenesis, and testicular cancer may be causally related to common genetic and environment perturbations and are components of the testicular dysgenesis syndrome.
Acquired Testicular Defects Viral orchitis may be caused by the mumps virus, echovirus, lymphocytic choriomeningitis virus, and group B arboviruses. Orchitis occurs in as many as one-fourth of adult men with mumps; the orchitis is unilateral in about two-thirds and bilateral in the remainder. Orchitis usually develops a few days after the onset of parotitis but may precede it. The testis may return to normal size and function or undergo atrophy. Semen analysis returns to normal for three-fourths of men with unilateral involvement but for only one-third of men with bilateral orchitis. Trauma, including testicular torsion, can also cause secondary atrophy of the testes. The exposed position of the testes in the scrotum renders them susceptible to both thermal and physical trauma, particularly in men with hazardous occupations.
The testes are sensitive to radiation damage. Doses >200 mGy (20 rad) are associated with increased FSH and LH levels and damage to the spermatogonia. After ~800 mGy (80 rad), oligospermia or azoospermia develops, and higher doses may obliterate the germinal epithelium. Permanent androgen deficiency in adult men is uncommon after therapeutic radiation; however, most boys given direct testicular radiation therapy for acute lymphoblastic leukemia have permanently low testosterone levels. Sperm banking should be considered before patients undergo radiation treatment or chemotherapy.
Drugs interfere with testicular function by several mechanisms, including inhibition of testosterone synthesis (e.g., ketoconazole), blockade of androgen action (e.g., spironolactone), increased estrogen (e.g., marijuana), or direct inhibition of spermatogenesis (e.g., chemotherapy).
Combination chemotherapy for acute leukemia, Hodgkin’s disease, and testicular and other cancers may impair Leydig cell function and cause infertility. The degree of gonadal dysfunction depends on the type of chemotherapeutic agent and the dose and duration of therapy. Because of high response rates and the young age of these men, infertility and androgen deficiency have emerged as important long-term complications of cancer chemotherapy. Cyclophosphamide and combination regimens containing procarbazine are particularly toxic to germ cells. Thus, 90% of men with Hodgkin’s lymphoma receiving MOPP (mechlorethamine, vincristine, procarbazine, prednisone) therapy develop azoospermia or extreme oligozoospermia; newer regimens that do not include procarbazine, such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine), are less toxic to germ cells.
Alcohol, when consumed in excess for prolonged periods, decreases testosterone, independent of liver disease or malnutrition. Elevated estradiol and decreased testosterone levels may occur in men taking digitalis.
The occupational and recreational history should be carefully evaluated in all men with infertility because of the toxic effects of many chemical agents on spermatogenesis. Known environmental hazards include pesticides (e.g., vinclozolin, dicofol, atrazine), sewage contaminants (e.g., ethinyl estradiol in birth control pills, surfactants such as octylphenol, nonyphenol), plasticizers (e.g., pthalates), flame retardants (e.g., polychlorinated biphenyls, polybrominated diphenol ethers), industrial pollutants (e.g., heavy metals cadmium and lead, dioxins, polycyclic aromatic hydrocarbons), microwaves, and ultrasound. In some populations, sperm density is said to have declined by as much as 40% in the past 50 years. Environmental estrogens or antiandrogens may be partly responsible.
Testicular failure also occurs as a part of polyglandular autoimmune insufficiency (Chap. 408). Sperm antibodies can cause isolated male infertility. In some instances, these antibodies are secondary phenomena resulting from duct obstruction or vasectomy. Granulomatous diseases can affect the testes, and testicular atrophy occurs in 10–20% of men with lepromatous leprosy because of direct tissue invasion by the mycobacteria. The tubules are involved initially, followed by endarteritis and destruction of Leydig cells.
