[level-membership-for-internal-medicine-category]
369 |
Diseases of the Gallbladder and Bile Ducts |
PHYSIOLOGY OF BILE PRODUCTION AND FLOW
BILE SECRETION AND COMPOSITION
Bile formed in the hepatic lobules is secreted into a complex network of canaliculi, small bile ductules, and larger bile ducts that run with lymphatics and branches of the portal vein and hepatic artery in portal tracts situated between hepatic lobules. These interlobular bile ducts coalesce to form larger septal bile ducts that join to form the right and left hepatic ducts, which in turn, unite to form the common hepatic duct. The common hepatic duct is joined by the cystic duct of the gallbladder to form the common bile duct (CBD), which enters the duodenum (often after joining the main pancreatic duct) through the ampulla of Vater.
Hepatic bile is an isotonic fluid with an electrolyte composition resembling blood plasma. The electrolyte composition of gallbladder bile differs from that of hepatic bile because most of the inorganic anions, chloride and bicarbonate, have been removed by reabsorption across the gallbladder epithelium. As a result of water reabsorption, total solute concentration of bile increases from 3–4 g/dL in hepatic bile to 10–15 g/dL in gallbladder bile.
Major solute components of bile by moles percent include bile acids (80%), lecithin and traces of other phospholipids (16%), and unesterified cholesterol (4.0%). In the lithogenic state, the cholesterol value can be as high as 8–10%. Other constituents include conjugated bilirubin; proteins (all immunoglobulins, albumin, metabolites of hormones, and other proteins metabolized in the liver); electrolytes; mucus; and, often, drugs and their metabolites.
The total daily basal secretion of hepatic bile is ~500–600 mL. Many substances taken up or synthesized by the hepatocyte are secreted into the bile canaliculi. The canalicular membrane forms microvilli and is associated with microfilaments of actin, microtubules, and other contractile elements. Prior to their secretion into the bile, many substances are taken up into the hepatocyte, while others, such as phospholipids, a portion of primary bile acids, and some cholesterol, are synthesized de novo in the hepatocyte. Three mechanisms are important in regulating bile flow: (1) active transport of bile acids from hepatocytes into the bile canaliculi, (2) active transport of other organic anions, and (3) cholangiocellular secretion. The last is a secretin-mediated and cyclic AMP–dependent mechanism that results in the secretion of a sodium- and bicarbonate-rich fluid into the bile ducts.
Active vectorial secretion of biliary constituents from the portal blood into the bile canaliculi is driven by a set of polarized transport systems at the basolateral (sinusoidal) and the canalicular apical plasma membrane domains of the hepatocyte. Two sinusoidal bile salt uptake systems have been cloned in humans, the Na+/taurocholate cotransporter (NTCP, SLC10A1) and the organic anion–transporting proteins (OATPs), which also transport a large variety of non-bile salt organic anions. Several ATP-dependent canalicular transport systems, “export pumps,” (ATP-binding cassette transport proteins, also known as ABC transporters) have been identified, the most important of which are: the bile salt export pump (BSEP, ABCB11); the anionic conjugate export pump (MRP2, ABCC2), which mediates the canalicular excretion of various amphiphilic conjugates formed by phase II conjugation (e.g., bilirubin mono- and diglucuronides and drugs); the multidrug export pump (MDR1, ABCB1) for hydrophobic cationic compounds; and the phospholipid export pump (MDR3, ABCB4). Two hemitransporters ABCG5/G8, functioning as a couple, constitute the canalicular cholesterol and phytosterol transporter. F1C1 (ATP8B1) is an aminophospholipid transferase (“flippase”) essential for maintaining the lipid asymmetry of the canalicular membrane. The canalicular membrane also contains ATP-independent transport systems such as the Cl/HCO3 anion exchanger isoform 2 (AE2, SLC4A2) for canalicular bicarbonate secretion. For most of these transporters, genetic defects have been identified that are associated with various forms of cholestasis or defects of biliary excretion. F1C1 is defective in progressive familial intrahepatic cholestasis type 1 (PFIC1) and benign recurrent intrahepatic cholestasis type 1 (BRIC1) and results in ablation of all other ATP-dependent transporter functions. BSEP is defective in PFIC2 and BRIC2. Mutations of MRP2 (ABCC2) cause the Dubin-Johnson syndrome, an inherited form of conjugated hyperbilirubinemia (Chap. 359). A defective MDR3 (ABCB4) results in PFIC3. ABCG5/G8, the canalicular half transporters for cholesterol and other neutral sterols, are defective in sitosterolemia. The cystic fibrosis transmembrane regulator (CFTR, ABCC7) located on bile duct epithelial cells but not on canalicular membranes is defective in cystic fibrosis, which is associated with impaired cholangiocellular pH regulation during ductular bile formation and chronic cholestatic liver disease, occasionally resulting in biliary cirrhosis.
THE BILE ACIDS
The primary bile acids, cholic acid and chenodeoxycholic acid (CDCA), are synthesized from cholesterol in the liver, conjugated with glycine or taurine, and secreted into the bile. Secondary bile acids, including deoxycholate and lithocholate, are formed in the colon as bacterial metabolites of the primary bile acids. However, lithocholic acid is much less efficiently absorbed from the colon than deoxycholic acid. Another secondary bile acid, found in low concentration, is ursodeoxycholic acid (UDCA), a stereoisomer of CDCA. In healthy subjects, the ratio of glycine to taurine conjugates in bile is ~3:1.
Bile acids are detergent-like molecules that in aqueous solutions and above a critical concentration of about 2 mM form molecular aggregates called micelles. Cholesterol alone is sparingly soluble in aqueous environments, and its solubility in bile depends on both the total lipid concentration and the relative molar percentages of bile acids and lecithin. Normal ratios of these constituents favor the formation of solubilizing mixed micelles, while abnormal ratios promote the precipitation of cholesterol crystals in bile via an intermediate liquid crystal phase.
In addition to facilitating the biliary excretion of cholesterol, bile acids facilitate the normal intestinal absorption of dietary fats, mainly cholesterol and fat-soluble vitamins, via a micellar transport mechanism (Chap. 349). Bile acids also serve as a major physiologic driving force for hepatic bile flow and aid in water and electrolyte transport in the small bowel and colon.
ENTEROHEPATIC CIRCULATION
Bile acids are efficiently conserved under normal conditions. Unconjugated, and to a lesser degree also conjugated, bile acids are absorbed by passive diffusion along the entire gut. Quantitatively much more important for bile salt recirculation, however, is the active transport mechanism for conjugated bile acids in the distal ileum (Chap. 349). The reabsorbed bile acids enter the portal bloodstream and are taken up rapidly by hepatocytes, reconjugated, and resecreted into bile (enterohepatic circulation).
The normal bile acid pool size is approximately 2–4 g. During digestion of a meal, the bile acid pool undergoes at least one or more enterohepatic cycles, depending on the size and composition of the meal. Normally, the bile acid pool circulates ~5–10 times daily. Intestinal reabsorption of the pool is about 95% efficient; therefore, fecal loss of bile acids is in the range of 0.2–0.4 g/d. In the steady state, this fecal loss is compensated by an equal daily synthesis of bile acids by the liver, and, thus, the size of the bile acid pool is maintained. Bile acids in the intestine release fibroblast growth factor 19 (FGF19) into the circulation, which is transported to the liver where it suppresses synthesis of bile acids from cholesterol by inhibiting the rate-limiting enzyme cytochrome P450 7A1 (CYP7A1) and also promotes gallbladder relaxation. While the loss of bile salts in stool is usually matched by increased hepatic synthesis, the maximum rate of synthesis is ~5 g/d, which may be insufficient to replete the bile acid pool size when there is pronounced impairment of intestinal bile salt reabsorption.
The expression of ABC transporters in the enterohepatic circulation and of the rate-limiting enzymes of bile acid and cholesterol synthesis are regulated in a coordinated fashion by nuclear receptors, which are ligand-activated transcription factors. The hepatic BSEP (ABCB11) is upregulated by the farnesoid × receptor (FXR), a bile acid sensor that also represses bile acid synthesis. The expression of the cholesterol transporter, ABCG5/G8, is upregulated by the liver × receptor (LXR), which is an oxysterol sensor.
GALLBLADDER AND SPHINCTERIC FUNCTIONS
In the fasting state, the sphincter of Oddi offers a high-pressure zone of resistance to bile flow from the CBD into the duodenum. Its tonic contraction serves to (1) prevent reflux of duodenal contents into the pancreatic and bile ducts and (2) promote filling of the gallbladder. The major factor controlling the evacuation of the gallbladder is the peptide hormone cholecystokinin (CCK), which is released from the duodenal mucosa in response to the ingestion of fats and amino acids. CCK produces (1) powerful contraction of the gallbladder, (2) decreased resistance of the sphincter of Oddi, and (3) enhanced flow of biliary contents into the duodenum.
Hepatic bile is “concentrated” within the gallbladder by energy-dependent transmucosal absorption of water and electrolytes. Almost the entire bile acid pool may be sequestered in the gallbladder following an overnight fast for delivery into the duodenum with the first meal of the day. The normal capacity of the gallbladder is ~30 mL of bile.
DISEASES OF THE GALLBLADDER
CONGENITAL ANOMALIES
Anomalies of the biliary tract are not uncommon and include abnormalities in number, size, and shape (e.g., agenesis of the gallbladder, duplications, rudimentary or oversized “giant” gallbladders, and diverticula). Phrygian cap is a clinically innocuous entity in which a partial or complete septum (or fold) separates the fundus from the body. Anomalies of position or suspension are not uncommon and include left-sided gallbladder, intrahepatic gallbladder, retrodisplacement of the gallbladder, and “floating” gallbladder. The latter condition predisposes to acute torsion, volvulus, or herniation of the gallbladder.
GALLSTONES
Epidemiology and Pathogenesis Gallstones are quite prevalent in most Western countries. Gallstone formation increases after age 50. In the United States, the third National Health and Nutrition Examination Survey (NHANES III) has revealed an overall prevalence of gallstones of 7.9% in men and 16.6% in women. The prevalence was high in Mexican Americans (8.9% in men, 26.7% in women), intermediate for non-Hispanic whites (8.6% in men, 16.6% in women), and low for African Americans (5.3% in men, 13.9% in women).
Gallstones are formed because of abnormal bile composition. They are divided into two major types: cholesterol stones and pigment stones. Cholesterol stones account for more than 90% of all gallstones in Western industrialized countries. Cholesterol gallstones usually contain >50% cholesterol monohydrate plus an admixture of calcium salts, bile pigments, proteins, and fatty acids. Pigment stones are composed primarily of calcium bilirubinate; they contain <20% cholesterol and are classified into “black” and “brown” types, the latter forming secondary to chronic biliary infection.
CHOLESTEROL STONES AND BILIARY SLUDGE Cholesterol is essentially water insoluble and requires aqueous dispersion into either micelles or vesicles, both of which require the presence of a second lipid to solubilize the cholesterol. Cholesterol and phospholipids are secreted into bile as unilamellar bilayered vesicles, which are converted into mixed micelles consisting of bile acids, phospholipids, and cholesterol by the action of bile acids. If there is an excess of cholesterol in relation to phospholipids and bile acids, unstable, cholesterol-rich vesicles remain, which aggregate into large multilamellar vesicles from which cholesterol crystals precipitate (Fig. 369-1).
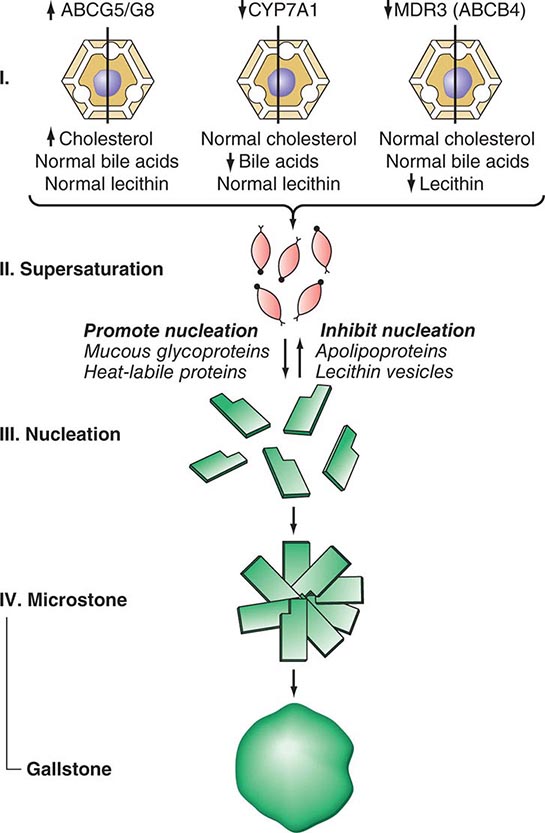
FIGURE 369-1 Scheme showing pathogenesis of cholesterol gallstone formation. Conditions or factors that increase the ratio of cholesterol to bile acids and phospholipids (lecithin) favor gallstone formation. ABCB4, ATP-binding cassette transporter; ABCG5/8, ATP-binding cassette (ABC) transporter G5/G8; CYP7A1, cytochrome P450 7A1; MDR3, multidrug resistance protein 3, also called phospholipid export pump.
There are several important mechanisms in the formation of lithogenic (stone-forming) bile. The most important is increased biliary secretion of cholesterol. This may occur in association with obesity, the metabolic syndrome, high-caloric and cholesterol-rich diets, or drugs (e.g., clofibrate) and may result from increased activity of hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme of hepatic cholesterol synthesis, and increased hepatic uptake of cholesterol from blood. In patients with gallstones, dietary cholesterol increases biliary cholesterol secretion. This does not occur in non-gallstone patients on high-cholesterol diets. In addition to environmental factors such as high-caloric and cholesterol-rich diets, genetic factors play an important role in gallstone disease. A large study of symptomatic gallstones in Swedish twins provided strong evidence for a role of genetic factors in gallstone pathogenesis. Genetic factors accounted for 25%, shared environmental factors for 13%, and individual environmental factors for 62% of the phenotypic variation among monozygotic twins. A single nucleotide polymorphism of the gene encoding the hepatic cholesterol transporter ABCG5/G8 has been found in 21% of patients with gallstones, but only in 9% of the general population. It is thought to cause a gain of function of the cholesterol transporter and to contribute to cholesterol hypersecretion. A high prevalence of gallstones is found among first-degree relatives of gallstone carriers and in certain ethnic populations such as American Indians, Chilean Indians, and Chilean Hispanics. A common genetic trait has been identified for some of these populations by mitochondrial DNA analysis. In some patients, impaired hepatic conversion of cholesterol to bile acids may also occur, resulting in an increase of the lithogenic cholesterol/bile acid ratio. Although most cholesterol stones have a polygenic basis, there are rare monogenic (Mendelian) causes. Recently, a mutation in the CYP7A1 gene has been described that results in a deficiency of the enzyme cholesterol 7-hydroxylase, which catalyzes the initial step in cholesterol catabolism and bile acid synthesis. The homozygous state is associated with hypercholesterolemia and gallstones. Because the phenotype is expressed in the heterozygote state, mutations in the CYP7A1 gene may contribute to the susceptibility to cholesterol gallstone disease in the population. Mutations in the MDR3 (ABCB4) gene, which encodes the phospholipid export pump in the canalicular membrane of the hepatocyte, may cause defective phospholipid secretion into bile, resulting in cholesterol supersaturation of bile and formation of cholesterol gallstones in the gallbladder and in the bile ducts. Thus, an excess of biliary cholesterol in relation to bile acids and phospholipids is primarily due to hypersecretion of cholesterol, but hyposecretion of bile acids or phospholipids may contribute. An additional disturbance of bile acid metabolism that is likely to contribute to supersaturation of bile with cholesterol is enhanced conversion of cholic acid to deoxycholic acid, with replacement of the cholic acid pool by an expanded deoxycholic acid pool. It may result from enhanced dehydroxylation of cholic acid and increased absorption of newly formed deoxycholic acid. An increased deoxycholate secretion is associated with hypersecretion of cholesterol into bile.
While supersaturation of bile with cholesterol is an important prerequisite for gallstone formation, it is generally not sufficient by itself to produce cholesterol precipitation in vivo. Most individuals with supersaturated bile do not develop stones because the time required for cholesterol crystals to nucleate and grow is longer than the time bile remains in the gallbladder.
An important mechanism is nucleation of cholesterol monohydrate crystals, which is greatly accelerated in human lithogenic bile. Accelerated nucleation of cholesterol monohydrate in bile may be due to either an excess of pronucleating factors or a deficiency of antinucleating factors. Mucin and certain nonmucin glycoproteins, principally immunoglobulins, appear to be pronucleating factors, while apolipoproteins A-I and A-II and other glycoproteins appear to be antinucleating factors. Pigment particles may possibly play a role as nucleating factors. In a genome-wide analysis of serum bilirubin levels, the uridine diphosphate-glucuronyltransferase 1A1 (UGT1A1) Gilbert’s syndrome gene variant was associated with the presence of gallstone disease. Because most gallstones associated with the UGT1A1 variant were cholesterol stones, this finding points to the role of pigment particles in the pathogenesis of gallbladder stones. Cholesterol monohydrate crystal nucleation and crystal growth probably occur within the mucin gel layer. Vesicle fusion leads to liquid crystals, which, in turn, nucleate into solid cholesterol monohydrate crystals. Continued growth of the crystals occurs by direct nucleation of cholesterol molecules from supersaturated unilamellar or multilamellar biliary vesicles.
A third important mechanism in cholesterol gallstone formation is gallbladder hypomotility. If the gallbladder emptied all supersaturated or crystal-containing bile completely, stones would not be able to grow. A high percentage of patients with gallstones exhibit abnormalities of gallbladder emptying. Ultrasonographic studies show that gallstone patients display an increased gallbladder volume during fasting and also after a test meal (residual volume) and that fractional emptying after gallbladder stimulation is decreased. The incidence of gallstones is increased in conditions associated with infrequent or impaired gallbladder emptying such as fasting, parenteral nutrition, or pregnancy and in patients using drugs that inhibit gallbladder motility.
Biliary sludge is a thick, mucous material that, upon microscopic examination, reveals lecithin-cholesterol liquid crystals, cholesterol monohydrate crystals, calcium bilirubinate, and mucin gels. Biliary sludge typically forms a crescent-like layer in the most dependent portion of the gallbladder and is recognized by characteristic echoes on ultrasonography (see below). The presence of biliary sludge implies two abnormalities: (1) the normal balance between gallbladder mucin secretion and elimination has become deranged, and (2) nucleation of biliary solutes has occurred. That biliary sludge may be a precursor form of gallstone disease is evident from several observations. In one study, 96 patients with gallbladder sludge were followed prospectively by serial ultrasound studies. In 18%, biliary sludge disappeared and did not recur for at least 2 years. In 60%, biliary sludge disappeared and reappeared; in 14%, gallstones (8% asymptomatic, 6% symptomatic) developed; and in 6%, severe biliary pain with or without acute pancreatitis occurred. In 12 patients, cholecystectomies were performed, 6 for gallstone-associated biliary pain and 3 in symptomatic patients with sludge but without gallstones who had prior attacks of pancreatitis; the latter did not recur after cholecystectomy. It should be emphasized that biliary sludge can develop with disorders that cause gallbladder hypomotility; i.e., surgery, burns, total parenteral nutrition, pregnancy, and oral contraceptives—all of which are associated with gallstone formation. However, the presence of biliary sludge implies supersaturation of bile with either cholesterol or calcium bilirubinate.
Two other conditions are associated with cholesterol-stone or biliary-sludge formation: pregnancy and rapid weight reduction through a very-low-calorie diet. There appear to be two key changes during pregnancy that contribute to a “cholelithogenic state”: (1) a marked increase in cholesterol saturation of bile during the third trimester and (2) sluggish gallbladder contraction in response to a standard meal, resulting in impaired gallbladder emptying. That these changes are related to pregnancy per se is supported by several studies that show reversal of these abnormalities quite rapidly after delivery. During pregnancy, gallbladder sludge develops in 20–30% of women and gallstones in 5–12%. Although biliary sludge is a common finding during pregnancy, it is usually asymptomatic and often resolves spontaneously after delivery. Gallstones, which are less common than sludge and frequently associated with biliary colic, may also disappear after delivery because of spontaneous dissolution related to bile becoming unsaturated with cholesterol postpartum.
Approximately 10–20% of persons with rapid weight reduction achieved through very-low-calorie dieting develop gallstones. In a study involving 600 patients who completed a 3-month, 520-kcal/d diet, UDCA in a dosage of 600 mg/d proved highly effective in preventing gallstone formation; gallstones developed in only 3% of UDCA recipients, compared to 28% of placebo-treated patients. In obese patients treated by gastric banding, 500 mg/d of UDCA reduced the risk of gallstone formation from 30% to 8% within a follow-up of 6 months.
To summarize, cholesterol gallstone disease occurs because of several defects, which include (1) bile supersaturation with cholesterol, (2) nucleation of cholesterol monohydrate with subsequent crystal retention and stone growth, and (3) abnormal gallbladder motor function with delayed emptying and stasis. Other important factors known to predispose to cholesterol-stone formation are summarized in Table 369-1.
|
PREDISPOSING FACTORS FOR CHOLESTEROL AND PIGMENT GALLSTONE FORMATION |
PIGMENT STONES Black pigment stones are composed of either pure calcium bilirubinate or polymer-like complexes with calcium and mucin glycoproteins. They are more common in patients who have chronic hemolytic states (with increased conjugated bilirubin in bile), liver cirrhosis, Gilbert’s syndrome, or cystic fibrosis. Gallbladder stones in patients with ileal diseases, ileal resection, or ileal bypass generally are also black pigment stones. Enterohepatic recycling of bilirubin in ileal disease states contributes to their pathogenesis. Brown pigment stones are composed of calcium salts of unconjugated bilirubin with varying amounts of cholesterol and protein. They are caused by the presence of increased amounts of unconjugated, insoluble bilirubin in bile that precipitates to form stones. Deconjugation of an excess of soluble bilirubin mono- and diglucuronides may be mediated by endogenous β-glucuronidase but may also occur by spontaneous hydrolysis. Sometimes, the enzyme is also produced when bile is chronically infected by bacteria, and such stones are brown. Pigment stone formation is frequent in Asia and is often associated with infections in the gallbladder and biliary tree (Table 369-1).
