ACID-NEUTRALIZING/INHIBITORY DRUGS
Antacids Before we understood the important role of histamine in stimulating parietal cell activity, neutralization of secreted acid with antacids constituted the main form of therapy for peptic ulcers. They are now rarely, if ever, used as the primary therapeutic agent but instead are often used by patients for symptomatic relief of dyspepsia. The most commonly used agents are mixtures of aluminum hydroxide and magnesium hydroxide. Aluminum hydroxide can produce constipation and phosphate depletion; magnesium hydroxide may cause loose stools. Many of the commonly used antacids (e.g., Maalox, Mylanta) have a combination of both aluminum and magnesium hydroxide in order to avoid these side effects. The magnesium-containing preparation should not be used in chronic renal failure patients because of possible hypermagnesemia, and aluminum may cause chronic neurotoxicity in these patients.
Calcium carbonate and sodium bicarbonate are potent antacids with varying levels of potential problems. The long-term use of calcium carbonate (converts to calcium chloride in the stomach) can lead to milk-alkali syndrome (hypercalcemia, hyperphosphatemia with possible renal calcinosis and progression to renal insufficiency). Sodium bicarbonate may induce systemic alkalosis.
H2 Receptor Antagonists Four of these agents are presently available (cimetidine, ranitidine, famotidine, and nizatidine), and their structures share homology with histamine. Although each has different potency, all will significantly inhibit basal and stimulated acid secretion to comparable levels when used at therapeutic doses. Moreover, similar ulcer-healing rates are achieved with each drug when used at the correct dosage. Presently, this class of drug is often used for treatment of active ulcers (4–6 weeks) in combination with antibiotics directed at eradicating H. pylori (see below).
Cimetidine was the first H2 receptor antagonist used for the treatment of acid peptic disorders. The initial recommended dosing profile for cimetidine was 300 mg qid. Subsequent studies have documented the efficacy of using 800 mg at bedtime for treatment of active ulcer, with healing rates approaching 80% at 4 weeks. Cimetidine may have weak antiandrogenic side effects resulting in reversible gynecomastia and impotence, primarily in patients receiving high doses for prolonged periods of time (months to years, as in ZES). In view of cimetidine’s ability to inhibit cytochrome P450, careful monitoring of drugs such as warfarin, phenytoin, and theophylline is indicated with long-term usage. Other rare reversible adverse effects reported with cimetidine include confusion and elevated levels of serum aminotransferases, creatinine, and serum prolactin. Ranitidine, famotidine, and nizatidine are more potent H2 receptor antagonists than cimetidine. Each can be used once a day at bedtime for ulcer prevention, which was commonly done before the discovery of H. pylori and the development of proton pump inhibitors (PPIs). Patients may develop tolerance to H2 blockers, a rare event with PPIs (see below). Comparable nighttime dosing regimens are ranitidine 300 mg, famotidine 40 mg, and nizatidine 300 mg.
Additional rare, reversible systemic toxicities reported with H2 receptor antagonists include pancytopenia, neutropenia, anemia, and thrombocytopenia, with a prevalence rate varying from 0.01–0.2%. Cimetidine and ranitidine (to a lesser extent) can bind to hepatic cytochrome P450; famotidine and nizatidine do not.
Proton Pump (H+,K+-ATPase) Inhibitors Omeprazole, esomeprazole, lansoprazole, rabeprazole, and pantoprazole are substituted benzimidazole derivatives that covalently bind and irreversibly inhibit H+,K+-ATPase. Esomeprazole, one of the newest members of this drug class, is the S-enantiomer of omeprazole, which is a racemic mixture of both S- and R-optical isomers. The R-isomer of lansoprazole, dexlansoprazole, is the most recent PPI approved for clinical use. Its reported advantage is a dual delayed-release system, aimed at improving treatment of gastroesophageal reflux disease (GERD). These are the most potent acid inhibitory agents available. Omeprazole and lansoprazole are the PPIs that have been used for the longest time. Both are acid-labile and are administered as enteric-coated granules in a sustained-release capsule that dissolves within the small intestine at a pH of 6. Lansoprazole is available in an orally disintegrating tablet that can be taken with or without water, an advantage for individuals who have significant dysphagia. Absorption kinetics are similar to the capsule. In addition, a lansoprazole-naproxen combination preparation that has been made available is targeted at decreasing NSAID-related GI injury (see below). Omeprazole is available as nonenteric-coated granules mixed with sodium bicarbonate in a powder form that can be administered orally or via gastric tube. The sodium bicarbonate has two purposes: to protect the omeprazole from acid degradation and to promote rapid gastric alkalinization and subsequent proton pump activation, which facilitates rapid action of the PPI. Pantoprazole and rabeprazole are available as enteric-coated tablets. Pantoprazole is also available as a parenteral formulation for intravenous use. These agents are lipophilic compounds; upon entering the parietal cell, they are protonated and trapped within the acid environment of the tubulovesicular and canalicular system. These agents potently inhibit all phases of gastric acid secretion. Onset of action is rapid, with a maximum acid inhibitory effect between 2 and 6 h after administration and duration of inhibition lasting up to 72–96 h. With repeated daily dosing, progressive acid inhibitory effects are observed, with basal and secretagogue-stimulated acid production being inhibited by >95% after 1 week of therapy. The half-life of PPIs is ~18 h; thus, it can take between 2 and 5 days for gastric acid secretion to return to normal levels once these drugs have been discontinued. Because the pumps need to be activated for these agents to be effective, their efficacy is maximized if they are administered before a meal (except for the immediate-release formulation of omeprazole) (e.g., in the morning before breakfast). Mild to moderate hypergastrinemia has been observed in patients taking these drugs. Carcinoid tumors developed in some animals given the drugs preclinically; however, extensive experience has failed to demonstrate gastric carcinoid tumor development in humans. Serum gastrin levels return to normal levels within 1–2 weeks after drug cessation. Rebound gastric acid hypersecretion has been described in H. pylori–negative individuals after discontinuation of PPIs. It occurs even after relatively short-term usage (2 months) and may last for up to 2 months after the PPI has been discontinued. The mechanism involves gastrin-induced hyperplasia and hypertrophy of histamine-secreting ECL cells. The clinical relevance of this observation is that individuals may have worsening symptoms of GERD or dyspepsia upon stopping the PPI. Gradual tapering of the PPI and switching to an H2 receptor antagonist may prevent this from occurring. H. pylori–induced inflammation and concomitant decrease in acid production may explain why this does not occur in H. pylori–positive patients. IF production is also inhibited, but vitamin B12-deficiency anemia is uncommon, probably because of the large stores of the vitamin. As with any agent that leads to significant hypochlorhydria, PPIs may interfere with absorption of drugs such as ketoconazole, ampicillin, iron, and digoxin. Hepatic cytochrome P450 can be inhibited by the earlier PPIs (omeprazole, lansoprazole). Rabeprazole, pantoprazole, and esomeprazole do not appear to interact significantly with drugs metabolized by the cytochrome P450 system. The overall clinical significance of this observation is not definitely established. Caution should be taken when using theophylline, warfarin, diazepam, atazanavir, and phenytoin concomitantly with PPIs. Long-term acid suppression, especially with PPIs, has been associated with a higher incidence of community-acquired pneumonia as well as community and hospital acquired Clostridium difficile–associated disease. These observations require confirmation but should alert the practitioner to take caution when recommending these agents for long-term use, especially in elderly patients at risk for developing pneumonia or C. difficile infection. A population-based study revealed that long-term use of PPIs was associated with the development of hip fractures in older women. The absolute risk of fracture remained low despite an observed increase associated with the dose and duration of acid suppression. The mechanism for this observation is not clear, and this finding must be confirmed before making broad recommendations regarding the discontinuation of these agents in patients who benefit from them. Long-term use of PPIs has also been implicated in the development of iron and magnesium deficiency, but here again, the studies are limited and inconclusive. PPIs may exert a negative effect on the antiplatelet effect of clopidogrel. Although the evidence is mixed and inconclusive, a small increase in mortality and readmission rate for coronary events was seen in patients receiving a PPI while on clopidogrel in earlier studies. Subsequently, three meta-analyses reported an inverse correlation between clopidogrel and PPI use; therefore, the influence of this drug interaction on mortality is not clearly established. The mechanism involves the competition of the PPI and clopidogrel with the same cytochrome P450 (CYP2C19). Whether this is a class effect of PPIs is unclear; there appears to be at least a theoretical advantage of pantoprazole over the other PPIs, but this has not been confirmed. This drug interaction is particularly relevant in light of the common use of aspirin and clopidogrel for prevention of coronary events and the efficacy of PPIs in preventing GI bleeding in these patients. The FDA has made several recommendations while awaiting further evidence to clarify the impact of PPI therapy on clopidogrel use. Health care providers should continue to prescribe clopidogrel to patients who require it and should reevaluate the need for starting or continuing treatment with a PPI. From a practical standpoint, additional recommendations to consider include the following: Patients taking clopidogrel with aspirin, especially with other GI risk factors for bleeding, should receive GI protective therapy. Although high-dose H2 blockers have been considered an option, these do not appear to be as effective as PPIs. If PPIs are to be given, some have recommended that there be a 12-h separation between administration of the PPI and clopidogrel to minimize competition of the two agents with the involved cytochrome P450. One option is to give the PPI 30 min before breakfast and the clopidogrel at bedtime. Insufficient data are available to firmly recommend one PPI over another. Patients 65 years of age or older have a higher risk for some of the long-term side effects of PPIs highlighted above, in part due to the higher prevalence of concomitant chronic diseases. It is therefore important to carefully select individuals, especially among the elderly, who need long-term PPI therapy and discontinue it in those individuals who do not need it.
Two new formulations of acid inhibitory agents are being developed. Tenatoprazole is a PPI containing an imidazopyridine ring instead of a benzimidazole ring, which promotes irreversible proton pump inhibition. This agent has a longer half-life than the other PPIs and may be beneficial for inhibiting nocturnal acid secretion, which has significant relevance in GERD. A second new class of agents is the potassium-competitive acid pump antagonists (P-CABs). These compounds inhibit gastric acid secretion via potassium competitive binding of the H+,K+-ATPase.
CYTOPROTECTIVE AGENTS
Sucralfate Sucralfate is a complex sucrose salt in which the hydroxyl groups have been substituted by aluminum hydroxide and sulfate. This compound is insoluble in water and becomes a viscous paste within the stomach and duodenum, binding primarily to sites of active ulceration. Sucralfate may act by several mechanisms: serving as a physicochemical barrier, promoting a trophic action by binding growth factors such as EGF, enhancing prostaglandin synthesis, stimulating mucus and bicarbonate secretion, and enhancing mucosal defense and repair. Toxicity from this drug is rare, with constipation being most common (2–3%). It should be avoided in patients with chronic renal insufficiency to prevent aluminum-induced neurotoxicity. Hypophosphatemia and gastric bezoar formation have also been reported rarely. Standard dosing of sucralfate is 1 g qid.
Bismuth-Containing Preparations Sir William Osler considered bismuth-containing compounds the drug of choice for treating PUD. The resurgence in the use of these agents is due to their effect against H. pylori. Colloidal bismuth subcitrate (CBS) and bismuth subsalicylate (BSS, Pepto-Bismol) are the most widely used preparations. The mechanism by which these agents induce ulcer healing is unclear. Adverse effects with short-term use include black stools, constipation, and darkening of the tongue. Long-term use with high doses, especially with the avidly absorbed CBS, may lead to neurotoxicity. These compounds are commonly used as one of the agents in an anti-H. pylori regimen (see below).
Prostaglandin Analogues In view of their central role in maintaining mucosal integrity and repair, stable prostaglandin analogues were developed for the treatment of PUD. The mechanism by which this rapidly absorbed drug provides its therapeutic effect is through enhancement of mucosal defense and repair. The most common toxicity noted with this drug is diarrhea (10–30% incidence). Other major toxicities include uterine bleeding and contractions; misoprostol is contraindicated in women who may be pregnant, and women of childbearing age must be made clearly aware of this potential drug toxicity. The standard therapeutic dose is 200 μg qid.
Miscellaneous Drugs A number of drugs including anticholinergic agents and tricyclic antidepressants were used for treating acid peptic disorders, but in light of their toxicity and the development of potent antisecretory agents, these are rarely, if ever, used today.
THERAPY OF H. PYLORI
The physician’s goal in treating PUD is to provide relief of symptoms (pain or dyspepsia), promote ulcer healing, and ultimately prevent ulcer recurrence and complications. The greatest influence of understanding the role of H. pylori in peptic disease has been the ability to prevent recurrence. Documented eradication of H. pylori in patients with PUD is associated with a dramatic decrease in ulcer recurrence to <10–20% as compared to 59% in GU patients and 67% in DU patients when the organism is not eliminated. Eradication of the organism may lead to diminished recurrent ulcer bleeding. The effect of its eradication on ulcer perforation is unclear.
Extensive effort has been made in determining who of the many individuals with H. pylori infection should be treated. The common conclusion arrived at by multiple consensus conferences around the world is that H. pylori should be eradicated in patients with documented PUD. This holds true independent of time of presentation (first episode or not), severity of symptoms, presence of confounding factors such as ingestion of NSAIDs, or whether the ulcer is in remission. Some have advocated treating patients with a history of documented PUD who are found to be H. pylori–positive by serology or breath testing. Over one-half of patients with gastric MALT lymphoma experience complete remission of the tumor in response to H. pylori eradication. The Maastricht IV/Florence Consensus Report recommends a test-and-treat approach for patients with uninvestigated dyspepsia if the local incidence of H. pylori is greater than 20%. In addition, recommendations from this consensus report include testing and eradicating H. pylori in patients who will be using NSAIDs (including low-dose aspirin) on a long-term basis, especially if there is a prior history of PUD. These individuals will require continued PPI treatment as well as eradication treatment, because eradication of the organism alone does not eliminate the risk of gastroduodenal ulcers in patients already receiving long-term NSAIDs. Treating patients with NUD to prevent gastric cancer or patients with GERD requiring long-term acid suppression remains controversial. Guidelines from the American College of Gastroenterology suggest eradication of H. pylori in patients who have undergone resection of early gastric cancer. The Maastricht IV/Florence Consensus Report also evaluated H. pylori treatment in gastric cancer prevention and recommends that eradication should be considered in the following situations: first-degree relatives of family members with gastric cancer; patients with previous gastric neoplasm treated by endoscopic or subtotal resection; individuals with a risk of gastritis (severe pangastritis or body-predominant gastritis) or severe atrophy; patients with gastric acid inhibition for more than 1 year; individuals with strong environmental risk factors for gastric cancer (heavy smoking; high exposure to dust, coal, quartz, or cement; and/or work in quarries); and H. pylori–positive patients with a fear of gastric cancer.
Multiple drugs have been evaluated in the therapy of H. pylori. No single agent is effective in eradicating the organism. Combination therapy for 14 days provides the greatest efficacy, although regimens based on sequential administration of antibiotics also appear promising (see below). A shorter administration course (7–10 days), although attractive, has not proved as successful as the 14-day regimens. The agents used with the greatest frequency include amoxicillin, metronidazole, tetracycline, clarithromycin, and bismuth compounds.
Suggested treatment regimens for H. pylori are outlined in Table 348-4. Choice of a particular regimen will be influenced by several factors, including efficacy, patient tolerance, existing antibiotic resistance, and cost of the drugs. The aim for initial eradication rates should be 85–90%. Dual therapy (PPI plus amoxicillin, PPI plus clarithromycin, ranitidine bismuth citrate [Tritec] plus clarithromycin) is not recommended in view of studies demonstrating eradication rates of <80–85%. The combination of bismuth, metronidazole, and tetracycline was the first triple regimen found effective against H. pylori. The combination of two antibiotics plus either a PPI, H2 blocker, or bismuth compound has comparable success rates. Addition of acid suppression assists in providing early symptom relief and enhances bacterial eradication.
|
REGIMENS RECOMMENDED FOR ERADICATION OF H. PYLORI INFECTION |
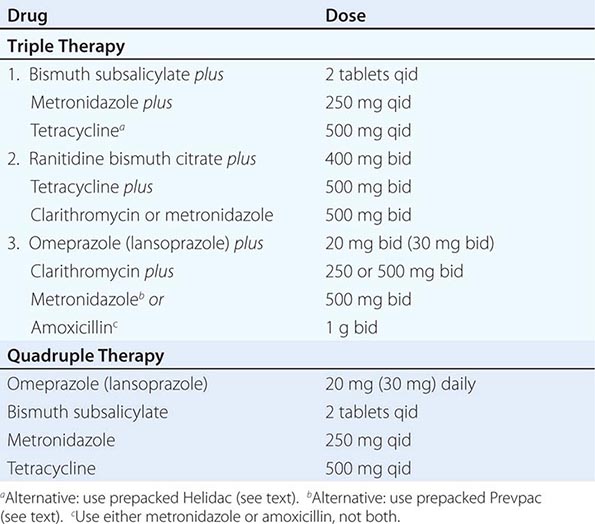
Triple therapy, although effective, has several drawbacks, including the potential for poor patient compliance and drug-induced side effects. Compliance is being addressed by simplifying the regimens so that patients can take the medications twice a day. Simpler (dual therapy) and shorter regimens (7 and 10 days) are not as effective as triple therapy for 14 days. Two anti-H. pylori regimens are available in prepackaged formulation: Prevpac (lansoprazole, clarithromycin, and amoxicillin) and Helidac (BSS, tetracycline, and metronidazole). The contents of the Prevpac are to be taken twice per day for 14 days, whereas Helidac constituents are taken four times per day with an antisecretory agent (PPI or H2 blocker), also for at least 14 days. Clarithromycin-based triple therapy should be avoided in settings where H. pylori resistance to this agent exceeds 15–20%.
Side effects have been reported in up to 20–30% of patients on triple therapy. Bismuth may cause black stools, constipation, or darkening of the tongue. The most feared complication with amoxicillin is pseudomembranous colitis, but this occurs in <1–2% of patients. Amoxicillin can also lead to antibiotic-associated diarrhea, nausea, vomiting, skin rash, and allergic reaction. Concomitant use of probiotics may ameliorate some of the antibiotic side effects (see below). Tetracycline has been reported to cause rashes and, very rarely, hepatotoxicity and anaphylaxis.
One important concern with treating patients who may not need therapy is the potential for development of antibiotic-resistant strains. The incidence and type of antibiotic-resistant H. pylori strains vary worldwide. Strains resistant to metronidazole, clarithromycin, amoxicillin, and tetracycline have been described, with the latter two being uncommon. Antibiotic-resistant strains are the most common cause for treatment failure in compliant patients. Unfortunately, in vitro resistance does not predict outcome in patients. Culture and sensitivity testing of H. pylori is not performed routinely. Although resistance to metronidazole has been found in as many as 30% of isolates in North America and 80% in developing countries, triple therapy is effective in eradicating the organism in >50% of patients infected with a resistant strain. Clarithromycin resistance is seen in 13% of individuals in the United States, with resistance to amoxicillin being <1% and resistance to both metronidazole and clarithromycin in the 5% range.
Failure of H. pylori eradication with triple therapy in a compliant patient is usually due to infection with a resistant organism. Quadruple therapy (Table 348-4), where clarithromycin is substituted for metronidazole (or vice versa), should be the next step. The combination of pantoprazole, amoxicillin, and rifabutin for 10 days has also been used successfully (86% cure rate) in patients infected with resistant strains. Additional regimens considered for second-line therapy include levofloxacin-based triple therapy (levofloxacin, amoxicillin, PPI) for 10 days and furazolidone-based triple therapy (furazolidone, amoxicillin, PPI) for 14 days. Unfortunately, there is no universally accepted treatment regimen recommended for patients who have failed two courses of antibiotics. If eradication is still not achieved in a compliant patient, then culture and sensitivity of the organism should be considered. Additional factors that may lower eradication rates include the patient’s country of origin (higher in Northeast Asia than other parts of Asia or Europe) and cigarette smoking. In addition, meta-analysis suggests that even the most effective regimens (quadruple therapy including PPI, bismuth, tetracycline, and metronidazole and triple therapy including PPI, clarithromycin, and amoxicillin) may have suboptimal eradication rates (<80%), thus demonstrating the need for the development of more efficacious treatments.
In view of the observation that 15–25% of patients treated with first-line therapy may still remain infected with the organism, new approaches to treatment have been explored. One promising approach is sequential therapy. Regimens examined consist of 5 days of amoxicillin and a PPI, followed by an additional 5 days of PPI plus tinidazole and clarithromycin or levofloxacin. One promising regimen that has the benefit of being shorter in duration, easier to take, and less expensive is 5 days of concomitant therapy (PPI twice daily, amoxicillin 1 g twice daily, levofloxacin 500 mg twice daily, and tinidazole 500 mg twice daily). Initial studies have demonstrated eradication rates of >90% with good patient tolerance. Confirmation of these findings and applicability of this approach in the United States are needed, although some experts are recommending abandoning clarithromycin-based triple therapy in the United States for the concomitant therapy or the alternative sequential therapies highlighted above.
Innovative non–antibiotic-mediated approaches have been explored in an effort to improve eradication rates of H. pylori. Pretreatment of patients with N-acetylcysteine as a mucolytic agent to destroy the H. pylori biofilm and therefore impair antibiotic resistance has been examined, but more studies are needed to confirm the applicability of this approach. In vitro studies suggest that certain probiotics like Lactobacillus or its metabolites can inhibit H. pylori. Administration of probiotics has been attempted in several clinical studies in an effort to maximize antibiotic-mediated eradication with varying results. Overall, it appears that the use of certain probiotics, such as Lactobacillus spp., Saccharomyces spp., Bifidobacterium spp., and Bacillus clausii, did not alter eradication rates but importantly decreased antibiotic-associated side effects including nausea, dysgeusia, diarrhea, and abdominal discomfort/pain, resulting in enhanced tolerability of H. pylori therapies. Additional studies are needed to confirm the potential benefits of probiotics in this setting.
Reinfection after successful eradication of H. pylori is rare in the United States (<1% per year). If recurrent infection occurs within the first 6 months after completing therapy, the most likely explanation is recrudescence as opposed to reinfection.
THERAPY OF NSAID-RELATED GASTRIC OR DUODENAL INJURY
Medical intervention for NSAID-related mucosal injury includes treatment of an active ulcer and primary prevention of future injury. Recommendations for the treatment and primary prevention of NSAID-related mucosal injury are listed in Table 348-5. Ideally, the injurious agent should be stopped as the first step in the therapy of an active NSAID-induced ulcer. If that is possible, then treatment with one of the acid inhibitory agents (H2 blockers, PPIs) is indicated. Cessation of NSAIDs is not always possible because of the patient’s severe underlying disease. Only PPIs can heal GUs or DUs, independent of whether NSAIDs are discontinued.
|
RECOMMENDATIONS FOR TREATMENT OF NSAID-RELATED MUCOSAL INJURY |
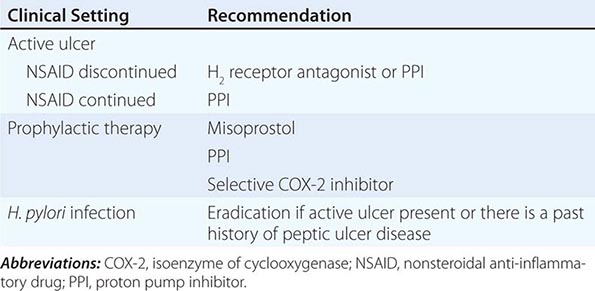
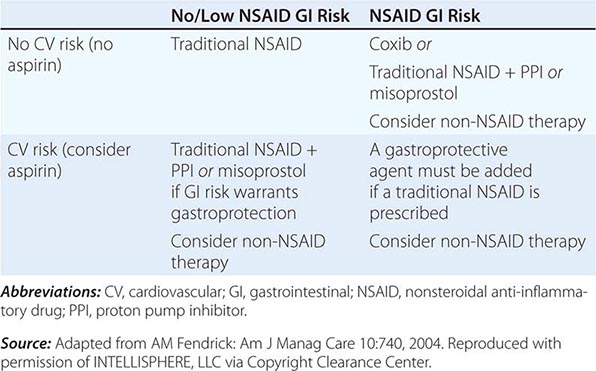
The approach to primary prevention has included avoiding the agent, using the lowest possible dose of the agent, using NSAIDs that are theoretically less injurious, using newer topical NSAID preparations, and/or using concomitant medical therapy to prevent NSAID-induced injury. Several nonselective NSAIDs that are associated with a lower likelihood of GI toxicity include diclofenac, aceclofenac, and ibuprofen, although the beneficial effect may be eliminated if higher dosages of the agents are used. Primary prevention of NSAID-induced ulceration can be accomplished by misoprostol (200 μg qid) or a PPI. High-dose H2 blockers (famotidine, 40 mg bid) have also shown some promise in preventing endoscopically documented ulcers, although PPIs are superior. The highly selective COX-2 inhibitors, celecoxib and rofecoxib, are 100 times more selective inhibitors of COX-2 than standard NSAIDs, leading to gastric or duodenal mucosal injury that is comparable to placebo; their utilization led to an increase in cardiovascular events and withdrawal from the market. Additional caution was engendered when the CLASS study demonstrated that the advantage of celecoxib in preventing GI complications was offset when low-dose aspirin was used simultaneously. Therefore, gastric protection therapy is required in individuals taking COX-2 inhibitors and aspirin prophylaxis. Finally, much of the work demonstrating the benefit of COX-2 inhibitors and PPIs on GI injury has been performed in individuals of average risk; it is unclear if the same level of benefit will be achieved in high-risk patients. For example, concomitant use of warfarin and a COX-2 inhibitor was associated with rates of GI bleeding similar to those observed in patients taking nonselective NSAIDs. A combination of factors, including withdrawal of the majority of COX-2 inhibitors from the market, the observation that low-dose aspirin appears to diminish the beneficial effect of COX-2 selective inhibitors, and the growing use of aspirin for prophylaxis of cardiovascular events, have significantly altered the approach to gastric protective therapy during the use of NSAIDs. A set of guidelines for the approach to the use of NSAIDs was published by the American College of Gastroenterology and is shown in Table 348-6. Individuals who are not at risk for cardiovascular events, do not use aspirin, and are without risk for GI complications can receive nonselective NSAIDs without gastric protection. In those without cardiovascular risk factors but with a high potential risk (prior GI bleeding or multiple GI risk factors) for NSAID-induced GI toxicity, cautious use of a selective COX-2 inhibitor and co-therapy with misoprostol or high-dose PPI are recommended. Individuals at moderate GI risk without cardiac risk factors can be treated with a COX-2 inhibitor alone or with a nonselective NSAID with misoprostol or a PPI. Individuals with cardiovascular risk factors, who require low-dose aspirin and have low potential for NSAID-induced toxicity, should be considered for a non-NSAID agent or use of a traditional NSAID in combination with gastric protection, if warranted. Finally, individuals with cardiovascular and GI risks who require aspirin must be considered for non-NSAID therapy, but if that is not an option, then gastric protection with any type of NSAID must be considered. Any patient, regardless of risk status, who is being considered for long-term traditional NSAID therapy, should also be considered for H. pylori testing and treatment if positive. Assuring the use of GI protective agents with NSAIDs is difficult, even in high-risk patients. This is in part due to underprescribing of the appropriate protective agent; other times the difficulty is related to patient compliance. The latter may be due to patients forgetting to take multiple pills or preferring not to take the extra pill, especially if they have no GI symptoms. Several NSAID gastroprotective-containing combination pills are now commercially available, including double-dose famotidine with ibuprofen, diclofenac with misoprostol, and naproxen with esomeprazole. Although initial studies suggested improved compliance and a cost advantage when taking these combination drugs, their clinical benefit over the use of separate pills has not been established. Efforts continue toward developing safer NSAIDs, including NO–releasing NSAIDs, hydrogen sulfide–releasing NSAIDs, dual COX/5-LOX inhibitors, NSAID prodrugs, or agents that can effectively sequester unbound NSAIDs without interfering with their efficacy.
|
GUIDE TO NSAID THERAPY |
APPROACH AND THERAPY: SUMMARY
Controversy continues regarding the best approach to the patient who presents with dyspepsia (Chap. 54). The discovery of H. pylori and its role in pathogenesis of ulcers has added a new variable to the equation. Previously, if a patient <50 years of age presented with dyspepsia and without alarming signs or symptoms suggestive of an ulcer complication or malignancy, an empirical therapeutic trial with acid suppression was commonly recommended. Although this approach is practiced by some today, an approach presently gaining approval for the treatment of patients with dyspepsia is outlined in Fig. 348-12. The referral to a gastroenterologist is for the potential need of endoscopy and subsequent evaluation and treatment if the endoscopy is negative.
