111 Disorders of Plasma Potassium Concentration
Dyskalemias are common electrolyte disorders in the critical care setting that may predispose a patient to serious cardiac arrhythmias.1 The pathophysiology of these electrolyte disturbances can be more easily understood if examined in the context of the major concept for the transport of potassium ions (K+) across membranes. This process has two components, an open channel for K+ in the cell membrane and a force to cause K+ to move across cell membranes.
 Potassium Channels
Potassium Channels
Clinical Examples
Sulfonylurea drugs stimulate the release of insulin.2 They act by diminishing the open probability of the KATP channels. When fewer K+ ions exit from pancreatic β cells, the ICF voltage becomes less negative. This causes voltage-gated calcium ion (Ca2+) channels to open, and thereby the concentration of Ca2+ in the ICF rises, which provides a signal for the release of insulin from these cells. By virtue of a similar cascade of events, sulfonylurea drugs can cause vasoconstriction by raising the concentration of Ca2+ in the ICF in vascular smooth muscle and hence can be used to improve hemodynamics in patients with septic shock3 (Figure 111-1).
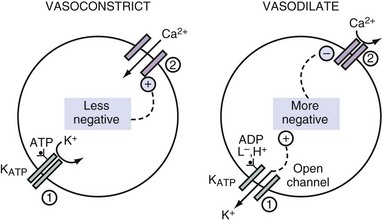
 Driving Forces
Driving Forces
K+ will move into a compartment that has a more negative voltage when K+ channels are open. To create this negative voltage in cells, cations are exported at a faster rate than anions. The cations are usually sodium ions (Na+) because of their abundance and the presence of a means to cause their movement out of cells, the activity of the electrogenic Na+/K+-ATPase. This ion pump is electrogenic because it exports 3 Na+ while importing only 2 K+ (Figure 111-2). Because Na+ movement is not accompanied by movement of ICF anions (because macromolecular phosphates such as RNA, DNA, and phospholipids are impermeable), a negative intracellular voltage is generated. Open KCHJ10 K+ ion channels in the immediate vicinity of the Na+/K+-ATPase in the plasma membrane serve the purpose of providing K+ to the K+ binding site of the Na+/K+-ATPase to permit continuing function of this critical electrogenic system (see Figure 111-2).
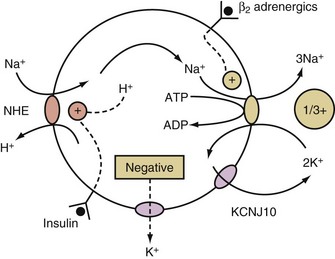
 Regulation of Potassium Homeostasis
Regulation of Potassium Homeostasis
Distribution of Potassium Between Extracellular and Intracellular Fluid Compartments
Electroneutral Entry of Sodium into Cells
This occurs when Na+ enters cells in exchange for hydrogen ions (H+) via the Na+/H+ exchanger (NHE) (see Figure 111-2).4 The NHE is normally inactive in cell membranes, as can be deduced from the fact that it catalyzes an electroneutral exchange and that the concentrations of its substrates (Na+ in the ECF and H+ in the ICF compartment) are considerably higher than that of its products (Na+ in the ICF and H+ in the ECF compartment) in steady state. The two major activators of NHE are insulin and a higher concentration of H+ in the ICF compartment (Figure 111-2).
Hormones that Affect the Distribution of Potassium
Catecholamines
β2-Adrenergic agonists activate the Na+/K+-ATPase via a cAMP-dependent mechanism that leads to phosphorylation of this ion pump5 and the export of preexisting intracellular Na+. Therefore, hypokalemia may develop in conditions where there is a surge of catecholamines (e.g., patients with a subarachnoid hemorrhage, myocardial ischemia, and/or an extreme degree of anxiety). β2-Agonists may be used to cause a shift of K+ into cells in the emergency treatment of patients with hyperkalemia. On the other hand, non-selective β-blockers have been used in the treatment of patients with thyrotoxic hypokalemic periodic paralysis and are a potential therapy for other conditions of acute hypokalemia due to shift of K+ into cells owing to a surge of catecholamines.
Insulin
The effect of insulin to shift K+ into cells is due primarily to an augmentation of the electroneutral entry of Na+ into cells via NHE.4 This, in conjunction with stimulating the electrogenic Na+/K+-ATPase, causes the voltage in cells to become more negative (see Figure 111-2). This effect of insulin is utilized clinically in the emergency treatment of patients with hyperkalemia.6,7
Acid-Base Influences
Acids That May Cause a Shift of Potassium Into Cells
When an acid is added to the body, most of its H+ are buffered in the ICF compartment.8 Only monocarboxylic acids, however, can enter cells via a specific transporter, and this is an electroneutral process.9 Once a monocarboxylic acid such as L-lactic acid enters cells on this transporter, its H+ are released, and if this occurs in close approximation to NHE in the cell membrane, it becomes activated, and the net result is the electroneutral entry of Na+ into these cells, which causes a rise in their intracellular concentration of Na+. This in turn causes more Na+ and positive voltage to exit from cells. The net result is the generation of a more negative voltage, which causes the retention of K+ in these cells (Figure 111-3).10
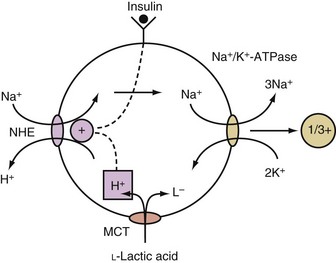
Acids That May Cause a Shift of Potassium Out of Cells
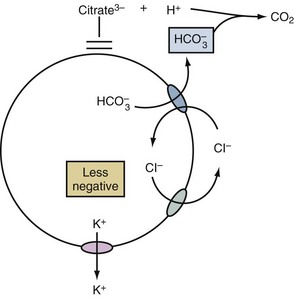
A shift of K+ out of cells may occur in patients with metabolic acidosis due to acids that are not substrates for the monocarboxylic acid transporter (e.g., HCl, citric acid). In this setting, the mechanism begins with the net exit of bicarbonate ions (HCO3−) from cells.11 This exit is an electroneutral process because it occurs on the Cl−/HCO3− anion exchanger (AE) (Figure 111-4). Nevertheless, the process becomes electrogenic because it results in a rise in the concentration of Cl− in the ICF compartment. Since virtually all cells have Cl− channels in their cell membranes,12 the usual negative voltage forces some of these Cl− to exit cells in an electrogenic fashion. As a result of the less negative voltage inside these cells, more K+ will exit.13
Clinical Pearls
Although the addition of inorganic acids (e.g., HCl) causes a shift of K+ out of cells, patients with chronic hyperchloremic metabolic acidosis (e.g., patients with chronic diarrhea or those with renal tubular acidosis [RTA]) usually have a low PK because of excessive loss of K+ in the diarrhea fluid14 or in the urine.15 Although hypokalemia is a common finding in patients with metabolic alkalosis,16 this usually reflects renal K+ wasting for the most part due to the underlying disorder (e.g., vomiting, diuretic use, primary hyperaldosteronism) rather than the small effect of alkalemia to shift K+ into cells. Respiratory acid-base disorders cause only small changes in the PK, because there is little movement of Na+ across cell membranes in these disorders.17
Tissue Anabolism/Catabolism
Hypokalemia may develop in conditions with rapid cell growth if insufficient K+ is given. Examples include the use of total parenteral nutrition (TPN), rapidly growing malignancies, and during treatment of DKA or pernicious anemia. On the other hand, hyperkalemia may be seen in patients with crush injury or tumor lysis syndrome.18 In these patients, factors that compromise the kidney’s ability to excrete K+ are usually present. In patients with DKA, there is total body K+ depletion,19 but hyperkalemia is present because there is a shift of K+ from cells secondary to a lack of insulin. The corollary is that during therapy, complete replacement of the deficit of K+ must await the provision of cellular constituents (phosphate, amino acids, Mg2+, etc.) and the presence of anabolic signals.
