[level-membership-for-pediatrics-category]
70 Disorders of the Adrenal Gland
Adrenal Gland Physiology
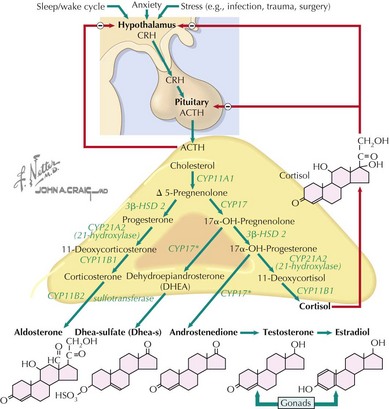
The adrenal cortex consists of three distinct zones, the glomerulosa, the fasciculata, and the reticularis. The fasciculata is the principal component of the hypothalamic–pituitary–adrenal axis. Glucocorticoid (cortisol) secretion from the fasciculata is regulated by adrenocorticotropic hormone (ACTH). ACTH is synthesized from pre-pro-opiomelanocortin (pre-POMC). The removal of the signal peptide during translation produces the 267 amino acid polypeptide POMC, which undergoes a series of posttranslational modifications to yield various polypeptide fragments with varying physiological activity. These fragments include the 39 amino acid polypeptide ACTH, as well as β-lipotropin, γ-lipotropin, melanocyte-stimulating hormone (α-MSH), and β-endorphin. POMC, ACTH and β-lipotropin are secreted from corticotropes in the anterior lobe of the pituitary gland in response to the hormone corticotropin-releasing hormone (CRH) released by the hypothalamus (Figure 70-1).
Functions of Adrenal Hormones
Cortisol plays important roles in cardiovascular stability (maintaining blood pressure by increasing the sensitivity of the vasculature to the vasoconstrictive effects of epinephrine and norepinephrine), metabolism (increasing gluconeogenesis to prevent hypoglycemia), and fluid and electrolyte balance (sodium retention and potassium excretion). It also inhibits bone formation and is a potent antiinflammatory agent (Figure 70-2).
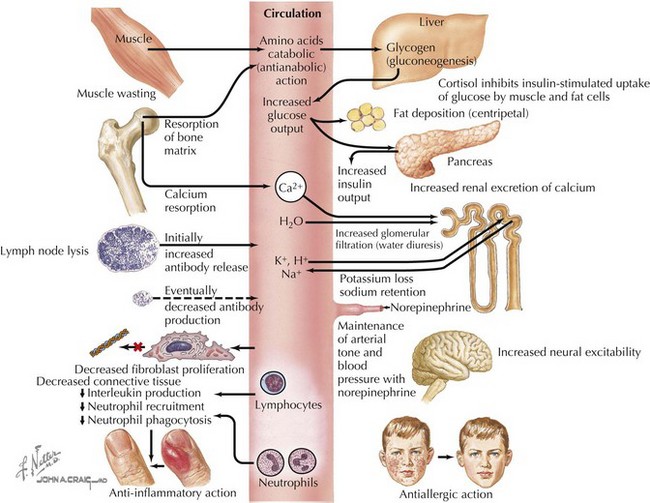
Aldosterone acts on the distal tubules and collecting ducts of the kidney to increase conservation of sodium and water, secrete potassium, and increase blood pressure. Adrenal androgens produced in the zona reticularis play a role in pubarche and the development of secondary sexual characteristics (i.e., pubic hair) in both males and females during puberty (see Chapter 67).