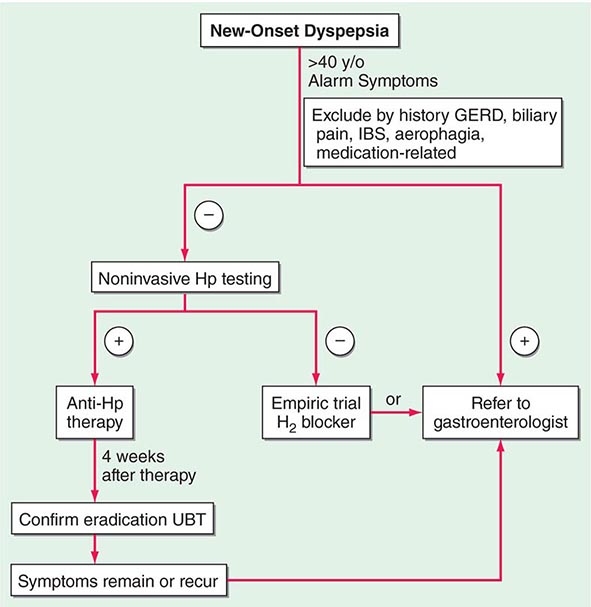
FIGURE 348-12 Overview of new-onset dyspepsia. GERD, gastroesophageal reflux disease; Hp, Helicobacter pylori; IBS, irritable bowel syndrome; UBT, urea breath test. (Adapted from BS Anand and DY Graham: Endoscopy 31:215, 1999.)
Once an ulcer (GU or DU) is documented, the main issue at stake is whether H. pylori or an NSAID is involved. With H. pylori present, independent of the NSAID status, triple therapy is recommended for 14 days, followed by continued acid-suppressing drugs (H2 receptor antagonist or PPIs) for a total of 4–6 weeks. Selection of patients for documentation of H. pylori eradication (organisms gone at least 4 weeks after completing antibiotics) is an area of some debate. The test of choice for documenting eradication is the laboratory-based validated monoclonal stool antigen test or a urea breath test (UBT). The patient must be off antisecretory agents when being tested for eradication of H. pylori with UBT or stool antigen. Serologic testing is not useful for the purpose of documenting eradication because antibody titers fall slowly and often do not become undetectable. Two approaches toward documentation of eradication exist: (1) Test for eradication only in individuals with a complicated course or in individuals who are frail or with multisystem disease who would do poorly with an ulcer recurrence, and (2) test all patients for successful eradication. Some recommend that patients with complicated ulcer disease, or who are frail, should be treated with long-term acid suppression, thus making documentation of H. pylori eradication a moot point. In view of this discrepancy in practice, it would be best to discuss with the patient the different options available.
Several issues differentiate the approach to a GU versus a DU. GUs, especially of the body and fundus, have the potential of being malignant. Multiple biopsies of a GU should be taken initially; even if these are negative for neoplasm, repeat endoscopy to document healing at 8–12 weeks should be performed, with biopsy if the ulcer is still present. About 70% of GUs eventually found to be malignant undergo significant (usually incomplete) healing. Repeat endoscopy is warranted in patients with DU if symptoms persist despite medical therapy or a complication is suspected.
The majority (>90%) of GUs and DUs heal with the conventional therapy outlined above. A GU that fails to heal after 12 weeks and a DU that does not heal after 8 weeks of therapy should be considered refractory. Once poor compliance and persistent H. pylori infection have been excluded, NSAID use, either inadvertent or surreptitious, must be excluded. In addition, cigarette smoking must be eliminated. For a GU, malignancy must be meticulously excluded. Next, consideration should be given to a gastric acid hypersecretory state such as ZES (see “Zollinger-Ellison Syndrome,” below) or the idiopathic form, which can be excluded with gastric acid analysis. Although a subset of patients have gastric acid hypersecretion of unclear etiology as a contributing factor to refractory ulcers, ZES should be excluded with a fasting gastrin or secretin stimulation test (see below). More than 90% of refractory ulcers (either DUs or GUs) heal after 8 weeks of treatment with higher doses of PPI (omeprazole, 40 mg/d; lansoprazole 30–60 mg/d). This higher dose is also effective in maintaining remission. Surgical intervention may be a consideration at this point; however, other rare causes of refractory ulcers must be excluded before recommending surgery. Rare etiologies of refractory ulcers that may be diagnosed by gastric or duodenal biopsies include ischemia, Crohn’s disease, amyloidosis, sarcoidosis, lymphoma, eosinophilic gastroenteritis, or infection (cytomegalovirus [CMV], tuberculosis, or syphilis).
Surgical intervention in PUD can be viewed as being either elective, for treatment of medically refractory disease, or as urgent/emergent, for the treatment of an ulcer-related complication. The development of pharmacologic and endoscopic approaches for the treatment of peptic disease and its complications has led to a substantial decrease in the number of operations needed for this disorder with a drop of over 90% for elective ulcer surgery over the last four decades. Refractory ulcers are an exceedingly rare occurrence. Surgery is more often required for treatment of an ulcer-related complication.
Hemorrhage is the most common ulcer-related complication, occurring in ~15–25% of patients. Bleeding may occur in any age group but is most often seen in older patients (sixth decade or beyond). The majority of patients stop bleeding spontaneously, but endoscopic therapy (Chap. 345) is necessary in some. Parenterally and orally administered PPIs also decrease ulcer rebleeding in patients who have undergone endoscopic therapy. Patients unresponsive or refractory to endoscopic intervention will require surgery (~5% of transfusion-requiring patients).
Free peritoneal perforation occurs in ~2–3% of DU patients. As in the case of bleeding, up to 10% of these patients will not have antecedent ulcer symptoms. Concomitant bleeding may occur in up to 10% of patients with perforation, with mortality being increased substantially. Peptic ulcer can also penetrate into adjacent organs, especially with a posterior DU, which can penetrate into the pancreas, colon, liver, or biliary tree.
Pyloric channel ulcers or DUs can lead to gastric outlet obstruction in ~2–3% of patients. This can result from chronic scarring or from impaired motility due to inflammation and/or edema with pylorospasm. Patients may present with early satiety, nausea, vomiting of undigested food, and weight loss. Conservative management with nasogastric suction, intravenous hydration/nutrition, and antisecretory agents is indicated for 7–10 days with the hope that a functional obstruction will reverse. If a mechanical obstruction persists, endoscopic intervention with balloon dilation may be effective. Surgery should be considered if all else fails.
SPECIFIC OPERATIONS FOR DUODENAL ULCERS
Surgical treatment was originally designed to decrease gastric acid secretion. Operations most commonly performed include (1) vagotomy and drainage (by pyloroplasty, gastroduodenostomy, or gastrojejunostomy), (2) highly selective vagotomy (which does not require a drainage procedure), and (3) vagotomy with antrectomy. The specific procedure performed is dictated by the underlying circumstances: elective versus emergency, the degree and extent of duodenal ulceration, the etiology of the ulcer (H. pylori, NSAIDs, malignancy), and the expertise of the surgeon. Moreover, the trend has been toward a dramatic decrease in the need for surgery for treatment of refractory PUD, and when needed, minimally invasive and anatomy-preserving operations are preferred.
Vagotomy is a component of each of these procedures and is aimed at decreasing acid secretion through ablating cholinergic input to the stomach. Unfortunately, both truncal and selective vagotomy (preserves the celiac and hepatic branches) result in gastric atony despite successful reduction of both basal acid output (BAO; decreased by 85%) and maximal acid output (MAO; decreased by 50%). Drainage through pyloroplasty or gastroduodenostomy is required in an effort to compensate for the vagotomy-induced gastric motility disorder. This procedure has an intermediate complication rate and a 10% ulcer recurrence rate. To minimize gastric dysmotility, highly selective vagotomy (also known as parietal cell, super-selective, or proximal vagotomy) was developed. Only the vagal fibers innervating the portion of the stomach that contains parietal cells is transected, thus leaving fibers important for regulating gastric motility intact. Although this procedure leads to an immediate decrease in both BAO and stimulated acid output, acid secretion recovers over time. By the end of the first postoperative year, basal and stimulated acid output are ~30 and 50%, respectively, of preoperative levels. Ulcer recurrence rates are higher with highly selective vagotomy (≥10%), although the overall complication rates are the lowest of the three procedures.
The procedure that provides the lowest rates of ulcer recurrence (1%) but has the highest complication rate is vagotomy (truncal or selective) in combination with antrectomy. Antrectomy is aimed at eliminating an additional stimulant of gastric acid secretion, gastrin. Two principal types of reanastomoses are used after antrectomy: gastroduodenostomy (Billroth I) or gastrojejunostomy (Billroth II) (Fig. 348-13). Although Billroth I is often preferred over II, severe duodenal inflammation or scarring may preclude its performance. Prospective, randomized studies confirm that partial gastrectomy followed by Roux-en-Y reconstruction leads to a significantly better clinical, endoscopic, and histologic outcome than Billroth II reconstruction.
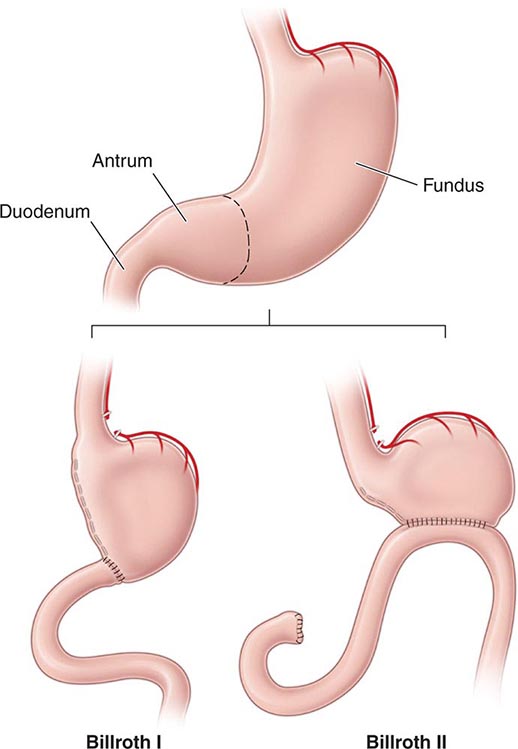
FIGURE 348-13 Schematic representation of Billroth I and II procedures.
Of these procedures, highly selective vagotomy may be the one of choice in the elective setting, except in situations where ulcer recurrence rates are high (prepyloric ulcers and those refractory to medical therapy). Selection of vagotomy and antrectomy may be more appropriate in these circumstances.
These procedures have been traditionally performed by standard laparotomy. The advent of laparoscopic surgery has led several surgical teams to successfully perform highly selective vagotomy, truncal vagotomy/pyloroplasty, and truncal vagotomy/antrectomy through this approach. An increase in the number of laparoscopic procedures for treatment of PUD has occurred. Laparoscopic repair of perforated peptic ulcers is safe, feasible for the experienced surgeon and is associated with decreased postoperative pain, although it does take longer than an open approach. Moreover, no difference between the two approaches is noted in postoperative complications or length of hospital stay.
Specific Operations for Gastric Ulcers The location and the presence of a concomitant DU dictate the operative procedure performed for a GU. Antrectomy (including the ulcer) with a Billroth I anastomosis is the treatment of choice for an antral ulcer. Vagotomy is performed only if a DU is present. Although ulcer excision with vagotomy and drainage procedure has been proposed, the higher incidence of ulcer recurrence makes this a less desirable approach. Ulcers located near the esophagogastric junction may require a more radical approach, a subtotal gastrectomy with a Roux-en-Y esophagogastrojejunostomy (Csendes’ procedure). A less aggressive approach, including antrectomy, intraoperative ulcer biopsy, and vagotomy (Kelling-Madlener procedure), may be indicated in fragile patients with a high GU. Ulcer recurrence approaches 30% with this procedure.
Surgery-Related Complications Complications seen after surgery for PUD are related primarily to the extent of the anatomic modification performed. Minimal alteration (highly selective vagotomy) is associated with higher rates of ulcer recurrence and less GI disturbance. More aggressive surgical procedures have a lower rate of ulcer recurrence but a greater incidence of GI dysfunction. Overall, morbidity and mortality related to these procedures are quite low. Morbidity associated with vagotomy and antrectomy or pyloroplasty is ≤5%, with mortality ~1%. Highly selective vagotomy has lower morbidity and mortality rates of 1 and 0.3%, respectively.
In addition to the potential early consequences of any intraabdominal procedure (bleeding, infection, thromboembolism), gastroparesis, duodenal stump leak, and efferent loop obstruction can be observed.
RECURRENT ULCERATION The risk of ulcer recurrence is directly related to the procedure performed. Ulcers that recur after partial gastric resection tend to develop at the anastomosis (stomal or marginal ulcer). Epigastric abdominal pain is the most frequent presenting complaint (>90%). Severity and duration of pain tend to be more progressive than observed with DUs before surgery.
Ulcers may recur for several reasons, including incomplete vagotomy, inadequate drainage, retained antrum, and, less likely, persistent or recurrent H. pylori infection. ZES should have been excluded preoperatively. Surreptitious use of NSAIDs is an important reason for recurrent ulcers after surgery, especially if the initial procedure was done for an NSAID-induced ulcer. Once H. pylori and NSAIDs have been excluded as etiologic factors, the question of incomplete vagotomy or retained gastric antrum should be explored. For the latter, fasting plasma gastrin levels should be determined. If elevated, retained antrum or ZES (see below) should be considered. Incomplete vagotomy can be ruled out by gastric acid analysis coupled with sham feeding. In this test, gastric acid output is measured while the patient sees, smells, and chews a meal (without swallowing). The cephalic phase of gastric secretion, which is mediated by the vagus, is being assessed with this study. An increase in gastric acid output in response to sham feeding is evidence that the vagus nerve is intact. A rise in serum pancreatic polypeptide >50% within 30 min of sham feeding is also suggestive of an intact vagus nerve.
Medical therapy with H2 blockers will heal postoperative ulceration in 70–90% of patients. The efficacy of PPIs has not been fully assessed in this group, but one may anticipate greater rates of ulcer healing compared to those obtained with H2 blockers. Repeat operation (complete vagotomy, partial gastrectomy) may be required in a small subgroup of patients who have not responded to aggressive medical management.
AFFERENT LOOP SYNDROMES Although rarely seen today as a result of the decrease in the performance of Billroth II anastomosis, two types of afferent loop syndrome can occur in patients who have undergone this type of partial gastric resection. The more common of the two is bacterial overgrowth in the afferent limb secondary to stasis. Patients may experience postprandial abdominal pain, bloating, and diarrhea with concomitant malabsorption of fats and vitamin B12. Cases refractory to antibiotics may require surgical revision of the loop. The less common afferent loop syndrome can present with severe abdominal pain and bloating that occur 20–60 min after meals. Pain is often followed by nausea and vomiting of bile-containing material. The pain and bloating may improve after emesis. The cause of this clinical picture is theorized to be incomplete drainage of bile and pancreatic secretions from an afferent loop that is partially obstructed. Cases refractory to dietary measures may need surgical revision or conversion of the Billroth II anastomosis to a Roux-en-Y gastrojejunostomy.
DUMPING SYNDROME Dumping syndrome consists of a series of vasomotor and GI signs and symptoms and occurs in patients who have undergone vagotomy and drainage (especially Billroth procedures). Two phases of dumping, early and late, can occur. Early dumping takes place 15–30 min after meals and consists of crampy abdominal discomfort, nausea, diarrhea, belching, tachycardia, palpitations, diaphoresis, light-headedness, and, rarely, syncope. These signs and symptoms arise from the rapid emptying of hyperosmolar gastric contents into the small intestine, resulting in a fluid shift into the gut lumen with plasma volume contraction and acute intestinal distention. Release of vasoactive GI hormones (vasoactive intestinal polypeptide, neurotensin, motilin) is also theorized to play a role in early dumping.
The late phase of dumping typically occurs 90 min to 3 h after meals. Vasomotor symptoms (light-headedness, diaphoresis, palpitations, tachycardia, and syncope) predominate during this phase. This component of dumping is thought to be secondary to hypoglycemia from excessive insulin release.
Dumping syndrome is most noticeable after meals rich in simple carbohydrates (especially sucrose) and high osmolarity. Ingestion of large amounts of fluids may also contribute. Up to 50% of postvagotomy and drainage patients will experience dumping syndrome to some degree early on. Signs and symptoms often improve with time, but a severe protracted picture can occur in up to 1% of patients.
Dietary modification is the cornerstone of therapy for patients with dumping syndrome. Small, multiple (six) meals devoid of simple carbohydrates coupled with elimination of liquids during meals is important. Antidiarrheals and anticholinergic agents are complementary to diet. Guar and pectin, which increase the viscosity of intraluminal contents, may be beneficial in more symptomatic individuals. Acarbose, an α-glucosidase inhibitor that delays digestion of ingested carbohydrates, has also been shown to be beneficial in the treatment of the late phases of dumping. The somatostatin analogue octreotide has been successful in diet-refractory cases. This drug is administered subcutaneously (50 μg tid), titrated according to clinical response. A long-acting depot formulation of octreotide can be administered once every 28 days and provides symptom relief comparable to the short-acting agent. In addition, patient weight gain and quality of life appear to be superior with the long-acting form.
POSTVAGOTOMY DIARRHEA Up to 10% of patients may seek medical attention for the treatment of postvagotomy diarrhea. This complication is most commonly observed after truncal vagotomy, which is rarely performed today. Patients may complain of intermittent diarrhea that occurs typically 1–2 h after meals. Occasionally the symptoms may be severe and relentless. This is due to a motility disorder from interruption of the vagal fibers supplying the luminal gut. Other contributing factors may include decreased absorption of nutrients (see below), increased excretion of bile acids, and release of luminal factors that promote secretion. Diphenoxylate or loperamide is often useful in symptom control. The bile salt–binding agent cholestyramine may be helpful in severe cases. Surgical reversal of a 10-cm segment of jejunum may yield a substantial improvement in bowel frequency in a subset of patients.
BILE REFLUX GASTROPATHY A subset of post–partial gastrectomy patients who present with abdominal pain, early satiety, nausea, and vomiting will have mucosal erythema of the gastric remnant as the only finding. Histologic examination of the gastric mucosa reveals minimal inflammation but the presence of epithelial cell injury. This clinical picture is categorized as bile or alkaline reflux gastropathy/gastritis. Although reflux of bile is implicated as the reason for this disorder, the mechanism is unknown. Prokinetic agents, cholestyramine, and sucralfate have been somewhat effective treatments. Severe refractory symptoms may require using either nuclear scanning with 99mTc-HIDA to document reflux or an alkaline challenge test, where 0.1 N NaOH is infused into the stomach in an effort to reproduce the patient’s symptoms. Surgical diversion of pancreaticobiliary secretions away from the gastric remnant with a Roux-en-Y gastrojejunostomy consisting of a long (50–60 cm) Roux limb has been used in severe cases. Bilious vomiting improves, but early satiety and bloating may persist in up to 50% of patients.
MALDIGESTION AND MALABSORPTION Weight loss can be observed in up to 60% of patients after partial gastric resection. Patients can experience a 10% loss of body weight, which stabilizes 3 months postoperatively. A significant component of this weight reduction is due to decreased oral intake. However, mild steatorrhea can also develop. Reasons for maldigestion/malabsorption include decreased gastric acid production, rapid gastric emptying, decreased food dispersion in the stomach, reduced luminal bile concentration, reduced pancreatic secretory response to feeding, and rapid intestinal transit.
Decreased serum vitamin B12 levels can be observed after partial gastrectomy. This is usually not due to deficiency of IF, since a minimal amount of parietal cells (source of IF) are removed during antrectomy. Reduced vitamin B12 may be due to competition for the vitamin by bacterial overgrowth or inability to split the vitamin from its protein-bound source due to hypochlorhydria.
Iron-deficiency anemia may be a consequence of impaired absorption of dietary iron in patients with a Billroth II gastrojejunostomy. Absorption of iron salts is normal in these individuals; thus, a favorable response to oral iron supplementation can be anticipated. Folate deficiency with concomitant anemia can also develop in these patients. This deficiency may be secondary to decreased absorption or diminished oral intake.
Malabsorption of vitamin D and calcium resulting in osteoporosis and osteomalacia is common after partial gastrectomy and gastrojejunostomy (Billroth II). Osteomalacia can occur as a late complication in up to 25% of post–partial gastrectomy patients. Bone fractures occur twice as commonly in men after gastric surgery as in a control population. It may take years before x-ray findings demonstrate diminished bone density. Elevated alkaline phosphatase, reduced serum calcium, bone pain, and pathologic fractures may be seen in patients with osteomalacia. The high incidence of these abnormalities in this subgroup of patients justifies treating them with vitamin D and calcium supplementation indefinitely. Therapy is especially important in females. Copper deficiency has also been reported in patients undergoing surgeries that bypass the duodenum, where copper is primarily absorbed. Patients may present with a rare syndrome that includes ataxia, myelopathy, and peripheral neuropathy.
GASTRIC ADENOCARCINOMA The incidence of adenocarcinoma in the gastric stump is increased 15 years after resection. Some have reported a four- to fivefold increase in gastric cancer 20–25 years after resection. The pathogenesis is unclear but may involve alkaline reflux, bacterial proliferation, or hypochlorhydria. The role of endoscopic screening is not clear, and most guidelines do not support its use.
ADDITIONAL COMPLICATIONS Reflux esophagitis and a higher incidence of gallstones and cholecystitis have been reported to patients undergoing subtotal gastrectomy. The latter is thought to be due to decreased gallbladder contractility associated with vagotomy and bypass of the duodenum, leading to decreased postprandial release of cholecystokinin.
RELATED CONDITIONS
ZOLLINGER–ELLISON SYNDROME
Severe peptic ulcer diathesis secondary to gastric acid hypersecretion due to unregulated gastrin release from a non-β cell endocrine tumor (gastrinoma) defines the components of ZES. Initially, ZES was typified by aggressive and refractory ulceration in which total gastrectomy provided the only chance for enhancing survival. Today it can be cured by surgical resection in up to 40% of patients.
Epidemiology The incidence of ZES varies from 0.1–1% of individuals presenting with PUD. Males are more commonly affected than females, and the majority of patients are diagnosed between ages 30 and 50. Gastrinomas are classified into sporadic tumors (more common) and those associated with multiple endocrine neoplasia (MEN) type 1 (see below). The widespread availability and use of PPIs has led to a decreased patient referral for gastrinoma evaluation, delay in diagnosis, and an increase in false-positive diagnoses of ZES. In fact, diagnosis may be delayed for 6 or more years after symptoms consistent with ZES are displayed.
Pathophysiology Hypergastrinemia originating from an autonomous neoplasm is the driving force responsible for the clinical manifestations in ZES. Gastrin stimulates acid secretion through gastrin receptors on parietal cells and by inducing histamine release from ECL cells. Gastrin also has a trophic action on gastric epithelial cells. Long-standing hypergastrinemia leads to markedly increased gastric acid secretion through both parietal cell stimulation and increased parietal cell mass. The increased gastric acid output leads to peptic ulcer diathesis, erosive esophagitis, and diarrhea.
Tumor Distribution Although early studies suggested that the vast majority of gastrinomas occurred within the pancreas, a significant number of these lesions are extrapancreatic. Over 80% of these tumors are found within the hypothetical gastrinoma triangle (confluence of the cystic and common bile ducts superiorly, junction of the second and third portions of the duodenum inferiorly, and junction of the neck and body of the pancreas medially). Duodenal tumors constitute the most common nonpancreatic lesion; between 50 and 75% of gastrinomas are found here. Duodenal tumors are smaller, slower growing, and less likely to metastasize than pancreatic lesions. Less common extrapancreatic sites include stomach, bones, ovaries, heart, liver, and lymph nodes. More than 60% of tumors are considered malignant, with up to 30–50% of patients having multiple lesions or metastatic disease at presentation. Histologically, gastrin-producing cells appear well-differentiated, expressing markers typically found in endocrine neoplasms (chromogranin, neuron-specific enolase).
Clinical Manifestations Gastric acid hypersecretion is responsible for the signs and symptoms observed in patients with ZES. Peptic ulcer is the most common clinical manifestation, occurring in >90% of gastrinoma patients. Initial presentation and ulcer location (duodenal bulb) may be indistinguishable from common PUD. Clinical situations that should create suspicion of gastrinoma are ulcers in unusual locations (second part of the duodenum and beyond), ulcers refractory to standard medical therapy, ulcer recurrence after acid-reducing surgery, ulcers presenting with frank complications (bleeding, obstruction, and perforation), or ulcers in the absence of H. pylori or NSAID ingestion. Symptoms of esophageal origin are present in up to two-thirds of patients with ZES, with a spectrum ranging from mild esophagitis to frank ulceration with stricture and Barrett’s mucosa.
Diarrhea, the next most common clinical manifestation, is found in up to 50% of patients. Although diarrhea often occurs concomitantly with acid peptic disease, it may also occur independent of an ulcer. Etiology of the diarrhea is multifactorial, resulting from marked volume overload to the small bowel, pancreatic enzyme inactivation by acid, and damage of the intestinal epithelial surface by acid. The epithelial damage can lead to a mild degree of maldigestion and malabsorption of nutrients. The diarrhea may also have a secretory component due to the direct stimulatory effect of gastrin on enterocytes or the co-secretion of additional hormones from the tumor such as vasoactive intestinal peptide.