 Long-Term Regulation of Potassium Homeostasis
Long-Term Regulation of Potassium Homeostasis
Control of the renal excretion of K+ maintains overall daily K+ balance. Although the usual intake of K+ in adults eating a typical western diet is close to 1 mmol/kg body weight, K+ excretion can decline to a nadir of 10 to 15 mmol/d when there is virtually no K+ intake,20 whereas the rate of excretion of K+ can match an intake of more than 200 mmol/d with only a minor rise in the PK.
Control of K+ excretion occurs primarily in the late distal convoluted tubule up to the end of the cortical collecting duct (the abbreviation CCD will be used in this chapter to indicate all of these nephron segments).19 There are two components that affect the rate of excretion of K+: the flow rate in the CCD and the net secretion of K+ by principal cells in the CCD. It is the latter which adjusts the luminal concentration of K+ ([K+]CCD) and thereby regulates the rate of excretion of K+:
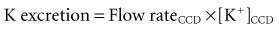
Flow Rate in the LATE CORTICAL DISTAL NEPHRON
When vasopressin acts, the flow rate in the CCD is determined by the rate of delivery of osmoles, because the osmolality of fluid in the terminal CCD is fixed (equal to the plasma osmolality (Posm)21:
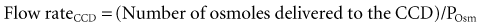
The major osmoles in the lumen of CCD are Na+, Cl−, and urea. Owing to urea recycling within the nephron, almost 75% of osmoles delivered to the CCD are urea (see Reference 22 for more detailed information).
Clinical Example
A patient with HIV and pneumocystis carinii pneumonia is treated with trimethoprim and develops hyperkalemia.23 Because his dietary intake is poor, the rate of delivery of osmoles (mainly urea) to the CCD is low, which means that the flow rate in his CCD is also diminished. This increases the concentration of trimethoprim in the lumen of the CCD (same quantity of trimethoprim is now contained in a smaller volume). Hence the ability of trimethoprim to block epithelial Na+ channels (ENaC) in principal cells in the CCD will be enhanced.24 Furthermore, in the presence of diminished ability to secrete K+ in the CCD owing to a less negative TE luminal voltage, the low flow rate in CCD will further compromise the ability to excrete K+. Increasing the rate of delivery of Na+ and Cl− with a loop diuretic can help augment the rate of excretion of K+ by increasing the flow rate in the CCD. Of greater importance, it will lower the concentration of trimethoprim in the luminal fluid in the CCD, and hence trimethoprim becomes less effective in blocking ENaC.25
Potassium CONCENTRATION in the Lumen of the LATE Cortical DISTAL NEPHRON
The secretory process for K+ in principal cells has two elements. First, a lumen negative voltage must be generated via electrogenic reabsorption of Na+ via ENaC. Actions of aldosterone increase the number of open ENaC. The steps for aldosterone action include its binding to the cytoplasmic aldosterone receptor in principal cells, entry of this hormone-receptor complex into the nucleus, and then the synthesis of new proteins including the serum and glucocorticoid regulated kinase (SGK).26 SGK phosphorylates and inactivates Nedd4-2 (Figure 111-5). As a result, this increases the number of open ENaC units in the luminal membrane of principal cells in the CCD. Second, open K+ channels must be present in the luminal membranes of principal cells in the CCD. K+ channels (ROMK) are abundant and have a high open probability in the absence of hypokalemia, and therefore they do not seem to be rate limiting for net secretion of K+ in most patients.
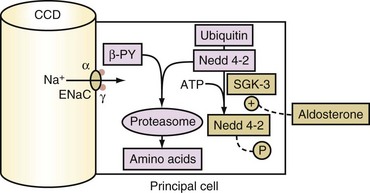
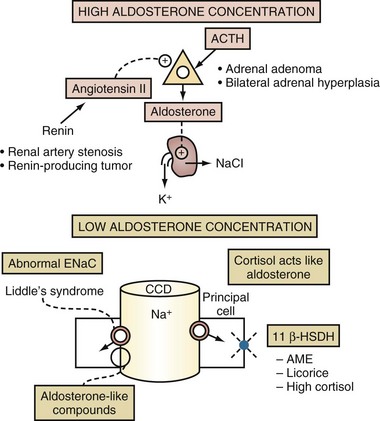
Glucocorticoids do not usually stimulate the secretion of K+ in the CCD because principal cells have a pair of enzymes called 11β-hydroxysteroid dehydrogenase (11β-HSDH). These enzymes convert cortisol to a metabolite (cortisone) that does not bind to the mineralocorticoid receptor (see Figure 111-5). Cortisol, however, can exert a mineralocorticoid effect if the activity of 11β-HSDH is decreased or if it is overwhelmed by an abundance of cortisol.
Under most circumstances, variations in the concentration of Na+ in the luminal fluid in the CCD does not regulate the secretion of K+.27 The reabsorption of Na+ in the CCD can be electroneutral or electrogenic, depending on whether the same quantity of Cl− (electroneutral) or a smaller quantity of Cl− (electrogenic) is reabsorbed as compared to Na+. The pathway(s) for the reabsorption of Cl− in the CCD is (are) not well defined, but it is likely that paracellular pathways play an important role.28,29
Reabsorbing less Cl− than Na+ in the CCD can occur for three reasons, as depicted in Figure 111-6. First, Na+ is delivered to the CCD with little Cl−. A key finding in these patients is a Cl−-poor urine.30 Second, reabsorption of Cl− in the CCD may be inhibited; this mechanism is suspected when the urine is not Cl−-poor. It appears that HCO3− and/or an alkaline luminal pH in the CCD may inhibit Cl− reabsorption31 (see Figure 111-6, middle panel). Third, a greater lumen-negative voltage in the CCD could develop when the delivery of Na+ and Cl− are very high and if the capacity for Cl− reabsorption is less than that for Na+. This requires a stimulated reabsorption of Na+ via ENaC in the CCD (see Figure 111-6, right panel).
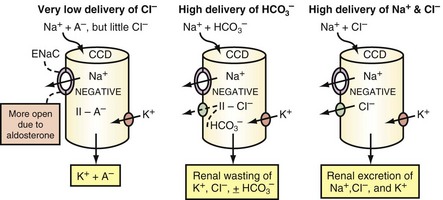
If there are near-equal rates of absorption of Na+ and Cl− in the CCD, an appreciably greater lumen-negative voltage cannot be generated, and hyperkalemia will develop if the intake of K+ remains high.28,32
 Tools to Assess Control of Renal Excretion of Potassium
Tools to Assess Control of Renal Excretion of Potassium
Examine Rate of Excretion Of Potassium
To assess the renal response in a patient with hypokalemia or hyperkalemia, we use the expected rate of K+ excretion when these electrolyte abnormalities are due to nonrenal causes. With a K+ deficit, the expected response is to excrete less than 15 mmol of K+/d.20,45 With a surfeit of K+, the expected response is to excrete greater than 200 mmol/d, values observed in response to a K+ load with a minor increase in PK.33
To assess the rate of excretion of K+, a 24-hour urine collection is not necessary. One can use the UK/UCreatinine ratio in a spot urine sample even though there is a diurnal variation in K+ excretion,21 because creatinine is excreted at a near-constant rate throughout the day.34 Moreover, the UK/UCreatinine in spot urine samples provides more relevant information because it can be evaluated relative to the PK at that time. The expected UK/UCreatinine ratio in a patient with hypokalemia is less than 1 mmol K+/mmol creatinine (less than 10 mmol K+/g creatinine), whereas in a patient with hyperkalemia, the expected UK/UCreatinine ratio is greater than 15 mmol K+/mmol creatinine (greater than 150 mmol K+/g creatinine).
