456 |
Diseases of the Spinal Cord |
Diseases of the spinal cord are frequently devastating. They produce quadriplegia, paraplegia, and sensory deficits far beyond the damage they would inflict elsewhere in the nervous system because the spinal cord contains, in a small cross-sectional area, almost the entire motor output and sensory input of the trunk and limbs. Many spinal cord diseases are reversible if recognized and treated at an early stage (Table 456-1); thus, they are among the most critical of neurologic emergencies. The efficient use of diagnostic procedures, guided by knowledge of the anatomy and the clinical features of spinal cord diseases, is required to maximize the likelihood of a successful outcome.
|
TREATABLE SPINAL CORD DISORDERS |
Abbreviations: CMV, cytomegalovirus; HSV, herpes simplex virus; HTLV, human T cell lymphotropic virus; VZV, varicella-zoster virus.
APPROACH TO THE PATIENT:
Spinal Cord Disease
SPINAL CORD ANATOMY RELEVANT TO CLINICAL SIGNS
The spinal cord is a thin, tubular extension of the central nervous system contained within the bony spinal canal. It originates at the medulla and continues caudally to the conus medullaris at the lumbar level; its fibrous extension, the filum terminale, terminates at the coccyx. The adult spinal cord is ~46 cm (18 in.) long, oval in shape, and enlarged in the cervical and lumbar regions, where neurons that innervate the upper and lower extremities, respectively, are located. The white matter tracts containing ascending sensory and descending motor pathways are located peripherally, whereas nerve cell bodies are clustered in an inner region of gray matter shaped like a four-leaf clover that surrounds the central canal (anatomically an extension of the fourth ventricle). The membranes that cover the spinal cord—the pia, arachnoid, and dura—are continuous with those of the brain, and the cerebrospinal fluid is contained within the subarachnoid space between the pia and arachnoid.
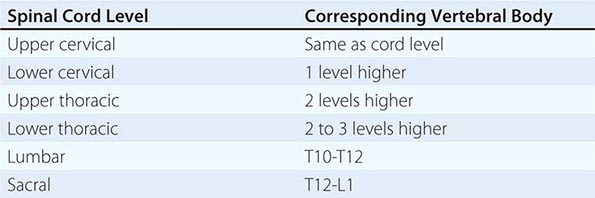
The spinal cord has 31 segments, each defined by an exiting ventral motor root and entering dorsal sensory root. During embryologic development, growth of the cord lags behind that of the vertebral column, and the mature spinal cord ends at approximately the first lumbar vertebral body. The lower spinal nerves take an increasingly downward course to exit via intervertebral foramina. The first seven pairs of cervical spinal nerves exit above the same-numbered vertebral bodies, whereas all the subsequent nerves exit below the same-numbered vertebral bodies because of the presence of eight cervical spinal cord segments but only seven cervical vertebrae. The relationship between spinal cord segments and the corresponding vertebral bodies is shown in Table 456-2. These relationships assume particular importance for localization of lesions that cause spinal cord compression. Sensory loss below the circumferential level of the umbilicus, for example, corresponds to the T10 cord segment but indicates involvement of the cord adjacent to the seventh or eighth thoracic vertebral body (see Figs. 31-2 and 31-3). In addition, at every level, the main ascending and descending tracts are somatotopically organized with a laminated distribution that reflects the origin or destination of nerve fibers.
|
SPINAL CORD LEVELS RELATIVE TO THE VERTEBRAL BODIES |
Determining the Level of the Lesion The presence of a horizontally defined level below which sensory, motor, and autonomic function is impaired is a hallmark of a lesion of the spinal cord. This sensory level is sought by asking the patient to identify a pinprick or cold stimulus applied to the proximal legs and lower trunk and successively moved up toward the neck on each side. Sensory loss below this level is the result of damage to the spinothalamic tract on the opposite side, one to two segments higher in the case of a unilateral spinal cord lesion, and at the level of a bilateral lesion. The discrepancy in the level of a unilateral lesion is the result of the course of the second-order sensory fibers, which originate in the dorsal horn, and ascend for one or two levels as they cross anterior to the central canal to join the opposite spinothalamic tract. Lesions that transect the descending corticospinal and other motor tracts cause paraplegia or quadriplegia with heightened deep tendon reflexes, Babinski signs, and eventual spasticity (the upper motor neuron syndrome). Transverse damage to the cord also produces autonomic disturbances consisting of absent sweating below the implicated cord level and bladder, bowel, and sexual dysfunction.
The uppermost level of a spinal cord lesion can also be localized by attention to the segmental signs corresponding to disturbed motor or sensory innervation by an individual cord segment. A band of altered sensation (hyperalgesia or hyperpathia) at the upper end of the sensory disturbance, fasciculations or atrophy in muscles innervated by one or several segments, or a muted or absent deep tendon reflex may be noted at this level. These signs also can occur with focal root or peripheral nerve disorders; thus, they are most useful when they occur together with signs of long tract damage. With severe and acute transverse lesions, the limbs initially may be flaccid rather than spastic. This state of “spinal shock” lasts for several days, rarely for weeks, and may be mistaken for extensive damage to the anterior horn cells over many segments of the cord or for an acute polyneuropathy.
The main features of transverse damage at each level of the spinal cord are summarized below.
CERVICAL CORD Upper cervical cord lesions produce quadriplegia and weakness of the diaphragm. The uppermost level of weakness and reflex loss with lesions at C5-C6 is in the biceps; at C7, in finger and wrist extensors and triceps; and at C8, finger and wrist flexion. Horner’s syndrome (miosis, ptosis, and facial hypohidrosis) may accompany a cervical cord lesion at any level.
THORACIC CORD Lesions here are localized by the sensory level on the trunk and, if present, by the site of midline back pain. Useful markers of the sensory level on the trunk are the nipples (T4) and umbilicus (T10). Leg weakness and disturbances of bladder and bowel function accompany the paralysis. Lesions at T9-T10 paralyze the lower—but not the upper—abdominal muscles, resulting in upward movement of the umbilicus when the abdominal wall contracts (Beevor’s sign).
LUMBAR CORD Lesions at the L2-L4 spinal cord levels paralyze flexion and adduction of the thigh, weaken leg extension at the knee, and abolish the patellar reflex. Lesions at L5-S1 paralyze only movements of the foot and ankle, flexion at the knee, and extension of the thigh, and abolish the ankle jerks (S1).
SACRAL CORD/CONUS MEDULLARIS The conus medullaris is the tapered caudal termination of the spinal cord, comprising the sacral and single coccygeal segments. The distinctive conus syndrome consists of bilateral saddle anesthesia (S3-S5), prominent bladder and bowel dysfunction (urinary retention and incontinence with lax anal tone), and impotence. The bulbocavernosus (S2-S4) and anal (S4-S5) reflexes are absent (Chap. 437). Muscle strength is largely preserved. By contrast, lesions of the cauda equina, the nerve roots derived from the lower cord, are characterized by low back and radicular pain, asymmetric leg weakness and sensory loss, variable areflexia in the lower extremities, and relative sparing of bowel and bladder function. Mass lesions in the lower spinal canal often produce a mixed clinical picture with elements of both cauda equina and conus medullaris syndromes. Cauda equina syndromes are also discussed in Chap. 22.
Special Patterns of Spinal Cord Disease The location of the major ascending and descending pathways of the spinal cord are shown in Fig. 456-1. Most fiber tracts—including the posterior columns and the spinocerebellar and pyramidal tracts—are situated on the side of the body they innervate. However, afferent fibers mediating pain and temperature sensation ascend in the spinothalamic tract contralateral to the side they supply. The anatomic configurations of these tracts produce characteristic syndromes that provide clues to the underlying disease process.
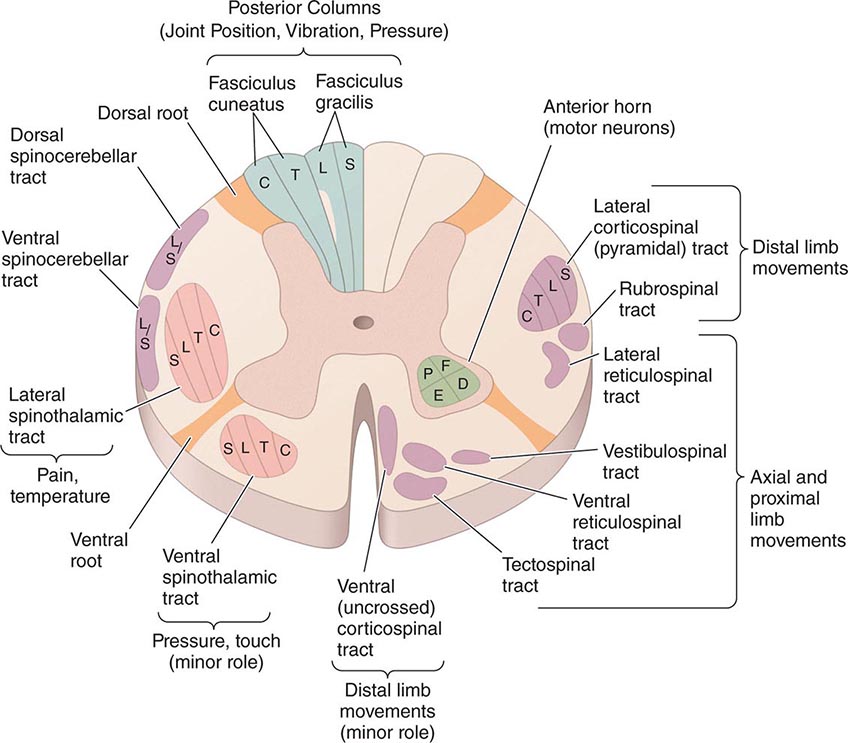
FIGURE 456-1 Transverse section through the spinal cord, composite representation, illustrating the principal ascending (left) and descending (right) pathways. The lateral and ventral spinothalamic tracts ascend contralateral to the side of the body that is innervated. C, cervical; D, distal; E, extensors; F, flexors; L, lumbar; P, proximal; S, sacral; T, thoracic.
BROWN-SEQUARD HEMICORD SYNDROME This consists of ipsilateral weakness (corticospinal tract) and loss of joint position and vibratory sense (posterior column), with contralateral loss of pain and temperature sense (spinothalamic tract) one or two levels below the lesion. Segmental signs, such as radicular pain, muscle atrophy, or loss of a deep tendon reflex, are unilateral. Partial forms are more common than the fully developed syndrome.
CENTRAL CORD SYNDROME This syndrome results from selective damage to the gray matter nerve cells and crossing spinothalamic tracts surrounding the central canal. In the cervical cord, the central cord syndrome produces arm weakness out of proportion to leg weakness and a “dissociated” sensory loss, meaning loss of pain and temperature sensations over the shoulders, lower neck, and upper trunk (cape distribution), in contrast to preservation of light touch, joint position, and vibration sense in these regions. Spinal trauma, syringomyelia, and intrinsic cord tumors are the main causes.
ANTERIOR SPINAL ARTERY SYNDROME Infarction of the cord is generally the result of occlusion or diminished flow in this artery. The result is bilateral tissue destruction at several contiguous levels that spares the posterior columns. All spinal cord functions—motor, sensory, and autonomic—are lost below the level of the lesion, with the striking exception of retained vibration and position sensation.
FORAMEN MAGNUM SYNDROME Lesions in this area interrupt decussating pyramidal tract fibers destined for the legs, which cross caudal to those of the arms, resulting in weakness of the legs (crural paresis). Compressive lesions near the foramen magnum may produce weakness of the ipsilateral shoulder and arm followed by weakness of the ipsilateral leg, then the contralateral leg, and finally the contralateral arm, an “around the clock” pattern that may begin in any of the four limbs. There is typically suboccipital pain spreading to the neck and shoulders.
INTRAMEDULLARY AND EXTRAMEDULLARY SYNDROMES It is useful to differentiate intramedullary processes, arising within the substance of the cord, from extramedullary ones that lie outside the cord and compress the spinal cord or its vascular supply. The differentiating features are only relative and serve as clinical guides. With extramedullary lesions, radicular pain is often prominent, and there is early sacral sensory loss and spastic weakness in the legs with incontinence due to the superficial location of the corresponding sensory and motor fibers in the spinothalamic and corticospinal tracts (Fig. 456-1). Intramedullary lesions tend to produce poorly localized burning pain rather than radicular pain and to spare sensation in the perineal and sacral areas (“sacral sparing”), reflecting the laminated configuration of the spinothalamic tract with sacral fibers outermost; corticospinal tract signs appear later. Regarding extramedullary lesions, a further distinction is made between extradural and intradural masses, as the former are generally malignant and the latter benign (neurofibroma being a common cause). Consequently, a long duration of symptoms favors an intradural origin.
ACUTE AND SUBACUTE SPINAL CORD DISEASES
The initial symptoms of structural diseases of the cord that evolve over days or weeks are focal neck or back pain, followed by various combinations of paresthesias, sensory loss, motor weakness, and sphincter disturbance. There may be only mild sensory symptoms or a devastating functional transection of the cord. Partial lesions selectively involve the posterior columns or anterior spinothalamic tracts or are limited to one side of the cord. Paresthesias or numbness typically begins in the feet and ascend symmetrically or asymmetrically. These symptoms simulate a polyneuropathy, but a sharply demarcated spinal cord level indicates the myelopathic nature of the process.
In severe and abrupt cases, areflexia reflecting spinal shock may be present, but hyperreflexia supervenes over days or weeks; persistent areflexic paralysis with a sensory level usually indicates necrosis over multiple segments of the spinal cord.
COMPRESSIVE MYELOPATHIES
Neoplastic Spinal Cord Compression In adults, most neoplasms are epidural in origin, resulting from metastases to the adjacent vertebral column. The propensity of solid tumors to metastasize to the vertebral column probably reflects the high proportion of bone marrow located in the axial skeleton. Almost any malignant tumor can metastasize to the spinal column, with breast, lung, prostate, kidney, lymphoma, and myeloma being particularly frequent. The thoracic spinal column is most commonly involved; exceptions are metastases from prostate and ovarian cancer, which occur disproportionately in the sacral and lumbar vertebrae, probably from spread through Batson’s plexus, a network of veins along the anterior epidural space. Retroperitoneal neoplasms (especially lymphomas or sarcomas) enter the spinal canal laterally through the intervertebral foramina and produce radicular pain with signs of weakness that corresponds to the level of involved nerve roots.
Pain is usually the initial symptom of spinal metastasis; it may be aching and localized or sharp and radiating in quality and typically worsens with movement, coughing, or sneezing and characteristically awakens patients at night. A recent onset of persistent back pain, particularly if in the thoracic spine (which is uncommonly involved by spondylosis), should prompt consideration of vertebral metastasis. Rarely, pain is mild or absent. Plain radiographs of the spine and radionuclide bone scans have a limited role in diagnosis because they do not identify 15–20% of metastatic vertebral lesions and fail to detect paravertebral masses that reach the epidural space through the intervertebral foramina. MRI provides excellent anatomic resolution of the extent of spinal tumors (Fig. 456-2) and is able to distinguish between malignant lesions and other masses—epidural abscess, tuberculoma, lipoma, or epidural hemorrhage, among others—that present in a similar fashion. Vertebral metastases are usually hypointense relative to a normal bone marrow signal on T1-weighted MRI; after the administration of gadolinium, contrast enhancement may deceptively “normalize” the appearance of the tumor by increasing its intensity to that of normal bone marrow. Infections of the spinal column (osteomyelitis and related disorders) are distinctive in that, unlike tumor, they often cross the disk space to involve the adjacent vertebral body.
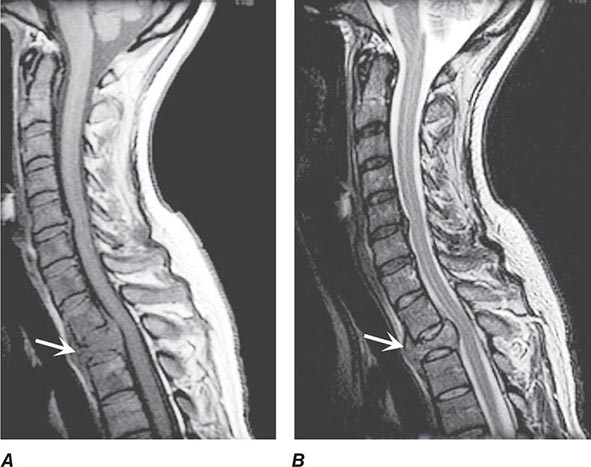
FIGURE 456-2 Epidural spinal cord compression due to breast carcinoma. Sagittal T1-weighted (A) and T2-weighted (B) magnetic resonance imaging scans through the cervicothoracic junction reveal an infiltrated and collapsed second thoracic vertebral body with posterior displacement and compression of the upper thoracic spinal cord. The low-intensity bone marrow signal in A signifies replacement by tumor.
If spinal cord compression is suspected, imaging should be obtained promptly. If there are radicular symptoms but no evidence of myelopathy, it may be safe to defer imaging for 24–48 h. Up to 40% of patients who present with cord compression at one level are found to have asymptomatic epidural metastases elsewhere; thus, the length of the spine is often imaged when epidural malignancy is in question.
In contrast to tumors of the epidural space, most intradural mass lesions are slow-growing and benign. Meningiomas and neurofibromas account for most of these, with occasional cases caused by chordoma, lipoma, dermoid, or sarcoma. Meningiomas (Fig. 456-3) are often located posterior to the thoracic cord or near the foramen magnum, although they can arise from the meninges anywhere along the spinal canal. Neurofibromas are benign tumors of the nerve sheath that typically arise from the posterior root; when multiple, neurofibromatosis is the likely etiology. Symptoms usually begin with radicular sensory symptoms followed by an asymmetric, progressive spinal cord syndrome. Therapy is surgical resection.
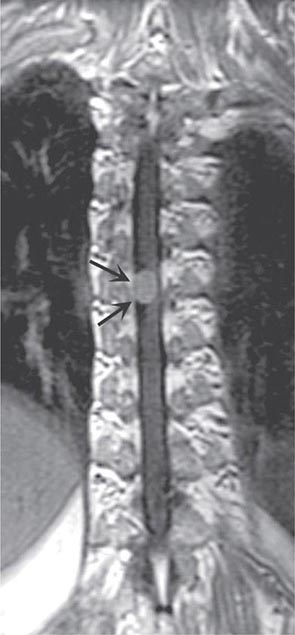
FIGURE 456-3 Magnetic resonance imaging of a thoracic meningioma. Coronal T1-weighted postcontrast image through the thoracic spinal cord demonstrates intense and uniform enhancement of a well-circumscribed extramedullary mass (arrows) that displaces the spinal cord to the left.
Primary intramedullary tumors of the spinal cord are uncommon. They present as central cord or hemicord syndromes, often in the cervical region. There may be poorly localized burning pain in the extremities and sparing of sacral sensation. In adults, these lesions are ependymomas, hemangioblastomas, or low-grade astrocytomas (Fig. 456-4). Complete resection of an intramedullary ependymoma is often possible with microsurgical techniques. Debulking of an intramedullary astrocytoma can also be helpful, as these are often slowly growing lesions; the value of adjunctive radiotherapy and chemotherapy is uncertain. Secondary (metastatic) intramedullary tumors also occur, especially in patients with advanced metastatic disease (Chap. 118), although these are not nearly as frequent as brain metastases.
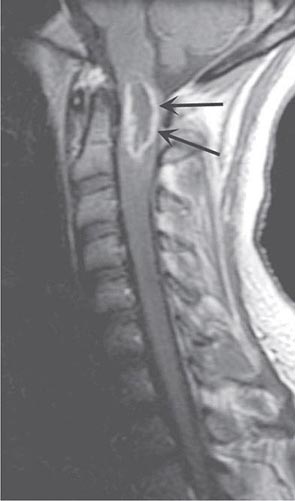
FIGURE 456-4 Magnetic resonance imaging of an intramedullary astrocytoma. Sagittal T1-weighted postcontrast image through the cervical spine demonstrates expansion of the upper cervical spine by a mass lesion emanating from within the spinal cord at the cervicomedullary junction. Irregular peripheral enhancement occurs within the mass (arrows).
Spinal Epidural Abscess Spinal epidural abscess presents with midline back or neck pain, fever, and progressive limb weakness. Prompt recognition of this distinctive process may prevent permanent sequelae. Aching pain is almost always present, either over the spine or in a radicular pattern. The duration of pain prior to presentation is generally ≤2 weeks but may on occasion be several months or longer. Fever is typically but not invariably present, accompanied by elevated white blood cell count, sedimentation rate, and C-reactive protein. As the abscess expands, further spinal cord damage results from venous congestion and thrombosis. Once weakness and other signs of myelopathy appear, progression may be rapid and irreversible. A more chronic sterile granulomatous form of abscess is also known, usually after treatment of an acute epidural infection.