Gastrinomas can develop in the presence of MEN 1 syndrome (Chaps. 113 and 408) in ~25% of patients. This autosomal dominant disorder involves primarily three organ sites: the parathyroid glands (80–90%), pancreas (40–80%), and pituitary gland (30–60%). The syndrome is caused by inactivating mutations of the MEN1 tumor suppressor gene found on the long arm of chromosome 11q13. The gene encodes for Menin, which has an important role in DNA replication and transcriptional regulation. A genetic diagnosis is obtained by sequencing of the MEN1 gene, which can reveal mutations in 70–90% of typical MEN 1 cases. A family may have an unknown mutation, making a genetic diagnosis impossible, and therefore certain individuals will require a clinical diagnosis, which is determined by whether a patient has tumors in two of the three endocrine organs (parathyroid, pancreas/duodenum, or pituitary) or has a family history of MEN 1 and one of the endocrine organ tumors. In view of the stimulatory effect of calcium on gastric secretion, the hyperparathyroidism and hypercalcemia seen in MEN 1 patients may have a direct effect on ulcer disease. Resolution of hypercalcemia by parathyroidectomy reduces gastrin and gastric acid output in gastrinoma patients. An additional distinguishing feature in ZES patients with MEN 1 is the higher incidence of gastric carcinoid tumor development (as compared to patients with sporadic gastrinomas). ZES presents and is diagnosed earlier in MEN 1 patients, and they have a more indolent course as compared to patients with sporadic gastrinoma. Gastrinomas tend to be smaller, multiple, and located in the duodenal wall more often than is seen in patients with sporadic ZES. Establishing the diagnosis of MEN 1 is critical in order to provide genetic counseling to the patient and his or her family and also to determine the recommended surgical approach.
Diagnosis Biochemical measurements of gastrin and acid secretion in patients suspected of ZES play an important role is establishing this rare diagnosis. Often, patients suspected of having ZES will be treated with a PPI in an effort to ameliorate symptoms and decrease the likelihood of possible acid-related complications. The presence of the PPI, which will lower acid secretion and potentially elevate fasting gastrin levels in normal individuals, will make the diagnostic approach in these individuals somewhat difficult. Significant morbidity related to peptic diathesis has been described when stopping PPIs in gastrinoma patients; therefore, a systematic approach in stopping these agents is warranted (see below). The first step in the evaluation of a patient suspected of having ZES is to obtain a fasting gastrin level. A list of clinical scenarios that should arouse suspicion regarding this diagnosis is shown in Table 348-7. Fasting gastrin levels obtained using a dependable assay are usually <150 pg/mL. A normal fasting gastrin, on two separate occasions, especially if the patient is on a PPI, virtually excludes this diagnosis. Virtually all gastrinoma patients will have a gastrin level >150–200 pg/mL. Measurement of fasting gastrin should be repeated to confirm the clinical suspicion. Some of the commercial biochemical assays used for measuring serum gastrin may be inaccurate. Variable specificity of the antibodies used have led to both false-positive and false-negative fasting gastrin levels, placing in jeopardy the ability to make an accurate diagnosis of ZES.
|
WHEN TO OBTAIN A FASTING SERUM GASTRIN LEVEL |
Multiple processes can lead to an elevated fasting gastrin level, the most frequent of which are gastric hypochlorhydria and achlorhydria, with or without pernicious anemia. Gastric acid induces feedback inhibition of gastrin release. A decrease in acid production will subsequently lead to failure of the feedback inhibitory pathway, resulting in net hypergastrinemia. Gastrin levels will thus be high in patients using antisecretory agents for the treatment of acid peptic disorders and dyspepsia. H. pylori infection can also cause hypergastrinemia. Additional causes of elevated gastrin include retained gastric antrum; G cell hyperplasia; gastric outlet obstruction; renal insufficiency; massive small-bowel obstruction; and conditions such as rheumatoid arthritis, vitiligo, diabetes mellitus, and pheochromocytoma. Although a fasting gastrin >10 times normal is highly suggestive of ZES, two-thirds of patients will have fasting gastrin levels that overlap with levels found in the more common disorders outlined above, especially if a PPI is being taken by the patient. The effect of the PPI on gastrin levels and acid secretion will linger several days after stopping the PPI; therefore, it should be stopped for a minimum of 7 days before testing. During this period, the patient should be placed on a histamine H2 antagonist, such as famotidine, twice to three times per day. Although this type of agent has a short-term effect on gastrin and acid secretion, it needs to be stopped 24 h before repeating fasting gastrin levels or performing some the tests highlighted below. The patient may take antacids for the final day, stopping them approximately 12 h before testing is performed. Heightened awareness of complications related to gastric acid hypersecretion during the period of PPI cessation is critical.
The next step in establishing a biochemical diagnosis of gastrinoma is to assess acid secretion. Nothing further needs to be done if decreased acid output in the absence of a PPI is observed. A pH can be measured on gastric fluid obtained either during endoscopy or through nasogastric aspiration; a pH <3 is suggestive of a gastrinoma, but a pH >3 is not helpful in excluding the diagnosis. In those situations where the pH is >3, formal gastric acid analysis should be performed if available. Normal BAO in nongastric surgery patients is typically <5 meq/h. A BAO >15 meq/h in the presence of hypergastrinemia is considered pathognomonic of ZES, but up to 12% of patients with common PUD may have elevated BAO to a lesser degree that can overlap with levels seen in ZES patients. In an effort to improve the sensitivity and specificity of gastric secretory studies, a BAO/MAO ratio was established using pentagastrin infusion as a way to maximally stimulate acid production, with a BAO/MAO ratio >0.6 being highly suggestive of ZES. Pentagastrin is no longer available in the United States, making measurement of MAO virtually impossible. An endoscopic method for measuring gastric acid output has been developed but requires further validation.
Gastrin provocative tests have been developed in an effort to differentiate between the causes of hypergastrinemia and are especially helpful in patients with indeterminate acid secretory studies. The tests are the secretin stimulation test and the calcium infusion study. The most sensitive and specific gastrin provocative test for the diagnosis of gastrinoma is the secretin study. An increase in gastrin of ≥120 pg within 15 min of secretin injection has a sensitivity and specificity of >90% for ZES. PPI-induced hypochlorhydria or achlorhydria may lead to a false-positive secretin test; thus, this agent must be stopped for 1 week before testing.
The calcium infusion study is less sensitive and specific than the secretin test, which, coupled with it being a more cumbersome study with greater potential for adverse effects, relegates it to rare utilization in the cases where the patient’s clinical characteristics are highly suggestive of ZES but the secretin stimulation is inconclusive.
Tumor Localization Once the biochemical diagnosis of gastrinoma has been confirmed, the tumor must be located. Multiple imaging studies have been used in an effort to enhance tumor localization (Table 348-8). The broad range of sensitivity is due to the variable success rates achieved by the different investigative groups. Endoscopic ultrasound (EUS) permits imaging of the pancreas with a high degree of resolution (<5 mm). This modality is particularly helpful in excluding small neoplasms within the pancreas and in assessing the presence of surrounding lymph nodes and vascular involvement, but it is not very sensitive for finding duodenal lesions. Several types of endocrine tumors express cell-surface receptors for somatostatin. This permits the localization of gastrinomas by measuring the uptake of the stable somatostatin analogue111 In-pentreotide (OctreoScan) with sensitivity and specificity rates of >85%.
|
SENSITIVITY OF IMAGING STUDIES IN ZOLLINGER-ELLISON SYNDROME |
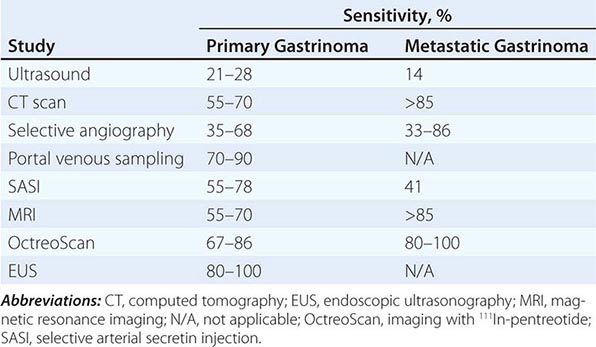
Up to 50% of patients have metastatic disease at diagnosis. Success in controlling gastric acid hypersecretion has shifted the emphasis of therapy toward providing a surgical cure. Detecting the primary tumor and excluding metastatic disease are critical in view of this paradigm shift. Once a biochemical diagnosis has been confirmed, the patient should first undergo an abdominal computed tomography (CT) scan, magnetic resonance imaging (MRI), or OctreoScan (depending on availability) to exclude metastatic disease. In addition, the positron emitter 68Ga has been used to label somatostatin analogues for positron emission tomography (PET) with some success. In addition, hybrid scanners combining CT scan with PET scan are also available in certain specialized centers. Once metastatic disease has been excluded, an experienced endocrine surgeon may opt for exploratory laparotomy with intraoperative ultrasound or transillumination. In other centers, careful examination of the peripancreatic area with EUS, accompanied by endoscopic exploration of the duodenum for primary tumors, will be performed before surgery. Selective arterial secretin injection may be a useful adjuvant for localizing tumors in a subset of patients. The extent of the diagnostic and surgical approach must be carefully balanced with the patient’s overall physiologic condition and the natural history of a slow-growing gastrinoma.
STRESS-RELATED MUCOSAL INJURY
Patients suffering from shock, sepsis, massive burns, severe trauma, or head injury can develop acute erosive gastric mucosal changes or frank ulceration with bleeding. Classified as stress-induced gastritis or ulcers, injury is most commonly observed in the acid-producing (fundus and body) portions of the stomach. The most common presentation is GI bleeding, which is usually minimal but can occasionally be life threatening. Respiratory failure requiring mechanical ventilation and underlying coagulopathy are risk factors for bleeding, which tends to occur 48–72 h after the acute injury or insult.
Histologically, stress injury does not contain inflammation or H. pylori; thus, “gastritis” is a misnomer. Although elevated gastric acid secretion may be noted in patients with stress ulceration after head trauma (Cushing’s ulcer) and severe burns (Curling’s ulcer), mucosal ischemia, breakdown of the normal protective barriers of the stomach, systemic release of cytokines, poor GI motility, and oxidative stress also play an important role in the pathogenesis. Acid must contribute to injury in view of the significant drop in bleeding noted when acid inhibitors are used as prophylaxis for stress gastritis.
Improvement in the general management of intensive care unit patients has led to a significant decrease in the incidence of GI bleeding due to stress ulceration. The estimated decrease in bleeding is from 20–30% to <5%. This improvement has led to some debate regarding the need for prophylactic therapy. The high mortality associated with stress-induced clinically important GI bleeding (>40%) and the limited benefit of medical (endoscopic, angiographic) and surgical therapy in a patient with hemodynamically compromising bleeding associated with stress ulcer/gastritis support the use of preventive measures in high-risk patients (mechanically ventilated, coagulopathy, multiorgan failure, or severe burns). Maintenance of gastric pH >3.5 with continuous infusion of H2 blockers or liquid antacids administered every 2–3 h are viable options. Tolerance to the H2 blocker is likely to develop; thus, careful monitoring of the gastric pH and dose adjustment are important if H2 blockers are used. Sucralfate slurry (1 g every 4–6 h) has also been somewhat successful but requires a gastric tube and may lead to constipation and aluminum toxicity. Sucralfate use in endotracheal intubated patients has also been associated with aspiration pneumonia. Meta-analysis comparing H2 blockers with PPIs for the prevention of stress-associated clinically important and overt GI bleeding demonstrates superiority of the latter without increasing the risk of nosocomial infections, increasing mortality, or prolonging intensive care unit length of stay. Therefore, PPIs are the treatment of choice for stress prophylaxis. Oral PPI is the best option if the patient can tolerate enteral administration. Pantoprazole is available as an intravenous formulation for individuals in whom enteral administration is not possible. If bleeding occurs despite these measures, endoscopy, intraarterial vasopressin, and embolization are options. If all else fails, then surgery should be considered. Although vagotomy and antrectomy may be used, the better approach would be a total gastrectomy, which has an exceedingly high mortality rate in this setting.
GASTRITIS
The term gastritis should be reserved for histologically documented inflammation of the gastric mucosa. Gastritis is not the mucosal erythema seen during endoscopy and is not interchangeable with “dyspepsia.” The etiologic factors leading to gastritis are broad and heterogeneous. Gastritis has been classified based on time course (acute vs chronic), histologic features, and anatomic distribution or proposed pathogenic mechanism (Table 348-9).
|
CLASSIFICATION OF GASTRITIS |
The correlation between the histologic findings of gastritis, the clinical picture of abdominal pain or dyspepsia, and endoscopic findings noted on gross inspection of the gastric mucosa is poor. Therefore, there is no typical clinical manifestation of gastritis.
Acute Gastritis The most common causes of acute gastritis are infectious. Acute infection with H. pylori induces gastritis. However, H. pylori acute gastritis has not been extensively studied. It is reported as presenting with sudden onset of epigastric pain, nausea, and vomiting, and limited mucosal histologic studies demonstrate a marked infiltrate of neutrophils with edema and hyperemia. If not treated, this picture will evolve into one of chronic gastritis. Hypochlorhydria lasting for up to 1 year may follow acute H. pylori infection.
Bacterial infection of the stomach or phlegmonous gastritis is a rare, potentially life-threatening disorder characterized by marked and diffuse acute inflammatory infiltrates of the entire gastric wall, at times accompanied by necrosis. Elderly individuals, alcoholics, and AIDS patients may be affected. Potential iatrogenic causes include polypectomy and mucosal injection with India ink. Organisms associated with this entity include streptococci, staphylococci, Escherichia coli, Proteus, and Haemophilus species. Failure of supportive measures and antibiotics may result in gastrectomy.
Other types of infectious gastritis may occur in immunocompromised individuals such as AIDS patients. Examples include herpetic (herpes simplex) or CMV gastritis. The histologic finding of intranuclear inclusions would be observed in the latter.
Chronic Gastritis Chronic gastritis is identified histologically by an inflammatory cell infiltrate consisting primarily of lymphocytes and plasma cells, with very scant neutrophil involvement. Distribution of the inflammation may be patchy, initially involving superficial and glandular portions of the gastric mucosa. This picture may progress to more severe glandular destruction, with atrophy and metaplasia. Chronic gastritis has been classified according to histologic characteristics. These include superficial atrophic changes and gastric atrophy. The association of atrophic gastritis with the development of gastric cancer has led to the development of endoscopic and serologic markers of severity. Some of these include gross inspection and classification of mucosal abnormalities during standard endoscopy, magnification endoscopy, endoscopy with narrow band imaging and/or autofluorescence imaging, and measurement of several serum biomarkers including pepsinogen I and II levels, gastrin-17, and anti-H. pylori serologies. The clinical utility of these tools is currently being explored.
The early phase of chronic gastritis is superficial gastritis. The inflammatory changes are limited to the lamina propria of the surface mucosa, with edema and cellular infiltrates separating intact gastric glands. The next stage is atrophic gastritis. The inflammatory infiltrate extends deeper into the mucosa, with progressive distortion and destruction of the glands. The final stage of chronic gastritis is gastric atrophy. Glandular structures are lost, and there is a paucity of inflammatory infiltrates. Endoscopically, the mucosa may be substantially thin, permitting clear visualization of the underlying blood vessels.
Gastric glands may undergo morphologic transformation in chronic gastritis. Intestinal metaplasia denotes the conversion of gastric glands to a small intestinal phenotype with small-bowel mucosal glands containing goblet cells. The metaplastic changes may vary in distribution from patchy to fairly extensive gastric involvement. Intestinal metaplasia is an important predisposing factor for gastric cancer (Chap. 109).
Chronic gastritis is also classified according to the predominant site of involvement. Type A refers to the body-predominant form (autoimmune), and type B is the antral-predominant form (H. pylori–related). this classification is artificial in view of the difficulty in distinguishing between these two entities. The term AB gastritis has been used to refer to a mixed antral/body picture.
TYPE A GASTRITIS The less common of the two forms involves primarily the fundus and body, with antral sparing. Traditionally, this form of gastritis has been associated with pernicious anemia (Chap. 128) in the presence of circulating antibodies against parietal cells and IF; thus, it is also called autoimmune gastritis. H. pylori infection can lead to a similar distribution of gastritis. The characteristics of an autoimmune picture are not always present.
Antibodies to parietal cells have been detected in >90% of patients with pernicious anemia and in up to 50% of patients with type A gastritis. The parietal cell antibody is directed against H+,K+-ATPase. T cells are also implicated in the injury pattern of this form of gastritis. A subset of patients infected with H. pylori develop antibodies against H+,K+-ATPase, potentially leading to the atrophic gastritis pattern seen in some patients infected with this organism. The mechanism is thought to involve molecular mimicry between H. pylori LPS and H+,K+-ATPase.
Parietal cell antibodies and atrophic gastritis are observed in family members of patients with pernicious anemia. These antibodies are observed in up to 20% of individuals over age 60 and in ~20% of patients with vitiligo and Addison’s disease. About one-half of patients with pernicious anemia have antibodies to thyroid antigens, and about 30% of patients with thyroid disease have circulating antiparietal cell antibodies. Anti-IF antibodies are more specific than parietal cell antibodies for type A gastritis, being present in ~40% of patients with pernicious anemia. Another parameter consistent with this form of gastritis being autoimmune in origin is the higher incidence of specific familial histocompatibility haplotypes such as HLA-B8 and HLA-DR3.
The parietal cell–containing gastric gland is preferentially targeted in this form of gastritis, and achlorhydria results. Parietal cells are the source of IF, the lack of which will lead to vitamin B12 deficiency and its sequelae (megaloblastic anemia, neurologic dysfunction).
Gastric acid plays an important role in feedback inhibition of gastrin release from G cells. Achlorhydria, coupled with relative sparing of the antral mucosa (site of G cells), leads to hypergastrinemia. Gastrin levels can be markedly elevated (>500 pg/mL) in patients with pernicious anemia. ECL cell hyperplasia with frank development of gastric carcinoid tumors may result from gastrin trophic effects. Hypergastrinemia and achlorhydria may also be seen in nonpernicious anemia–associated type A gastritis.
TYPE B GASTRITIS Type B, or antral-predominant, gastritis is the more common form of chronic gastritis. H. pylori infection is the cause of this entity. Although described as “antral-predominant,” this is likely a misnomer in view of studies documenting the progression of the inflammatory process toward the body and fundus of infected individuals. The conversion to a pangastritis is time-dependent and estimated to require 15–20 years. This form of gastritis increases with age, being present in up to 100% of persons over age 70. Histology improves after H. pylori eradication. The number of H. pylori organisms decreases dramatically with progression to gastric atrophy, and the degree of inflammation correlates with the level of these organisms. Early on, with antral-predominant findings, the quantity of H. pylori is highest and a dense chronic inflammatory infiltrate of the lamina propria is noted, accompanied by epithelial cell infiltration with polymorphonuclear leukocytes (Fig. 348-14).
FIGURE 348-14 Chronic gastritis and H. pylori organisms. Steiner silver stain of superficial gastric mucosa showing abundant darkly stained microorganisms layered over the apical portion of the surface epithelium. Note that there is no tissue invasion.
Multifocal atrophic gastritis, gastric atrophy with subsequent metaplasia, has been observed in chronic H. pylori–induced gastritis. This may ultimately lead to development of gastric adenocarcinoma (Fig. 348-8; Chap. 109). H. pylori infection is now considered an independent risk factor for gastric cancer. Worldwide epidemiologic studies have documented a higher incidence of H. pylori infection in patients with adenocarcinoma of the stomach as compared to control subjects. Seropositivity for H. pylori is associated with a three- to sixfold increased risk of gastric cancer. This risk may be as high as ninefold after adjusting for the inaccuracy of serologic testing in the elderly. The mechanism by which H. pylori infection leads to cancer is unknown, but it appears to be related to the chronic inflammation induced by the organism. Eradication of H. pylori as a general preventative measure for gastric cancer is being evaluated but is not yet recommended.
Infection with H. pylori is also associated with development of a low-grade B cell lymphoma, gastric MALT lymphoma (Chap. 134). The chronic T cell stimulation caused by the infection leads to production of cytokines that promote the B cell tumor. The tumor should be initially staged with a CT scan of the abdomen and EUS. Tumor growth remains dependent on the presence of H. pylori, and its eradication is often associated with complete regression of the tumor. The tumor may take more than a year to regress after treating the infection. Such patients should be followed by EUS every 2–3 months. If the tumor is stable or decreasing in size, no other therapy is necessary. If the tumor grows, it may have become a high-grade B cell lymphoma. When the tumor becomes a high-grade aggressive lymphoma histologically, it loses responsiveness to H. pylori eradication.
Miscellaneous Forms of Gastritis Lymphocytic gastritis is characterized histologically by intense infiltration of the surface epithelium with lymphocytes. The infiltrative process is primarily in the body of the stomach and consists of mature T cells and plasmacytes. The etiology of this form of chronic gastritis is unknown. It has been described in patients with celiac sprue, but whether there is a common factor associating these two entities is unknown. No specific symptoms suggest lymphocytic gastritis. A subgroup of patients have thickened folds noted on endoscopy. These folds are often capped by small nodules that contain a central depression or erosion; this form of the disease is called varioliform gastritis. H. pylori probably plays no significant role in lymphocytic gastritis. Therapy with glucocorticoids or sodium cromoglycate has obtained unclear results.
Marked eosinophilic infiltration involving any layer of the stomach (mucosa, muscularis propria, and serosa) is characteristic of eosinophilic gastritis. Affected individuals will often have circulating eosinophilia with clinical manifestation of systemic allergy. Involvement may range from isolated gastric disease to diffuse eosinophilic gastroenteritis. Antral involvement predominates, with prominent edematous folds being observed on endoscopy. These prominent antral folds can lead to outlet obstruction. Patients can present with epigastric discomfort, nausea, and vomiting. Treatment with glucocorticoids has been successful.
Several systemic disorders may be associated with granulomatous gastritis. Gastric involvement has been observed in Crohn’s disease. Involvement may range from granulomatous infiltrates noted only on gastric biopsies to frank ulceration and stricture formation. Gastric Crohn’s disease usually occurs in the presence of small-intestinal disease. Several rare infectious processes can lead to granulomatous gastritis, including histoplasmosis, candidiasis, syphilis, and tuberculosis. Other unusual causes of this form of gastritis include sarcoidosis, idiopathic granulomatous gastritis, and eosinophilic granulomas involving the stomach. Establishing the specific etiologic agent in this form of gastritis can be difficult, at times requiring repeat endoscopy with biopsy and cytology. Occasionally, a surgically obtained full-thickness biopsy of the stomach may be required to exclude malignancy.
Russell body gastritis (RBG) is a mucosal lesion of unknown etiology that has a pseudotumoral endoscopic appearance. Histologically, it is defined by the presence of numerous plasma cells containing Russell bodies (RBs) that express kappa and lambda light chains. Only 10 cases have been reported, and 7 of these have been associated with H. pylori infection. The lesion can be confused with a neoplastic process, but it is benign in nature, and the natural history of the lesion is not known. There have been cases of resolution of the lesion when H. pylori was eradicated.
MÉNÉTRIER’S DISEASE
Ménétrier’s disease (MD) is a very rare gastropathy characterized by large, tortuous mucosal folds. MD has an average age of onset of 40–60 years with a male predominance. The differential diagnosis of large gastric folds includes ZES, malignancy (lymphoma, infiltrating carcinoma), infectious etiologies (CMV, histoplasmosis, syphilis, tuberculosis), gastritis polyposa profunda, and infiltrative disorders such as sarcoidosis. MD is most commonly confused with large or multiple gastric polyps (prolonged PPI use) or familial polyposis syndromes. The mucosal folds in MD are often most prominent in the body and fundus, sparing the antrum. Histologically, massive foveolar hyperplasia (hyperplasia of surface and glandular mucous cells) and a marked reduction in oxyntic glands and parietal cells and chief cells are noted. This hyperplasia produces the prominent folds observed. The pits of the gastric glands elongate and may become extremely dilated and tortuous. Although the lamina propria may contain a mild chronic inflammatory infiltrate including eosinophils and plasma cells, MD is not considered a form of gastritis. The etiology of this unusual clinical picture in children is often CMV, but the etiology in adults is unknown. Overexpression of the growth factor TGF-α has been demonstrated in patients with MD. The overexpression of TGF-α in turn results in overstimulation of the epidermal growth factor receptor (EGFR) pathway and increased proliferation of mucus cells, resulting in the observed foveolar hyperplasia.
The clinical presentation in adults is usually insidious and progressive. Epigastric pain, nausea, vomiting, anorexia, peripheral edema, and weight loss are signs and symptoms in patients with MD. Occult GI bleeding may occur, but overt bleeding is unusual and, when present, is due to superficial mucosal erosions. In fact, bleeding is more often seen in one of the common mimics of MD, gastric polyposis. Twenty to 100% of patients (depending on time of presentation) develop a protein-losing gastropathy due to hypersecretion of gastric mucus accompanied by hypoalbuminemia and edema. Gastric acid secretion is usually reduced or absent because of the decreased parietal cells. Large gastric folds are readily detectable by either radiographic (barium meal) or endoscopic methods. Endoscopy with deep mucosal biopsy, preferably full thickness with a snare technique, is required to establish the diagnosis and exclude other entities that may present similarly. A nondiagnostic biopsy may lead to a surgically obtained full-thickness biopsy to exclude malignancy. Although MD is considered premalignant by some, the risk of neoplastic progression is not defined. Complete blood count, serum gastrin, serum albumin, CMV and H. pylori serology, and pH testing of gastric aspirate during endoscopy should be included as part of the initial evaluation of patients with large gastric folds.
349 |
Disorders of Absorption |
Disorders of absorption constitute a broad spectrum of conditions with multiple etiologies and varied clinical manifestations. Almost all of these clinical problems are associated with diminished intestinal absorption of one or more dietary nutrients and are often referred to as the malabsorption syndrome. This term is not ideal as it represents a pathophysiologic state, does not provide an etiologic explanation for the underlying problem, and should not be considered an adequate final diagnosis. The only clinical conditions in which absorption is increased are hemochromatosis and Wilson’s disease, in which absorption of iron and copper, respectively, is elevated.
Most malabsorption syndromes are associated with steatorrhea, an increase in stool fat excretion to >6% of dietary fat intake. Some malabsorption disorders are not associated with steatorrhea: primary lactase deficiency, a congenital absence of the small-intestinal brush border disaccharidase enzyme lactase, is associated with lactose “malabsorption,” and pernicious anemia is associated with a marked decrease in intestinal absorption of cobalamin (vitamin B12) due to an absence of gastric parietal-cell intrinsic factor, which is required for cobalamin absorption.