Adrenal Insufficiency
Etiology and Pathogenesis
Primary Adrenal Insufficiency
Primary adrenal insufficiency is caused by congenital or acquired dysfunction of the adrenal cortex or the hormone-producing steroidogenic pathway (Table 70-1). The most common cause in the developed world is autoimmune adrenalitis, also known as Addison’s disease. In developing countries, tuberculosis remains the most prominent cause. Destruction of the gland leads to deficiencies in all adrenal cortex hormones, but this process may be metasynchronous, and not all hormones are lacking in all patients. Enzyme defects can cause cortisol deficiency with an excess of precursor hormones. Loss of negative feedback from low cortisol levels leads to high ACTH in these disorders.
| Cause | Associations or Pathogenesis | Diagnosis |
|---|---|---|
| Acquired | ||
| Addison’s disease | Other autoimmune disease | Adrenal antibodies |
| Autoimmune polyglandular syndrome | Type 1: hypoparathyroidism and mucocutaneous candidiasis Type 2: type 1 diabetes, thyroiditis, other autoimmune |
AIRE gene mutation in type 1 |
| Infiltration or infection | TB, fungal, cancer, amyloidosis, sarcoid, hemochromatosis, CMV (HIV patients) | PPD, cultures, imaging, biopsy, ELISA or Western blot |
| Waterhouse-Friderichsen syndrome | Meningococcemia leading to adrenal hemorrhage | Cultures |
| Bilateral hemorrhage | Trauma, anticoagulants | Imaging |
| Medications: mitotane, ketoconazole | Destruction of gland, enzyme blockage | History |
| Congenital | ||
| CAH | Autosomal recessive; mutation of 21-hydroxylase and others | Adrenal steroid profiles, genetic testing CYP21 |
| Adrenoleukodystrophy; adrenomyeloneuropathy | X-linked; buildup of VLCFAs in adrenals and cerebral or spinal cord involvement; neuromuscular disease | Serum VLCFAs; ALD gene |
| X-linked congenital adrenal hypoplasia | Delayed puberty, contiguous gene mutations (Duchenne’s muscular dystrophy, glycerol kinase) | DAX1 gene testing |
| Triple A syndrome | Autosomal recessive; achalasia, alacrima, adrenal insufficiency | AAAS gene at 12q13 |
| Other syndromes: IMAGE, Smith-Lemli-Opitz | Vary | Genetic testing |
| ACTH resistance | ACTH receptor or melanocortin 2 receptor accessory protein (MRAP) gene mutations | Genotyping of receptor or MRAP genes |
ACTH, adrenocorticotropic hormone; CAH, congenital adrenal hyper-plasia; CMV, cytomegalovirus; ELISA, enzyme-linked immunosorbent assay; HIV, human immunodeficiency virus; IMAGE, intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, genital abnormalities; PPD, purified protein derivative; TB, tuberculosis; VLCFA, very long chain fatty acid.
Clinical Presentation
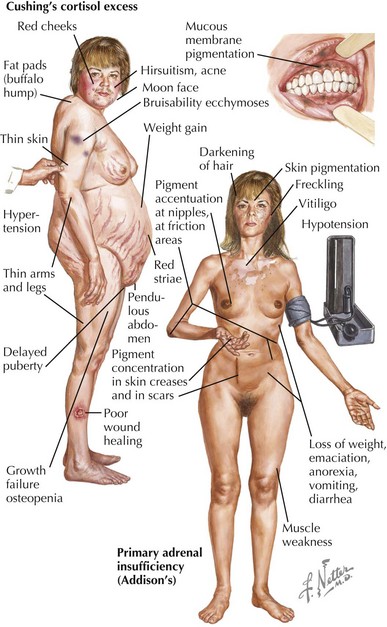
Chronic symptoms of adrenal insufficiency are vague and often unrecognized. Children experience fatigue, malaise, poor weight gain and growth, and anorexia. In primary adrenal insufficiency, pituitary secretion of ACTH is markedly increased and is often associated with hyperplasia of corticotrophic cells in the pituitary. Generalized hyperpigmentation of the skin and buccal mucosa may occur because of generation of α-MSH from ACTH (Figure 70-3).