Risk factors include an impaired immune status (HIV, diabetes mellitus, renal failure, alcoholism, malignancy), intravenous drug abuse, and infections of the skin or other tissues. Two-thirds of epidural infections result from hematogenous spread of bacteria from the skin (furunculosis), soft tissue (pharyngeal or dental abscesses; sinusitis), or deep viscera (bacterial endocarditis). The remainder arises from direct extension of a local infection to the subdural space; examples of local predisposing conditions are vertebral osteomyelitis, decubitus ulcers, lumbar puncture, epidural anesthesia, or spinal surgery. Most cases are due to Staphylococcus aureus; gram-negative bacilli, Streptococcus, anaerobes, and fungi can also cause epidural abscesses. Tuberculosis from an adjacent vertebral source (Pott’s disease) remains an important cause in the developing world.
MRI (Fig. 456-5) localizes the abscess and excludes other causes of myelopathy. Blood cultures are positive in more than half of cases, but direct aspiration of the abscess at surgery is often required for a microbiologic diagnosis. Lumbar puncture is only required if encephalopathy or other clinical signs raise the question of associated meningitis, a feature that is found in <25% of cases. The level of the puncture should be planned to minimize the risk of meningitis due to passage of the needle through infected tissue. A high cervical tap is sometimes the safest approach. Cerebrospinal fluid (CSF) abnormalities in epidural and subdural abscess consist of pleocytosis with a preponderance of polymorphonuclear cells, an elevated protein level, and a reduced glucose level, but the responsible organism is not cultured unless there is associated meningitis.
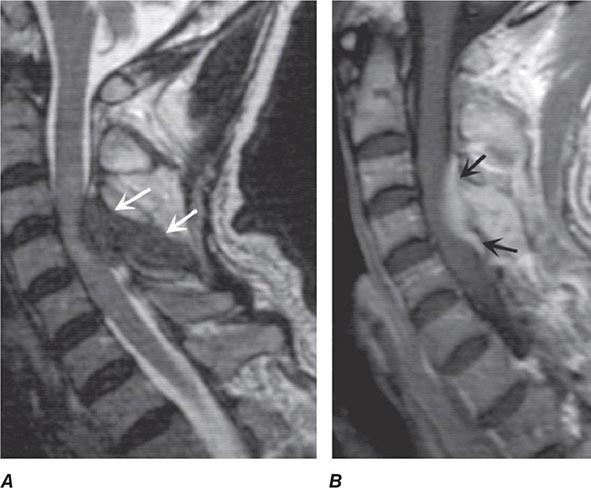
FIGURE 456-5 Magnetic resonance (MR) imaging of a spinal epidural abscess due to tuberculosis. A. Sagittal T2-weighted free spin-echo MR sequence. A hypointense mass replaces the posterior elements of C3 and extends epidurally to compress the spinal cord (arrows). B. Sagittal T1-weighted image after contrast administration reveals a diffuse enhancement of the epidural process (arrows) with extension into the epidural space.
Spinal Epidural Hematoma Hemorrhage into the epidural (or subdural) space causes acute focal or radicular pain followed by variable signs of a spinal cord or conus medullaris disorder. Therapeutic anticoagulation, trauma, tumor, or blood dyscrasias are predisposing conditions. Rare cases complicate lumbar puncture or epidural anesthesia. MRI and computed tomography (CT) confirm the clinical suspicion and can delineate the extent of the bleeding. Treatment consists of prompt reversal of any underlying clotting disorder and surgical decompression. Surgery may be followed by substantial recovery, especially in patients with some preservation of motor function preoperatively. Because of the risk of hemorrhage, lumbar puncture should be avoided whenever possible in patients with severe thrombocytopenia or other coagulopathies.
Hematomyelia Hemorrhage into the substance of the spinal cord is a rare result of trauma, intraparenchymal vascular malformation (see below), vasculitis due to polyarteritis nodosa or systemic lupus erythematosus (SLE), bleeding disorders, or a spinal cord neoplasm. Hematomyelia presents as an acute painful transverse myelopathy. With large lesions, extension into the subarachnoid space results in subarachnoid hemorrhage (Chap. 330). Diagnosis is by MRI or CT. Therapy is supportive, and surgical intervention is generally not useful. An exception is hematomyelia due to an underlying vascular malformation, for which spinal angiography and endovascular occlusion may be indicated, or surgery to evacuate the clot and remove the underlying vascular lesion.
NONCOMPRESSIVE MYELOPATHIES
The most frequent causes of noncompressive acute transverse myelopathy are spinal cord infarction; systemic inflammatory disorders, including SLE and sarcoidosis; demyelinating diseases, including multiple sclerosis (MS); neuromyelitis optica (NMO); postinfectious or idiopathic transverse myelitis, which is presumed to be an immune condition related to acute disseminated encephalomyelitis (Chap. 458); and infectious (primarily viral) causes. After spinal cord compression is excluded, the evaluation generally requires a lumbar puncture and a search for underlying systemic disease (Table 456-3).
|
EVALUATION OF ACUTE TRANSVERSE MYELOPATHY |
Abbreviations: ANA, antinuclear antibodies; CMV, cytomegalovirus; CSF, cerebrospinal fluid; CT, computed tomography; EBV, Epstein-Barr virus; ENA, epithelial neutrophil-activating peptide; ESR, erythrocyte sedimentation rate; HHV, human herpes virus; HSV, herpes simplex virus; HTLV, human T cell leukemia/lymphoma virus; MRI, magnetic resonance imaging; O&P, ova and parasites; p-ANCA, perinuclear antineutrophilic cytoplasmic antibodies; PCR, polymerase chain reaction; RPR, rapid plasma reagin (test); VDRL, Venereal Disease Research Laboratory; VZV, varicella-zoster virus.
Spinal Cord Infarction The cord is supplied by three arteries that course vertically over its surface: a single anterior spinal artery and paired posterior spinal arteries. The anterior spinal artery originates in paired branches of the vertebral arteries at the cranciocervical junction and is fed by additional radicular vessels that arise at C6, at an upper thoracic level, and, most consistently, at T11-L2 (artery of Adamkiewicz). At each spinal cord segment, paired penetrating vessels branch from the anterior spinal artery to supply the anterior two-thirds of the cord; the posterior spinal arteries, which often become less distinct below the midthoracic level, supply the posterior columns.
Spinal cord ischemia can occur at any level; however, the presence of the artery of Adamkiewicz below, and the anterior spinal artery circulation above, creates a region of marginal blood flow in the upper thoracic segments. With hypotension or cross-clamping of the aorta, cord infarction typically occurs at the level of T3-T4, and also at boundary zones between the anterior and posterior spinal artery territories. The latter may result in a rapidly progressive syndrome over hours of weakness and spasticity with little sensory change.
Acute infarction in the territory of the anterior spinal artery produces paraplegia or quadriplegia, dissociated sensory loss affecting pain and temperature sense but sparing vibration and position sense, and loss of sphincter control (“anterior cord syndrome”). Onset may be sudden but more typically is progressive over minutes or a few hours, quite unlike stroke in the cerebral hemispheres. Sharp midline or radiating back pain localized to the area of ischemia is frequent. Areflexia due to spinal shock is often present initially; with time, hyperreflexia and spasticity appear. Less common is infarction in the territory of the posterior spinal arteries, resulting in loss of posterior column function either on one side or bilaterally.
Causes of spinal cord infarction include aortic atherosclerosis, dissecting aortic aneurysm, vertebral artery occlusion or dissection in the neck, aortic surgery, or profound hypotension from any cause. A “surfer’s myelopathy” in the cervical region is probably vascular in origin. Cardiogenic emboli, vasculitis (Chap. 385), and collagen vascular disease (particularly SLE [Chap. 378], Sjögren’s syndrome [Chap. 383], and the antiphospholipid antibody syndrome [Chap. 379]) are other etiologies. Occasional cases develop from embolism of nucleus pulposus material into spinal vessels, usually from local spine trauma. In a substantial number of cases, no cause can be found, and thromboembolism in arterial feeders is suspected. MRI may fail to demonstrate infarctions of the cord, especially in the first day, but often the imaging becomes abnormal at the affected level.
In cord infarction due to presumed thromboembolism, acute anticoagulation is not indicated, with the possible exception of the unusual transient ischemic attack or incomplete infarction with a stuttering or progressive course. The antiphospholipid antibody syndrome is treated with anticoagulation (Chap. 379). Lumbar drainage of spinal fluid has reportedly been successful in some cases of cord infarction and has been used prophylactically during aortic surgery, but it has not been studied systematically.
Inflammatory and Immune Myelopathies (Myelitis) This broad category includes the demyelinating conditions MS, NMO, and postinfectious myelitis, as well as sarcoidosis and systemic autoimmune disease. In approximately one-quarter of cases of myelitis, no underlying cause can be identified. Some will later manifest additional symptoms of an immune-mediated disease. Recurrent episodes of myelitis are usually due to one of the immune-mediated diseases or to infection with herpes simplex virus (HSV) type 2 (below).
MULTIPLE SCLEROSIS MS may present with acute myelitis, particularly in individuals of Asian or African ancestry. In Caucasians, MS attacks rarely cause a transverse myelopathy (i.e., attacks of bilateral sensory disturbances, unilateral or bilateral weakness, and bladder or bowel symptoms), but it is among the most common causes of a partial cord syndrome. MRI findings in MS-associated myelitis typically consist of mild swelling of the cord and diffuse or multifocal “shoddy” areas of abnormal signal on T2-weighted sequences. Contrast enhancement, indicating disruption in the blood-brain barrier associated with inflammation, is present in many acute cases. A brain MRI is most helpful in gauging the likelihood that a case of myelitis represents an initial attack of MS. A normal scan indicates that the risk of evolution to MS is low, ~10–15% over 5 years; in contrast, the finding of multiple periventricular T2-bright lesions indicates a much higher risk, >50% over 5 years and >90% by 14 years. The CSF may be normal, but more often there is a mild mononuclear cell pleocytosis, with normal or mildly elevated CSF protein levels; the presence of oligoclonal bands is variable, but when they are found, a diagnosis of MS is more likely.
There are no adequate trials of therapy for MS-associated transverse myelitis. Intravenous methylprednisolone (500 mg qd for 3 days) followed by oral prednisone (1 mg/kg per day for several weeks, then gradual taper) has been used as initial treatment. A course of plasma exchange may be indicated for severe cases if glucocorticoids are ineffective. MS is discussed in Chap. 458.
NEUROMYELITIS OPTICA NMO is an immune-mediated demyelinating disorder consisting of a severe myelopathy that is typically longitudinally extensive, meaning that the lesion spans three or more vertebral segments. NMO is associated with optic neuritis that is often bilateral and may precede or follow myelitis by weeks or months, and also by brainstem and, in some cases, hypothalamic involvement. Recurrent myelitis without optic nerve involvemement can also occur in NMO; affected individuals are usually female and often of Asian ancestry. CSF studies reveal a variable mononuclear pleocytosis of up to several hundred cells per microliter; unlike MS, oligoclonal bands are generally absent. Diagnostic serum autoantibodies against the water channel protein aquaporin-4 are present in 60–70% of patients with NMO. NMO has also been associated with SLE and antiphospholipid antibodies (see below) as well as with other systemic autoimmune diseases; rare cases are paraneoplastic in origin. Treatment is with glucocorticoids and, for refractory cases, plasma exchange (as for MS, above). Preliminary studies suggest that treatment with azathioprine, mycophenylate, or anti-CD20 (anti–B cell) monoclonal antibody may protect against subsequent relapses; treatment for 5 years or longer is generally recommended. NMO is discussed in Chap. 458.
SYSTEMIC IMMUNE-MEDIATED DISORDERS Myelitis occurs in a small number of patients with SLE, many cases of which are associated with antibodies to antiphospholipids and/or to aquaporin-4. Patients with aquaporin-4 antibodies are likely to have longitudinally extensive myelitis by MRI, are considered to have an NMO-spectrum disorder, and are at high risk of developing future episodes of myelitis and/or optic neuritis. The CSF in SLE myelitis is usually normal or shows a mild lymphocytic pleocytosis; oligoclonal bands are a variable finding. Although there are no systematic trials of therapy for SLE myelitis, based on limited data, high-dose glucocorticoids followed by cyclophosphamide have been recommended. Acute severe episodes of transverse myelitis that do not initially respond to glucocorticoids are often treated with a course of plasma exchange. Sjögren’s syndrome (Chap. 383) can also be associated with NMO spectrum disorder and also with cases of acute transverse or chronic progressive myelopathy. Other immune-mediated myelitides include antiphospholipid antibody syndrome (Chap. 379), mixed connective tissue disease (Chap. 382), Behçet’s syndrome (Chap. 387), and vasculitis related to polyarteritis nodosa, perinuclear antineutrophilic cytoplasmic (p-ANCA) antibodies, or primary central nervous system vasculitis (Chap. 385).
Another important consideration in this group is sarcoid myelopathy that may present as a slowly progressive or relapsing disorder. MRI reveals an edematous swelling of the spinal cord that may mimic tumor; there is almost always gadolinium enhancement of active lesions and in some cases nodular enhancement of the adjacent surface of the cord; lesions may be single or multiple, and on axial images, enhancement of the central cord is usually present. The typical CSF profile consists of a mild lymphocytic pleocytosis and mildly elevated protein level; in a minority of cases, reduced glucose and oligoclonal bands are found. The diagnosis is particularly difficult when systemic manifestations of sarcoid are minor or absent (nearly 50% of cases) or when other typical neurologic manifestations of the disease—such as cranial neuropathy, hypothalamic involvement, or meningeal enhancement visualized by MRI—are lacking. A slit-lamp examination of the eye to search for uveitis, chest x-ray and CT to assess pulmonary involvement and mediastinal lymphadenopathy, serum or CSF angiotensin-converting enzyme (ACE; CSF values elevated in only a minority of cases), serum calcium, and a gallium scan may assist in the diagnosis. The usefulness of spinal fluid ACE is uncertain. Initial treatment is with oral glucocorticoids; immunosuppressant drugs, including the tumor necrosis factor α inhibitor infliximab, have been used for resistant cases. Sarcoidosis is discussed in Chap. 390.
POSTINFECTIOUS MYELITIS Many cases of myelitis, termed postinfectious or postvaccinal, follow an infection or vaccination. Numerous organisms have been implicated, including Epstein-Barr virus (EBV), cytomegalovirus (CMV), mycoplasma, influenza, measles, varicella, rubeola, and mumps. As in the related disorder acute disseminated encephalomyelitis (Chap. 458), postinfectious myelitis often begins as the patient appears to be recovering from an acute febrile infection, or in the subsequent days or weeks, but an infectious agent cannot be isolated from the nervous system or CSF. The presumption is that the myelitis represents an autoimmune disorder triggered by infection and is not due to direct infection of the spinal cord. No randomized controlled trials of therapy exist; treatment is usually with glucocorticoids or, in fulminant cases, plasma exchange.
ACUTE INFECTIOUS MYELITIS Many viruses have been associated with an acute myelitis that is infectious in nature rather than postinfectious. Nonetheless, the two processes are often difficult to distinguish. Herpes zoster is the best characterized viral myelitis, but HSV types 1 and 2, EBV, CMV, and rabies virus are other well-described causes. HSV-2 (and less commonly HSV-1) produces a distinctive syndrome of recurrent sacral cauda equina neuritis in association with outbreaks of genital herpes (Elsberg’s syndrome). Poliomyelitis is the prototypic viral myelitis, but it is more or less restricted to the anterior gray matter of the cord containing the spinal motoneurons. A polio-like syndrome can also be caused by a large number of enteroviruses (including enterovirus 71 and coxsackie), and with West Nile virus and other flaviviruses. Recently, cases of paralysis in children and adolescents were associated with enterovirus D-68 infection but a causal role for this virus has not been established. Chronic viral myelitic infections, such as those due to HIV or human T cell lymphotropic virus type 1 (HTLV-1), are discussed below.
Bacterial and mycobacterial myelitis (most are essentially abscesses) are less common than viral causes and much less frequent than cerebral bacterial abscess. Almost any pathogenic species may be responsible, including Borrelia burgdorferi (Lyme disease), Listeria monocytogenes, Mycobacterium tuberculosis, and Treponema pallidum (syphilis). Mycoplasma pneumoniae may be a cause of myelitis, but its status is uncertain because many cases are more properly classified as postinfectious.
Schistosomiasis (Chap. 259) is an important cause of parasitic myelitis in endemic areas. The process is intensely inflammatory and granulomatous, caused by a local response to tissue-digesting enzymes from the ova of the parasite, typically Schistosoma mansoni. Toxoplasmosis (Chap. 253) can occasionally cause a focal myelopathy, and this diagnosis should especially be considered in patients with AIDS (Chap. 226).
In cases of suspected viral myelitis, it may be appropriate to begin specific therapy pending laboratory confirmation. Herpes zoster, HSV, and EBV myelitis are treated with intravenous acyclovir (10 mg/kg q8h) or oral valacyclovir (2 g tid) for 10–14 days; CMV is treated with ganciclovir (5 mg/kg IV bid) plus foscarnet (60 mg/kg IV tid) or cidofovir (5 mg/kg per week for 2 weeks).
High-Voltage Electrical Injury Spinal cord injuries are prominent following electrocution from lightning strikes or other accidental electrical exposures. The syndrome consists of transient weakness acutely (often with an altered sensorium and focal cerebral disturbances), followed several days or even weeks later by a myelopathy that can be severe and permanent. This is a rare injury type, and limited data incriminate a vascular pathology involving the anterior spinal artery and its branches in some cases. Therapy is supportive.
CHRONIC MYELOPATHIES
SPONDYLOTIC MYELOPATHY
Spondylotic myelopathy is one of the most common causes of chronic cord compression and of gait difficulty in the elderly. Neck and shoulder pain with stiffness are early symptoms; impingement of bone and soft tissue overgrowth on nerve roots results in radicular arm pain, most often in a C5 or C6 distribution. Compression of the cervical cord, which occurs in fewer than one-third of cases, produces a slowly progressive spastic paraparesis, at times asymmetric and often accompanied by paresthesias in the feet and hands. Vibratory sense is diminished in the legs, there is a Romberg sign, and occasionally there is a sensory level for vibration or pinprick on the upper thorax. In some cases, coughing or straining produces leg weakness or radiating arm or shoulder pain. Dermatomal sensory loss in the arms, atrophy of intrinsic hand muscles, increased deep-tendon reflexes in the legs, and extensor plantar responses are common. Urinary urgency or incontinence occurs in advanced cases, but there are many alternative causes of these problems in older individuals. A tendon reflex in the arms is often diminished at some level; most often at the biceps (C5-C6). In individual cases, radicular, myelopathic, or combined signs may predominate. The diagnosis should be considered in appropriate cases of progressive cervical myelopathy, paresthesias of the feet and hands, or wasting of the hands.
Diagnosis is usually made by MRI and may be suspected from CT images; plain x-rays are less helpful. Extrinsic cord compression and deformation are appreciated on axial MRI views, and T2-weighted sequences may reveal areas of high signal intensity within the cord adjacent to the site of compression. A cervical collar may be helpful in milder cases, but definitive therapy consists of surgical decompression. Posterior laminectomy or an anterior approach with resection of the protruded disk and bony material may be required. Cervical spondylosis and related degenerative diseases of the spine are discussed in Chap. 22.
VASCULAR MALFORMATIONS OF THE CORD AND DURA
Vascular malformations of the cord and overlying dura are treatable causes of progressive myelopathy. Most common are fistulas located within the dura or posteriorly along the surface of the cord. Most dural arteriovenous (AV) fistulas are located at or below the midthoracic level, usually consisting of a direct connection between a radicular feeding artery in the nerve root sleeve with dural veins. The typical presentation is a middle-aged man with a progressive myelopathy that worsens slowly or intermittently and may have periods of remission, sometimes mimicking MS. Acute deterioration due to hemorrhage into the spinal cord (hematomyelia) or subarachnoid space may also occur but is rare. A saltatory progression is most common and appears to be the result of local ischemia and edema from venous congestion. Most patients have incomplete sensory, motor, and bladder disturbances. The motor disorder may predominate and produce a mixture of upper and restricted lower motor neuron signs, simulating amyotrophic lateral sclerosis (ALS). Pain over the dorsal spine, dysesthesias, or radicular pain may be present. Other symptoms suggestive of AV malformation (AVM) or dural fistula include intermittent claudication; symptoms that change with posture, exertion, Valsalva maneuver, or menses; and fever.
Less commonly, AVM disorders are intramedullary rather than dural. One unusual disorder is a progressive thoracic myelopathy with paraparesis developing over weeks or months, characterized pathologically by abnormally thick, hyalinized vessels within the cord (subacute necrotic myelopathy, or Foix-Alajouanine syndrome).