Disorders of absorption must be included in the differential diagnosis of diarrhea (Chap. 55). First, diarrhea is frequently associated with and/or is a consequence of the diminished absorption of one or more dietary nutrients. The diarrhea may be secondary either to the intestinal process that is responsible for the steatorrhea or to steatorrhea per se. Thus, celiac disease (see below) is associated with both extensive morphologic changes in the small-intestinal mucosa and reduced absorption of several dietary nutrients; in contrast, the diarrhea of steatorrhea is the result of the effect of nonabsorbed dietary fatty acids on intestinal (usually colonic) ion transport. For example, oleic acid and ricinoleic acid (a bacterially hydroxylated fatty acid that is also the active ingredient in castor oil, a widely used laxative) induce active colonic Cl ion secretion, most likely secondary to increasing intracellular Ca. In addition, diarrhea per se may result in mild steatorrhea (<11 g of fat excretion while on a 100-g fat diet). Second, most patients will indicate that they have diarrhea, not that they have fat malabsorption. Third, many intestinal disorders that have diarrhea as a prominent symptom (e.g., ulcerative colitis, traveler’s diarrhea secondary to an enterotoxin produced by Escherichia coli) do not necessarily have diminished absorption of any dietary nutrient.
Diarrhea as a symptom (i.e., when the term is used by patients to describe their bowel movement pattern) may reflect a decrease in stool consistency, an increase in stool volume, an increase in number of bowel movements, or any combination of these three changes. In contrast, diarrhea as a sign is a quantitative increase in stool water or weight of >200–225 mL or g per 24 h when a Western-type diet is consumed. Individuals consuming a diet with higher fiber content may normally have a stool weight of up to 400 g/24 h. Thus, the clinician must clarify what an individual patient means by diarrhea. Some 10% of patients referred to gastroenterologists for further evaluation of unexplained diarrhea do not have an increase in stool water when this variable is determined quantitatively. Such patients may have small, frequent, somewhat loose bowel movements with stool urgency that is indicative of proctitis but do not have an increase in stool weight or volume.
It is also critical to establish whether a patient’s diarrhea is secondary to diminished absorption of one or more dietary nutrients rather than being due to small- and/or large-intestinal fluid and electrolyte secretion. The former has often been termed osmotic diarrhea, while the latter has been referred to as secretory diarrhea. Unfortunately, both secretory and osmotic elements can be present simultaneously in the same disorder; thus, this distinction is not always precise. Nonetheless, two studies—determination of stool electrolytes and observation of the effect of a fast on stool output—can help make this distinction.
The demonstration of the effect of prolonged (>24 h) fasting on stool output can suggest that a dietary nutrient is responsible for the individual’s diarrhea. Secretory diarrhea associated with enterotoxin-induced traveler’s diarrhea would not be affected by prolonged fasting, as enterotoxin-induced stimulation of intestinal fluid and electrolyte secretion is not altered by eating. In contrast, diarrhea secondary to lactose malabsorption in primary lactase deficiency would undoubtedly cease during a prolonged fast. Thus, a substantial decrease in stool output by a fasting patient during quantitative stool collection lasting at least 24 h is presumptive evidence that the diarrhea is related to malabsorption of a dietary nutrient. The persistence of stool output during fasting indicates that the diarrhea is likely secretory and that its cause is not a dietary nutrient. Either a luminal (e.g., E. coli enterotoxin) or a circulating (e.g., vasoactive intestinal peptide) secretagogue could be responsible for unaltered persistence of a patient’s diarrhea during a prolonged fast. The observed effects of fasting can be compared and correlated with stool electrolyte and osmolality determinations.
Measurement of stool electrolytes and osmolality requires comparison of Na+ and K+ concentrations in liquid stool with the osmolality of the stool in order to determine the presence or absence of a so-called stool osmotic gap. The following formula is used:
2 × (stool [Na+] + stool [K+]) ≤ stool osmolality
The cation concentrations are doubled to estimate stool anion concentrations. The presence of a significant osmotic gap suggests the presence in stool water of a substance (or substances) other than Na/K/anions that is presumably responsible for the patient’s diarrhea. Originally, stool osmolality was measured, but it is almost invariably greater than the required 290–300 mosmol/kg H2O, reflecting bacterial degradation of nonabsorbed carbohydrate either immediately before defecation or in the stool jar while specimen awaits chemical analysis, even when the stool is refrigerated. As a result, the stool osmolality should be assumed to be 300 mosmol/kg H2O. A low stool osmolality (<290 mosmol/kg H2O) reflects the addition of either dilute urine or water, indicating either collection of urine and stool together or so-called factitious diarrhea, a form of Münchausen’s syndrome. When the calculated difference in the formula above is >50, an osmotic gap exists; its presence suggests that the diarrhea is due to a nonabsorbed dietary nutrient—e.g., a fatty acid and/or a carbohydrate. When this difference is <25, it is presumed that a dietary nutrient is not responsible for the diarrhea. Since elements of both osmotic diarrhea (i.e., due to malabsorption of a dietary nutrient) and secretory diarrhea may be present, this distinction at times is less clear-cut at the bedside than when used as a teaching example. Ideally, the presence of an osmotic gap will be associated with a marked decrease in stool output during a prolonged fast, while an osmotic gap will likely be absent in an individual whose stool output is not reduced substantially during a period of fasting.
NUTRIENT DIGESTION AND ABSORPTION
The lengths of the small intestine and the colon are ~300 cm and ~80 cm, respectively. However, the effective functional surface area is ~600-fold greater than that of a hollow tube as a result of folds, villi (in the small intestine), and microvilli. The functional surface area of the small intestine is somewhat greater than that of a doubles tennis court. In addition to nutrient digestion and absorption, the intestinal epithelia have several other functions:
1. Barrier and immune defense. The intestine is exposed to a large number of potential antigens and enteric and invasive microorganisms, and it is extremely effective at preventing the entry of almost all of these agents. The intestinal mucosa also synthesizes and secretes secretory IgA.
2. Fluid and electrolyte absorption and secretion. The intestine absorbs ~7–8 L of fluid daily, a volume comprising dietary fluid intake (1–2 L/d) and salivary, gastric, pancreatic, biliary, and intestinal fluid (6–7 L/d). Several stimuli, especially bacteria and bacterial enterotoxins, induce fluid and electrolyte secretion that may lead to diarrhea (Chap. 160).
3. Synthesis and secretion of several proteins. The intestinal mucosa is a major site for the production of proteins, including apolipoproteins.
4. Production of several bioactive amines and peptides. The intestine is one of the largest endocrine organs in the body and produces several amines (e.g., 5-hydroxytryptophan) and peptides that serve as paracrine and hormonal mediators of intestinal function.
The small and large intestines are distinct anatomically (villi are present in the small intestine but are absent in the colon) and functionally (nutrient digestion and absorption take place in the small intestine but not in the colon). No precise anatomic characteristics separate duodenum, jejunum, and ileum, although certain nutrients are absorbed exclusively in specific areas of the small intestine. However, villous cells in the small intestine (surface epithelial cells in the colon) and crypt cells have distinct anatomic and functional characteristics. Intestinal epithelial cells are continuously renewed; new proliferating epithelial cells at the base of the crypt migrate over 48–72 h to the tip of the villus (or surface of the colon), where they exist as well-developed epithelial cells with digestive and absorptive function. This high rate of cell turnover explains the relatively rapid resolution of diarrhea and other digestive-tract side effects during chemotherapy as new cells not exposed to these toxic agents are produced. Equally important is the paradigm of separation of villous/surface cell and crypt cell functions. Digestive hydrolytic enzymes are present primarily in the brush border of villous epithelial cells. Absorptive and secretory functions are also separate: villous/surface cells are primarily, but not exclusively, the site for absorptive function, while secretory function is located in crypts of both the small and large intestines.
Nutrients, minerals, and vitamins are absorbed by one or more active-transport mechanisms. These mechanisms are energy dependent and are mediated by membrane transport proteins. These processes will result in the net movement of a substance against or in the absence of an electrochemical concentration gradient. Intestinal absorption of amino acids and monosaccharides (e.g., glucose) is also a specialized form of active transport—secondary active transport. The movement of actively transported nutrients against a concentration gradient is Na+ dependent and is due to a Na+ gradient across the apical membrane. The Na+ gradient is maintained by Na+,K+-adenosine triphosphatase (ATPase), the so-called Na+ pump located on the basolateral membrane, which extrudes Na+ and maintains low intracellular [Na] as well as the Na+ gradient across the apical membrane. As a result, active glucose absorption and glucose-stimulated Na+ absorption require both the apical membrane transport protein SGLT1 and the basolateral Na+,K+-ATPase. In addition to exhibiting Na+ for its absorption, glucose stimulates Na+ and fluid absorption; this effect is the physiologic basis of oral rehydration therapy for the treatment of diarrhea (Chap. 55). The mechanisms of intestinal fluid and electrolyte absorption and secretion are discussed in Chap. 55.
Although the intestinal epithelial cells are crucial mediators of absorption and of ion and water flow, the several cell types in the lamina propria (e.g., mast cells, macrophages, myofibroblasts) and the enteric nervous system interact with the epithelium to regulate mucosal cell function. Intestinal function results from the integrated responses and interactions of intestinal epithelial cells and intestinal muscle.
ENTEROHEPATIC CIRCULATION OF BILE ACIDS
Bile acids are not present in the diet but are synthesized in the liver by a series of enzymatic steps that also represent cholesterol catabolism. Indeed, interruption of the enterohepatic circulation of bile acids can reduce serum cholesterol levels by 10% before a new steady state is established. Bile acids are either primary or secondary. Primary bile acids are synthesized in the liver from cholesterol, and secondary bile acids are synthesized from primary bile acids in the intestine by colonic bacterial enzymes. The two primary bile acids in humans are cholic acid and chenodeoxycholic acid; the two most abundant secondary bile acids are deoxycholic acid and lithocholic acid. The liver synthesizes ~500 mg of bile acids daily; the bile acids are conjugated to either taurine or glycine (to form tauroconjugated and glycoconjugated bile acids, respectively) and are secreted into the duodenum in bile. The primary functions of bile acids are (1) to promote bile flow, (2) to solubilize cholesterol and phospholipid in the gallbladder by mixed micelle formation, and (3) to enhance dietary lipid digestion and absorption by forming mixed micelles in the proximal small intestine.
Bile acids are primarily absorbed by an active, Na+-dependent process that takes place exclusively in the ileum; to a lesser extent, they are absorbed by non-carrier-mediated transport processes in the jejunum, ileum, and colon. Conjugated bile acids that enter the colon are deconjugated by colonic bacterial enzymes. The unconjugated bile acids are rapidly absorbed by nonionic diffusion. Colonic bacterial enzymes also dehydroxylate bile acids to secondary bile acids.
Bile acids absorbed from the intestine return to the liver via the portal vein and are then re-secreted (Fig. 349-1). Bile acid synthesis is largely autoregulated by 7α-hydroxylase, the initial enzyme in cholesterol degradation. A decrease in the volume of bile acids returning to the liver from the intestine is associated with an increase in bile acid synthesis/cholesterol catabolism, which helps keep the bile-acid pool size relatively constant. However, the capacity to increase bile acid synthesis is limited to ~2- to 2.5-fold (see below). The bile-acid pool size is ~4 g. The pool is circulated via the enterohepatic circulation about twice during each meal, or six to eight times during a 24-h period. A relatively small quantity of bile acids is not absorbed and is excreted in stool daily; this fecal loss is matched by hepatic bile-acid synthesis.
FIGURE 349-1 Schematic representation of the enterohepatic circulation of bile acids. Bile acid synthesis is cholesterol catabolism and occurs in the liver. Bile acids are secreted in bile and are stored in the gallbladder between meals and at night. Food in the duodenum induces the release of cholecystokinin, a potent stimulus for gallbladder contraction resulting in bile acid entry into the duodenum. Bile acids are primarily absorbed via an Na-dependent transport process that is located only in the ileum. A relatively small quantity of bile acids (~500 mg) is not absorbed in a 24-h period and is lost in stool. Fecal bile acid losses are matched by bile acid synthesis. The bile acid pool (the total amount of bile acids in the body) is ~4 g and is circulated twice during each meal or six to eight times in a 24-h period.
Defects in any of the steps in enterohepatic circulation of bile acids can result in a decrease in the duodenal concentration of conjugated bile acids and consequently in the development of steatorrhea. Thus, steatorrhea can be caused by abnormalities in bile acid synthesis and excretion, their physical state in the intestinal lumen, and reabsorption (Table 349-1).
|
DEFECTS IN ENTEROHEPATIC CIRCULATION OF BILE ACIDS |
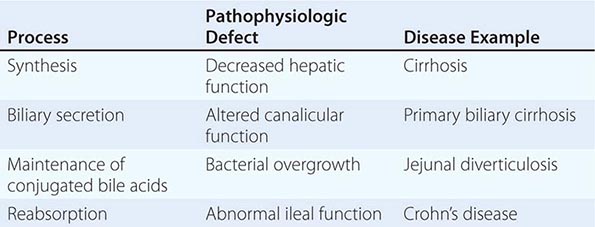
Synthesis Decreased bile acid synthesis and steatorrhea have been demonstrated in chronic liver disease, but steatorrhea often is not a major component of illness in these patients.
Secretion Although bile acid secretion may be reduced or absent in biliary obstruction, steatorrhea is rarely a significant medical problem in these patients. In contrast, primary biliary cirrhosis represents a defect in canalicular excretion of organic anions, including bile acids, and not infrequently is associated with steatorrhea and its consequences (e.g., chronic bone disease). Thus, the osteopenia/osteomalacia and other chronic bone abnormalities often present in patients with primary biliary cirrhosis and other cholestatic syndromes are secondary to steatorrhea that then leads to calcium and vitamin D malabsorption as well as to the effects of cholestasis (e.g., bile acids and inflammatory cytokines).
Maintenance of Conjugated Bile Acids In bacterial overgrowth syndromes associated with diarrhea, steatorrhea, and macrocytic anemia, a colonic type of bacterial flora is increased in the small intestine. Steatorrhea is primarily a result of the decrease in conjugated bile acids secondary to their deconjugation by colonic-type bacteria. Two complementary explanations account for the resulting impairment of micelle formation: (1) Unconjugated bile acids are rapidly absorbed in the jejunum by nonionic diffusion, and the result is a reduced concentration of duodenal bile acids. (2) The critical micellar concentration (CMC) of unconjugated bile acids is higher than that of conjugated bile acids; therefore, unconjugated bile acids are less effective than conjugated bile acids in micelle formation.
Reabsorption Ileal dysfunction caused by either Crohn’s disease or surgical resection results in a decrease in bile acid reabsorption in the ileum and an increase in the delivery of bile acids to the large intestine. The resulting clinical consequences—diarrhea with or without steatorrhea—are determined by the degree of ileal dysfunction and the response of the enterohepatic circulation to bile acid losses (Table 349-2). Patients with limited ileal disease or resection often have diarrhea but not steatorrhea. The diarrhea, a result of stimulation of active Cl secretion by bile acids in the colon, has been called bile acid diarrhea or choleretic enteropathy and responds promptly to cholestyramine, an anion-binding resin. Steatorrhea does not develop because hepatic synthesis of bile acids increases to compensate for the rate of fecal bile-acid losses, resulting in maintenance of both the bile-acid pool size and the intraduodenal concentrations of bile acids. In contrast, patients with greater degrees of ileal disease and/or resection often have diarrhea and steatorrhea that do not respond to cholestyramine. In this situation, ileal disease is also associated with increased volumes of bile acids entering the colon; however, hepatic synthesis can no longer increase sufficiently to maintain the bile-acid pool size. As a consequence, the intraduodenal concentration of bile acids is reduced to less than the CMC, and the result is impaired micelle formation and steatorrhea. This second situation is often called fatty acid diarrhea. Cholestyramine may not be effective (and may even exacerbate the diarrhea by further depleting the intraduodenal bile-acid concentration); however, a low-fat diet to reduce fatty acid entry into the colon can be effective. Two clinical features—the length of the ileal section removed and the degree of steatorrhea—can predict whether an individual patient will respond to cholestyramine. Unfortunately, these predictors are imperfect, and a therapeutic trial of cholestyramine is often necessary to establish whether an individual patient will benefit from cholestyramine. Table 349-2 contrasts the characteristics of bile acid diarrhea (small ileal dysfunction) and fatty acid diarrhea (large ileal dysfunction).
|
COMPARISON OF BILE ACID AND FATTY ACID DIARRHEA |
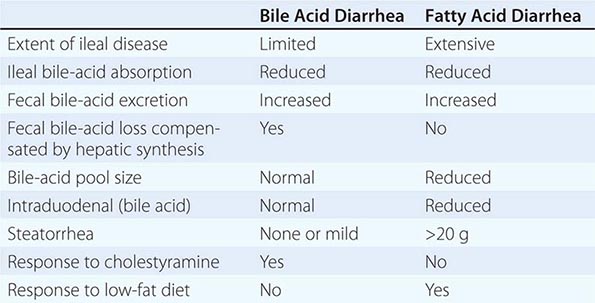
Bile acid diarrhea can also occur in the absence of ileal inflammation and/or resection and is characterized by an abnormal 75SeHCAT retention study and reduced ileal release of fibroblast growth factor 19, a negative regulator of bile acid synthesis, with a consequent increase in bile acid synthesis and secretion that exceeds ileal bile-acid absorption. The diarrhea in these patients also responds to cholestyramine.
LIPIDS
Steatorrhea is caused by one or more defects in the digestion and absorption of dietary fat. The average intake of dietary fat in the United States is ~120–150 g/d, and fat absorption is linear to dietary fat intake. The total load of fat presented to the small intestine is considerably greater, as substantial amounts of lipid are secreted in bile each day (see “Enterohepatic Circulation of Bile Acids,” above). Three types of fatty acids compose fats: long-chain fatty acids (LCFAs), medium-chain fatty acids (MCFAs), and short-chain fatty acids (SCFAs) (Table 349-3). Dietary fat is exclusively composed of long-chain triglycerides (LCTs)—i.e., glycerol that is bound via ester linkages to three LCFAs. While the majority of dietary LCFAs have carbon chain lengths of 16 or 18, all fatty acids of carbon chain length >12 are metabolized in the same manner; saturated and unsaturated fatty acids are handled identically.
|
COMPARISON OF DIFFERENT TYPES OF FATTY ACIDS |
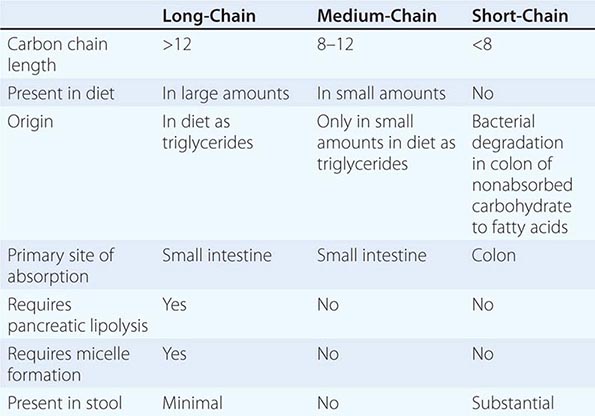
Assimilation of dietary lipid requires three integrated processes: (1) an intraluminal, or digestive, phase; (2) a mucosal, or absorptive, phase; and (3) a delivery, or postabsorptive, phase. An abnormality at any site involved in these processes can cause steatorrhea (Table 349-4). Therefore, it is essential that any patient with steatorrhea be evaluated to identify the specific physiologic defect in overall lipid digestion/absorption, as therapy will be determined by the specific etiology.
|
DEFECTS IN LIPID DIGESTION AND ABSORPTION IN STEATORRHEA |
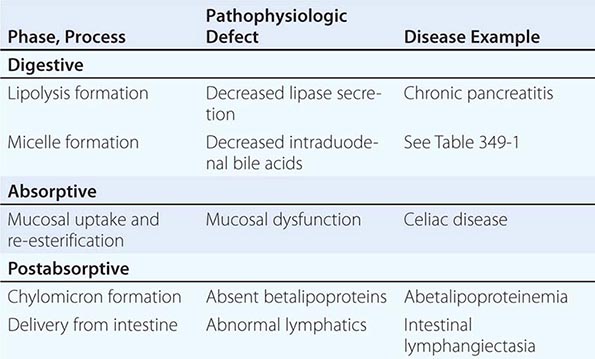
The digestive phase has two components, lipolysis and micelle formation. Although dietary lipid is in the form of LCTs, the intestinal mucosa does not absorb triglycerides; they must first be hydrolyzed (Fig. 349-2). The initial step in lipid digestion is the formation of emulsions of finely dispersed lipid, which is accomplished by mastication and gastric contractions. Lipolysis, the hydrolysis of triglycerides to free fatty acids, monoglycerides, and glycerol by lipase, is initiated in the stomach by lingual and gastric lipases that have a pH optimum of 4.5–6.0. About 20–30% of total lipolysis occurs in the stomach. Lipolysis is completed in the duodenum and jejunum by pancreatic lipase, which is inactivated by a pH <7.0. Pancreatic lipolysis is greatly enhanced by the presence of a second pancreatic enzyme, colipase, which facilitates the movement of lipase to the triglyceride.
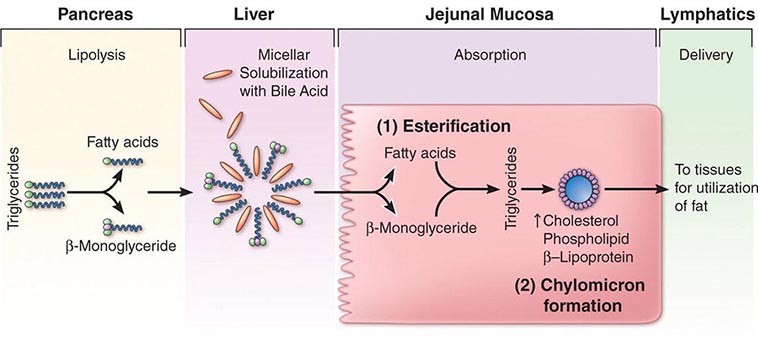
FIGURE 349-2 Schematic representation of lipid digestion and absorption. Dietary lipid is in the form of long-chain triglycerides. The overall process can be divided into (1) a digestive phase that includes both lipolysis and micelle formation requiring pancreatic lipase and conjugated bile acids, respectively, in the duodenum; (2) an absorptive phase for mucosal uptake and re-esterification; and (3) a postabsorptive phase that includes chylomicron formation and exit from the intestinal epithelial cell via lymphatics. (Courtesy of John M. Dietschy, MD; with permission.)
Impaired lipolysis can lead to steatorrhea and can occur in the presence of pancreatic insufficiency due to chronic pancreatitis in adults or cystic fibrosis in children and adolescents. Normal lipolysis can be maintained by ~5% of maximal pancreatic lipase secretion; thus, steatorrhea is a late manifestation of these disorders. A reduction in intraduodenal pH can also result in altered lipolysis, as pancreatic lipase is inactivated at pH <7. Thus, ~15% of patients who have gastrinoma (Chap. 348), with substantial increases in gastric acid secretion from ectopic production of gastrin (usually from an islet cell adenoma), have diarrhea, and some have steatorrhea believed to be secondary to acid inactivation of pancreatic lipase. Similarly, patients who have chronic pancreatitis (with reduced lipase secretion) often have a decrease in pancreatic bicarbonate secretion, which will also result in a lowering of intraduodenal pH and inactivation of endogenous pancreatic lipase or of therapeutically administered lipase.
Overlying the microvillus membrane of the small intestine is the so-called unstirred water layer, a relatively stagnant aqueous phase that must be traversed by the products of lipolysis that are primarily water insoluble. Water-soluble mixed micelles provide a mechanism by which the water-insoluble products of lipolysis can reach the luminal plasma membrane of villous epithelial cells—the site for lipid absorption. Mixed micelles are molecular aggregates composed of fatty acids, monoglycerides, phospholipids, cholesterol, and conjugated bile acids. These mixed micelles are formed when the concentration of conjugated bile acids is greater than its CMC, which differs among the several bile acids present in the small-intestinal lumen. Conjugated bile acids, synthesized in the liver and excreted into the duodenum in bile, are regulated by the enterohepatic circulation (see above). Steatorrhea can result from impaired movement of fatty acids across the unstirred aqueous fluid layer in two situations: (1) an increase in the relative thickness of the unstirred water layer that occurs in bacterial overgrowth syndromes (see below) secondary to functional stasis (e.g., scleroderma); and (2) a decrease in the duodenal concentration of conjugated bile acids below the CMC, resulting in impaired micelle formation. Thus, steatorrhea can be caused by one or more defects in the enterohepatic circulation of bile acids.
Uptake and re-esterification constitute the absorptive phase of lipid digestion/absorption. Although passive diffusion has been thought to be responsible, a carrier-mediated process may mediate fatty acid and monoglyceride uptake. Regardless of the uptake process, fatty acids and monoglycerides are re-esterified by a series of enzymatic steps in the endoplasmic reticulum to form triglycerides, in which lipid exits from the intestinal epithelial cell. Impaired lipid absorption as a result of mucosal inflammation (e.g., celiac disease) and/or intestinal resection can also lead to steatorrhea.
The re-esterified triglycerides require the formation of chylomicrons to permit their exit from the small-intestinal epithelial cell and their delivery to the liver via the lymphatics. Chylomicrons are composed of β-lipoprotein and contain triglycerides, cholesterol, cholesterol esters, and phospholipids and enter the lymphatics, not the portal vein. Defects in the postabsorptive phase of lipid digestion/absorption can also result in steatorrhea, but these disorders are uncommon. Abetalipoproteinemia, or acanthocytosis, is a rare disorder of impaired synthesis of β-lipoprotein associated with abnormal erythrocytes (acanthocytes), neurologic problems, and steatorrhea (Chap. 421). Lipolysis, micelle formation, and lipid uptake are all normal in patients with abetalipoproteinemia, but the re-esterified triglyceride cannot exit the epithelial cell because of the failure to produce chylomicrons. Small-intestinal biopsy samples obtained from these rare patients in the postprandial state reveal lipid-laden small-intestinal epithelial cells that become perfectly normal in appearance after a 72- to 96-h fast. Similarly, abnormalities of intestinal lymphatics (e.g., intestinal lymphangiectasia) may also be associated with steatorrhea as well as protein loss (see below). Steatorrhea can result from defects at any of the several steps in lipid digestion/absorption.
The mechanism of lipid digestion/absorption outlined above is limited to dietary lipid, which is almost exclusively in the form of LCTs (Table 349-3). Medium-chain triglycerides (MCTs), composed of fatty acids with carbon chain lengths of 8–12, are present in large amounts in coconut oil and are used as a nutritional supplement. MCTs can be digested and absorbed by a pathway different from that involved in LCT digestion and absorption; at one time, MCTs held promise as an important treatment for steatorrhea of almost all etiologies. Unfortunately, they have been less therapeutically effective than expected because, for reasons that are not completely understood, their use often is not associated with an increase in body weight.