Cushing’s Syndrome
Clinical Presentation
The signs and symptoms of Cushing’s syndrome include weight gain, growth failure, fatigue, hypertension, glucose intolerance, and delayed puberty (see Figure 70-3). Osteopenia, acne, plethora, hirsutism, a dorsocervical fat pad (“buffalo hump”), and striae can occur. Hyperandrogenism and virilization can indicate an adrenal carcinoma.
Congenital Adrenal Hyperplasia
Etiology and Pathogenesis
Congenital adrenal hyperplasia (CAH) is a group of disorders characterized by defective adrenal steroid synthesis; accumulation of androgenic steroid intermediates; and variably, cortisol and mineralocorticoid deficiency. The most common form of CAH is caused by homozygous mutation of the CYP21 gene that encodes 21-hydroxylase, the enzyme that catalyzes the conversion of 17-hydroxyprogesterone to 11-deoxycortisol and the conversion of progesterone to deoxycorticosterone (see Figure 70-1). The absence (or severe deficiency) of 21-hydroxylase results in a deficiency of cortisol and aldosterone.
Clinical Presentation
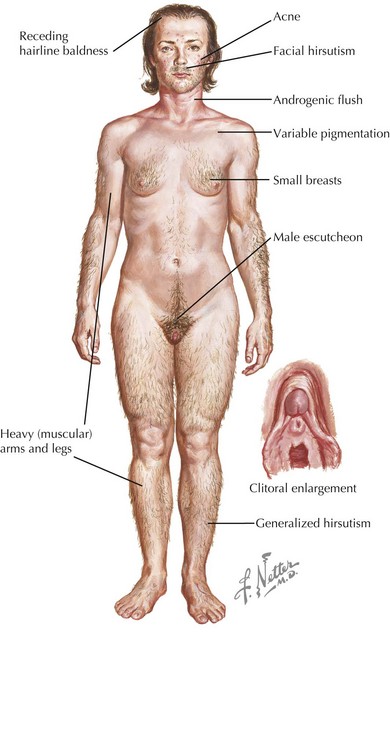
Deficiency of 21-hydroxylase activity classically presents as a salt-wasting adrenal crisis during the second week of life; 46 XX females can have varying degrees of virilization with genital ambiguity (Figure 70-4). Affected children can also present later in childhood with premature development of pubic hair, penile enlargement in boys, or as infertility or a polycystic ovary syndrome– (PCOS-) like syndrome in women. Newborn screening programs now include measurement of 17-hydroxyprogesterone as a screen for 21-hydroxylase deficiency, as well as less common 11-hydroxylase deficiency. Both conditions are virilizing, but 21-hydroxylase deficiency is associated with salt wasting and low blood pressure, and 11-β-hydroxylase deficiency (CYP11 gene) is associated with hypertension.
Clayton PE, Miller WL, Oberfield SE, et alESPE/ LWPES CAH Working Group. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Joint ESPE/LWPES CAH working group. Horm Res. 2002;58:188-195.
Shulman DI, Palmert MR, Kemp SF, Lawson Wilkins Drug and Therapeutics Committee. Adrenal insufficiency: still a cause of morbidity and death in childhood. Pediatrics. 2007;119(2):e484-e494.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
70 Disorders of the Adrenal Gland
Adrenal Gland Physiology
The adrenal cortex consists of three distinct zones, the glomerulosa, the fasciculata, and the reticularis. The fasciculata is the principal component of the hypothalamic–pituitary–adrenal axis. Glucocorticoid (cortisol) secretion from the fasciculata is regulated by adrenocorticotropic hormone (ACTH). ACTH is synthesized from pre-pro-opiomelanocortin (pre-POMC). The removal of the signal peptide during translation produces the 267 amino acid polypeptide POMC, which undergoes a series of posttranslational modifications to yield various polypeptide fragments with varying physiological activity. These fragments include the 39 amino acid polypeptide ACTH, as well as β-lipotropin, γ-lipotropin, melanocyte-stimulating hormone (α-MSH), and β-endorphin. POMC, ACTH and β-lipotropin are secreted from corticotropes in the anterior lobe of the pituitary gland in response to the hormone corticotropin-releasing hormone (CRH) released by the hypothalamus (Figure 70-1).