Spinal bruits are infrequent but may be sought at rest and after exercise in suspected cases. A vascular nevus on the overlying skin may indicate an underlying vascular malformation as occurs with Klippel-Trenaunay-Weber syndrome. High-resolution MRI with contrast administration detects the draining vessels of many but not all AVMs (Fig. 456-6). An uncertain proportion may be visualized by CT myelography as enlarged vessels along the surface of the cord. Definitive diagnosis requires selective spinal angiography, which defines the feeding vessels and the extent of the malformation. Endovascular embolization of the major feeding vessels may stabilize a progressive neurologic deficit or allow for gradual recovery. Some lesions, especially small dural fistulas, can be resected surgically.
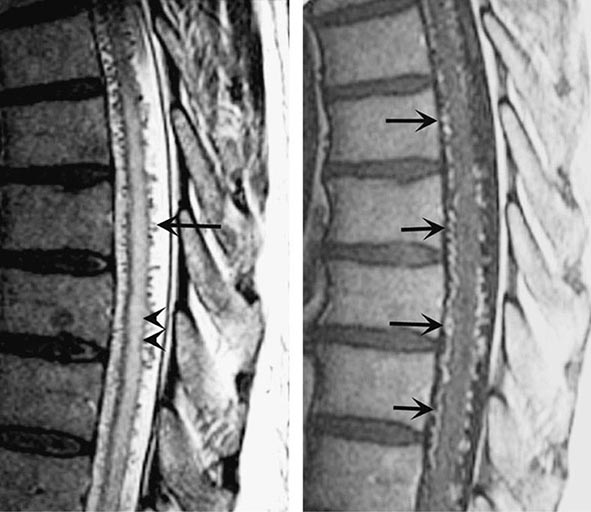
FIGURE 456-6 Arteriovenous malformation. Sagittal magnetic resonance scans of the thoracic spinal cord: T2 fast spin-echo technique (left) and T1 postcontrast image (right). On the T2-weighted image (left), abnormally high signal intensity is noted in the central aspect of the spinal cord (arrowheads). Numerous punctate flow voids indent the dorsal and ventral spinal cord (arrow). These represent the abnormally dilated venous plexus supplied by a dural arteriovenous fistula. After contrast administration (right), multiple, serpentine, enhancing veins (arrows) on the ventral and dorsal aspect of the thoracic spinal cord are visualized, diagnostic of arteriovenous malformation. This patient was a 54-year-old man with a 4-year history of progressive paraparesis.
RETROVIRUS-ASSOCIATED MYELOPATHIES
The myelopathy associated with HTLV-1, formerly called tropical spastic paraparesis, is a slowly progressive spastic syndrome with variable sensory and bladder disturbance. Approximately half of patients have mild back or leg pain. The neurologic signs may be asymmetric, often lacking a well-defined sensory level; the only sign in the arms may be hyperreflexia after several years of illness. The onset is insidious, and the illness is slowly progressive at a variable rate; most patients are unable to walk within 10 years of onset. This presentation may resemble primary progressive MS or a thoracic AVM. Diagnosis is made by demonstration of HTLV-1-specific antibody in serum by enzyme-linked immunosorbent assay (ELISA), confirmed by radioimmunoprecipitation or Western blot analysis. Especially in endemic areas, a finding of HTLV-1 seropositivity in a patient with myelopathy does not necessarily prove that HTLV-1 is causative. The CSF/serum antibody index may provide support by establishing intrathecal synthesis of antibodies favoring HTVL-1 myelopathy over asymptomatic carriage. Measuring proviral DNA by polymerase chain reaction (PCR) in serum and CSF cells can be useful as an ancillary part of diagnosis, because proviral DNA levels may be higher in patients with myelopathy. The myelopathy appears to result from an immune-mediated attack on the spinal cord rather than the result of direct viral infection. There is no effective treatment, but symptomatic therapy for spasticity and bladder symptoms may be helpful.
A progressive myelopathy may also result from HIV infection (Chap. 226). It is characterized by vacuolar degeneration of the posterior and lateral tracts, resembling subacute combined degeneration (see below).
SYRINGOMYELIA
Syringomyelia is a developmental cavity of the cervical cord that may enlarge and produce progressive myelopathy or may remain asymptomatic. Symptoms begin insidiously in adolescence or early adulthood, progress irregularly, and may undergo spontaneous arrest for several years. Many young patients acquire a cervical-thoracic scoliosis. More than half of all cases are associated with Chiari type 1 malformations in which the cerebellar tonsils protrude through the foramen magnum and into the cervical spinal canal. The pathophysiology of syrinx expansion is controversial, but some interference with the normal flow of CSF seems likely, perhaps by the Chiari malformation. Acquired cavitations of the cord in areas of necrosis are also termed syrinx cavities; these follow trauma, myelitis, necrotic spinal cord tumors, and chronic arachnoiditis due to tuberculosis and other etiologies.
The presentation is a central cord syndrome consisting of a regional dissociated sensory loss (loss of pain and temperature sensation with sparing of touch and vibration) and areflexic weakness in the upper limbs. The sensory deficit has a distribution that is “suspended” over the nape of the neck, shoulders, and upper arms (cape distribution) or in the hands. Most cases begin asymmetrically with unilateral sensory loss in the hands that leads to injuries and burns that are not appreciated by the patient. Muscle wasting in the lower neck, shoulders, arms, and hands with asymmetric or absent reflexes in the arms reflects expansion of the cavity in the gray matter of the cord. As the cavity enlarges and compresses the long tracts, spasticity and weakness of the legs, bladder and bowel dysfunction, and a Horner’s syndrome appear. Some patients develop facial numbness and sensory loss from damage to the descending tract of the trigeminal nerve (C2 level or above). In cases with Chiari malformations, cough-induced headache and neck, arm, or facial pain may be reported. Extension of the syrinx into the medulla, syringobulbia, causes palatal or vocal cord paralysis, dysarthria, horizontal or vertical nystagmus, episodic dizziness or vertigo, and tongue weakness with atrophy.
MRI accurately identifies developmental and acquired syrinx cavities and their associated spinal cord enlargement (Fig. 456-7). Images of the brain and the entire spinal cord should be obtained to delineate the full longitudinal extent of the syrinx, assess posterior fossa structures for the Chiari malformation, and determine whether hydrocephalus is present.
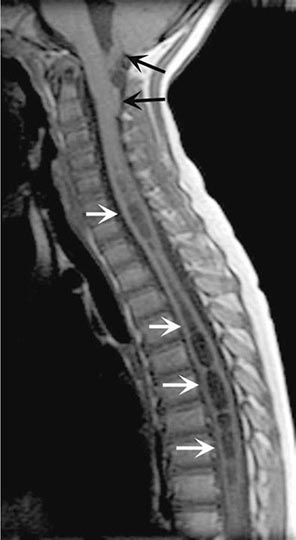
FIGURE 456-7 Magnetic resonance imaging of syringomyelia associated with a Chiari malformation. Sagittal T1-weighted image through the cervical and upper thoracic spine demonstrates descent of the cerebellar tonsils below the level of the foramen magnum (black arrows). Within the substance of the cervical and thoracic spinal cord, a cerebrospinal fluid collection dilates the central canal (white arrows).
CHRONIC MYELOPATHY OF MULTIPLE SCLEROSIS
A chronic progressive myelopathy is the most frequent cause of disability in both primary progressive and secondary progressive forms of MS. Involvement is typically bilateral but asymmetric and produces motor, sensory, and bladder/bowel disturbances. Fixed motor disability appears to result from extensive loss of axons in the corticospinal tracts. Diagnosis is facilitated by identification of earlier attacks such as optic neuritis. MRI, CSF, and evoked response testing are confirmatory. Disease-modifying therapy is indicated for patients with progressive myelopathy who also have coexisting MS relapses. Therapy is sometimes offered to patients who have a progressive course without relapses but with “active” MRI scans (e.g., the presence of new focal demyelinating lesions) despite the lack of evidence supporting the value of treatment in this setting. MS is discussed in Chap. 458.
SUBACUTE COMBINED DEGENERATION (VITAMIN B12 DEFICIENCY)
This treatable myelopathy presents with subacute paresthesias in the hands and feet, loss of vibration and position sensation, and a progressive spastic and ataxic weakness. Loss of reflexes due to an associated peripheral neuropathy in a patient who also has Babinski signs is an important diagnostic clue. Optic atrophy and irritability or other cognitive changes may be prominent in advanced cases and are occasionally the presenting symptoms. The myelopathy of subacute combined degeneration tends to be diffuse rather than focal; signs are generally symmetric and reflect predominant involvement of the posterior and lateral tracts, including Romberg’s sign. The diagnosis is confirmed by the finding of macrocytic red blood cells, a low serum B12 concentration, elevated serum levels of homocysteine and methylmalonic acid, and in uncertain cases, testing for anti–parietal cell antibodies and a Schilling test. Treatment is by replacement therapy, beginning with 1000 μg of intramuscular vitamin B12 repeated at regular intervals or by subsequent oral treatment (Chap. 128).
HYPOCUPRIC MYELOPATHY
This myelopathy is similar to subacute combined degeneration (described above), except there is no neuropathy, and explains cases with normal serum levels of B12. Low levels of serum copper are found, and often there is also a low level of serum ceruloplasmin. Some cases follow gastrointestinal procedures, particularly bariatric surgery, that result in impaired copper absorption; others have been associated with excess zinc from health food supplements or, until recently, zinc-containing denture creams, all of which impair copper absorption via induction of metallothionein, a copper-binding protein. Many cases are idiopathic. Improvement or at least stabilization may be expected with reconstitution of copper stores by oral supplementation. There is microcytic or macrocytic anemia. The pathophysiology and pathology of the idiopathic form are not known.
TABES DORSALIS
The classic syphilitic syndromes of tabes dorsalis and meningovascular inflammation of the spinal cord are now less frequent than in the past but must be considered in the differential diagnosis of spinal cord disorders. The characteristic symptoms of tabes are fleeting and repetitive lancinating pains, primarily in the legs or less often in the back, thorax, abdomen, arms, and face. Ataxia of the legs and gait due to loss of position sense occurs in half of patients. Paresthesias, bladder disturbances, and acute abdominal pain with vomiting (visceral crisis) occur in 15–30% of patients. The cardinal signs of tabes are loss of reflexes in the legs; impaired position and vibratory sense; Romberg’s sign; and, in almost all cases, bilateral Argyll Robertson pupils, which fail to constrict to light but accommodate. Diabetic polyradiculopathy may simulate tabes.
FAMILIAL SPASTIC PARAPLEGIA
Many cases of slowly progressive myelopathy are genetic in origin (Chap. 452). More than 30 different causative loci have been identified, including autosomal dominant, autosomal recessive, and X-linked forms. Especially for the recessive and X-linked forms, a family history of myelopathy may be lacking. Most patients present with almost imperceptibly progressive spasticity and weakness in the legs, usually but not always symmetrical. Sensory symptoms and signs are absent or mild, but sphincter disturbances may be present. In some families, additional neurologic signs are prominent, including nystagmus, ataxia, or optic atrophy. The onset may be as early as the first year of life or as late as middle adulthood. Only symptomatic therapies are available.
ADRENOMYELONEUROPATHY
This X-linked disorder is a variant of adrenoleukodystrophy. Most affected males have a history of adrenal insufficiency and then develop a progressive spastic (or ataxic) paraparesis beginning in early or sometimes middle adulthood; some patients also have a mild peripheral neuropathy. Female heterozygotes may develop a slower, insidiously progressive spastic myelopathy beginning later in adulthood and without adrenal insufficiency. Diagnosis is usually made by demonstration of elevated levels of very-long-chain fatty acids in plasma and in cultured fibroblasts. The responsible gene encodes the adrenoleukodystrophy protein (ADLP), a peroxisomal membrane transporter involved in carrying long-chain fatty acids to peroxisomes for degradation. Corticosteroid replacement is indicated if hypoadrenalism is present, and bone marrow transplantation and nutritional supplements have been attempted for this condition without clear evidence of efficacy.
OTHER CHRONIC MYELOPATHIES
Primary lateral sclerosis (Chap. 452) is a degenerative disorder characterized by progressive spasticity with weakness, eventually accompanied by dysarthria and dysphonia; bladder symptoms occur in approximately half of patients. Sensory function is spared. The disorder resembles ALS and is considered a variant of the motor neuron degenerations, but without the characteristic lower motor neuron disturbance. Some cases may represent familial spastic paraplegia, particularly autosomal recessive or X-linked varieties in which a family history may be absent.
Tethered cord syndrome is a developmental disorder of the lower spinal cord and nerve roots that rarely presents in adulthood as low back pain accompanied by a progressive lower spinal cord and/or nerve root syndrome. Some patients have a small leg or foot deformity indicating a long-standing process, and in others, a dimple, patch of hair, or sinus tract on the skin overlying the lower back is the clue to a congenital lesion. Diagnosis is made by MRI, which demonstrates a low-lying conus medullaris and thickened filum terminale. The MRI may also reveal diastematomyelia (division of the lower spinal cord into two halves), lipomas, cysts, or other congenital abnormalities of the lower spine coexisting with the tethered cord. Treatment is with surgical release.
There are a number of rare toxic causes of spastic myelopathy, including lathyrism due to ingestion of chickpeas containing the excitotoxin β-N-oxalylamino-L-alanine (BOAA), seen primarily in the developing world, and nitrous oxide inhalation producing a myelopathy identical to subacute combined degeneration. SLE, Sjögren’s syndrome, and sarcoidosis may each cause a myelopathy without overt evidence of systemic disease. Cancer-related causes of chronic myelopathy, besides the common neoplastic compressive myelopathy discussed earlier, include radiation injury (Chap. 118) and rare paraneoplastic myelopathies. The last of these are most often associated with lung or breast cancer and anti-Hu antibodies (Chap. 122) or with lymphoma that causes a syndrome of destruction of anterior horn cells; NMO (Chap. 458) can also rarely be paraneoplastic in origin. Metastases to the cord are probably more common than either of these in patients with cancer. Often, a cause of intrinsic myelopathy can be identified only through periodic reassessment.
REHABILITATION OF SPINAL CORD DISORDERS
The prospects for recovery from an acute destructive spinal cord lesion fade after ~6 months. There are currently no effective means to promote repair of injured spinal cord tissue; promising but entirely experimental approaches include the use of factors that influence reinnervation by axons of the corticospinal tract, nerve and neural sheath graft bridges, forms of electrical stimulation at the site of injury, and the local introduction of stem cells. The disability associated with irreversible spinal cord damage is determined primarily by the level of the lesion and by whether the disturbance in function is complete or incomplete (Table 456-4). Even a complete high cervical cord lesion may be compatible with a productive life. The primary goals are development of a rehabilitation plan framed by realistic expectations and attention to the neurologic, medical, and psychological complications that commonly arise.
|
EXPECTED NEUROLOGIC FUNCTION FOLLOWING COMPLETE CORD LESIONS |

Many of the usual symptoms associated with medical illnesses, especially somatic and visceral pain, may be lacking because of the destruction of afferent pain pathways. Unexplained fever, worsening of spasticity, or deterioration in neurologic function should prompt a search for infection, thrombophlebitis, or an intraabdominal pathology. The loss of normal thermoregulation and inability to maintain normal body temperature can produce recurrent fever (quadriplegic fever), although most episodes of fever are due to infection of the urinary tract, lung, skin, or bone.
Bladder dysfunction generally results from loss of supraspinal innervation of the detrusor muscle of the bladder wall and the sphincter musculature. Detrusor spasticity is treated with anticholinergic drugs (oxybutynin, 2.5–5 mg qid) or tricyclic antidepressants with anticholinergic properties (imipramine, 25–200 mg/d). Failure of the sphincter muscle to relax during bladder emptying (urinary dyssynergia) may be managed with the α-adrenergic blocking agent terazosin hydrochloride (1–2 mg tid or qid), with intermittent catheterization, or, if that is not feasible, by use of a condom catheter in men or a permanent indwelling catheter. Surgical options include the creation of an artificial bladder by isolating a segment of intestine that can be catheterized intermittently (enterocystoplasty) or can drain continuously to an external appliance (urinary conduit). Bladder areflexia due to acute spinal shock or conus lesions is best treated by catheterization. Bowel regimens and disimpaction are necessary in most patients to ensure at least biweekly evacuation and avoid colonic distention or obstruction.
Patients with acute cord injury are at risk for venous thrombosis and pulmonary embolism. Use of calf-compression devices and anticoagulation with low-molecular-weight heparin is recommended. In cases of persistent paralysis, anticoagulation should probably be continued for 3 months.
Prophylaxis against decubitus ulcers should involve frequent changes in position in a chair or bed, the use of special mattresses, and cushioning of areas where pressure sores often develop, such as the sacral prominence and heels. Early treatment of ulcers with careful cleansing, surgical or enzyme debridement of necrotic tissue, and appropriate dressing and drainage may prevent infection of adjacent soft tissue or bone.
Spasticity is aided by stretching exercises to maintain mobility of joints. Drug treatment is effective but may result in reduced function, as some patients depend on spasticity as an aid to stand, transfer, or walk. Baclofen (up to 240 mg/d in divided doses) is effective; it acts by facilitating γ-aminobutyric acid–mediated inhibition of motor reflex arcs. Diazepam acts by a similar mechanism and is useful for leg spasms that interrupt sleep (2–4 mg at bedtime). Tizanidine (2–8 mg tid), an α2 adrenergic agonist that increases presynaptic inhibition of motor neurons, is another option. For nonambulatory patients, the direct muscle inhibitor dantrolene (25–100 mg qid) may be used, but it is potentially hepatotoxic. In refractory cases, intrathecal baclofen administered via an implanted pump, botulinum toxin injections, or dorsal rhizotomy may be required to control spasticity.
Despite the loss of sensory function, many patients with spinal cord injury experience chronic pain sufficient to diminish their quality of life. Randomized controlled studies indicate that gabapentin or pregabalin is useful in this setting. Epidural electrical stimulation and intrathecal infusion of pain medications have been tried with some success. Management of chronic pain is discussed in Chap. 18.
A paroxysmal autonomic hyperreflexia may occur following lesions above the major splanchnic sympathetic outflow at T6. Headache, flushing, and diaphoresis above the level of the lesion, as well as hypertension with bradycardia or tachycardia, are the major symptoms. The trigger is typically a noxious stimulus—for example, bladder or bowel distention, a urinary tract infection, or a decubitus ulcer—below the level of the cord lesion. Treatment consists of removal of offending stimuli; ganglionic blocking agents (mecamylamine, 2.5–5 mg) or other short-acting antihypertensive drugs are useful in some patients.
Attention to these details allows longevity and a productive life for patients with complete transverse myelopathies.
457e |
Concussion and Other Traumatic Brain Injuries |
Almost 10 million head injuries occur annually in the United States, about 20% of which are serious enough to cause brain damage. Among men <35 years, accidents, usually motor vehicle collisions, are the chief cause of death and >70% of these involve head injury. Furthermore, minor head injuries are so common that almost all physicians will be called upon to provide immediate care or to see patients who are suffering from various sequelae.
Medical personnel caring for head injury patients should be aware that (1) spinal injury often accompanies head injury, and care must be taken in handling the patient to prevent compression of the spinal cord due to instability of the spinal column; (2) intoxication is frequently associated with traumatic brain injury, and thus testing for drugs and alcohol should be carried out when appropriate; and (3) additional injuries, including rupture of abdominal organs, may produce vascular collapse, shock, or respiratory distress that requires immediate attention.
TYPES OF HEAD INJURIES
CONCUSSION
This form of minor head injury had in the past referred to an immediate and transient loss of consciousness that was associated with a short period of amnesia. Many patients, however, do not lose consciousness after a minor head injury but instead are dazed or confused, or feel stunned or “star struck,” and the term concussion is now applied to all such cognitive and perceptual changes experienced after a blow to the head. Severe concussion may precipitate a brief convulsion or autonomic signs such as facial pallor, bradycardia, faintness with mild hypotension, or sluggish pupillary reaction, but most patients quickly return to a neurologically normal state.
The mechanics of a typical concussion involve sudden deceleration of the head when hitting a blunt stationary object. This creates an anterior-posterior movement of the brain within the skull due to inertia and rotation of the cerebral hemispheres on the fulcrum of the relatively fixed upper brainstem. Loss of consciousness in concussion is believed to result from a transient electrophysiologic dysfunction of the reticular activating system in the upper midbrain that is at the site of rotation (Chap. 328). The transmission of a wave of kinetic energy throughout the brain is an alternative explanation for the disruption in consciousness.