Diagnosis Procedures of potential use in the diagnosis of cholelithiasis and other diseases of the gallbladder are detailed in Table 369-2. Ultrasonography of the gallbladder is very accurate in the identification of cholelithiasis and has replaced oral cholecystography (Fig. 369-2A). Stones as small as 1.5 mm in diameter may be confidently identified provided that firm criteria are used (e.g., acoustic “shadowing” of opacities that are within the gallbladder lumen and that change with the patient’s position [by gravity]). In major medical centers, the false-negative and false-positive rates for ultrasound in gallstone patients are ~2–4%. Biliary sludge is material of low echogenic activity that typically forms a layer in the most dependent position of the gallbladder. This layer shifts with postural changes but fails to produce acoustic shadowing; these two characteristics distinguish sludges from gallstones. Ultrasound can also be used to assess the emptying function of the gallbladder.
|
DIAGNOSTIC EVALUATION OF THE GALLBLADDER |
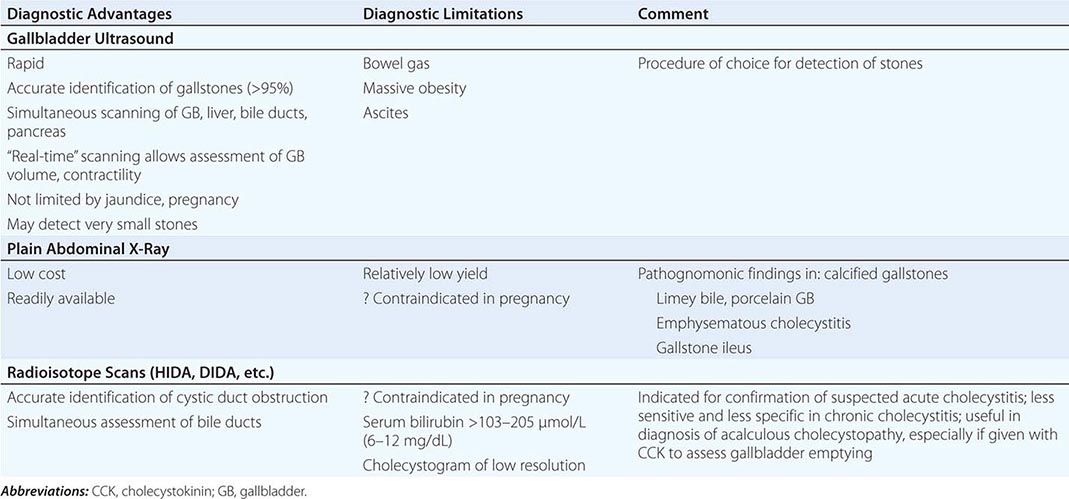
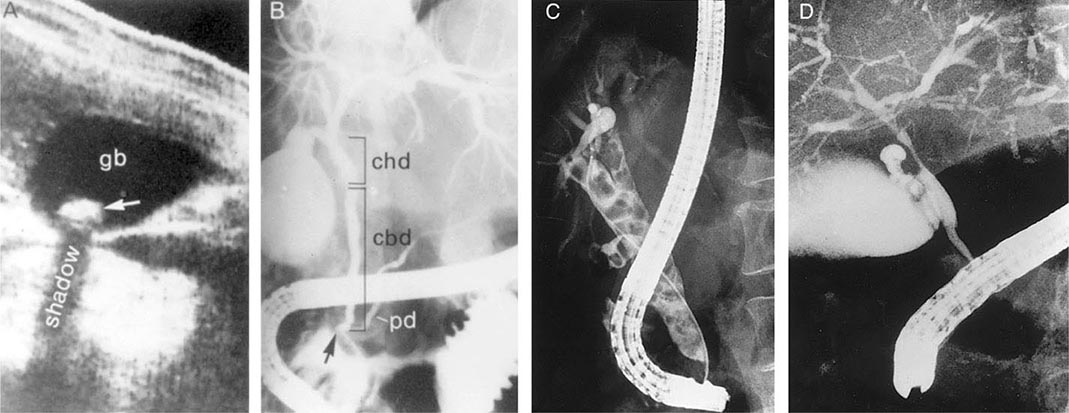
FIGURE 369-2 Examples of ultrasound and radiologic studies of the biliary tract. A. An ultrasound study showing a distended gallbladder (GB) containing a single large stone (arrow), which casts an acoustic shadow. B. Endoscopic retrograde cholangiopancreatogram (ERCP) showing normal biliary tract anatomy. In addition to the endoscope and large vertical gallbladder filled with contrast dye, the common hepatic duct (CHD), common bile duct (CBD), and pancreatic duct (PD) are shown. The arrow points to the ampulla of Vater. C. Endoscopic retrograde cholangiogram (ERC) showing choledocholithiasis. The biliary tract is dilated and contains multiple radiolucent calculi. D. ERCP showing sclerosing cholangitis. The common bile duct shows areas that are strictured and narrowed.
The plain abdominal film may detect gallstones containing sufficient calcium to be radiopaque (10–15% of cholesterol and ~50% of pigment stones). Plain radiography may also be of use in the diagnosis of emphysematous cholecystitis, porcelain gallbladder, limey bile, and gallstone ileus.
Oral cholecystography (OCG) has historically been a useful procedure for the diagnosis of gallstones but has been replaced by ultrasound and is regarded as obsolete. It may be used to assess the patency of the cystic duct and gallbladder emptying function. Further, OCG can also delineate the size and number of gallstones and determine whether they are calcified.
Radiopharmaceuticals such as 99mTc-labeled N-substituted iminodiacetic acids (HIDA, DIDA, DISIDA, etc.) are rapidly extracted from the blood and are excreted into the biliary tree in high concentration even in the presence of mild to moderate serum bilirubin elevations. Failure to image the gallbladder in the presence of biliary ductal visualization may indicate cystic duct obstruction, acute or chronic cholecystitis, or surgical absence of the organ. Such scans have some application in the diagnosis of acute cholecystitis.
Symptoms of Gallstone Disease Gallstones usually produce symptoms by causing inflammation or obstruction following their migration into the cystic duct or CBD. The most specific and characteristic symptom of gallstone disease is biliary colic that is a constant and often long-lasting pain (see below). Obstruction of the cystic duct or CBD by a stone produces increased intraluminal pressure and distention of the viscus that cannot be relieved by repetitive biliary contractions. The resultant visceral pain is characteristically a severe, steady ache or fullness in the epigastrium or right upper quadrant (RUQ) of the abdomen with frequent radiation to the interscapular area, right scapula, or shoulder.
Biliary colic begins quite suddenly and may persist with severe intensity for 30 min to 5 h, subsiding gradually or rapidly. It is steady rather than intermittent, as would be suggested by the word colic, which must be regarded as a misnomer, although it is in widespread use. An episode of biliary pain persisting beyond 5 h should raise the suspicion of acute cholecystitis (see below). Nausea and vomiting frequently accompany episodes of biliary pain. An elevated level of serum bilirubin and/or alkaline phosphatase suggests a common duct stone. Fever or chills (rigors) with biliary pain usually imply a complication, i.e., cholecystitis, pancreatitis, or cholangitis. Complaints of short-lasting, vague epigastric fullness, dyspepsia, eructation, or flatulence, especially following a fatty meal, should not be confused with biliary pain. Such symptoms are frequently elicited from patients with or without gallstone disease but are not specific for biliary calculi. Biliary colic may be precipitated by eating a fatty meal, by consumption of a large meal following a period of prolonged fasting, or by eating a normal meal; it is frequently nocturnal, occurring within a few hours of retiring.
Natural History Gallstone disease discovered in an asymptomatic patient or in a patient whose symptoms are not referable to cholelithiasis is a common clinical problem. Sixty to 80% of persons with asymptomatic gallstones remain asymptomatic over follow-up periods of up to 25 years. The probability of developing symptoms within 5 years after diagnosis is 2–4% per year and decreases in the years thereafter to 1–2%. The yearly incidence of complications is about 0.1–0.3%. Patients remaining asymptomatic for 15 years were found to be unlikely to develop symptoms during further follow-up, and most patients who did develop complications from their gallstones experienced prior warning symptoms. Similar conclusions apply to diabetic patients with silent gallstones. Decision analysis has suggested that (1) the cumulative risk of death due to gallstone disease while on expectant management is small, and (2) prophylactic cholecystectomy is not warranted.
Complications requiring cholecystectomy are much more common in gallstone patients who have developed symptoms of biliary pain. Patients found to have gallstones at a young age are more likely to develop symptoms from cholelithiasis than are patients >60 years at the time of initial diagnosis. Patients with diabetes mellitus and gallstones may be somewhat more susceptible to septic complications, but the magnitude of risk of septic biliary complications in diabetic patients is incompletely defined.
ACUTE AND CHRONIC CHOLECYSTITIS
Acute Cholecystitis Acute inflammation of the gallbladder wall usually follows obstruction of the cystic duct by a stone. Inflammatory response can be evoked by three factors: (1) mechanical inflammation produced by increased intraluminal pressure and distention with resulting ischemia of the gallbladder mucosa and wall, (2) chemical inflammation caused by the release of lysolecithin (due to the action of phospholipase on lecithin in bile) and other local tissue factors, and (3) bacterial inflammation, which may play a role in 50–85% of patients with acute cholecystitis. The organisms most frequently isolated by culture of gallbladder bile in these patients include Escherichia coli, Klebsiella spp., Streptococcus spp., and Clostridium spp.
Acute cholecystitis often begins as an attack of biliary pain that progressively worsens. Approximately 60–70% of patients report having experienced prior attacks that resolved spontaneously. As the episode progresses, however, the pain of acute cholecystitis becomes more generalized in the right upper abdomen. As with biliary colic, the pain of cholecystitis may radiate to the interscapular area, right scapula, or shoulder. Peritoneal signs of inflammation such as increased pain with jarring or on deep respiration may be apparent. The patient is anorectic and often nauseated. Vomiting is relatively common and may produce symptoms and signs of vascular and extracellular volume depletion. Jaundice is unusual early in the course of acute cholecystitis but may occur when edematous inflammatory changes involve the bile ducts and surrounding lymph nodes.
A low-grade fever is characteristically present, but shaking chills or rigors are not uncommon. The RUQ of the abdomen is almost invariably tender to palpation. An enlarged, tense gallbladder is palpable in 25–50% of patients. Deep inspiration or cough during subcostal palpation of the RUQ usually produces increased pain and inspiratory arrest (Murphy’s sign). Localized rebound tenderness in the RUQ is common, as are abdominal distention and hypoactive bowel sounds from paralytic ileus, but generalized peritoneal signs and abdominal rigidity are usually lacking, in the absence of perforation.
The diagnosis of acute cholecystitis is usually made on the basis of a characteristic history and physical examination. The triad of sudden onset of RUQ tenderness, fever, and leukocytosis is highly suggestive. Typically, leukocytosis in the range of 10,000–15,000 cells per microliter with a left shift on differential count is found. The serum bilirubin is mildly elevated (<85.5 μmol/L [5 mg/dL]) in fewer than half of patients, whereas about one-fourth have modest elevations in serum aminotransferases (usually less than a fivefold elevation). Ultrasound will demonstrate calculi in 90–95% of cases and is useful for detection of signs of gallbladder inflammation including thickening of the wall, pericholecystic fluid, and dilatation of the bile duct. The radionuclide (e.g., HIDA) biliary scan may be confirmatory if bile duct imaging is seen without visualization of the gallbladder.
Approximately 75% of patients treated medically have remission of acute symptoms within 2–7 days following hospitalization. In 25%, however, a complication of acute cholecystitis will occur despite conservative treatment (see below). In this setting, prompt surgical intervention is required. Of the 75% of patients with acute cholecystitis who undergo remission of symptoms, ~25% will experience a recurrence of cholecystitis within 1 year, and 60% will have at least one recurrent bout within 6 years. In view of the natural history of the disease, acute cholecystitis is best treated by early surgery whenever possible.
Mirizzi’s syndrome is a rare complication in which a gallstone becomes impacted in the cystic duct or neck of the gallbladder causing compression of the CBD, resulting in CBD obstruction and jaundice. Ultrasound shows gallstone(s) lying outside the hepatic duct. Endoscopic retrograde cholangiopancreatography (ERCP) (Fig. 369-2B), percutaneous transhepatic cholangiography (PTC), or magnetic resonance cholangiopancreatography (MRCP) will usually demonstrate the characteristic extrinsic compression of the CBD. Surgery consists of removing the cystic duct, diseased gallbladder, and the impacted stone. The preoperative diagnosis of Mirizzi’s syndrome is important to avoid CBD injury.
ACALCULOUS CHOLECYSTITIS In 5–10% of patients with acute cholecystitis, calculi obstructing the cystic duct are not found at surgery. In >50% of such cases, an underlying explanation for acalculous inflammation is not found. An increased risk for the development of acalculous cholecystitis is especially associated with serious trauma or burns, with the postpartum period following prolonged labor, and with orthopedic and other nonbiliary major surgical operations in the postoperative period. It may possibly complicate periods of prolonged parenteral hyperalimentation. For some of these cases, biliary sludge in the cystic duct may be responsible. Other precipitating factors include vasculitis, obstructing adenocarcinoma of the gallbladder, diabetes mellitus, torsion of the gallbladder, “unusual” bacterial infections of the gallbladder (e.g., Leptospira, Streptococcus, Salmonella, or Vibrio cholerae), and parasitic infestation of the gallbladder. Acalculous cholecystitis may also be seen with a variety of other systemic disease processes (e.g., sarcoidosis, cardiovascular disease, tuberculosis, syphilis, actinomycosis).
Although the clinical manifestations of acalculous cholecystitis are indistinguishable from those of calculous cholecystitis, the setting of acute gallbladder inflammation complicating severe underlying illness is characteristic of acalculous disease. Ultrasound or computed tomography (CT) examinations demonstrating a large, tense, static gallbladder without stones and with evidence of poor emptying over a prolonged period may be diagnostically useful in some cases. The complication rate for acalculous cholecystitis exceeds that for calculous cholecystitis. Successful management of acute acalculous cholecystitis appears to depend primarily on early diagnosis and surgical intervention, with meticulous attention to postoperative care.
ACALCULOUS CHOLECYSTOPATHY Disordered motility of the gallbladder can produce recurrent biliary pain in patients without gallstones. Infusion of an octapeptide of CCK can be used to measure the gallbladder ejection fraction during cholescintigraphy. The surgical findings have included abnormalities such as chronic cholecystitis, gallbladder muscle hypertrophy, and/or a markedly narrowed cystic duct. Some of these patients may well have had antecedent gallbladder disease. The following criteria can be used to identify patients with acalculous cholecystopathy: (1) recurrent episodes of typical RUQ pain characteristic of biliary tract pain, (2) abnormal CCK cholescintigraphy demonstrating a gallbladder ejection fraction of <40%, and (3) infusion of CCK reproducing the patient’s pain. An additional clue would be the identification of a large gallbladder on ultrasound examination. Finally, it should be noted that sphincter of Oddi dysfunction can also give rise to recurrent RUQ pain and CCK-scintigraphic abnormalities.
EMPHYSEMATOUS CHOLECYSTITIS So-called emphysematous cholecystitis is thought to begin with acute cholecystitis (calculous or acalculous) followed by ischemia or gangrene of the gallbladder wall and infection by gas-producing organisms. Bacteria most frequently cultured in this setting include anaerobes, such as Clostridium welchii or Clostridium perfringens, and aerobes, such as E. coli. This condition occurs most frequently in elderly men and in patients with diabetes mellitus. The clinical manifestations are essentially indistinguishable from those of nongaseous cholecystitis. The diagnosis is usually made on plain abdominal film by finding gas within the gallbladder lumen, dissecting within the gallbladder wall to form a gaseous ring, or in the pericholecystic tissues. The morbidity and mortality rates with emphysematous cholecystitis are considerable. Prompt surgical intervention coupled with appropriate antibiotics is mandatory.
Chronic Cholecystitis Chronic inflammation of the gallbladder wall is almost always associated with the presence of gallstones and is thought to result from repeated bouts of subacute or acute cholecystitis or from persistent mechanical irritation of the gallbladder wall by gallstones. The presence of bacteria in the bile occurs in >25% of patients with chronic cholecystitis. The presence of infected bile in a patient with chronic cholecystitis undergoing elective cholecystectomy probably adds little to the operative risk. Chronic cholecystitis may be asymptomatic for years, may progress to symptomatic gallbladder disease or to acute cholecystitis, or may present with complications (see below).
Complications of Cholecystitis • EMPYEMA AND HYDROPS Empyema of the gallbladder usually results from progression of acute cholecystitis with persistent cystic duct obstruction to superinfection of the stagnant bile with a pus-forming bacterial organism. The clinical picture resembles that of cholangitis with high fever; severe RUQ pain; marked leukocytosis; and often, prostration. Empyema of the gallbladder carries a high risk of gram-negative sepsis and/or perforation. Emergency surgical intervention with proper antibiotic coverage is required as soon as the diagnosis is suspected.
Hydrops or mucocele of the gallbladder may also result from prolonged obstruction of the cystic duct, usually by a large solitary calculus. In this instance, the obstructed gallbladder lumen is progressively distended, over a period of time, by mucus (mucocele) or by a clear transudate (hydrops) produced by mucosal epithelial cells. A visible, easily palpable, nontender mass sometimes extending from the RUQ into the right iliac fossa may be found on physical examination. The patient with hydrops of the gallbladder frequently remains asymptomatic, although chronic RUQ pain may also occur. Cholecystectomy is indicated, because empyema, perforation, or gangrene may complicate the condition.
GANGRENE AND PERFORATION Gangrene of the gallbladder results from ischemia of the wall and patchy or complete tissue necrosis. Underlying conditions often include marked distention of the gallbladder, vasculitis, diabetes mellitus, empyema, or torsion resulting in arterial occlusion. Gangrene usually predisposes to perforation of the gallbladder, but perforation may also occur in chronic cholecystitis without premonitory warning symptoms. Localized perforations are usually contained by the omentum or by adhesions produced by recurrent inflammation of the gallbladder. Bacterial superinfection of the walled-off gallbladder contents results in abscess formation. Most patients are best treated with cholecystectomy, but some seriously ill patients may be managed with cholecystostomy and drainage of the abscess. Free perforation is less common but is associated with a mortality rate of ~30%. Such patients may experience a sudden transient relief of RUQ pain as the distended gallbladder decompresses; this is followed by signs of generalized peritonitis.
FISTULA FORMATION AND GALLSTONE ILEUS Fistula formation into an adjacent organ adherent to the gallbladder wall may result from inflammation and adhesion formation. Fistulas into the duodenum are most common, followed in frequency by those involving the hepatic flexure of the colon, stomach or jejunum, abdominal wall, and renal pelvis. Clinically “silent” biliary-enteric fistulas occurring as a complication of acute cholecystitis have been found in up to 5% of patients undergoing cholecystectomy. Asymptomatic cholecystoenteric fistulas may sometimes be diagnosed by finding gas in the biliary tree on plain abdominal films. Barium contrast studies or endoscopy of the upper gastrointestinal tract or colon may demonstrate the fistula. Treatment in the symptomatic patient usually consists of cholecystectomy, CBD exploration, and closure of the fistulous tract.
Gallstone ileus refers to mechanical intestinal obstruction resulting from the passage of a large gallstone into the bowel lumen. The stone customarily enters the duodenum through a cholecystoenteric fistula at that level. The site of obstruction by the impacted gallstone is usually at the ileocecal valve, provided that the more proximal small bowel is of normal caliber. The majority of patients do not give a history of either prior biliary tract symptoms or complaints suggestive of acute cholecystitis or fistula formation. Large stones, >2.5 cm in diameter, are thought to predispose to fistula formation by gradual erosion through the gallbladder fundus. Diagnostic confirmation may occasionally be found on the plain abdominal film (e.g., small-intestinal obstruction with gas in the biliary tree and a calcified, ectopic gallstone) or following an upper gastrointestinal series (cholecystoduodenal fistula with small-bowel obstruction at the ileocecal valve). Laparotomy with stone extraction (or propulsion into the colon) remains the procedure of choice to relieve obstruction. Evacuation of large stones within the gallbladder should also be performed. In general, the gallbladder and its attachment to the intestines should be left alone.
LIMEY (MILK OF CALCIUM) BILE AND PORCELAIN GALLBLADDER Calcium salts in the lumen of the gallbladder in sufficient concentration may produce calcium precipitation and diffuse, hazy opacification of bile or a layering effect on plain abdominal roentgenography. This so-called limey bile, or milk of calcium bile, is usually clinically innocuous, but cholecystectomy is recommended, especially when it occurs in a hydropic gallbladder. In the entity called porcelain gallbladder, calcium salt deposition within the wall of a chronically inflamed gallbladder may be detected on the plain abdominal film. Cholecystectomy is advised in all patients with porcelain gallbladder because in a high percentage of cases this finding appears to be associated with the development of carcinoma of the gallbladder.
Postcholecystectomy Complications Early complications following cholecystectomy include atelectasis and other pulmonary disorders, abscess formation (often subphrenic), external or internal hemorrhage, biliary-enteric fistula, and bile leaks. Jaundice may indicate absorption of bile from an intraabdominal collection following a biliary leak or mechanical obstruction of the CBD by retained calculi, intraductal blood clots, or extrinsic compression.
Overall, cholecystectomy is a very successful operation that provides total or near-total relief of preoperative symptoms in 75–90% of patients. The most common cause of persistent postcholecystectomy symptoms is an overlooked symptomatic nonbiliary disorder (e.g., reflux esophagitis, peptic ulceration, pancreatitis, or—most often—irritable bowel syndrome). In a small percentage of patients, however, a disorder of the extrahepatic bile ducts may result in persistent symptomatology. These so-called postcholecystectomy syndromes may be due to (1) biliary strictures, (2) retained biliary calculi, (3) cystic duct stump syndrome, (4) stenosis or dyskinesia of the sphincter of Oddi, or (5) bile salt–induced diarrhea or gastritis.