In contrast to LCTs, MCTs do not require pancreatic lipolysis as they can be absorbed intact by the intestinal epithelial cell. Further, micelle formation is not necessary for the absorption of MCTs (or MCFAs, if hydrolyzed by pancreatic lipase). MCTs are absorbed more efficiently than LCTs for the following reasons: (1) The rate of absorption is greater for MCTs than for LCFAs. (2) After absorption, MCFAs are not re-esterified. (3) After absorption, MCTs are hydrolyzed to MCFAs. (4) MCTs do not require chylomicron formation to exit intestinal epithelial cells. (5) The route of MCT exit is via the portal vein and not via lymphatics. Thus, the absorption of MCTs is greater than that of LCTs in pancreatic insufficiency, conditions with reduced intraduodenal bile acid concentrations, small-intestinal mucosal disease, abetalipoproteinemia, and intestinal lymphangiectasia.
SCFAs are not dietary lipids but are synthesized by colonic bacterial enzymes from nonabsorbed carbohydrate and are the anions present at the highest concentration in stool (80–130 mM). The SCFAs in stool are primarily acetate, propionate, and butyrate, whose carbon chain lengths are 2, 3, and 4, respectively. Butyrate is the primary nutrient for colonic epithelial cells, and its deficiency can be associated with one or more colitides. SCFAs conserve calories and carbohydrate: carbohydrates that are not completely absorbed in the small intestine will not be absorbed in the large intestine because of the absence of both disaccharidases and SGLT1, the transport protein that mediates monosaccharide absorption. In contrast, SCFAs are rapidly absorbed and stimulate colonic NaCl and fluid absorption. Most antibiotic-associated diarrhea not caused by Clostridium difficile is due to antibiotic suppression of the colonic microbiota, with a resulting decrease in SCFA production. As C. difficile accounts for only ~15–20% of all antibiotic-associated diarrhea, a relative decrease in colonic production of SCFA is likely the cause of most antibiotic-associated diarrhea.
The clinical manifestations of steatorrhea are a consequence both of the underlying disorder responsible for its development and of steatorrhea per se. Depending on the degree of steatorrhea and the level of dietary intake, significant fat malabsorption may lead to weight loss. Steatorrhea per se can be responsible for diarrhea; if the primary cause of the steatorrhea has not been identified, a low-fat diet can often ameliorate the diarrhea by decreasing fecal fat excretion. Steatorrhea is commonly associated with fat-soluble vitamin deficiency, which requires replacement with water-soluble preparations of these vitamins.
Disorders of absorption may also be associated with malabsorption of other dietary nutrients— most often carbohydrates—with or without a decrease in dietary lipid digestion and absorption. Therefore, knowledge of the mechanisms of digestion and absorption of carbohydrates, proteins, and other minerals and vitamins is useful in the evaluation of patients with altered intestinal nutrient absorption.
CARBOHYDRATES
Carbohydrates in the diet are present in the form of starch, disaccharides (sucrose and lactose), and glucose. Carbohydrates are absorbed only in the small intestine and only in the form of monosaccharides. Therefore, before their absorption, starch and disaccharides must first be digested by pancreatic amylase and intestinal brush border disaccharidases to monosaccharides. Monosaccharide absorption occurs by a Na-dependent process mediated by the brush border transport protein SGLT1.
Lactose malabsorption is the only clinically important disorder of carbohydrate absorption. Lactose, the disaccharide present in milk, requires digestion by brush border lactase to its two constituent monosaccharides, glucose and galactose. Lactase is present in almost all species in the postnatal period but then disappears throughout the animal kingdom, except in humans. Lactase activity persists in many individuals throughout life. Two different types of lactase deficiency exist—primary and secondary. In primary lactase deficiency, a genetically determined decrease or absence of lactase is noted, while all other aspects of both intestinal absorption and brush border enzymes are normal. In a number of nonwhite groups, primary lactase deficiency is common in adulthood. In fact, Northern European and North American whites are the only groups to maintain small-intestinal lactase activity throughout adult life. Table 349-5 presents the incidence of primary lactase deficiency in several ethnic groups. Lactase persistence in adults is an abnormality due to a defect in the regulation of its maturation. In contrast, secondary lactase deficiency occurs in association with small-intestinal mucosal disease, with abnormalities in both structure and function of other brush border enzymes and transport processes. Secondary lactase deficiency is often seen in celiac disease.
|
PRIMARY LACTASE DEFICIENCY IN ADULT ETHNIC GROUPS |
As lactose digestion is rate-limiting compared to glucose/galactose absorption, lactase deficiency is associated with significant lactose malabsorption. Some individuals with lactose malabsorption develop symptoms such as diarrhea, abdominal pain, cramps, and/or flatus. Most individuals with primary lactase deficiency do not have symptoms. Since lactose intolerance may be associated with symptoms suggestive of irritable bowel syndrome, persistence of such symptoms in an individual who exhibits lactose intolerance while on a strict lactose-free diet suggests that the person’s symptoms were related to irritable bowel syndrome.
The development of symptoms of lactose intolerance is related to several factors:
1. Amount of lactose in the diet.
2. Rate of gastric emptying. Symptoms are more likely when gastric emptying is rapid than when it is slower. Therefore, skim milk is more likely to be associated with symptoms of lactose intolerance than whole milk, as the rate of gastric emptying after skim milk intake is more rapid. Similarly, diarrhea following subtotal gastrectomy is often a result of lactose intolerance, as gastric emptying is accelerated in patients with a gastrojejunostomy.
3. Small-intestinal transit time. Although the small and large intestines both contribute to the development of symptoms, many symptoms of lactase deficiency are related to the interaction of colonic bacteria and nonabsorbed lactose. More rapid small-intestinal transit makes symptoms more likely.
4. Colonic compensation by production of SCFAs from nonabsorbed lactose. Reduced levels of colonic microflora, which can follow antibiotic use, are associated with increased symptoms after lactose ingestion, especially in a lactase-deficient individual.
Glucose-galactose or monosaccharide malabsorption may also be associated with diarrhea and is due to a congenital absence of SGLT1. Diarrhea develops when individuals with this disorder ingest carbohydrates that contain actively transported monosaccharides (e.g., glucose, galactose) but not when they ingest monosaccharides that are not actively transported (e.g., fructose). Fructose is absorbed by the brush border transport protein GLUT 5, a facilitated diffusion process that is not Na-dependent and is distinct from SGLT1. In contrast, some individuals develop diarrhea as a result of the consumption of large quantities of sorbitol, a sugar used in diabetic candy; sorbitol is only minimally absorbed because of the absence of an intestinal absorptive transport mechanism for this sugar.
PROTEINS
Protein is present in food almost exclusively as polypeptides and requires extensive hydrolysis to di- and tripeptides and amino acids before absorption. Proteolysis occurs in both the stomach and the small intestine; it is mediated by pepsin, which is secreted as pepsinogen by gastric chief cells, and by trypsinogen and other peptidases from pancreatic acinar cells. The proenzymes pepsinogen and trypsinogen must be activated to pepsin (by pepsin at a pH <5) and to trypsin (by the intestinal brush border enzyme enterokinase and subsequently by trypsin), respectively. Proteins are absorbed by separate transport systems for di- and tripeptides and for different types of amino acids—e.g., neutral and dibasic. Alterations in either protein or amino acid digestion and absorption are rarely observed clinically, even in the presence of extensive small-intestinal mucosal inflammation. However, three rare genetic disorders involve protein digestion/absorption: (1) Enterokinase deficiency is due to an absence of the brush border enzyme that converts the proenzyme trypsinogen to trypsin and is associated with diarrhea, growth retardation, and hypoproteinemia. (2) Hartnup’s syndrome, a defect in neutral amino acid transport, is characterized by a pellagra-like rash and neuropsychiatric symptoms. (3) Cystinuria, a defect in dibasic amino acid transport, is associated with renal calculi and chronic pancreatitis.
APPROACH TO THE PATIENT:
Malabsorption
The clues provided by the history, symptoms, and initial preliminary observations will serve to limit extensive, ill-focused, and expensive laboratory and imaging studies. For example, a clinician evaluating a patient who has symptoms suggestive of malabsorption and who has recently undergone extensive small-intestinal resection for mesenteric ischemia should direct the initial assessment almost exclusively to defining whether a short-bowel syndrome might explain the entire clinical picture. Similarly, the development of a pattern of bowel movements suggestive of steatorrhea in a patient with long-standing alcohol abuse and chronic pancreatitis should prompt an assessment of pancreatic exocrine function.
The classic picture of malabsorption is rarely seen today in most parts of the United States. As a consequence, diseases with malabsorption must be suspected in individuals who have less severe symptoms and signs and subtle evidence of the altered absorption of only a single nutrient rather than obvious evidence of the malabsorption of multiple nutrients.
Although diarrhea can be caused by changes in fluid and electrolyte movement in either the small or the large intestine, dietary nutrients are absorbed almost exclusively in the small intestine. therefore, the demonstration of diminished absorption of a dietary nutrient provides unequivocal evidence for small-intestinal disease, although colonic dysfunction may also be present (e.g., Crohn’s disease may involve both the small and large intestines). Dietary nutrient absorption may be segmental or diffuse along the small intestine and is site specific. Thus, for example, calcium, iron, and folic acid are exclusively absorbed by active-transport processes in the proximal small intestine, especially the duodenum; in contrast, the active-transport mechanisms for both cobalamin and bile acids are operative only in the ileum. Therefore, in an individual who years previously has had an intestinal resection, the details of which are not presently available, a presentation with evidence of calcium, folic acid, and/or iron malabsorption but without cobalamin deficiency makes it likely that the duodenum and proximal jejunum, but not the ileum, were resected.
Some nutrients—e.g., glucose, amino acids, and lipids—are absorbed throughout the small intestine, although their rate of absorption is greater in the proximal than in the distal segments. However, after segmental resection of the small intestine, the remaining segments undergo both morphologic and functional “adaptation” to enhance absorption. Such adaptation is secondary to the presence of luminal nutrients and hormonal stimuli and may not be complete in humans for several months after resection. Adaptation is critical for the survival of individuals who have undergone massive resection of the small intestine and/or colon.
Establishing the diagnosis of steatorrhea and identifying its specific cause are often quite difficult. The “gold standard” remains a timed, quantitative stool-fat determination. From a practical standpoint, stool collections are invariably difficult and often incomplete, as nobody wants to handle stool. A qualitative test—Sudan III staining—has long been available to document an increase in stool fat. This test is rapid and inexpensive but, as a qualitative test, does not establish the degree of fat malabsorption and is best used as a preliminary screening study. Many of the blood, breath, and isotopic tests that have been developed (1) do not directly measure fat absorption; (2) exhibit excellent sensitivity when steatorrhea is obvious and severe but poor sensitivity when steatorrhea is mild (e.g., assays for stool chymotrypsin and elastase, which can potentially distinguish pancreatic from nonpancreatic etiologies of steatorrhea); or (3) have not survived the transition from the research laboratory to commercial application.
Nevertheless, routine laboratory studies (i.e., complete blood count, prothrombin time, serum protein determination, alkaline phosphatase) may suggest dietary nutrient depletion, especially deficiencies of iron, folate, cobalamin, and vitamins D and K. Additional studies include measurement of serum carotene, cholesterol, albumin, iron, folate, and cobalamin levels. The serum carotene level can also be reduced if the patient’s dietary intake of leafy vegetables is poor.
If steatorrhea and/or altered absorption of other nutrients is suspected, then history, clinical observations, and laboratory testing can help detect deficiency of a nutrient, especially of a fat-soluble vitamin (A, D, E, or K). Thus, evidence of metabolic bone disease with elevated alkaline phosphatase concentrations and/or reduced serum calcium levels suggests vitamin D malabsorption. A deficiency of vitamin K is suggested by an elevated prothrombin time in an individual without liver disease who is not taking anticoagulants. Macrocytic anemia leads to an evaluation for possible cobalamin or folic acid malabsorption. Iron-deficiency anemia in the absence of occult bleeding from the gastrointestinal tract in either a male patient or a nonmenstruating female patient requires an evaluation for iron malabsorption and the exclusion of celiac disease, as iron is absorbed exclusively in the proximal small intestine.
At times, however, a timed (72-h) quantitative stool collection, preferably while the patient is on a defined diet, must be undertaken in order to determine stool fat content and establish the diagnosis of steatorrhea. The presence of steatorrhea then requires further assessment to identify the pathophysiologic process(es) responsible for the defect in dietary lipid digestion/absorption (Table 349-4). Other studies include the Schilling test (Chap. 350e), the D-xylose test, duodenal mucosal biopsy, small-intestinal radiologic examination, and tests of pancreatic exocrine function.
THE SCHILLING TEST
This test (Chap. 350e) is performed to determine the cause of cobalamin malabsorption. An understanding of the physiology and pathophysiology of cobalamin absorption is very valuable, enhancing comprehension of aspects of gastric, pancreatic, and ileal function. Unfortunately, the Schilling test has not been available commercially in the United States for the past few years.
URINARY D-XYLOSE TEST
The urinary D-xylose test for carbohydrate absorption provides an assessment of proximal small-intestinal mucosal function. D-Xylose, a pentose, is absorbed almost exclusively in the proximal small intestine. The D-xylose test is usually performed by administering 25 g of D-xylose and collecting urine for 5 h. An abnormal test (excretion of <4.5 g) primarily reflects duodenal/jejunal mucosal disease. The D-xylose test can also be abnormal in patients with blind loop syndrome (as a consequence primarily of an abnormal intestinal mucosa) and, as a false-positive study, in patients with large collections of fluid in a third space (i.e., ascites, pleural fluid). The ease of obtaining a mucosal biopsy of the small intestine by endoscopy and the false-negative rate of the D-xylose test have led to its diminished use. When small-intestinal mucosal disease is suspected, a small-intestinal mucosal biopsy should be performed.
RADIOLOGIC EXAMINATION
Radiologic examination of the small intestine using barium contrast (small-bowel series or study) can provide important information in the evaluation of the patient with presumed or suspected malabsorption. This study is most often performed in conjunction with an examination of the esophagus, stomach, and duodenal bulb. Because insufficient barium is given to the patient to permit an adequate examination of the small-intestinal mucosa, especially in the ileum, many gastrointestinal radiologists alter the procedure by performing either a small-bowel series in which a large amount of barium is given by mouth, without concurrent examination of the esophagus and stomach, or an enteroclysis study in which a large amount of barium is introduced into the duodenum via a fluoroscopically placed tube. In addition, many of the diagnostic features initially described by radiologists to denote the presence of small-intestinal disease (e.g., flocculation, segmentation) are rarely seen with current barium suspensions. Nonetheless, in skilled hands, barium contrast examination of the small intestine can yield important information. For example, with extensive mucosal disease, intestinal dilation can be seen as a dilution of barium from increased intestinal fluid secretion (Fig. 349-3). A normal barium contrast study does not exclude the possibility of small-intestinal disease. However, a small-bowel series remains useful in the search for anatomic abnormalities, such as strictures and fistulas (as in Crohn’s disease) or blind loop syndrome (e.g., multiple jejunal diverticula) and to define the extent of a previous surgical resection. Other imaging studies that assess the integrity of small-intestinal morphology are CT enterography and magnetic resonance enterography. Capsule endoscopy and double-balloon enteroscopy are other useful aids in the diagnostic assessment of small-intestinal pathology.
FIGURE 349-3 Barium contrast small-intestinal radiologic examinations. A. Normal individual. B. Celiac sprue. C. Jejunal diverticulosis. D. Crohn’s disease. (Courtesy of Morton Burrell, MD, Yale University; with permission.)
BIOPSY OF SMALL-INTESTINAL MUCOSA
A small-intestinal mucosal biopsy is essential in the evaluation of a patient with documented steatorrhea or chronic diarrhea (i.e., that lasting >3 weeks) (Chap. 55). The ready availability of endoscopic equipment to examine the stomach and duodenum has led to its almost uniform use as the preferred method of obtaining histologic material from the proximal small-intestinal mucosa. The primary indications for a small-intestinal biopsy are evaluation of a patient (1) either with documented or suspected steatorrhea or with chronic diarrhea, and (2) with diffuse or focal abnormalities of the small intestine defined on a small-intestinal series. Lesions seen on small-bowel biopsy can be classified into three categories (Table 349-6):
|
DISEASES THAT CAN BE DIAGNOSED BY SMALL-INTESTINAL MUCOSAL BIOPSIES |
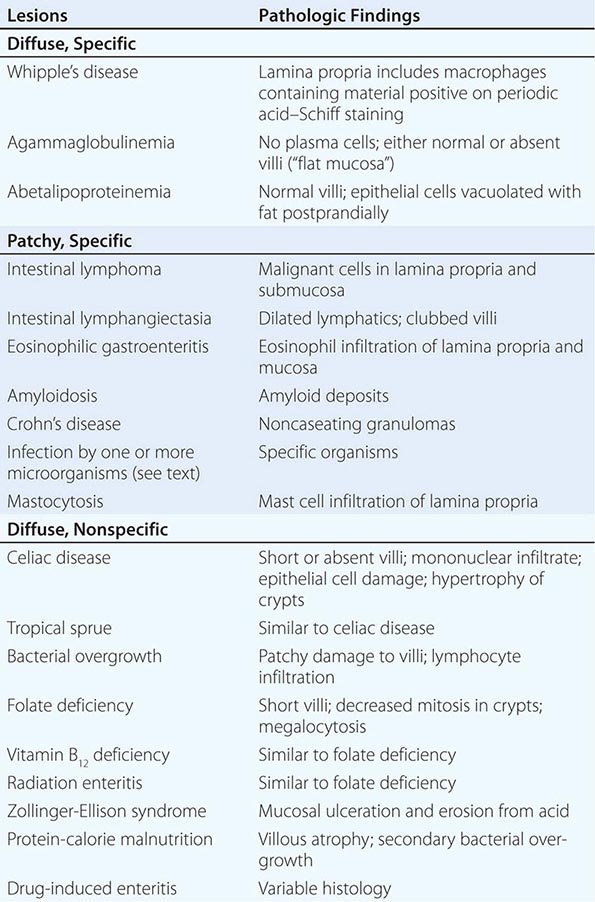
1. Diffuse, specific lesions. Relatively few diseases associated with altered nutrient absorption have specific histopathologic abnormalities on small-intestinal mucosal biopsy, and these diseases are uncommon. Whipple’s disease is characterized by the presence of periodic acid–Schiff (PAS)–positive macrophages in the lamina propria; the bacilli that are also present may require electron microscopic examination for identification (Fig. 349-4). Abetalipoproteinemia is characterized by a normal mucosal appearance except for the presence of mucosal absorptive cells that contain lipid postprandially and disappear after a prolonged period of either fat-free intake or fasting. Immune globulin deficiency is associated with a variety of histopathologic findings on small-intestinal mucosal biopsy. The characteristic feature is the absence of or substantial reduction in the number of plasma cells in the lamina propria; the mucosal architecture may be either perfectly normal or flat (i.e., villous atrophy). As patients with immune globulin deficiency are often infected with Giardia lamblia, Giardia trophozoites may also be seen in the biopsy.
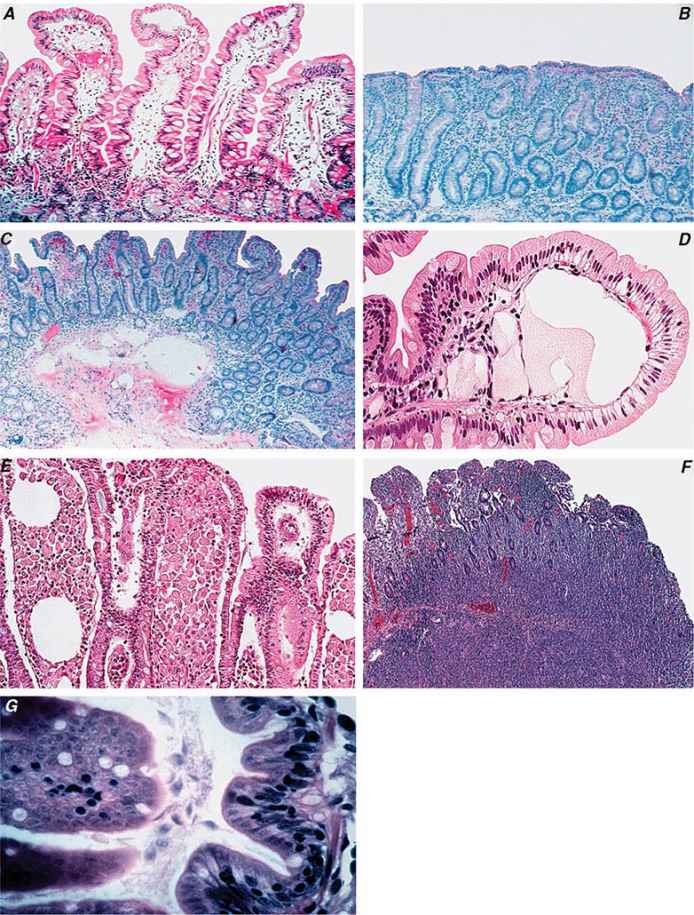
FIGURE 349-4 Small-intestinal mucosal biopsies. A. Normal individual. B. Untreated celiac sprue. C. Treated celiac sprue. D. Intestinal lymphangiectasia. E. Whipple’s disease. F. Lymphoma. G. Giardiasis. (Courtesy of Marie Robert, MD, Yale University; with permission.)
2. Patchy, specific lesions. Several diseases feature an abnormal small-intestinal mucosa with a patchy distribution. As a result, biopsy samples obtained randomly or in the absence of endoscopically visualized abnormalities may not reveal diagnostic features. Intestinal lymphoma can at times be diagnosed on mucosal biopsy by the identification of malignant lymphoma cells in the lamina propria and submucosa (Chap. 134). Dilated lymphatics in the submucosa and sometimes in the lamina propria indicate lymphangiectasia associated with hypoproteinemia secondary to protein loss into the intestine. Eosinophilic gastroenteritis comprises a heterogeneous group of disorders with a spectrum of presentations and symptoms, with an eosinophilic infiltrate of the lamina propria, and with or without peripheral eosinophilia. The patchy nature of the infiltrate and its presence in the submucosa often lead to an absence of histopathologic findings on mucosal biopsy. As the involvement of the duodenum in Crohn’s disease is also submucosal and not necessarily continuous, mucosal biopsies are not the most direct approach to the diagnosis of duodenal Crohn’s disease (Chap. 351). Amyloid deposition can be identified by Congo Red staining in some patients with amyloidosis involving the duodenum (Chap. 136).
3. Diffuse, nonspecific lesions. Celiac disease presents with a characteristic mucosal appearance on duodenal/proximal jejunal mucosal biopsy that is not diagnostic of the disease. The diagnosis of celiac disease is established by clinical, histologic, and immunologic responses to a gluten-free diet. Tropical sprue (see below) is associated with histologic findings similar to those of celiac disease after a tropical or subtropical exposure but does not respond to gluten restriction; most often symptoms improve with antibiotics and folate administration.
Several microorganisms can be identified in small-intestinal biopsy samples, establishing a correct diagnosis. At times, the biopsy is performed specifically to diagnose infection (e.g., Whipple’s disease or giardiasis). In most other instances, the infection is detected incidentally during the workup for diarrhea or other abdominal symptoms. Many of these infections occur in immunocompromised patients with diarrhea; the etiologic agents include Cryptosporidium, Isospora belli, microsporidia, Cyclospora, Toxoplasma, cytomegalovirus, adenovirus, Mycobacterium avium-intracellulare, and G. lamblia. In immunocompromised patients, when Candida, Aspergillus, Cryptococcus, or Histoplasma organisms are seen on duodenal biopsy, their presence generally reflects systemic infection. Apart from Whipple’s disease and infections in the immunocompromised host, small-bowel biopsy is seldom used as the primary mode of diagnosis of infection. Even giardiasis is more easily diagnosed by stool antigen studies and/or duodenal aspiration than by duodenal biopsy.
Patients with steatorrhea require assessment of pancreatic exocrine function, which is often abnormal in chronic pancreatitis. The secretin test that collects pancreatic secretions by duodenal intubation following intravenous administration of secretin is the only test that directly measures pancreatic exocrine function but is available only at a few specialized centers. Endoscopic approaches (endoscopic retrograde cholangiopancreatography, endoscopic ultrasound) provide an excellent assessment of pancreatic duct anatomy but do not assess exocrine function (Chap. 370).
Table 349-7 summarizes the results of the D-xylose test, the Schilling test, and small-intestinal mucosal biopsy in patients with steatorrhea of various etiologies.
|
RESULTS OF DIAGNOSTIC STUDIES IN STEATORRHEA OF VARIOUS ETIOLOGIES |
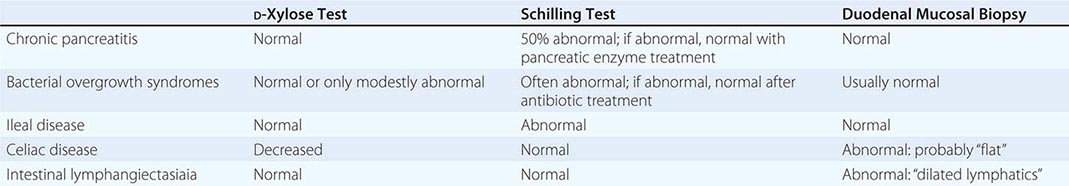
SPECIFIC DISEASE ENTITIES
CELIAC DISEASE
![]() Celiac disease is a common cause of malabsorption of one or more nutrients. Although celiac disease was originally considered largely a disease of white individuals, especially persons of European descent, recent observations have established that it is a common disease with protean manifestations, a worldwide distribution, and an estimated incidence in the United States that is as high as 1 in 113 people. Its incidence has increased over the past 50 years. Celiac disease has had several other names, including nontropical sprue, celiac sprue, adult celiac disease, and gluten-sensitive enteropathy. The etiology of celiac disease is not known, but environmental, immunologic, and genetic factors are important. Celiac disease is considered an “iceberg” disease. A small number of individuals have classical symptoms and manifestations related to nutrient malabsorption along with a varied natural history; the onset of symptoms can occur at all points from the first year of life through the eighth decade. A much larger number of individuals have “atypical celiac disease”, with manifestations that are not obviously related to intestinal malabsorption (e.g., anemia, osteopenia, infertility, and neurologic symptoms). Finally, an even larger number of persons have “silent celiac disease”; they are essentially asymptomatic despite abnormal small-intestinal histopathology and serologies (see below).
Celiac disease is a common cause of malabsorption of one or more nutrients. Although celiac disease was originally considered largely a disease of white individuals, especially persons of European descent, recent observations have established that it is a common disease with protean manifestations, a worldwide distribution, and an estimated incidence in the United States that is as high as 1 in 113 people. Its incidence has increased over the past 50 years. Celiac disease has had several other names, including nontropical sprue, celiac sprue, adult celiac disease, and gluten-sensitive enteropathy. The etiology of celiac disease is not known, but environmental, immunologic, and genetic factors are important. Celiac disease is considered an “iceberg” disease. A small number of individuals have classical symptoms and manifestations related to nutrient malabsorption along with a varied natural history; the onset of symptoms can occur at all points from the first year of life through the eighth decade. A much larger number of individuals have “atypical celiac disease”, with manifestations that are not obviously related to intestinal malabsorption (e.g., anemia, osteopenia, infertility, and neurologic symptoms). Finally, an even larger number of persons have “silent celiac disease”; they are essentially asymptomatic despite abnormal small-intestinal histopathology and serologies (see below).