Gross and light-microscopic changes in the brain are usually absent following concussion, but biochemical and ultrastructural changes, such as mitochondrial ATP depletion and local disruption of the blood-brain barrier, may be transient abnormalities. Computed tomography (CT) and magnetic resonance imaging (MRI) scans are usually normal; however, a small number of patients will be found to have a skull fracture, an intracranial hemorrhage, or a brain contusion.
A brief period of both retrograde and anterograde amnesia is characteristic of concussion, and it recedes rapidly in alert patients. Memory loss spans the moments before impact but may encompass the previous days or weeks (rarely months). With severe injuries, the extent of retrograde amnesia roughly correlates with the severity of injury. Memory is regained erratically from the most distant to more recent memories, with islands of amnesia occasionally remaining. The mechanism of amnesia is not known. Hysterical posttraumatic amnesia is not uncommon after head injury and should be suspected when inexplicable behavioral abnormalities occur, such as recounting events that cannot be recalled on later testing, a bizarre affect, forgetting one’s own name, or a persistent anterograde deficit that is excessive in comparison with the degree of injury. Amnesia is discussed in Chap. 36.
A single, uncomplicated concussion only infrequently produces permanent neurobehavioral changes in patients who are free of preexisting psychiatric and neurologic problems. Nonetheless, residual problems in memory and concentration may have an anatomic correlate in microscopic cerebral lesions (see below).
The mechanisms by which a blast injury affects the brain and causes symptoms that are associated with concussion, a problem mainly in military medicine, are not known. The energy of a blast wave can enter the cranium through the openings of the orbits, auditory canals, and foramen magnum. There are not consistent changes in cerebral imaging studies but more subtle indications of tissue disruption have been found, comparable to those of mild concussion. It has been difficult to separate the direct effects of the blast from the consequences of being thrown against fixed objects or injured by flying debris.
CONTUSION, BRAIN HEMORRHAGE, AND AXONAL SHEARING LESIONS
These pathologic changes are the result of severe cranial trauma. A surface bruise of the brain, or contusion, consists of varying degrees of petechial hemorrhage, edema, and tissue destruction. Contusions and deeper hemorrhages result from mechanical forces that displace and compress the hemispheres forcefully and by deceleration of the brain against the inner skull, either under a point of impact (coup lesion) or, as the brain swings back, in the antipolar area (contrecoup lesion). Trauma sufficient to cause prolonged unconsciousness usually produces some degree of contusion. Blunt deceleration impact, as occurs against an automobile dashboard or from falling forward onto a hard surface, causes contusions on the orbital surfaces of the frontal lobes and the anterior and basal portions of the temporal lobes. With lateral forces, as from impact on an automobile door frame, contusions are situated on the lateral convexity of the hemisphere. The clinical signs of contusion are determined by the location and size of the lesion; often, there are no focal neurologic abnormalities, but these injured regions are later the sites of gliotic scars that may produce seizures. A hemiparesis or gaze preference is fairly typical of moderately sized contusions. Large bilateral contusions produce stupor with extensor posturing, while those limited to the frontal lobes cause a taciturn state. Contusions in the temporal lobe may cause delirium or an aggressive, combative syndrome.
Acute contusions are easily visible on CT and MRI scans, appearing as inhomogeneous hyperdensities on CT and as hyperintensities on T2 and fluid-attenuated inversion recovery (FLAIR) MRI sequences; there is usually surrounding localized brain edema (Fig. 457e-1) and some subarachnoid bleeding. Blood in the cerebrospinal fluid (CSF) due to trauma may provoke a mild inflammatory reaction. Over a few days, contusions acquire a surrounding contrast enhancement and edema that may be mistaken for tumor or abscess. Glial and macrophage reactions result in chronic, scarred, hemosiderin-stained depressions on the cortex (plaques jaunes) that are the main source of posttraumatic epilepsy.
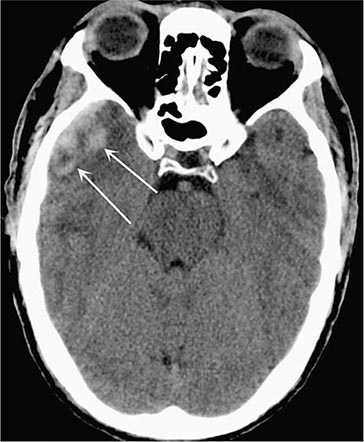
FIGURE 457e-1 Traumatic cerebral contusion. Noncontrast computed tomography scan demonstrating a hyperdense hemorrhagic region in the anterior temporal lobe.
Torsional or shearing forces within the brain cause hemorrhages of the basal ganglia and other deep regions. Large hemorrhages after minor trauma suggest that there is a bleeding diathesis or cerebrovascular amyloidosis. For unexplained reasons, deep cerebral hemorrhages may not develop until several days after injury. Sudden neurologic deterioration in a comatose patient or a sudden rise in intracranial pressure (ICP) suggests this complication has occurred and should therefore prompt investigation with a CT scan.
A special type of deep white matter lesion consists of widespread mechanical disruption, or shearing, of axons at the time of impact. Most characteristic are small areas of tissue injury in the corpus callosum and dorsolateral pons. The presence of widespread multifocal axonal damage in both hemispheres, a state called diffuse axonal injury (DAI), has been proposed to explain persistent coma and the vegetative state after closed head injury (Chap. 328), but small ischemic-hemorrhagic lesions in the midbrain and thalamus are an alternative explanation. Only severe shearing lesions that contain blood are visualized by CT, usually in the corpus callosum and centrum semiovale (Fig. 457e-2); however, special MRI sequences that detect small amounts of blood and diffusion tensor imaging can demonstrate numerous such lesions throughout the white matter.
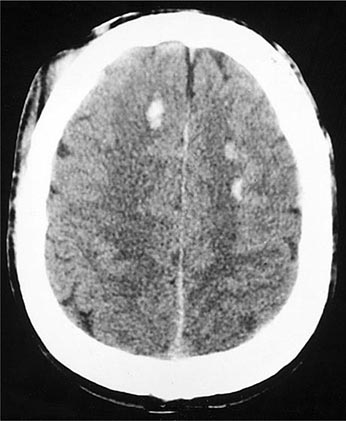
FIGURE 457e-2 Multiple small areas of hemorrhage and tissue disruption in the white matter of the frontal lobes on noncontrast computed tomography scan. These appear to reflect an extreme type of the diffuse axonal shearing lesions that occur with closed head injury.
SKULL FRACTURES
A blow to the skull that exceeds the elastic tolerance of the bone causes a fracture. Intracranial lesions accompany roughly two-thirds of skull fractures, and the presence of a fracture increases many-fold the chances of an underlying subdural or epidural hematoma. Consequently, fractures are primarily markers of the site and severity of injury. If the underlying arachnoid membrane has been torn, fractures also provide potential pathways for entry of bacteria to the CSF with a risk of meningitis and for leakage of CSF outward through the dura. If there is leakage of CSF, severe orthostatic headache results from lowered pressure in the spinal fluid compartment.
Most fractures are linear and extend from the point of impact toward the base of the skull. Basilar skull fractures are often extensions of adjacent linear fractures over the convexity of the skull but may occur independently owing to stresses on the floor of the middle cranial fossa or occiput. Basilar fractures are usually parallel to the petrous bone or along the sphenoid bone and directed toward the sella turcica and ethmoidal groove. Although most basilar fractures are uncomplicated, they can cause CSF leakage, pneumocephalus, and delayed cavernous-carotid fistulas. Hemotympanum (blood behind the tympanic membrane), ecchymosis over the mastoid process (Battle sign), and periorbital ecchymosis (“raccoon sign”) are associated with basilar fractures. Because routine x-ray examination may fail to disclose basilar fractures, they should be suspected if these clinical signs are present.
CSF may leak through the cribriform plate or the adjacent sinus and cause CSF rhinorrhea (a watery discharge from the nose). Persistent rhinorrhea and recurrent meningitis usually require surgical repair of torn dura underlying the fracture. The site of the leak is often difficult to determine, but useful diagnostic tests include the instillation of water-soluble contrast into the CSF followed by CT with the patient in various positions, or injection of radionuclide compounds or fluorescein into the CSF and the insertion of absorptive nasal pledgets. The location of an intermittent leak is infrequently delineated, and many resolve spontaneously.
Sellar fractures, even those associated with serious neuroendocrine dysfunction, may be radiologically occult or evident only by an air-fluid level in the sphenoid sinus. Fractures of the dorsum sella cause sixth or seventh nerve palsies or optic nerve damage.
Petrous bone fractures, especially those oriented along the long axis of the bone, may be associated with facial palsy, disruption of ear ossicles, and CSF otorrhea. Transverse petrous fractures are less common; they almost always damage the cochlea or labyrinths and often the facial nerve as well. External bleeding from the ear is usually from local abrasion of the external canal but can also result from petrous fracture.
Fractures of the frontal bone are usually depressed, involving the frontal and paranasal sinuses and the orbits. Depressed skull fractures are typically compound, but they may be asymptomatic because the impact energy is dissipated in breaking the bone; some have underlying brain contusions. Debridement and exploration of compound fractures are required in order to avoid infection; simple fractures usually do not require surgery.
CRANIAL NERVE INJURIES
The cranial nerves most often injured with head trauma are the olfactory, optic, oculomotor, and trochlear; the first and second branches of the trigeminal nerve; and the facial and auditory nerves. Anosmia and an apparent loss of taste (actually a loss of perception of aromatic flavors, with retained elementary taste perception) occur in ~10% of persons with serious head injuries, particularly from falls on the back of the head. This is the result of displacement of the brain and shearing of the fine olfactory nerve filaments that course through the cribriform bone. At least partial recovery of olfactory and gustatory function is expected, but if bilateral anosmia persists for several months, the prognosis is poor. Partial optic nerve injuries from closed trauma result in blurring of vision, central or paracentral scotomas, or sector defects. Direct orbital injury may cause short-lived blurred vision for close objects due to reversible iridoplegia. Diplopia limited to downward gaze and corrected when the head is tilted away from the side of the affected eye indicates trochlear (fourth nerve) nerve damage. It occurs frequently as an isolated problem after minor head injury or may develop for unknown reasons after a delay of several days. Facial nerve injury caused by a basilar fracture is present immediately in up to 3% of severe injuries; it may also be delayed for 5–7 days. Fractures through the petrous bone, particularly the less common transverse type, are liable to produce facial palsy. Delayed facial palsy occurring up to a week after injury, the mechanism of which is unknown, has a good prognosis. Injury to the eighth cranial nerve from a fracture of the petrous bone causes loss of hearing, vertigo, and nystagmus immediately after injury. Deafness from eighth nerve injury is rare and must be distinguished from blood in the middle ear or disruption of the middle ear ossicles. Dizziness, tinnitus, and high-tone hearing loss occur from cochlear concussion, most typically after blast injury.
SEIZURES
Convulsions are surprisingly uncommon immediately after a head injury, but a brief period of tonic extensor posturing or a few clonic movements of the limbs just after the moment of impact can occur. However, the cortical scars that evolve from contusions are highly epileptogenic and may later manifest as seizures, even after many months or years (Chap. 445). The severity of injury roughly determines the risk of future seizures. It has been estimated that 17% of individuals with brain contusion, subdural hematoma, or prolonged loss of consciousness will develop a seizure disorder and that this risk extends for an indefinite period of time, whereas the risk is ≤2% after mild injury. The majority of convulsions in the latter group occur within 5 years of injury but may be delayed for decades. Penetrating injuries have a much higher rate of subsequent epilepsy.
SUBDURAL AND EPIDURAL HEMATOMAS
Hemorrhages beneath the dura (subdural) or between the dura and skull (epidural) have characteristic clinical and imaging features. They are sometimes associated with underlying contusions and other injuries, often making it difficult to determine the relative contribution of each component to the clinical state. The mass effect and raised ICP caused by these hematomas can be life threatening, making it imperative to identify them rapidly by CT or MRI scan and to remove them when appropriate.
Acute Subdural Hematoma (Fig. 457e-3) Direct cranial trauma may be minor and is not required for acute subdural hemorrhage to occur, especially in the elderly and those taking anticoagulant medications. Acceleration forces alone, as from whiplash, are sometimes sufficient to produce subdural hematoma. Up to one-third of patients have a lucid interval lasting minutes to hours before coma supervenes, but most are drowsy or comatose from the moment of injury. A unilateral headache and slightly enlarged pupil on the side of the hematoma are frequently, but not invariably, present. Stupor or coma, hemiparesis, and unilateral pupillary enlargement are signs of larger hematomas. In an acutely deteriorating patient, burr (drainage) holes or an emergency craniotomy are required. Small subdural hematomas may be asymptomatic and usually do not require evacuation if they do not enlarge.
FIGURE 457e-3 Acute subdural hematoma. Noncontrast computed tomography scan reveals a hyperdense clot that has an irregular border with the brain and causes more horizontal displacement (mass effect) than might be expected from its thickness. The disproportionate mass effect is the result of the large rostral-caudal extent of these hematomas. Compare to Fig. 457e-4.
A subacutely evolving syndrome due to subdural hematoma occurs days or weeks after injury with drowsiness, headache, confusion, or mild hemiparesis, usually in alcoholics and in the elderly and often after only minor trauma. On imaging studies, subdural hematomas appear as crescentic collections over the convexity of one or both hemispheres, most commonly in the frontotemporal region, and less often in the inferior middle fossa or over the occipital poles (Fig. 457e-3). Interhemispheric, posterior fossa, or bilateral convexity hematomas are less frequent and are difficult to diagnose clinically, although drowsiness and the neurologic signs expected from damage in each region can usually be detected. The bleeding that causes larger hematomas is primarily venous in origin, although additional arterial bleeding sites are sometimes found at operation, and a few large hematomas have a purely arterial origin.
Epidural Hematoma (Fig. 457e-4) These usually evolve more rapidly than subdural hematomas and are correspondingly more treacherous. They occur in up to 10% of cases of severe head injury but are associated with underlying cortical damage less often than for subdural hematomas. Most patients are unconscious when first seen. A “lucid interval” of several minutes to hours before coma supervenes is most characteristic of epidural hemorrhage, but it is still uncommon, and epidural hemorrhage is not the only cause of this temporal sequence. Rapid surgical evacuation and ligation or cautery of the damaged vessel is indicated, usually the middle meningeal artery that has been lacerated by an overlying skull fracture.
FIGURE 457e-4 Acute epidural hematoma. The tightly attached dura is stripped from the inner table of the skull, producing a characteristic lenticular-shaped hemorrhage on noncontrast computed tomography scan. Epidural hematomas are usually caused by tearing of the middle meningeal artery following fracture of the temporal bone.
Chronic Subdural Hematoma (Fig. 457e-5) A subacutely evolving syndrome due to subdural hematoma occurs days or weeks after injury with drowsiness, headache, confusion, or mild hemiparesis, usually in alcoholics and in the elderly and often after only minor or unnoticed trauma. On imaging studies, chronic subdural hematomas appear as crescentic clots over the convexity of one or both hemispheres, most commonly in the frontotemporal region (Fig. 457e-3). A history of trauma may or may not be elicited in relation to chronic subdural hematoma; the injury may have been trivial and forgotten, particularly in the elderly and those with clotting disorders. Headache is common but not invariable. Additional features that may appear weeks later include slowed thinking, vague change in personality, seizure, or a mild hemiparesis. Headache fluctuates in severity, sometimes with changes in head position.
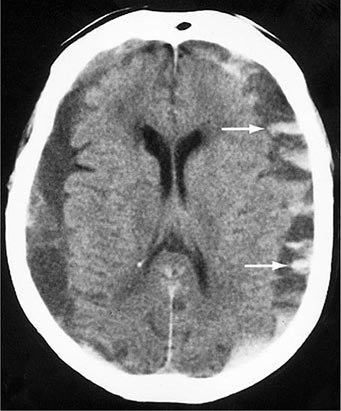
FIGURE 457e-5 Computed tomography scan of chronic bilateral subdural hematomas of different ages. The collections began as acute hematomas and have become hypodense in comparison to the adjacent brain after a period during which they were isodense and difficult to appreciate. Some areas of resolving blood are contained on the more recently formed collection on the left (arrows).
Bilateral chronic subdural hematomas produce perplexing clinical syndromes, and the initial clinical impression may be of a stroke, brain tumor, drug intoxication, depression, or a dementing illness. Drowsiness, inattentiveness, and incoherence of thought are generally more prominent than focal signs such as hemiparesis. Rarely, chronic hematomas cause brief episodes of hemiparesis or aphasia that are indistinguishable from transient ischemic attacks. Patients with undetected bilateral subdural hematomas have a low tolerance for surgery, anesthesia, and drugs that depress the nervous system; drowsiness or confusion persists for long periods postoperatively.
CT without contrast initially shows a low-density mass over the convexity of the hemisphere (Fig. 457e-5). Between 2 and 6 weeks after the initial bleeding, the clot becomes isodense compared to adjacent brain and may be inapparent. Many subdural hematomas that are several weeks in age contain areas of blood and intermixed serous fluid. Bilateral chronic hematomas may fail to be detected because of the absence of lateral tissue shifts; this circumstance in an older patient is suggested by a “hypernormal” CT scan with fullness of the cortical sulci and small ventricles. Infusion of contrast material demonstrates enhancement of the vascular fibrous capsule surrounding the collection. MRI reliably identifies subacute and chronic hematomas.
Clinical observation coupled with serial imaging is a reasonable approach to patients with few symptoms, such as headache alone, and in those with small chronic subdural collections. Treatment of minimally symptomatic chronic subdural hematoma with glucocorticoids is favored by some clinicians, but surgical evacuation is more often successful. The fibrous membranes that grow from the dura and encapsulate the collection require removal to prevent recurrent fluid accumulation. Small hematomas are resorbed, leaving only the organizing membranes. On imaging studies, very chronic subdural hematomas are difficult to distinguish from hygromas, which are collections of CSF from a rent in the arachnoid membrane.
CLINICAL SYNDROMES AND TREATMENT OF HEAD INJURY
MINOR INJURY
The patient who has briefly lost consciousness or been stunned after a minor head injury usually becomes fully alert and attentive within minutes but may complain of headache, dizziness, faintness, nausea, a single episode of emesis, difficulty with concentration, a brief amnestic period, or slight blurring of vision. This typical concussion syndrome has a good prognosis with little risk of subsequent deterioration. Children are particularly prone to drowsiness, vomiting, and irritability, symptoms that are sometimes delayed for several hours after apparently minor injuries. Vasovagal syncope that follows injury may cause undue concern. Generalized or frontal headache is common in the following days. It may be migrainous (throbbing and hemicranial) in nature or aching and bilateral. After several hours of observation, patients with minor injury may be accompanied home and observed for a day by a family member or friend, with written instructions to return if symptoms worsen.
Persistent severe headache and repeated vomiting in the context of normal alertness and no focal neurologic signs is usually benign, but CT should be obtained and a longer period of observation is appropriate. The decision to perform imaging tests also depends on clinical signs that indicate that the impact was severe (e.g., persistent confusion, periorbital or mastoid hematoma, repeated vomiting, palpable skull fracture), on the seriousness of other bodily injuries, and on the degree of surveillance that can be anticipated after discharge. Two studies have indicated that older age, two or more episodes of vomiting, >30 min of retrograde or persistent anterograde amnesia, seizure, and concurrent drug or alcohol intoxication are sensitive (but not specific) indicators of intracranial hemorrhage that justify CT scanning. It may be appropriate to be more liberal in obtaining CT scans in children because a small number, even without loss of consciousness, will have intracranial traumatic lesions but this exposes the child to radiation.
Concussion in Sports In the current absence of adequate data, a common sense approach to athletic concussion has been to remove the individual from play immediately and avoid contact sports for at least several days after a mild injury and for a longer period if there are more severe injuries or if there are protracted neurologic symptoms such as headache and difficulty concentrating. No individual should return to play unless all symptoms have resolved and an assessment has been made by a health care professional who has experience with treatment of concussion. Once cleared, the individual can then begin a graduated program of increasing activity. Younger athletes are particularly likely to experience protracted concussive symptoms, and a slower return to play in this age group may be reasonable. These guidelines are designed in part to avoid a perpetuation of symptoms but also to prevent the rare second impact syndrome, in which diffuse and fatal cerebral swelling follows a second minor head injury.
In the past, mental decline in boxers late in their careers had been called dementia pugilistica. There is some evidence that repeated concussions from other sports are associated with a similar delayed and progressive cognitive disorder that is due mainly to the deposition of tau protein in cortical neurons. The brains of these patients display deposition of tau protein in the superficial cortical layers, and particularly in the depths of sulci within the frontal cortices, a pattern named chronic traumatic encephalopathy (CTE) that is quite unlike other degenerative conditions. CTE is an intensively studied and provocative entity. Its contribution, if any, to late-life dementia and parkinsonism in former athletes, soldiers, or others who have sustained repeated concussive injuries is unknown. CTE is also discussed in Chap. 444e.
