[level-membership-for-pediatrics-category]
Disorders of Sexual Differentiation
Normal Gender and Sexual Differentiation
The most commonly accepted paradigm, described by Jost, involves a stepwise process to gender and sexual development.1 The primary determinant is the chromosomal gender, which is established at fertilization when the sperm provides an X or Y chromosome to the ovum’s X chromosome. Chromosomal gender determines gonadal gender, with XX resulting in ovarian development and XY resulting in testicular formation. Finally, the gonadal function determines the phenotypic gender. Although this paradigm is a helpful cascade to explain gender development, the simple Y = male, no Y = female equations are not always valid.
The testis determining factor (TDF) is located on the short arm of the Y chromosome near the centromere at the distal aspect of the Y-unique region.2 TDF is a 35 kb pair sequence on the 11.3 sub-band of the gender-determining region of the Y chromosome (SRY). Interestingly, SRY appears to be expressed by the somatic cells from the urogenital ridge and not from germ cells.
Many other genes play a role in gender development. Despite the presence of a functional SRY gene, the absence of the SOX9 gene results in a female phenotype in the majority of chromosomal males.3,4 The Wilms tumor gene (WT1) appears to play a key role not only in renal development, but also in testicular development. Early alteration of WT1 function results in testicular agenesis and later dysfunction results in aberrant testicular development (streak gonad or dysgerminomas). This tumor suppressor gene has been implicated in Denys–Drash syndrome involving testicular (mixed gonadal dysgenesis) and renal (Wilms tumor) abnormalities.5,6
Fushi–Tarzu factor-1 (FTZ-F1) exerts its effect on gonadal development through its regulation of steroidogenic factor-1 (SF-1). The SF1 gene is involved with steroid hormone production and the production of Müllerian-inhibiting substance (MIS) by the Sertoli cells of the testis that causes regression of the Müllerian ductal system. Although FTZ-F1 and SF-1 are also expressed in ovarian tissues, the timing and intensity of their effect are critical for normal gonadal development.5,7
Finally, the lack of an SRY gene alone does not impart normal female phenotypic and gonadal development. The DAX1 gene appears to be essential for the development of the ovary. The DAX1 gene product appears to compete with the SRY gene product for a steroidogenic regulatory protein (StAR). A dosage-sensitive element is also important. Normally, the single SRY gene has a greater impact than a single DAX1 gene and causes upregulation of StAR. However, in those chromosomal abnormalities in which more than one DAX1 gene is present, downregulation of StAR occurs, testicular development is inhibited, and ovarian development is promoted. As in the case of Turner syndrome, these primordial ovaries develop into streak gonads. Likely, other genes are also important for normal ovarian development.8–10
Development of the internal ductal structures is dependent on hormone secretion by the developing gonads (Table 62-1). In the absence of functioning testicular tissue, the female internal Müllerian duct structures develop. The presence of a functioning testis results in male internal Wolffian duct development. This differentiation is mediated by the production of testosterone from the testis. Testosterone promotes Wolffian duct development along with MIS, which results in regression of the Müllerian duct structures. This is a paracrine effect and therefore results in gonad specific ipsilateral ductal differentiation. This effect is likely dependent on high concentrations of androgen produced by the physically proximate gonad. Decreased levels of MIS by an abnormal testis or streak gonad result in ipsilateral Müllerian development. This occurs despite regression of the Müllerian ducts on the contralateral side with normal testicular MIS production. Conversely, systemic administration of androgen does not result in male ductal development in a female fetus.
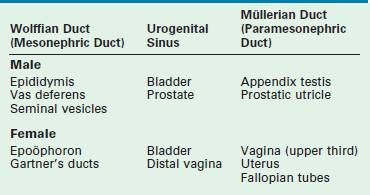
Produced by Sertoli cells, MIS functions as a suppressor of Müllerian duct development and is a specific marker for functioning testicular tissue in infancy. In its absence, the Müllerian structures develop. The concentration and timing of MIS secretion appear to be critical. Normally, secretion occurs during week 7 of gestation. By week 9, the Müllerian ducts become insensitive to MIS.9
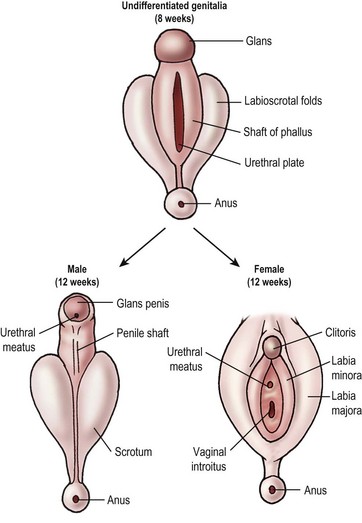
External genital development follows a similar path (Fig. 62-1). In the absence of the testosterone metabolite dihydrotestosterone (DHT), the external genitalia develop into the female phenotype. The male and female phenotypes are identical until week 7. In the male, testosterone production by the testicular Leydig cells surges at 7 weeks and remains elevated until week 14 of gestation. Testosterone is converted to DHT by 5α-reductase in the tissues of the genital skin and urogenital sinus. The testosterone-binding receptor has much higher affinity for DHT than testosterone and serves to amplify the effect of testosterone on the developing external genitalia. In the absence of 5α-reductase, the internal Wolffian ducts are preserved, but the external structures are feminized.
Aberrant Gender Development
Incidence
In the Americas and Western Europe, congenital adrenal hyperplasia (CAH) is the most common cause of neonatal ambiguous genitalia, accounting for approximately 70% of cases.11,12 The overall incidence is approximately 1 in 15,000 live births. The rate is much higher in stillborns and in certain regional populations (Yupic Eskimos and the people of La Réunion, France).13 In the United States, mixed gonadal dysgenesis is the next most common intersex disorder, with ovotesticular DSD (true hermaphroditism) being the third most common.
Classification
Until recently, the most commonly used classification system was the one proposed by Allen in 1976 and was based primarily on gonadal histology.14 This system categorized the most common DSD well, but did not accommodate less common syndromes easily. Also, the older ‘hermaphrodite’ terminology has become offensive to some. A newer classification released by the International Consensus Conference on Intersex has largely replaced Allen’s system. This newer system incorporates an evolving understanding of the molecular basis of these disorders and replaces more offensive gender-based labels. The new terminology is used primarily in this text.15
46,XX DSD (Female Pseudohermaphrodite)
The majority of neonates with external genital ambiguity fall into this category. All patients have a 46,XX genotype and exclusively ovarian tissue in nonpalpable gonads. Simplistically, the cause of the gender ambiguity is an excess of androgen. More than 95% are due to CAH, with the remainder due to maternal androgen exposure. These patients have a normal female Müllerian ductal system with an upper vagina, uterus, and fallopian tubes (see Table 62-1). They also have normal regression of the Wolffian ducts. The level of virilization is largely dependent on the timing and magnitude of androgen exposure to the external genitalia. The phenotype can range from mild clitoromegaly to a normal male appearance.
