[level-membership-for-hematology-oncology-and-palliative-medicine-category]35
Disorders of platelet function and vascular purpuras
Laboratory testing of platelet function
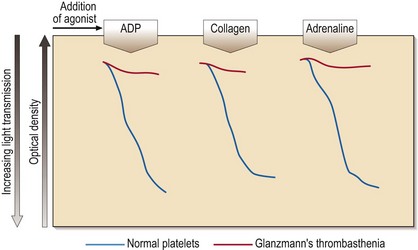
Platelet aggregation studies assess the ability of platelets to aggregate in response to the addition of a variety of agonists (e.g. ADP, adrenaline (epinephrine), collagen, arachidonic acid, ristocetin). The tracings produced (Fig 35.1) require expert interpretation. The methodology remains the gold standard with the response to agonists giving characteristic patterns in inherited disorders. Other tests of platelet function include flow cytometry for the quantitation of glycoprotein receptor density, and the measurement of total and/or released adenine nucleotides. The latter tests may confirm the findings from platelet aggregation studies (e.g. in Bernard–Soulier syndrome) or reveal abnormalities where aggregation studies are normal or equivocal (e.g. in a storage pool disease or release defect).
Inherited disorders of platelet function
Glanzmann’s thrombasthenia
This rare autosomal recessive disease is also caused by loss or dysfunction of a platelet glycoprotein complex – GP IIb/IIIa. This normally acts as a receptor for adhesive proteins such as fibrinogen and von Willebrand factor. Platelet numbers and morphology are normal but the platelets fail to aggregate with all agonists except ristocetin (see Fig 35.1). Clinical manifestations are variable but there is typically onset in the neonatal period and subsequent cutaneous and gastrointestinal bleeding, and menorrhagia. Platelet transfusions are indicated where local haemostatic measures fail. If there is platelet refractoriness, recombinant factor VIIa can be used.
Acquired disorders of platelet function
These disorders are common. Causes include foods, drugs, systemic disorders and diseases of the blood (Table 35.1).
Table 35.1
Causes of abnormal platelet function
| Inherited | Bernard–Soulier syndrome |
| Glanzmann’s thrombasthenia | |
| Storage pool disorders | |
| Release defects | |
| Other (e.g. von Willebrand disease) | |
| Acquired | Drugs (e.g. aspirin) |
| Foods (e.g. garlic) | |
| Chronic renal failure | |
| Cirrhosis | |
| Cardiopulmonary bypass surgery | |
| Blood diseases: acute myeloid leukaemia, myelodysplastic syndromes, myeloproliferative disorders, myeloma | |
| Various systemic disorders1 |
1These include disseminated intravascular coagulation (DIC) and thrombotic thrombocytopenic purpura (TTP).
Vascular purpuras
A bleeding tendency caused by a local or general vascular abnormality is referred to as a vascular purpura (Table 35.2). Diagnosis of these diseases is made mainly on clinical grounds with laboratory exclusion of other haemostatic defects.
Table 35.2
| Inherited | Hereditary haemorrhagic telangiectasia1 |
| Connective tissue diseases | |
| Ehlers–Danlos syndrome | |
| Pseudoxanthoma elasticum | |
| Marfan syndrome | |
| Acquired | Henoch–Schönlein purpura |
| Various infections | |
| Drug reactions (allergic purpuras) | |
| Senility | |
| Prolonged corticosteroid treatment | |
| Scurvy | |
| Mechanical |
Inherited disorders
Hereditary haemorrhagic telangiectasia (HHT)
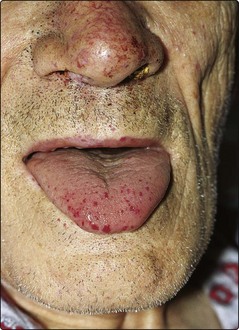
The hallmark of this rare autosomal dominant disease is the development of multiple ateriovenous malformations (AVMs). Small AVMs are referred to as telangiectasia. Close to the surface of the skin and mucous membranes, they often rupture and bleed. Two mutated genes, endoglin and ALK1, have been implicated. Clinical problems include recurrent epistaxes (90% of cases), gastrointestinal haemorrhage, haematuria and larger pulmonary arteriovenous malformations (PAVMs). Chronic bleeding from the gut causes iron deficiency anaemia. On examination there are the characteristic telangiectasia (Fig 35.2). Management includes local control of bleeding (e.g. laser treatment of telangiectasia), iron supplements and embolisation of PAVMs.
Acquired disorders
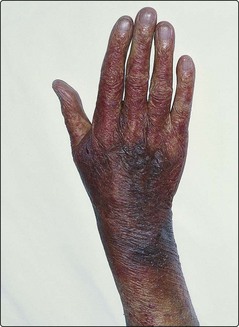
This is a very heterogeneous group. Henoch–Schönlein purpura is a syndrome usually seen in childhood where an itchy purpuric rash typically follows an infection. Spontaneous remission is the rule but renal failure may result. Other causes of acquired purpuric rashes include infections, drug reactions, scurvy, trauma, prolonged steroid therapy and simple old age (senile purpura, Fig 35.3).





[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]35
Disorders of platelet function and vascular purpuras
Laboratory testing of platelet function
Platelet aggregation studies assess the ability of platelets to aggregate in response to the addition of a variety of agonists (e.g. ADP, adrenaline (epinephrine), collagen, arachidonic acid, ristocetin). The tracings produced (Fig 35.1) require expert interpretation. The methodology remains the gold standard with the response to agonists giving characteristic patterns in inherited disorders. Other tests of platelet function include flow cytometry for the quantitation of glycoprotein receptor density, and the measurement of total and/or released adenine nucleotides. The latter tests may confirm the findings from platelet aggregation studies (e.g. in Bernard–Soulier syndrome) or reveal abnormalities where aggregation studies are normal or equivocal (e.g. in a storage pool disease or release defect).
Inherited disorders of platelet function
Glanzmann’s thrombasthenia
This rare autosomal recessive disease is also caused by loss or dysfunction of a platelet glycoprotein complex – GP IIb/IIIa. This normally acts as a receptor for adhesive proteins such as fibrinogen and von Willebrand factor. Platelet numbers and morphology are normal but the platelets fail to aggregate with all agonists except ristocetin (see Fig 35.1). Clinical manifestations are variable but there is typically onset in the neonatal period and subsequent cutaneous and gastrointestinal bleeding, and menorrhagia. Platelet transfusions are indicated where local haemostatic measures fail. If there is platelet refractoriness, recombinant factor VIIa can be used.