CYSTIC DUCT STUMP SYNDROME In the absence of cholangiographically demonstrable retained stones, symptoms resembling biliary pain or cholecystitis in the postcholecystectomy patient have frequently been attributed to disease in a long (>1 cm) cystic duct remnant (cystic duct stump syndrome). Careful analysis, however, reveals that postcholecystectomy complaints are attributable to other causes in almost all patients in whom the symptom complex was originally thought to result from the existence of a long cystic duct stump. Accordingly, considerable care should be taken to investigate the possible role of other factors in the production of postcholecystectomy symptoms before attributing them to cystic duct stump syndrome.
PAPILLARY DYSFUNCTION, PAPILLARY STENOSIS, SPASM OF THE SPHINCTER OF ODDI, AND BILIARY DYSKINESIA Symptoms of biliary colic accompanied by signs of recurrent, intermittent biliary obstruction may be produced by acalculous cholecystopathy, papillary stenosis, papillary dysfunction, spasm of the sphincter of Oddi, and biliary dyskinesia. Papillary stenosis is thought to result from acute or chronic inflammation of the papilla of Vater or from glandular hyperplasia of the papillary segment. Five criteria have been used to define papillary stenosis: (1) upper abdominal pain, usually RUQ or epigastric; (2) abnormal liver tests; (3) dilatation of the CBD upon ERCP examination; (4) delayed (>45 min) drainage of contrast material from the duct; and (5) increased basal pressure of the sphincter of Oddi, a finding that may be of only minor significance. An alternative to ERCP is magnetic resonance cholangiography (MRC) if ERCP and/or biliary manometry are either unavailable or not feasible. After exclusion of acalculous cholecystopathy, treatment consists of endoscopic or surgical sphincteroplasty to ensure wide patency of the distal portions of both the bile and pancreatic ducts. The greater the number of the preceding criteria present, the greater is the likelihood that a patient does have a degree of papillary stenosis sufficient to justify correction. The factors usually considered as indications for sphincterotomy include (1) prolonged duration of symptoms, (2) lack of response to symptomatic treatment, (3) presence of severe disability, and (4) the patient’s choice of sphincterotomy over surgery (given a clear understanding on his or her part of the risks involved in both procedures).
Criteria for diagnosing dyskinesia of the sphincter of Oddi are even more controversial than those for papillary stenosis. Proposed mechanisms include spasm of the sphincter, denervation sensitivity resulting in hypertonicity, and abnormalities of the sequencing or frequency rates of sphincteric-contraction waves. When thorough evaluation has failed to demonstrate another cause for the pain, and when cholangiographic and manometric criteria suggest a diagnosis of biliary dyskinesia, medical treatment with nitrites or anticholinergics to attempt pharmacologic relaxation of the sphincter has been proposed. Endoscopic biliary sphincterotomy (EBS) or surgical sphincteroplasty may be indicated in patients who fail to respond to a 2- to 3-month trial of medical therapy, especially if basal sphincter of Oddi pressures are elevated. EBS has become the procedure of choice for removing bile duct stones and for other biliary and pancreatic problems.
BILE SALT–INDUCED DIARRHEA AND GASTRITIS Postcholecystectomy patients may develop symptoms of dyspepsia, which have been attributed to duodenogastric reflux of bile. However, firm data linking these symptoms to bile gastritis after surgical removal of the gallbladder are lacking. Cholecystectomy induces persistent changes in gut transit, and these changes effect a noticeable modification of bowel habits. Cholecystectomy shortens gut transit time by accelerating passage of the fecal bolus through the colon with marked acceleration in the right colon, thus causing an increase in colonic bile acid output and a shift in bile acid composition toward the more diarrheagenic secondary bile acids, i.e. deoxycholic acid. Diarrhea that is severe enough, i.e., three or more watery movements per day, can be classified as postcholecystectomy diarrhea, and this occurs in 5–10% of patients undergoing elective cholecystectomy. Treatment with bile acid–sequestering agents such as cholestyramine or colestipol is often effective in ameliorating troublesome diarrhea.
THE HYPERPLASTIC CHOLECYSTOSES
The term hyperplastic cholecystoses is used to denote a group of disorders of the gallbladder characterized by excessive proliferation of normal tissue components.
Adenomyomatosis is characterized by a benign proliferation of gallbladder surface epithelium with glandlike formations, extramural sinuses, transverse strictures, and/or fundal nodule (“adenoma” or “adenomyoma”) formation.
Cholesterolosis is characterized by abnormal deposition of lipid, especially cholesteryl esters, within macrophages in the lamina propria of the gallbladder wall. In its diffuse form (“strawberry gallbladder”), the gallbladder mucosa is brick red and speckled with bright yellow flecks of lipid. The localized form shows solitary or multiple “cholesterol polyps” studding the gallbladder wall. Cholesterol stones of the gallbladder are found in nearly half the cases. Cholecystectomy is indicated in both adenomyomatosis and cholesterolosis when symptomatic or when cholelithiasis is present.
The prevalence of gallbladder polyps in the adult population is ~5%, with a marked male predominance. Few significant changes have been found over a 5-year period in asymptomatic patients with gallbladder polyps <10 mm in diameter. Cholecystectomy is recommended in symptomatic patients, as well as in asymptomatic patients >50 years of age, or in those whose polyps are >10 mm in diameter or associated with gallstones or polyp growth on serial ultrasonography.
DISEASES OF THE BILE DUCTS
CONGENITAL ANOMALIES
Biliary Atresia and Hypoplasia Atretic and hypoplastic lesions of the extrahepatic and large intrahepatic bile ducts are the most common biliary anomalies of clinical relevance encountered in infancy. The clinical picture is one of severe obstructive jaundice during the first month of life, with pale stools. When biliary atresia is suspected on the basis of clinical, laboratory, and imaging findings, the diagnosis is confirmed by surgical exploration and operative cholangiography. Approximately 10% of cases of biliary atresia are treatable with Roux-en-Y choledochojejunostomy, with the Kasai procedure (hepatic portoenterostomy) being attempted in the remainder in an effort to restore some bile flow. Most patients, even those having successful biliary-enteric anastomoses, eventually develop chronic cholangitis, extensive hepatic fibrosis, and portal hypertension.
Choledochal Cysts Cystic dilatation may involve the free portion of the CBD, i.e., choledochal cyst, or may present as diverticulum formation in the intraduodenal segment. In the latter situation, chronic reflux of pancreatic juice into the biliary tree can produce inflammation and stenosis of the extrahepatic bile ducts leading to cholangitis or biliary obstruction. Because the process may be gradual, ~50% of patients present with onset of symptoms after age 10. The diagnosis may be made by ultrasound, abdominal CT, MRC, or cholangiography. Only one-third of patients show the classic triad of abdominal pain, jaundice, and an abdominal mass. Ultrasonographic detection of a cyst separate from the gallbladder should suggest the diagnosis of choledochal cyst, which can be confirmed by demonstrating the entrance of extrahepatic bile ducts into the cyst. Surgical treatment involves excision of the “cyst” and biliary-enteric anastomosis. Patients with choledochal cysts are at increased risk for the subsequent development of cholangiocarcinoma.
Congenital Biliary Ectasia Dilatation of intrahepatic bile ducts may involve either the major intrahepatic radicles (Caroli’s disease), the inter- and intralobular ducts (congenital hepatic fibrosis), or both. In Caroli’s disease, clinical manifestations include recurrent cholangitis, abscess formation in and around the affected ducts, and, often, brown pigment gallstone formation within portions of ectatic intrahepatic biliary radicles. Ultrasound, MRC, and CT are of great diagnostic value in demonstrating cystic dilatation of the intrahepatic bile ducts. Treatment with ongoing antibiotic therapy is usually undertaken in an effort to limit the frequency and severity of recurrent bouts of cholangitis. Progression to secondary biliary cirrhosis with portal hypertension, extrahepatic biliary obstruction, cholangiocarcinoma, or recurrent episodes of sepsis with hepatic abscess formation is common.
CHOLEDOCHOLITHIASIS
Pathophysiology and Clinical Manifestations Passage of gallstones into the CBD occurs in ~10–15% of patients with cholelithiasis. The incidence of common duct stones increases with increasing age of the patient, so that up to 25% of elderly patients may have calculi in the common duct at the time of cholecystectomy. Undetected duct stones are left behind in ~1–5% of cholecystectomy patients. The overwhelming majority of bile duct stones are cholesterol stones formed in the gallbladder, which then migrate into the extrahepatic biliary tree through the cystic duct. Primary calculi arising de novo in the ducts are usually brown pigment stones developing in patients with (1) hepatobiliary parasitism or chronic, recurrent cholangitis; (2) congenital anomalies of the bile ducts (especially Caroli’s disease); (3) dilated, sclerosed, or strictured ducts; or (4) an MDR3 (ABCB4) gene defect leading to impaired biliary phospholipids secretion (low phospholipid–associated cholesterol cholelithiasis). Common duct stones may remain asymptomatic for years, may pass spontaneously into the duodenum, or (most often) may present with biliary colic or a complication.
Complications • CHOLANGITIS Cholangitis may be acute or chronic, and symptoms result from inflammation, which usually is caused by at least partial obstruction to the flow of bile. Bacteria are present on bile culture in ~75% of patients with acute cholangitis early in the symptomatic course. The characteristic presentation of acute cholangitis involves biliary pain, jaundice, and spiking fevers with chills (Charcot’s triad). Blood cultures are frequently positive, and leukocytosis is typical. Nonsuppurative acute cholangitis is most common and may respond relatively rapidly to supportive measures and to treatment with antibiotics. In suppurative acute cholangitis, however, the presence of pus under pressure in a completely obstructed ductal system leads to symptoms of severe toxicity—mental confusion, bacteremia, and septic shock. Response to antibiotics alone in this setting is relatively poor, multiple hepatic abscesses are often present, and the mortality rate approaches 100% unless prompt endoscopic or surgical relief of the obstruction and drainage of infected bile are carried out. Endoscopic management of bacterial cholangitis is as effective as surgical intervention. ERCP with endoscopic sphincterotomy is safe and the preferred initial procedure for both establishing a definitive diagnosis and providing effective therapy.
OBSTRUCTIVE JAUNDICE Gradual obstruction of the CBD over a period of weeks or months usually leads to initial manifestations of jaundice or pruritus without associated symptoms of biliary colic or cholangitis. Painless jaundice may occur in patients with choledocholithiasis, but is much more characteristic of biliary obstruction secondary to malignancy of the head of the pancreas, bile ducts, or ampulla of Vater.
In patients whose obstruction is secondary to choledocholithiasis, associated chronic calculous cholecystitis is very common, and the gallbladder in this setting may be unable to distend. The absence of a palpable gallbladder in most patients with biliary obstruction from duct stones is the basis for Courvoisier’s law, i.e., that the presence of a palpably enlarged gallbladder suggests that the biliary obstruction is secondary to an underlying malignancy rather than to calculous disease. Biliary obstruction causes progressive dilatation of the intrahepatic bile ducts as intrabiliary pressures rise. Hepatic bile flow is suppressed, and reabsorption and regurgitation of conjugated bilirubin into the bloodstream lead to jaundice accompanied by dark urine (bilirubinuria) and light-colored (acholic) stools.
CBD stones should be suspected in any patient with cholecystitis whose serum bilirubin level is >85.5 μmol/L (5 mg/dL). The maximum bilirubin level is seldom >256.5 μmol/L (15.0 mg/dL) in patients with choledocholithiasis unless concomitant hepatic or renal disease or another factor leading to marked hyperbilirubinemia exists. Serum bilirubin levels ≥342.0 μmol/L (20 mg/dL) should suggest the possibility of neoplastic obstruction. The serum alkaline phosphatase level is almost always elevated in biliary obstruction. A rise in alkaline phosphatase often precedes clinical jaundice and may be the only abnormality in routine liver function tests. There may be a two- to tenfold elevation of serum aminotransferases, especially in association with acute obstruction. Following relief of the obstructing process, serum aminotransferase elevations usually return rapidly to normal, while the serum bilirubin level may take 1–2 weeks to return to normal. The alkaline phosphatase level usually falls slowly, lagging behind the decrease in serum bilirubin.
PANCREATITIS The most common associated entity discovered in patients with nonalcoholic acute pancreatitis is biliary tract disease. Biochemical evidence of pancreatic inflammation complicates acute cholecystitis in 15% of cases and choledocholithiasis in >30%, and the common factor appears to be the passage of gallstones through the common duct. Coexisting pancreatitis should be suspected in patients with symptoms of cholecystitis who develop (1) back pain or pain to the left of the abdominal midline, (2) prolonged vomiting with paralytic ileus, or (3) a pleural effusion, especially on the left side. Surgical treatment of gallstone disease is usually associated with resolution of the pancreatitis.
SECONDARY BILIARY CIRRHOSIS Secondary biliary cirrhosis may complicate prolonged or intermittent duct obstruction with or without recurrent cholangitis. Although this complication may be seen in patients with choledocholithiasis, it is more common in cases of prolonged obstruction from stricture or neoplasm. Once established, secondary biliary cirrhosis may be progressive even after correction of the obstructing process, and increasingly severe hepatic cirrhosis may lead to portal hypertension or to hepatic failure and death. Prolonged biliary obstruction may also be associated with clinically relevant deficiencies of the fat-soluble vitamins A, D, E, and K.
Diagnosis and Treatment The diagnosis of choledocholithiasis is usually made by cholangiography (Table 369-3), either preoperatively by endoscopic retrograde cholangiogram (ERC) (Fig. 369-2C) or MRCP or intraoperatively at the time of cholecystectomy. As many as 15% of patients undergoing cholecystectomy will prove to have CBD stones. When CBD stones are suspected prior to laparoscopic cholecystectomy, preoperative ERCP with endoscopic papillotomy and stone extraction is the preferred approach. It not only provides stone clearance but also defines the anatomy of the biliary tree in relationship to the cystic duct. CBD stones should be suspected in gallstone patients who have any of the following risk factors: (1) a history of jaundice or pancreatitis, (2) abnormal tests of liver function, and (3) ultrasonographic or MRCP evidence of a dilated CBD or stones in the duct. Alternatively, if intraoperative cholangiography reveals retained stones, postoperative ERCP can be carried out. The need for preoperative ERCP is expected to decrease further as laparoscopic techniques for bile duct exploration improve.
|
DIAGNOSTIC EVALUATION OF THE BILE DUCTS |
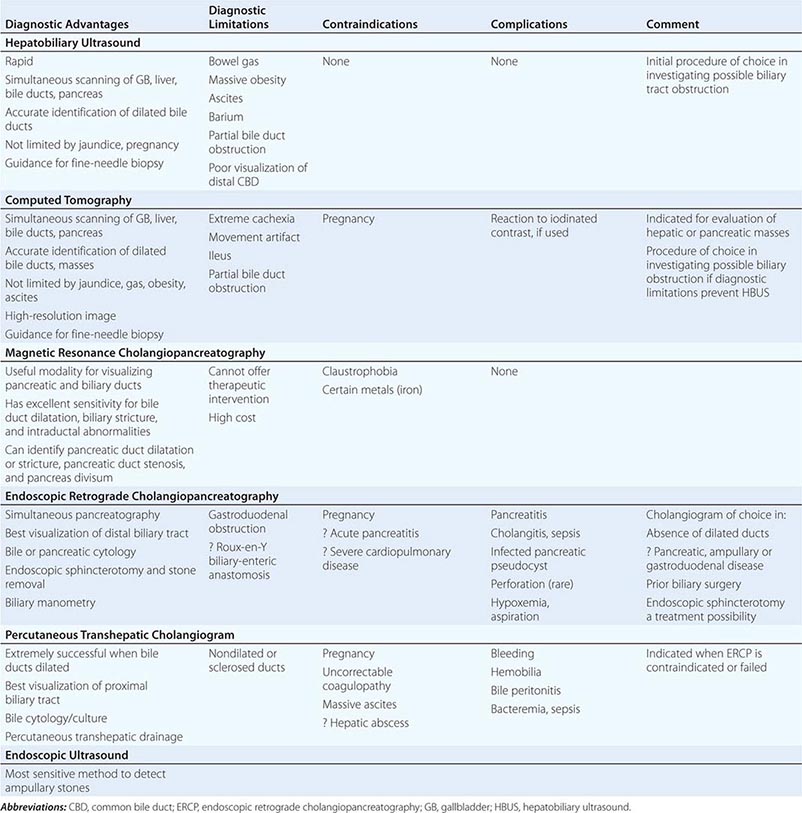
The widespread use of laparoscopic cholecystectomy and ERCP has decreased the incidence of complicated biliary tract disease and the need for choledocholithotomy and T-tube drainage of the bile ducts. EBS followed by spontaneous passage or stone extraction is the treatment of choice in the management of patients with common duct stones, especially in elderly or poor-risk patients.
TRAUMA, STRICTURES, AND HEMOBILIA
Most benign strictures of the extrahepatic bile ducts result from surgical trauma and occur in about 1 in 500 cholecystectomies. Strictures may present with bile leak or abscess formation in the immediate postoperative period or with biliary obstruction or cholangitis as long as 2 years or more following the inciting trauma. The diagnosis is established by percutaneous or endoscopic cholangiography. Endoscopic brushing of biliary strictures may be helpful in establishing the nature of the lesion and is more accurate than bile cytology alone. When positive exfoliative cytology is obtained, the diagnosis of a neoplastic stricture is established. This procedure is especially important in patients with primary sclerosing cholangitis (PSC) who are predisposed to the development of cholangiocarcinomas. Successful operative correction of non-PSC bile duct strictures by a skillful surgeon with duct-to-bowel anastomosis is usually possible, although mortality rates from surgical complications, recurrent cholangitis, or secondary biliary cirrhosis are high.
Hemobilia may follow traumatic or operative injury to the liver or bile ducts, intraductal rupture of a hepatic abscess or aneurysm of the hepatic artery, biliary or hepatic tumor hemorrhage, or mechanical complications of choledocholithiasis or hepatobiliary parasitism. Diagnostic procedures such as liver biopsy, PTC, and transhepatic biliary drainage catheter placement may also be complicated by hemobilia. Patients often present with a classic triad of biliary pain, obstructive jaundice, and melena or occult blood in the stools. The diagnosis is sometimes made by cholangiographic evidence of blood clot in the biliary tree, but selective angiographic verification may be required. Although minor episodes of hemobilia may resolve without operative intervention, surgical ligation of the bleeding vessel is frequently required.
EXTRINSIC COMPRESSION OF THE BILE DUCTS
Partial or complete biliary obstruction may be produced by extrinsic compression of the ducts. The most common cause of this form of obstructive jaundice is carcinoma of the head of the pancreas. Biliary obstruction may also occur as a complication of either acute or chronic pancreatitis or involvement of lymph nodes in the porta hepatis by lymphoma or metastatic carcinoma. The latter should be distinguished from cholestasis resulting from massive replacement of the liver by tumor.
HEPATOBILIARY PARASITISM
Infestation of the biliary tract by adult helminths or their ova may produce a chronic, recurrent pyogenic cholangitis with or without multiple hepatic abscesses, ductal stones, or biliary obstruction. This condition is relatively rare but does occur in inhabitants of southern China and elsewhere in Southeast Asia. The organisms most commonly involved are trematodes or flukes, including Clonorchis sinensis, Opisthorchis viverrini or Opisthorchis felineus, and Fasciola hepatica. The biliary tract also may be involved by intraductal migration of adult Ascaris lumbricoides from the duodenum or by intrabiliary rupture of hydatid cysts of the liver produced by Echinococcus spp. The diagnosis is made by cholangiography and the presence of characteristic ova on stool examination. When obstruction is present, the treatment of choice is laparotomy under antibiotic coverage, with common duct exploration and a biliary drainage procedure.
SCLEROSING CHOLANGITIS
Primary or idiopathic sclerosing cholangitis is characterized by a progressive, inflammatory, sclerosing, and obliterative process affecting the extrahepatic and/or the intrahepatic bile ducts. The disorder occurs up to 75% in association with inflammatory bowel disease, especially ulcerative colitis. It may also be associated with autoimmune pancreatitis; multifocal fibrosclerosis syndromes such as retroperitoneal, mediastinal, and/or periureteral fibrosis; Riedel’s struma; or pseudotumor of the orbit.
Immunoglobulin G4 (IgG4)–associated cholangitis is a recently described biliary disease of unknown etiology that presents with biochemical and cholangiographic features indistinguishable from PSC, is often associated with autoimmune pancreatitis and other fibrosing conditions, and is characterized by elevated serum IgG4 and infiltration of IgG4-positive plasma cells in bile ducts and liver tissue. In contrast to PSC, it is not associated with inflammatory bowel disease and should be suspected if associated with increased serum IgG4 and unexplained pancreatic disease. Glucocorticoids are regarded as the initial treatment of choice. Relapse is common after steroid withdrawal, especially with proximal strictures. Long-term treatment with glucocorticoids and/or azathioprine may be needed after relapse or for inadequate response (Chap. 371).
Patients with primary sclerosing cholangitis often present with signs and symptoms of chronic or intermittent biliary obstruction: RUQ abdominal pain, pruritus, jaundice, or acute cholangitis. Late in the course, complete biliary obstruction, secondary biliary cirrhosis, hepatic failure, or portal hypertension with bleeding varices may occur. The diagnosis is usually established by finding multifocal, diffusely distributed strictures with intervening segments of normal or dilated ducts, producing a beaded appearance on cholangiography (Fig. 369-2D). The cholangiographic techniques of choice in suspected cases are MRCP and ERCP. When a diagnosis of sclerosing cholangitis has been established, a search for associated diseases, especially for chronic inflammatory bowel disease, should be carried out.