Virilization in CAH is due to the inability of the adrenal gland to form cortisol. The precursors above the enzymatic defect are shunted into the mineralocorticoid or sex-steroid pathways. Also, the end products generally have some, albeit weak, glucocorticoid function. The lack of cortisol for negative feedback inhibition of adrenocorticotropic hormone (ACTH) production by the pituitary leaves this pathway unchecked. Excess androgen is produced and is responsible for the virilization. The corticosteroid synthetic and alternative pathways are shown in Figure 62-2. The most common form of CAH is 21-hydroxylase deficiency (21-OHD), which accounts for more than 90% of CAH.16 21-OHD has been mapped to the short arm of chromosome 6. The variable location of the adrenal defect and relative function of the gene results in salt-wasting and nonsalt-wasting forms.17–19 Type 1 results in virilization but no salt wasting. The gene defect affects only the fasciculata zone of the adrenal, resulting in blocking cortisol production. However, the gene is normally expressed in the glomerulosa zone with preservation of mineralocorticoid production. In type 2, also called the classic type, the gene abnormality affects both adrenal zones. Salt wasting results in dehydration or vascular collapse, and hyperkalemia develops because of the block in mineralocorticoid production.
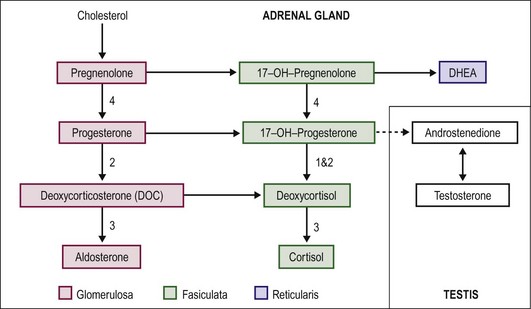
FIGURE 62-2 Pathways of steroid biosynthesis. The numbers correspond to CAH type and the location of the enzyme defect (see the text).
Virilization of the female fetus can be caused by exogenous androgen exposure from the mother. This occurs primarily with the use of progesterone, commonly used as an adjunct to assist with fertility and in-vitro fertilization. Endogenous androgen exposure due to virilizing maternal ovarian tumors also has been reported, but these tumors are usually virilizing to the mother with the fetus being unaffected.20
Treatment
As all forms of CAH are inherited in an autosomal recessive manner, genetic counseling is recommended. Families with a history of CAH should consider maternal treatment with dexamethasone before week 10 of gestation to eliminate or improve the level of fetal virilization.21 Postnatally, cortisol replacement with hydrocortisone is the mainstay of therapy, with the addition of fluorohydrocortisone if salt wasting is present. Supportive management of fluid and electrolyte abnormalities is best provided in a neonatal intensive care unit.
With regard to gender identity, the vast majority of patients with CAH identify as female and gender assignment is generally female given the 46,XX karyotype. The ovaries are normal and have fertility potential.22 Surgical reconstruction requires a feminizing genitoplasty, and involves clitoroplasty, monsplasty, and vaginoplasty.
46,XY DSD (Male Pseudohermaphrodite)
Defects in 17,20-desmolase and 17β-hydroxysteroid oxidoreductase act at the testicular level to convert androstenedione to testosterone. Because the adrenal is unaffected, CAH does not occur. The phenotype can be quite variable, but those with complete feminization can escape detection at birth and be reared as female. Progressive virilization is related to excess gonadotropin production at puberty, which may partially compensate for the lack of testosterone synthesis. Phallic growth and the development of male secondary sex characteristics create a conundrum with regard to gender reassignment when the diagnosis is made later in life.23
The extreme is normal female external genitalia resulting from complete androgen insensitivity syndrome (CAIS, also termed testicular feminization). The incidence of this syndrome is approximately 1 in 40,000. It usually results from a point mutation in the androgen receptor gene, located on the X chromosome.24,25
Partial androgen insensitivity is associated with a large spectrum of phenotypic variation (e.g., Gilbert–Dreyfus, Lub, and Reifenstein syndromes). It can be a sporadic or inherited condition, and gender assignment and treatment are individualized.26
Diagnosis
Metabolically, the diagnosis of CAH is made similarly to the 46,XX DSD patient, noting excess steroid levels above the enzymatic block and elevated levels of ACTH. The physical examination confirms absence of a cervix on rectal examination, and bronzing of the skin may be present. Palpation of a cryptorchid or descended testis is possible. The genitogram and ultrasound mirror these findings, but a prominent utricle may be present that lacks a cervical impression at its apex. In CAIS, testosterone levels are elevated postpubertally, but the diagnosis in the prepubertal child may require human chorionic gonadotropin (hCG) stimulation and genital skin fibroblast androgen receptor studies. Receptor assays can delineate a quantitative versus qualitative receptor defect. LH levels are elevated, related to the loss of testosterone feedback inhibition, which requires normal receptor hormone interaction. 5α-reductase deficiency is confirmed by an elevated testosterone-to-DHT ratio and an abnormal 5α-reductase type 2 gene.27
Treatment
In CAIS, the gender assignment is always female. CAIS patients who are assigned as female in infancy later identify themselves as female.28 Because the androgen receptor defect is ubiquitous, virilization of the brain does not occur. Orchiectomy is required, given the risk of malignant degeneration, but is often deferred until after puberty.29 The testis synthesizes estradiol, facilitating feminine development at puberty. Orchiectomy before puberty necessitates hormone replacement for normal pubertal development.
Gender assignment in partial androgen insensitivity syndrome (PAIS) is largely based on the response of the external genitalia to exogenous testosterone. A significant virilization response argues for the male gender. If there is no response, the female gender is favored. This subgroup is the most variable and has the least consensus with regard to gender assignment. There are reports of gender reassignment at puberty.30,31 Dissatisfaction with the gender of rearing occurs in approximately 25% of PAIS patients, whether raised male or female.32 In 5α-reductase deficiency syndrome, the brain is normally virilized and these individuals identify with the male gender. Thus, male gender assignment is recommended.33
Müllerian-Inhibitory Substance Deficiency (Hernia Uterine Inguinale)
MIS is produced by the Sertoli cells in the testis and causes regression of the Müllerian ductal structures. In this rare syndrome of abnormal MIS production or MIS-receptor abnormality, Wolffian ductal development is unimpaired, but the Müllerian ducts also persist. Because the infant has a normal male phenotype, this syndrome is rarely encountered in the neonatal period. The most common presentation to the surgeon is that of finding a fallopian tube adjacent to an undescended testis in the hernia sac at the time of orchiopexy.34
If this scenario is encountered, a biopsy of the gonad should be performed, the hernia should be repaired, and all structures left intact until completion of a full evaluation with karyotype and MIS levels. Apparent males can also present with bilateral nonpalpable testes, and Müllerian structures are found at laparoscopy (Fig. 62-3). Abnormal MIS-receptor gene assays also can be helpful for verifying the diagnosis in those with a normal MIS level. Subsequent management is primarily orchiopexy. This, however, can be difficult because the vas deferens can be closely adherent or ectopic to the fallopian tube or uterus. Excision of discordant ductal structures may be attempted, but given the relatively low risk associated with leaving these structures, the risk of damage to the vas during this dissection outweighs the benefit of removal. Despite normal testosterone levels, the patient often has impaired spermatogenesis.35–37
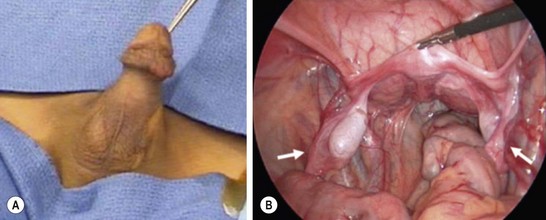
FIGURE 62-3 A 2-year-old boy presented with nonpalpable testes. A karyotype showed XY. (A) External genitalia were male. (B) Laparoscopy revealed bilateral gonads that were testes on longitudinal biopsy. Müllerian structures (arrows) were also seen. Note the rudimentary uterus (being held by the grasping forceps). This patient had abnormal MIS-receptor function.