Establish Basis for Abnormal Rate of Excretion of Potassium
In a patient with hypokalemia, a higher than expected rate of excretion of K+ implies that the lumen-negative voltage is abnormally more negative and that open luminal K+ channels (likely ROMK) are present in the luminal membranes of the CCD.39 The greater lumen negative voltage is due to reabsorbing more Na+ than Cl− per unit time in the CCD. The converse is true in a patient with hyperkalemia where there is a lower than expected rate of excretion of K+.
The clinical indices that help in the differential diagnosis of the pathophysiology of the abnormal rate of electrogenic reabsorption of Na+ in CCD are an assessment of the ECF volume and the ability to conserve Na+ and Cl− in response to a contracted effective arterial blood volume. The measurement of the activity of renin (PRenin) and the level of aldosterone in plasma (PAldosterone) are also helpful in this setting (Box 111-1).35
Box 111-1
Plasma Renin and Aldosterone Values to Assess the Basis of Hypokalemia or Hyperkalemia
| Lesions That Cause Hypokalemia | ||
| Renin | Aldosterone | |
| Adrenal Gland | ||
| Primary hyperaldosteronism | Low | High |
| Glucocorticoid remediable hyperaldosteronism | Low | High |
| Kidney | ||
| Renal artery stenosis | High | High |
| Malignant hypertension | High | High |
| Renin-secreting tumor | High | High |
| Liddle’s syndrome | Low | Low |
| Disorders involving 11β-HSDH | Low | Low |
| Lesions That Cause Hyperkalemia | ||
| Adrenal Gland | ||
| Addison’s disease | High | Low |
| Kidney | ||
| Pseudohypoaldosteronism type 1 | High | High |
| Hyporeninemic hypoaldosteronism | Low | Low |
Hyperkalemia
 Therapy of Hyperkalemia
Therapy of Hyperkalemia
Medical Emergencies
The major danger of a severe degree of hyperkalemia is a cardiac arrhythmia. Because mild electrocardiographic (ECG) changes may progress rapidly to a dangerous arrhythmia, any patient with an ECG abnormality related to hyperkalemia should be considered as a medical emergency. We would aggressively treat patients with a PK greater than 7.0 mmol/L, even in the absence of ECG changes—the exceptions include those who develop hyperkalemia after extreme exercise (the super-marathon47).
Induce a Shift of Potassium Into the Intracellular Fluid
Insulin
A number of studies support the use of insulin to treat acute hyperkalemia (reviewed in Reference 6). Large doses of insulin (20 units of regular insulin) are needed to have high enough levels of insulin in plasma for a maximal shift of K+ into cells. Give enough glucose, and monitor PGlucose closely to avoid hypoglycemia.
β2-Adrenergic Agonists
Although a number of studies suggest that β2-agonists (e.g., 20 mg of nebulized albuterol) is effective treatment to lower PK rapidly, we do not use these agents as a primary treatment of emergency hyperkalemia for two reasons. First, in a number of studies it was noted that 20% to 40% of patients with end-stage renal disease are resistant to this therapy, and it is not possible to predict who the non-responders will be. Second, we are concerned about the safety of these drugs in the doses used for the treatment of hyperkalemia; these doses, which are 4 to 8 times those prescribed for the treatment of acute asthma. The combination of nebulized β2-agonists and insulin was reported to produce a greater fall in PK (1.2 mmol/L) compared with either drug alone (~0.65 mmol/L).48 One should note, however, that only 10 units of regular insulin were given in this study, and the magnitude of the fall in PK was lower than that observed in other studies using higher doses of insulin.49 Thus, it remains uncertain whether β2-agonists have a PK-lowering effect that is additive to that of higher doses of insulin.
Sodium Bicarbonate
A number of studies have found NaHCO3 therapy to be ineffective, as the sole treatment of hyperkalemia.49–51 Notwithstanding, these studies were performed in stable hemodialysis patients who did not have significant acidemia. Studies that examined the combined use of NaHCO3 with insulin also have yielded conflicting results.52,53 Thus the question remains, Would NaHCO3 be effective in patients with a more significant degree of acidemia? There are no data in the literature to answer this question definitively (for review, see Reference 6). Given this uncertainty, we only use NaHCO3 in addition to other therapies to treat emergency hyperkalemia in patients with a significant degree of acidemia. Caution is warranted because an excessive administration of NaHCO3 has the risk of inducing hypernatremia, ECF volume expansion, carbon dioxide retention, and acute hypocalcemia.
 Clinical Approach
Clinical Approach
It is imperative to recognize when hyperkalemia represents a medical emergency because therapy must take precedence over diagnosis (Figure 111-7). A step-by-step approach to diagnosis of hyperkalemia is illustrated in Figures 111-8 and 111-9.
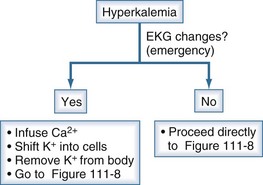
Figure 111-7 Initial clinical approach for the patient with hyperkalemia.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission ref 74.)
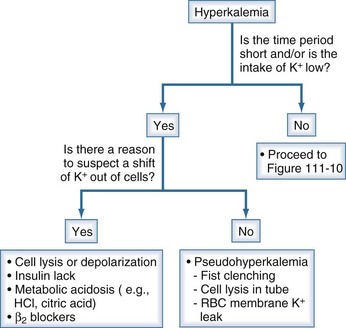
Figure 111-8 Initial steps in the clinical diagnosis of the cause of hyperkalemia.
The most important issue is to determine if a shift of K+ out of cells is likely; this is done by assessing the time course for the rise in the PK and whether there was little intake of K+. If that was the case and there is no reason to suspect a shift of K+ out of cells, pseudohyperkalemia should be ruled out. In this latter setting, there should not be ECG changes related to hyperkalemia. If this is ruled out, proceed to Figure 111-9, and examine the rate of excretion of K+.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission ref 74.)
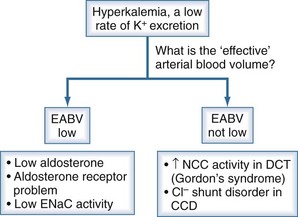
Figure 111-9 Basis for the low rate of excretion of K+.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission, ref 74.)
In Box 111-2, we provide a list of causes of hyperkalemia and of hypokalemia based on the presence or absence of hypertension.
Box 111-2
Dyskalemias and Blood Pressure*
Hyperkalemia
 Specific Causes of Hyperkalemia
Specific Causes of Hyperkalemia
A list of the causes of hyperkalemia based on their possible underlying pathophysiology is provided in Box 111-3.
Box 111-3
Causes of Hyperkalemia
Shift of Potassium Out of Cells
Addison’s Disease
Patients with chronic primary adrenal insufficiency may present with chronic malaise, fatigue, anorexia, and weight loss. In most patients, the blood pressure is low, and postural symptoms of dizziness and syncope are common. The PK is usually close to 5.5 mmol/L unless a significant degree of intravascular volume depletion diminishes the flow rate in CCD. Nevertheless, hyperkalemia is not seen on presentation in approximately a third of cases.38 The diagnosis can be established by finding a low PAldosterone and cortisol levels, high PRenin (see Box 111-1), and a blunted cortisol response to the administration of ACTH. Both glucocorticoid and mineralocorticoid replacement are required.