A recent study describes the natural history and outcome for 305 patients of Swedish descent with primary sclerosing cholangitis; 134 (44%) of the patients were asymptomatic at the time of diagnosis and, not surprisingly, had a significantly higher survival rate. The independent predictors of a bad prognosis were advanced age, serum bilirubin concentration, and liver histologic changes. Cholangiocarcinoma was found in 24 patients (8%). Inflammatory bowel disease was closely associated with primary sclerosing cholangitis and had a prevalence of 81% in this study population.
Small duct PSC is defined by the presence of chronic cholestasis and hepatic histology consistent with PSC but with normal findings on cholangiography. Small duct PSC is found in ~5% of patients with PSC and may represent an earlier stage of PSC associated with a significantly better long-term prognosis. However, such patients may progress to classic PSC and/or end-stage liver disease with consequent necessity of liver transplantation.
In patients with AIDS, cholangiopancreatography may demonstrate a broad range of biliary tract changes as well as pancreatic duct obstruction and occasionally pancreatitis (Chap. 226). Further, biliary tract lesions in AIDS include infection and cholangiopancreatographic changes similar to those of PSC. Changes noted include: (1) diffuse involvement of intrahepatic bile ducts alone, (2) involvement of both intra- and extrahepatic bile ducts, (3) ampullary stenosis, (4) stricture of the intrapancreatic portion of the CBD, and (5) pancreatic duct involvement. Associated infectious organisms include Cryptosporidium, Mycobacterium avium-intracellulare, cytomegalovirus, Microsporidia, and Isospora. In addition, acalculous cholecystitis occurs in up to 10% of patients. ERCP sphincterotomy, while not without risk, provides significant pain reduction in patients with AIDS-associated papillary stenosis. Secondary sclerosing cholangitis may occur as a long-term complication of choledocholithiasis, cholangiocarcinoma, operative or traumatic biliary injury, or contiguous inflammatory processes.
SECTION 3 |
DISORDERS OF THE PANCREAS |
370 |
Approach to the Patient with Pancreatic Disease |
GENERAL CONSIDERATIONS
As emphasized in Chap. 371, the etiologies as well as clinical manifestations of pancreatitis are quite varied. Although it is well-appreciated that pancreatitis is frequently secondary to biliary tract disease and alcohol abuse, it can also be caused by drugs, genetic mutations, trauma, and viral infections and is associated with metabolic and connective tissue disorders. In ~30% of patients with acute pancreatitis and 25–40% of patients with chronic pancreatitis, the etiology initially can be obscure.
The incidence of acute pancreatitis is about 5–35/100,000 new cases per year worldwide, with a mortality rate of about 3%. The incidence of chronic pancreatitis is about 4–8 new cases per 100,000 per year with a prevalence of 26–42 cases per 100,000. The number of patients admitted to the hospital who suffer with both acute and chronic pancreatitis in the United States is largely increasing and is now estimated to be 274,119 for acute pancreatitis and 19,724 for chronic pancreatitis. Acute pancreatitis is now the most common gastrointestinal diagnosis requiring hospitalization in the United States. Acute and chronic pancreatic disease costs an estimated 3 billion dollars annually in health care expenditures. These numbers may underestimate the true incidence and prevalence, because non–alcohol-induced pancreatitis has been largely ignored. At autopsy, the prevalence of chronic pancreatitis ranges from 0.04 to 5%.
The diagnosis of acute pancreatitis is generally clearly defined based on a combination of laboratory, imaging, and clinical symptoms. The diagnosis of chronic pancreatitis, especially in mild disease, is hampered by the relative inaccessibility of the pancreas to direct examination and the nonspecificity of the abdominal pain associated with chronic pancreatitis. Many patients with chronic pancreatitis do not have elevated blood amylase or lipase levels. Some patients with chronic pancreatitis develop signs and symptoms of pancreatic exocrine insufficiency, and thus, objective evidence for pancreatic disease can be demonstrated. However, there is a very large reservoir of pancreatic exocrine function. More than 90% of the pancreas must be damaged before maldigestion of fat and protein is manifested. Noninvasive, indirect tests of pancreatic exocrine function (fecal elastase) are much more likely to give abnormal results in patients with obvious advanced pancreatic disease (i.e., pancreatic calcification, steatorrhea, or diabetes mellitus) than in patients with occult disease. Invasive, direct tests of pancreatic secretory function (secretin tests) are the most sensitive and specific tests to detect early chronic pancreatic disease when imaging is equivocal or normal.
TESTS USEFUL IN THE DIAGNOSIS OF PANCREATIC DISEASE
Several tests have proved of value in the evaluation of pancreatic disease. Examples of specific tests and their usefulness in the diagnosis of acute and chronic pancreatitis are summarized in Table 370-1 and Fig. 370-1. At some institutions, pancreatic function tests are available and performed if the diagnosis of chronic pancreatic disease remains a possibility after noninvasive tests (ultrasound, computed tomography [CT], magnetic resonance cholangiopancreatography [MRCP]) or invasive tests (endoscopic retrograde cholangiopancreatography [ERCP], endoscopic ultrasonography [EUS]) have given normal or inconclusive results. In this regard, tests using direct stimulation of the pancreas with secretin are the most sensitive.
|
TESTS USEFUL IN THE DIAGNOSIS OF ACUTE AND CHRONIC PANCREATITIS AND PANCREATIC TUMORS |
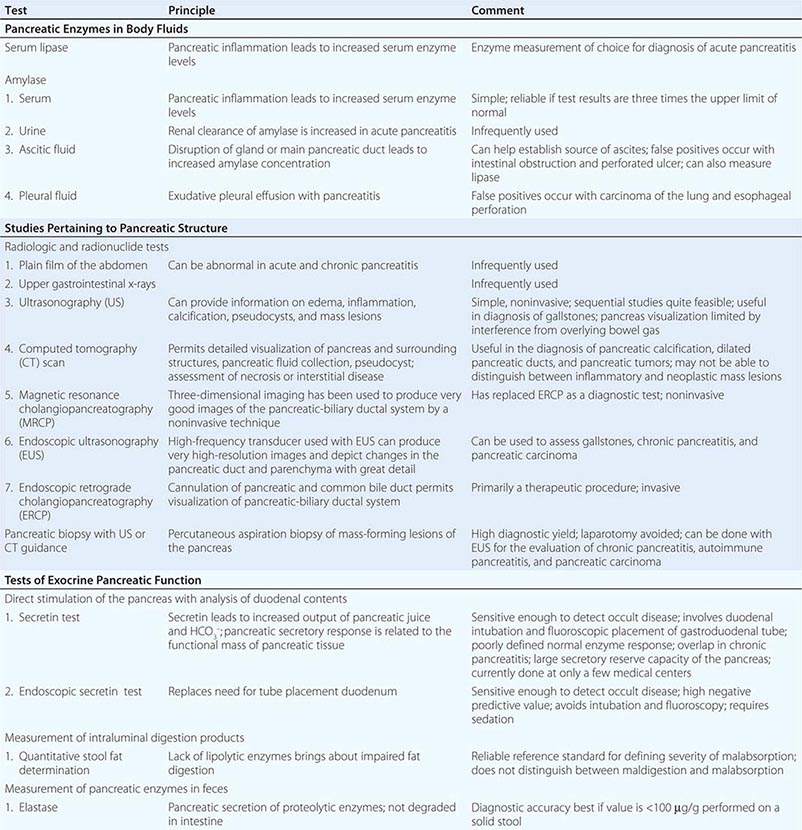
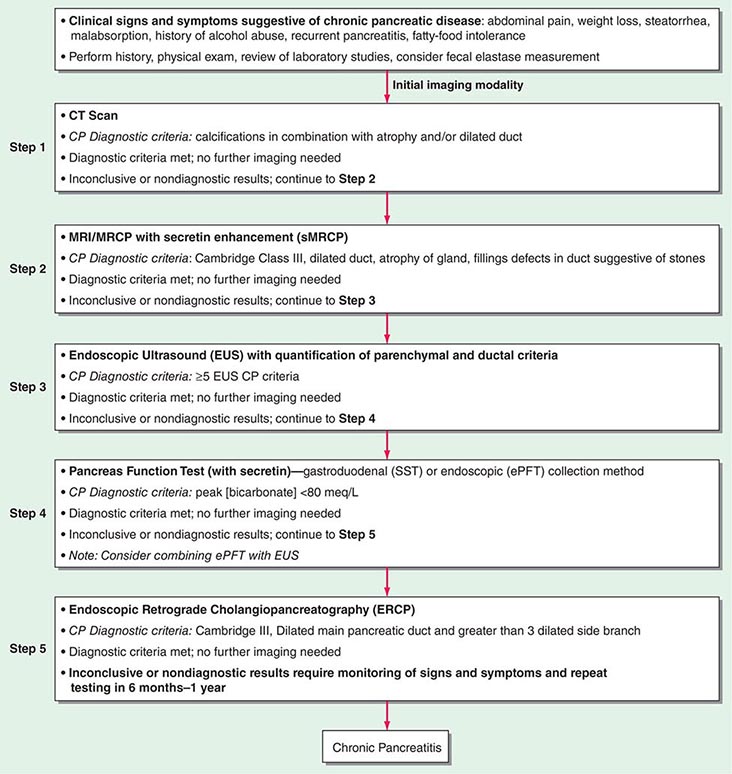
FIGURE 370-1 A stepwise diagnostic approach to the patient with suspected chronic pancreatitis (CP). Endoscopic ultrasonography (EUS) and magnetic resonance cholangiopancreatography (sMRCP/MRCP) are appropriate diagnostic alternatives to endoscopic retrograde cholangiopancreatography (ERCP). CT, computed tomography.
Pancreatic Enzymes in Body Fluids The serum amylase and lipase levels are widely used as screening tests for acute pancreatitis in the patient with acute abdominal pain or back pain. Values greater than three times the upper limit of normal in combination with epigastric pain strongly suggest the diagnosis if gut perforation or infarction is excluded. In acute pancreatitis, the serum amylase and lipase are usually elevated within 24 h of onset and remain so for 3–7 days. Levels usually return to normal within 7 days unless there is pancreatic ductal disruption, ductal obstruction, or pseudocyst formation. Approximately 85% of patients with acute pancreatitis have a threefold or greater elevated serum lipase and amylase levels. The values may be normal if (1) there is a delay (of 2–5 days) before blood samples are obtained, (2) the underlying disorder is chronic pancreatitis rather than acute pancreatitis, or (3) hypertriglyceridemia is present. Patients with hypertriglyceridemia and proven pancreatitis have been found to have spuriously low levels of amylase and perhaps lipase activity. In the absence of objective evidence of pancreatitis by abdominal ultrasound, CT scan, MRCP, or EUS, mild to moderate elevations of amylase and/or lipase are not helpful in making a diagnosis of chronic pancreatitis.
The serum amylase can be elevated in other conditions (Table 370-2), in part because the enzyme is found in many organs. In addition to the pancreas and salivary glands, small quantities of amylase are found in the tissues of the fallopian tubes, lung, thyroid, and tonsils and can be produced by various tumors (carcinomas of the lung, esophagus, breast, and ovary). Isoamylase determinations do not accurately distinguish elevated blood amylase levels due to bona fide pancreatitis from elevated blood amylase levels due to a nonpancreatic source of amylase, especially when the blood amylase level is only moderately elevated. In patients with unexplained hyperamylasemia, measurement of macroamylase can avoid numerous tests in patients with this rare disorder.
|
CAUSES OF HYPERAMYLASEMIA AND HYPERAMYLASURIA |
Elevation of ascitic fluid amylase occurs in acute pancreatitis as well as in (1) ascites due to disruption of the main pancreatic duct or a leaking pseudocyst and (2) other abdominal disorders that simulate pancreatitis (e.g., intestinal obstruction, intestinal infarction, or perforated peptic ulcer). Elevation of pleural fluid amylase can occur in acute pancreatitis, chronic pancreatitis, carcinoma of the lung, and esophageal perforation. Lipase is the single best enzyme to measure for the diagnosis of acute pancreatitis. No single blood test is reliable for the diagnosis of acute pancreatitis in patients with renal failure. Pancreatic enzyme elevations are usually less than three times the upper limit of normal. Determining whether a patient with renal failure and abdominal pain has pancreatitis remains a difficult clinical problem. One study found that serum amylase levels were elevated in patients with renal dysfunction only when creatinine clearance was <0.8 mL/s (<50 mL/min). In such patients, the serum amylase level was invariably <500 IU/L in the absence of objective evidence of acute pancreatitis. In that study, serum lipase and trypsin levels paralleled serum amylase values. With these limitations in mind, the recommended screening test for acute pancreatitis in renal disease is serum lipase.
Studies Pertaining to Pancreatic Structure • RADIOLOGIC TESTS Plain films of the abdomen, which once provided useful information in patients with acute and chronic pancreatitis, have been superseded by other more detailed imaging procedures (ultrasound, EUS, CT, MRCP).
Ultrasonography (US) can provide important information in patients with acute pancreatitis, chronic pancreatitis, pseudocysts, and pancreatic carcinoma. Echographic appearances can indicate the presence of edema, inflammation, and calcification (not obvious on plain films of the abdomen), as well as pseudocysts, mass lesions, and gallstones. In acute pancreatitis, the pancreas is characteristically enlarged. In pancreatic pseudocyst, the usual appearance is primarily that of smooth, round fluid collection. Pancreatic carcinoma distorts the usual landmarks, and mass lesions >3.0 cm are usually detected as localized, solid lesions. US is often the initial investigation for most patients with suspected pancreatic disease. However, obesity and excess small- and large-bowel gas can interfere with pancreatic imaging by US studies.
Computed tomography (CT) is the best imaging study for initial evaluation of a suspected pancreatic disorder and for the assessment of complications of acute and chronic pancreatitis. It is especially useful in the detection of pancreatic and peripancreatic acute fluid collections, fluid-containing lesions such as pseudocysts, walled-off necrosis, calcium deposits (see Chap. 371, Figs. 371-1, 371-2, and 371-4), and pancreatic neoplasms. Acute pancreatitis is characterized by (1) enlargement of the pancreatic outline, (2) distortion of the pancreatic contour, and/or (3) a pancreatic fluid that has a different attenuation coefficient than normal pancreas. Oral, water-soluble contrast agents are used to opacify the stomach and duodenum during CT scans; this strategy permits more precise delineation of various organs as well as mass lesions. Dynamic CT (using rapid IV administration of contrast) is useful in estimating the extent of pancreatic necrosis and in predicting morbidity and mortality. CT provides clear images much more rapidly and essentially negates artifact caused by patient movement. If acute pancreatitis is confirmed with serology and physical examination findings, CT scan in the first 3 days is not recommended to avoid overuse and minimize costs.
Endoscopic ultrasonography (EUS) produces high-resolution images of the pancreatic parenchyma and pancreatic duct with a transducer fixed to an endoscope that can be directed onto the surface of the pancreas through the stomach or duodenum. EUS and MRCP have largely replaced ERCP for diagnostic purposes in many centers. EUS allows one to obtain information about the pancreatic duct as well as the parenchyma and has few procedure-related complications associated with it, in contrast to the 5–10% of post-ERCP pancreatitis observed. EUS is also helpful in detecting common bile duct stones in acute pancreatitis. Pancreatic masses can also be biopsied via EUS in cases with suspected pancreas cancer, and one can deliver nerve-blocking agents through EUS fine-needle injection in patients suffering from pancreatic pain from chronic pancreatitis or cancer. EUS has been studied as a diagnostic modality for chronic pancreatitis. Criteria for abnormalities on EUS in severe chronic pancreatic disease have been developed. There is general agreement that the presence of five or more of the nine criteria listed in Table 370-3 is highly predictive of chronic pancreatitis. Recent studies comparing EUS and ERCP to the secretin test in patients with unexplained abdominal pain suspected of having chronic pancreatitis show similar diagnostic accuracy in detecting early changes of chronic pancreatitis. The exact role of EUS versus CT, ERCP, or function testing in the early diagnosis of chronic pancreatitis has yet to be clearly defined.
|
ENDOSCOPIC ULTRASONOGRAPHIC CRITERIA FOR CHRONIC PANCREATITIS (TOTAL CRITERIA = 9) |
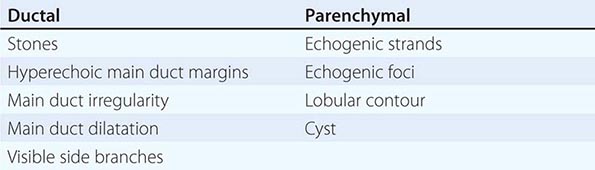
Magnetic resonance imaging (MRI) and magnetic resonance cholangiopancreatography (MRCP) are now being used to view the bile ducts, pancreatic duct, and the pancreas parenchyma in both acute pancreatitis and chronic pancreatitis. For diagnostic imaging in chronic pancreatitis, non-breath-holding and three-dimensional turbo spin-echo techniques are being used to produce superb MRCP images. The main pancreatic duct and common bile duct can be seen well, but there is still a question as to whether changes can be detected consistently in the secondary ducts. The secondary ducts are not visualized in a normal pancreas. Secretin-enhanced MRCP is currently under investigation but is emerging as a method to better evaluate ductal changes. In anteroposterior imaging, T2 imaging of fluid collections can differentiate necrotic debris from fluid in suspected walled-off necrosis, and T1 imaging can diagnose hemorrhage in suspected pseudoaneurysm rupture.
Both EUS and MRCP have largely replaced ERCP in the diagnostic evaluation of pancreatic disease. As these techniques become more refined, especially with the administration of secretin, they may well be the diagnostic tests of choice to evaluate the pancreatic duct. ERCP is still needed for treatment of bile duct and pancreatic duct lesions. ERCP is primarily of therapeutic value after CT, EUS, or MRCP has detected abnormalities requiring invasive endoscopic treatment. ERCP can also be helpful at clarification of equivocal findings discovered with other imaging techniques (see Chap. 371, Fig. 371-1). Pancreatic carcinoma is characterized by stenosis or obstruction of either the pancreatic duct or the common bile duct; both ductal systems are often abnormal (double-duct sign). In chronic pancreatitis, ERCP abnormalities in the main pancreatic duct and side branches have been outlined by the Cambridge classification. The presence of ductal stenosis and irregularity can make it difficult to distinguish chronic pancreatitis from carcinoma. It is important to be aware that ERCP changes interpreted as indicating chronic pancreatitis actually may be due to the effects of aging on the pancreatic duct or sequelae of a recent attack of acute pancreatitis. Although aging may cause impressive ductal alterations, it does not affect the results of pancreatic function tests (i.e., the secretin test). Elevated serum amylase levels after ERCP have been reported in the majority of patients, and clinical pancreatitis in 5–10% of patients. Recent data suggest that pancreatic duct stenting and rectal indomethacin can decrease the incidence of ERCP-induced pancreatitis. ERCP should rarely be done for diagnostic purposes and should especially be avoided in high-risk patients.
PANCREATIC BIOPSY WITH RADIOLOGIC GUIDANCE Percutaneous aspiration biopsy or a trucut biopsy of a pancreatic mass often distinguishes a pancreatic inflammatory mass from a pancreatic neoplasm.
TESTS OF EXOCRINE PANCREATIC FUNCTION
Pancreatic function tests (Table 370-1) can be divided into the following:
1. Direct stimulation of the pancreas by IV infusion of secretin followed by collection and measurement of duodenal contents
The secretin test, used to detect diffuse pancreatic disease, is based on the physiologic principle that the pancreatic secretory response is directly related to the functional mass of pancreatic tissue. In the standard assay, secretin is given IV in a dose of 0.2 μg/kg of synthetic human secretin as a bolus. Normal values for the standard secretin test are (1) volume output >2 mL/kg per hour, (2) bicarbonate (HCO3–) concentration >80 mmol/L, and (3) HCO3– output >10 mmol/L in 1 h. The most reproducible measurement, giving the highest level of discrimination between normal subjects and patients with chronic pancreatic exocrine insufficiency, appears to be the maximal bicarbonate concentration. A cutoff point below 80 mmol/L is considered abnormal and suggestive of abnormal secretory function that is most commonly observed in early chronic pancreatitis.
There may be a dissociation between the results of the secretin test and other tests of absorptive function. For example, patients with chronic pancreatitis often have abnormally low outputs of HCO3– after secretin but have normal fecal fat excretion. Thus the secretin test measures the secretory capacity of ductular epithelium, whereas fecal fat excretion indirectly reflects intraluminal lipolytic activity. Steatorrhea does not occur until intraluminal levels of lipase are markedly reduced, underscoring the fact that only small amounts of enzymes are necessary for intraluminal digestive activities. It must be emphasized that an abnormal secretin test result suggests only that chronic pancreatic damage is present.
2. Measurement of fecal pancreatic enzymes such as elastase
Measurement of intraluminal digestion products (i.e., undigested muscle fibers, stool fat, and fecal nitrogen) is discussed in Chap. 349. The amount of human elastase in stool reflects the pancreatic output of this proteolytic enzyme. Decreased elastase-1 activity (FE-1) in stool is an excellent test to detect severe pancreatic exocrine insufficiency (PEI) in patients with chronic pancreatitis and cystic fibrosis. FE-1 levels >200 μg/g are normal; levels of 100–200 μg/g are considered mild, and levels <100 μg/g are severe for PEI. Although the test is simple and noninvasive, it can give false-positive results and has a low sensitivity. Fecal levels <50 μg/g are definitive for PEI provided that the stool specimen is solid.
Tests useful in the diagnosis of exocrine pancreatic insufficiency and the differential diagnosis of malabsorption are also discussed in Chaps. 349 and 371.
371 |
Acute and Chronic Pancreatitis |
BIOCHEMISTRY AND PHYSIOLOGY OF PANCREATIC EXOCRINE SECRETION
GENERAL CONSIDERATIONS
The pancreas secretes 1500–3000 mL of isosmotic alkaline (pH >8) fluid per day containing about 20 enzymes. The pancreatic secretions provide the enzymes and bicarbonate needed to affect the major digestive activity of the gastrointestinal tract and provide an optimal pH for the function of these enzymes.