Systemic disease can cause primary testis dysfunction in addition to suppressing gonadotropin production. In cirrhosis, a combined testicular and pituitary abnormality leads to decreased testosterone production independent of the direct toxic effects of ethanol. Impaired hepatic extraction of adrenal androstenedione leads to extraglandular conversion to estrone and estradiol, which partially suppresses LH. Testicular atrophy and gynecomastia are present in approximately one-half of men with cirrhosis. In chronic renal failure, androgen synthesis and sperm production decrease despite elevated gonadotropins. The elevated LH level is due to reduced clearance, but it does not restore normal testosterone production. About one-fourth of men with renal failure have hyperprolactinemia. Improvement in testosterone production with hemodialysis is incomplete, but successful renal transplantation may return testicular function to normal. Testicular atrophy is present in one-third of men with sickle cell anemia. The defect may be at either the testicular or the hypothalamic-pituitary level. Sperm density can decrease temporarily after acute febrile illness in the absence of a change in testosterone production. Infertility in men with celiac disease is associated with a hormonal pattern typical of androgen resistance, namely elevated testosterone and LH levels.
Neurologic diseases associated with altered testicular function include myotonic dystrophy, spinobulbar muscular atrophy, and paraplegia. In myotonic dystrophy, small testes may be associated with impairment of both spermatogenesis and Leydig cell function. Spinobulbar muscular atrophy is caused by an expansion of the glutamine repeat sequences in the amino-terminal region of the AR; this expansion impairs function of the AR, but it is unclear how the alteration is related to the neurologic manifestations. Men with spinobulbar muscular atrophy often have undervirilization and infertility as a late manifestation. Spinal cord lesions that cause paraplegia can lead to a temporary decrease in testosterone levels and may cause persistent defects in spermatogenesis; some patients retain the capacity for penile erection and ejaculation.
ANDROGEN INSENSITIVITY SYNDROMES
Mutations in the AR cause resistance to the action of testosterone and DHT. These X-linked mutations are associated with variable degrees of defective male phenotypic development and undervirilization (Chap. 410). Although not technically hormone-insensitivity syndromes, two genetic disorders impair testosterone conversion to active sex steroids. Mutations in the SRD5A2 gene, which encodes 5α-reductase type 2, prevent the conversion of testosterone to DHT, which is necessary for the normal development of the male external genitalia. Mutations in the CYP19 gene, which encodes aromatase, prevent testosterone conversion to estradiol. Males with CYP19 mutations have delayed epiphyseal fusion, tall stature, eunuchoid proportions, and osteoporosis, consistent with evidence from an estrogen receptor–deficient individual that these testosterone actions are mediated indirectly via estrogen.
GYNECOMASTIA
Gynecomastia refers to enlargement of the male breast. It is caused by excess estrogen action and is usually the result of an increased estrogen-to-androgen ratio. True gynecomastia is associated with glandular breast tissue that is >4 cm in diameter and often tender. Glandular tissue enlargement should be distinguished from excess adipose tissue: glandular tissue is firmer and contains fibrous-like cords. Gynecomastia occurs as a normal physiologic phenomenon in the newborn (due to transplacental transfer of maternal and placental estrogens), during puberty (high estrogen-to-androgen ratio in early stages of puberty), and with aging (increased fat tissue and increased aromatase activity), but it can also result from pathologic conditions associated with androgen deficiency or estrogen excess. The prevalence of gynecomastia increases with age and body mass index (BMI), likely because of increased aromatase activity in adipose tissue. Medications that alter androgen metabolism or action may also cause gynecomastia. The relative risk of breast cancer is increased in men with gynecomastia, although the absolute risk is relatively small.
PATHOLOGIC GYNECOMASTIA
Any cause of androgen deficiency can lead to gynecomastia, reflecting an increased estrogen-to-androgen ratio, because estrogen synthesis still occurs by aromatization of residual adrenal and gonadal androgens. Gynecomastia is a characteristic feature of Klinefelter’s syndrome (Chap. 410). Androgen insensitivity disorders also cause gynecomastia. Excess estrogen production may be caused by tumors, including Sertoli cell tumors in isolation or in association with Peutz-Jeghers syndrome or Carney complex. Tumors that produce hCG, including some testicular tumors, stimulate Leydig cell estrogen synthesis. Increased conversion of androgens to estrogens can be a result of increased availability of substrate (androstenedione) for extraglandular estrogen formation (CAH, hyperthyroidism, and most feminizing adrenal tumors) or of diminished catabolism of androstenedione (liver disease) so that estrogen precursors are shunted to aromatase in peripheral sites. Obesity is associated with increased aromatization of androgen precursors to estrogens. Extraglandular aromatase activity can also be increased in tumors of the liver or adrenal gland or rarely as an inherited disorder. Several families with increased peripheral aromatase activity inherited as an autosomal dominant or as an X-linked disorder have been described. In some families with this disorder, an inversion in chromosome 15q21.2-3 causes the CYP19 gene to be activated by the regulatory elements of contiguous genes, resulting in excessive estrogen production in the fat and other extragonadal tissues. Drugs can cause gynecomastia by acting directly as estrogenic substances (e.g., oral contraceptives, phytoestrogens, digitalis) or by inhibiting androgen synthesis (e.g., ketoconazole) or action (e.g., spironolactone).