The hallmark of celiac disease is an abnormal small-intestinal biopsy (Fig. 349-4) and the response of the condition (including symptoms and histologic changes on small-intestinal biopsy) to the elimination of gluten from the diet. The histologic changes have a proximal-to-distal intestinal distribution of severity, which probably reflects the exposure of the intestinal mucosa to varied amounts of dietary gluten. The symptoms do not necessarily correlate with histologic changes, especially as many newly diagnosed patients with celiac disease may be asymptomatic or only minimally symptomatic (often with no gastrointestinal symptoms).
The symptoms of celiac disease may appear with the introduction of cereals into an infant’s diet, although spontaneous remissions often occur during the second decade of life that may be either permanent or followed by the reappearance of symptoms over several years. Alternatively, the symptoms of celiac disease may first become evident at almost any age throughout adulthood. In many patients, frequent spontaneous remissions and exacerbations occur. The symptoms range from significant malabsorption of multiple nutrients, with diarrhea, steatorrhea, weight loss, and the consequences of nutrient depletion (i.e., anemia and metabolic bone disease), to the absence of gastrointestinal symptoms despite evidence of the depletion of a single nutrient (e.g., iron or folate deficiency, osteomalacia, edema from protein loss). Asymptomatic relatives of patients with celiac disease have been identified as having this disease either by small-intestinal biopsy or by serologic studies (e.g., antiendomysial antibodies, tissue transglutaminase [tTG], deamidated gliadin peptide). The availability of these “celiac serologies” has led to a substantial increase in the frequency of diagnosis of celiac disease, and the diagnosis is now being made primarily in patients without “classic” symptoms but with atypical and subclinical presentations.
Etiology The etiology of celiac disease is not known, but environmental, immunologic, and genetic factors all appear to contribute to the disease. One environmental factor is the clear association of the disease with gliadin, a component of gluten that is present in wheat, barley, and rye. In addition to the role of gluten restriction in treatment, the instillation of gluten into both the normal-appearing rectum and the distal ileum of patients with celiac disease results in morphologic changes within hours.
An immunologic component in the pathogenesis of celiac disease is critical and involves both adaptive and innate immune responses. Serum antibodies—IgA antigliadin, antiendomysial, and anti-tTG antibodies—are present, but it is not known whether such antibodies are primary or secondary to the tissue damage. The presence of antiendomysial antibody is 90–95% sensitive and 90–95% specific; the antigen recognized by antiendomysial antibody is tTG, which deaminates gliadin, which is presented to HLA-DQ2 or HLA-DQ8 (see below). Antibody studies are frequently used to identify patients with celiac disease; patients with these antibodies should undergo duodenal biopsy. This autoantibody has not been linked to a pathogenetic mechanism (or mechanisms) responsible for celiac disease. Nonetheless, this antibody is useful in establishing the true prevalence of celiac disease in the general population. A 4-week course of treatment with prednisolone induces a remission in a patient with celiac disease who continues to eat gluten and converts the “flat” abnormal duodenal biopsy to a more normal-appearing one. In addition, gliadin peptides interact with gliadin-specific T cells that mediate tissue injury and induce the release of one or more cytokines (e.g., interferon γ) that cause tissue injury.
![]() Genetic factor(s) are also involved in celiac disease. The incidence of symptomatic celiac disease varies widely in different population groups (high among whites, low among blacks and Asians) and is 10% among first-degree relatives of celiac disease patients. However, serologic studies provide clear evidence that celiac disease is present worldwide. Furthermore, all patients with celiac disease express the HLA-DQ2 or HLA-DQ8 allele, although only a minority of people expressing DQ2/DQ8 have celiac disease. Absence of DQ2/DQ8 excludes the diagnosis of celiac disease.
Genetic factor(s) are also involved in celiac disease. The incidence of symptomatic celiac disease varies widely in different population groups (high among whites, low among blacks and Asians) and is 10% among first-degree relatives of celiac disease patients. However, serologic studies provide clear evidence that celiac disease is present worldwide. Furthermore, all patients with celiac disease express the HLA-DQ2 or HLA-DQ8 allele, although only a minority of people expressing DQ2/DQ8 have celiac disease. Absence of DQ2/DQ8 excludes the diagnosis of celiac disease.
Diagnosis A small-intestinal biopsy is required to establish a diagnosis of celiac disease (Fig. 349-4). A biopsy should be performed when patients have symptoms and laboratory findings suggestive of nutrient malabsorption and/or deficiency as well as a positive tTG antibody test. Since the presentation of celiac disease is often subtle, without overt evidence of malabsorption or nutrient deficiency, a relatively low threshold for biopsy performance is important. It is more prudent to perform a biopsy than another test of intestinal absorption that can never completely exclude or establish this diagnosis.
The diagnosis of celiac disease requires the detection of characteristic histologic changes on small-intestinal biopsy together with a prompt clinical and histologic response after the institution of a gluten-free diet. If IgA antiendomysial or tTG antibodies have been detected in serologic studies, they too should disappear after a gluten-free diet is started. With the increase in the number of patients diagnosed with celiac disease (mostly by serologic studies), the spectrum of histologic changes seen on duodenal biopsy has increased and includes findings that are not as severe as the classic changes shown in Fig. 349-4. The classic changes seen on duodenal/jejunal biopsy are restricted to the mucosa and include (1) an increase in the number of intraepithelial lymphocytes; (2) absence or a reduced height of villi, which causes a flat appearance with increased crypt cell proliferation resulting in crypt hyperplasia and loss of villous structure, with consequent villous, but not mucosal, atrophy; (3) a cuboidal appearance and nuclei that are no longer oriented basally in surface epithelial cells; and (4) increased numbers of lymphocytes and plasma cells in the lamina propria (Fig. 349-4B). Although these features are characteristic of celiac disease, they are not diagnostic because a similar appearance can develop in tropical sprue, eosinophilic enteritis, and milk-protein intolerance in children and occasionally in lymphoma, bacterial overgrowth, Crohn’s disease, and gastrinoma with acid hypersecretion. However, a characteristic histologic appearance that reverts toward normal after the initiation of a gluten-free diet establishes the diagnosis of celiac disease (Fig. 349-4C). Readministration of gluten, with or without an additional small-intestinal biopsy, is not necessary.
A number of patients exhibit gluten sensitivity; i.e., they have gastrointestinal symptoms that respond to gluten restriction but do not have celiac disease. The basis for such gluten sensitivity is not known.
Failure to Respond to Gluten Restriction The most common cause of persistent symptoms in a patient who fulfills all the criteria for the diagnosis of celiac disease is continued intake of gluten. Gluten is ubiquitous, and a significant effort must be made to exclude all gluten from the diet. Use of rice flour in place of wheat flour is very helpful, and several support groups provide important aid to patients with celiac disease and to their families. More than 90% of patients who have the characteristic findings of celiac disease respond to complete dietary gluten restriction. The remainder constitute a heterogeneous group (whose condition is often called refractory celiac disease or refractory sprue) that includes some patients who (1) respond to restriction of other dietary protein (e.g., soy); (2) respond to glucocorticoid treatment; (3) are “temporary” (i.e., whose clinical and morphologic findings disappear after several months or years); or (4) fail to respond to all measures and have a fatal outcome, with or without documented complications of celiac disease, such as the development of intestinal T cell lymphoma or autoimmune enteropathy.
Therapeutic approaches that do not include a gluten-free diet are being developed and include the use of peptidases to inactivate toxic gliadin peptides and of small molecules to block toxic peptide uptake across intestinal tight junctions.
Mechanism of Diarrhea The diarrhea in celiac disease has several pathogenetic mechanisms. Diarrhea may be secondary to (1) steatorrhea, which is primarily a result of changes in jejunal mucosal function; (2) secondary lactase deficiency, a consequence of changes in jejunal brush border enzymatic function; (3) bile acid malabsorption resulting in bile acid–induced fluid secretion in the colon (in cases with more extensive disease involving the ileum); and (4) endogenous fluid secretion resulting from crypt hyperplasia. Celiac disease patients with more severe involvement may improve temporarily with dietary lactose and fat restriction while awaiting the full effects of total gluten restriction, which constitutes primary therapy.
Associated Diseases Celiac disease is associated with dermatitis herpetiformis (DH), but this association has not been explained. Patients with DH have characteristic papulovesicular lesions that respond to dapsone. Almost all patients with DH have histologic changes in the small intestine consistent with celiac disease, although usually much milder and less diffuse in distribution. Most patients with DH have mild or no gastrointestinal symptoms. In contrast, relatively few patients with celiac disease have DH.
Celiac disease is also associated with diabetes mellitus type 1, IgA deficiency, Down syndrome, and Turner’s syndrome. The clinical importance of the association with diabetes is that, although severe watery diarrhea without evidence of malabsorption is most often diagnosed as “diabetic diarrhea” (Chap. 417), assay of antiendomysial antibodies and/or a small-intestinal biopsy must be considered to exclude celiac disease.
Complications The most important complication of celiac disease is the development of cancer. The incidences of both gastrointestinal and nongastrointestinal neoplasms as well as intestinal lymphoma are elevated among patients with celiac disease. For unexplained reasons, the frequency of lymphoma in patients with celiac disease is higher in Ireland and the United Kingdom than in the United States. The possibility of lymphoma must be considered whenever a patient with celiac disease who has previously done well on a gluten-free diet is no longer responsive to gluten restriction or a patient who presents with clinical and histologic features consistent with celiac disease does not respond to a gluten-free diet. Other complications of celiac disease include the development of intestinal ulceration independent of lymphoma and so-called refractory sprue (see above) and collagenous sprue. In collagenous sprue, a layer of collagen-like material is present beneath the basement membrane; patients with collagenous sprue generally do not respond to a gluten-free diet and often have a poor prognosis.
TROPICAL SPRUE
![]() Tropical sprue is a poorly understood syndrome that affects both expatriates and natives in certain but not all tropical areas and is manifested by chronic diarrhea, steatorrhea, weight loss, and nutritional deficiencies, including those of both folate and cobalamin. This disease affects 5–10% of the population in some tropical areas.
Tropical sprue is a poorly understood syndrome that affects both expatriates and natives in certain but not all tropical areas and is manifested by chronic diarrhea, steatorrhea, weight loss, and nutritional deficiencies, including those of both folate and cobalamin. This disease affects 5–10% of the population in some tropical areas.
Chronic diarrhea in a tropical environment is most often caused by infectious agents, including G. lamblia, Yersinia enterocolitica, C. difficile, Cryptosporidium parvum, and Cyclospora cayetanensis. Tropical sprue should not be entertained as a possible diagnosis until the presence of cysts and trophozoites has been excluded in three stool samples. Chronic infections of the gastrointestinal tract and diarrhea in patients with or without AIDS are discussed in Chaps. 160, 161, and 226.
The small-intestinal mucosa of individuals living in tropical areas is not identical to that of individuals who reside in temperate climates. In residents of tropical areas, biopsies reveal a mild alteration of villous architecture with a modest increase in mononuclear cells in the lamina propria, which on occasion can be as severe as that seen in celiac disease. These changes are observed both in native residents and in expatriates living in tropical regions and are usually associated with mild decreases in absorptive function, but they revert to “normal” when an individual moves or returns to a temperate area. Some have suggested that the changes seen in tropical enteropathy and in tropical sprue represent different ends of the spectrum of a single entity, but convincing evidence to support this concept is lacking.
Etiology Because tropical sprue responds to antibiotics, the consensus is that it may be caused by one or more infectious agents. Nonetheless, the etiology and pathogenesis of tropical sprue are uncertain. First, its occurrence is not evenly distributed in all tropical areas; rather, it is found in specific locations, including southern India, the Philippines, and several Caribbean islands (e.g., Puerto Rico, Haiti), but is rarely observed in Africa, Jamaica, or Southeast Asia. Second, an occasional individual does not develop symptoms of tropical sprue until long after having left an endemic area. For this reason, celiac disease (often referred to as celiac sprue) was originally called nontropical sprue to distinguish it from tropical sprue. Third, multiple microorganisms have been identified in jejunal aspirates, with relatively little consistency among studies. Klebsiella pneumoniae, Enterobacter cloacae, and E. coli have been implicated in some studies of tropical sprue, while other studies have favored a role for a toxin produced by one or more of these bacteria. Fourth, the incidence of tropical sprue appears to have decreased substantially during the past two or three decades, perhaps in relation to improved sanitation in many tropical countries during this time. Some have speculated that the reduced occurrence is attributable to the wider use of antibiotics in acute diarrhea, especially in travelers to tropical areas from temperate countries. Fifth, the role of folic acid deficiency in the pathogenesis of tropical sprue requires clarification. Folic acid is absorbed exclusively in the duodenum and proximal jejunum, and most patients with tropical sprue have evidence of folate malabsorption and depletion. Although folate deficiency can cause changes in small-intestinal mucosa that are corrected by folate replacement, several earlier studies reporting that tropical sprue could be cured by folic acid did not provide an explanation for the “insult” that was initially responsible for folate malabsorption.
The clinical pattern of tropical sprue varies in different areas of the world (e.g., India vs. Puerto Rico). Not infrequently, individuals in southern India initially report the occurrence of acute enteritis before the development of steatorrhea and malabsorption. In contrast, in Puerto Rico, a more insidious onset of symptoms and a more dramatic response to antibiotics are seen than in some other locations. Tropical sprue in different areas of the world may not be the same disease, and similar clinical entities may have different etiologies.
Diagnosis The diagnosis of tropical sprue is best based on an abnormal small-intestinal mucosal biopsy in an individual with chronic diarrhea and evidence of malabsorption who is either residing or has recently lived in a tropical country. The small-intestinal biopsy in tropical sprue does not reveal pathognomonic features but resembles, and can often be indistinguishable from, that seen in celiac disease (Fig. 349-4). The biopsy sample in tropical sprue has less villous architectural alteration and more mononuclear cell infiltrate in the lamina propria. In contrast to those of celiac disease, the histologic features of tropical sprue manifest with a similar degree of severity throughout the small intestine, and a gluten-free diet does not result in either clinical or histologic improvement in tropical sprue.
SHORT-BOWEL SYNDROME
Short-bowel syndrome is a descriptive term for the myriad clinical problems that follow resection of various lengths of small intestine or, on rare occasions, are congenital (e.g., microvillous inclusion disease). The factors that determine both the type and degree of symptoms include (1) the specific segment (jejunum vs. ileum) resected, (2) the length of the resected segment, (3) the integrity of the ileocecal valve, (4) whether any large intestine has also been removed, (5) residual disease in the remaining small and/or large intestine (e.g., Crohn’s disease, mesenteric artery disease), and (6) the degree of adaptation in the remaining intestine. Short-bowel syndrome can occur in persons of any age, from neonates to the elderly.
Three different situations in adults mandate intestinal resection: (1) mesenteric vascular disease, including atherosclerosis, thrombotic phenomena, and vasculitides; (2) primary mucosal and submucosal disease (e.g., Crohn’s disease); and (3) operations without preexisting small-intestinal disease (e.g., after trauma).
After resection of the small intestine, the residual intestine undergoes adaptation of both structure and function that may last for up to 6–12 months. Continued intake of dietary nutrients and calories is required to stimulate adaptation via direct contact with the intestinal mucosa, the release of one or more intestinal hormones, and pancreatic and biliary secretions. Thus, enteral nutrition with calorie administration must be maintained, especially in the early postoperative period, even if an extensive intestinal resection requiring parenteral nutrition (PN) has been performed. The subsequent ability of such patients to absorb nutrients will not be known for several months, until adaptation is complete.
Multiple factors besides the absence of intestinal mucosa (required for lipid, fluid, and electrolyte absorption) contribute to diarrhea and steatorrhea in these patients. Removal of the ileum, and especially the ileocecal valve, is often associated with more severe diarrhea than jejunal resection. Without part or all of the ileum, diarrhea can be caused by an increase in bile acids entering the colon; these acids stimulate colonic fluid and electrolyte secretion. Absence of the ileocecal valve is also associated with a decrease in intestinal transit time and bacterial overgrowth from the colon. The presence of the colon (or a major portion) is associated with substantially less diarrhea and a lower likelihood of intestinal failure (an inability to maintain nutrition without parenteral support) as a result of fermentation of nonabsorbed carbohydrates to SCFAs. The latter are absorbed in the colon and stimulate Na and water absorption, improving overall fluid balance. Lactose intolerance as a result of the removal of lactase-containing mucosa as well as gastric hypersecretion may also contribute to the diarrhea.
In addition to diarrhea and/or steatorrhea, a range of nonintestinal symptoms is observed in some patients. The frequency of renal calcium oxalate calculi increases significantly in patients with a small-intestinal resection and an intact colon; this greater frequency is due to an increase in oxalate absorption by the large intestine, with subsequent enteric hyperoxaluria. Two possible mechanisms for the increase in oxalate absorption in the colon have been suggested: (1) increased bile acids and fatty acids that augment colonic mucosal permeability, resulting in enhanced oxalate absorption; and (2) increased fatty acids that bind calcium, resulting in an enhanced amount of soluble oxalate that is then absorbed. Since oxalate is high in relatively few foods (e.g., spinach, rhubarb, tea), dietary restrictions alone do not constitute adequate treatment. Cholestyramine (an anion-binding resin) and calcium have proved useful in reducing hyperoxaluria. Similarly, an increase in cholesterol gallstones is related to a decrease in the bile-acid pool size, which results in the generation of cholesterol supersaturation in gallbladder bile. Gastric hypersecretion of acid occurs in many patients after large resections of the small intestine. The etiology is unclear but may be related to either reduced hormonal inhibition of acid secretion or increased gastrin levels due to reduced small-intestinal catabolism of circulating gastrin. The resulting gastric acid secretion may be an important factor contributing to diarrhea and steatorrhea. A reduced pH in the duodenum can inactivate pancreatic lipase and/or precipitate duodenal bile acids, thereby increasing steatorrhea, and an increase in gastric secretion can create a volume overload relative to the reduced small-intestinal absorptive capacity. Inhibition of gastric acid secretion with proton pump inhibitors can help reduce diarrhea and steatorrhea, but only for the first 6 months.
BACTERIAL OVERGROWTH SYNDROMES
Bacterial overgrowth syndromes comprise a group of disorders with diarrhea, steatorrhea, and macrocytic anemia whose common feature is the proliferation of colonic-type bacteria within the small intestine. This bacterial proliferation is due to stasis caused by impaired peristalsis (functional stasis), changes in intestinal anatomy (anatomic stasis), or direct communication between the small and large intestine. These conditions have also been referred to as stagnant bowel syndrome or blind loop syndrome.
Pathogenesis The manifestations of bacterial overgrowth syndromes are a direct consequence of the presence of increased amounts of a colonic-type bacterial flora, such as E. coli or Bacteroides, in the small intestine. Macrocytic anemia is due to cobalamin—not folate—deficiency. Most bacteria require cobalamin for growth, and increasing concentrations of bacteria use up the relatively small amounts of dietary cobalamin. Steatorrhea is due to impaired micelle formation as a consequence of a reduced intraduodenal concentration of conjugated bile acids and the presence of unconjugated bile acids. Certain bacteria, including Bacteroides, deconjugate conjugated bile acids to unconjugated bile acids. Unconjugated bile acids are absorbed more rapidly than conjugated bile acids; as a result, the intraduodenal concentration of bile acids is reduced. In addition, the CMC of unconjugated bile acids is higher than that of conjugated bile acids, and the result is a decrease in micelle formation. Diarrhea is due, at least in part, to steatorrhea, when it is present. However, some patients manifest diarrhea without steatorrhea, and it is assumed that the colonic-type bacteria in these patients are producing one or more bacterial enterotoxins that are responsible for fluid secretion and diarrhea.
Etiology The etiology of these different disorders is bacterial proliferation in the small-intestinal lumen secondary to anatomic or functional stasis or to a communication between the relatively sterile small intestine and the colon, with its high levels of aerobic and anaerobic bacteria. Several examples of anatomic stasis have been identified: (1) one or more diverticula (both duodenal and jejunal) (Fig. 294-3C); (2) fistulas and strictures related to Crohn’s disease (Fig. 349-3D); (3) a proximal duodenal afferent loop following subtotal gastrectomy and gastrojejunostomy; (4) a bypass of the intestine (e.g., a jejunoileal bypass for obesity); and (5) dilation at the site of a previous intestinal anastomosis. These anatomic derangements are often associated with the presence of a segment (or segments) of intestine out of continuity of propagated peristalsis, with consequent stasis and bacterial proliferation. Bacterial overgrowth syndromes can also occur in the absence of an anatomic blind loop when functional stasis is present. Impaired peristalsis and bacterial overgrowth in the absence of a blind loop occur in scleroderma, where motility abnormalities exist in both the esophagus and the small intestine (Chap. 382). Functional stasis and bacterial overgrowth can also develop in association with diabetes mellitus and in the small intestine when a direct connection exists between the small and large intestines, including an ileocolonic resection, or occasionally after an enterocolic anastomosis that permits entry of bacteria into the small intestine as a result of bypassing the ileocecal valve.
Diagnosis The diagnosis may be suspected from the combination of a low serum cobalamin level and an elevated serum folate level, as enteric bacteria frequently produce folate compounds that are absorbed in the duodenum. Ideally, the bacterial overgrowth syndromes are diagnosed by the demonstration of increased levels of aerobic and/or anaerobic colonic-type bacteria in a jejunal aspirate obtained by intubation. However, this specialized test is rarely available. Breath hydrogen testing with administration of lactulose (a nondigestible disaccharide) has also been used to detect bacterial overgrowth. The Schilling test can diagnose bacterial overgrowth (see Chap. 350e) but is not available routinely. Often the diagnosis is suspected clinically and confirmed by the response to treatment.
WHIPPLE’S DISEASE
Whipple’s disease is a chronic multisystemic disease associated with diarrhea, steatorrhea, weight loss, arthralgia, and central nervous system (CNS) and cardiac problems; it is caused by the bacterium Tropheryma whipplei. Until the identification of T. whipplei by polymerase chain reaction, the hallmark of Whipple’s disease had been the presence of PAS-positive macrophages in the small intestine (Fig. 349-4E) and other organs with evidence of disease.
Etiology T. whipplei, a small (50–500 nm) gram-positive bacillus in the group Actinobacteria, has low virulence but high infectivity. Symptoms of Whipple’s disease are relatively minimal compared to the bacterial burden in multiple tissues.
Clinical presentation The onset of Whipple’s disease is insidious and is characterized by diarrhea, steatorrhea, abdominal pain, weight loss, migratory large-joint arthropathy, and fever as well as ophthalmologic and CNS symptoms. Dementia is a relatively late symptom and an extremely poor prognostic sign, especially in patients who experience relapse after the induction of a remission with antibiotics. For unexplained reasons, the disease occurs primarily in middle-aged white men. The steatorrhea in these patients is generally believed to be secondary to both small-intestinal mucosal injury and lymphatic obstruction due to the increased number of PAS-positive macrophages in the lamina propria of the small intestine.
Diagnosis The diagnosis of Whipple’s disease is suggested by a multisystemic disease in a patient with diarrhea and steatorrhea. Tissue biopsy of the small intestine and/or other organs that may be involved (e.g., liver, lymph nodes, heart, eyes, CNS, or synovial membranes), given the patient’s symptoms, is the primary approach. The presence of PAS-positive macrophages containing the characteristic small bacilli is suggestive of this diagnosis. However, T. whipplei–containing macrophages can be confused with PAS-positive macrophages containing M. avium complex, which may be a cause of diarrhea in AIDS. The presence of the T. whipplei bacillus outside of macrophages is a more important indicator of active disease than is their presence within the macrophages. T. whipplei has now been successfully grown in culture.
PROTEIN-LOSING ENTEROPATHY
Protein-losing enteropathy is not a specific disease but rather a group of gastrointestinal and nongastrointestinal disorders with hypoproteinemia and edema in the absence of either proteinuria or defects in protein synthesis (e.g., chronic liver disease). These diseases are characterized by excess protein loss into the gastrointestinal tract. Normally, ~10% of total protein catabolism occurs via the gastrointestinal tract. Evidence of increased protein loss into the gastrointestinal tract is found in more than 65 different diseases, which can be classified into three groups: (1) mucosal ulceration, such that the protein loss primarily represents exudation across damaged mucosa (e.g., ulcerative colitis, gastrointestinal carcinomas, and peptic ulcer); (2) nonulcerated mucosa, but with evidence of mucosal damage so that the protein loss represents loss across epithelia with altered permeability (e.g., celiac disease and Ménétrier’s disease in the small intestine and stomach, respectively); and (3) lymphatic dysfunction, representing either primary lymphatic disease or lymphatic disease secondary to partial lymphatic obstruction that may occur as a result of enlarged lymph nodes or cardiac disease.
Diagnosis The diagnosis of protein-losing enteropathy is suggested by peripheral edema and low serum albumin and globulin levels in the absence of renal and hepatic disease. An individual with protein-losing enteropathy only rarely has selective loss of only albumin or only globulins. Therefore, marked reduction of serum albumin with normal serum globulins should not initiate an evaluation for protein-losing enteropathy but should suggest renal and/or hepatic disease. Likewise, reduced serum globulins with normal serum albumin levels are more likely a result of reduced globulin synthesis rather than enhanced globulin loss into the intestine. An increase in protein loss into the gastrointestinal tract has been documented by the administration of one of several radiolabeled proteins and its quantitation in stool during a 24- or 48-h period. Unfortunately, none of these radiolabeled proteins is available for routine clinical use. α1-Antitrypsin, a protein that accounts for ~4% of total serum proteins and is resistant to proteolysis, can be used to detect enhanced rates of serum protein loss into the intestinal tract but cannot be used to assess gastric protein loss because of its degradation in an acid milieu. α1-Antitrypsin clearance is measured by determining stool volume as well as both stool and plasma α1-antitrypsin concentrations. In addition to the loss of protein via abnormal and distended lymphatics, peripheral lymphocytes may be lost via lymphatics, with consequent relative lymphopenia. Thus, lymphopenia in a patient with hypoproteinemia indicates increased loss of protein into the gastrointestinal tract.
Patients with increased protein loss into the gastrointestinal tract from lymphatic obstruction often have steatorrhea and diarrhea. The steatorrhea is a result of altered lymphatic flow as lipid-containing chylomicrons exit from intestinal epithelial cells via intestinal lymphatics (Table 349-4; Fig. 349-4). In the absence of mechanical or anatomic lymphatic obstruction, intrinsic intestinal lymphatic dysfunction—with or without lymphatic dysfunction in the peripheral extremities—has been designated intestinal lymphangiectasia. Similarly, ~50% of individuals with intrinsic peripheral lymphatic disease (Milroy’s disease) also have intestinal lymphangiectasia and hypoproteinemia. Other than steatorrhea and enhanced protein loss into the gastrointestinal tract, all other aspects of intestinal absorptive function are normal in intestinal lymphangiectasia.