Leydig Cell Abnormalities
As the Leydig cell is responsible for testosterone production in the testes, impaired testosterone production can also manifest from Leydig cell hypoplasia, agenesis, or abnormal Leydig cell gonadotropin receptors. These disorders are rare. Although the karyotype is 46,XY, the phenotype tends to be female, with a blind-ending vaginal pouch and absence of internal Müllerian structures. These patients usually are seen initially around puberty with amenorrhea and, therefore, are reared as female. Management is similar to that for CAIS, with orchiectomy and estrogen replacement.38,39
Ovotesticular DSD (True Hermaphrodite)
Ovotesticular DSD exists when both ovarian and testicular tissue are present. The gonadal configuration also can be quite variable, with the ovary/ovotestis combination being most common in the United States, but any combination can occur. Ovotestes are usually polar with an ovary at one end and a testis at the other, but the distribution can be longitudinal, requiring deep longitudinal biopsy to sample the gonad adequately. Because of the paracrine effect of the gonad, the ipsilateral internal duct structures correlate with the type of gonad present. Ovotestes are associated with a variable duct structure, but usually fallopian tubes prevail. A decisively Müllerian or Wolffian duct structure is usually found rather than an ipsilateral combination.40
The phenotype covers the entire spectrum, with ambiguity and asymmetry the rule, but with a tendency toward masculinization. Although it is unusual for ovaries to be found in the labioscrotum, testes and ovotestes are often palpable. Fertility has been described in those raised as female, but testicular fibrosis makes this unlikely in those raised as male.25,41
Treatment
Gender assignment in ovotesticular DSD is quite variable and should be based on the functional potential of the phenotype. In either case, the discordant gonads should be removed early in life. Retained testicular tissue will cause virilization in the female. In males, the testicular tissue is preserved and orchiopexy is performed. A 1–10% incidence of testicular tumors are found in males, predominantly gonadoblastomas and dysgerminomas, so long-term surveillance is needed.42 Hypospadias repair also is required in the male, and feminizing genitoplasty is performed in the female. Males tend to require hormonal replacement because of the progressive testicular fibrosis, but females usually do not. Fertility is possible in those raised as female. Females should, however, be screened for testosterone levels, which can signal inadequate resection of testicular tissue.43
Mixed Gonadal Dysgenesis
Mixed gonadal dysgenesis (MGD) is the second most common form of neonatal ambiguous genitalia. The patient will have a testis on one side and a streak gonad on the other, characterized microscopically by normal ovarian stroma without oocytes (Fig. 62-4). The internal duct structure mirrors the ipsilateral gonad, with the streak associated with a fallopian tube and uterus resulting from the lack of MIS. The karyotype is generally a mosaic of 45,XO/46,XY, and the stigmata of Turner syndrome are variably present. The phenotype is ambiguous, but masculinized, and the testis may be descended, but more commonly is not.44
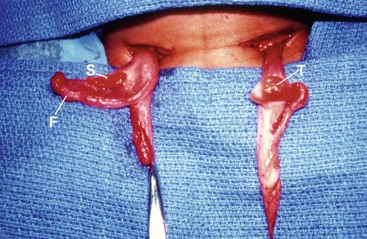
FIGURE 62-4 Mixed gonadal dysgenesis. The left testis was descended, and the right streak gonad was intra-abdominal. T, testis; F, fallopian tube; S, streak gonad. (Photo taken from foot of table.)
The risk of a gonadal tumor, usually gonadoblastoma, is as high as 20%, and tumors can develop in either the testis or streak gonad.45 An increased risk of Wilms tumor also is present in MGD. The Denys–Drash syndrome occurs in approximately 5% of patients with MGD and is classically described as ambiguous genitalia, Wilms tumor, and glomerulopathy, which is often associated with hypertension.46
Treatment
The majority of patients with MGD have been raised as female because of the short stature conferred by Turner syndrome and the malignant risk of the retained testis. Females undergo early gonadectomy and feminizing genitoplasty. Males require early excision of the streak gonad, orchiopexy or orchiectomy, and hypospadias repair. Infertility is the rule despite adequate testicular endocrine function. Because of the increasing awareness regarding testosterone imprinting on the brain, more masculinized patients are being raised as male. If individuals are raised as male, close surveillance of the testis is necessary, unless elective orchiectomy and hormone replacement are chosen. Testicular biopsy at the time of puberty to exclude dysgenetic elements is generally recommended.42 If carcinoma in situ is identified, low-dose radiation therapy is curative.
Other Syndromes of Aberrant Sexual Differentiation
Several syndromes worth mentioning do not neatly fit into the described classification systems.
Vanishing testis syndrome is characterized by a 46,XY karyotype, but absent testes bilaterally. This generally results in virilization to the point of normal external genitalia and internal duct structure, but absent testes. The testes were thought to have produced androgen at some point, resulting in masculinization, but subsequently vanished related to torsion or regression. Patients are generally raised as boys, and hormonal supplementation at puberty is required.47
Klinefelter syndrome is characterized by a male karyotype containing two or more X chromosomes (47,XXY, 48,XXXY, etc.). Although phenotypically male prepubertally, these patients acquire abnormal male secondary sexual characteristics (tall stature with disproportionately long legs, sparse facial hair, decreased muscle mass, and a feminine fat distribution) and infertility. The testes are small and hard, with decreased androgen production and elevated estradiol levels related to primary hypergonadotropic hypogonadism. Gynecomastia often occurs with an increased risk of breast cancer.48 Fertility has been reported but requires assisted means, such as intracytoplasmic sperm injection.49
46,XX Testicular DSD (XX sex reversal) is characterized by a male phenotype with a 46,XX karyotype. Most commonly, this occurs from translocation of Y chromosomal material to the X chromosome, but it also can occur from mutation of the X chromosome or from mosaicism. The phenotype and management are similar to those of Klinefelter syndrome, with the exception of shorter stature.50
Mayer–Rokitansky–Küster–Hauser syndrome is characterized by a 46,XX karyotype with normal female external genitalia but a short, blind-ending vagina. Normal ovaries and fallopian tubes are present, but the uterus is generally rudimentary. Patients are seen initially with primary amenorrhea, but may have cyclical pain related to functioning endometrium. Treatment is geared toward vaginal reconstruction to allow menses or intercourse, or both.51
Evaluation of the Newborn with Ambiguous Genitalia
The diagnosis of ambiguous genitalia is extremely disconcerting to the family and should be addressed as a medical emergency. Usually, genital ambiguity is obvious, but the finding of any degree of hypospadias, particularly in association with a nonpalpable testis, merits a DSD evaluation. In this population, a high rate of intersex conditions is found despite the absence of classic ambiguity.52 Table 62-2 indicates other abnormal physical examination findings that warrant consideration for DSD. The family history may reveal maternal hormone exposure, previous fetal death, or a history of genital ambiguity.
TABLE 62-2
Physical Examination Findings that Warrant Consideration for DSD
| Apparent Female | Unsure | Apparent Male |
| Clitoral hypertrophy Fused labia Palpable gonad |
Ambiguous | Impalpable testes Severe hypospadias Hypospadias and cryptorchidism |
The initial metabolic evaluation should include a karyotype or fluorescent in situ hybridization to identify X and Y chromosomes. 17-OH progesterone levels should be obtained after 3 or 4 days of life, by which time spurious elevations resulting from the stress related to birth have subsided. Electrolyte levels should bemonitored closely in the interim to identify salt wasting with CAH. Testosterone and DHT levels are important for evaluating 5α-reductase deficiency. An elevated LH level and a low MIS level suggest testis dysgenesis or absence. ACTH or hCG stimulation tests can be performed but are more controversial.34 Genetic testing for nearly all known defective genes is now available, but the list is extensive and nondirected testing is not cost effective.53
Early imaging studies include pelvic ultrasound, which should identify a uterus if one is present. Although a gonad may be seen, ultrasound is not useful in differentiating a testis, ovotestis, or ovary. A genitogram performed by retrograde contrast injection into the urogenital sinus is helpful in identifying the level of confluence of a vagina and urethra and its relation to the urethral sphincter (Figs 62-5 and 62-6).