REGULATION OF PANCREATIC SECRETION
The exocrine pancreas is influenced by intimately interacting hormonal and neural systems. Gastric acid is the stimulus for the release of secretin from the duodenal mucosa (S cells), which stimulates the secretion of water and electrolytes from pancreatic ductal cells. Release of cholecystokinin (CCK) from the duodenal and proximal jejunal mucosa (Ito cells) is largely triggered by long-chain fatty acids, essential amino acids (tryptophan, phenylalanine, valine, methionine), and gastric acid itself. CCK evokes an enzyme-rich secretion from acinar cells in the pancreas. The parasympathetic nervous system (via the vagus nerve) exerts significant control over pancreatic secretion. Secretion evoked by secretin and CCK depends on permissive roles of vagal afferent and efferent pathways. This is particularly true for enzyme secretion, whereas water and bicarbonate secretions are heavily dependent on the hormonal effects of secretin and to a lesser extent CCK. Also, vagal stimulation affects the release of vasoactive intestinal peptide (VIP), a secretin agonist. Pancreatic exocrine secretion is also influenced by inhibitory neuropeptides such as somatostatin, pancreatic polypeptide, peptide YY, neuropeptide Y, enkephalin, pancreastatin, calcitonin gene–related peptides, glucagon, and galanin. Although pancreatic polypeptide and peptide YY may act primarily on nerves outside the pancreas, somatostatin acts at multiple sites. Nitric oxide (NO) is also an important neurotransmitter.
WATER AND ELECTROLYTE SECRETION
Bicarbonate is the ion of primary physiologic importance within pancreatic secretion. The ductal cells secrete bicarbonate predominantly derived from plasma (93%) more than from intracellular metabolism (7%). Bicarbonate enters the duct lumen through the sodium bicarbonate cotransporter with depolarization caused by chloride efflux through the cystic fibrosis transmembrane conductance regulator (CFTR). Secretin and VIP bind at the basolateral surface and cause an increase in secondary messenger intracellular cyclic AMP, and act on the apical surface of the ductal cells opening the CFTR in promoting secretion. CCK, acting as a neuromodulator, markedly potentiates the stimulatory effects of secretin. Acetylcholine also plays an important role in ductal cell secretion. Intraluminal bicarbonate secreted from the ductal cells helps neutralize gastric acid and creates the appropriate pH for the activity of pancreatic enzymes and bile salts on ingested food.
ENZYME SECRETION
The acinar cell is highly compartmentalized and is concerned with the secretion of pancreatic enzymes. Proteins synthesized by the rough endoplasmic reticulum are processed in the Golgi and then targeted to the appropriate site, whether that be zymogen granules, lysosomes, or other cell compartments. The zymogen granules migrate to the apical region of the acinar cell awaiting the appropriate neural or hormonal stimulatory response. The pancreas secretes amylolytic, lipolytic, and proteolytic enzymes into the duct lumen. Amylolytic enzymes, such as amylase, hydrolyze starch to oligosaccharides and to the disaccharide maltose. The lipolytic enzymes include lipase, phospholipase A2, and cholesterol esterase. Bile salts inhibit lipase in isolation, but colipase, another constituent of pancreatic secretion, binds to lipase and prevents this inhibition. Bile salts activate phospholipase A and cholesterol esterase. Proteolytic enzymes include endopeptidases (trypsin, chymotrypsin), which act on internal peptide bonds of proteins and polypeptides; exopeptidases (carboxypeptidases, aminopeptidases), which act on the free carboxyl- and amino-terminal ends of peptides, respectively; and elastase. The proteolytic enzymes are secreted as inactive zymogen precursors. Ribonucleases (deoxyribonucleases, ribonuclease) are also secreted. Enterokinase, an enzyme found in the duodenal mucosa, cleaves the lysine-isoleucine bond of trypsinogen to form trypsin. Trypsin then activates the other proteolytic zymogens and phospholipase A2 in a cascade phenomenon. All pancreatic enzymes have pH optima in the alkaline range. The nervous system initiates pancreatic enzyme secretion. The neurologic stimulation is cholinergic, involving extrinsic innervation by the vagus nerve and subsequent innervation by intrapancreatic cholinergic nerves. The stimulatory neurotransmitters are acetylcholine and gastrin-releasing peptides. These neurotransmitters activate calcium-dependent secondary messenger systems, resulting in the release of zymogens into the pancreas duct. VIP is present in intrapancreatic nerves and potentiates the effect of acetylcholine. In contrast to other species, there are no CCK receptors on acinar cells in humans. CCK in physiologic concentrations stimulates pancreatic secretion by stimulating afferent vagal and intrapancreatic nerves.
AUTOPROTECTION OF THE PANCREAS
Autodigestion of the pancreas is prevented by (1) the packaging of pancreatic proteases in precursor (proenzyme) form, (2) intracellular calcium homeostasis (low intracellular calcium in the cytosol of the acinar cell promotes the destruction of spontaneously activated trypsin), (3) acid-base balance, and (4) the synthesis of protective protease inhibitors (pancreatic secretory trypsin inhibitor [PSTI] or SPINK1), which can bind and inactivate about 20% of intracellular trypsin activity. Chymotrypsin C can also lyse and inactivate trypsin. These protease inhibitors are found in the acinar cell, the pancreatic secretions, and the α1– and α2-globulin fractions of plasma. Loss of any of these four protective mechanisms leads to premature enzyme activation, autodigestion, and acute pancreatitis.
ENTEROPANCREATIC AXIS AND FEEDBACK INHIBITION
Pancreatic enzyme secretion is controlled, at least in part, by a negative feedback mechanism induced by the presence of active serine proteases in the duodenum. To illustrate, perfusion of the duodenal lumen with phenylalanine (stimulates early digestion) causes a prompt increase in plasma CCK levels as well as increased secretion of chymotrypsin and other pancreatic enzymes. However, simultaneous perfusion with trypsin (stimulates late digestion) blunts both responses. Conversely, perfusion of the duodenal lumen with protease inhibitors actually leads to enzyme hypersecretion. The available evidence supports the concept that the duodenum contains a peptide called CCK-releasing factor (CCK-RF) that is involved in stimulating CCK release. It appears that serine proteases inhibit pancreatic secretion by inactivating a CCK-releasing peptide in the lumen of the small intestine. Thus, the integrative result of both bicarbonate and enzyme secretion depends on a feedback process for both bicarbonate and pancreatic enzymes. Acidification of the duodenum releases secretin, which stimulates vagal and other neural pathways to activate pancreatic duct cells, which secrete bicarbonate. This bicarbonate then neutralizes the duodenal acid, and the feedback loop is completed. Dietary proteins bind proteases, thereby leading to an increase in free CCK-RF. CCK is then released into the blood in physiologic concentrations, acting primarily through the neural pathways (vagal-vagal). This leads to acetylcholine-mediated pancreatic enzyme secretion. Proteases continue to be secreted from the pancreas until the protein within the duodenum is digested. At this point, pancreatic protease secretion is reduced to basic levels, thus completing this step in the feedback process.
ACUTE PANCREATITIS
GENERAL CONSIDERATIONS
Recent U.S. estimates from the National Inpatient Sample report that acute pancreatitis is the most common inpatient principal gastrointestinal diagnosis. The incidence of acute pancreatitis also varies in different countries and depends on cause (e.g., alcohol, gallstones, metabolic factors, drugs [Table 371-1]). The annual incidence ranges from 13–45/100,000 persons. Acute pancreatitis results in >250,000 hospitalizations per year. The median length of hospital stay is 4 days, with a median hospital cost of $6,096 and a mortality of 1%. The estimated cost annually approaches $2.6 billion. Hospitalization rates increase with age, are 88% higher among blacks, and are higher among males than females. The age-adjusted rate of hospital discharges with an acute pancreatitis diagnosis increased 62% between 1988 and 2004. From 2000 to 2009, the rate increased 30%. Thus, acute pancreatitis is increasing and is a significant burden on health care costs and resource utilization.
|
CAUSES OF ACUTE PANCREATITIS |
ETIOLOGY AND PATHOGENESIS
There are many causes of acute pancreatitis (Table 371-1), but the mechanisms by which these conditions trigger pancreatic inflammation have not been fully elucidated. Gallstones continue to be the leading cause of acute pancreatitis in most series (30–60%). The risk of acute pancreatitis in patients with at least one gallstone <5 mm in diameter is fourfold greater than that in patients with larger stones. Alcohol is the second most common cause, responsible for 15–30% of cases in the United States. The incidence of pancreatitis in alcoholics is surprisingly low (5/100,000), indicating that in addition to the amount of alcohol ingested, other factors affect a person’s susceptibility to pancreatic injury such as cigarette smoking. Acute pancreatitis occurs in 5–10% of patients following endoscopic retrograde cholangiopancreatography (ERCP). Use of a prophylactic pancreatic duct stent and rectal nonsteroidal anti-inflammatory drugs (NSAIDs) has been shown to reduce pancreatitis after ERCP. Risk factors for post-ERCP pancreatitis include minor papilla sphincterotomy, sphincter of Oddi dysfunction, prior history of post-ERCP pancreatitis, age <60 years, >2 contrast injections into the pancreatic duct, and endoscopic trainee involvement.
Hypertriglyceridemia is the cause of acute pancreatitis in 1.3–3.8% of cases; serum triglyceride levels are usually >11.3 mmol/L (>1000 mg/dL). Most patients with hypertriglyceridemia, when subsequently examined, show evidence of an underlying derangement in lipid metabolism, probably unrelated to pancreatitis. Such patients are prone to recurrent episodes of pancreatitis. Any factor (e.g., drugs or alcohol) that causes an abrupt increase in serum triglycerides can precipitate a bout of acute pancreatitis. Patients with a deficiency of apolipoprotein CII have an increased incidence of pancreatitis; apolipoprotein CII activates lipoprotein lipase, which is important in clearing chylomicrons from the bloodstream. Patients with diabetes mellitus who have developed ketoacidosis and patients who are on certain medications such as oral contraceptives may also develop high triglyceride levels. Approximately 0.1–2% of cases of acute pancreatitis are drug related. Drugs cause pancreatitis either by a hypersensitivity reaction or by the generation of a toxic metabolite, although in some cases, it is not clear which of these mechanisms is operative (Table 371-1).
Pathologically, acute pancreatitis varies from interstitial pancreatitis (pancreas blood supply maintained), which is generally self-limited to necrotizing pancreatitis (pancreas blood supply interrupted), in which the extent of necrosis may correlate with the severity of the attack and its systemic complications. Autodigestion is a currently accepted pathogenic theory; according to this theory, pancreatitis results when proteolytic enzymes (e.g., trypsinogen, chymotrypsinogen, proelastase, and lipolytic enzymes such as phospholipase A2) are activated in the pancreas acinar cell rather than in the intestinal lumen. A number of factors (e.g., endotoxins, exotoxins, viral infections, ischemia, oxidative stress, lysosomal calcium, and direct trauma) are believed to facilitate premature activation of trypsin. Activated proteolytic enzymes, especially trypsin, not only digest pancreatic and peripancreatic tissues but also can activate other enzymes, such as elastase and phospholipase A2. Spontaneous activation of trypsin also can occur.
ACTIVATION OF PANCREATIC ENZYMES IN THE PATHOGENESIS OF ACUTE PANCREATITIS
Several recent studies have suggested that pancreatitis is a disease that evolves in three phases. The initial phase is characterized by intrapancreatic digestive enzyme activation and acinar cell injury. Trypsin activation appears to be mediated by lysosomal hydrolases such as cathepsin B that become colocalized with digestive enzymes in intracellular organelles; it is currently believed that acinar cell injury is the consequence of trypsin activation. The second phase of pancreatitis involves the activation, chemoattraction, and sequestration of leukocytes and macrophages in the pancreas, resulting in an enhanced intrapancreatic inflammatory reaction. Neutrophil depletion induced by prior administration of an antineutrophil serum has been shown to reduce the severity of experimentally induced pancreatitis. There is also evidence to support the concept that neutrophils can activate trypsinogen. Thus, intrapancreatic acinar cell activation of trypsinogen could be a two-step process (i.e., an early neutrophil-independent and a later neutrophil-dependent phase). The third phase of pancreatitis is due to the effects of activated proteolytic enzymes and cytokines, released by the inflamed pancreas, on distant organs. Activated proteolytic enzymes, especially trypsin, not only digest pancreatic and peripancreatic tissues but also activate other enzymes such as elastase and phospholipase A2. The active enzymes and cytokines then digest cellular membranes and cause proteolysis, edema, interstitial hemorrhage, vascular damage, coagulation necrosis, fat necrosis, and parenchymal cell necrosis. Cellular injury and death result in the liberation of bradykinin peptides, vasoactive substances, and histamine that can produce vasodilation, increased vascular permeability, and edema with profound effects on many organs. The systemic inflammatory response syndrome (SIRS) and acute respiratory distress syndrome (ARDS), as well as multiorgan failure, may occur as a result of this cascade of local and distant effects.
A number of genetic factors can increase the susceptibility and/or modify the severity of pancreatic injury in acute pancreatitis, recurrent pancreatitis, and chronic pancreatitis. All of the major genetic susceptibility factors center on the control of trypsin activity within the pancreatic acinar cell, in part because they were identified as candidate genes linked to intrapancreatic trypsin control. Five genetic variants have been identified as being associated with susceptibility to pancreatitis. The genes that have been identified include (1) cationic trypsinogen gene (PRSS1), (2) pancreatic secretory trypsin inhibitor (SPINK1), (3) the cystic fibrosis transmembrane conductance regulator gene (CFTR), (4) the chymotrypsin C gene (CTRC), and (5) the calcium-sensing receptor (CASR). Investigations of other genetic variants are currently under way, and new genes will be added to this list in the future. Multiple medical, ethical, and psychological issues arise when these genes are discovered, and referral to genetic counselors is recommended.
LABORATORY DATA
Serum amylase and lipase values threefold or more above normal virtually clinch the diagnosis if gut perforation, ischemia, and infarction are excluded. Serum lipase is the preferred test. However, it should be noted that there is no correlation between the severity of pancreatitis and the degree of serum lipase and amylase elevations. After 3–7 days, even with continuing evidence of pancreatitis, total serum amylase values tend to return toward normal. However, pancreatic isoamylase and lipase levels may remain elevated for 7–14 days. It should be recognized that amylase elevations in serum and urine occur in many conditions other than pancreatitis (see Chap. 370, Table 370-2). Importantly, patients with acidemia (arterial pH ≤7.32) may have spurious elevations in serum amylase. This finding explains why patients with diabetic ketoacidosis may have marked elevations in serum amylase without any other evidence of acute pancreatitis. Serum lipase activity increases in parallel with amylase activity and is more specific than amylase. A serum lipase measurement can be instrumental in differentiating a pancreatic or nonpancreatic cause for hyperamylasemia. Leukocytosis (15,000–20,000 leukocytes/μL) occurs frequently. Patients with more severe disease may show hemoconcentration with hematocrit values >44% and/or prerenal azotemia with a blood urea nitrogen (BUN) level >22 mg/dL resulting from loss of plasma into the retroperitoneal space and peritoneal cavity.
|
REVISED ATLANTA DEFINITIONS OF MORPHOLOGIC FEATURES OF ACUTE PANCREATITIS |
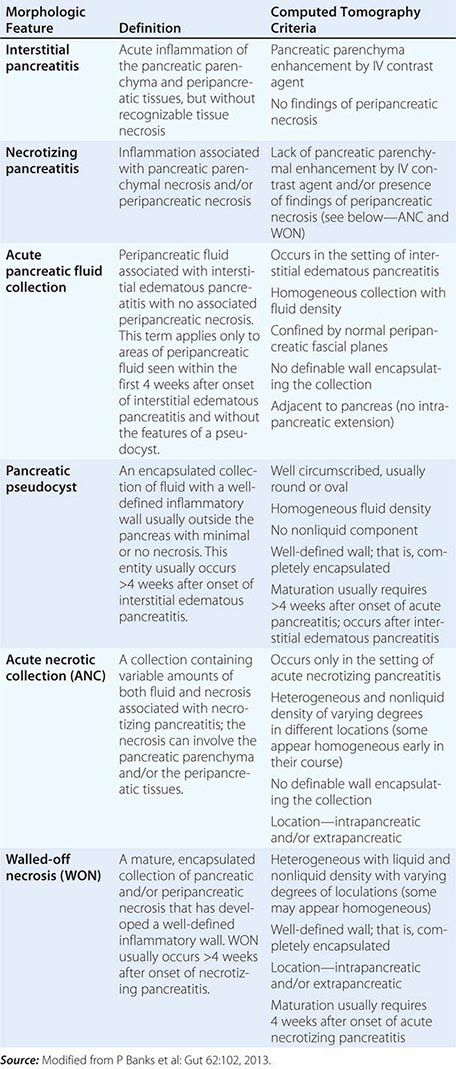
Hemoconcentration may be the harbinger of more severe disease (i.e., pancreatic necrosis), whereas azotemia is a significant risk factor for mortality. Hyperglycemia is common and is due to multiple factors, including decreased insulin release, increased glucagon release, and an increased output of adrenal glucocorticoids and catecholamines. Hypocalcemia occurs in ~25% of patients, and its pathogenesis is incompletely understood. Although earlier studies suggested that the response of the parathyroid gland to a decrease in serum calcium is impaired, subsequent observations have failed to confirm this phenomenon. Intraperitoneal saponification of calcium by fatty acids in areas of fat necrosis occurs occasionally, with large amounts (up to 6.0 g) dissolved or suspended in ascitic fluid. Such “soap formation” may also be significant in patients with pancreatitis, mild hypocalcemia, and little or no obvious ascites. Hyperbilirubinemia (serum bilirubin >68 mmoL or >4.0 mg/dL) occurs in ~10% of patients. However, jaundice is transient, and serum bilirubin levels return to normal in 4–7 days. Serum alkaline phosphatase and aspartate aminotransferase levels are also transiently elevated, and they parallel serum bilirubin values and may point to gallbladder-related disease or inflammation in the pancreatic head. Hypertriglyceridemia occurs in 5–10% of patients, and serum amylase levels in these individuals are often spuriously normal (Chap. 370). Approximately 5–10% of patients have hypoxemia (arterial PO2 ≤60 mmHg), which may herald the onset of ARDS. Finally, the electrocardiogram is occasionally abnormal in acute pancreatitis with ST-segment and T-wave abnormalities simulating myocardial ischemia.
An abdominal ultrasound is recommended in the emergency ward as the initial diagnostic imaging modality and is most useful to evaluate for gallstone disease and the pancreatic head.
The revised Atlanta criteria have clearly outlined the morphologic features of acute pancreatitis on computed tomography (CT) scan as follows: (1) interstitial pancreatitis, (2) necrotizing pancreatitis, (3) acute pancreatic fluid collection, (4) pancreatic pseudocyst, (5) acute necrotic collection (ANC), and (6) walled-off pancreatic necrosis (WON) (Table 371-2 and Fig. 371-1). Radiologic studies useful in the diagnosis of acute pancreatitis are discussed in Chap. 370 and listed in Table 370-1.
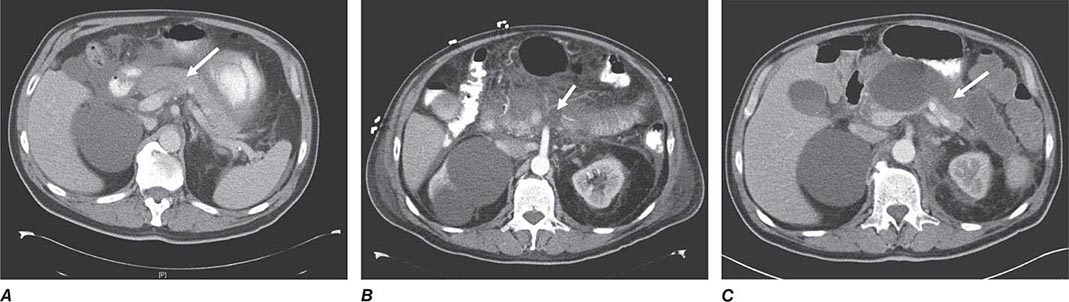
FIGURE 371-1 Acute pancreatitis: computed tomography (CT) evolution. A. Contrast-enhanced CT scan of the abdomen performed on admission for a patient with clinical and biochemical parameters suggestive of acute pancreatitis. Note the abnormal enhancement of the pancreatic parenchyma (arrow) suggestive of interstitial pancreatitis. B. Contrast-enhanced CT scan of the abdomen performed on the same patient 6 days later for persistent fever and systemic inflammatory response syndrome. The pancreas now demonstrates significant areas of nonenhancement consistent with development of necrosis, particularly in the body and neck region (arrow). Note that an early CT scan obtained within the first 48 h of hospitalization may underestimate or miss necrosis. C. Contrast-enhanced CT scan of the abdomen performed on the same patient 2 months after the initial episode of acute pancreatitis. CT now demonstrates evidence of a fluid collection consistent with walled-off pancreatic necrosis (arrow). (Courtesy of Dr. KJ Mortele, Brigham and Women’s Hospital; with permission.)
DIAGNOSIS
Any severe acute pain in the abdomen or back should suggest the possibility of acute pancreatitis. The diagnosis is established by two of the following three criteria: (1) typical abdominal pain in the epigastrium that may radiate to the back, (2) threefold or greater elevation in serum lipase and/or amylase, and (3) confirmatory findings of acute pancreatitis on cross-sectional abdominal imaging. Patients also have associated nausea, emesis, fever, tachycardia, and abnormal findings on abdominal examination. Laboratory studies may reveal leukocytosis, hypocalcemia, and hyperglycemia. Although not required for diagnosis, markers of severity may include hemoconcentration (hematocrit >44%), admission azotemia (BUN >22 mg/dL), SIRS, and signs of organ failure (Table 371-3).
|
SEVERE ACUTE PANCREATITIS |
Abbreviations: APACHE II, Acute Physiology and Chronic Health Evaluation II; BMI, body mass index; BISAP, Bedside Index of Severity in Acute Pancreatitis; BP, blood pressure; BUN, blood urea nitrogen; SIRS, systemic inflammatory response syndrome.