INJURY OF INTERMEDIATE SEVERITY
Patients who are not fully alert or have persistent confusion, behavioral changes, extreme dizziness, or focal neurologic signs such as hemiparesis should be admitted to the hospital and have cerebral imaging. A cerebral contusion or hematoma will usually be found. Common syndromes include: (1) delirium with a disinclination to be examined or moved, expletive speech, and resistance if disturbed (anterior temporal lobe contusions); (2) a quiet, disinterested, slowed mental state (abulia) alternating with irascibility (inferior frontal and frontopolar contusions); (3) a focal deficit such as aphasia or mild hemiparesis (due to subdural hematoma or convexity contusion or, less often, carotid artery dissection); (4) confusion and inattention, poor performance on simple mental tasks, and fluctuating orientation (associated with several types of injuries, including those described above, and with medial frontal contusions and interhemispheric subdural hematoma); (5) repetitive vomiting, nystagmus, drowsiness, and unsteadiness (labyrinthine concussion, but occasionally due to a posterior fossa subdural hematoma or vertebral artery dissection); and (6) diabetes insipidus (damage to the median eminence or pituitary stalk). Injuries of this degree are often complicated by drug or alcohol intoxication, and clinically inapparent cervical spine injury may be present. Blast injuries are often accompanied by rupture of the tympanic membranes.
After surgical removal of hematomas, most patients in this category improve over weeks. During the first week, the state of alertness, memory, and other cognitive functions often fluctuate, and agitation and somnolence are common. Behavioral changes tend to be worse at night, as with many other encephalopathies, and may be treated with small doses of antipsychotic medications. Subtle abnormalities of attention, intellect, spontaneity, and memory return toward normal weeks or months after the injury, sometimes abruptly. Persistent cognitive problems are discussed below.
SEVERE INJURY
Patients who are comatose from the moment of injury require immediate neurologic attention and resuscitation. After intubation, with care taken to immobilize the cervical spine, the depth of coma, pupillary size and reactivity, limb movements, and Babinski responses are assessed. As soon as vital functions permit and cervical spine x-rays and a CT scan have been obtained, the patient should be transported to a critical care unit. Hypoxia should be reversed, and normal saline used as the resuscitation fluid in preference to albumin. The finding of an epidural or subdural hematoma or large intracerebral hemorrhage is usually an indication for prompt surgery and intracranial decompression in an otherwise salvageable patient. Measurement of ICP with a ventricular catheter or fiberoptic device in order to guide treatment has been favored by many units but has not improved outcome. Hyperosmolar intravenous solutions are used in various regimens to limit intracranial pressure. The inherently appealing approach of removing portions of the skull in order to decompress the intracranial contents, as has been successful for brain swelling after cerebral infarction, has so far not proven effective for traumatic brain injury. The use of prophylactic antiepileptic medications has been recommended, but there is little supportive data. Management of raised ICP, a frequent feature of severe head injury, is discussed in Chap. 330.
GRADING AND PROGNOSIS
In severe head injury, the clinical features of eye opening, motor responses of the limbs, and verbal output have been found to be generally predictive of outcome. These three responses are assessed by the Glasgow Coma Scale; a score between 3 and 15 is assigned (Table 457e-1). Over 85% of patients with aggregate scores of <5 die within 24 h. However, a number of patients with slightly higher scores, including a few without pupillary light responses, survive, suggesting that an initially aggressive approach is justified in most patients. Patients <20 years old, particularly children, may make remarkable recoveries after having grave early neurologic signs. In one large study of severe head injury, 55% of children had a good outcome at 1 year, compared with 21% of adults. Older age, increased ICP, early hypoxia or hypotension, compression of the brainstem on CT or MRI, and a delay in the evacuation of large intracranial hemorrhages are indicators of a poor prognosis.
|
GLASGOW COMA SCALE FOR HEAD INJURY |
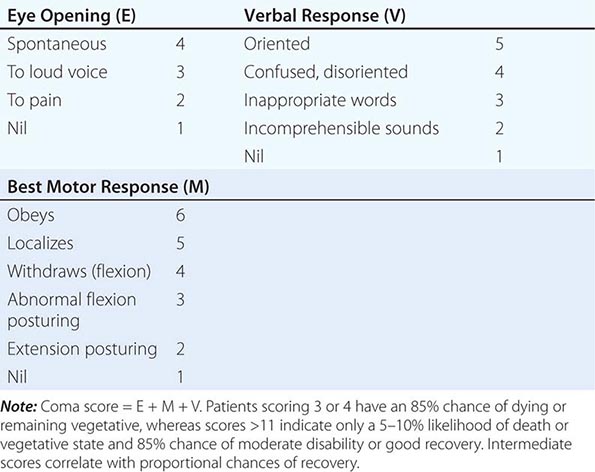
POSTCONCUSSION SYNDROME
The postconcussion syndrome refers to a state following minor head injury consisting of combinations of fatigue, dizziness, headache, and difficulty in concentration. The syndrome simulates asthenia and anxious depression. Based on experimental models, it has been proposed that subtle axonal shearing lesions or as yet undefined biochemical alterations account for the cognitive symptoms. In moderate and severe trauma, neuropsychological changes such as difficulty with attention and memory and other cognitive deficits are undoubtedly present, sometimes severe, but many problems identified by formal testing do not affect daily functioning. Test scores tend to improve rapidly during the first 6 months after injury and then more slowly for years.
Management of the postconcussive syndrome requires the identification and treatment of each separate element of depression, sleeplessness, anxiety, persistent headache, and dizziness. A clear explanation of the problems that may follow concussion has been shown to reduce subsequent complaints. Care is taken to avoid prolonged use of drugs that produce dependence. Headache may initially be treated with acetaminophen and small doses of amitriptyline. Vestibular exercises (Chap. 28) and small doses of vestibular suppressants such as promethazine (Phenergan) may be helpful when dizziness is the main problem. Patients who after minor or moderate injury have difficulty with memory or with complex cognitive tasks at work may be reassured that these problems usually improve over 6–12 months, and workload may be reduced in the interim. It is sometimes helpful to obtain serial and quantified neuropsychological testing in order to adjust the work environment to the patient’s abilities and to document improvement over time. Whether cognitive exercises are useful in contrast to rest and a reduction in mental challenges is uncertain. Previously energetic and resilient individuals usually have the best recoveries. In patients with persistent symptoms, the possibility exists of malingering or prolongation as a result of litigation.
458 |
Multiple Sclerosis and Other Demyelinating Diseases |
Demyelinating disorders are immune-mediated conditions characterized by preferential destruction of central nervous system (CNS) myelin. The peripheral nervous system (PNS) is spared, and most patients have no evidence of an associated systemic illness. Multiple sclerosis, the most common disease in this category, is second only to trauma as a cause of neurologic disability beginning in early to middle adulthood.
MULTIPLE SCLEROSIS
Multiple sclerosis (MS) is an autoimmune disease of the CNS characterized by chronic inflammation, demyelination, gliosis (scarring), and neuronal loss; the course can be relapsing-remitting or progressive. Lesions of MS typically develop at different times and in different CNS locations (i.e., MS is said to be disseminated in time and space). Approximately 350,000 individuals in the United States and 2.5 million individuals worldwide are affected. The clinical course can be extremely variable, ranging from a benign condition to a rapidly evolving and incapacitating disease requiring profound lifestyle adjustments.
PATHOGENESIS
Pathology New MS lesions begin with perivenular cuffing by inflammatory mononuclear cells, predominantly T cells and macrophages, which also infiltrate the surrounding white matter. At sites of inflammation, the blood-brain barrier (BBB) is disrupted, but unlike vasculitis, the vessel wall is preserved. Involvement of the humoral immune system is also evident; small numbers of B lymphocytes also infiltrate the nervous system, myelin-specific autoantibodies are present on degenerating myelin sheaths, and complement is activated. Demyelination is the hallmark of the pathology, and evidence of myelin degeneration is found at the earliest time points of tissue injury. A remarkable feature of MS plaques is that oligodendrocyte precursor cells survive—and in many lesions are present in even greater numbers than in normal tissue—but these cells fail to differentiate into mature myelin-producing cells. In some lesions, surviving oligodendrocytes or those that differentiate from precursor cells partially remyelinate the surviving naked axons, producing so-called shadow plaques. As lesions evolve, there is prominent astrocytic proliferation (gliosis). Over time, ectopic lymphocyte follicle-like structures, consisting of aggregates of T and B cells resembling secondary lymphoid tissue, appear in the meninges and especially overlying deep cortical sulci and also in perivascular spaces. Although relative sparing of axons is typical of MS, partial or total axonal destruction can also occur, especially within highly inflammatory lesions. Thus MS is not solely a disease of myelin, and neuronal pathology is increasingly recognized as a major contributor to irreversible neurologic disability. Inflammation, demyelination, and plaque formation are also present in the cerebral cortex, and significant axon loss indicating death of neurons is widespread, especially in advanced cases (see “Neurodegeneration,” below).
Physiology Nerve conduction in myelinated axons occurs in a saltatory manner, with the nerve impulse jumping from one node of Ranvier to the next without depolarization of the axonal membrane underlying the myelin sheath between nodes (Fig. 458-1). This produces considerably faster conduction velocities (~70 m/s) than the slow velocities (~1 m/s) produced by continuous propagation in unmyelinated nerves. Conduction block occurs when the nerve impulse is unable to traverse the demyelinated segment. This can happen when the resting axon membrane becomes hyperpolarized due to the exposure of voltage-dependent potassium channels that are normally buried underneath the myelin sheath. A temporary conduction block often follows a demyelinating event before sodium channels (originally concentrated at the nodes) redistribute along the naked axon (Fig. 458-1). This redistribution ultimately allows continuous propagation of nerve action potentials through the demyelinated segment. Conduction block may be incomplete, affecting high- but not low-frequency volleys of impulses. Variable conduction block can occur with raised body temperature or metabolic alterations and may explain clinical fluctuations that vary from hour to hour or appear with fever or exercise. Conduction slowing occurs when the demyelinated segments of the axonal membrane are reorganized to support continuous (slow) nerve impulse propagation.
FIGURE 458-1 Nerve conduction in myelinated and demyelinated axons. A. Saltatory nerve conduction in myelinated axons occurs with the nerve impulse jumping from one node of Ranvier to the next. Sodium channels (shown as breaks in the solid black line) are concentrated at the nodes where axonal depolarization occurs. B. Following demyelination, additional sodium channels are redistributed along the axon itself, thereby allowing continuous propagation of the nerve action potential despite the absence of myelin.
Epidemiology MS is approximately threefold more common in women than men. The age of onset is typically between 20 and 40 years (slightly later in men than in women), but the disease can present across the lifespan. Approximately 10% of cases begin before age 18 years of age, and a small percentage of cases begin before the age of 10 years.
Geographical gradients have been repeatedly observed in MS, with the highest known prevalence for MS (250 per 100,000) in the Orkney Islands, located north of Scotland. In other temperate zone areas (e.g., northern North America, northern Europe, southern Australia, and southern New Zealand), the prevalence of MS is 0.1–0.2%. By contrast, in the tropics (e.g., Asia, equatorial Africa, and the Middle East), the prevalence is often 10- to 20-fold less.
The prevalence of MS has increased steadily (and dramatically) in several regions around the world over the past half-century, presumably reflecting the impact of some environmental shift. Moreover, the fact that this increase has occurred primarily (or exclusively) in women indicates that women are more responsive to this environmental change.
Well-established risk factors for MS include vitamin D deficiency, exposure to Epstein-Barr virus (EBV) after early childhood, and cigarette smoking.
Vitamin D deficiency is associated with an increase in MS risk, and data suggest that ongoing deficiency may also increase disease activity after MS begins. Immunoregulatory effects of vitamin D could explain these apparent relationships. Exposure of the skin to ultraviolet-B (UVB) radiation from the sun is essential for the biosynthesis of vitamin D, and this endogenous production is the most important source of vitamin D in most individuals; a diet rich in fatty fish represents another source of vitamin D. At high latitudes, the amount of UVB radiation reaching the earth’s surface is often insufficient, particularly during winter months, and consequently, low serum levels of vitamin D are common in temperate zones. The common practice to avoid direct sun exposure and the widespread use of sun block, which (at sun protection factor [SPF] 15) blocks 94% of the incoming UVB radiation, would be expected to exacerbate any population-wide vitamin D deficiency.
Evidence of a remote EBV infection playing some role in MS is supported by numerous epidemiologic and laboratory studies. A higher risk of infectious mononucleosis (associated with relatively late EBV infection) and higher antibody titers to latency-associated EBV nuclear antigen have been repeatedly associated with MS risk, although a causal role for EBV has not been established.
A history of cigarette smoking has also been associated with MS risk. Interestingly, in an animal model of MS, the lung was identified as a critical site for activation of pathogenic T lymphocytes responsible for autoimmune demyelination.
Recent data in MS models have also shown that high levels of dietary sodium activate pathogenic autoreactive T lymphocytes, suggesting that consumption of a high-salt diet, now widespread in the Western world, might be part of the explanation for the observed increase in the prevalence of MS in recent years.
GENETIC CONSIDERATIONS
![]() Whites are inherently at higher risk for MS than Africans or Asians, even when residing in a similar environment. MS also aggregates within some families, and adoption, half-sibling, twin, and spousal studies indicate that familial aggregation is due to genetic, and not environmental, factors (Table 458-1).
Whites are inherently at higher risk for MS than Africans or Asians, even when residing in a similar environment. MS also aggregates within some families, and adoption, half-sibling, twin, and spousal studies indicate that familial aggregation is due to genetic, and not environmental, factors (Table 458-1).
|
RISK OF DEVELOPING MULTIPLE SCLEROSIS (MS) |

Susceptibility to MS is polygenic, with each gene contributing a relatively small amount to the overall risk. The strongest susceptibility signal in genome-wide studies maps to the HLA-DRB1 gene in the class II region of the major histocompatibility complex (MHC), and this association accounts for approximately 10% of the disease risk. This HLA association, which was first described several decades ago, suggests that MS, at its core, is an antigen-specific autoimmune disease. Whole-genome association studies have now identified approximately 110 other MS susceptibility variants, each of which individually has only a modest effect on MS risk. Most of these MS-associated genes have known roles in the adaptive immune system, for example the genes for the interleukin (IL) 7 receptor (CD127), IL-2 receptor (CD25), and T cell costimulatory molecule LFA-3 (CD58); some variants also influence susceptibility to other autoimmune diseases in addition to MS. The variants identified so far all lack specificity and sensitivity for MS; thus, at present, they are not useful for diagnosis or to predict the future course of the disease.
IMMUNOLOGY A proinflammatory autoimmune response directed against a component of CNS myelin, and perhaps other neural elements as well, remains the cornerstone of current concepts of MS pathogenesis.
AUTOREACTIVE T LYMPHOCYTES Myelin basic protein (MBP), an intracellular protein involved in myelin compaction, is an important T cell antigen in experimental allergic encephalomyelitis (EAE), a laboratory model, and probably also in human MS. Activated MBP-reactive T cells have been identified in the blood, in cerebrospinal fluid (CSF), and within MS lesions. Moreover, DRB1*15:01 may influence the autoimmune response because it binds with high affinity to a fragment of MBP (spanning amino acids 89–96), stimulating T cell responses to this self-protein. Two different populations of proinflammatory T cells are likely to mediate autoimmunity in MS. T-helper type 1 (TH1) cells producing interferon γ (IFN-γ) are one key effector population, and more recently, a role for highly proinflammatory TH17 T cells has been established. TH17 cells are induced by transforming growth factor β (TGF-β) and IL-6 and are amplified by IL-21 and IL-23. TH17 cells, and levels of their corresponding cytokine IL-17, are increased in MS lesions and also in the circulation of people with active MS. High circulating levels of IL-17 may also be a marker of a more severe course of MS. TH1 cytokines, including IL-2, tumor necrosis factor (TNF)-α, and IFN-γ, also play key roles in activating and maintaining autoimmune responses, and TNF-α and IFN-γ may directly injure oligodendrocytes or the myelin membrane.
HUMORAL AUTOIMMUNITY B cell activation and antibody responses also appear to be necessary for the full development of demyelinating lesions to occur, both in experimental models and in human MS. Clonally restricted populations of activated, antigen-experienced, memory B cells and plasma cells are present in MS lesions, in lymphoid follicle-like structures in the meninges overlying the cerebral cortex, and in the CSF. Similar or identical clonal populations are found in each compartment, indicating that a highly focused B cell response is occurring locally within the CNS in MS. Myelin-specific autoantibodies, some directed against an extracellular myelin protein, myelin oligodendrocyte glycoprotein (MOG), have been detected bound to vesiculated myelin debris in MS plaques. In the CSF, elevated levels of locally synthesized immunoglobulins and oligoclonal antibodies, derived from clonally restricted CNS B cells and plasma cells, are also characteristic of MS. The pattern of oligoclonal banding is unique to each individual, and attempts to identify the targets of these antibodies have been largely unsuccessful.
TRIGGERS Serial magnetic resonance imaging (MRI) studies in early relapsing-remitting MS reveal that bursts of focal inflammatory disease activity occur far more frequently than would have been predicted by the frequency of relapses. Thus, early in MS, most disease activity is clinically silent. Although the triggers causing these bursts are unknown, molecular mimicry between environmental agents, presumably pathogens, and myelin antigens activating pathogenic T cells may be responsible (Chap. 377e).
Neurodegeneration Axonal damage occurs in every newly formed MS lesion, and cumulative axonal loss is considered to be one important cause of irreversible neurologic disability in MS. As many as 70% of axons are lost from the lateral corticospinal (e.g., motor) tracts in patients with advanced paraparesis from MS, and longitudinal MRI studies suggest there is progressive axonal loss over time within established, inactive lesions. Demyelination can result in reduced trophic support for axons, redistribution of ion channels, and destabilization of action potential membrane potentials. Axons can adapt initially to these injuries, but over time, distal and retrograde degeneration often occurs. Therefore, promoting remyelination remains an important therapeutic goal.
In progressive MS, a key unresolved question is whether the primary neurodegenerative process occurs primarily in the cerebral cortex, the white matter, or in some combination of the two sites. As noted above, meningeal infiltrates of B and T cells are particularly prominent in progressive MS cases, and these “lymphoid follicles” are associated with underlying microglial activation, gray matter plaques, and loss of cortical neurons. White matter lesions may also contribute to late progressive MS; inactive plaques are often noninflammatory at the center, but at the edges, microglia and macrophages and evidence of ongoing axonal injury can be found. This suggests that a simmering, and possibly concentrically expanding, axonopathy may be present, even in the most chronic cases. In addition, a diffuse low-grade inflammation across large areas of white matter may be present, associated with reduced myelin staining and axonal injury (“dirty white matter”). Another characteristic of progressive MS is that inflammation is often present without a concomitant disruption of the BBB; possibly, this feature might explain the failure of immunotherapies not capable of crossing the BBB to benefit patients with progressive MS.
Evidence supports a role of one, or more likely several, of the following mechanisms in progressive MS. Axonal and neuronal death may result from glutamate-mediated excitotoxicity, oxidative injury, iron accumulation, and/or mitochondrial failure either occurring as a consequence of free-radical damage or due to accumulation of deletions in mitochondrial DNA.
CLINICAL MANIFESTATIONS
The onset of MS may be abrupt or insidious. Symptoms may be severe or seem so trivial that a patient may not seek medical attention for months or years. Indeed, at autopsy, approximately 0.1% of individuals who were asymptomatic during life will be found, unexpectedly, to have pathologic evidence of MS. Similarly, in the modern era, an MRI scan obtained for an unrelated reason may show evidence of asymptomatic MS. Symptoms of MS are extremely varied and depend on the location and severity of lesions within the CNS (Table 458-2). Examination often reveals evidence of neurologic dysfunction, often in asymptomatic locations. For example, a patient may present with symptoms in one leg but signs in both.
|
INITIAL SYMPTOMS OF MS |
Weakness of the limbs may manifest as loss of strength, speed, or dexterity, as fatigue, or as a disturbance of gait. Exercise-induced weakness is a characteristic symptom of MS. The weakness is of the upper motor neuron type (Chap. 30) and is usually accompanied by other pyramidal signs such as spasticity, hyperreflexia, and Babinski signs. Occasionally a tendon reflex may be lost (simulating a lower motor neuron lesion) if an MS lesion disrupts the afferent reflex fibers in the spinal cord (see Fig. 30-2).