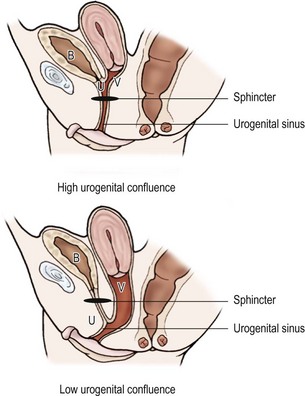
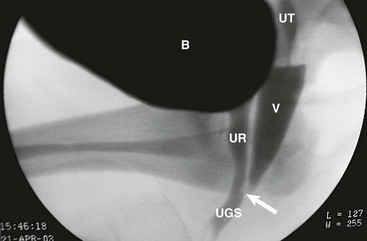
FIGURE 62-6 Genitogram, lower confluence. White arrow, level of the confluence of urethra and vagina to become the urogenital sinus. B, Bladder; UT, uterus; V, vagina; UR, urethra; UGS, urogenital sinus.
Gonadal biopsy is often required for diagnostic purposes, but the diagnosis of CAH can be made by metabolic evaluation alone. Endoscopy is not usually required for diagnosis, but is essential in characterizing the internal duct structure, level of confluence of the urogenital sinus, and planning for and performing the reconstructive procedures.25
A gender-assignment team should include a pediatric urologist/surgeon, endocrinologist, geneticist, neonatologist, psychologist, and social worker who together evaluate all newborns with ambiguous genitalia. This information is synthesized by the team and presented to the parents in a combined care conference. The goals of gender assignment and management should include preservation of sexual function and any reproductive potential with the least number of operations, appropriate gender appearance with a stable gender identity, and psychosocial well-being.54
Reconstructive Genital Surgical Procedures
Controversies and Considerations
For more than 20 years, largely based on the work of John Money and the ‘John/Joan Case,’ the overwhelming bias was that gender identity was largely inducible and loosely dependent on chromosomal constitution.55 The focus was on one of two twin boys who was reassigned to the female gender early in life after a demasculinizing circumcision injury. The child reportedly developed normally from a psychosocial standpoint and adapted well to life as a girl. Only with extended follow-up into adulthood was it discovered that the individual converted back to the male gender after severe dissatisfaction with a female identity (including attempted suicide).56 This rattled the fundamental concepts on which gender assignment had been based for decades and brought to the forefront a tremendous controversy regarding the appropriate management of children with ambiguous genitalia and gender reassignment.
As reconstruction is rarely done in response to any life-threatening issues, support groups for individuals with DSD have advocated delaying any reconstruction until the child can express his or her wishes regarding gender assignment.57 Although this would decrease the likelihood of a mismatch between physical and psychological gender, the period of genital ambiguity could be quite challenging for child and family in our society. Despite the controversy, the International Consensus Panel on Intersex stated that the evidence is currently insufficient to abandon the practice of early genital reconstruction.15 However, this must be discussed thoroughly with the family before embarking on any reconstructive operations.
In general, if genital reconstruction is thought to be appropriate, it is planned in the first 3 to 6 months of life. For feminizing reconstruction, the vaginal tissue is thicker as a result of maternal hormonal influence and the distance from the vagina to perineum is shorter at this age. Because parents have a great degree of anxiety surrounding the gender of their child, earlier repair may help reduce this anxiety and encourage parent/child bonding.58
Male Gender Assignment
Reconstructive efforts for the male gender of rearing include orchiopexy or orchiectomy, when appropriate, and hypospadias repair. These techniques are described elsewhere in this book. It bears mentioning again that orchiopexy may be extremely difficult because the vas deferens can be closely adherent to Müllerian duct remnants, such as a fallopian tube or uterus. A portion of these structures may be left in situ if the risk of damage to the vas deferens or testicular vasculature is significant, but this adherence may severely limit mobility of the testis and preclude orchiopexy. Methods of total penile reconstruction in cases of aphallia, demasculinizing penile trauma, or female-to-male gender reassignment by using local skin flaps, a radial forearm flap, or osteocutaneous fibula flap have been described with reasonable success.59–61
Female Gender Assignment
Feminizing genitoplasty includes three major components: monsplasty, clitoroplasty, and vaginoplasty (Figs 62-7 and 62-8). The timing of the vaginoplasty depends on the level of confluence of the urogenital sinus. For a low confluence, the vaginoplasty is performed in the neonatal period with the monsplasty and clitoroplasty. Even with a high confluence, the vaginoplasty can be performed with the monsplasty and clitoroplasty in the newborn. However, because vaginal dilation is often necessary after repair, it may better to defer the vaginoplasty until the patient is peripubertal and more capable and interested in this requirement. Cystoscopy is invaluable for this assessment. We generally insert a Fogarty balloon catheter in the vagina and a catheter in the urethra and bladder to define the confluence during the dissection.
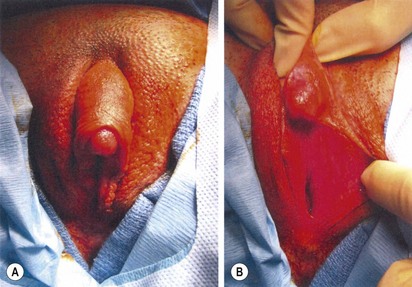
FIGURE 62-7 Ambiguous genitalia. The patient has congenital adrenal hyperplasia, 21-hydroxylase deficiency.
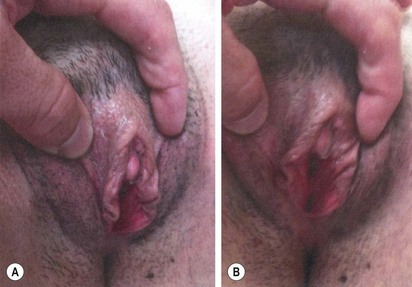
FIGURE 62-8 Same patient as in Figure 63-7. Appearance of genitalia six months after undergoing a feminizing genitoplasty.
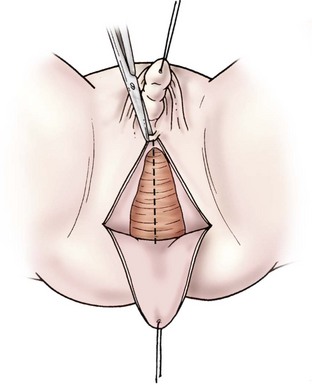
The procedure is initiated by placing a traction suture in the glans. A dorsolateral circumcising incision is made, leaving a 4–5 mm distal preputial cuff, much like is done in a hypospadias repair. The lateral borders of the mucosalized plate are incised, taking this back adjacent to the urogenital sinus. The shaft of the phallus is then degloved of skin superficial to Buck’s fascia. Fascial incisions are then made lateral to the neurovascular bundles. A plane is created just beneath Buck’s fascia from the level just proximal to the glans back to the pubic symphysis. The mucosalized plate is elevated on the ventrum and preserved to fill naturally the void between the urethral meatus and clitoris. The dorsal pedicles, including the neurovascular bundles, are preserved. The base of the corporeal bodies are suture ligated at the level of the pubic symphysis, and the distal corporeal tissue is excised. An alternative technique preserving the corporeal bodies has been described to maintain the potential for reversibility, but long-term functional outcome is pending.62
No clitoral reduction is performed to avoid nerve injury. It can be recessed and has a quite normal appearance in adulthood.63 The clitoris is anchored to the corporal stumps to secure its position, being sure not to compromise the dorsal neurovascular pedicle.