The differential diagnosis should include the following disorders: (1) perforated viscus, especially peptic ulcer; (2) acute cholecystitis and biliary colic; (3) acute intestinal obstruction; (4) mesenteric vascular occlusion; (5) renal colic; (6) inferior myocardial infarction; (7) dissecting aortic aneurysm; (8) connective tissue disorders with vasculitis; (9) pneumonia; and (10) diabetic ketoacidosis. It may be difficult to differentiate acute cholecystitis from acute pancreatitis, because an elevated serum amylase may be found in both disorders. Pain of biliary tract origin is more right sided or epigastric than periumbilical or left upper quadrant and can be more severe; ileus is usually absent. Ultrasound is helpful in establishing the diagnosis of cholelithiasis and cholecystitis. Intestinal obstruction due to mechanical factors can be differentiated from pancreatitis by the history of crescendo-decrescendo pain, findings on abdominal examination, and CT of the abdomen showing changes characteristic of mechanical obstruction. Acute mesenteric vascular occlusion is usually suspected in elderly debilitated patients with brisk leukocytosis, abdominal distention, and bloody diarrhea, confirmed by CT or magnetic resonance angiography. Vasculitides secondary to systemic lupus erythematosus and polyarteritis nodosa may be confused with pancreatitis, especially because pancreatitis may develop as a complication of these diseases. Diabetic ketoacidosis is often accompanied by abdominal pain and elevated total serum amylase levels, thus closely mimicking acute pancreatitis. However, the serum lipase level is not elevated in diabetic ketoacidosis.
CLINICAL COURSE, DEFINITIONS, AND CLASSIFICATIONS
The Revised Atlanta Classification (1) defines phases of acute pancreatitis, (2) defines severity of acute pancreatitis, and (3) clarifies imaging definitions as outlined below.
Phases of Acute Pancreatitis Two phases of acute pancreatitis have been defined, early (<2 weeks) and late (>2 weeks), which primarily describes the hospital course of the disease. In the early phase of acute pancreatitis, which lasts 1–2 weeks, severity is defined by clinical parameters rather than morphologic findings. Most patients exhibit SIRS, and if this persists, patients are predisposed to organ failure. Three organ systems should be assessed to define organ failure: respiratory, cardiovascular, and renal. Organ failure is defined as a score of 2 or more for one of these three organ systems using the modified Marshall scoring system. Persistent organ failure (>48 h) is the most important clinical finding in regard to severity of the acute pancreatitis episode. Organ failure that affects more than one organ is considered multisystem organ failure. CT imaging is usually not needed or recommended during the first 48 h of admission in acute pancreatitis.
The late phase is characterized by a protracted course of illness and may require imaging to evaluate for local complications. The important clinical parameter of severity, as in the early phase, is persistent organ failure. These patients may require supportive measures such as renal dialysis, ventilator support, or need for supplemental nutrition via the nasojejunal or parenteral route. The radiographic feature of greatest importance to recognize in this phase is the development of necrotizing pancreatitis on CT imaging. Necrosis generally prolongs hospitalization and, if infected, may require operative, endoscopic, or percutaneous intervention.
Severity of Acute Pancreatitis Three severity classifications have also been defined: mild, moderately severe, and severe. Mild acute pancreatitis is without local complications or organ failure. Most patients with interstitial acute pancreatitis have mild pancreatitis. In mild acute pancreatitis, the disease is self-limited and subsides spontaneously, usually within 3–7 days after treatment is instituted. Oral intake can be resumed if the patient is hungry, has normal bowel function, and is without nausea and vomiting. Typically, a clear or full liquid diet has been recommended for the initial meal; however, a low-fat solid diet is a reasonable choice following recovery from mild acute pancreatitis.
Moderately severe acute pancreatitis is characterized by transient organ failure (resolves in <48 h) or local or systemic complications in the absence of persistent organ failure. These patients may or may not have necrosis, but may develop a local complication such as a fluid collection that requires a prolonged hospitalization greater than 1 week.
Severe acute pancreatitis is characterized by persistent organ failure (>48 h). Organ failure can be single or multiple. A CT scan or magnetic resonance imaging (MRI) should be obtained to assess for necrosis and/or complications. If a local complication is encountered, management is dictated by clinical symptoms, evidence of infection, maturity of fluid collection, and clinical stability of the patient. Prophylactic antibiotics are not recommended.
Imaging in Acute Pancreatitis Two types of pancreatitis are recognized on imaging as interstitial or necrotizing based on pancreatic perfusion. CT imaging is best evaluated 3–5 days into hospitalization when patients are not responding to supportive care to look for local complications such as necrosis. Recent studies report the overutilization of CT imaging in acute pancreatitis and its inability to be better than clinical judgment in the early days of acute pancreatitis management. The revised criteria also outline the terminology for local complications and fluid collections along with a CT imaging template to guide reporting of findings. Local morphologic features are summarized in Table 371-1. Interstitial pancreatitis occurs in 90–95% of admissions for acute pancreatitis and is characterized by diffuse gland enlargement, homogenous contrast enhancement, and mild inflammatory changes or peripancreatic stranding. Symptoms generally resolve with a week of hospitalization. Necrotizing pancreatitis occurs in 5–10% of acute pancreatitis admissions and does not evolve until several days of hospitalization. It is characterized by lack of pancreatic parenchymal enhancement by intravenous contrast agent and/or presence of findings of peripancreatic necrosis. According to the revised Atlanta criteria, the natural history of pancreatic and peripancreatic necrosis is variable because it may remain solid or liquefy, remain sterile or become infected, and persist or disappear over time. CT identification of local complications, particularly necrosis, is critical in patients who are not responding to therapy because patients with infected and sterile necrosis are at greatest risk of mortality (Figs. 371-1B, 371-2, and 371-3). The median prevalence of organ failure is 54% in necrotizing pancreatitis. The prevalence of organ failure is perhaps slightly higher in infected versus sterile necrosis. With single-organ system failure, the mortality is 3–10% but increases to 47% with multisystem organ failure.
FIGURE 371-2 A. Acute necrotizing pancreatitis: computed tomography (CT) scan. Contrast-enhanced CT scan showing acute pancreatitis with necrosis. Arrow shows partially enhancing body/tail of pancreas surrounded by fluid with decreased enhancement in the neck/body of the pancreas. B. Acute fluid collection: CT scan. Contrast-enhanced CT scan showing fluid collection in the retroperitoneum (arrow) compressing the air-filled stomach arising from the pancreas in a patient with asparaginase-induced acute necrotizing pancreatitis. C. Walled-off pancreatic necrosis: CT scan. CT scan showing marked walled-off necrosis of the pancreas and peripancreatic area (arrow) in a patient with necrotizing pancreatitis. Addendum: In past years, both of these CT findings (Figs. 371-2B and 371-2C) would have been misinterpreted as pseudocysts. D. Spiral CT showing a pseudocyst (small arrow) with a pseudoaneurysm (light area in pseudocyst). Note the demonstration of the main pancreatic duct (big arrow), even though this duct is minimally dilated by endoscopic retrograde cholangiopancreatography. (A, B, C, courtesy of Dr. KJ Mortele, Brigham and Women’s Hospital; D, courtesy of Dr. PR Ros, Brigham and Women’s Hospital; with permission.)
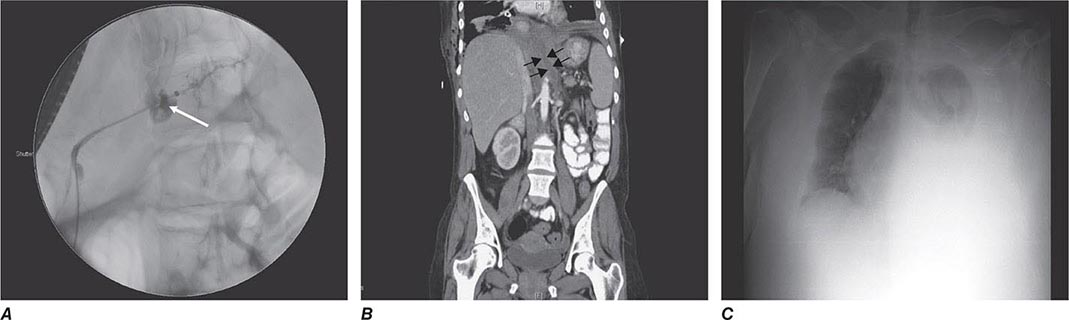
FIGURE 371-3 A. Pancreaticopleural fistula: pancreatic duct leak on endoscopic retrograde cholangiopancreatography. Pancreatic duct leak (arrow) demonstrated at the time of retrograde pancreatogram in a patient with acute exacerbation of alcohol-induced acute or chronic pancreatitis. B. Pancreaticopleural fistula: computed tomography (CT) scan. Contrast-enhanced CT scan (coronal view) with arrows showing fistula tract from pancreatic duct disruption in the pancreatic pleural fistula. C. Pancreaticopleural fistula: chest x-ray. Large pleural effusion in the left hemithorax from a disrupted pancreatic duct. Analysis of pleural fluid revealed elevated amylase concentration. (Courtesy of Dr. KJ Mortele, Brigham and Women’s Hospital; with permission.)
ACUTE PANCREATITIS MANAGEMENT
We will briefly describe the management of patients with acute pancreatitis from the time of diagnosis in the emergency ward to ongoing hospital admission and, finally, to time of discharge, highlighting salient features based on severity and complications. It is important to note that 85–90% of cases of acute pancreatitis are self-limited and subside spontaneously, usually within 3–7 days after initiation of treatment, and do not exhibit organ failure or local complications.
The management of acute pancreatitis begins in the emergency ward. After a diagnosis has been confirmed, aggressive fluid resuscitation is initiated, intravenous analgesics are administered, severity is assessed, and a search for etiologies that may impact acute care is begun. Patients who do not respond to aggressive fluid resuscitation in the emergency ward should be considered for admission to a step-down or intensive care unit for aggressive fluid resuscitation, hemodynamic monitoring, and management of necrosis or organ failure.
Fluid Resuscitation and Monitoring Response to Therapy The most important treatment intervention for acute pancreatitis is safe, aggressive intravenous fluid resuscitation. The patient is made NPO to rest the pancreas and is given intravenous narcotic analgesics to control abdominal pain and supplemental oxygen (2 L) via nasal cannula.
Intravenous fluids of lactated Ringer’s or normal saline are initially bolused at 15–20 cc/kg (1050–1400 mL), followed by 3 mg/kg per hour (200–250 mL/h), to maintain urine output >0.5 cc/kg per hour. Serial bedside evaluations are required every 6–8 h to assess vital signs, oxygen saturation, and change in physical examination. Lactated Ringer’s solution has been shown to decrease systemic inflammation and may be a better crystalloid than normal saline. A targeted resuscitation strategy with measurement of hematocrit and BUN every 8–12 h is recommended to ensure adequacy of fluid resuscitation and monitor response to therapy, noting less aggressive resuscitation strategy may be needed in milder forms of pancreatitis. A rising BUN during hospitalization is not only associated with inadequate hydration but also higher in-hospital mortality.
A decrease in hematocrit and BUN during the first 12–24 h is strong evidence that sufficient fluids are being administered. Serial measurements and bedside assessment for fluid overload are continued, and fluid rates are maintained at the current rate. Adjustments in fluid resuscitation may be required in patients with cardiac, pulmonary, or renal disease. A rise in hematocrit or BUN during serial measurement should be treated with a repeat volume challenge with a 2-L crystalloid bolus followed by increasing the fluid rate by 1.5 mg/kg per hour. If the BUN or hematocrit fails to respond (i.e., remains elevated or does not decrease) to this bolus challenge and increase in fluid rate, consideration of transfer to an intensive care unit is strongly recommended for hemodynamic monitoring.
Assessment of Severity and Hospital Triage Severity of acute pancreatitis should be determined in the emergency ward to assist in patient triage to a regular hospital ward or step-down unit or direct admission to an intensive care unit. The Bedside Index of Severity in Acute Pancreatitis (BISAP) incorporates five clinical and laboratory parameters obtained within the first 24 h of hospitalization (Table 371-3)—BUN >25 mg/dL, impaired mental status (Glasgow coma score <15), SIRS, age >60 years, and pleural effusion on radiography—that can be useful in assessing severity. Presence of three or more of these factors was associated with substantially increased risk for in-hospital mortality among patients with acute pancreatitis. In addition, an elevated hematocrit >44% and admission BUN >22 mg/dL are also associated with more severe acute pancreatitis. Incorporating these indices with the overall patient response to initial fluid resuscitation in the emergency ward can be useful at triaging patients to the appropriate hospital acute care setting.
In general, patients with lower BISAP scores, hematocrits, and admission BUNs tend to respond to initial management and are triaged to a regular hospital ward for ongoing care. If SIRS is not present at 24 h, the patient is unlikely to develop organ failure or necrosis. Therefore, patients with persistent SIRS at 24 h or underlying comorbid illnesses (e.g., chronic obstructive pulmonary disease, congestive heart failure) should be considered for a step-down unit setting if available. Patients with higher BISAP scores and elevations in hematocrit and admission BUN that do not respond to initial fluid resuscitation and exhibit evidence of respiratory failure, hypotension, or organ failure should be considered for direct admission to an intensive care unit.
Special Considerations Based on Etiology A careful history, review of medications, selected laboratory studies (liver profile, serum triglycerides, serum calcium), and an abdominal ultrasound are recommended in the emergency ward to assess for etiologies that may impact acute management. An abdominal ultrasound is the initial imaging modality of choice and will evaluate the gallbladder and common duct and assess the pancreatic head.
GALLSTONE PANCREATITIS Patients with evidence of ascending cholangitis (rising white blood cell count, increasing liver enzymes) should undergo ERCP within 24–48 h of admission. Patients with gallstone pancreatitis are at increased risk of recurrence, and consideration should be given to performing a cholecystectomy during the same admission or within 4–6 weeks of discharge. An alternative for patients who are not surgical candidates would be to perform an endoscopic biliary sphincterotomy before discharge.
HYPERTRIGLYCERIDEMIA Serum triglycerides >1000 mg/dL are associated with acute pancreatitis. Initial therapy may include insulin, heparin, or plasmapheresis. Outpatient therapies include control of diabetes if present, administration of lipid-lowering agents, weight loss, and avoidance of drugs that elevate lipid levels.
Other potential etiologies that may impact acute hospital care include hypercalcemia, autoimmune pancreatitis, post-ERCP pancreatitis, and drug-induced pancreatitis. Treatment of hyperparathyroidism or malignancy is effective at reducing serum calcium. Autoimmune pancreatitis is responsive to glucocorticoid administration. Pancreatic duct stenting and rectal indomethacin administration are effective at decreasing pancreatitis after ERCP. Drugs that cause pancreatitis should be discontinued. Multiple drugs have been implicated, but only about 30 have been challenged (Class 1A) and found to be causative.
Nutritional Therapy A low-fat solid diet can be administered to subjects with mild acute pancreatitis after the abdominal pain has resolved. Enteral nutrition should be considered 2–3 days after admission in subjects with more severe pancreatitis instead of total parenteral nutrition (TPN). Enteral feeding maintains gut barrier integrity, limits bacterial translocation, is less expensive, and has fewer complications than TPN. The choice of gastric versus nasojejunal enteral feeding is currently under investigation.
Management of Local Complications (Table 371-4) Patients exhibiting signs of clinical deterioration despite aggressive fluid resuscitation and hemodynamic monitoring should be assessed for local complications, which may include necrosis, pseudocyst formation, pancreas duct disruption, peripancreatic vascular complications, and extrapancreatic infections. A multidisciplinary team approach is recommended including gastroenterology, surgery, interventional radiology, and intensive care specialists, and consideration should also be made for transfer to a pancreas center.
|
COMPLICATIONS OF ACUTE PANCREATITIS |
NECROSIS The management of necrosis requires a multidisciplinary team approach. Percutaneous aspiration of necrosis with Gram stain and culture should be performed if there are ongoing signs of possible pancreatic infection such as sustained leukocytosis, fever, or organ failure. There is currently no role for prophylactic antibiotics in necrotizing pancreatitis. It is reasonable to start broad-spectrum antibiotics in a patient who appears septic while awaiting the results of Gram stain and cultures. If cultures are negative, the antibiotics should be discontinued to minimize the risk of developing opportunistic or fungal superinfection. Repeated fine-needle aspiration and Gram stain with culture of pancreatic necrosis may be done every 5–7 days in the presence of persistent fever. Repeated CT or MRI imaging should also be considered with any change in clinical course to monitor for complications (e.g., thromboses, hemorrhage, abdominal compartment syndrome).
In general, sterile necrosis is most often managed conservatively unless complications arise. Once a diagnosis of infected necrosis is established and an organism identified, targeted antibiotics should be instituted. Pancreatic debridement (necrosectomy) should be considered for definitive management of infected necrosis, but clinical decisions are generally influenced by response to antibiotic treatment and overall clinical condition. Symptomatic local complications as outlined in the revised Atlanta criteria may require definitive therapy.
A step-up approach (percutaneous or endoscopic transgastric drainage followed, if necessary, by open necrosectomy) has been successfully reported by some pancreatic centers. One-third of the patients successfully treated with the step-up approach did not require major abdominal surgery. A recent randomized trial reported advantages to an initial endoscopic approach compared to an initial surgical necrosectomy approach in select patients requiring intervention for symptomatic WON. Taken together, a more conservative approach to the management of infected pancreatic necrosis has evolved over the years under the close supervision of a multidisciplinary team. If conservative therapy can be safely implemented for 4–6 weeks, to allow the pancreatic collections to resolve or “wall-off,” surgical or endoscopic intervention is generally much safer and better tolerated by the patient.
PSEUDOCYST The incidence of pseudocyst is low, and most acute collections resolve over time. Less than 10% of patients have persistent fluid collections after 6 weeks that would meet the definition of a pseudocyst. Only symptomatic collections should be drained with surgery or endoscopy or by percutaneous route.
PANCREATIC DUCT DISRUPTION Pancreatic duct disruption may present with symptoms of increasing abdominal pain or shortness of breath in the setting of an enlarging fluid collection. Diagnosis can be confirmed on magnetic resonance cholangiopancreatography (MRCP) or ERCP. Placement of a bridging pancreatic stent for at least 6 weeks is >90% effective at resolving the leak. Nonbridging stents are less effective.
PERIVASCULAR COMPLICATIONS Perivascular complications may include splenic vein thrombosis with gastric varices and pseudoaneurysms. Gastric varices bleed less than 5% of the time. Life-threatening bleeding from a ruptured pseudoaneurysm can be diagnosed and treated with mesenteric angiography and embolization.
EXTRAPANCREATIC INFECTIONS Hospital-acquired infections occur in up to 20% of patients with acute pancreatitis. Patients should be continually monitored for the development pneumonia, urinary tract infection, and line infection. Continued culturing of urine, monitoring of chest x-rays, and routine changing of intravenous lines are important during hospitalization.
Follow-Up Care Hospitalizations for moderately severe and severe acute pancreatitis can last weeks to months and often involve a period of intensive care unit admission and outpatient rehabilitation or subacute nursing care. Follow-up evaluation should assess for development of diabetes, exocrine insufficiency, recurrent cholangitis, or development of infected fluid collections. As mentioned previously, cholecystectomy should be performed within 4–6 weeks of discharge if possible for patients with uncomplicated gallstone pancreatitis.
RECURRENT PANCREATITIS
Approximately 25% of patients who have had an attack of acute pancreatitis have a recurrence. The two most common etiologic factors are alcohol and cholelithiasis. In patients with recurrent pancreatitis without an obvious cause, the differential diagnosis should encompass occult biliary tract disease including microlithiasis, hypertriglyceridemia, drugs, pancreatic cancer, pancreas divisum, and cystic fibrosis (Table 371-1). In one series of 31 patients diagnosed initially as having idiopathic or recurrent acute pancreatitis, 23 were found to have occult gallstone disease. Thus, approximately two-thirds of patients with recurrent acute pancreatitis without an obvious cause actually have occult gallstone disease due to microlithiasis. Genetic defects as in hereditary pancreatitis and cystic fibrosis mutations can result in recurrent pancreatitis. Other diseases of the biliary tree and pancreatic ducts that can cause acute pancreatitis include choledochocele; ampullary tumors; pancreas divisum; and pancreatic duct stones, stricture, and tumor. Approximately 2–4% of patients with pancreatic carcinoma present with acute pancreatitis.
PANCREATITIS IN PATIENTS WITH AIDS
The incidence of acute pancreatitis is increased in patients with AIDS for two reasons: (1) the high incidence of infections involving the pancreas such as infections with cytomegalovirus, Cryptosporidium, and the Mycobacterium avium complex; and (2) the frequent use by patients with AIDS of medications such as didanosine, pentamidine, trimethoprim-sulfamethoxazole, and protease inhibitors. Incidence has been markedly reduced due to advances in therapy (Chap. 226).
CHRONIC PANCREATITIS AND PANCREATIC EXOCRINE INSUFFICIENCY
PATHOPHYSIOLOGY
Chronic pancreatitis is a disease process characterized by irreversible damage to the pancreas as distinct from the reversible changes noted in acute pancreatitis (Table 371-4). The events that initiate and then perpetuate the inflammatory process in the pancreas are becoming more clearly understood. Irrespective of the mechanism of injury, it is becoming apparent that stellate cell activation that results in cytokine expression and production of extracellular matrix proteins cause acute and chronic inflammation and collagen deposition in the pancreas. Thus, the condition is defined by the presence of histologic abnormalities, including chronic inflammation, fibrosis, and progressive destruction of both exocrine and eventually endocrine tissue (atrophy). A number of etiologies have been associated with chronic pancreatitis resulting in the cardinal manifestations of the disease such as abdominal pain, steatorrhea, weight loss, and diabetes mellitus (Table 371-5).
|
CHRONIC PANCREATITIS AND PANCREATIC EXOCRINE INSUFFICIENCY: TIGAR-O CLASSIFICATION SYSTEM |
Abbreviations: DBTC, dibutylin dichloride; TIGAR-O, toxic-metabolic, idiopathic, genetic, autoimmune, recurrent and severe acute pancreatitis, obstructive.