Functions of Adrenal Hormones
Cortisol plays important roles in cardiovascular stability (maintaining blood pressure by increasing the sensitivity of the vasculature to the vasoconstrictive effects of epinephrine and norepinephrine), metabolism (increasing gluconeogenesis to prevent hypoglycemia), and fluid and electrolyte balance (sodium retention and potassium excretion). It also inhibits bone formation and is a potent antiinflammatory agent (Figure 70-2).
Aldosterone acts on the distal tubules and collecting ducts of the kidney to increase conservation of sodium and water, secrete potassium, and increase blood pressure. Adrenal androgens produced in the zona reticularis play a role in pubarche and the development of secondary sexual characteristics (i.e., pubic hair) in both males and females during puberty (see Chapter 67).
Adrenal Insufficiency
Etiology and Pathogenesis
Primary Adrenal Insufficiency
Primary adrenal insufficiency is caused by congenital or acquired dysfunction of the adrenal cortex or the hormone-producing steroidogenic pathway (Table 70-1). The most common cause in the developed world is autoimmune adrenalitis, also known as Addison’s disease. In developing countries, tuberculosis remains the most prominent cause. Destruction of the gland leads to deficiencies in all adrenal cortex hormones, but this process may be metasynchronous, and not all hormones are lacking in all patients. Enzyme defects can cause cortisol deficiency with an excess of precursor hormones. Loss of negative feedback from low cortisol levels leads to high ACTH in these disorders.
| Cause | Associations or Pathogenesis | Diagnosis |
|---|---|---|
| Acquired | ||
| Addison’s disease | Other autoimmune disease | Adrenal antibodies |
| Autoimmune polyglandular syndrome | Type 1: hypoparathyroidism and mucocutaneous candidiasis Type 2: type 1 diabetes, thyroiditis, other autoimmune |
AIRE gene mutation in type 1 |
| Infiltration or infection | TB, fungal, cancer, amyloidosis, sarcoid, hemochromatosis, CMV (HIV patients) | PPD, cultures, imaging, biopsy, ELISA or Western blot |
| Waterhouse-Friderichsen syndrome | Meningococcemia leading to adrenal hemorrhage | Cultures |
| Bilateral hemorrhage | Trauma, anticoagulants | Imaging |
| Medications: mitotane, ketoconazole | Destruction of gland, enzyme blockage | History |
| Congenital | ||
| CAH | Autosomal recessive; mutation of 21-hydroxylase and others | Adrenal steroid profiles, genetic testing CYP21 |
| Adrenoleukodystrophy; adrenomyeloneuropathy | X-linked; buildup of VLCFAs in adrenals and cerebral or spinal cord involvement; neuromuscular disease | Serum VLCFAs; ALD gene |
| X-linked congenital adrenal hypoplasia | Delayed puberty, contiguous gene mutations (Duchenne’s muscular dystrophy, glycerol kinase) | DAX1 gene testing |
| Triple A syndrome | Autosomal recessive; achalasia, alacrima, adrenal insufficiency | AAAS gene at 12q13 |
| Other syndromes: IMAGE, Smith-Lemli-Opitz | Vary | Genetic testing |
| ACTH resistance | ACTH receptor or melanocortin 2 receptor accessory protein (MRAP) gene mutations | Genotyping of receptor or MRAP genes |
ACTH, adrenocorticotropic hormone; CAH, congenital adrenal hyper-plasia; CMV, cytomegalovirus; ELISA, enzyme-linked immunosorbent assay; HIV, human immunodeficiency virus; IMAGE, intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, genital abnormalities; PPD, purified protein derivative; TB, tuberculosis; VLCFA, very long chain fatty acid.
[/not-level-membership-for-pediatrics-category]