Adrenal crisis is an emergency that requires immediate restoration of the intravascular volume with the administration of intravenous (IV) saline and correction of the cortisol deficiency (administer dexamethasone or hydrocortisone). Beware of raising the PNa too rapidly if hyponatremia is present because of the risk of osmotic demyelination in a catabolic patient.39
Pseudohypoaldosteronism Type I
The underlying pathophysiology is a fewer number of open ENaC units in the CCD. In the autosomal recessive form, most mutations are in the α subunit of ENaC.40 These patients usually present in the neonatal period with renal salt wasting, hyperkalemia, metabolic acidosis, weight loss, and failure to thrive. ENaC activity is also impaired in the lung, leading to excessive airway fluid and recurrent lower respiratory tract infections. The autosomal dominant form this disorder is due to mutations involving the mineralocorticoid receptor.41 The clinical disorder is usually milder and may remit with time.
Syndrome of Hyporeninemic Hypoaldosteronism
These patients represent a heterogeneous group with regard to the pathophysiology of their disorder.
Group 2: Patients with Low Stimulus to Produce Renin
Subgroup One
Patients in this category have Gordon syndrome, a disorder where there is a low delivery of Na+ and Cl− to the CCD due to their enhanced reabsorption in the early distal convoluted tubule. The activity of the thiazide-sensitive NCC is increased in this disorder.32 Hypertension and hyperkalemia are common presenting features. The PRenin is suppressed, and the PAldosterone is inappropriately low considering that hyperkalemia is present (see Box 111-1). Thiazide diuretics are particularly helpful in these patients in treating both the hypertension and the hyperkalemia.42
The molecular basis involves mutations in the family of WNK (meaning with no lysine, where K is the single letter symbol for lysine) kinases (Figure 111-10). Major deletions in the genes encoding for WNK kinase 1 and WNK kinase 4 were reported in these patients. WNK kinase 4 normally causes a decrease in luminal NCC activity.32 Therefore if WNK kinase 4 were deleted, reabsorption of Na+ and Cl− by NCC in the early distal convoluted tubule will be augmented. The molecular defect in WNK kinase 1 is the removal of intron bases that leads to a gain of function. WNK kinase 1 normally inactivates WNK kinase 4, hence a gain in WNK kinase 1 function leads to the presence of more open NCC units in the luminal membranes of the early distal convoluted tubule.
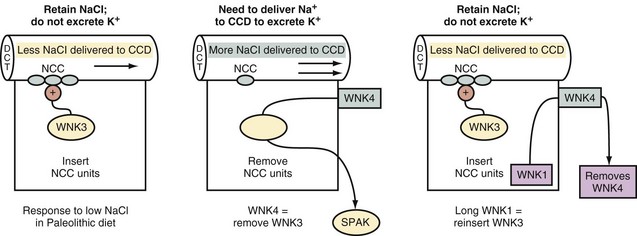
Subgroup Two
Hyperkalemia is due to less electrogenic reabsorption of Na+ in the CCD. The underlying pathophysiology may involve augmented reabsorption of Na+ and Cl− in the early distal convoluted tubule, or some of these patients may have a Cl− shunt disorder in CCD. There is no known molecular basis for the latter, and the most common setting is in patients with diabetic nephropathy. The PRenin is suppressed, and the PAldosterone is inappropriately low considering that hyperkalemia is present (see Box 111-1). In patients with a Cl− shunt disorder, there is a significant increase in the rate of excretion of K+ when bicarbonaturia is induced by the administration of acetazolamide.25,43,44
Drugs Associated with Hyperkalemia
Drugs that Inhibit Release of Renin
Hyperkalemic Periodic Paralysis
This syndrome has an autosomal dominant inheritance and is the result of a mutation in the α-subunit of the skeletal muscle Na+ channel gene.46 This leads to failure to completely close these voltage-gated Na+ channels when the concentration of K+ in the ECF is raised—hence there is a diminished electrical excitability of skeletal muscle cells. Symptoms of weakness and ultimately paralysis in association with hyperkalemia usually follow bouts of exercise. Acetazolamide seems to be effective in preventing these episodes, although its mechanism of action is not clear.
No Medical Emergency
Removal of Potassium from the Body
It is important to appreciate that very much less K+ loss is needed to lower the PK from 7.0 to 6.0 mmol/L than to lower it from 6.0 to 5.0 mmol/L.54 Hence creating a relatively small K+ loss can be very important when there is a severe degree of hyperkalemia.
Cation Exchange Resins for Treatment of Hyperkalemia
A cation exchange resin can exchange bound Na+ (Kayexalate) or Ca2+ (calcium resonium) for cations including K+. Kayexalate contains 4 mEq of Na+ per gram. The only favorable location for the exchange of Na+ for K+ is in the lumen of the colon. Based on data from patients with ileostomy, the amount of K+ delivered to the colon that would be available for this exchange is close to 5 mmol/d. Furthermore, other cations such as NH4+, Ca2+, and Mg2+ may exchange for resin-bound Na+ apart from K+. One possible theoretical benefit of using cation exchange resins is if they were to lower the concentration of K+ in luminal water in the lower intestinal tract and thereby enhance the secretion of K+ by the rectosigmoid colon. Even if more K+ were secreted, the low stool volume would limit the total K+ loss. For example, if the lumen-negative transepithelial voltage were −90 millivolts, and the PK were 5 mmol/L, the concentration of K+ in stool water would be 75 mmol/L. With a usual stool volume of 125 mL, of which 75% is water, only close to 7 mmol of K+ would be lost by this route. Hence we feel that there is virtually no theoretical benefit to using resins for acute hyperkalemia and little benefit to adding resins to cathartics in the setting of chronic hyperkalemia.6
Hypokalemia
 Clinical Approach
Clinical Approach
A list of causes of hypokalemia is provided in Box 111-4.
Box 111-4
Causes of Hypokalemia
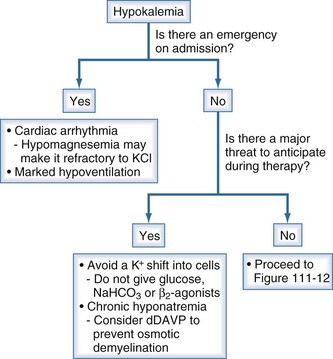
Figure 111-11 Initial clinical approach for the patient with hypokalemia.
The steps are to deal with emergencies and anticipate and prevent dangers during therapy.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission ref 74.)
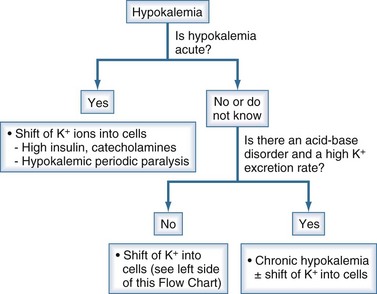
Figure 111-12 Initial steps in the clinical diagnosis of hypokalemia.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission.)
The most important initial step is to establish whether the duration of illness is short. The following characteristics should be present if the basis of hypokalemia is a shift of K+ into cells. The most important etiology is an adrenergic surge that lasts for many hours (e.g., post myocardial infarction, head trauma55) or the presence of hyperthyroidism in Asian patients with acute hypokalemia and extreme weakness.56 There should be a minimum rate of excretion of K+. A significant degree of metabolic acidosis or metabolic alkalosis should not be present.
3-What is the Rate of Excretion of Potassium?
To assess the renal response to hypokalemia, we use the expected rate of K+ excretion when hypokalemia was due to nonrenal causes—i.e., less than 10-15 mmol/d or close to 1 mmol K+/mmol creatinine.20 The rate of renal excretion of K+ may be low in a patient with chronic hypokalemia due to extrarenal loss of K+ or a renal loss of K+ in the recent past.