Although alcohol has been believed to be the primary cause of chronic pancreatitis, other factors contribute to the disease because not all heavy consumers of alcohol develop pancreatic disease. There is also a strong association between smoking and chronic pancreatitis. Cigarette smoke leads to an increased susceptibility to pancreatic autodigestion and predisposes to dysregulation of duct cell CFTR function. Smoking is an independent, dose-dependent risk factor for chronic pancreatitis and recurrent acute pancreatitis. Both continued alcohol and smoking exposure are associated with pancreatic fibrosis, calcifications, and progression of disease
Recent characterization of pancreatic stellate cells (PSCs) has added insight into the underlying cellular responses behind development of chronic pancreatitis. Specifically, PSCs are believed to play a role in maintaining normal pancreatic architecture that can shift toward fibrogenesis in the case of chronic pancreatitis. The sentinel acute pancreatitis event (SAPE) hypothesis uniformly describes the events in the pathogenesis of chronic pancreatitis. It is believed that alcohol or additional stimuli lead to matrix metalloproteinase–mediated destruction of normal collagen in pancreatic parenchyma, which later allows for pancreatic remodeling. Proinflammatory cytokines, tumor necrosis factor α (TNF-α), interleukin 1 (IL-1), and interleukin 6 (IL-6), as well as oxidant complexes, are able to induce PSC activity with subsequent new collagen synthesis. In addition to being stimulated by cytokines, oxidants, or growth factors, PSCs also possess transforming growth factor β (TGF-β)–mediated self-activating autocrine pathways that may explain disease progression in chronic pancreatitis even after removal of noxious stimuli.
ETIOLOGIC CONSIDERATIONS
Among adults in the United States, alcoholism is the most common cause of clinically apparent chronic pancreatitis, whereas cystic fibrosis is the most frequent cause in children. As many as 25% of adults in the United States with chronic pancreatitis have the idiopathic form. Recent investigations have indicated that up to 15% of patients with idiopathic pancreatitis may have pancreatitis due to genetic defects (Table 371-5).
Whitcomb and associates studied several large families with hereditary chronic pancreatitis and were able to identify a genetic defect that affects the gene encoding for trypsinogen. Several additional defects of this gene have also been described. The defect prevents the destruction of prematurely activated trypsin and allows it to be resistant to the intracellular protective effect of trypsin inhibitor. It is hypothesized that this continual activation of digestive enzymes within the gland leads to acute injury and, finally, chronic pancreatitis. Since the initial discovery of the PRSS1 mutation defect, other genetic diseases have been detected (Table 371-5).
Several other groups of investigators have documented mutations of CFTR. This gene functions as a cyclic AMP–regulated chloride channel. In patients with cystic fibrosis, the high concentration of macromolecules can block the pancreatic ducts. It must be appreciated, however, that there is a great deal of heterogeneity in relationship to the CFTR gene defect. More than 1000 putative mutations of the CFTR gene have been identified. Attempts to elucidate the relationship between the genotype and pancreatic manifestations have been hampered by the number of mutations. The ability to detect CFTR mutations has led to the recognition that the clinical spectrum of the disease is broader than previously thought. Two studies have clarified the association between mutations of the CFTR gene and another monosymptomatic form of cystic fibrosis (i.e., chronic pancreatitis). It is estimated that in patients with idiopathic pancreatitis, the frequency of a single CFTR mutation is 11 times the expected frequency and the frequency of two mutant alleles is 80 times the expected frequency. In these studies, the patients were adults when the diagnosis of pancreatitis was made; none had any clinical evidence of pulmonary disease, and sweat test results were not diagnostic of cystic fibrosis. The prevalence of such mutations is unclear, and further studies are certainly needed. In addition, the therapeutic and prognostic implication of these findings with respect to managing pancreatitis remains to be determined. Long-term follow-up of affected patients is needed. CFTR mutations are common in the general population. It is unclear whether the CFTR mutation alone can lead to pancreatitis as an autosomal recessive disease. A study evaluated 39 patients with idiopathic chronic pancreatitis to assess the risk associated with these mutations. Patients with two CFTR mutations (compound heterozygotes) demonstrated CFTR function at a level between that seen in typical cystic fibrosis and cystic fibrosis carriers and had a 40-fold increased risk of pancreatitis. The presence of an N34S SPINK1 mutation increased the risk 20-fold. A combination of two CFTR mutations and an N34S SPINK1 mutation increased the risk of pancreatitis 900-fold. Knowledge of the genetic defects and downstream alterations in protein expression has led to the development of novel genetic therapy in cystic fibrosis children that potentiates the CFTR channel resulting in improvement in lung function, quality of life, and weight gain. Table 371-5 lists recognized causes of chronic pancreatitis and pancreatic exocrine insufficiency.
AUTOIMMUNE PANCREATITIS (TABLE 371-6)
|
CLINICAL FEATURES OF AUTOIMMUNE PANCREATITIS (AIP) |
Abbreviations: ERCP, endoscopic retrograde cholangiopancreatography; MRCP, magnetic resonance cholangiopancreatography.
Autoimmune pancreatitis (AIP) is an uncommon disorder of presumed autoimmune causation with characteristic laboratory, histologic, and morphologic findings. In type 1 AIP, the pancreas is involved as part of an IgG4 systemic disease (Chap. 391e) and meets HISORt criteria as defined below. The characteristic pancreatic histopathologic findings include lymphoplasmacytic infiltrate, storiform fibrosis, and abundant IgG4 cells. AIP type 2 is histologically confirmed idiopathic duct centric pancreatitis with granulocytic infiltration of the duct wall (termed GEL), but without IgG4 positive cells and systemic involvement. Although AIP was initially described as a primary pancreatic disorder, it is now recognized that it is associated with other disorders of presumed autoimmune etiology, and this has been termed IgG4 systemic disease (Chap. 391e). The clinical features include IgG4-associated cholangitis, rheumatoid arthritis, Sjögren’s syndrome, ulcerative colitis, mediastinal fibrosis and adenopathy, autoimmune thyroiditis, tubulointerstitial nephritis, retroperitoneal fibrosis, chronic periaortitis, chronic sclerosing sialadenitis, and Mikulicz’s disease. Mild symptoms, usually abdominal pain, and recurrent acute pancreatitis are unusual. Furthermore, AIP is not a common cause of idiopathic recurrent pancreatitis.
Weight loss and new onset of diabetes may also occur. An obstructive pattern on liver tests is common (i.e., disproportionately elevated serum alkaline phosphatase and minimally elevated serum aminotransferases). Elevated serum levels of IgG4 provide a marker for the disease, particularly in Western populations. Serum IgG4 normally accounts for only 5–6% of the total IgG4 in healthy patients but is elevated to values >280 mg/dL in those with AIP. CT scans reveal abnormalities in the majority of patients and include diffuse enlargement, focal enlargement, and a distinct enlargement at the head of the pancreas. ERCP or MRCP reveals strictures in the bile duct in more than one-third of patients with AIP; these may include common bile duct strictures, intrahepatic bile duct strictures, or proximal bile duct strictures, with accompanying narrowing of the pancreatic portion of the bile duct. This has been termed autoimmune IgG4 cholangitis. Characteristic histologic findings include extensive lymphoplasmacytic infiltrates with dense fibrosis around pancreatic ducts, as well as a lymphoplasmacytic infiltration, resulting in an obliterative phlebitis.
The Mayo Clinic HISORt criteria indicate that AIP can be diagnosed by the presence of at least two of the following: (1) histology; (2) imaging; (3) serology (elevated serum IgG4 levels); (4) other organ involvement; and (5) response to glucocorticoid therapy, with improvement in pancreatic and extrapancreatic manifestations.
Glucocorticoids have shown efficacy in alleviating symptoms, decreasing the size of the pancreas, and reversing histopathologic features in patients with AIP. Patients may respond dramatically to glucocorticoid therapy within a 2- to 4-week period. Prednisone is usually administered at an initial dose of 40 mg/d for 4 weeks followed by a taper of the daily dosage by 5 mg/wk based on monitoring of clinical parameters. Relief of symptoms, serial changes in abdominal imaging of the pancreas and bile ducts, decreased serum γ-globulin and IgG4 levels, and improvements in liver tests are parameters to follow. A poor response to glucocorticoids over a 2- to 4-week period should raise suspicion of pancreatic cancer or other forms of chronic pancreatitis. A recent multicenter international report reviewed 1064 patients with AIP. Clinical remission was achieved in 99% of type I and 92% of type II AIP patients with steroids. However, disease relapse occurred in 31% of type I and 9% of type II AIP patients. For treatment of disease relapse in type 1 AIP, glucocorticoids were successful in 201 of 295 (68%) patients, and azathioprine was successful in 52 of 58 patients (85%). A small number of patients responded favorably to 6-mercaptapurine, rituximab, cyclosporine, and cyclophosphamide. Types 1 and 2 AIP are highly responsive to initial glucocorticoid treatment. Relapse is common in type 1 patients, especially those with biliary tract strictures. Most relapses occur after glucocorticoids are discontinued. Patients with refractory symptoms and strictures generally require immunomodulator therapy as noted above. Appearance of interval cancers following a diagnosis of AIP is uncommon.
Clinical Features of Chronic Pancreatitis Patients with chronic pancreatitis seek medical attention predominantly because of two symptoms: abdominal pain or maldigestion and weight loss. The abdominal pain may be quite variable in location, severity, and frequency. The pain can be constant or intermittent with frequent pain-free intervals. Eating may exacerbate the pain, leading to a fear of eating with consequent weight loss. The spectrum of abdominal pain ranges from mild to quite severe, with narcotic dependence as a frequent consequence. Maldigestion is manifested as chronic diarrhea, steatorrhea, weight loss, and fatigue. Patients with chronic abdominal pain may or may not progress to maldigestion, and ~20% of patients will present with symptoms of maldigestion without a history of abdominal pain. Patients with chronic pancreatitis have significant morbidity and mortality and use appreciable amounts of societal resources. Despite steatorrhea, clinically apparent deficiencies of fat-soluble vitamins are surprisingly uncommon. Physical findings in these patients are usually unimpressive, so that there is a disparity between the severity of abdominal pain and the physical signs that usually consist of some mild tenderness.
The diagnosis of early or mild chronic pancreatitis can be challenging because there is no biomarker for the disease. In contrast to acute pancreatitis, the serum amylase and lipase levels are usually not strikingly elevated in chronic pancreatitis. Elevation of serum bilirubin and alkaline phosphatase may indicate cholestasis secondary to common bile duct stricture caused by chronic inflammation. Many patients have impaired glucose tolerance with elevated fasting blood glucose levels. The fecal elastase-1 and small-bowel biopsy are useful in the evaluation of patients with suspected pancreatic steatorrhea. The fecal elastase level will be abnormal and small-bowel histology will be normal in such patients. A decrease of fecal elastase level to <100 μg per gram of stool strongly suggests severe pancreatic exocrine insufficiency.
The radiographic evaluation of a patient with suspected chronic pancreatitis usually proceeds from a noninvasive to more invasive approach. Abdominal CT imaging (Fig. 371-4A,B) is the initial modality of choice, followed by MRI (Fig. 371-4C), endoscopic ultrasound, and pancreas function testing. In addition to excluding a pseudocyst and pancreatic cancer, CT may show calcification, dilated ducts, or an atrophic pancreas. Although abdominal CT scanning and MRCP greatly aid in the diagnosis of pancreatic disease, the diagnostic test with the best sensitivity and specificity is the hormone stimulation test using secretin. The secretin test becomes abnormal when ≥60% of the pancreatic exocrine function has been lost. This usually correlates well with the onset of chronic abdominal pain. The role of endoscopic ultrasonography (EUS) in diagnosing early chronic pancreatitis is still being defined. A total of nine endosonographic features have been described in chronic pancreatitis. The presence of five or more features is considered diagnostic of chronic pancreatitis. EUS is not a sensitive enough test for detecting early chronic pancreatitis alone (Chap. 370) and may show positive features in patients who have dyspepsia or even normal aging individuals. Recent data suggest that EUS can be combined with endoscopic pancreatic function testing (EUS-ePFT) during a single endoscopy to screen for chronic pancreatitis in patients with chronic abdominal pain. Diffuse calcifications noted on plain film of the abdomen usually indicate significant damage to the pancreas and are pathognomic for chronic pancreatitis (Fig. 371-4A). Although alcohol is by far the most common cause of pancreatic calcification, such calcification may also be noted in hereditary pancreatitis, posttraumatic pancreatitis, hypercalcemic pancreatitis, idiopathic chronic pancreatitis, and tropical pancreatitis.
FIGURE 371-4 A. Chronic pancreatitis and pancreatic calculi: computed tomography (CT) scan. In this contrast-enhanced CT scan of the abdomen, there is evidence of an atrophic pancreas with multiple calcifications and stones in the parenchyma and dilated pancreatic duct (arrow). B. In this contrast-enhanced CT scan of the abdomen, there is evidence of an atrophic pancreas with multiple calcifications (arrows). Note the markedly dilated pancreatic duct seen in this section through the body and tail (open arrows). C. Chronic pancreatitis on magnetic resonance cholangiopancreatography (MRCP): dilated duct with filling defects. Gadolinium-enhanced magnetic resonance imaging/MRCP reveals a dilated pancreatic duct (arrow) in chronic pancreatitis with multiple filling defects suggestive of pancreatic duct calculi. (A, C, courtesy of Dr. KJ Mortele, Brigham and Women’s Hospital; with permission.)
Complications of Chronic Pancreatitis The complications of chronic pancreatitis are protean and are listed in Table 371-7. Although most patients have impaired glucose tolerance, diabetic ketoacidosis and diabetic coma are uncommon. Likewise, end-organ damage (retinopathy, neuropathy, nephropathy) is also uncommon. A nondiabetic retinopathy may be due to either vitamin A and/or zinc deficiency. Gastrointestinal bleeding may occur from peptic ulceration, gastritis, a pseudocyst eroding into the duodenum, arterial bleeding into the pancreatic duct (hemosuccus pancreaticus), or ruptured varices secondary to splenic vein thrombosis due to chronic inflammation of the tail of the pancreas. Jaundice, cholestasis, and biliary cirrhosis may occur from the chronic inflammatory reaction around the intrapancreatic portion of the common bile duct. Twenty years after the diagnosis of calcific chronic pancreatitis, the cumulative risk of pancreatic carcinoma is 4%. Patients with hereditary pancreatitis are at a 10-fold higher risk for pancreatic cancer.
|
COMPLICATIONS OF CHRONIC PANCREATITIS |
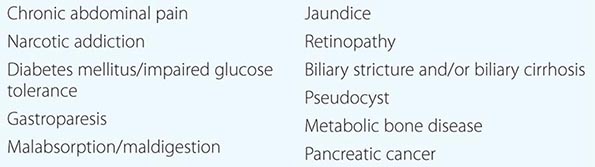
|
TREATMENT |
CHRONIC PANCREATITIS |
STEATORRHEA
The treatment of steatorrhea with pancreatic enzymes is straightforward even though complete correction of steatorrhea is unusual. Enzyme therapy usually brings diarrhea under control and restores absorption of fat to an acceptable level and affects weight gain. Thus, pancreatic enzyme replacement has been the cornerstone of therapy. In treating steatorrhea, it is important to use a potent pancreatic formulation that will deliver sufficient lipase into the duodenum to correct maldigestion and decrease steatorrhea. In an attempt to standardize the enzyme activity, potency, and bioavailability, the U.S. Food and Drug Administration (FDA) required that all pancreas enzyme drugs in the United States obtain a New Drug Application (NDA) by April 2008. Table 371-8 lists frequently used formulations, but availability will be based on compliance with the FDA mandate. Recent data suggest that dosages up to 80,000–100,000 units of lipase taken during the meal may be necessary to normalize nutritional parameters in malnourished chronic pancreatitis patients, and some may require acid suppression with proton pump inhibitors.
|
FDA-APPROVED PANCREATIC ENZYME (PANCRELIPASE) PREPARATIONS |
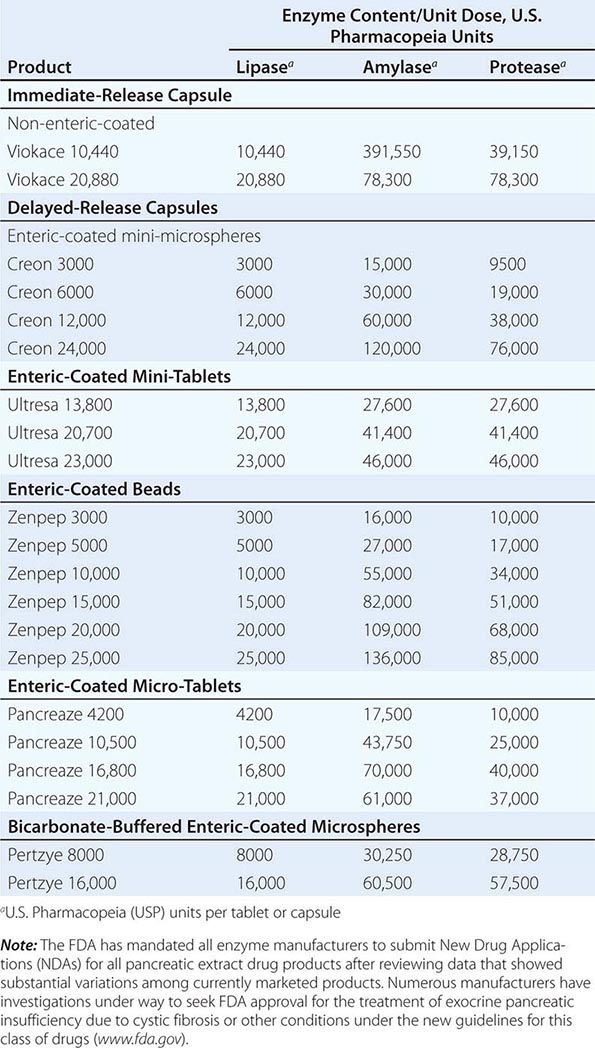
ABDOMINAL PAIN
The management of pain in patients with chronic pancreatitis is problematic.
Recent meta-analyses have shown no consistent benefit of enzyme therapy at reducing pain in chronic pancreatitis. In some patients with idiopathic chronic pancreatitis, conventional non-enteric-coated enzyme preparations containing high concentrations of serine proteases may relieve mild abdominal pain or discomfort. The pain relief experienced by these patients actually may be due to improvements in the dyspepsia from maldigestion.
Gastroparesis is also quite common in patients with chronic pancreatitis. It is important to recognize and treat with prokinetic drugs because treatment with enzymes may fail simply because gastric dysmotility is interfering with the delivery of enzymes into the upper intestine. A recent prospective study reported that pregabalin can improve pain in chronic pancreatitis and lower pain medication requirement.
Endoscopic treatment of chronic pancreatitis pain may involve sphincterotomy, stenting, stone extraction, and drainage of a pancreatic pseudocyst. Therapy directed to the pancreatic duct would seem to be most appropriate in the setting of a dominant stricture, especially if a ductal stone has led to obstruction. The use of endoscopic stenting for patients with chronic pain, but without a dominant stricture, has not been subjected to any controlled trials. It is now appreciated that significant complications can occur from stenting (i.e., bleeding, cholangitis, stent migration, pancreatitis, and stent clogging). In patients with large-duct disease usually from alcohol-induced chronic pancreatitis, ductal decompression with surgical therapy has been the therapy of choice. Among such patients, 80% seem to obtain immediate relief; however, at the end of 3 years, one-half of the patients have recurrence of pain. Two randomized prospective trials comparing endoscopic to surgical therapy for chronic pancreatitis demonstrated that surgical therapy was superior to endoscopy at decreasing pain and improving quality of life in selected patients with dilated ducts and abdominal pain. This would suggest that chronic pancreatitis patients with dilated ducts and pain should be considered for surgical intervention. The role of preoperative stenting prior to surgery as a predictor of response has yet to be proven.
A Whipple procedure, total pancreatectomy, and autologous islet cell transplantation have been used in selected patients with chronic pancreatitis and abdominal pain refractory to conventional therapy. The patients who have benefited the most from total pancreatectomy have chronic pancreatitis without prior pancreatic surgery or evidence of islet cell insufficiency. The role of this procedure remains to be fully defined but may be an option in lieu of ductal decompression surgery or pancreatic resection in patients with intractable, painful small-duct disease, particularly as the standard surgical procedures tend to decrease islet cell yield. Celiac plexus block has not resulted in long-lasting pain relief.
HEREDITARY PANCREATITIS
Hereditary pancreatitis is a rare disease that is similar to chronic pancreatitis except for an early age of onset and evidence of hereditary factors. A genomewide search using genetic linkage analysis identified the hereditary pancreatitis gene on chromosome 7. Mutations in ion codons 29 (exon 2) and 122 (exon 3) of the cationic trypsinogen gene cause autosomal dominant forms of hereditary pancreatitis. The codon 122 mutations lead to a substitution of the corresponding arginine with another amino acid, usually histidine. This substitution, when it occurs, eliminates a fail-safe trypsin self-destruction site necessary to eliminate trypsin that is prematurely activated within the acinar cell. These patients have recurring attacks of severe abdominal pain that may last from a few days to a few weeks. The serum amylase and lipase levels may be elevated during acute attacks but are usually normal. Patients frequently develop pancreatic calcification, diabetes mellitus, and steatorrhea; in addition, they have an increased incidence of pancreatic carcinoma, with the cumulative incidence being as high as 40% by age 70 years. A recent natural history study of hereditary pancreatitis in more than 200 patients from France reported that abdominal pain started in childhood at age 10 years, steatorrhea developed at age 29 years, diabetes at age 38 years, and pancreatic carcinoma at age 55 years. Such patients often require surgical ductal decompression for pain relief. Abdominal complaints in relatives of patients with hereditary pancreatitis should raise the question of pancreatic disease.