A posterior inverted U-shaped flap is then made from the level of the ischial tuberosities to just posterior to the urogenital meatus. For a very low confluence, dissection is carried along the posterior aspect of the urogenital sinus to the level of the confluence, the posterior wall is incised until the vaginal introitus is normal in caliber, and the U-flap is advanced to complete the posterior vaginal wall (Fig. 62-9). With higher confluences, total urogenital mobilization is favored.64–66
For total urogenital mobilization, the urogenital sinus is incised circumferentially and mobilized as one unit to the level of the confluence (Fig. 62-10). At this point, the vagina can be carefully separated from the urethra under direct vision and the defect in the urethra is closed. The urogenital sinus can be incised in the ventral and dorsal midline and rotated posterolaterally to lengthen the distal vagina. Using this technique, a long distance to the perineum can be bridged.
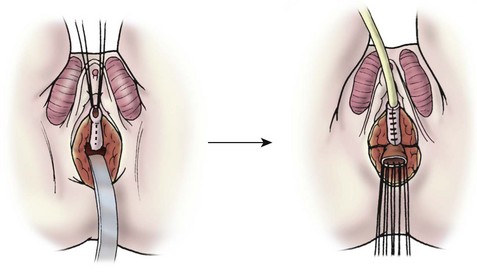
FIGURE 62-10 Total urogenital mobilization. The urogenital sinus is mobilized as a unit, bringing the confluence toward the perineum. Once visualized, the vagina is then detached and the urethral defect is closed.
Total urogenital mobilization is attractive as one can approach even the high urogenital confluences in the neonatal period without vaginal substitution or grafting, but the family must be appropriately cautioned. Although early results are favorable, descent of the bladder neck is counterintuitive when considering our knowledge of adult female stress incontinence and long-term continence could be an issue. To complete the monsplasty, the dorsal phallic shaft skin is incised vertically, a preputial hood is formed for the clitoris, and the majority of this tissue is used to construct the labia minor with V-shaped advancement flaps (Fig. 62-11). Other techniques for the high urogenital sinus include a posterior approach that requires incising the rectum and an anterior transvesical approach.67,68
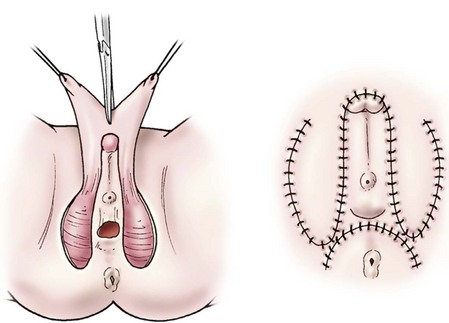
FIGURE 62-11 Monsplasty. Dorsal shaft skin is degloved from the phallus and incised. These flaps are then advanced to become the labia minora. (Shown before and after excision of the corporeal tissues and clitoropexy.)
In some patients with a high urogenital confluence, vaginal reconstruction is delayed until the peripubertal period, just before menarche. The techniques described are still used, but in some patients, especially those who are obese, these methods are insufficient. In such cases, vaginal substitution with a colonic segment is preferred, but ileal substitutions and split-thickness skin grafts also have been used. The benefit of vascularized bowel substitution is less vaginal stenosis and the natural formation of lubricating mucus, but this may require wearing a pad if there is excessive mucus production. Colonic segments appear to have a lower rate of stenosis than do ileal segments.69 Conversely, skin grafts have a tendency to become stenotic and may require frequent dilation and revision, but long-term satisfaction also has been reported.70 The barrier function to sexually transmitted diseases is likely superior with skin grafts when compared with intestine.71 If a rudimentary vagina or depression exists, sequential dilation with the technique described by Frank and modified to a dilating seat by Ingram may be successful.72,73
References
1. Jost, A, Vigier, B, Prepin, J, et al. Studies on sex differentiation in mammals. Recent Prog Horm Res. 1973; 29:1–41.
2. Lukusa, T, Fryns, JP, van der Berghe, H. The role of the Y-chromosome in sex determination. Genet Couns. 1992; 3:1–11.
3. Clarkson, MJ, Harley, VR. Sex with two SOX on: SRY and SOX9 in testis development. Trends Endocrinol Metab. 2002; 13:106–111.
4. Moog, U, Jansen, NJ, Scherer, G, et al. Acampomelic campomelic syndrome. Am J Med Genet. 2001; 104:239–245.
5. Parker, KL, Schimmer, BP, Schedl, A. Genes essential for early events in gonadal development. EXS. 2001; 11–24.
6. Schedl, A, Hastie, N. Multiple roles for the Wilms’ tumour suppressor gene, WT1 in genitourinary development. Mol Cell Endocrinol. 1998; 140:65–69.
7. Nordqvist, K. Sex differentiation—gonadogenesis and novel genes. Int J Dev Biol. 1995; 39:727–736.
8. Goodfellow, PN, Camerino, G. DAX-1, an ‘antitestis’ gene. Cell Mol Life Sci. 1999; 55:857–863.
9. Taguchi, O, Cunha, GR, Lawrence, WD, et al. Timing and irreversibility of Mullerian duct inhibition in the embryonic reproductive tract of the human male. Dev Biol. 1984; 106:394–398.
10. Tajima, K, Dantes, A, Yao, Z, et al. Down-regulation of steroidogenic response to gonadotropins in human and rat preovulatory granulosa cells involves mitogen-activated protein kinase activation and modulation of DAX-1 and steroidogenic factor-1. J Clin Endocrinol Metab. 2003; 88:2288–2299.
11. Menon, PS, Harinarayan, CV, Forest, MG. Congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Indian Pediatr. 1992; 29:98–103.
12. Pellerin, D, Nihoul-Fekete, C, Lortat-Jacob, S. [Surgery of sexual ambiguity: Experience of 298 cases]. Bull Acad Natl Med. 1989; 173:555–562.
13. Pang, SY, Wallace, MA, Hofman, L, et al. Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics. 1988; 81:866–874.
14. Allen, TD. Disorders of sexual differentiation. Urology. 1976; 7:1–32.
15. Lee, PA, Houk, CP, Ahmed, SF, et al. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006; 118:e488–e500.
16. Dacou-Voutetakis, C, Maniati-Christidi, M, Dracopoulou-Vabouli, M. Genetic aspects of congenital adrenal hyperplasia. J Pediatr Endocrinol Metab. 2001; 14:1303–1308.
17. Reindollar, RH, Tho, SP, McDonough, PG. Abnormalities of sexual differentiation: Evaluation and management. Clin Obstet Gynecol. 1987; 30:697–713.
18. Laue, L, Rennert, OM. Congenital adrenal hyperplasia: molecular genetics and alternative approaches to treatment. Adv Pediatr. 1995; 42:113–143.
19. Wilson, RC, Mercado, AB, Cheng, KC, et al. Steroid 21-hydroxylase deficiency: Genotype may not predict phenotype. J Clin Endocrinol Metab. 1995; 80:2322–2329.
20. Calaf, J, Prats, J, Esteban-Altirriba, J. Female pseudohermaphroditism caused by maternal hyperandrogenism. In: J M-M, ed. Intersexual States: Disorders of Sex Differentiation. Barcelona: Ediciones Doyma; 1994:187–197.