4-What is the acid-base status?
Patients with chronic hypokalemia can then be divided into two groups based on their metabolic acid-base disorder (Box 111-5).
Box 111-5
Plasma Acid-Base Status and Hypokalemia
Patients with Hyperchloremic Metabolic Acidosis
Patients with Metabolic Alkalosis
Patients with Chronic Hypokalemia and Metabolic Acidosis
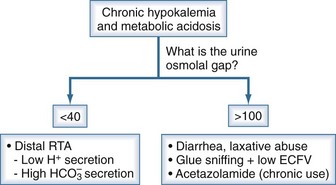
These patients can be divided into two further categories based on their rate of excretion NH4+ (Figure 111-13). The rate of excretion of NH4+ can be estimated from the calculation of the urine osmolal gap.
Patients with Chronic Hypokalemia and Metabolic Alkalosis
These patients can be classified into two groups based on whether the loss of K+ is nonrenal or renal with the use of the UK/UCreatinine ratio in a spot urine sample (Figure 111-14).
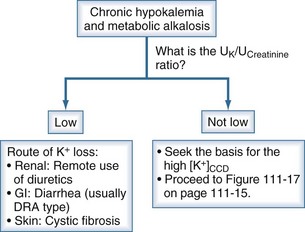
Figure 111-14 Chronic hypokalemia and metabolic alkalosis.
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission.)
Patients with Chronic Hypokalemia, Metabolic Alkalosis, and High Renal Excretion of Potassium
These patients have a high [K+]CCD in the presence of hypokalemia. The most common cause of high [K+]CCD is a more negative voltage in the lumen of the CCD. This higher lumen negative voltage may be due to disorders that cause more reabsorption of Na+ than Cl− in the CCD or disorders that may cause less reabsorption of Cl− than Na+ in the CCD. These two types of disorders can be differentiated with assessment of effective arterial blood volume and measurement of blood pressure (see Box 111-2 and Figure 111-15).
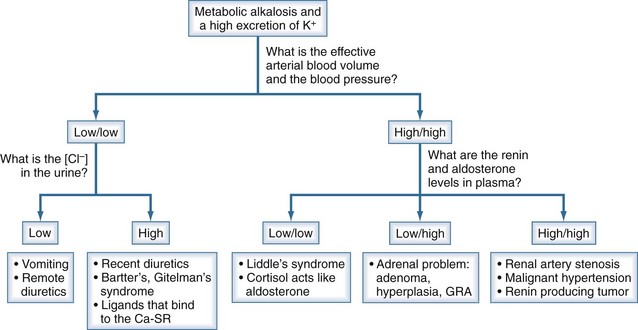
Figure 111-15 Chronic hypokalemia, metabolic alkalosis, and a high K/UCreatinine.
The goal is to determine why the [K+]CCD is higher than expected in a patient with hypokalemia, metabolic alkalosis, and a UK/UCreatinine that is definitely not low. Major clues are an estimate of the effective arterial blood volume and blood pressure. If both are low (left side of figure), the next step is to examine the concentration of Cl− in the urine. On the other hand, if the effective arterial blood volume is not low and the blood pressure is high, the differential diagnosis is based primarily on the PRenin and PAldosterone (see Box 111-1).
(From Halperin ML. The ACID truth and BASIC facts—with a sweet touch, an enLYTEnment. 5th ed. Toronto: RossMark Medical Publishers; 2003. Reproduced with permission, ref 74.)
Disorders with More Reabsorption of Sodium than Chloride in the Cortical Distal Nephron
Patients with these types of disorders are expected to have hypertension and an effective arterial blood volume that is not contracted. Based on measurement of PAldosterone, these patients can be classified into two groups: those with conditions in which the PAldosterone is high and those with conditions in which the actions of aldosterone are mimicked and hence the PAldosterone is low (see Figure 111-15 and Figure 111-16).
Disorders with Less Reabsorption of Chloride than Sodium in the Cortical Collecting Duct
These patients are expected to have a contracted effective arterial blood volume and the absence of hypertension (unless patients are given diuretics for treatment of hypertension; see Figure 111-15). The most common causes are protracted vomiting or the use of diuretics. The use of urine electrolytes in the differential diagnosis in patients with hypokalemia and a contracted effective arterial blood volume is illustrated in Box 111-6.
Box 111-6
Urine Electrolytes* in the Differential Diagnosis of Hypokalemia
| Condition | Urine Electrolyte | |
|---|---|---|
| Na+ | Cl− | |
| Vomiting | ||
| Recent | High† | Low‡ |
| Remote | Low | Low |
| Diuretics | ||
| Recent | High | High |
| Remote | Low | Low |
| Diarrhea or Laxative Abuse | Low | High |
| Bartter’s or Gitelman’s Syndrome | High | High |
 Specific Causes of Hypokalemia
Specific Causes of Hypokalemia
Hypokalemia and a Low Extracellular Fluid Volume
Diuretic-Induced Hypokalemia
Two factors contribute to the development of hypokalemia in patients receiving diuretics: a high flow rate in the CCD and an increased secretion of K+ in these nephron segments. The latter requires an enhanced electrogenic reabsorption of Na+ via ENaC due to effects of aldosterone. Hypokalemia is usually modest in degree; a PK less than 3 mmol/L is observed in less than 10% of patients and usually within the first 2 weeks of therapy.57
Diuretic abuse should be considered if little Na+ and Cl− are found in a single urine collection, as this reflects the normal renal response to a low effective arterial blood volume (see Box 111-6). The urine should be screened for diuretics, the assay should be performed on a urine sample that contains abundant Na+ and Cl− (i.e., a urine sample that reflects the action of a diuretic).
Vomiting-Induced Hypokalemia
Since the K+ concentration in gastric fluid is usually less than 15 mmol/L,14 hypokalemia in patients with vomiting or nasogastric suction results primarily from the loss of K+ in the urine due to a higher rate of electrogenic reabsorption of Na+ in the CCD. This is due to actions of aldosterone released in response to decreased effective arterial blood volume, along with distal delivery of Na+ with nonabsorbable anions (SO42− anions from metabolism of sulfur-containing amino acids in the early phase of vomiting, organic anions in the later phase of vomiting).31 To a lesser extent, hypokalemia may be the result of a shift of K+ into the ICF compartment due to the alkalemia. Key diagnostic elements are a history of vomiting or a strong concern about body weight, a significant degree of hypokalemia, metabolic alkalosis, and especially a very low UCl (see Box 111-6). In a patient with recent vomiting, the urine may contain a considerable amount of Na+ despite ECF volume contraction, because the excretion of HCO3− obligates the excretion of Na+. Other causes of hypokalemia with a low effective arterial blood volume must be considered (see Box 111-6).
Therapy must deal with the underlying cause of vomiting and the administration of KCl.16,58 If the patient has a contracted effective arterial blood volume, NaCl should be administered as needed.
Hypokalemia in Patients with Hyperchloremic Metabolic Acidosis
Rare causes of excessive excretion of K+ and metabolic acidosis include distal RTA due to a low rate of secretion of H+ in the distal nephron15 and inhibition of renal carbonic anhydrase (see Box 111-5). Hypokalemia is also seen in patients who sniff glue and overproduce hippuric acid.59 Excessive excretion of K+ in this setting is due to an open ENaC in the CCD owing to the effect of aldosterone released in response to a contracted effective arterial blood volume and the distal delivery of Na+ with hippurate anions instead of Cl−.