Because up to two-thirds of pubertal boys and half of hospitalized men have palpable glandular tissue that is benign, detailed investigation or intervention is not indicated in all men presenting with gynecomastia (Fig. 411-5). In addition to the extent of gynecomastia, recent onset, rapid growth, tender tissue, and occurrence in a lean subject should prompt more extensive evaluation. This should include a careful drug history, measurement and examination of the testes, assessment of virilization, evaluation of liver function, and hormonal measurements including testosterone, estradiol, and androstenedione, LH, and hCG. A karyotype should be obtained in men with very small testes to exclude Klinefelter’s syndrome. Despite extensive evaluation, the etiology is established in fewer than one-half of patients.
FIGURE 411-5 Evaluation of gynecomastia. E2, 17β-estradiol; hCGβ, human chorionic gonadotropin β; T, testosterone.
|
TREATMENT |
GYNECOMASTIA |
When the primary cause can be identified and corrected, breast enlargement usually subsides over several months. However, if gynecomastia is of long duration, surgery is the most effective therapy. Indications for surgery include severe psychological and/or cosmetic problems, continued growth or tenderness, or suspected malignancy. In patients who have painful gynecomastia and in whom surgery cannot be performed, treatment with antiestrogens such as tamoxifen (20 mg/d) can reduce pain and breast tissue size in over half the patients. Estrogen receptor antagonists, tamoxifen and raloxifene, have been reported in small trials to reduce breast size in men with pubertal gynecomastia, although complete regression of breast enlargement is unusual with the use of estrogen receptor antagonists. Aromatase inhibitors can be effective in the early proliferative phase of the disorder. However, in a randomized trial in men with established gynecomastia, anastrozole proved no more effective than placebo in reducing breast size. Tamoxifen is effective in the prevention and treatment of breast enlargement and breast pain in men with prostate cancer who are receiving antiandrogen therapy.
AGING-RELATED CHANGES IN MALE REPRODUCTIVE FUNCTION
A number of cross-sectional and longitudinal studies (e.g., The Baltimore Longitudinal Study of Aging, the Framingham Heart Study, the Massachusetts Male Aging Study, and the European Male Aging Study) have established that testosterone concentrations decrease with advancing age. This age-related decline starts in the third decade of life and progresses slowly; the rate of decline in testosterone concentrations is greater in obese men, men with chronic illness, and those taking medications than in healthy older men. Because SHBG concentrations are higher in older men than in younger men, free or bioavailable testosterone concentrations decline with aging to a greater extent than total testosterone concentrations. The age-related decline in testosterone is due to defects at all levels of the hypothalamic-pituitary-testicular axis: pulsatile GnRH secretion is attenuated, LH response to GnRH is reduced, and testicular response to LH is impaired. However, the gradual rise of LH with aging suggests that testis dysfunction is the main cause of declining androgen levels. The term andropause has been used to denote age-related decline in testosterone concentrations; this term is a misnomer because there is no discrete time when testosterone concentrations decline abruptly. The approach to evaluating hypogonadism is summarized in Fig. 411-6.
FIGURE 411-6 Evaluation of hypogonadism. GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; T, testosterone.