Other Causes Patients who appear to have idiopathic protein-losing enteropathy without evidence of gastrointestinal disease should be examined for cardiac disease—especially right-sided valvular disease and chronic pericarditis (Chaps. 284 and 288). On occasion, hypoproteinemia can be the only presenting manifestation in these two types of heart disease. Ménétrier’s disease (also called hypertrophic gastropathy) is an uncommon entity that involves the body and fundus of the stomach and is characterized by large gastric folds, reduced gastric acid secretion, and, at times, enhanced protein loss into the stomach.
SUMMARY
The many conditions that can produce malabsorption are classified by their pathophysiology in Table 349-8. The pathophysiology of the various clinical manifestations of malabsorption is summarized in Table 349-9.
|
CLASSIFICATION OF MALABSORPTION SYNDROMES |

|
PATHOPHYSIOLOGY OF CLINICAL MANIFESTATIONS OF MALABSORPTION DISORDERS |
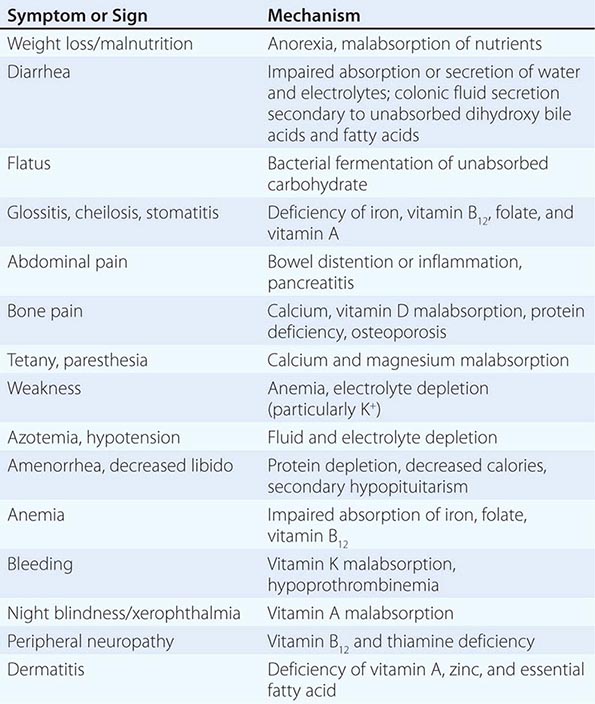
350e |
The Schilling Test |
The Schilling test is performed to determine the cause of cobalamin malabsorption. Unfortunately, this test has not been available commercially in the United States for the last few years. Since an understanding of the physiology and pathophysiology of cobalamin absorption is very valuable in enhancing one’s understanding of aspects of gastric, pancreatic, and ileal function, discussion of the Schilling test is provided as supplemental information to Chap. 349. Because cobalamin absorption requires multiple steps, including gastric, pancreatic, and ileal processes, the Schilling test also can be used to assess the integrity of the organs involved in those processes (Chap. 128).
Cobalamin is present primarily in meat. Except in strict vegans, dietary cobalamin deficiency is exceedingly uncommon. Dietary cobalamin is bound in the stomach to a glycoprotein called R-binder protein, which is synthesized in both the stomach and the salivary glands. This cobalamin–R binder complex is formed in the acid milieu of the stomach. Cobalamin absorption has an absolute requirement for intrinsic factor, another glycoprotein synthesized and released by gastric parietal cells, to promote its uptake by specific cobalamin receptors on the brush border of ileal enterocytes. Pancreatic protease enzymes split the cobalamin–R binder complex to release cobalamin in the proximal small intestine, where cobalamin then is bound by intrinsic factor.
As a consequence, cobalamin absorption may be abnormal in the following conditions:
1. Pernicious anemia. In this disease, immunologically mediated atrophy of gastric parietal cells leads to an absence of both gastric acid and intrinsic factor secretion.
2. Chronic pancreatitis can result from a deficiency of pancreatic proteases to split the cobalamin–R binder complex. Although 50% of patients with chronic pancreatitis reportedly have an abnormal Schilling test that is corrected by pancreatic enzyme replacement, cobalamin-responsive macrocytic anemia in chronic pancreatitis is extremely rare. Although this probably reflects a difference in the digestion/absorption of cobalamin in food versus that in a crystalline form, the Schilling test still can be used to assess pancreatic exocrine function.
3. Achlorhydria is the absence of hydrochloric acid; intrinsic factor is also secreted with acid which is responsible for splitting cobalamin away from the proteins in food to which it is bound. Up to one-third of individuals >60 years of age have marginal vitamin B12 absorption because of an inability to release cobalamin from food; these people have no defects in the absorption of crystalline vitamin B12.
4. Bacterial overgrowth syndromes, which are most often secondary to stasis in the small intestine, lead to bacterial utilization of cobalamin (often referred to as stagnant bowel syndrome; see below).
5. Ileal dysfunction (as a result of either inflammation or prior intestinal resection) is due to impaired function of the mechanism of cobalamin–intrinsic factor uptake by ileal intestinal epithelial cells.
In the Schilling test, 58Co-labeled cobalamin is administered orally, and urine is collected for 24 h. The test is dependent on normal renal and bladder function. Urinary excretion of cobalamin reflects cobalamin absorption, provided that intrahepatic binding sites for cobalamin are fully occupied. To ensure saturation of these binding sites so that all absorbed radiolabeled cobalamin will be excreted in urine, 1 mg of cobalamin is administered intramuscularly 1 h after ingestion of the radiolabeled cobalamin. The Schilling test may yield an abnormal result (usually defined as <10% excretion in 24 h) in pernicious anemia, chronic pancreatitis, blind loop syndrome, and ileal disease (Table 350e-1). Therefore, whenever an abnormal Schilling result is obtained, 58Co-labeled cobalamin should be administered on another occasion, this time bound to intrinsic factor, with pancreatic enzymes, or after a 5-day course of antibiotic treatment (often with tetracycline). A variation of the Schilling test can detect failure to split cobalamin from food proteins. The labeled cobalamin is cooked together with a scrambled egg and administered orally. People with achlorydria excrete <10% of the labeled cobalamin in the urine. In addition to establishing the etiology for cobalamin deficiency, the Schilling test can help delineate the pathologic process responsible for steatorrhea by assessing ileal, pancreatic, and small-intestinal luminal function. Unfortunately, the Schilling test is performed infrequently because of the unavailability of human intrinsic factor.
|
DIFFERENTIAL RESULTS OF THE SCHILLING TEST IN SEVERAL DISEASES ASSOCIATED WITH COBALAMIN MALABSORPTION |
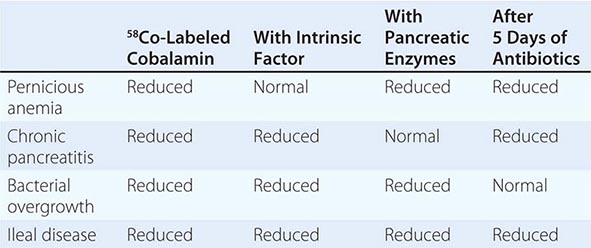
351 |
Inflammatory Bowel Disease |
Inflammatory bowel disease (IBD) is an immune-mediated chronic intestinal condition. Ulcerative colitis (UC) and Crohn’s disease (CD) are the two major types of IBD.
GLOBAL CONSIDERATIONS: EPIDEMIOLOGY
![]() The incidence and prevalence of IBD are highest in Westernized nations, with UC incidence estimates ranging from 0.6 to 24.3 per 100,000 in Europe, 0 to 19.2 per 100,000 in North America, and 0.1 to 6.3 per 100,000 in the Middle East and Asia and CD estimates ranging from 0.3 to 12.7 per 100,000 in Europe, 0 to 20.2 per 100,000 in North America, and 0.04 to 5.0 per 100,000 in the Middle East and Asia (Table 351-1). For prevalence rates, the UC estimates range from 4.9 to 505 per 100,000 in Europe, 37.5 to 248.6 per 100,000 in North America, and 4.9 to 168.3 per 100,000 in the Middle East and Asia, and the CD estimates range from 0.6 to 322 per 100,000 in Europe, 16.7 to 318.5 per 100,000 in North America, and 0.88 to 67.9 per 100,000 in Asia and the Middle East. The highest reported incidence rates are in Canada (19.2 per 100,000 for UC and 20.2 per 100,000 for CD), with approximately 0.6% of the Canadian population having IBD. Countries in the Pacific, including New Zealand and Australia, which share many possible environmental risk factors and similar genetic background as northwest Europe and North America, have high incidence rates of IBD.
The incidence and prevalence of IBD are highest in Westernized nations, with UC incidence estimates ranging from 0.6 to 24.3 per 100,000 in Europe, 0 to 19.2 per 100,000 in North America, and 0.1 to 6.3 per 100,000 in the Middle East and Asia and CD estimates ranging from 0.3 to 12.7 per 100,000 in Europe, 0 to 20.2 per 100,000 in North America, and 0.04 to 5.0 per 100,000 in the Middle East and Asia (Table 351-1). For prevalence rates, the UC estimates range from 4.9 to 505 per 100,000 in Europe, 37.5 to 248.6 per 100,000 in North America, and 4.9 to 168.3 per 100,000 in the Middle East and Asia, and the CD estimates range from 0.6 to 322 per 100,000 in Europe, 16.7 to 318.5 per 100,000 in North America, and 0.88 to 67.9 per 100,000 in Asia and the Middle East. The highest reported incidence rates are in Canada (19.2 per 100,000 for UC and 20.2 per 100,000 for CD), with approximately 0.6% of the Canadian population having IBD. Countries in the Pacific, including New Zealand and Australia, which share many possible environmental risk factors and similar genetic background as northwest Europe and North America, have high incidence rates of IBD.
|
EPIDEMIOLOGY OF IBD |
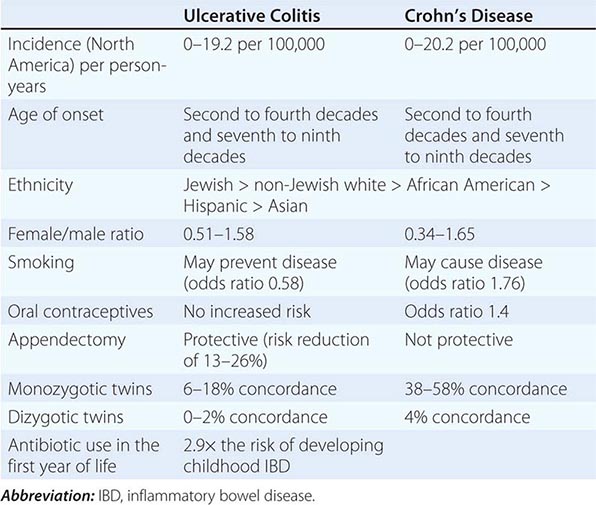
In countries that are becoming more Westernized, including China, South Korea, India, Lebanon, Iran, Thailand, and countries in the French West Indies and North Africa, IBD appears to be emerging, emphasizing the importance of environmental factors in disease pathogenesis. In Japan, the prevalence of CD has risen rapidly from 2.9 cases per 100,000 in 1986 to 13.5 per 100,000 in 1998, whereas in South Korea, the prevalence of UC has quadrupled from 7.6 per 100,000 in 1997 to 30.9 per 100,000 in 2005. In Hong Kong, the prevalence of UC almost tripled from 2.3 in 1997 to 6.3 per 100,000 over a 9-year period. In Singapore, the prevalence of CD increased from 1.3 in 1990 to 7.2 per 100,000 in 2004. In China the number of cases of UC has increased by fourfold between 1981–1990 and 1991–2000.
Increasing immigration to Western societies also has an impact on the incidence and prevalence of IBD. The prevalence of UC among southern Asians who immigrated to the United Kingdom (UK) was higher in comparison to the European UK population (17 cases per 100,000 persons vs 7 per 100,000). Spanish patients who emigrated within Europe, but not those who immigrated to Latin America, developed IBD more frequently than controls. Individuals who have immigrated to Westernized countries and then returned to their country of birth also continue to demonstrate an increased risk of developing IBD.
Peak incidence of UC and CD is in the second to fourth decades, with 78% of CD studies and 51% of UC studies reporting the highest incidence among those age 20–29 years old. A second modest rise in incidence occurs between the seventh and ninth decades of life. The female-to-male ratio ranges from 0.51 to 1.58 for UC studies and 0.34 to 1.65 for CD studies, suggesting that the diagnosis of IBD is not gender specific. The greatest incidence of IBD is among white and Jewish people, but the incidence of IBD in Hispanic and Asian people is increasing, as noted above. Urban areas have a higher prevalence of IBD than rural areas, and high socioeconomic classes have a higher prevalence than lower socioeconomic classes.
Epidemiologic studies have identified a number of potential environmental factors that are associated with disease risk (Fig. 351-1). In Caucasian populations, smoking is an important risk factor in IBD with opposite effects on UC (odds ratio [OR] 0.58) and CD (OR 1.76), whereas in other ethnic groups with different genetic susceptibility, smoking may play a lesser role. There is a protective effect of previous appendectomy with confirmed appendicitis (reduction of 13–26%), particularly at a young age, on the development of UC across different geographical regions and populations. There is a modest association with the development of CD. Oral contraceptive use is associated with the risk of CD (OR 1.4). The association between oral contraceptive use and UC is limited to women with a history of smoking. There is an association between antibiotic use and the development of childhood IBD with children who received one or more dispensations of antibiotics during the first year of life having a 2.9-fold increase in the risk of developing IBD during childhood. Breastfeeding may also protect against the development of IBD. These factors are consistent with the rapid increase in IBD incidence recently noted during the first decade of life. Infectious gastroenteritis with pathogens (e.g., Salmonella, Shigella, Campylobacter spp., Clostridium difficile) increases IBD risk by two- to threefold. Diets high in animal protein, sugars, sweets, oils, fish and shellfish, and dietary fat, especially ω-6 fatty acids, and low in ω-3 fatty acids have been implicated in increasing the risk of IBD.
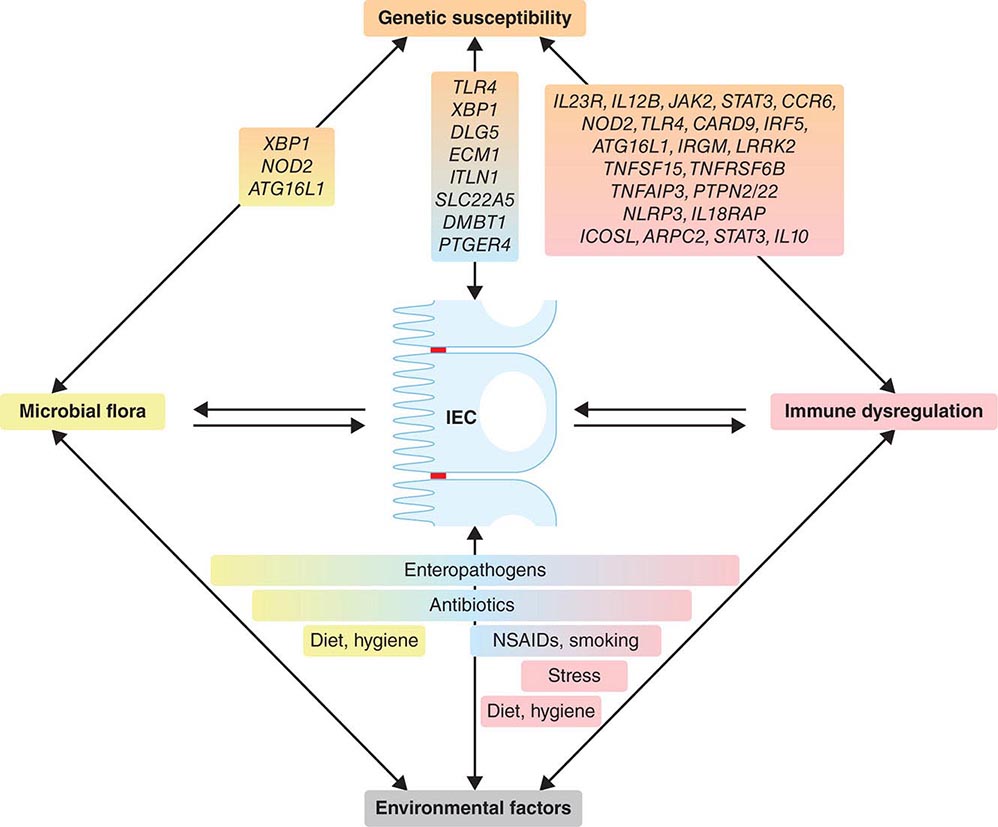
FIGURE 351-1 Pathogenesis of inflammatory bowel disease (IBD). In IBD, the tridirectional relationship between the commensal flora (microbiota), intestinal epithelial cells (IEC), and mucosal immune system is dysregulated, leading to chronic inflammation. Each of these three factors is affected by genetic and environmental factors that determine risk for the disease. NSAIDs, nonsteroidal anti-inflammatory drugs. (Adapted from A Kaser et al: Annu Rev Immunol 28:573, 2010.)
IBD is a familial disease in 5–10% of patients (Fig. 351-2). Some of these patients may exhibit early-onset disease during the first decade of life and, in CD, a concordance of anatomic site and clinical type within families. In the remainder of patients, IBD is observed in the absence of a family history (i.e., sporadic disease). If a patient has IBD, the lifetime risk that a first-degree relative will be affected is ~10%. If two parents have IBD, each child has a 36% chance of being affected. In twin studies, 38–58% of monozygotic twins are concordant for CD and 6–18% are concordant for UC, whereas 4% of dizygotic twins are concordant for CD and 0–2% are concordant for UC in Swedish and Danish cohorts. The risks of developing IBD are higher in first-degree relatives of Jewish versus non-Jewish patients: 7.8% versus 5.2% for CD and 4.5% versus 1.6% for UC.
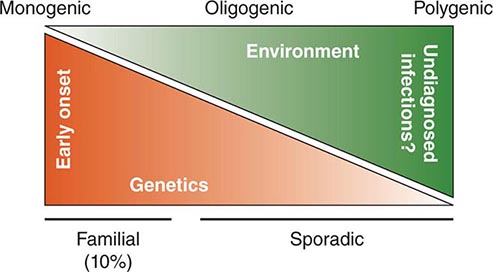
FIGURE 351-2 A model for the syndromic nature of inflammatory bowel disease. Genetic and environmental factors variably influence the development and phenotypic manifestations of IBD. At the one extreme, IBD is a exemplified as a simple Mendelian disorder as observed in “early-onset IBD” due to single gene defects such as IL10, IL10RA, and IL10RB; and at the other extreme, it may be exemplified by as yet to be described emerging infectious diseases. (Adapted from A Kaser et al: Dig Dis 28:395, 2010.)
GLOBAL CONSIDERATIONS: IBD PHENOTYPES
![]() There are racial differences in IBD location and behavior that may reflect underlying genetic variations and have important implications for diagnosis and management of disease. For example, African-American patients are more likely than non-Hispanic whites to develop esophagogastroduodenal CD, colorectal disease, and perianal disease and are less likely to have ileal involvement. They are also at higher risk for uveitis and sacroiliitis. Hispanics have a higher prevalence of perianal CD and erythema nodosum and a more proximal extent of disease. Fistulizing CD has been reported in nearly one-third of Hispanic patients, up to one-quarter of African-American patients, and up to one-half of Asian patients. Both African-American and Hispanic CD patients, but not UC patients, had a lower prevalence of family history of IBD than their white counterparts. There are few data on all aspects of disease in Hispanics, in the incidence and prevalence of IBD in African Americans, and in Asians with IBD outside Asia. These ethnic variations implicate the importance of different genetic and/or environmental factors in the pathogenesis of this disorder.
There are racial differences in IBD location and behavior that may reflect underlying genetic variations and have important implications for diagnosis and management of disease. For example, African-American patients are more likely than non-Hispanic whites to develop esophagogastroduodenal CD, colorectal disease, and perianal disease and are less likely to have ileal involvement. They are also at higher risk for uveitis and sacroiliitis. Hispanics have a higher prevalence of perianal CD and erythema nodosum and a more proximal extent of disease. Fistulizing CD has been reported in nearly one-third of Hispanic patients, up to one-quarter of African-American patients, and up to one-half of Asian patients. Both African-American and Hispanic CD patients, but not UC patients, had a lower prevalence of family history of IBD than their white counterparts. There are few data on all aspects of disease in Hispanics, in the incidence and prevalence of IBD in African Americans, and in Asians with IBD outside Asia. These ethnic variations implicate the importance of different genetic and/or environmental factors in the pathogenesis of this disorder.
ETIOLOGY AND PATHOGENESIS
Under physiologic conditions, homeostasis normally exists between the commensal microbiota, epithelial cells that line the interior of the intestines (intestinal epithelial cells [IECs]) and immune cells within the tissues (Fig. 351-1). A consensus hypothesis is that each of these three major host compartments that function together as an integrated “supraorganism” (microbiota, IECs, and immune cells) are affected by specific environmental (e.g., smoking, antibiotics, enteropathogens) and genetic factors that, in a susceptible host, cumulatively and interactively disrupt homeostasis, which in so doing culminates in a chronic state of dysregulated inflammation; that is IBD. Although chronic activation of the mucosal immune system may represent an appropriate response to an infectious agent, a search for such an agent has thus far been unrewarding in IBD. As such, IBD is currently considered an inappropriate immune response to the endogenous (autocthonous) commensal microbiota within the intestines, with or without some component of autoimmunity. Importantly, the normal, uninflamed intestines contain a large number of immune cells that are in a unique state of activation, in which the gut is restrained from full immunologic responses to the commensal microbiota and dietary antigens by very powerful regulatory pathways that function within the immune system (e.g., T regulatory cells that express the FoxP3 transcription factor and suppress inflammation). During the course of infections or other environmental stimuli in the normal host, full activation of the gut-associated lymphoid tissues occurs but is rapidly superseded by dampening of the immune response and tissue repair. In IBD such processes may not be regulated normally.
GENETIC CONSIDERATIONS
![]() The genetic underpinning of IBD is known from its occurrence in the context of several genetic syndromes and the development of severe, refractory IBD in early life in the setting of single gene defects that affect the immune system (Table 351-2). In addition, IBD has a familial origin in at least 10% of afflicted individuals (Fig. 351-2). In the majority of patients, IBD is considered to be a polygenic disorder that gives rise to multiple clinical subgroups within UC and CD. A variety of genetic approaches including candidate gene studies, linkage analysis, and genome-wide association studies (GWASs) that focus on the identification of disease-associated, single-nucleotide polymorphisms (SNPs) within the human genome and, more recently, whole-genome sequencing have elucidated many of the genetic factors that affect risk for these diseases. GWASs have, to date, identified 163 genetic loci with 100 of these loci observed to be associated with both disease phenotypes (Table 351-3). The remainder are specific for either CD (30 loci) or UC (20 loci). These genetic similarities account for the overlapping immunopathogenesis and consequently epidemiologic observations of both diseases in the same families and similarities in response to therapies. Because the specific causal variants for each identified gene or locus are largely unknown, it is not clear whether the similarities in the genetic risk factors associated with CD and UC that are observed are shared at structural or functional levels. The risk conferred by each identified gene or locus is unequal and generally small, such that only ~20% of the genetic variance is considered to be explained by the current genetic information. Further, many of the genetic risk factors identified are also observed to be associated with risk for other immune-mediated diseases, suggesting that related immunogenetic pathways are involved in the pathogenesis of multiple different disorders accounting for the common responsiveness to similar types of biologic therapies (e.g., anti–tumor necrosis factor therapies) and possibly the simultaneous occurrence of these disorders. The diseases and the genetic risk factors that are shared with IBD include rheumatoid arthritis (TNFAIP3), psoriasis (IL23R, IL12B), ankylosing spondylitis (IL23R), type 1 diabetes mellitus (IL10, PTPN2), asthma (ORMDL3), and systemic lupus erythematosus (TNFAIP3, IL10) among others.