In patients with a secretory type of diarrhea (e.g., cholera), much K+ can be lost in K+-rich colonic fluids.14 Nevertheless, despite the large K+ deficit, hypokalemia is usually not present on presentation because the severe degree of intravascular volume depletion leads to an α-adrenergic surge, which inhibits the release of insulin. Hypokalemia becomes evident after therapy is initiated and the effective arterial blood volume is expanded. Patients with diarrhea due to a defect that leads to diminished reabsorption of Na+ and Cl− in the colon usually have a low PK but only a modest deficit of K+ unless there is also a reason for increased delivery of Na+ and Cl− to the colon (intake of certain types of laxatives). The low PK in these patients likely reflects a shift of K+ into cells due to a β2-adrenergic response to the mild degree of contraction of effective arterial blood volume.
Abuse of laxatives may be denied, so measurement of urine electrolytes may provide helpful clues (see Box 111-6). The UNa will be low if the effective arterial blood volume is contracted, but the UCl is characteristically high, reflecting the high rate of excretion of NH4+ in response to metabolic acidosis and/or hypokalemia. At times, one might have to rely on measurements of stool electrolytes and other evidence for laxatives in the stool to confirm the diagnosis.60
Gitelman’s Syndrome
Gitelman’s syndrome is a disease of young adults for the most part. The main clinical symptoms are tetany and weakness.61 Mutations that cause Gitelman’s syndrome have been identified in three separate genes that affect NaCl transport in distal convoluted tubule. Most patients have mutations in the gene encoding for the NaCl cotransporter in the early distal convoluted tubule. Other mutations involve the basolateral Cl− channel or the γ subunit of Na+/K+-ATPase in the basolateral membrane. One can anticipate other molecular causes that enhance WNK 4 kinase or lower WNK 1 kinase activity. The clinical picture is dominated by effective arterial blood volume contraction, while hypokalemia, renal wasting of Na+, Cl−, and K+, as well as metabolic alkalosis are the major laboratory findings. Because the thick ascending limb of the loop of Henle is not abnormal, patients can have a high Uosm when vasopressin acts. There is little calcium excretion in these patients (very low urine calcium/creatinine ratio). Hypomagnesemia is a common finding in patients with longer-standing Gitelman’s syndrome.62
Hypokalemia Due to Cationic Drugs Like Gentamicin and Tobramycin
Gentamicin and tobramycin are cationic antibiotics that bind to the calcium-sensing receptor on the basolateral aspect of cells of the loop of Henle.63 This leads to inhibition of the luminal ROMK channel and thereby to “Lasix-like” effects.
Hypokalemia and a Normal or High Extracellular Fluid Volume
Primary Hyperaldosteronism
Hypersecretion of aldosterone may be due to an adrenal adenoma or bilateral adrenal hyperplasia. This diagnosis should be suspected in patients with hypertension and unexplained hypokalemia with renal K+ wasting. Nevertheless, a significant proportion of these patients do not have hypokalemia and/or hypertension.64 An elevated PAldosterone and a very low PRenin are characteristic findings (see Box 111-2). A high PAldosterone-to-PRenin ratio in a random blood sample is usually a sufficient screening test. Primary hyperaldosteronism must be confirmed by finding of a non-suppressible high PAldosterone or 24-hour urinary aldosterone excretion during salt loading. A computed tomography (CT) scan is the best imaging test to detect an adrenal adenoma. If surgery to remove the adenoma is an option, adrenal vein sampling should be done to confirm that the lesion detected on CT is a functioning adenoma.
The finding of very low PRenin with high PAldosterone separates patients with primary hyperaldosteronism from those with other causes of hypertension and hypokalemia (see Box 111-1). The differential diagnosis includes patients with glucocorticoid-remediable aldosteronism (GRA). These latter patients have elevated PAldosterone and suppressed PRenin, but they are unique because of suppression of aldosterone with the administration of dexamethasone.65
ACTH-Producing Tumor or Severe Cushing’s Syndrome
The clinical picture is similar to primary hyperaldosteronism, but the level of aldosterone in plasma is low. Because of an overabundance of cortisol, the activity of 11β-HSDH is insufficient to inactivate all the cortisol that enters principal cells (Figure 111-17). As a result, cortisol binds to the mineralocorticoid receptor and exerts mineralocorticoid activity.
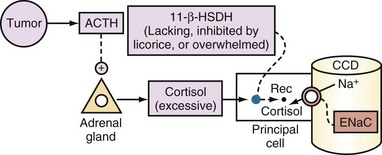
Syndrome of Apparent Mineralocorticoid Excess
The clinical picture is of hyperaldosteronism, but the level of aldosterone in plasma is low. Because of decreased activity of the enzyme 11β-HSDH, cortisol binds to the mineralocorticoid receptors and exerts mineralocorticoid activity (see Figure 111-17).66 PAldosterone and PRenin are both suppressed (see Box 111-1). The diagnosis is confirmed by finding an elevated urinary cortisol-to-cortisone ratio. Blood pressure control and correction of hypokalemia are achieved with administration of aldosterone receptor blocker or an ENaC blocker (e.g., amiloride with the same caveat noted earlier for the need for salt restriction).
A similar clinical picture can be induced with chronic ingestion of licorice or other compounds that contain glycyrrhetinic acid.67
Liddle’s Syndrome
The clinical picture is of hyperaldosteronism, but the level of aldosterone is low (see Box 111-2 and Figure 111-16). The pathophysiology of this disorder is one of a constitutively active ENaC in the CCD.68 Several mutations in the genes encoding for the β or γ subunits of ENaC have been described in patients with this syndrome69,70 (see Figure 111-5). One finds an autosomal dominant inherited disorder with early onset of severe hypertension and hypokalemia. Interestingly, a number of patients with this disorder, however, do not have hypokalemia. A positive family history of early-onset hypertension and hypokalemia and very low PAldosterone and PRenin are key elements in the diagnosis. There is no excess secretion of cortisol, and the urine cortisol-to-cortisone ratio is not elevated. Control of hypertension and correction of hypokalemia can be achieved by the administration of large doses of ENaC blockers (e.g., amiloride) but not with mineralocorticoid receptor antagonists (e.g., spironolactone).
Hypokalemic Periodic Paralysis
This disorder is characterized by episodes of a transient shift of K+ from the ECF to the ICF compartment of skeletal muscle. Thyrotoxic hypokalemic paralysis is more common in Asian and Hispanic males, and the first attack typically occurs between 20 and 50 years of age.56 A familial nonthyrotoxic variety is more common in Caucasian males younger than 20 and is inherited as an autosomal dominant disorder. Genetic analyses have suggested that the abnormality in these patients is linked to the gene that encodes for the dihydropyridine-sensitive Ca2+ channel in skeletal muscles; it is not clear how this leads to hypokalemia. While it is stated that these attacks can be provoked by a large carbohydrate meal (release of insulin) or strenuous exercise (adrenergic surge), this association is not impressive when large groups of patients are studied.
Laboratory findings are very helpful to differentiate this acute hypokalemia from an acute shift of K+ into cells in a patient with chronic hypokalemia.56 First, there is an absence of acid-base disorders. Second, one should anticipate a low rate of excretion of K+ as manifested by a low UK/UCreatinine. Patients with hypokalemic periodic paralysis usually need far less KCl to normalize their PK than do patients who have a chronic K+-wasting disease together with a reason to shift K+ acutely into cells (~1 versus > 3 mmol KCl/kg body weight).
An acute attack is treated with the administration of KCl. There is, however, the risk of posttreatment hyperkalemia when K+ moves back into the ECF compartment. Patients with the thyrotoxic variety of hypokalemic periodic paralysis can be treated with a nonselective beta-blocker and a much smaller administration of KCl.71
Therapy is largely symptomatic or empirical. Hyperthyroidism, if present, is treated in the usual fashion. Patients are advised to avoid carbohydrate-rich meals and vigorous exercise. Nonselective beta-blockers may reduce the number of attacks of paralysis, with little effect on the degree of fall in the PK.72 Acetazolamide, 250 to 750 mg per day, has been used successfully in patients with the familial form of hypokalemic periodic paralysis, although the basis of its beneficial effect is unclear.
