[level-membership-for-pediatrics-category]
50 Disorders of Red Blood Cells
Etiology and Pathogenesis
Congenital Red Blood Cell Disorders
Thalassemias
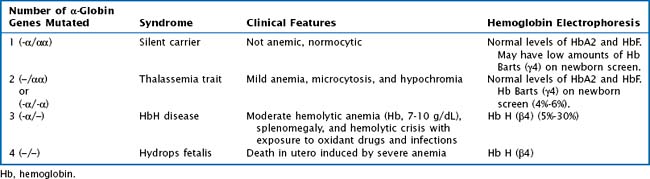
Whereas α -thalassemia is more commonly found in Southeast Asia, β-thalassemia is more common in Mediterranean countries. There are two α-globin genes located on chromosome 16. α-Thalassemias are usually the result of large gene deletions, causing a reduction in α-globin production. The severity of disease is directly related to the number of genes involved (Table 50-1).
Hemoglobinopathies
The hemoglobinopathies are a group of autosomal recessive inherited disorders characterized by synthesis of abnormal Hb molecules (i.e., S, and C). The most common and severe hemoglobinopathy is sickle cell disease, in which only HbS is produced. Chapter 53 of this book is dedicated to the in-depth discussion of sickle cell disease.
Acquired Red Blood Cell Disorders
Transient Red Blood Cell Aplasia from Parvovirus B19
Infection with parvovirus causes a reticulocytopenia for approximately 7 to 10 days. Patients with congenital RBC disorders with increased RBC turnover and decreased RBC life span are at risk for developing a significant anemia during the period of acquired reticulocytopenia (see Chapter 53). Clinically, patients can present with pallor, headache, and a marked decrease in Hb level. The hallmark is a low reticulocyte count, indicating suppression of bone marrow activity. Blood transfusion is indicated in patients with significant anemia who are symptomatic.
Clinical Presentation and Differential Diagnosis
The differential diagnosis of anemia is diverse. Physiologically, anemia can be divided into three categories: decreased or ineffective RBC production, premature RBC destruction, and blood loss. Causes of anemia can be further subdivided based on a morphologic approach using the reticulocyte index (see Diagnostic Approach) and the mean corpuscular volume (MCV) (Figure 50-1).
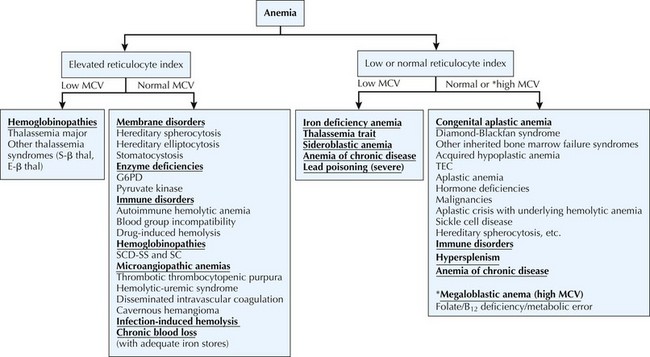
Diagnostic Approach
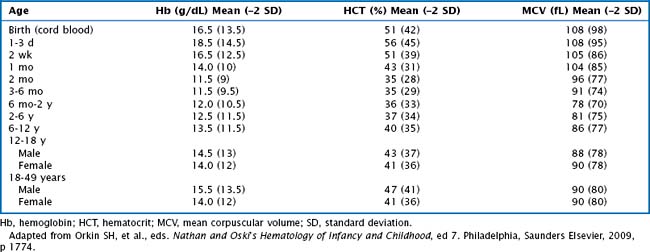
Anemia is defined as a decrease in the Hb concentration more than 2 standard deviations below the mean for age (Table 50-2). The MCV and reticulocyte count are helpful measurements in categorizing anemia. The MCV provides a quick, accurate, and readily available method of distinguishing the microcytic anemias (iron deficiency, thalassemia syndromes) from the normocytic (membrane disorders, enzyme deficiencies, AIHA, most hemoglobinopathies) or macrocytic (bone marrow or stem cell failure, disorders of vitamin B12, and folic acid absorption or metabolism) anemias. The MCV varies with age, necessitating the use of age-adjusted normal values (see Table 50-2).
After the cause of the anemia has been further categorized into broad disease categories, specific tests can be drawn. Table 50-3 lists specific diagnostic tests and their associated conditions.
| Diagnostic Test | Disease |
|---|---|
| DAT or Coombs test | AIHA |
| Hemoglobin electrophoresis | Sickle cell disease |
| Thalassemia | |
| RBC enzyme assays | G6PD deficiency |
| PK deficiency | |
| Osmotic fragility test | Hereditary spherocytosis |
| Iron studies | Iron-deficiency anemia |
| Folate, vitamin B12 | Macrocytic or megaloblastic anemia |
| Bone marrow aspiration and biopsy | Myelodysplastic syndrome |
| Aplastic anemia | |
| Malignancy | |
| Diamond-Blackfan anemia | |
| ADAMTS13 activity and inhibitor level | TTP |
| Chromosomal breakage analysis | Fanconi’s anemia |
AIHA, autoimmune hemolytic anemia; DAT, direct antiglobulin; G6PD, glucose-6-phosphate dehydrogenase; PK, pyruvate kinase; RBC, red blood cell; TTP, thrombotic thrombocytopenic purpura.
Brugnara C, Oski FA, Nathan DG. Diagnostic approach to the anemic patient. In: Orkin SH, Nathan DG, Ginsburg D, et al, editors. Hematology of Infancy and Childhood. Philadelphia: Saunders; 2009:455-466.
Rund D, Rachmilewitz E. β-Thalassemia. N Engl J Med. 2005;353(11):1135-1145.
Segel GB. Anemia. Pediatr Rev. 1988;10(3):77-88.
Shah S, Vega R. Hereditary spherocytosis. Pediatr Rev. 2004;25(5):168-172.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
50 Disorders of Red Blood Cells
Etiology and Pathogenesis
Congenital Red Blood Cell Disorders
Thalassemias
Whereas α -thalassemia is more commonly found in Southeast Asia, β-thalassemia is more common in Mediterranean countries. There are two α-globin genes located on chromosome 16. α-Thalassemias are usually the result of large gene deletions, causing a reduction in α-globin production. The severity of disease is directly related to the number of genes involved (Table 50-1).
Hemoglobinopathies
The hemoglobinopathies are a group of autosomal recessive inherited disorders characterized by synthesis of abnormal Hb molecules (i.e., S, and C). The most common and severe hemoglobinopathy is sickle cell disease, in which only HbS is produced. Chapter 53 of this book is dedicated to the in-depth discussion of sickle cell disease.