In epidemiologic surveys, low total and bioavailable testosterone concentrations have been associated with decreased appendicular skeletal muscle mass and strength, decreased self-reported physical function, higher visceral fat mass, insulin resistance, and increased risk of coronary artery disease and mortality, although the associations are weak. An analysis of signs and symptoms in older men in the European Male Aging Study revealed a syndromic association of sexual symptoms with total testosterone levels below 320 ng/dL and free testosterone levels below 64 pg/mL in community-dwelling older men. In systematic reviews of randomized controlled trials, testosterone therapy of healthy older men with low or low-normal testosterone levels was associated with greater increments in lean body mass, grip strength, and self-reported physical function compared with placebo. Testosterone therapy also induced greater improvement in vertebral but not femoral bone mineral density. Testosterone therapy of older men with sexual dysfunction and unequivocally low testosterone levels improves libido, but testosterone effects on erectile function and response to selective phosphodiesterase inhibitors have been inconsistent. Testosterone therapy has not been shown to improve depression scores, fracture risk, cognitive function, response to phosphodiesterase inhibitors, or clinical outcomes in older men. Furthermore, neither the long-term risks nor clinical benefits of testosterone therapy in older men have been demonstrated in adequately powered trials. Although there is no evidence that testosterone causes prostate cancer, there is concern that testosterone therapy might cause subclinical prostate cancers to grow. Testosterone therapy is associated with increased risk of detection of prostate events (Fig. 411-7).
FIGURE 411-7 Meta-analyses of cardiovascular and prostate adverse events associated with testosterone therapy. A. A meta-analysis of cardiovascular-related events in randomized testosterone trials of 12 weeks or longer in duration. Randomization to testosterone was associated with a significantly increased risk of cardiovascular-related event (odds ratio [OR] 1.54). (Modified with permission from L Xu et al: Testosterone therapy and cardiovascular events among men: a systematic review and meta-analysis of placebo-controlled randomized trials BMC Med 11:108, 2013.) B. The relative risk of prostate events and the associated 95% confidence intervals (CIs) in a meta-analysis of randomized testosterone trials. PSA, prostate-specific antigen. (Data were derived from a meta-analysis by MM Fernández-Balsells et al: J Clin Endocrinol Metab 95:2560, 2010, and the figure was reproduced with permission from M Spitzer et al: Nat Rev Endocrinol 9:414, 2013.)
One randomized testosterone trial in older men with mobility limitation and high burden of chronic conditions, such as diabetes, heart disease, hypertension, and hyperlipidemia, reported a greater number of cardiovascular events in men randomized to the testosterone arm of the study than in those randomized to the placebo arm. Since then, two large retrospective analyses of patient databases have reported higher frequency of cardiovascular events, including myocardial infarction, in older men with preexisting heart disease (Fig. 411-7).
Population screening of all older men for low testosterone levels is not recommended, and testing should be restricted to men who have symptoms or physical features attributable to androgen deficiency. Testosterone therapy is not recommended for all older men with low testosterone levels. In older men with significant symptoms of androgen deficiency who have testosterone levels below 200 ng/dL, testosterone therapy may be considered on an individualized basis and should be instituted after careful discussion of the risks and benefits (see “Testosterone Replacement,” below).
Testicular morphology, semen production, and fertility are maintained up to a very old age in men. Although concern has been expressed about age-related increases in germ cell mutations and impairment of DNA repair mechanisms, there is no clear evidence that the frequency of chromosomal aneuploidy is increased in the sperm of older men. However, the incidence of autosomal dominant diseases, such as achondroplasia, polyposis coli, Marfan’s syndrome, and Apert’s syndrome, increases in the offspring of men who are advanced in age, consistent with transmission of sporadic missense mutations. Advanced paternal age may be associated with increased rates of de novo mutations, which may contribute to an increased risk of neurodevelopmental diseases such as schizophrenia and autism. The somatic mutations in male germ cells that enhance the proliferation of germ cells could lead to within-testis expansion of mutant clonal lines, thus favoring the propagation of germ cells carrying these pathogenic mutations and increasing the risk of mutations in the offspring of older fathers (the “selfish spermatogonial selection” hypothesis).
|
TREATMENT |
ANDROGEN DEFICIENCY |
GONADOTROPINS
[/not-level-membership-for-internal-medicine-category]