The genetic underpinning of IBD is known from its occurrence in the context of several genetic syndromes and the development of severe, refractory IBD in early life in the setting of single gene defects that affect the immune system (Table 351-2). In addition, IBD has a familial origin in at least 10% of afflicted individuals (Fig. 351-2). In the majority of patients, IBD is considered to be a polygenic disorder that gives rise to multiple clinical subgroups within UC and CD. A variety of genetic approaches including candidate gene studies, linkage analysis, and genome-wide association studies (GWASs) that focus on the identification of disease-associated, single-nucleotide polymorphisms (SNPs) within the human genome and, more recently, whole-genome sequencing have elucidated many of the genetic factors that affect risk for these diseases. GWASs have, to date, identified 163 genetic loci with 100 of these loci observed to be associated with both disease phenotypes (Table 351-3). The remainder are specific for either CD (30 loci) or UC (20 loci). These genetic similarities account for the overlapping immunopathogenesis and consequently epidemiologic observations of both diseases in the same families and similarities in response to therapies. Because the specific causal variants for each identified gene or locus are largely unknown, it is not clear whether the similarities in the genetic risk factors associated with CD and UC that are observed are shared at structural or functional levels. The risk conferred by each identified gene or locus is unequal and generally small, such that only ~20% of the genetic variance is considered to be explained by the current genetic information. Further, many of the genetic risk factors identified are also observed to be associated with risk for other immune-mediated diseases, suggesting that related immunogenetic pathways are involved in the pathogenesis of multiple different disorders accounting for the common responsiveness to similar types of biologic therapies (e.g., anti–tumor necrosis factor therapies) and possibly the simultaneous occurrence of these disorders. The diseases and the genetic risk factors that are shared with IBD include rheumatoid arthritis (TNFAIP3), psoriasis (IL23R, IL12B), ankylosing spondylitis (IL23R), type 1 diabetes mellitus (IL10, PTPN2), asthma (ORMDL3), and systemic lupus erythematosus (TNFAIP3, IL10) among others.
|
PRIMARY GENETIC DISORDERS ASSOCIATED WITH IBD |
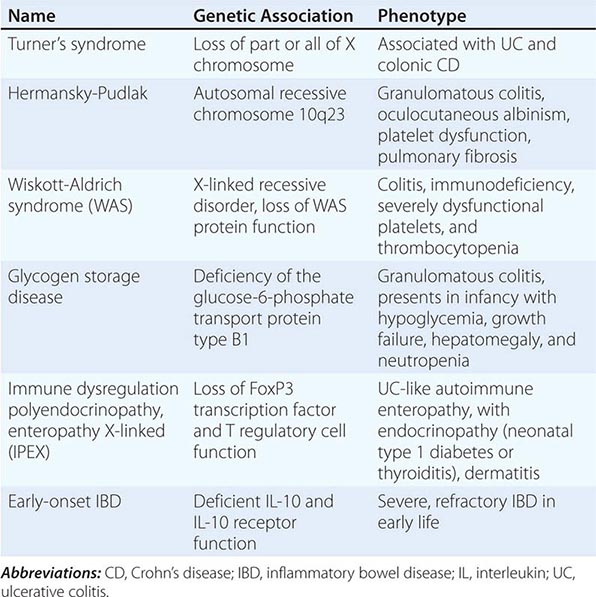
|
EXAMPLES OF GENETIC LOCI ASSOCIATED WITH CD AND/OR UC |
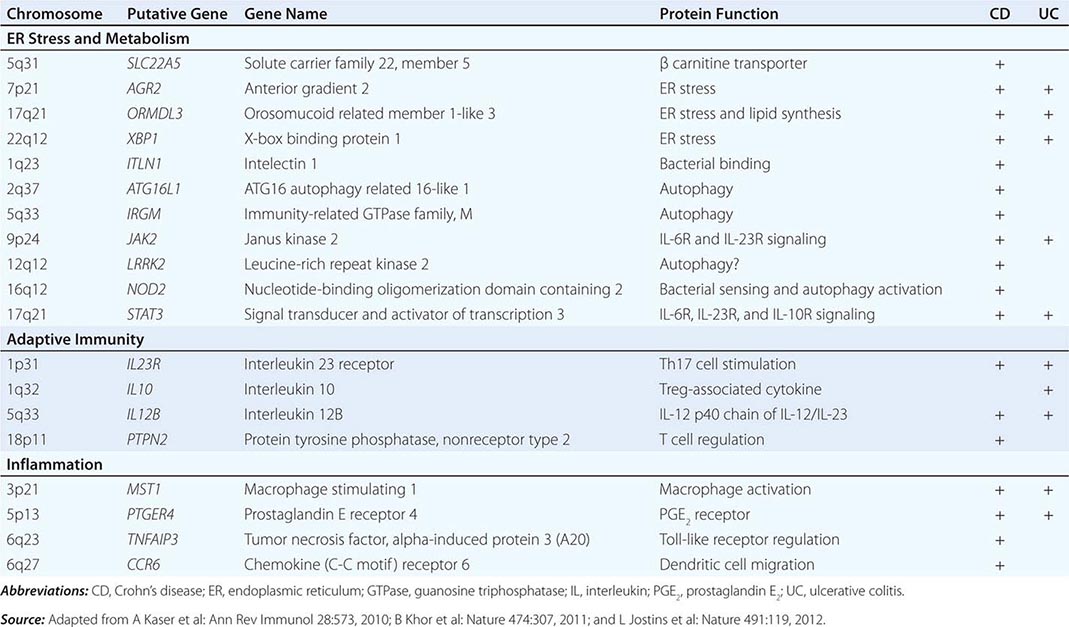
The genetic factors defined to date that are recognized to mediate risk for IBD have highlighted the importance of several common mechanisms of disease (Table 351-3). These include the following: those genes that are associated with fundamental cell biologic processes such as endoplasmic reticulum (ER) and metabolic stress (e.g., XBP1, ORMDL3, OCTN), which serve to regulate the secretory activity of cells involved in responses to the commensal microbiota such as Paneth and goblet cells and the manner in which intestinal cells respond to the metabolic products of bacteria; those associated with innate immunity and autophagy (e.g., NOD2, ATG16L1, IRGM, JAK2, STAT3) that function in innate immune cells (both parenchymal and hematopoietic) to respond to and effectively clear bacteria, mycobacteria, and viruses; those that are associated with the regulation of adaptive immunity (e.g., IL23R, IL12B, IL10, PTPN2), which regulate the balance between inflammatory and anti-inflammatory (regulatory) cytokines; and, finally, those that are involved in the development and resolution of inflammation (e.g., MST1, CCR6, TNFAIP3, PTGER4) and ultimately leukocyte recruitment and inflammatory mediator production. Some of these loci are associated with specific subtypes of disease such as the association between NOD2 polymorphisms and fibrostenosing CD or ATG16L1 and fistulizing disease, especially within the ileum. However, the clinical utility of these genetic risk factors for the diagnosis or determination of prognosis and therapeutic responses remains to be defined.
COMMENSAL MICROBIOTA AND IBD
The endogenous commensal microbiota within the intestines plays a central role in the pathogenesis of IBD. Humans are born sterile and acquire their commensal microbiota initially from the mother during egress through the birth canal and subsequently from environmental sources. A stable configuration of up to 1000 species of bacteria that achieves a biomass of approximately 1012 colony-forming units per gram of feces is achieved by 3 years of age, which likely persists into adult life, with each individual human possessing a unique combination of species. In addition, the intestines contain other microbial life forms including archae, viruses, and protists. The microbiota is thus considered as a critical and sustaining component of the organism. The establishment and maintenance of the intestinal microbiota composition and function is under the control of host (e.g., immune and epithelial responses), environmental (e.g., diet and antibiotics), and likely genetic (e.g., NOD2) factors (Fig. 351-1). In turn, the microbiota, through its structural components and metabolic activity, has major influences on the epithelial and immune function of the host, which, through epigenetic effects, may have durable consequences. During early life when the commensal microbiota is being established, these microbial effects on the host may be particularly important in determining later life risk for IBD. Specific components of the microbiota can promote or protect from disease. The commensal microbiota in patients with both UC and CD is demonstrably different from nonafflicted individuals, a state of dysbiosis, suggesting the presence of microorganisms that drive disease (e.g., Proteobacteria such as enteroinvasive and adherent Escherichia coli) and to which the immune response is directed and/or the loss of microorganisms that hinder inflammation (e.g., Firmicutes such as Faecalibacterium prausnitzii). Many of the changes in the commensal microbiota occur as a consequence of the inflammation. In addition, agents that alter the intestinal microbiota such as metronidazole, ciprofloxacin, and elemental diets, may improve CD. CD may also respond to fecal diversion, demonstrating the ability of luminal contents to exacerbate disease.
DEFECTIVE IMMUNE REGULATION IN IBD
The mucosal immune system is normally unreactive to luminal contents due to oral (mucosal) tolerance. When soluble antigens are administered orally rather than subcutaneously or intramuscularly, antigen-specific nonresponsiveness is induced. Multiple mechanisms are involved in the induction of oral tolerance and include deletion or anergy of antigen-reactive T cells or induction of CD4+ T cells that suppress gut inflammation (e.g., T regulatory cells expressing the FoxP3 transcription factor) that secrete anti-inflammatory cytokines such as interleukin (IL) 10, IL-35, and transforming growth factor β (TGF-β). Oral tolerance may be responsible for the lack of immune responsiveness to dietary antigens and the commensal microbiota in the intestinal lumen. In IBD this suppression of inflammation is altered, leading to uncontrolled inflammation. The mechanisms of this regulated immune suppression are incompletely known.
Gene knockout (–/–) or transgenic (Tg) mouse models of IBD, which include those that are directed at genes demonstrated to be associated with risk for the human disease, have revealed that deleting specific cytokines (e.g., IL-2, IL-10, TGF-β) or their receptors, deleting molecules associated with T cell antigen recognition (e.g., T cell antigen receptors), or interfering with IEC barrier function and the regulation of responses to commensal bacteria (e.g., XBP1, N-cadherin, mucus glycoprotein, or nuclear factor-κB [NF-κB]) leads to spontaneous colitis or enteritis. In the majority of circumstances, intestinal inflammation in these animal models requires the presence of the commensal microbiota. Thus, a variety of specific alterations can lead to immune activation by commensal microbiota and inflammation directed at the intestines in mice. How these relate to human IBD remains to be defined, but they are consistent with inappropriate responses of the genetically susceptible host to the commensal microbiota.
In both UC and CD, an inflammatory pathway thus likely emerges from the genetic predisposition that is associated with inappropriate innate immune and epithelial sensing and reactivity to commensal bacteria that secrete inflammatory mediators together with inadequate regulatory pathways that lead to activated CD4+ and CD8+ T cells within the epithelium and lamina propria that altogether secrete excessive quantities of inflammatory cytokines relative to anti-inflammatory cytokines. Some cytokines activate other inflammatory cells (macrophages and B cells), and others act indirectly to recruit other lymphocytes, inflammatory leukocytes, and mononuclear cells from the bloodstream into the gut through interactions between homing receptors on leukocytes (e.g., α4β7 integrin) and addressins on vascular endothelium (e.g., MadCAM1). Consistent with this, neutralization of tumor necrosis factor (TNF) or α4β7 integrin demonstrate therapeutic efficacy in IBD. CD4+ T helper (TH) cells that promote inflammation are of three major types, all of which may be associated with colitis in animal models and perhaps humans: TH1 cells (secrete interferon [IFN] γ), TH2 cells (secrete IL-4, IL-5, IL-13), and TH17 cells (secrete IL-17, IL-21). TH1 cells induce transmural granulomatous inflammation that resembles CD; TH2 cells, and related natural killer T cells that secrete IL-13, induce superficial mucosal inflammation resembling UC in animal models; and TH17 cells may be responsible for neutrophilic recruitment. However, neutralization of the cytokines produced by these cells, such as IFN-γ or IL-17, has yet to show efficacy in therapeutic trials. Each of these T cell subsets cross-regulate each other. The TH1 cytokine pathway is initiated by IL-12, a key cytokine in the pathogenesis of experimental models of mucosal inflammation. IL-4 and IL-23, together with IL-6 and TGF-β, induce TH2 and TH17 cells, respectively, and IL-23 inhibits the suppressive function of regulatory T cells. Activated macrophages secrete TNF and IL-6. These characteristics of the immune response in IBD explain the beneficial therapeutic effects of antibodies to block proinflammatory cytokines or the signaling by their receptors (e.g., anti-TNF, anti-IL-12, anti-IL-23, anti-IL-6, or Janus kinase [JAK] inhibitors) or molecules associated with leukocyte recruitment (e.g., anti-α4β7), or the use of cytokines that inhibit inflammation and promote regulatory T cells (e.g., IL-10) or promote intestinal barrier function and may be beneficial to humans with intestinal inflammation.
THE INFLAMMATORY CASCADE IN IBD
Once initiated in IBD by abnormal innate immune sensing of bacteria by parenchymal cells (e.g., IECs) and hematopoietic cells (e.g., dendritic cells), the immune inflammatory response is perpetuated by T cell activation. A sequential cascade of inflammatory mediators extends the response; each step is a potential target for therapy. Inflammatory cytokines such as IL-1, IL-6, and TNF have diverse effects on tissues. They promote fibrogenesis, collagen production, activation of tissue metalloproteinases, and the production of other inflammatory mediators; they also activate the coagulation cascade in local blood vessels (e.g., increased production of von Willebrand’s factor). These cytokines are normally produced in response to infection but are usually turned off or inhibited at the appropriate time to limit tissue damage. In IBD their activity is not regulated, resulting in an imbalance between the proinflammatory and anti-inflammatory mediators. Therapies such as the 5-aminosalicylic acid (5-ASA) compounds and glucocorticoids are potent inhibitors of these inflammatory mediators through inhibition of transcription factors such as NF-κB that regulate their expression.
PATHOLOGY
ULCERATIVE COLITIS: MACROSCOPIC FEATURES
UC is a mucosal disease that usually involves the rectum and extends proximally to involve all or part of the colon. About 40–50% of patients have disease limited to the rectum and rectosigmoid, 30–40% have disease extending beyond the sigmoid but not involving the whole colon, and 20% have a total colitis. Proximal spread occurs in continuity without areas of uninvolved mucosa. When the whole colon is involved, the inflammation extends 2–3 cm into the terminal ileum in 10–20% of patients. The endoscopic changes of backwash ileitis are superficial and mild and are of little clinical significance. Although variations in macroscopic activity may suggest skip areas, biopsies from normal-appearing mucosa are usually abnormal. Thus, it is important to obtain multiple biopsies from apparently uninvolved mucosa, whether proximal or distal, during endoscopy. One caveat is that effective medical therapy can change the appearance of the mucosa such that either skip areas or the entire colon can be microscopically normal.
With mild inflammation, the mucosa is erythematous and has a fine granular surface that resembles sandpaper. In more severe disease, the mucosa is hemorrhagic, edematous, and ulcerated (Fig. 351-3). In long-standing disease, inflammatory polyps (pseudopolyps) may be present as a result of epithelial regeneration. The mucosa may appear normal in remission, but in patients with many years of disease it appears atrophic and featureless, and the entire colon becomes narrowed and shortened. Patients with fulminant disease can develop a toxic colitis or megacolon where the bowel wall thins and the mucosa is severely ulcerated; this may lead to perforation.
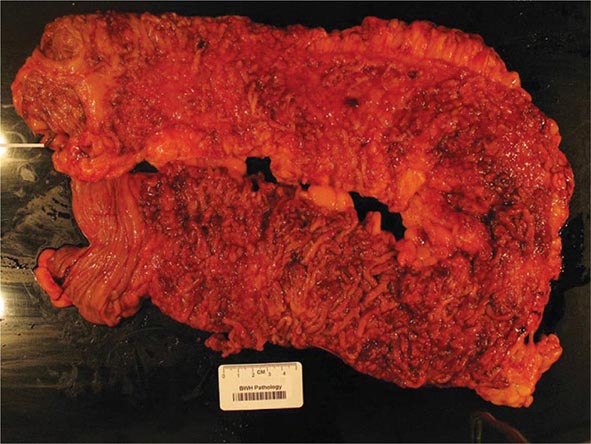
FIGURE 351-3 Ulcerative colitis. Diffuse (nonsegmental) mucosal disease, with broad areas of ulceration. The bowel wall is not thickened, and there is no cobblestoning. (Courtesy of Dr. R. Odze, Division of Gastrointestinal Pathology, Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts; with permission.)
ULCERATIVE COLITIS: MICROSCOPIC FEATURES
Histologic findings correlate well with the endoscopic appearance and clinical course of UC. The process is limited to the mucosa and superficial submucosa, with deeper layers unaffected except in fulminant disease. In UC, two major histologic features suggest chronicity and help distinguish it from infectious or acute self-limited colitis. First, the crypt architecture of the colon is distorted; crypts may be bifid and reduced in number, often with a gap between the crypt bases and the muscularis mucosae. Second, some patients have basal plasma cells and multiple basal lymphoid aggregates. Mucosal vascular congestion, with edema and focal hemorrhage, and an inflammatory cell infiltrate of neutrophils, lymphocytes, plasma cells, and macrophages may be present. The neutrophils invade the epithelium, usually in the crypts, giving rise to cryptitis and, ultimately, to crypt abscesses (Fig. 351-4). Ileal changes in patients with backwash ileitis include villous atrophy and crypt regeneration with increased inflammation, increased neutrophil and mononuclear inflammation in the lamina propria, and patchy cryptitis and crypt abscesses.
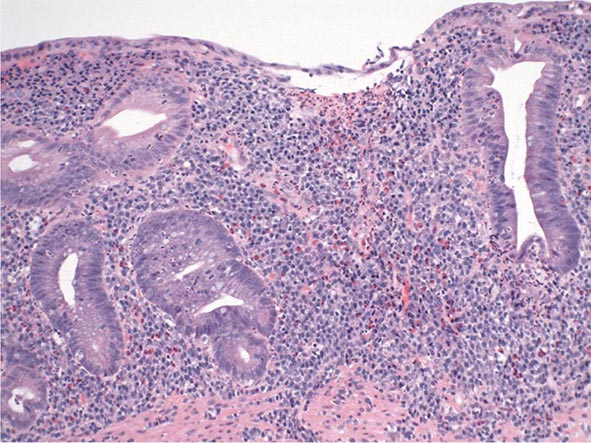
FIGURE 351-4 Medium-power view of colonic mucosa in ulcerative colitis showing diffuse mixed inflammation, basal lymphoplasmacytosis, crypt atrophy and irregularity, and superficial erosion. These features are typical of chronic active ulcerative colitis. (Courtesy of Dr. R. Odze, Division of Gastrointestinal Pathology, Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts; with permission.)
CROHN’S DISEASE: MACROSCOPIC FEATURES
CD can affect any part of the gastrointestinal (GI) tract from the mouth to the anus. Some 30–40% of patients have small bowel disease alone, 40–55% have disease involving both the small and large intestines, and 15–25% have colitis alone. In the 75% of patients with small intestinal disease, the terminal ileum is involved in 90%. Unlike UC, which almost always involves the rectum, the rectum is often spared in CD. CD is segmental with skip areas in the midst of diseased intestine (Fig. 351-5). Perirectal fistulas, fissures, abscesses, and anal stenosis are present in one-third of patients with CD, particularly those with colonic involvement. Rarely, CD may also involve the liver and the pancreas.
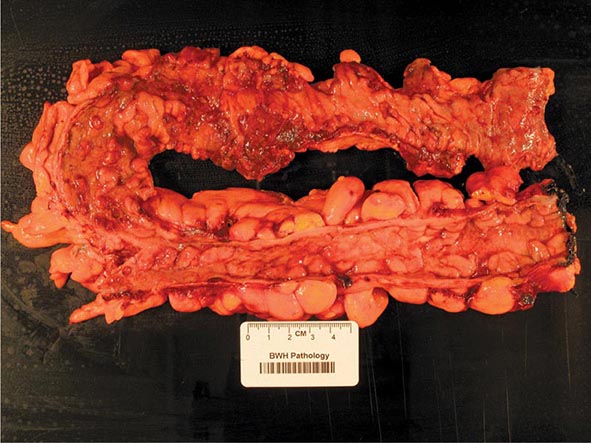
FIGURE 351-5 Crohn’s disease of the colon showing thickening of the wall, with stenosis, linear serpiginous ulcers and cobblestoning of the mucosa. (Courtesy of Dr. R Odze, Division of Gastrointestinal Pathology, Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts; with permission.)
Unlike UC, CD is a transmural process. Endoscopically, aphthous or small superficial ulcerations characterize mild disease; in more active disease, stellate ulcerations fuse longitudinally and transversely to demarcate islands of mucosa that frequently are histologically normal. This “cobblestone” appearance is characteristic of CD, both endoscopically and by barium radiography. As in UC, pseudopolyps can form in CD.
Active CD is characterized by focal inflammation and formation of fistula tracts, which resolve by fibrosis and stricturing of the bowel. The bowel wall thickens and becomes narrowed and fibrotic, leading to chronic, recurrent bowel obstructions. Projections of thickened mesentery encase the bowel (“creeping fat”), and serosal and mesenteric inflammation promotes adhesions and fistula formation.
CROHN’S DISEASE: MICROSCOPIC FEATURES
The earliest lesions are aphthoid ulcerations and focal crypt abscesses with loose aggregations of macrophages, which form noncaseating granulomas in all layers of the bowel wall (Fig. 351-6). Granulomas can be seen in lymph nodes, mesentery, peritoneum, liver, and pancreas. Although granulomas are a pathognomonic feature of CD, they are rarely found on mucosal biopsies. Surgical resection reveals granulomas in about one-half of cases. Other histologic features of CD include submucosal or subserosal lymphoid aggregates, particularly away from areas of ulceration, gross and microscopic skip areas, and transmural inflammation that is accompanied by fissures that penetrate deeply into the bowel wall and sometimes form fistulous tracts or local abscesses.
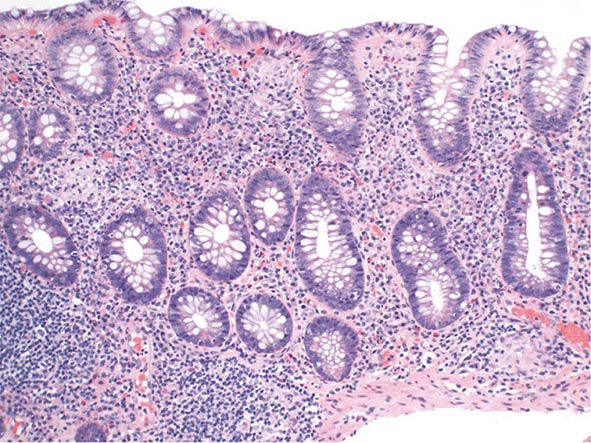
FIGURE 351-6 Medium-power view of Crohn’s colitis showing mixed acute and chronic inflammation, crypt atrophy, and multiple small epithelioid granulomas in the mucosa. (Courtesy of Dr. R Odze, Division of Gastrointestinal Pathology, Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts; with permission.)
CLINICAL PRESENTATION
ULCERATIVE COLITIS
Signs and Symptoms The major symptoms of UC are diarrhea, rectal bleeding, tenesmus, passage of mucus, and crampy abdominal pain. The severity of symptoms correlates with the extent of disease. Although UC can present acutely, symptoms usually have been present for weeks to months. Occasionally, diarrhea and bleeding are so intermittent and mild that the patient does not seek medical attention.
Patients with proctitis usually pass fresh blood or blood-stained mucus, either mixed with stool or streaked onto the surface of a normal or hard stool. They also have tenesmus, or urgency with a feeling of incomplete evacuation, but rarely have abdominal pain. With proctitis or proctosigmoiditis, proximal transit slows, which may account for the constipation commonly seen in patients with distal disease.
When the disease extends beyond the rectum, blood is usually mixed with stool or grossly bloody diarrhea may be noted. Colonic motility is altered by inflammation with rapid transit through the inflamed intestine. When the disease is severe, patients pass a liquid stool containing blood, pus, and fecal matter. Diarrhea is often nocturnal and/or postprandial. Although severe pain is not a prominent symptom, some patients with active disease may experience vague lower abdominal discomfort or mild central abdominal cramping. Severe cramping and abdominal pain can occur with severe attacks of the disease. Other symptoms in moderate to severe disease include anorexia, nausea, vomiting, fever, and weight loss.
Physical signs of proctitis include a tender anal canal and blood on rectal examination. With more extensive disease, patients have tenderness to palpation directly over the colon. Patients with a toxic colitis have severe pain and bleeding, and those with megacolon have hepatic tympany. Both may have signs of peritonitis if a perforation has occurred. The classification of disease activity is shown in Table 351-4.
|
ULCERATIVE COLITIS: DISEASE PRESENTATION |
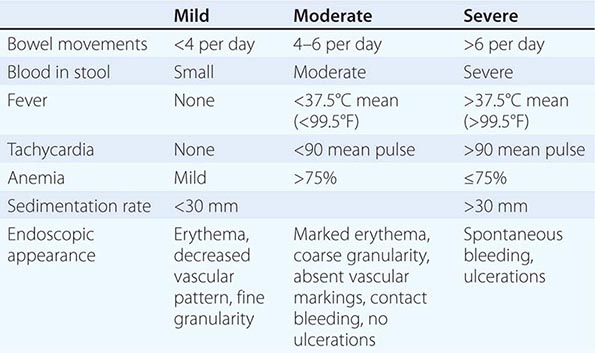
Laboratory, Endoscopic, and Radiographic Features Active disease can be associated with a rise in acute-phase reactants (C-reactive protein [CRP]), platelet count, and erythrocyte sedimentation rate (ESR), and a decrease in hemoglobin. Fecal lactoferrin is a highly sensitive and specific marker for detecting intestinal inflammation. Fecal calprotectin levels correlate well with histologic inflammation, predict relapses, and detect pouchitis. Both fecal lactoferrin and calprotectin are becoming an integral part of IBD management and are used frequently to rule out active inflammation versus symptoms of irritable bowel or bacterial overgrowth. In severely ill patients, the serum albumin level will fall rather quickly. Leukocytosis may be present but is not a specific indicator of disease activity. Proctitis or proctosigmoiditis rarely causes a rise in CRP. Diagnosis relies on the patient’s history; clinical symptoms; negative stool examination for bacteria, C. difficile toxin, and ova and parasites; sigmoidoscopic appearance (see Fig. 345-4A); and histology of rectal or colonic biopsy specimens.
Sigmoidoscopy is used to assess disease activity and is usually performed before treatment. If the patient is not having an acute flare, colonoscopy is used to assess disease extent and activity (Fig. 351-7). Endoscopically mild disease is characterized by erythema, decreased vascular pattern, and mild friability. Moderate disease is characterized by marked erythema, absent vascular pattern, friability and erosions, and severe disease by spontaneous bleeding and ulcerations. Histologic features change more slowly than clinical features but can also be used to grade disease activity.
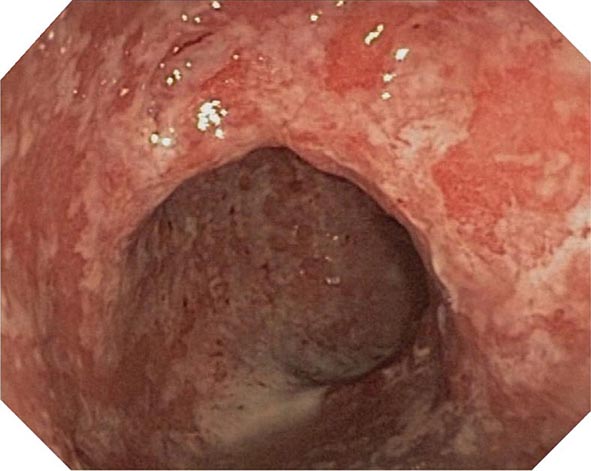
FIGURE 351-7 Colonoscopy with acute ulcerative colitis: severe colon inflammation with erythema, friability, and exudates. (Courtesy of Dr. M. Hamilton, Gastroenterology Division, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts; with permission.)
The earliest radiologic change of UC seen on single-contrast barium enema is a fine mucosal granularity. With increasing severity, the mucosa becomes thickened, and superficial ulcers are seen. Deep ulcerations can appear as “collar-button” ulcers, which indicate that the ulceration has penetrated the mucosa. Haustral folds may be normal in mild disease, but as activity progresses they become edematous and thickened. Loss of haustration can occur, especially in patients with long-standing disease. In addition, the colon becomes shortened and narrowed. Polyps in the colon may be postinflammatory polyps or pseudopolyps, adenomatous polyps, or carcinoma.
Computed tomography (CT) scanning or magnetic resonance imaging (MRI) is not as helpful as endoscopy in making the diagnosis of UC, but typical findings include mild mural thickening (<1.5 cm), inhomogeneous wall density, absence of small bowel thickening, increased perirectal and presacral fat, target appearance of the rectum, and adenopathy.
Complications Only 15% of patients with UC present initially with catastrophic illness. Massive hemorrhage occurs with severe attacks of disease in 1% of patients, and treatment for the disease usually stops the bleeding. However, if a patient requires 6–8 units of blood within 24–48 h, colectomy is indicated. Toxic megacolon is defined as a transverse or right colon with a diameter of >6 cm, with loss of haustration in patients with severe attacks of UC. It occurs in about 5% of attacks and can be triggered by electrolyte abnormalities and narcotics. About 50% of acute dilations will resolve with medical therapy alone, but urgent colectomy is required for those that do not improve. Perforation is the most dangerous of the local complications, and the physical signs of peritonitis may not be obvious, especially if the patient is receiving glucocorticoids. Although perforation is rare, the mortality rate for perforation complicating a toxic megacolon is about 15%. In addition, patients can develop a toxic colitis and such severe ulcerations that the bowel may perforate without first dilating.
Strictures occur in 5–10% of patients and are always a concern in UC because of the possibility of underlying neoplasia. Although benign strictures can form from the inflammation and fibrosis of UC, strictures that are impassable with the colonoscope should be presumed malignant until proven otherwise. A stricture that prevents passage of the colonoscope is an indication for surgery. UC patients occasionally develop anal fissures, perianal abscesses, or hemorrhoids, but the occurrence of extensive perianal lesions should suggest CD.