 Therapy of Hypokalemia
Therapy of Hypokalemia
Medical Emergencies
Clinical Example
A patient had an acute traumatic brain injury.55 Within the first few hours, his PK fell to a nadir of 1.3 mmol/L, and ventricular tachycardia developed. The basis for the fall in PK was a sudden and marked shift of K+ into cells secondary to the extreme adrenergic response and the administered adrenergic agents to maintain hemodynamics.
No Medical Emergencies
Hypokalemia Due to an Acute Shift of Potassium Into Cells
In the absence of a cardiac or respiratory emergency, small doses of KCl should be given in patients with hypokalemic periodic paralysis to minimize the risk of severe rebound hyperkalemia, because they do not have a large deficit of K+. If associated with hyperthyroidism or a condition in which there is a large adrenergic surge, a nonselective beta-blocker (propranolol 3 mg/kg) can provide effective therapy.72
Magnitude of the Potassium Deficit
There is no useful quantitative relationship between the PK and the total body K+ deficit, because there may also be a shift of K+ into cells.54 Hence, careful monitoring of PK during replacement of the K+ deficit is mandatory.
Risks of Therapy
With prolonged hypokalemia, the CCD may become temporarily hyporesponsive to the kaliuretic effect of aldosterone (reviewed in Reference 73). Hence, it is important to monitor the PK frequently during the treatment of hypokalemia. Hyperkalemia has been observed in about 4% of patients taking K+ supplements. The risk is highest in patients with renal failure and diabetes mellitus. The simultaneous use of ACE inhibitors, beta-blockers, or NSAIDs may also predispose to the development of hyperkalemia.
Kamel KS, Wei C. Controversial issues in treatment of hyperkalemia. Nephrol Dialysis Transplant. 2003;18:2215-2218.
Juel C, Halestrap AP. Lactate transport in skeletal muscle—role and regulation of the monocarboxylate transporter. J Physiol. 1999;517:633-642.
Halperin ML, Kamel KS, Oh MS. Mechanisms to concentrate the urine: an opinion. Curr Opin Nephrol Hypertens. 2008;17:416-422.
Carlisle EJF, Donnelly SM, Ethier J, Quaggin SE, Kaiser U, Vasuvattakul S, et al. Modulation of the secretion of potassium by accompanying anions in humans. Kidney Int. 1991;39:1206-1212.
Lin SH, Lin YF, Halperin ML. Hypokalemia and paralysis: clues on admission to help in the differential diagnosis. Quart J Med. 2001;94:133-139.
Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001;37:620-624.
1 Halperin ML, Kamel KS. Potassium. Lancet. 1998;352:135-142.
2 Wollheim CB. Beta-cell mitochondria in the regulation of insulin secretion: a new culprit in type II diabetes. Diabetologia. 2000;43:265-277.
3 Landry DW, Oliver JA. The pathogenesis of vasodilatory shock. N Engl J Med. 2001;345:588-595.
4 Counillon LL, Pouyssegur RJ. The members of the Na+/H+ exchanger gene family: Their structure, function, expression, and regulation. In: Seldin DW, Giebisch G, editors. The Kidney: Physiology & Pathophysiology, vol 1. Philadelphia PA: Lippincott Williams & Wilkins; 2000:223-234.
5 Clausen T. Regulation of active Na+-K+ transport in skeletal muscle. Physiol Rev. 1986;66:542-580.
6 Kamel KS, Wei C. Controversial issues in treatment of hyperkalemia. Nephrol Dialysis Transplant. 2003;18:2215-2218.
7 Kamel KS, Oh MS, Halperin ML. Treatment of hypokalemia and hyperkalemia. In: Brady HR, Wilcox CS, editors. Therapy in Nephrology and Hypertension: a companion to Brenner and Rector’s The Kidney. Philadelphia: WB Saunders; 2003:349-363.
8 Gowrishankar M, Kamel KS, Halperin ML. Buffering of a H+ load; A ‘brain-protein-centered’ view. J Amer Soc Nephrol. 2007;18:2278-2280.
9 Juel C, Halestrap AP. Lactate transport in skeletal muscle—role and regulation of the monocarboxylate transporter. J Physiol. 1999;517:633-642.
10 Cheema-Dhadli S, Kamel KS, Halperin ML. A defense against a severe degree of hyperkalemia during a sprint. J Am Soc Nephrol. 19, 2008. Abstract
11 Alper S. The band 3-related anion exchanger (AE) gene family. Ann Rev Physiol. 1991;53:549-564.
12 Waldegger S, Jentsch TJ. From tonus to tonicity: physiology of CIC chloride channels. J Am Soc Nephrol. 2000;11:1331-1339.
13 Davids MR, Edoute Y, Jungas RL, Halperin ML. Facilitating an understanding of integrative physiology: Emphasis on the composition of body fluid compartments. Can J Physiol Pharmacol. 2002;80:835-850.
14 Field M. Intestinal transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931-943.
15 Sebastian A, McSherry E, Morris RCJr. Renal potassium wasting in renal tubular acidosis (RTA): its occurrence in types 1 and 2 RTA despite sustained correction of systemic acidosis. J Clin Invest. 1971;50:667-678.
16 Kassirer JP, Schwartz WB. The response of normal man to selective depletion of hydrochloric acid. Am J Med. 1966;40:10-18.
17 Bercovici M, Chen C, Goldstein M, Stinebaugh B, Halperin M. Effect of acute changes in the PaCO2 on acid-base parameters in normal dogs and dogs with metabolic acidosis or alkalosis. Canadian J Physiol and Pharmacology. 1983;61:166-173.
18 Arrambide K, Toto RD. Tumor lysis syndrome. Seminars in Nephrol. 1993;13:273-280.
19 Giebisch G. Renal potassium transport: mechanisms and regulation. Am J Physiol. 1998;274:F817-F833.
20 Huth EJ, Squires RD, Elkinton JR. Experimental potassium depletion in normal human subjects. II. Renal and hormonal factors in the development of extracellular alkalosis during depletion. J Clin Invest. 1959;38:1149-1165.
21 Steele A, deVeber H, Quaggin SE, Scheich A, Ethier J, Halperin ML. What is responsible for the diurnal variation in potassium excretion? Am J Physiol. 1994;36:R554-R560.
22 Halperin ML, Kamel KS, Oh MS. Mechanisms to concentrate the urine: An opinion. Curr Opin Nephrol Hypertens. 2008;17:416-422.
23 Choi MJ, Fernandez PC, Patnaik A, Coupaye-Gerard B, D’Andrea D, Zerlip H, et al. Trimethoprim induced hyperkalemia in a patient with AIDS. N Engl J Med. 1993;328:703-706.
24 Schreiber M, Halperin ML. Urea excretion rate as a contributor to trimethoprim-induced hyperkalemia. Ann Intern Med. 1994;120:166-167.
25 Kamel KS, Quaggin S, Scheich A, Halperin ML. Disorders of potassium homeostasis: an approach based on pathophysiology. Am J Kidney Dis. 1994;24:597-613.
26 Rossier BC, Palmer LG. Mechanisms of aldosterone action on sodium and potassium transport. In: Seldin DW, Giebisch G, editors. The Kidney: Physiology and Pathophysiology. New York: Raven Press Ltd.; 1992:1373-1409.
27 Velazquez H, Wright FS, Good DW. Luminal influences on potassium secretion: chloride replacement with sulfate. Am J Physiol. 1982;242:F46-F55.