21. Trautman, PD, Meyer-Bahlburg, HF, Postelnek, J, et al. Mothers’ reactions to prenatal diagnostic procedures and dexamethasone treatment of congenital adrenal hyperplasia. J Psychosom Obstet Gynaecol. 1996; 17:175–181.
22. Berenbaum, SA, Bailey, JM. Effects on gender identity of prenatal androgens and genital appearance: Evidence from girls with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2003; 88:1102–1106.
23. Saez, JM, De Peretti, E, Morera, AM, et al. Familial male pseudohermaphroditism with gynecomastia due to a testicular 17-ketosteroid reductase defect. I. Studies in vivo. J Clin Endocrinol Metab. 1971; 32:604–610.
24. Quigley, CA, De Bellis, A, Marschke, KB, et al. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr Rev. 1995; 16:271–321.
25. Diamond, DA. Sexual differentiation: Normal and abnormal. In: Walsh PC, Retik AB, Vaughn ED, Jr., Wein AJ, eds. Campbell’s Urology. 8th ed. Philadelphia: WB Saunders; 2002:2395–2427.
26. Batch, JA, Davies, HR, Evans, BA, et al. Phenotypic variation and detection of carrier status in the partial androgen insensitivity syndrome. Arch Dis Child. 1993; 68:453–457.
27. Barthold, J, Gonzalez, R. Intersex states. In: Gonzales E, Bauer SB, eds. Pediatric urology practice. Philadelphia: Lippincott Williams & Wilkins; 1999:547.
28. Mazur, T. Gender dysphoria and gender change in androgen insensitivity or micropenis. Arch Sex Behav. 2005; 34:411–421.
29. Muller, J, Skakkebaek, NE. Testicular carcinoma in situ in children with the androgen insensitivity (testicular feminisation) syndrome. Br Med J (Clin Res Ed). 1984; 288:1419–1420.
30. Rosler, A, Kohn, G. Male pseudohermaphroditism due to 17 beta-hydroxysteroid dehydrogenase deficiency: Studies on the natural history of the defect and effect of androgens on gender role. J Steroid Biochem. 1983; 19:663–674.
31. Imperato-McGinley, J, Peterson, RE, Gautier, T, et al. Androgens and the evolution of male-gender identity among male pseudohermaphrodites with 5alpha-reductase deficiency. N Engl J Med. 1979; 300:1233–1237.
32. Migeon, CJ, Wisniewski, AB, Gearhart, JP, et al. Ambiguous genitalia with perineoscrotal hypospadias in 46,XY individuals: Long-term medical, surgical, and psychosexual outcome. Pediatrics. 2002; 110:e31.
33. Wilson, JD. Syndromes of androgen resistance. Biol Reprod. 1992; 46:168–173.
34. Huseman, DA, The genitalia intersex 4th ed. Gillenwater JY, Grayhack JT, Howards SS, Mitchell ME. Lippincott Williams & Wilkins, Philadelphia, 2002.
35. Loeff, DS, Imbeaud, S, Reyes, HM, et al. Surgical and genetic aspects of persistent mullerian duct syndrome. J Pediatr Surg. 1994; 29:61–65.
36. Fernandes, ET, Hollabaugh, RS, Young, JA, et al. Persistent mullerian duct syndrome. Urology. 1990; 36:516–518.
37. Gustafson, ML, Lee, MM, Asmundson, L, et al. Mullerian inhibiting substance in the diagnosis and management of intersex and gonadal abnormalities. J Pediatr Surg. 1993; 28:439–444.
38. Eil, C, Austin, RM, Sesterhenn, I, et al. Leydig cell hypoplasia causing male pseudohermaphroditism: Diagnosis 13 years after prepubertal castration. J Clin Endocrinol Metab. 1984; 58:441–448.
39. Lee, PA, Rock, JA, Brown, TR, et al. Leydig cell hypofunction resulting in male pseudohermaphroditism. Fertil Steril. 1982; 37:675–679.
40. Berkovitz, GD, Fechner, PY, Zacur, HW, et al. Clinical and pathologic spectrum of 46,XY gonadal dysgenesis: Its relevance to the understanding of sex differentiation. Medicine (Baltimore). 1991; 70:375–383.
41. Walker, AM, Walker, JL, Adams, S, et al. True hermaphroditism. J Paediatr Child Health. 2000; 36:69–73.
42. Verp, MS, Simpson, JL. Abnormal sexual differentiation and neoplasia. Cancer Genet Cytogenet. 1987; 25:191–218.
43. Hadjiathanasiou, CG, Brauner, R, Lortat-Jacob, S, et al. True hermaphroditism: Genetic variants and clinical management. J Pediatr. 1994; 125:738–744.
44. Davidoff, F, Federman, DD. Mixed gonadal dysgenesis. Pediatrics. 1973; 52:725–742.
45. Robboy, SJ, Miller, T, Donahoe, PK, et al. Dysgenesis of testicular and streak gonads in the syndrome of mixed gonadal dysgenesis: Perspective derived from a clinicopathologic analysis of twenty-one cases. Hum Pathol. 1982; 13:700–716.
46. Drash, A, Sherman, F, Hartmann, WH, Blizzard, RM. A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr. 1970; 76:585–593.
47. Edman, CD, Winters, AJ, Porter, JC, et al. Embryonic testicular regression. A clinical spectrum of XY agonadal individuals. Obstet Gynecol. 1977; 49:208–217.
48. Klinefelter, H, Jr., Reifenstein, E, Jr., Albright, F. Syndrome characterized by gynecomastia, aspermatogensis, without a-Leydigism and increased excretion of follicle stimulating hormone. J Clin Endocrinol Metab. 1942; 2:615.
49. Kitamura, M, Matsumiya, K, Koga, M, et al. Ejaculated spermatozoa in patients with non-mosaic Klinefelter’s syndrome. Int J Urol. 2000; 7:88–92.
50. Van Dyke, DC, Hanson, JW, Moore, JW, et al. Clinical management issues in males with sex chromosomal mosaicism and discordant phenotype/sex chromosomal patterns. Clin Pediatr. 1991; 30:15–21.
51. Griffin, JE, Edwards, C, Madden, JD, et al. Congenital absence of the vagina. The Mayer-Rokitansky-Kuster-Hauser syndrome. Ann Intern Med. 1976; 85:224–236.
52. Kaefer, M, Diamond, D, Hendren, WH, et al. The incidence of intersexuality in children with cryptorchidism and hypospadias: Stratification based on gonadal palpability and meatal position. J Urol. 1999; 162:1003–1006.
53. Achermann, JC, Ozisik, G, Meeks, JJ. Genetic causes of human reproductive disease. J Clin Endocrinol Metab. 2002; 87:2447–2454.
54. Meyer-Bahlburg, HF. Gender assignment and reassignment in intersexuality: Controversies, data, and guidelines for research. Adv Exp Med Biol. 2002; 511:199–223.
55. Money, J. Ablatio penis: Normal male infant sex-reassigned as a girl. Arch Sex Behav. 1975; 4:65–71.
56. Diamond, M, Sigmundson, HK. Sex reassignment at birth. Long-term review and clinical implications. Arch Pediatr Adolesc Med. 1997; 151:298–304.