Spasticity (Chap. 30) is commonly associated with spontaneous and movement-induced muscle spasms. More than 30% of MS patients have moderate to severe spasticity, especially in the legs. This is often accompanied by painful spasms interfering with ambulation, work, or self-care. Occasionally spasticity provides support for the body weight during ambulation, and in these cases, treatment of spasticity may actually do more harm than good.
Optic neuritis (ON) presents as diminished visual acuity, dimness, or decreased color perception (desaturation) in the central field of vision. These symptoms can be mild or may progress to severe visual loss. Rarely, there is complete loss of light perception. Visual symptoms are generally monocular but may be bilateral. Periorbital pain (aggravated by eye movement) often precedes or accompanies the visual loss. An afferent pupillary defect (Chap. 39) is usually present. Funduscopic examination may be normal or reveal optic disc swelling (papillitis). Pallor of the optic disc (optic atrophy) commonly follows ON. Uveitis is uncommon and should raise the possibility of alternative diagnoses such as sarcoid or lymphoma.
Visual blurring in MS may result from ON or diplopia (double vision); if the symptom resolves when either eye is covered, the cause is diplopia.
Diplopia may result from internuclear ophthalmoplegia (INO) or from palsy of the sixth cranial nerve (rarely the third or fourth). An INO consists of impaired adduction of one eye due to a lesion in the ipsilateral medial longitudinal fasciculus (Chaps. 41e and 42). Prominent nystagmus is often observed in the abducting eye, along with a small skew deviation. A bilateral INO is particularly suggestive of MS. Other common gaze disturbances in MS include (1) a horizontal gaze palsy, (2) a “one and a half” syndrome (horizontal gaze palsy plus an INO), and (3) acquired pendular nystagmus.
Sensory symptoms are varied and include both paresthesias (e.g., tingling, prickling sensations, formications, “pins and needles,” or painful burning) and hypesthesia (e.g., reduced sensation, numbness, or a “dead” feeling). Unpleasant sensations (e.g., feelings that body parts are swollen, wet, raw, or tightly wrapped) are also common. Sensory impairment of the trunk and legs below a horizontal line on the torso (a sensory level) indicates that the spinal cord is the origin of the sensory disturbance. It is often accompanied by a bandlike sensation of tightness around the torso. Pain is a common symptom of MS, experienced by >50% of patients. Pain can occur anywhere on the body and can change locations over time.
Ataxia usually manifests as cerebellar tremors (Chap. 450). Ataxia may also involve the head and trunk or the voice, producing a characteristic cerebellar dysarthria (scanning speech).
Bladder dysfunction is present in >90% of MS patients, and in a third of patients, dysfunction results in weekly or more frequent episodes of incontinence. During normal reflex voiding, relaxation of the bladder sphincter (α-adrenergic innervation) is coordinated with contraction of the detrusor muscle in the bladder wall (muscarinic cholinergic innervation). Detrusor hyperreflexia, due to impairment of suprasegmental inhibition, causes urinary frequency, urgency, nocturia, and uncontrolled bladder emptying. Detrusor sphincter dyssynergia, due to loss of synchronization between detrusor and sphincter muscles, causes difficulty in initiating and/or stopping the urinary stream, producing hesitancy, urinary retention, overflow incontinence, and recurrent infection.
Constipation occurs in >30% of patients. Fecal urgency or bowel incontinence is less common (<15%) but can be socially debilitating.
Cognitive dysfunction can include memory loss; impaired attention; difficulties in executive functioning, memory, and problem solving; slowed information processing; and problems shifting between cognitive tasks. Euphoria (elevated mood) was once thought to be characteristic of MS but is actually uncommon, occurring in <20% of patients. Cognitive dysfunction sufficient to impair activities of daily living is rare.
Depression, experienced by approximately half of patients, can be reactive, endogenous, or part of the illness itself and can contribute to fatigue.
Fatigue (Chap. 29) is experienced by 90% of patients; this symptom is the most common reason for work-related disability in MS. Fatigue can be exacerbated by elevated temperatures, depression, expending exceptional effort to accomplish basic activities of daily living, or sleep disturbances (e.g., from frequent nocturnal awakenings to urinate).
Sexual dysfunction may manifest as decreased libido, impaired genital sensation, impotence in men, and diminished vaginal lubrication or adductor spasms in women.
Facial weakness due to a lesion in the pons may resemble idiopathic Bell’s palsy (Chap. 455). Unlike Bell’s palsy, facial weakness in MS is usually not associated with ipsilateral loss of taste sensation or retroauricular pain.
Vertigo may appear suddenly from a brainstem lesion, superficially resembling acute labyrinthitis (Chap. 28). Hearing loss (Chap. 43) may also occur in MS but is uncommon.
Ancillary Symptoms Heat sensitivity refers to neurologic symptoms produced by an elevation of the body’s core temperature. For example, unilateral visual blurring may occur during a hot shower or with physical exercise (Uhthoff’s symptom). It is also common for MS symptoms to worsen transiently, sometimes dramatically, during febrile illnesses (see “Acute Attacks or Initial Demyelinating Episodes,” below). Such heat-related symptoms probably result from transient conduction block (see above).
Lhermitte’s symptom is an electric shock–like sensation (typically induced by flexion or other movements of the neck) that radiates down the back into the legs. Rarely, it radiates into the arms. It is generally self-limited but may persist for years. Lhermitte’s symptom can also occur with other disorders of the cervical spinal cord (e.g., cervical spondylosis).
Paroxysmal symptoms are distinguished by their brief duration (10 s to 2 min), high frequency (5–40 episodes per day), lack of any alteration of consciousness or change in background electroencephalogram during episodes, and a self-limited course (generally lasting weeks to months). They may be precipitated by hyperventilation or movement. These syndromes may include Lhermitte’s symptom; tonic contractions of a limb, face, or trunk (tonic seizures); paroxysmal dysarthria and ataxia; paroxysmal sensory disturbances; and several other less well-characterized syndromes. Paroxysmal symptoms probably result from spontaneous discharges, arising at the edges of demyelinated plaques and spreading to adjacent white matter tracts.
Trigeminal neuralgia, hemifacial spasm, and glossopharyngeal neuralgia (Chap. 455) can occur when the demyelinating lesion involves the root entry (or exit) zone of the fifth, seventh, and ninth cranial nerve, respectively. Trigeminal neuralgia (tic douloureux) is a very brief lancinating facial pain often triggered by an afferent input from the face or teeth. Most cases of trigeminal neuralgia are not MS related; however, atypical features such as onset before age 50 years, bilateral symptoms, objective sensory loss, or nonparoxysmal pain should raise the possibility that MS could be responsible.
Facial myokymia consists of either persistent rapid flickering contractions of the facial musculature (especially the lower portion of the orbicularis oculus) or a contraction that slowly spreads across the face. It results from lesions of the corticobulbar tracts or brainstem course of the facial nerve.
DISEASE COURSE
Four clinical types of MS exist (Fig. 458-2):
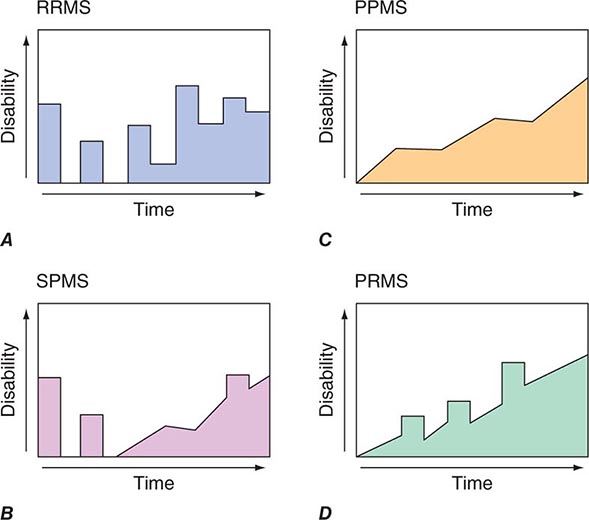
FIGURE 458-2 Clinical course of multiple sclerosis (MS). A. Relapsing/remitting MS (RRMS). B. Secondary progressive MS (SPMS). C. Primary progressive MS (PPMS). D. Progressive/relapsing MS (PRMS).
1. Relapsing/remitting MS (RRMS) accounts for 85% of MS cases at onset and is characterized by discrete attacks that generally evolve over days to weeks (rarely over hours). With initial attacks, there is often substantial or complete recovery over the ensuing weeks to months, but as attacks continue over time recovery may be less evident (Fig. 458-2A). Between attacks, patients are neurologically stable.
2. Secondary progressive MS (SPMS) always begins as RRMS (Fig. 458-2B). At some point, however, the clinical course changes so that the patient experiences a steady deterioration in function unassociated with acute attacks (which may continue or cease during the progressive phase). SPMS produces a greater amount of fixed neurologic disability than RRMS. For a patient with RRMS, the risk of developing SPMS is ~2% each year, meaning that the great majority of RRMS ultimately evolves into SPMS. SPMS appears to represent a late stage of the same underlying illness as RRMS.
3. Primary progressive MS (PPMS) accounts for ~15% of cases. These patients do not experience attacks but only a steady functional decline from disease onset (Fig. 458-2C). Compared to RRMS, the sex distribution is more even, the disease begins later in life (mean age ~40 years), and disability develops faster (at least relative to the onset of the first clinical symptom). Despite these differences, PPMS appears to represent the same underlying illness as RRMS.
4. Progressive/relapsing MS (PRMS) overlaps PPMS and SPMS and accounts for ~5% of MS patients. Like patients with PPMS, these patients experience a steady deterioration in their condition from disease onset. However, like SPMS patients, they experience occasional attacks superimposed upon their progressive course (Fig. 458-2D).
DIAGNOSIS
There is no definitive diagnostic test for MS. Diagnostic criteria for clinically definite MS require documentation of two or more episodes of symptoms and two or more signs that reflect pathology in anatomically noncontiguous white matter tracts of the CNS (Table 458-3). Symptoms must last for >24 h and occur as distinct episodes that are separated by a month or more. In patients who have only one of the two required signs on neurologic examination, the second may be documented by abnormal tests such as MRI or evoked potentials (EPs). Similarly, in the most recent diagnostic scheme, the second clinical event (in time) may be supported solely by MRI findings, consisting of either the development of new focal white matter lesions on MRI or the simultaneous presence of both an enhancing lesion and a nonenhancing lesion in an asymptomatic location. In patients whose course is progressive from onset for ≥6 months without superimposed relapses, documentation of intrathecal IgG synthesis may be used to support a diagnosis of PPMS.
|
DIAGNOSTIC CRITERIA FOR MULTIPLE SCLEROSIS (MS) |

DIAGNOSTIC TESTS
Magnetic Resonance Imaging MRI has revolutionized the diagnosis and management of MS (Fig. 458-3); characteristic abnormalities are found in >95% of patients, although more than 90% of the lesions visualized by MRI are asymptomatic. An increase in vascular permeability from a breakdown of the BBB is detected by leakage of intravenous gadolinium (Gd) into the parenchyma. Such leakage occurs early in the development of an MS lesion and serves as a useful marker of inflammation. Gd enhancement typically persists for approximately 1 month, and the residual MS plaque remains visible indefinitely as a focal area of hyperintensity (a lesion) on spin-echo (T2-weighted) and proton-density images. Lesions are frequently oriented perpendicular to the ventricular surface, corresponding to the pathologic pattern of perivenous demyelination (Dawson’s fingers). Lesions are multifocal within the brain, brainstem, and spinal cord. Lesions larger than 6 mm located in the corpus callosum, periventricular white matter, brainstem, cerebellum, or spinal cord are particularly helpful diagnostically. Current criteria for the use of MRI in the diagnosis of MS are shown in Table 458-3.
FIGURE 458-3 Magnetic resonance imaging findings in multiple sclerosis (MS). A. Axial first-echo image from T2-weighted sequence demonstrates multiple bright signal abnormalities in white matter, typical for MS. B. Sagittal T2-weighted fluid-attenuated inversion recovery (FLAIR) image in which the high signal of cerebrospinal fluid (CSF) has been suppressed. CSF appears dark, whereas areas of brain edema or demyelination appear high in signal as shown here in the corpus callosum (arrows). Lesions in the anterior corpus callosum are frequent in MS and rare in vascular disease. C. Sagittal T2-weighted fast spin echo image of the thoracic spine demonstrates a fusiform high-signal-intensity lesion in the midthoracic spinal cord. D. Sagittal T1-weighted image obtained after the intravenous administration of gadolinium DTPA reveals focal areas of blood-brain barrier disruption, identified as high-signal-intensity regions (arrows).
The total volume of T2-weighted signal abnormality (the “burden of disease”) shows a significant (albeit weak) correlation with clinical disability, as do measures of brain atrophy. Approximately one-third of T2-weighted lesions appear as hypointense lesions (black holes) on T1-weighted imaging. Black holes may be a marker of irreversible demyelination and axonal loss, although even this measure depends on the timing of the image acquisition (e.g., most acute Gd-enhancing T2 lesions are T1 dark).
Newer MRI methods such as magnetization transfer ratio (MTR) imaging and proton magnetic resonance spectroscopic imaging (MRSI) may ultimately serve as surrogate markers of clinical disability. MRSI can quantitate molecules such as N-acetyl aspartate, which is a marker of axonal integrity, and MTR may be able to distinguish demyelination from edema.
Evoked Potentials EP testing assesses function in afferent (visual, auditory, and somatosensory) or efferent (motor) CNS pathways. EPs use computer averaging to measure CNS electric potentials evoked by repetitive stimulation of selected peripheral nerves or of the brain. These tests provide the most information when the pathways studied are clinically uninvolved. For example, in a patient with a remitting and relapsing spinal cord syndrome with sensory deficits in the legs, an abnormal somatosensory EP following posterior tibial nerve stimulation provides little new information. By contrast, an abnormal visual EP in this circumstance would permit a diagnosis of clinically definite MS (Table 458-3). Abnormalities on one or more EP modalities occur in 80–90% of MS patients. EP abnormalities are not specific to MS, although a marked delay in the latency of a specific EP component (as opposed to a reduced amplitude or distorted wave-shape) is suggestive of demyelination.
Cerebrospinal Fluid CSF abnormalities found in MS include a mononuclear cell pleocytosis and an increased level of intrathecally synthesized IgG. The total CSF protein is usually normal. Various formulas distinguish intrathecally synthesized IgG from IgG that may have entered the CNS passively from the serum. One formula, the CSF IgG index, expresses the ratio of IgG to albumin in the CSF divided by the same ratio in the serum. The IgG synthesis rate uses serum and CSF IgG and albumin measurements to calculate the rate of CNS IgG synthesis. The measurement of oligoclonal bands (OCBs) by agarose gel electrophoresis in the CSF also assesses intrathecal production of IgG. Two or more discrete OCBs, not present in a paired serum sample, are found in >75% of patients with MS. OCBs may be absent at the onset of MS, and in individual patients, the number of bands may increase with time.
A mild CSF pleocytosis (>5 cells/μL) is present in ~25% of cases, usually in young patients with RRMS. A pleocytosis of >75 cells/μL, the presence of polymorphonuclear leukocytes, or a protein concentration >1 g/L (>100 mg/dL) in CSF should raise concern that the patient may not have MS.
DIFFERENTIAL DIAGNOSIS
No single clinical sign or test is diagnostic of MS. The diagnosis is readily made in a young adult with relapsing and remitting symptoms involving different areas of CNS white matter. The possibility of an alternative diagnosis should always be considered (Table 458-4), particularly when (1) symptoms are localized exclusively to the posterior fossa, craniocervical junction, or spinal cord; (2) the patient is <15 or >60 years of age; (3) the clinical course is progressive from onset; (4) the patient has never experienced visual, sensory, or bladder symptoms; or (5) laboratory findings (e.g., MRI, CSF, or EPs) are atypical. Similarly, uncommon or rare symptoms in MS (e.g., aphasia, parkinsonism, chorea, isolated dementia, severe muscular atrophy, peripheral neuropathy, episodic loss of consciousness, fever, headache, seizures, or coma) should increase concern about an alternative diagnosis. Diagnosis is also difficult in patients with a rapid or explosive (stroke-like) onset or with mild symptoms and a normal neurologic examination. Rarely, intense inflammation and swelling may produce a mass lesion that mimics a primary or metastatic tumor. In the current era, the disorders most likely to be mistaken for MS are neuromyelitis optica (see below), sarcoid, vascular disorders (including antiphospholipid syndrome and vasculitis), and rarely CNS lymphoma. The specific tests required to exclude alternative diagnoses will vary with each clinical situation; however, an erythrocyte sedimentation rate, serum B12 level, ANA, and treponemal antibody should probably be obtained in all patients with suspected MS.
|
DISORDERS THAT CAN MIMIC MULTIPLE SCLEROSIS (MS) |
Abbreviations: AV, arteriovenous; CNS, central nervous system; HTLV, human T cell lymphotropic virus.
PROGNOSIS
Most patients with clinically evident MS ultimately experience progressive neurologic disability. In older studies conducted mostly before disease-modifying therapies for MS were widely available, 15 years after onset, only 20% of patients had no functional limitation, and between one-third and one-half progressed to SPMS and required assistance with ambulation; furthermore, 25 years after onset, ~80% of MS patients reached this level of disability. The long-term prognosis for untreated MS appears to have improved in recent years, at least in part because of the development of therapies for the early relapsing form of the disease. Although the prognosis in an individual is difficult to establish, certain clinical features suggest a more favorable prognosis. These include ON or sensory symptoms at onset; fewer than two relapses in the first year of illness; and minimal impairment after 5 years. By contrast, patients with truncal ataxia, action tremor, pyramidal symptoms, or a progressive disease course are more likely to become disabled. Patients with a long-term favorable course are likely to have developed fewer MRI lesions during the early years of disease, and vice versa. Importantly, some MS patients have a benign variant of MS and never develop neurologic disability. The likelihood of having benign MS is thought to be <20%. Patients with benign MS 15 years after onset who have entirely normal neurologic examinations are likely to maintain their benign course
In patients with their first demyelinating event (i.e., a clinically isolated syndrome), the brain MRI provides prognostic information. With three or more typical T2-weighted lesions, the risk of developing MS after 20 years is ~80%. Conversely, with a normal brain MRI, the likelihood of developing MS is <20%. Similarly, the presence of two or more Gd-enhancing lesions at baseline is highly predictive of future MS, as is the appearance of either new T2-weighted lesions or new Gd enhancement ≥3 months after the initial episode.
Mortality as a direct consequence of MS is uncommon, although it has been estimated that the 25-year survival is only 85% of expected. Death can occur during an acute MS attack, although this is distinctly rare. More commonly, death occurs as a complication of MS (e.g., pneumonia in a debilitated individual). Death can also result from suicide. Early disease-modifying therapy seems to reduce this excess mortality.
Effect of Pregnancy Pregnant MS patients experience fewer attacks than expected during gestation (especially in the last trimester), but more attacks than expected in the first 3 months postpartum. When considering the pregnancy year as a whole (i.e., 9 months of pregnancy plus 3 months postpartum), the overall disease course is unaffected. Decisions about childbearing should thus be made based on (1) the mother’s physical state, (2) her ability to care for the child, and (3) the availability of social support. Disease-modifying therapy is generally discontinued during pregnancy, although the actual risk from the interferons and glatiramer acetate (see below) appears to be low.
|
TREATMENT |
MULTIPLE SCLEROSIS |
Therapy for MS can be divided into several categories: (1) treatment of acute attacks, (2) treatment with disease-modifying agents that reduce the biologic activity of MS, and (3) symptomatic therapy. Treatments that promote remyelination or neural repair do not currently exist, but several promising approaches are being actively investigated.
The Expanded Disability Status Score (EDSS) is a widely used measure of neurologic impairment in MS (Table 458-5). Most patients with EDSS scores <3.5 have RRMS, walk normally, and are generally not disabled; by contrast, patients with EDSS scores >5.5 have progressive MS (SPMS or PPMS), are gait-impaired, and, typically, are occupationally disabled.
|
SCORING SYSTEMS FOR MULTIPLE SCLEROSIS (MS) |

ACUTE ATTACKS OR INITIAL DEMYELINATING EPISODES
When patients experience acute deterioration, it is important to consider whether this change reflects new disease activity or a “pseudoexacerbation” resulting from an increase in ambient temperature, fever, or an infection. When the clinical change is thought to reflect a pseudoexacerbation, glucocorticoid treatment is inappropriate. Glucocorticoids are used to manage either first attacks or acute exacerbations. They provide short-term clinical benefit by reducing the severity and shortening the duration of attacks. Whether treatment provides any long-term benefit on the course of the illness is less clear. Therefore, mild attacks are often not treated. Physical and occupational therapy can help with mobility and manual dexterity.