28 Wilson FH, Disse-Nocodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107-1112.
29 Stokes JB. Ion transport by the collecting duct. Sem Nephrol. 1993;13:202-212.
30 Kamel KS, Ethier JH, Richardson RMA, Bear RA, Halperin ML. Urine electrolytes and osmolality: when and how to use them. Am J Nephrol. 1990;10:89-102.
31 Carlisle EJF, Donnelly SM, Ethier J, Quaggin SE, Kaiser U, Vasuvattakul S, et al. Modulation of the secretion of potassium by accompanying anions in humans. Kidney Int. 1991;39:1206-1212.
32 Yang C-L, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest. 2003;111:1039-1045.
33 Talbott JH, Schwab RS. Recent advances in the biochemistry and therapeutics of potassium salts. N Engl J Med. 1940;222:585-590.
34 Walser M. Creatinine excretion as a measure of protein nutrition in adults of varying age. J Parenter Enteral Nutr. 1987;11:73S-78S.
35 Halperin ML, Oh MS. The Dysnatremias: hyponatremia and hypernatremia. In: Glassock RJ, editor. Current Therapy in Nephrology and Hypertension. 4th ed. Philadelphia, PA: Mosby-Year Book Inc; 1998:1-9.
36 Iolascon A, Stewart GW, Ajetunmobi JF, Perrotta S, Delaunay J, Carella M, et al. Familial pseudohyperkalemia maps to the same locus as dehydrated hereditary stomatocytosis (hereditary xerocytosis). Blood. 1999;93:3120-3123.
37 Don BR, Sebastian A, Cheitlin M, Christiansen M, Schambelan M. Pseudohyperkalemia caused by fist clenching during phlebotomy. N Engl J Med. 1990;322:1290-1292.
38 Gagnon RF, Halperin ML. Possible mechanisms to explain the absence of hyperkalemia in a patient with Addison’s disease. Nephrol Dial Transplant. 2001;16:1280-1284.
39 Lin SH, Hsu Y-J ChiuJ-S, Chu S-J, Davids MR, Halperin ML. Osmotic demyelination syndrome: A potentially avoidable disaster. Quart J Med. 2003;96:935-947.
40 Schild L. The ENaC channel as the primary determinant of two human diseases: Liddle’s syndrome and pseudohypoaldosteronism. Neprhologie. 1996;17:395-400.
41 Bonny O, Rossier BC. Disturbances of Na/K balance: Pseudohypoaldosteronism revisited. J Am Soc Nephrol. 2002;13:2399-2414.
42 Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab. 2002;87:3248-3254.
43 Phelps KR, Lieberman RL, Oh MS, Carroll HJ. Pathophysiology of the syndrome of hyporeninemic hypoaldosteronism. Metabolism. 1980;29:186-199.
44 Schambelan M, Sebastian A, Rector FCJr. Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism): Role of increased renal chloride reabsorption. Kidney Int. 1981;19:716-727.
45 Kamel K, Ethier JH, Quaggin S, Levin A, Albert S, Carlisle EJF, Halperin ML. Studies to determine the basis for hyperkalemia in recipients of a renal transplant who are treated with cyclosporin. J Am Soc Nephrol. 1992;2:1279-1284.
46 Fontaine B, Khurana TS, Hoffman EP, et al. Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene. Science. 1990;250:1000-1002.
47 McKechnie JK, Leary WP, Joubert SM. Some electrocardiographic and biochemical changes recorded in marathon runners. South African Med J. 1967;41:722-725.
48 Allon M, Copkney C. Albuterol and insulin for treatment of hyperkalemia in hemodialysis patients. Kidney Int. 1990;38:869-872.
49 Blumberg A, Weidmann P, Shaw S, Gnadinger M. Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure. Am J Med. 1988;85:507-512.
50 Guttierez R, Schlessinger F, Oster JR, Rietberg B, Perez GO. Effect of hypertonic versus isotonic sodium bicarbonate on plasma potassium concentration in patients with end-stage renal disease. Miner Electrolyte Metab. 1991;17:297-302.
51 Blumberg A, Weidmann P, Ferrari P. Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure. Kidney Int. 1992;41:369-374.
52 Allon M, Shanklin N. Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol. Am J Kidney Dis. 1996;28:508-514.
53 Kim HJ. Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end-stage renal disease patients. Nephron. 1996;72:476-482.
54 Sterns RH, Guzzo J, Feig PU, Singer I. Internal potassium balance and the control of the plasma potassium concentration. Medicine. 1981;60:339-354.
55 Schaefer M, Link J, Hannemann L, Rudolph KH. Excessive hypokalemia and hyperkalemia following head injury. Intensive Care Med. 1995;21:235-237.
56 Lin SH, Lin YF, Halperin ML. Hypokalemia and Paralysis: Clues on admission to help in the differential diagnosis. Quart J Med. 2001;94:133-139.
57 Tannen RL. Diuretic-induced hypokalemia. Kidney Int. 1985;28:988-1000.
58 Halperin ML, Scheich A. Should we continue to recommend that a deficit of KCl be treated with NaCl? A fresh look at chloride-depletion metabolic alkalosis. Nephron. 1994;67:263-269.
59 Carlisle EJF, Donnelly SM, Vasuvattakul S, Kamel KS, Tobe S, Halperin ML. Glue-sniffing and distal renal tubular acidosis: sticking to the facts. J Am Soc Nephrol. 1991;1:1019-1027.
60 Emmett M, Hootkins RE, Fine KD. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology. 1995;108:752-760.
61 Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB. Gitelman’s syndrome revisited: An evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59:710-717.
62 Ring T, Knoers N, Oh MS, Halperin ML. Reevaluation of the criteria for the clinical diagnosis of Gitelman’s syndrome. Pediatr Nephrol. 2002;17:612-616.
63 Hebert SC. Extracellular calcium-sensing receptor: Implications for calcium and magnesium handling in the kidney. Kidney Int. 1996;50:2129-2139.
64 Lin SH, Lin YF, Tsai W-S, Davids MR, Halperin ML. Severe hypokalemia in a Chinese male. Quart J Med. 2002;95:695-704.
65 Lifton RP, Dluhy RG, Power M, Rich GM, Cook S, Ulick S, Lalouel JM. A chimaeric 11ß-hydroxylase/aldosterone synthetase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992;355:262-265.
66 Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: Target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583-585.
67 Edwards CRW. Lessons from licorice. New Engl J Med. 1991;24:1242-1243.
68 Palmer BF, Alpern RJ. Liddle’s syndrome. Am J Med. 1998;104:301-309.
69 Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, et al. Liddle’s syndrome: Heritable human hypertension caused by mutations in the ß subunit of the epithelial sodium channel. Cell. 1994;79:407-414.
70 Hiltunen TP, Hannila-Handelberg T, Petajaniemi N, Kantola I, Tikkanen I, Virtamo J, et al. Liddle’s syndrome associated with a point mutation in the extracellular domain of the epithelial sodium channel g subunit. J Hypertens. 2002;20:2383-2390.
71 Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001;37:620-624.
72 Conway MJ, Seibel JA, Eaton RP. Thyrotoxicosis and hypokalemic periodic paralysis: Improvement with beta blockade. Ann Int Med. 1974;81:332-336.
73 Ethier JH, Kamel KS, Magner PO, Lemann JJ, Halperin ML. The transtubular potassium concentration in patients with hypokalemia and hyperkalemia. Am J Kidney Dis. 1990;15:309-315.
74 Halperin ML. The ACID truth and BASIC facts—with a Sweet Touch, an enLYTEnment, 5th ed. Toronto: RossMark Medical Publishers; 2003.