57. Recommendations for Treatment: Intersex infants and children. Intersex Society of North America, 1995.
58. Hensle, TW, Bingham, J. Feminizing genitoplasty. Adv Exp Med Biol. 2002; 511:251–265.
59. Jordan, GH. Total phallic construction, option to gender reassignment. Adv Exp Med Biol. 2002; 511:275–280.
60. Sadove, RC, Sengezer, M, McRoberts, JW, et al. One-stage total penile reconstruction with a free sensate osteocutaneous fibula flap. Plast Reconstr Surg. 1993; 92:1314–1323.
61. De Castro, R, Merlini, E, Rigamonti, W, et al. Phalloplasty and urethroplasty in children with penile agenesis: Preliminary report. J Urol. 2007; 177:1112–1116.
62. Pippi Salle, JL, Braga, LP, Macedo, N, et al. Corporeal sparing dismembered clitoroplasty: An alternative technique for feminizing genitoplasty. J Urol. 2007; 178:1796–1800.
63. Minto, CL, Liao, LM, Woodhouse, CR, et al. The effect of clitoral surgery on sexual outcome in individuals who have intersex conditions with ambiguous genitalia: A cross-sectional study. Lancet. 2003; 361:1252–1257.
64. Pena, A. Total urogenital mobilization–an easier way to repair cloacas. J Pediatr Surg. 1997; 32:263–267.
65. Rink, RC, Pope, JC, Kropp, BP, et al. Reconstruction of the high urogenital sinus: Early perineal prone approach without division of the rectum. J Urol. 1997; 158:1293–1297.
66. Rink, RC, Adams, MC. Feminizing genitoplasty: State of the art. World J Urol. 1998; 16:212–218.
67. Pena, A, Filmer, B, Bonilla, E, et al. Transanorectal approach for the treatment of urogenital sinus: Preliminary report. J Pediatr Surg. 1992; 27:681–685.
68. Passerini-Glazel, G. A new 1-stage procedure for clitorovaginoplasty in severely masculinized female pseudohermaphrodites. J Urol. 1989; 142:565–568.
69. Hensle, TW, Dean, GE. Vaginal replacement in children. J Urol. 1992; 148:677–679.
70. Martinez-Mora, J, Isnard, R, Castellvi, A, et al. Neovagina in vaginal agenesis: Surgical methods and long-term results. J Pediatr Surg. 1992; 27:10–14.
71. Rink, R, Kaefer, M. Surgical management of intersexuality, cloacal malformations, and other abnormalities of the genitalia in girls. In: Walsh PC, Retk AB, Vaughn ED, Wein AJ, eds. Campbell’s Urology. 8th ed. Philadelphia: WB Saunders; 2002:2428–2468.
72. Frank, R. The formation of an artificial vagina without operation. Am J Obstet Gynecol. 1938; 35:1053.
73. Ingram, JM. The bicycle seat stool in the treatment of vaginal agenesis and stenosis: A preliminary report. Am J Obstet Gynecol. 1981; 140:867–873.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Disorders of Sexual Differentiation
Normal Gender and Sexual Differentiation
The most commonly accepted paradigm, described by Jost, involves a stepwise process to gender and sexual development.1 The primary determinant is the chromosomal gender, which is established at fertilization when the sperm provides an X or Y chromosome to the ovum’s X chromosome. Chromosomal gender determines gonadal gender, with XX resulting in ovarian development and XY resulting in testicular formation. Finally, the gonadal function determines the phenotypic gender. Although this paradigm is a helpful cascade to explain gender development, the simple Y = male, no Y = female equations are not always valid.
The testis determining factor (TDF) is located on the short arm of the Y chromosome near the centromere at the distal aspect of the Y-unique region.2 TDF is a 35 kb pair sequence on the 11.3 sub-band of the gender-determining region of the Y chromosome (SRY). Interestingly, SRY appears to be expressed by the somatic cells from the urogenital ridge and not from germ cells.
Many other genes play a role in gender development. Despite the presence of a functional SRY gene, the absence of the SOX9 gene results in a female phenotype in the majority of chromosomal males.3,4 The Wilms tumor gene (WT1) appears to play a key role not only in renal development, but also in testicular development. Early alteration of WT1 function results in testicular agenesis and later dysfunction results in aberrant testicular development (streak gonad or dysgerminomas). This tumor suppressor gene has been implicated in Denys–Drash syndrome involving testicular (mixed gonadal dysgenesis) and renal (Wilms tumor) abnormalities.5,6
Fushi–Tarzu factor-1 (FTZ-F1) exerts its effect on gonadal development through its regulation of steroidogenic factor-1 (SF-1). The SF1 gene is involved with steroid hormone production and the production of Müllerian-inhibiting substance (MIS) by the Sertoli cells of the testis that causes regression of the Müllerian ductal system. Although FTZ-F1 and SF-1 are also expressed in ovarian tissues, the timing and intensity of their effect are critical for normal gonadal development.5,7
Finally, the lack of an SRY gene alone does not impart normal female phenotypic and gonadal development. The DAX1 gene appears to be essential for the development of the ovary. The DAX1 gene product appears to compete with the SRY gene product for a steroidogenic regulatory protein (StAR). A dosage-sensitive element is also important. Normally, the single SRY gene has a greater impact than a single DAX1 gene and causes upregulation of StAR. However, in those chromosomal abnormalities in which more than one DAX1 gene is present, downregulation of StAR occurs, testicular development is inhibited, and ovarian development is promoted. As in the case of Turner syndrome, these primordial ovaries develop into streak gonads. Likely, other genes are also important for normal ovarian development.8–10
Development of the internal ductal structures is dependent on hormone secretion by the developing gonads (Table 62-1). In the absence of functioning testicular tissue, the female internal Müllerian duct structures develop. The presence of a functioning testis results in male internal Wolffian duct development. This differentiation is mediated by the production of testosterone from the testis. Testosterone promotes Wolffian duct development along with MIS, which results in regression of the Müllerian duct structures. This is a paracrine effect and therefore results in gonad specific ipsilateral ductal differentiation. This effect is likely dependent on high concentrations of androgen produced by the physically proximate gonad. Decreased levels of MIS by an abnormal testis or streak gonad result in ipsilateral Müllerian development. This occurs despite regression of the Müllerian ducts on the contralateral side with normal testicular MIS production. Conversely, systemic administration of androgen does not result in male ductal development in a female fetus.
Produced by Sertoli cells, MIS functions as a suppressor of Müllerian duct development and is a specific marker for functioning testicular tissue in infancy. In its absence, the Müllerian structures develop. The concentration and timing of MIS secretion appear to be critical. Normally, secretion occurs during week 7 of gestation. By week 9, the Müllerian ducts become insensitive to MIS.9
External genital development follows a similar path (Fig. 62-1). In the absence of the testosterone metabolite dihydrotestosterone (DHT), the external genitalia develop into the female phenotype. The male and female phenotypes are identical until week 7. In the male, testosterone production by the testicular Leydig cells surges at 7 weeks and remains elevated until week 14 of gestation. Testosterone is converted to DHT by 5α-reductase in the tissues of the genital skin and urogenital sinus. The testosterone-binding receptor has much higher affinity for DHT than testosterone and serves to amplify the effect of testosterone on the developing external genitalia. In the absence of 5α-reductase, the internal Wolffian ducts are preserved, but the external structures are feminized.
Aberrant Gender Development
Incidence
In the Americas and Western Europe, congenital adrenal hyperplasia (CAH) is the most common cause of neonatal ambiguous genitalia, accounting for approximately 70% of cases.11,12 The overall incidence is approximately 1 in 15,000 live births. The rate is much higher in stillborns and in certain regional populations (Yupic Eskimos and the people of La Réunion, France).13 In the United States, mixed gonadal dysgenesis is the next most common intersex disorder, with ovotesticular DSD (true hermaphroditism) being the third most common.