Glucocorticoid treatment is usually administered as intravenous methylprednisolone, 500–1000 mg/d for 3–5 days, either without a taper or followed by a course of oral prednisone beginning at a dose of 60–80 mg/d and gradually tapered over 2 weeks. Orally administered methylprednisolone or dexamethasone (in equivalent dosages) can be substituted for the intravenous portion of the therapy, although gastrointestinal complications are more common by this route. Outpatient treatment is almost always possible.
Side effects of short-term glucocorticoid therapy include fluid retention, potassium loss, weight gain, gastric disturbances, acne, and emotional lability. Concurrent use of a low-salt, potassium-rich diet and avoidance of potassium-wasting diuretics are advisable. Lithium carbonate (300 mg orally bid) may help to manage emotional lability and insomnia associated with glucocorticoid therapy. Patients with a history of peptic ulcer disease may require cimetidine (400 mg bid) or ranitidine (150 mg bid). Proton pump inhibitors such as pantoprazole (40 mg orally bid) may reduce the likelihood of gastritis, especially when large doses are administered orally. Plasma exchange (five to seven exchanges: 40–60 mL/kg per exchange, every other day for 14 days) may benefit patients with fulminant attacks of demyelination that are unresponsive to glucocorticoids. However, the cost is high, and conclusive evidence of efficacy is lacking.
DISEASE-MODIFYING THERAPIES FOR RELAPSING FORMS OF MS (RRMS, SPMS WITH EXACERBATIONS)
Ten such agents are approved by the U.S. Food and Drug Administration (FDA): (1) IFN-β-1a (Avonex), (2) IFN-β-1a (Rebif), (3) IFN-β-1b (Betaseron or Extavia), (4) glatiramer acetate (Copaxone), (5) natalizumab (Tysabri), (6) fingolimod (Gilenya), (7) dimethyl fumarate (Tecfidera), (8) teriflunomide (Aubagio), (9) mitoxantrone (Novantrone), and (10) alemtuzumab (Lemtrada). Several other promising agents are in varying stages of product development. Each of these treatments can also be used in SPMS patients who continue to experience attacks, both because SPMS can be difficult to distinguish from RRMS and because the available clinical trials, although not definitive, suggest that such patients may sometimes derive therapeutic benefit. Thus, in several phase 3 clinical trials, recipients of each of these agents experienced fewer clinical exacerbations and fewer new MRI lesions compared to placebo recipients (Table 458-6). Because of its potential toxicity as an immunosuppressant, mitoxantrone is generally reserved for patients with progressive disability who have failed other treatments. When considering the data in Table 458-6, however, it is important to note that the relative efficacy of the different agents cannot be determined by cross-trial comparisons. Relative efficacy can only be assessed from nonbiased head-to-head clinical trials.
|
TWO-YEAR OUTCOMES FOR FDA-APPROVED THERAPIES FOR MULTIPLE SCLEROSISa |
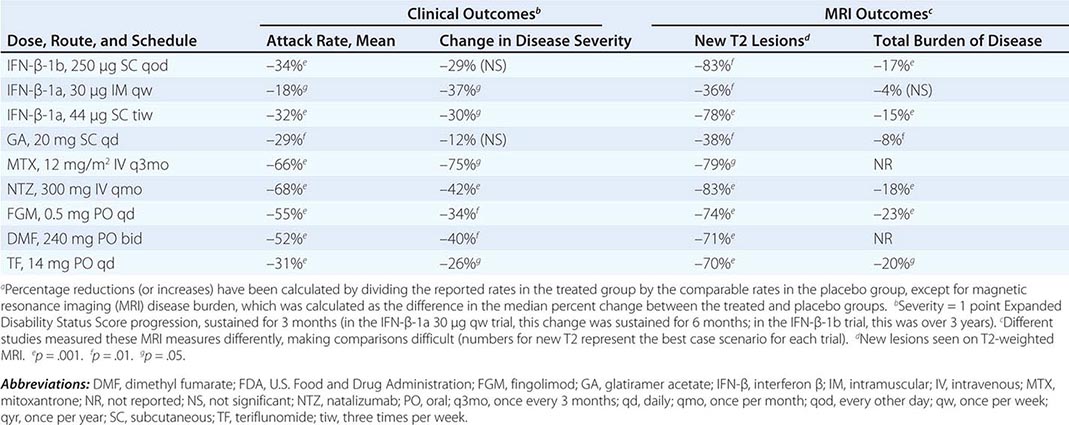
Interferon-β IFN-β is a class I interferon originally identified by its antiviral properties. Efficacy in MS probably results from immunomodulatory properties including (1) downregulating expression of MHC molecules on antigen-presenting cells, (2) reducing proinflammatory and increasing regulatory cytokine levels, (3) inhibiting T cell proliferation, and (4) limiting the trafficking of inflammatory cells in the CNS. IFN-β reduces the attack rate and improves disease severity measures such as EDSS progression and MRI-documented disease burden. IFN-β should be considered in patients with either RRMS or SPMS with superimposed relapses. In patients with SPMS but without relapses, efficacy has not been established. Head-to-head trials suggest that dosing IFN-β more frequently and at higher doses has better efficacy but is also more likely to induce neutralizing antibodies (see below). IFN-β-1a (Avonex), 30 μg, is administered by intramuscular injection once every week. IFN-β-1a (Rebif), 44 μg, is administered by subcutaneous injection three times per week. IFN-β-1b (Betaseron or Extavia), 250 μg, is administered by subcutaneous injection every other day.
Common side effects of IFN-β therapy include flulike symptoms (e.g., fevers, chills, and myalgias) and mild abnormalities on routine laboratory evaluation (e.g., elevated liver function tests or lymphopenia). Rarely, more severe hepatotoxicity may occur. Subcutaneous IFN-β also causes reactions at the injection site (e.g., pain, redness, induration, or, rarely, skin necrosis). Side effects can usually be managed with concomitant nonsteroidal anti-inflammatory medications. Depression, increased spasticity, and cognitive changes have been reported, although these symptoms can also be due to the underlying disease. In any event, side effects due to IFN-β therapy usually subside over time.
Approximately 2–10% of IFN-β-1a (Avonex) recipients, 15–25% of IFN-β-1a (Rebif) recipients, and 30–40% of IFN-β-1b (Betaseron/Extavia) recipients develop neutralizing antibodies to IFN-β, which may disappear over time. Two very large randomized trials (one with >2000 patients) provide unequivocal evidence that neutralizing antibodies reduce efficacy as determined by several MRI outcomes. Paradoxically, however, these same trials, despite abundant statistical power, failed to demonstrate any concomitant impact on the clinical outcomes of disability and relapse rate. The reason for this clinical-radiologic dissociation is unresolved. For a patient doing well on therapy, the presence of antibodies should not affect treatment. Conversely, for a patient doing poorly on therapy, alternative treatment should be considered, even if there are no detectable antibodies.
Glatiramer Acetate Glatiramer acetate is a synthetic, random polypeptide composed of four amino acids (L-glutamic acid, L-lysine, L-alanine, and L-tyrosine). Its mechanism of action may include (1) induction of antigen-specific suppressor T cells; (2) binding to MHC molecules, thereby displacing bound MBP; or (3) altering the balance between proinflammatory and regulatory cytokines. Glatiramer acetate reduces the attack rate (whether measured clinically or by MRI) in RRMS. Glatiramer acetate also benefits disease severity measures, although, for clinical disability, this is less well established than for IFN-β. Nevertheless, two very large head-to-head trials demonstrated that the impact of glatiramer acetate on clinical relapse rates and disability was comparable to high-dose, high-frequency IFN-β. Therefore, glatiramer acetate should be considered as an equally effective alternative to IFN-β in RRMS patients. Its usefulness in progressive disease is unknown. Glatiramer acetate is administered by subcutaneous injection of either 20 mg every day or 40 mg thrice weekly. Injection-site reactions also occur with glatiramer acetate. Initially, these were thought to be less severe than with IFN-β, although two recent head-to-head comparisons of high-dose, high-frequency IFN-β to daily glatiramer acetate did not bear out this impression. In addition, approximately 15% of patients experience one or more episodes of flushing, chest tightness, dyspnea, palpitations, and anxiety after injection. This systemic reaction is unpredictable, brief (duration <1 h), and tends not to recur. Finally, some patients experience lipoatrophy, which, on occasion, can be disfiguring and require cessation of treatment.
Natalizumab Natalizumab is a humanized monoclonal antibody directed against the α4 subunit of α4β1 integrin, a cellular adhesion molecule expressed on the surface of lymphocytes. It prevents lymphocytes from binding to endothelial cells, thereby preventing lymphocytes from penetrating the BBB and entering the CNS. Natalizumab is highly effective in reducing the attack rate and significantly improves all measures of disease severity in MS (both clinical and MRI). Moreover, it is well-tolerated, and the dosing schedule of monthly intravenous infusions makes it very convenient for patients. However, progressive multifocal leukoencephalopathy (PML), a life-threatening condition resulting from infection by the John Cunningham (JC) virus, has occurred in approximately 0.3% of patients treated with natalizumab. The incidence of PML is very low in the first year of treatment but then rises by the second year to reach a level of about 2 cases per 1000 patients per year. Nevertheless, the measurement of antibodies against the JC virus in the serum can be used to stratify this risk. Thus, in patients who do not have these antibodies, the risk of PML is either minimal or nonexistent (as long as they remain JC antibody free). Conversely, in patients who have these antibodies (especially those who have them in high titer), the risk may be as high as 0.6% or greater. The risk is also high in patients who have previously received immunosuppressive therapy. Natalizumab is currently recommended only for JC antibody–negative patients, unless they have failed alternative therapies or if they have a particularly aggressive disease course. Head-to-head data show that natalizumab is superior to low-dose (weekly) IFN-β-1a in RRMS. However, its relative efficacy compared to other agents has not been established conclusively.
Natalizumab, 300 mg, is administered by IV infusion each month. Treatment with natalizumab is, in general, well tolerated. A small percentage (<10%) of patients experience hypersensitivity reactions (including anaphylaxis), and ~6% develop neutralizing antibodies to the molecule (only half of which persist).
The major concern with long-term treatment is the risk of PML. Approximately half of the adult population is JC antibody positive, indicating that they experienced an asymptomatic infection with the JC virus at some time in the past. Nevertheless, because the risk is extremely low during the first year of treatment with natalizumab (regardless of antibody status), natalizumab can still be used safely in JC antibody–positive patients for a period of 12 months. After this time, in antibody-positive patients, a change to another disease-modifying therapy should be strongly considered. By contrast, persistently antibody-negative patients can be continued on treatment indefinitely. Up to 2% of seronegative MS patients undergoing treatment with natalizumab seroconvert annually; thus it is recommended that JC antibody status be assessed at 6-month intervals in all patients receiving treatment with this agent.
Fingolimod Fingolimod is a sphingosine-1-phosphate (S1P) inhibitor that prevents the egress of lymphocytes from the secondary lymphoid organs such as the lymph nodes and spleen. Its mechanism of action is probably due, in part, to the trapping of lymphocytes in the periphery and inhibiting their trafficking to the CNS. Fingolimod reduces the attack rate and significantly improves all measures of disease severity in MS. It is well tolerated, and the daily oral dosing schedule makes it very convenient for patients. A large head-to-head phase 3 randomized study demonstrated the superiority of fingolimod over low-dose (weekly) IFN-β-1a. However, its relative efficacy compared to other agents has not been established conclusively.
Fingolimod, 0.5 mg, is administered orally each day. Treatment with fingolimod is also, in general, well tolerated. Mild abnormalities on routine laboratory evaluation (e.g., elevated liver function tests or lymphopenia) are more common than in controls, sometimes requiring discontinuation of the medication. First- and second-degree heart block and bradycardia can also occur when fingolimod therapy is initiated. A 6-h period of observation (including electrocardiogram monitoring) is recommended for all patients receiving their first dose, and individuals with preexisting cardiac disease should probably not be treated with this agent. Other side effects include macular edema and, rarely, disseminated varicella-zoster virus (VZV) infection; prior to initiating therapy with fingolimod, an ophthalmic exam and VZV vaccination for seronegative individuals are indicated.
Dimethyl Fumarate (DMF) Although the precise mechanisms of action of DMF are not fully understood, it seems to have anti-inflammatory effects through its modulation of the expression of proinflammatory and anti-inflammatory cytokines. Also, DMF inhibits the ubiquitylation and degradation of nuclear factor E2-related factor 2 (Nrf2)—a transcription factor that binds to the antioxidant response elements (AREs) located on the DNA and thereby induces the transcription of several antioxidant proteins. DMF reduces the attack rate and significantly improves all measures of disease severity in MS patients. However, its twice-daily oral dosing schedule makes it somewhat less convenient for patients than daily oral therapies. In addition, compliance is likely to be less with a twice-daily dosing regimen—a factor that could be of concern given the observation (in a small clinical trial) that once-daily DMF lacks efficacy. A head-to-head trial provided evidence that DMF was superior to glatiramer acetate on some outcome measures.
DMF, 240 mg, is administered orally twice each day. Gastrointestinal side effects (abdominal discomfort, nausea, vomiting, flushing, and diarrhea) are common at the start of therapy but generally subside with continued administration. Other adverse events included mild decreases in neutrophil and lymphocyte counts and mild elevations in liver enzymes. Nevertheless, in general, treatment with DMF is well tolerated after an initial period of adjustment. Following the release of DMF, four cases of PML were reported in patients receiving other products (not Tecfidera) that contained DMF. Each of these patients was lymphocytopenic, and most had received previous immunosuppressant therapy so that the relationship of DMF to the PML (if any) in these cases is uncertain. Nevertheless, these reports underscore the fact, stated previously, that long-term safety can never be guaranteed by the results of short-term trials. In the case of DMF for MS, only time and experience will tell us whether or not there is any cause for concern.
Teriflunomide Teriflunomide inhibits the mitochondrial enzyme dihydro-orotate dehydrogenase, which is a key part of the pathway for de novo pyrimidine biosynthesis from carbamoyl phosphate and aspartate. It is the active metabolite of the drug leflunomide (FDA-approved for rheumatoid arthritis), and it exerts its anti-inflammatory effects by limiting the proliferation of rapidly dividing T and B cells. This enzyme is not involved in the so-called “salvage pathway,” by which existing pyrimidine pools are recycled for DNA and RNA synthesis in resting and homeostatically proliferating cells. Consequently, teriflunomide is considered to be cytostatic rather than cytotoxic. Teriflunomide reduces the attack rate and significantly improves all measures of disease severity in MS patients. It is well tolerated, and its daily oral dosing schedule makes it very convenient for patients. A head-to-head trial suggested the equivalence, but not superiority, of teriflunomide and high-dose (thrice-weekly) IFN-β-1a. Teriflunomide, either 7 or 14 mg, is administered orally each day. In the pivotal clinical trials, mild hair thinning and gastrointestinal symptoms (nausea and diarrhea) were more common than in controls, but in general, treatment with teriflunomide was well tolerated. As with any new agent, the long-term safety is not guaranteed by the results of short-term trials. A major limitation, especially in women of childbearing age, is its possible teratogenicity (pregnancy category X); teriflunomide can remain in the bloodstream for 2 years, and it is recommended that exposed men and women who wish to conceive receive cholestyramine or activated charcoal to eliminate residual drug.
Mitoxantrone Hydrochloride Mitoxantrone, an anthracenedione, exerts its antineoplastic action by (1) intercalating into DNA and producing both strand breaks and interstrand cross-links, (2) interfering with RNA synthesis, and (3) inhibiting topoisomerase II (involved in DNA repair). The FDA approved mitoxantrone on the basis of a single (relatively small) phase 3 clinical trial in Europe, in addition to an even smaller phase 2 study completed earlier. Mitoxantrone received (from the FDA) the broadest indication of any current treatment for MS. Thus, mitoxantrone is indicated for use in SPMS, in PRMS, and in patients with worsening RRMS (defined as patients whose neurologic status remains significantly abnormal between MS attacks). Despite this broad indication, however, the data supporting its efficacy are weaker than for other approved therapies.
Mitoxantrone can be cardiotoxic (e.g., cardiomyopathy, reduced left ventricular ejection fraction, and irreversible congestive heart failure). As a result, a cumulative dose <140 mg/m2 is not recommended. At currently approved doses (12 mg/m2 every 3 months), the maximum duration of therapy can be only 2–3 years. Furthermore, >40% of women will experience amenorrhea, which may be permanent. Finally, there is risk of acute leukemia from mitoxantrone, estimated as at least a 1% lifetime risk, and this complication has been reported in several mitoxantrone-treated MS patients.
Because of these risks, and a growing list of alternative therapies, mitoxantrone is now only rarely used for MS. It should not be used as a first-line agent in either RRMS or relapsing SPMS, but might be considered in selected patients with a progressive course who have failed other therapies.
Alemtuzumab Alemtuzumab is a humanized monoclonal antibody directed against the CD52 antigen, which is expressed on both monocytes and lymphocytes. It causes lymphocyte depletion (of both B and T cells) and a change in the composition of lymphocyte subsets. Both of these changes, particularly the impact on lymphocyte subsets, are long lasting. In preliminary trials, alemtuzumab markedly reduced the attack rate and significantly improved all measures of disease severity in MS patients. In two phase 3 trials, however, its impact on clinical disability was less convincing. Notably, both trials used the active comparator of thrice-weekly, high-dose IFN-β-1a. The European and Canadian drug agencies were the first to approve this agent for use in RRMS; the FDA has also approved alemtuzumab, but only after an appeal following initial disapproval. The reasons for the initial disapproval were based on a perceived lack of a convincing disability effect and concerns over potential toxicity. The toxicities of concern were the occurrence (during the trial or thereafter) of (1) autoimmune diseases including thyroiditis, Graves’ disease, thrombocytopenia, hemolytic anemia, pancytopenia, antiglomerular basement membrane disease, and membranous glomerulonephritis; (2) malignancies including thyroid cancer, melanoma, breast cancer, and human papillomavirus (HPV)–related cancers; (3) serious infections; and (4) infusion reactions.
Initiating and Changing Treatment Previously, most patients with relapsing forms of MS received injectable agents (IFN-β or glatiramer acetate) as first-line therapy. However, with the introduction of effective and probably safe oral agents, including DMF, fingolimod, and teriflunomide, this has begun to change. In addition, the monthly infusion therapy natalizumab, which is highly effective, well tolerated, and apparently safe in JC antibody–negative patients, provides an attractive option in many cases. As noted above, with the exception of the first-generation injectable agents, long-term safety data are not available, and for the most part, comparative data are lacking. The value of combination therapy is also largely unknown, although a recent clinical trial demonstrated no added benefit to the combination of glatiramer acetate with low-dose, once-weekly IFN-β-1a.
Despite these unknowns, clinicians need to make decisions based on the best available evidence, coupled with practical considerations. One reasonable approach stratifies initial decision-making based on two levels of disease aggressiveness (Fig. 458-4).
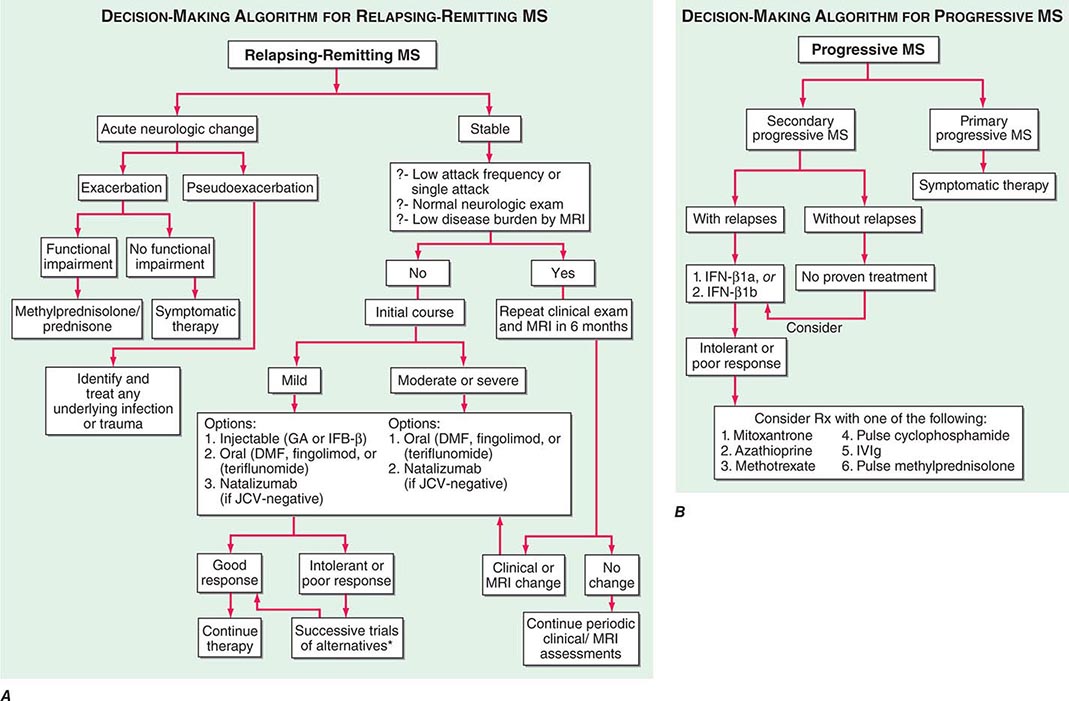
FIGURE 458-4 Therapeutic decision-making for multiple sclerosis (MS). *Can include trials of different preparations of interferon β (IFN-β, particularly advancing from once-weekly (Avonex) to a more frequent (e.g., Rebif, Betaseron/Extavia) dosing regimen. Options also include use of natalizumab in JC virus–positive patients. MRI, magnetic resonance imaging.