Classification
Until recently, the most commonly used classification system was the one proposed by Allen in 1976 and was based primarily on gonadal histology.14 This system categorized the most common DSD well, but did not accommodate less common syndromes easily. Also, the older ‘hermaphrodite’ terminology has become offensive to some. A newer classification released by the International Consensus Conference on Intersex has largely replaced Allen’s system. This newer system incorporates an evolving understanding of the molecular basis of these disorders and replaces more offensive gender-based labels. The new terminology is used primarily in this text.15
46,XX DSD (Female Pseudohermaphrodite)
The majority of neonates with external genital ambiguity fall into this category. All patients have a 46,XX genotype and exclusively ovarian tissue in nonpalpable gonads. Simplistically, the cause of the gender ambiguity is an excess of androgen. More than 95% are due to CAH, with the remainder due to maternal androgen exposure. These patients have a normal female Müllerian ductal system with an upper vagina, uterus, and fallopian tubes (see Table 62-1). They also have normal regression of the Wolffian ducts. The level of virilization is largely dependent on the timing and magnitude of androgen exposure to the external genitalia. The phenotype can range from mild clitoromegaly to a normal male appearance.
Virilization in CAH is due to the inability of the adrenal gland to form cortisol. The precursors above the enzymatic defect are shunted into the mineralocorticoid or sex-steroid pathways. Also, the end products generally have some, albeit weak, glucocorticoid function. The lack of cortisol for negative feedback inhibition of adrenocorticotropic hormone (ACTH) production by the pituitary leaves this pathway unchecked. Excess androgen is produced and is responsible for the virilization. The corticosteroid synthetic and alternative pathways are shown in Figure 62-2. The most common form of CAH is 21-hydroxylase deficiency (21-OHD), which accounts for more than 90% of CAH.16 21-OHD has been mapped to the short arm of chromosome 6. The variable location of the adrenal defect and relative function of the gene results in salt-wasting and nonsalt-wasting forms.17–19 Type 1 results in virilization but no salt wasting. The gene defect affects only the fasciculata zone of the adrenal, resulting in blocking cortisol production. However, the gene is normally expressed in the glomerulosa zone with preservation of mineralocorticoid production. In type 2, also called the classic type, the gene abnormality affects both adrenal zones. Salt wasting results in dehydration or vascular collapse, and hyperkalemia develops because of the block in mineralocorticoid production.
FIGURE 62-2 Pathways of steroid biosynthesis. The numbers correspond to CAH type and the location of the enzyme defect (see the text).
Virilization of the female fetus can be caused by exogenous androgen exposure from the mother. This occurs primarily with the use of progesterone, commonly used as an adjunct to assist with fertility and in-vitro fertilization. Endogenous androgen exposure due to virilizing maternal ovarian tumors also has been reported, but these tumors are usually virilizing to the mother with the fetus being unaffected.20
Treatment
As all forms of CAH are inherited in an autosomal recessive manner, genetic counseling is recommended. Families with a history of CAH should consider maternal treatment with dexamethasone before week 10 of gestation to eliminate or improve the level of fetal virilization.21 Postnatally, cortisol replacement with hydrocortisone is the mainstay of therapy, with the addition of fluorohydrocortisone if salt wasting is present. Supportive management of fluid and electrolyte abnormalities is best provided in a neonatal intensive care unit.
With regard to gender identity, the vast majority of patients with CAH identify as female and gender assignment is generally female given the 46,XX karyotype. The ovaries are normal and have fertility potential.22 Surgical reconstruction requires a feminizing genitoplasty, and involves clitoroplasty, monsplasty, and vaginoplasty.
46,XY DSD (Male Pseudohermaphrodite)
Defects in 17,20-desmolase and 17β-hydroxysteroid oxidoreductase act at the testicular level to convert androstenedione to testosterone. Because the adrenal is unaffected, CAH does not occur. The phenotype can be quite variable, but those with complete feminization can escape detection at birth and be reared as female. Progressive virilization is related to excess gonadotropin production at puberty, which may partially compensate for the lack of testosterone synthesis. Phallic growth and the development of male secondary sex characteristics create a conundrum with regard to gender reassignment when the diagnosis is made later in life.23
The extreme is normal female external genitalia resulting from complete androgen insensitivity syndrome (CAIS, also termed testicular feminization). The incidence of this syndrome is approximately 1 in 40,000. It usually results from a point mutation in the androgen receptor gene, located on the X chromosome.24,25
Partial androgen insensitivity is associated with a large spectrum of phenotypic variation (e.g., Gilbert–Dreyfus, Lub, and Reifenstein syndromes). It can be a sporadic or inherited condition, and gender assignment and treatment are individualized.26
Diagnosis
Metabolically, the diagnosis of CAH is made similarly to the 46,XX DSD patient, noting excess steroid levels above the enzymatic block and elevated levels of ACTH. The physical examination confirms absence of a cervix on rectal examination, and bronzing of the skin may be present. Palpation of a cryptorchid or descended testis is possible. The genitogram and ultrasound mirror these findings, but a prominent utricle may be present that lacks a cervical impression at its apex. In CAIS, testosterone levels are elevated postpubertally, but the diagnosis in the prepubertal child may require human chorionic gonadotropin (hCG) stimulation and genital skin fibroblast androgen receptor studies. Receptor assays can delineate a quantitative versus qualitative receptor defect. LH levels are elevated, related to the loss of testosterone feedback inhibition, which requires normal receptor hormone interaction. 5α-reductase deficiency is confirmed by an elevated testosterone-to-DHT ratio and an abnormal 5α-reductase type 2 gene.27
Treatment
In CAIS, the gender assignment is always female. CAIS patients who are assigned as female in infancy later identify themselves as female.28 Because the androgen receptor defect is ubiquitous, virilization of the brain does not occur. Orchiectomy is required, given the risk of malignant degeneration, but is often deferred until after puberty.29 The testis synthesizes estradiol, facilitating feminine development at puberty. Orchiectomy before puberty necessitates hormone replacement for normal pubertal development.
Gender assignment in partial androgen insensitivity syndrome (PAIS) is largely based on the response of the external genitalia to exogenous testosterone. A significant virilization response argues for the male gender. If there is no response, the female gender is favored. This subgroup is the most variable and has the least consensus with regard to gender assignment. There are reports of gender reassignment at puberty.30,31 Dissatisfaction with the gender of rearing occurs in approximately 25% of PAIS patients, whether raised male or female.32 In 5α-reductase deficiency syndrome, the brain is normally virilized and these individuals identify with the male gender. Thus, male gender assignment is recommended.33
Müllerian-Inhibitory Substance Deficiency (Hernia Uterine Inguinale)
MIS is produced by the Sertoli cells in the testis and causes regression of the Müllerian ductal structures. In this rare syndrome of abnormal MIS production or MIS-receptor abnormality, Wolffian ductal development is unimpaired, but the Müllerian ducts also persist. Because the infant has a normal male phenotype, this syndrome is rarely encountered in the neonatal period. The most common presentation to the surgeon is that of finding a fallopian tube adjacent to an undescended testis in the hernia sac at the time of orchiopexy.34
If this scenario is encountered, a biopsy of the gonad should be performed, the hernia should be repaired, and all structures left intact until completion of a full evaluation with karyotype and MIS levels. Apparent males can also present with bilateral nonpalpable testes, and Müllerian structures are found at laparoscopy (Fig. 62-3
[/not-level-membership-for-pediatrics-category]