MILD INITIAL COURSE In the case of recent onset, normal exam or minimal impairment (EDSS ≤2.5 or less), or low disease activity, either an injectable (IFN-β or glatiramer acetate) or an oral (DMF, fingolimod, or teriflunomide) agent is reasonable. Although head-to-head comparisons are not available, natalizumab is thought to be more effective than these other agents, and therefore, this therapy can be considered even in minimally affected, JCV antibody–seronegative patients. The injectable agents (IFN-β and glatiramer acetate) have a superb track record for safety but have a high nuisance factor due to the need for frequent injections, as well as bothersome side effects that contribute to noncompliance. Some of the oral agents (DMF and fingolimod) are probably more effective than the injectables, but long-term risks are mostly unknown; DMF produces bothersome gastrointestinal symptoms in many patients at least initially (can be mitigated by beginning at one-quarter strength and gradually advancing to full dose), and fingolimod can lead to bradycardia and other cardiac disturbances of unclear clinical significance. Teriflunomide may be less effective than the other oral agents, and there are concerns about its possible long-lasting pregnancy risks. Nevertheless, its long-term safety has likely been established because of its extensive human exposure as the active metabolite of leflunomide—a drug long approved by the FDA.
MODERATE OR SEVERE INITIAL COURSE In highly active disease or moderate impairment (EDSS >2.5), either a highly effective oral agent (DMF or fingolimod) or, if the patient is JC virus antibody seronegative, infusion therapy with natalizumab is recommended.
Regardless of which agent is chosen first, treatment should probably be changed in patients who continue to have relapses, progressive neurologic impairment or, arguably, ongoing evidence of subclinical MRI activity (Fig. 458-4).
The long-term impact of these treatments on the disease course remains controversial, although several recent studies have shown that these agents improve the long-term outcome of MS including a prolongation of the time to reach certain disability outcomes (e.g., SPMS and requiring assistance to ambulate) and reduction in MS-related mortality. These benefits seem most conspicuous when treatment begins early in the RRMS stage of the illness. Unfortunately, however, already established progressive symptoms do not respond well to treatment with these disease-modifying therapies. Because progressive symptoms are likely to result from accumulated axonal and neuronal loss, many experts now believe that very early treatment with a disease-modifying drug is appropriate for most MS patients. It may also be reasonable to delay initiating treatment in patients with (1) normal neurologic exams, (2) a single attack or a low attack frequency, and (3) a low burden of disease as assessed by brain MRI. Untreated patients, however, should be followed closely with periodic brain MRI scans; the need for therapy is reassessed if scans reveal evidence of ongoing, subclinical disease. Finally, vitamin D deficiency should be corrected in all patients with MS, and generally this requires oral supplementation with vitamin D3, 4000 to 5000 IU daily.
DISEASE-MODIFYING THERAPIES FOR PROGRESSIVE MS
SPMS High-dose IFN-β probably has a beneficial effect in patients with SPMS who are still experiencing acute relapses. IFN-β is probably ineffective in patients with SPMS who are not having acute attacks. All of the other agents have not yet been studied in this patient population. Although mitoxantrone has been approved for patients with progressive MS, this is not the population studied in the pivotal trial. Therefore no evidence-based recommendation can be made with regard to its use in this setting.
PPMS No therapies have been convincingly shown to modify the course of PPMS. A phase 3 clinical trial of glatiramer acetate in PPMS was stopped because of lack of efficacy. A phase 2/3 trial of the monoclonal antibody rituximab (anti-CD20) in PPMS was also negative, but in a preplanned secondary analysis, treatment appeared to modestly slow disability progression in patients with Gd-enhancing lesions at entry; the results of a follow-up trial with a fully humanized monoclonal anti-CD20 therapy (ocrelizumab) will soon be available.
OFF-LABEL TREATMENT OPTIONS FOR RRMS AND SPMS
Azathioprine (2–3 mg/kg per day) has been used primarily in SPMS. Meta-analysis of published trials suggests that azathioprine is marginally effective at lowering relapse rates, although a benefit on disability progression has not been demonstrated.
Methotrexate (7.5–20 mg/week) was shown in one study to slow the progression of upper extremity dysfunction in SPMS. Because of the possibility of developing irreversible liver damage, some experts recommend a blind liver biopsy after 2 years of therapy.
Cyclophosphamide (700 mg/m2, every other month) may be helpful for treatment-refractory patients who are (1) otherwise in good health, (2) ambulatory, and (3) <40 years of age. Because cyclophosphamide can be used for periods in excess of 3 years, it may be preferable to mitoxantrone in these circumstances.
Intravenous immunoglobulin (IVIg), administered in monthly pulses (up to 1 g/kg) for up to 2 years, appears to reduce annual exacerbation rates. However, its use is limited because of its high cost, questions about optimal dose, and uncertainty about its having any impact on long-term disability.
Methylprednisolone, administered in one study as monthly high-dose intravenous pulses, reduced disability progression (see above).
OTHER THERAPEUTIC CLAIMS
Many purported treatments for MS have never been subjected to scientific scrutiny. These include dietary therapies (e.g., the Swank diet, in addition to others), megadose vitamins, calcium orotate, bee stings, cow colostrum, hyperbaric oxygen, procarin (a combination of histamine and caffeine), chelation, acupuncture, acupressure, various Chinese herbal remedies, and removal of mercury-amalgam tooth fillings, among many others. Patients should avoid costly or potentially hazardous unproven treatments. Many such treatments lack biologic plausibility. For example, no reliable case of mercury poisoning resembling typical MS has ever been described.
Although potential roles for EBV, human herpesvirus (HHV) 6, or chlamydia have been suggested for MS, these reports are unconfirmed, and treatment with antiviral agents or antibiotics is not recommended.
Most recently, chronic cerebrospinal insufficiency (CCSVI) has been proposed as a cause of MS, and vascular-surgical intervention is recommended. However, the failure of independent investigators to even approximate the initial claims of 100% sensitivity and 100% specificity for the diagnostic procedure have raised considerable doubt that CCSVI is a real entity. Certainly, any potentially dangerous surgery should be avoided until more rigorous science is available.
SYMPTOMATIC THERAPY
For all patients, it is useful to encourage attention to a healthy lifestyle, including maintaining an optimistic outlook, a healthy diet, and regular exercise as tolerated (swimming is often well-tolerated because of the cooling effect of cold water). It is reasonable also to correct vitamin D deficiency with oral vitamin D and to recommend dietary supplementation with long-chain (omega-3) unsaturated fatty acids (present in oily fish such as salmon), because of their biologic plausibility for MS pathogenesis, safety, and general health benefits.
Ataxia/tremor is often intractable. Clonazepam, 1.5–20 mg/d; primidone, 50–250 mg/d; propranolol, 40–200 mg/d; or ondansetron, 8–16 mg/d, may help. Wrist weights occasionally reduce tremor in the arm or hand. Thalamotomy or deep-brain stimulation has been tried with mixed success.
Spasticity and spasms may improve with physical therapy, regular exercise, and stretching. Avoidance of triggers (e.g., infections, fecal impactions, bed sores) is extremely important. Effective medications include baclofen (20–120 mg/d), diazepam (2–40 mg/d), tizanidine (8–32 mg/d), dantrolene (25–400 mg/d), and cyclobenzaprine hydrochloride (10–60 mg/d). For severe spasticity, a baclofen pump (delivering medication directly into the CSF) can provide substantial relief.
Weakness can sometimes be improved with the use of potassium channel blockers such as 4-aminopyridine (10–40 mg/d) and 3,4-di-aminopyridine (40–80 mg/d), particularly in the setting where lower extremity weakness interferes with the patient’s ability to ambulate. The FDA has approved 4-aminopyridine (at 20 mg/d), and this can be obtained either as dalfampridine (Ampyra) or, more cheaply, through a compounding pharmacy. The principle concern with the use of these agents is the possibility of inducing seizures at high doses.
Pain is treated with anticonvulsants (carbamazepine, 100–1000 mg/d; phenytoin, 300–600 mg/d; gabapentin, 300–3600 mg/d; or pregabalin, 50–300 mg/d), antidepressants (amitriptyline, 25–150 mg/d; nortriptyline, 25–150 mg/d; desipramine, 100–300 mg/d; or venlafaxine, 75–225 mg/d), or antiarrhythmics (mexiletine, 300–900 mg/d). If these approaches fail, patients should be referred to a comprehensive pain management program.
Bladder dysfunction management is best guided by urodynamic testing. Evening fluid restriction or frequent voluntary voiding may help detrusor hyperreflexia. If these methods fail, propantheline bromide (10–15 mg/d), oxybutynin (5–15 mg/d), hyoscyamine sulfate (0.5–0.75 mg/d), tolterodine tartrate (2–4 mg/d), or solifenacin (5–10 mg/d) may help. Coadministration of pseudoephedrine (30–60 mg) is sometimes beneficial.
Detrusor/sphincter dyssynergia may respond to phenoxybenzamine (10–20 mg/d) or terazosin hydrochloride (1–20 mg/d). Loss of reflex bladder wall contraction may respond to bethanechol (30–150 mg/d). However, both conditions often require catheterization.
Urinary tract infections should be treated promptly. Patients with large postvoid residual urine volumes are predisposed to infections. Prevention by urine acidification (with cranberry juice or vitamin C) inhibits some bacteria. Prophylactic administration of antibiotics is sometimes necessary but may lead to colonization by resistant organisms. Intermittent catheterization may help to prevent recurrent infections.
Treatment of constipation includes high-fiber diets and fluids. Natural or other laxatives may help. Fecal incontinence may respond to a reduction in dietary fiber.
Depression should be treated. Useful drugs include the selective serotonin reuptake inhibitors (fluoxetine, 20–80 mg/d, or sertraline, 50–200 mg/d), the tricyclic antidepressants (amitriptyline, 25–150 mg/d; nortriptyline, 25–150 mg/d; or desipramine, 100–300 mg/d), and the nontricyclic antidepressants (venlafaxine, 75–225 mg/d).
Fatigue may improve with assistive devices, help in the home, or successful management of spasticity. Patients with frequent nocturia may benefit from anticholinergic medication at bedtime. Primary MS fatigue may respond to amantadine (200 mg/d), methylphenidate (5–25 mg/d), or modafinil (100–400 mg/d).
Cognitive problems may respond to the cholinesterase inhibitor donepezil hydrochloride (10 mg/d).
Paroxysmal symptoms respond dramatically to low-dose anticonvulsants (acetazolamide, 200–600 mg/d; carbamazepine, 50–400 mg/d; phenytoin, 50–300 mg/d; or gabapentin, 600–1800 mg/d).
Heat sensitivity may respond to heat avoidance, air-conditioning, or cooling garments.
Sexual dysfunction may be helped by lubricants to aid in genital stimulation and sexual arousal. Management of pain, spasticity, fatigue, and bladder/bowel dysfunction may also help. Sildenafil (50–100 mg), tadalafil (5–20 mg), or vardenafil (5–20 mg), taken 1–2 h before sex, is now the standard treatment for maintaining erections.
PROMISING EXPERIMENTAL THERAPIES
Numerous clinical trials are currently under way. These include studies on (1) monoclonal antibodies against CD20 to deplete B cells and against the IL-2 receptor; (2) selective oral sphingosine-1-phosphate receptor antagonists to sequester lymphocytes in secondary lymphoid organs; (3) estriol to induce a pregnancy-like state; (4) molecules to promote remyelination; and (4) bone marrow transplantation.
CLINICAL VARIANTS OF MS
Acute MS (Marburg’s variant) is a fulminant demyelinating process that in some cases progresses inexorably to death within 1–2 years. Typically, there are no remissions. When acute MS presents as a solitary, usually cavitary, lesion, a brain tumor is often suspected. In such cases, a brain biopsy is usually required to establish the diagnosis. An antibody-mediated process appears to be responsible for most cases. Marburg’s variant does not seem to follow infection or vaccination, and it is unclear whether this syndrome represents an extreme form of MS or another disease altogether. No controlled trials of therapy exist; high-dose glucocorticoids, plasma exchange, and cyclophosphamide have been tried, with uncertain benefit.
NEUROMYELITIS OPTICA
Neuromyelitis optica (NMO; Devic’s disease) is an aggressive inflammatory disorder characterized by recurrent attacks of ON and myelitis (Table 458-7). NMO is more frequent in women than men (>3:1), typically begins in childhood or early adulthood but can arise at any age, and is uncommon in whites compared with individuals of Asian and African ancestry. Attacks of ON can be bilateral (rare in MS) or unilateral; myelitis can be severe and transverse (rare in MS) and is typically longitudinally extensive, involving three or more contiguous vertebral segments. Also in contrast to MS, progressive symptoms do not occur in NMO. The brain MRI was earlier thought to be normal in NMO, but it is now recognized that in approximately half of cases, there are lesions involving the hypothalamus causing an endocrinopathy; the lower brainstem presenting as intractable hiccoughs or vomiting from involvement of the area postrema in the lower medulla; or the cerebral hemispheres producing focal symptoms, encephalopathy, or seizures. Large MRI lesions in the cerebral hemispheres can be asymptomatic, sometimes have a “cloud-like” appearance and, unlike MS lesions, are often not destructive, and can resolve completely. Spinal cord MRI lesions typically consist of focal enhancing areas of swelling and tissue destruction, extending over three or more spinal cord segments, and on axial sequences, these are centered on the gray matter of the cord. CSF findings include pleocytosis greater than that observed in MS, with neutrophils and eosinophils present in some cases; OCBs are uncommon, occurring in fewer than 30% of NMO patients. The pathology of NMO is a distinctive astrocytopathy with inflammation, a loss of astrocytes, and an absence of staining of the water channel protein aquaporin-4 by immunohistochemistry, plus thickened blood vessel walls, demyelination, and deposition of antibody and complement.
|
DIAGNOSTIC CRITERIA FOR NEUROMYELITIS OPTICA |
Source: Adapted from DM Wingerchuk et al: Neurology 66:1485, 2006.
NMO is best understood as a syndrome with diverse causes. Up to 40% of patients have a systemic autoimmune disorder, often systemic lupus erythematosus, Sjögren’s syndrome, perinuclear antineutrophil cytoplasmic antibody (p-ANCA)–associated vasculitis, myasthenia gravis, Hashimoto’s thyroiditis, or mixed connective tissue disease. In others, onset may be associated with acute infection with VZV, EBV, HIV, or tuberculosis. Rare cases appear to be paraneoplastic and associated with breast, lung, or other cancers. NMO is often idiopathic, however. NMO is usually disabling over time; in one series, respiratory failure from cervical myelitis was present in one-third of patients, and 8 years after onset, 60% of patients were blind and more than half had permanent paralysis of one or more limbs.
A highly specific autoantibody directed against aquaporin-4 is present in the sera of approximately two-thirds of patients with a clinical diagnosis of NMO. Seropositive patients have a very high risk for future relapses; more than half will relapse within 1 year if untreated. Aquaporin-4 is localized to the foot processes of astrocytes in close apposition to endothelial surfaces, as well as at paranodal regions near nodes of Ranvier. It is likely that aquaporin-4 antibodies are pathogenic, as passive transfer of antibodies from NMO patients into laboratory animals reproduce histologic features of the disease.
When MS affects individuals of African or Asian ancestry, there is a propensity for demyelinating lesions to involve predominantly the optic nerve and spinal cord, an MS subtype termed opticospinal MS. Interestingly, some individuals with opticospinal MS are seropositive for aquaporin-4 antibodies, suggesting that such cases represent an NMO spectrum disorder.
ACUTE DISSEMINATED ENCEPHALOMYELITIS
Acute disseminated encephalomyelitis (ADEM) has a monophasic course and is most frequently associated with an antecedent infection (postinfectious encephalomyelitis); approximately 5% of ADEM cases follow immunization (postvaccinal encephalomyelitis). ADEM is far more common in children than adults, and many adult cases initially thought to represent ADEM subsequently experience late relapses qualifying as either MS or another chronic inflammatory disorder such as vasculitis, sarcoid, or lymphoma. The hallmark of ADEM is the presence of widely scattered small foci of perivenular inflammation and demyelination, in contrast to larger confluent demyelinating lesions typical of MS. In the most explosive form of ADEM, acute hemorrhagic leukoencephalitis, the lesions are vasculitic and hemorrhagic, and the clinical course is devastating.
Postinfectious encephalomyelitis is most frequently associated with the viral exanthems of childhood. Infection with measles virus is the most common antecedent (1 in 1000 cases). Worldwide, measles encephalomyelitis is still common, although use of the live measles vaccine has dramatically reduced its incidence in developed countries. An ADEM-like illness rarely follows vaccination with live measles vaccine (1–2 in 106 immunizations). ADEM is now most frequently associated with varicella (chickenpox) infections (1 in 4000–10,000 cases). It may also follow infection with rubella, mumps, influenza, parainfluenza, EBV, HHV-6, HIV, other viruses, and Mycoplasma pneumoniae. Some patients may have a nonspecific upper respiratory infection or no known antecedent illness. In addition to measles, postvaccinal encephalomyelitis may also follow the administration of vaccines for smallpox (5 cases per million), the Semple rabies, and Japanese encephalitis. Modern vaccines that do not require viral culture in CNS tissue have reduced the ADEM risk.
All forms of ADEM presumably result from a cross-reactive immune response to the infectious agent or vaccine that then triggers an inflammatory demyelinating response. Autoantibodies to MBP and to other myelin antigens have been detected in the CSF from some patients with ADEM. Attempts to demonstrate direct viral invasion of the CNS have been unsuccessful.
CLINICAL MANIFESTATIONS
In severe cases, onset is abrupt and progression rapid (hours to days). In postinfectious ADEM, the neurologic syndrome generally begins late in the course of the viral illness as the exanthem is fading. Fever reappears, and headache, meningismus, and lethargy progressing to coma may develop. Seizures are common. Signs of disseminated neurologic disease are consistently present (e.g., hemiparesis or quadriparesis, extensor plantar responses, lost or hyperactive tendon reflexes, sensory loss, and brainstem involvement). In ADEM due to chickenpox, cerebellar involvement is often conspicuous. CSF protein is modestly elevated (0.5–1.5 g/L [50–150 mg/dL]). Lymphocytic pleocytosis, generally 200 cells/μL or greater, occurs in 80% of patients. Occasional patients have higher counts or a mixed polymorphonuclear-lymphocytic pattern during the initial days of the illness. Transient CSF oligoclonal banding has been reported. MRI usually reveals extensive changes in the brain and spinal cord, consisting of white matter hyperintensities on T2 and fluid-attenuated inversion recovery sequences with Gd enhancement on T1-weighted sequences.
DIAGNOSIS
The diagnosis is most reliably established when there is a history of recent vaccination or viral exanthematous illness. In severe cases with predominantly cerebral involvement, acute encephalitis due to infection with herpes simplex or other viruses including HIV may be difficult to exclude (Chap. 164); other considerations include hypercoagulable states including the antiphospholipid antibody syndrome, vasculitis, neurosarcoid, primary CNS lymphoma, or metastatic cancer. An explosive presentation of MS can mimic ADEM, and especially in adults, it may not be possible to distinguish these conditions at onset. The simultaneous onset of disseminated symptoms and signs is common in ADEM and rare in MS. Similarly, meningismus, drowsiness, coma, and seizures suggest ADEM rather than MS. Unlike MS, in ADEM, optic nerve involvement is generally bilateral and transverse myelopathy complete. MRI findings that favor ADEM include extensive and relatively symmetric white matter abnormalities, basal ganglia or cortical gray matter lesions, and Gd enhancement of all abnormal areas. By contrast, OCBs in the CSF are more common in MS. In one study of adult patients initially thought to have ADEM, 30% experienced additional relapses over a follow-up period of 3 years, and they were reclassified as having MS. Occasional patients with “recurrent ADEM” have also been reported, especially children; however, it is not possible to distinguish this entity from atypical MS.