PSTI, or SPINK1, is a 56-amino-acid peptide that specifically inhibits trypsin by physically blocking its active site. SPINK1 acts as the first line of defense against prematurely activated trypsinogen in the acinar cell. Recently, it has been shown that the frequency of SPINK1 mutations in patients with idiopathic chronic pancreatitis is markedly increased, suggesting that these mutations may be associated with pancreatitis.
PANCREATIC ENDOCRINE TUMORS
Pancreatic endocrine tumors are discussed in Chap. 113.
OTHER CONDITIONS
ANNULAR PANCREAS
When the ventral pancreatic anlage fails to migrate correctly to make contact with the dorsal anlage, the result may be a ring of pancreatic tissue encircling the duodenum. Such an annular pancreas may cause intestinal obstruction in the neonate or the adult. Symptoms of postprandial fullness, epigastric pain, nausea, and vomiting may be present for years before the diagnosis is entertained. The radiographic findings are symmetric dilation of the proximal duodenum with bulging of the recesses on either side of the annular band, effacement but not destruction of the duodenal mucosa, accentuation of the findings in the right anterior oblique position, and lack of change on repeated examinations. The differential diagnosis should include duodenal webs, tumors of the pancreas or duodenum, postbulbar peptic ulcer, regional enteritis, and adhesions. Patients with annular pancreas have an increased incidence of pancreatitis and peptic ulcer. Because of these and other potential complications, the treatment is surgical even if the condition has been present for years. Retrocolic duodenojejeunostomy is the procedure of choice, although some surgeons advocate Billroth II gastrectomy, gastroenterostomy, and vagotomy.
PANCREAS DIVISUM
Pancreas divisum is present in 7–10% of the population and occurs when the embryologic ventral and dorsal pancreatic anlagen fail to fuse, so that pancreatic drainage is accomplished mainly through the accessory papilla. Pancreas divisum is the most common congenital anatomic variant of the human pancreas. Current evidence indicates that this anomaly does not predispose to the development of pancreatitis in the great majority of patients who harbor it. However, the combination of pancreas divisum and a small accessory orifice could result in dorsal duct obstruction. The challenge is to identify this subset of patients with dorsal duct pathology. Cannulation of the dorsal duct by ERCP is not as easily done as is cannulation of the ventral duct. Patients with pancreatitis and pancreas divisum demonstrated by MRCP or ERCP should be treated with conservative measures. In many of these patients, pancreatitis is idiopathic and unrelated to the pancreas divisum. Endoscopic or surgical intervention is indicated only if pancreatitis recurs and no other cause can be found. If marked dilation of the dorsal duct can be demonstrated, surgical ductal decompression should be performed. It should be stressed that the ERCP/MRCP appearance of pancreas divisum (i.e., a small-caliber ventral duct with an arborizing pattern) may be mistaken as representing an obstructed main pancreatic duct secondary to a mass lesion.
MACROAMYLASEMIA
In macroamylasemia, amylase circulates in the blood in a polymer form too large to be easily excreted by the kidney. Patients with this condition demonstrate an elevated serum amylase value and a low urinary amylase value. The presence of macroamylase can be documented by chromatography of the serum. The prevalence of macroamylasemia is 1.5% of the nonalcoholic general adult hospital population. Usually macroamylasemia is an incidental finding and is not related to disease of the pancreas or other organs. Macrolipasemia has now been documented in a few patients with cirrhosis or non-Hodgkin’s lymphoma. In these patients, the pancreas appeared normal on ultrasound and CT examination. Lipase was shown to be complexed with immunoglobulin A. Thus, the possibility of both macroamylasemia and macrolipasemia should be considered in patients with elevated blood levels of these enzymes.
ACKNOWLEDGMENTS
This chapter represents a revised version of chapters by Drs. Norton J. Greenberger, Phillip P. Toskes, and Bechien Wu that were in previous editions of Harrison’s.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
369 |
Diseases of the Gallbladder and Bile Ducts |
PHYSIOLOGY OF BILE PRODUCTION AND FLOW
BILE SECRETION AND COMPOSITION
Bile formed in the hepatic lobules is secreted into a complex network of canaliculi, small bile ductules, and larger bile ducts that run with lymphatics and branches of the portal vein and hepatic artery in portal tracts situated between hepatic lobules. These interlobular bile ducts coalesce to form larger septal bile ducts that join to form the right and left hepatic ducts, which in turn, unite to form the common hepatic duct. The common hepatic duct is joined by the cystic duct of the gallbladder to form the common bile duct (CBD), which enters the duodenum (often after joining the main pancreatic duct) through the ampulla of Vater.
Hepatic bile is an isotonic fluid with an electrolyte composition resembling blood plasma. The electrolyte composition of gallbladder bile differs from that of hepatic bile because most of the inorganic anions, chloride and bicarbonate, have been removed by reabsorption across the gallbladder epithelium. As a result of water reabsorption, total solute concentration of bile increases from 3–4 g/dL in hepatic bile to 10–15 g/dL in gallbladder bile.
Major solute components of bile by moles percent include bile acids (80%), lecithin and traces of other phospholipids (16%), and unesterified cholesterol (4.0%). In the lithogenic state, the cholesterol value can be as high as 8–10%. Other constituents include conjugated bilirubin; proteins (all immunoglobulins, albumin, metabolites of hormones, and other proteins metabolized in the liver); electrolytes; mucus; and, often, drugs and their metabolites.
The total daily basal secretion of hepatic bile is ~500–600 mL. Many substances taken up or synthesized by the hepatocyte are secreted into the bile canaliculi. The canalicular membrane forms microvilli and is associated with microfilaments of actin, microtubules, and other contractile elements. Prior to their secretion into the bile, many substances are taken up into the hepatocyte, while others, such as phospholipids, a portion of primary bile acids, and some cholesterol, are synthesized de novo in the hepatocyte. Three mechanisms are important in regulating bile flow: (1) active transport of bile acids from hepatocytes into the bile canaliculi, (2) active transport of other organic anions, and (3) cholangiocellular secretion. The last is a secretin-mediated and cyclic AMP–dependent mechanism that results in the secretion of a sodium- and bicarbonate-rich fluid into the bile ducts.
Active vectorial secretion of biliary constituents from the portal blood into the bile canaliculi is driven by a set of polarized transport systems at the basolateral (sinusoidal) and the canalicular apical plasma membrane domains of the hepatocyte. Two sinusoidal bile salt uptake systems have been cloned in humans, the Na+/taurocholate cotransporter (NTCP, SLC10A1) and the organic anion–transporting proteins (OATPs), which also transport a large variety of non-bile salt organic anions. Several ATP-dependent canalicular transport systems, “export pumps,” (ATP-binding cassette transport proteins, also known as ABC transporters) have been identified, the most important of which are: the bile salt export pump (BSEP, ABCB11); the anionic conjugate export pump (MRP2, ABCC2), which mediates the canalicular excretion of various amphiphilic conjugates formed by phase II conjugation (e.g., bilirubin mono- and diglucuronides and drugs); the multidrug export pump (MDR1, ABCB1) for hydrophobic cationic compounds; and the phospholipid export pump (MDR3, ABCB4). Two hemitransporters ABCG5/G8, functioning as a couple, constitute the canalicular cholesterol and phytosterol transporter. F1C1 (ATP8B1) is an aminophospholipid transferase (“flippase”) essential for maintaining the lipid asymmetry of the canalicular membrane. The canalicular membrane also contains ATP-independent transport systems such as the Cl/HCO3 anion exchanger isoform 2 (AE2, SLC4A2) for canalicular bicarbonate secretion. For most of these transporters, genetic defects have been identified that are associated with various forms of cholestasis or defects of biliary excretion. F1C1 is defective in progressive familial intrahepatic cholestasis type 1 (PFIC1) and benign recurrent intrahepatic cholestasis type 1 (BRIC1) and results in ablation of all other ATP-dependent transporter functions. BSEP is defective in PFIC2 and BRIC2. Mutations of MRP2 (ABCC2) cause the Dubin-Johnson syndrome, an inherited form of conjugated hyperbilirubinemia (Chap. 359). A defective MDR3 (ABCB4) results in PFIC3. ABCG5/G8, the canalicular half transporters for cholesterol and other neutral sterols, are defective in sitosterolemia. The cystic fibrosis transmembrane regulator (CFTR, ABCC7) located on bile duct epithelial cells but not on canalicular membranes is defective in cystic fibrosis, which is associated with impaired cholangiocellular pH regulation during ductular bile formation and chronic cholestatic liver disease, occasionally resulting in biliary cirrhosis.
THE BILE ACIDS
The primary bile acids, cholic acid and chenodeoxycholic acid (CDCA), are synthesized from cholesterol in the liver, conjugated with glycine or taurine, and secreted into the bile. Secondary bile acids, including deoxycholate and lithocholate, are formed in the colon as bacterial metabolites of the primary bile acids. However, lithocholic acid is much less efficiently absorbed from the colon than deoxycholic acid. Another secondary bile acid, found in low concentration, is ursodeoxycholic acid (UDCA), a stereoisomer of CDCA. In healthy subjects, the ratio of glycine to taurine conjugates in bile is ~3:1.
Bile acids are detergent-like molecules that in aqueous solutions and above a critical concentration of about 2 mM form molecular aggregates called micelles. Cholesterol alone is sparingly soluble in aqueous environments, and its solubility in bile depends on both the total lipid concentration and the relative molar percentages of bile acids and lecithin. Normal ratios of these constituents favor the formation of solubilizing mixed micelles, while abnormal ratios promote the precipitation of cholesterol crystals in bile via an intermediate liquid crystal phase.
In addition to facilitating the biliary excretion of cholesterol, bile acids facilitate the normal intestinal absorption of dietary fats, mainly cholesterol and fat-soluble vitamins, via a micellar transport mechanism (Chap. 349). Bile acids also serve as a major physiologic driving force for hepatic bile flow and aid in water and electrolyte transport in the small bowel and colon.
ENTEROHEPATIC CIRCULATION
Bile acids are efficiently conserved under normal conditions. Unconjugated, and to a lesser degree also conjugated, bile acids are absorbed by passive diffusion along the entire gut. Quantitatively much more important for bile salt recirculation, however, is the active transport mechanism for conjugated bile acids in the distal ileum (Chap. 349). The reabsorbed bile acids enter the portal bloodstream and are taken up rapidly by hepatocytes, reconjugated, and resecreted into bile (enterohepatic circulation).
The normal bile acid pool size is approximately 2–4 g. During digestion of a meal, the bile acid pool undergoes at least one or more enterohepatic cycles, depending on the size and composition of the meal. Normally, the bile acid pool circulates ~5–10 times daily. Intestinal reabsorption of the pool is about 95% efficient; therefore, fecal loss of bile acids is in the range of 0.2–0.4 g/d. In the steady state, this fecal loss is compensated by an equal daily synthesis of bile acids by the liver, and, thus, the size of the bile acid pool is maintained. Bile acids in the intestine release fibroblast growth factor 19 (FGF19) into the circulation, which is transported to the liver where it suppresses synthesis of bile acids from cholesterol by inhibiting the rate-limiting enzyme cytochrome P450 7A1 (CYP7A1) and also promotes gallbladder relaxation. While the loss of bile salts in stool is usually matched by increased hepatic synthesis, the maximum rate of synthesis is ~5 g/d, which may be insufficient to replete the bile acid pool size when there is pronounced impairment of intestinal bile salt reabsorption.
The expression of ABC transporters in the enterohepatic circulation and of the rate-limiting enzymes of bile acid and cholesterol synthesis are regulated in a coordinated fashion by nuclear receptors, which are ligand-activated transcription factors. The hepatic BSEP (ABCB11) is upregulated by the farnesoid × receptor (FXR), a bile acid sensor that also represses bile acid synthesis. The expression of the cholesterol transporter, ABCG5/G8, is upregulated by the liver × receptor (LXR), which is an oxysterol sensor.
GALLBLADDER AND SPHINCTERIC FUNCTIONS
In the fasting state, the sphincter of Oddi offers a high-pressure zone of resistance to bile flow from the CBD into the duodenum. Its tonic contraction serves to (1) prevent reflux of duodenal contents into the pancreatic and bile ducts and (2) promote filling of the gallbladder. The major factor controlling the evacuation of the gallbladder is the peptide hormone cholecystokinin (CCK), which is released from the duodenal mucosa in response to the ingestion of fats and amino acids. CCK produces (1) powerful contraction of the gallbladder, (2) decreased resistance of the sphincter of Oddi, and (3) enhanced flow of biliary contents into the duodenum.
Hepatic bile is “concentrated” within the gallbladder by energy-dependent transmucosal absorption of water and electrolytes. Almost the entire bile acid pool may be sequestered in the gallbladder following an overnight fast for delivery into the duodenum with the first meal of the day. The normal capacity of the gallbladder is ~30 mL of bile.
DISEASES OF THE GALLBLADDER
CONGENITAL ANOMALIES
Anomalies of the biliary tract are not uncommon and include abnormalities in number, size, and shape (e.g., agenesis of the gallbladder, duplications, rudimentary or oversized “giant” gallbladders, and diverticula). Phrygian cap is a clinically innocuous entity in which a partial or complete septum (or fold) separates the fundus from the body. Anomalies of position or suspension are not uncommon and include left-sided gallbladder, intrahepatic gallbladder, retrodisplacement of the gallbladder, and “floating” gallbladder. The latter condition predisposes to acute torsion, volvulus, or herniation of the gallbladder.
GALLSTONES
Epidemiology and Pathogenesis Gallstones are quite prevalent in most Western countries. Gallstone formation increases after age 50. In the United States, the third National Health and Nutrition Examination Survey (NHANES III) has revealed an overall prevalence of gallstones of 7.9% in men and 16.6% in women. The prevalence was high in Mexican Americans (8.9% in men, 26.7% in women), intermediate for non-Hispanic whites (8.6% in men, 16.6% in women), and low for African Americans (5.3% in men, 13.9% in women).
Gallstones are formed because of abnormal bile composition. They are divided into two major types: cholesterol stones and pigment stones. Cholesterol stones account for more than 90% of all gallstones in Western industrialized countries. Cholesterol gallstones usually contain >50% cholesterol monohydrate plus an admixture of calcium salts, bile pigments, proteins, and fatty acids. Pigment stones are composed primarily of calcium bilirubinate; they contain <20% cholesterol and are classified into “black” and “brown” types, the latter forming secondary to chronic biliary infection.
CHOLESTEROL STONES AND BILIARY SLUDGE Cholesterol is essentially water insoluble and requires aqueous dispersion into either micelles or vesicles, both of which require the presence of a second lipid to solubilize the cholesterol. Cholesterol and phospholipids are secreted into bile as unilamellar bilayered vesicles, which are converted into mixed micelles consisting of bile acids, phospholipids, and cholesterol by the action of bile acids. If there is an excess of cholesterol in relation to phospholipids and bile acids, unstable, cholesterol-rich vesicles remain, which aggregate into large multilamellar vesicles from which cholesterol crystals precipitate (Fig. 369-1).
FIGURE 369-1 Scheme showing pathogenesis of cholesterol gallstone formation. Conditions or factors that increase the ratio of cholesterol to bile acids and phospholipids (lecithin) favor gallstone formation. ABCB4, ATP-binding cassette transporter; ABCG5/8, ATP-binding cassette (ABC) transporter G5/G8; CYP7A1, cytochrome P450 7A1; MDR3, multidrug resistance protein 3, also called phospholipid export pump.
There are several important mechanisms in the formation of lithogenic (stone-forming) bile. The most important is increased biliary secretion of cholesterol. This may occur in association with obesity, the metabolic syndrome, high-caloric and cholesterol-rich diets, or drugs (e.g., clofibrate) and may result from increased activity of hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme of hepatic cholesterol synthesis, and increased hepatic uptake of cholesterol from blood. In patients with gallstones, dietary cholesterol increases biliary cholesterol secretion. This does not occur in non-gallstone patients on high-cholesterol diets. In addition to environmental factors such as high-caloric and cholesterol-rich diets, genetic factors play an important role in gallstone disease. A large study of symptomatic gallstones in Swedish twins provided strong evidence for a role of genetic factors in gallstone pathogenesis. Genetic factors accounted for 25%, shared environmental factors for 13%, and individual environmental factors for 62% of the phenotypic variation among monozygotic twins. A single nucleotide polymorphism of the gene encoding the hepatic cholesterol transporter ABCG5/G8 has been found in 21% of patients with gallstones, but only in 9% of the general population. It is thought to cause a gain of function of the cholesterol transporter and to contribute to cholesterol hypersecretion. A high prevalence of gallstones is found among first-degree relatives of gallstone carriers and in certain ethnic populations such as American Indians, Chilean Indians, and Chilean Hispanics. A common genetic trait has been identified for some of these populations by mitochondrial DNA analysis. In some patients, impaired hepatic conversion of cholesterol to bile acids may also occur, resulting in an increase of the lithogenic cholesterol/bile acid ratio. Although most cholesterol stones have a polygenic basis, there are rare monogenic (Mendelian) causes. Recently, a mutation in the CYP7A1 gene has been described that results in a deficiency of the enzyme cholesterol 7-hydroxylase, which catalyzes the initial step in cholesterol catabolism and bile acid synthesis. The homozygous state is associated with hypercholesterolemia and gallstones. Because the phenotype is expressed in the heterozygote state, mutations in the CYP7A1 gene may contribute to the susceptibility to cholesterol gallstone disease in the population. Mutations in the MDR3 (ABCB4) gene, which encodes the phospholipid export pump in the canalicular membrane of the hepatocyte, may cause defective phospholipid secretion into bile, resulting in cholesterol supersaturation of bile and formation of cholesterol gallstones in the gallbladder and in the bile ducts. Thus, an excess of biliary cholesterol in relation to bile acids and phospholipids is primarily due to hypersecretion of cholesterol, but hyposecretion of bile acids or phospholipids may contribute. An additional disturbance of bile acid metabolism that is likely to contribute to supersaturation of bile with cholesterol is enhanced conversion of cholic acid to deoxycholic acid, with replacement of the cholic acid pool by an expanded deoxycholic acid pool. It may result from enhanced dehydroxylation of cholic acid and increased absorption of newly formed deoxycholic acid. An increased deoxycholate secretion is associated with hypersecretion of cholesterol into bile.
While supersaturation of bile with cholesterol is an important prerequisite for gallstone formation, it is generally not sufficient by itself to produce cholesterol precipitation in vivo. Most individuals with supersaturated bile do not develop stones because the time required for cholesterol crystals to nucleate and grow is longer than the time bile remains in the gallbladder.
An important mechanism is nucleation of cholesterol monohydrate crystals, which is greatly accelerated in human lithogenic bile. Accelerated nucleation of cholesterol monohydrate in bile may be due to either an excess of pronucleating factors or a deficiency of antinucleating factors. Mucin and certain nonmucin glycoproteins, principally immunoglobulins, appear to be pronucleating factors, while apolipoproteins A-I and A-II and other glycoproteins appear to be antinucleating factors. Pigment particles may possibly play a role as nucleating factors. In a genome-wide analysis of serum bilirubin levels, the uridine diphosphate-glucuronyltransferase 1A1 (UGT1A1) Gilbert’s syndrome gene variant was associated with the presence of gallstone disease. Because most gallstones associated with the UGT1A1 variant were cholesterol stones, this finding points to the role of pigment particles in the pathogenesis of gallbladder stones. Cholesterol monohydrate crystal nucleation and crystal growth probably occur within the mucin gel layer. Vesicle fusion leads to liquid crystals, which, in turn, nucleate into solid cholesterol monohydrate crystals. Continued growth of the crystals occurs by direct nucleation of cholesterol molecules from supersaturated unilamellar or multilamellar biliary vesicles.
A third important mechanism in cholesterol gallstone formation is gallbladder hypomotility. If the gallbladder emptied all supersaturated or crystal-containing bile completely, stones would not be able to grow. A high percentage of patients with gallstones exhibit abnormalities of gallbladder emptying. Ultrasonographic studies show that gallstone patients display an increased gallbladder volume during fasting and also after a test meal (residual volume) and that fractional emptying after gallbladder stimulation is decreased. The incidence of gallstones is increased in conditions associated with infrequent or impaired gallbladder emptying such as fasting, parenteral nutrition, or pregnancy and in patients using drugs that inhibit gallbladder motility.
Biliary sludge is a thick, mucous material that, upon microscopic examination, reveals lecithin-cholesterol liquid crystals, cholesterol monohydrate crystals, calcium bilirubinate, and mucin gels. Biliary sludge typically forms a crescent-like layer in the most dependent portion of the gallbladder and is recognized by characteristic echoes on ultrasonography (see below). The presence of biliary sludge implies two abnormalities: (1) the normal balance between gallbladder mucin secretion and elimination has become deranged, and (2) nucleation of biliary solutes has occurred. That biliary sludge may be a precursor form of gallstone disease is evident from several observations. In one study, 96 patients with gallbladder sludge were followed prospectively by serial ultrasound studies. In 18%, biliary sludge disappeared and did not recur for at least 2 years. In 60%, biliary sludge disappeared and reappeared; in 14%, gallstones (8% asymptomatic, 6% symptomatic) developed; and in 6%, severe biliary pain with or without acute pancreatitis occurred. In 12 patients, cholecystectomies were performed, 6 for gallstone-associated biliary pain and 3 in symptomatic patients with sludge but without gallstones who had prior attacks of pancreatitis; the latter did not recur after cholecystectomy. It should be emphasized that biliary sludge can develop with disorders that cause gallbladder hypomotility; i.e., surgery, burns, total parenteral nutrition, pregnancy, and oral contraceptives—all of which are associated with gallstone formation. However, the presence of biliary sludge implies supersaturation of bile with either cholesterol or calcium bilirubinate.
Two other conditions are associated with cholesterol-stone or biliary-sludge formation: pregnancy and rapid weight reduction through a very-low-calorie diet. There appear to be two key changes during pregnancy that contribute to a “cholelithogenic state”: (1) a marked increase in cholesterol saturation of bile during the third trimester and (2) sluggish gallbladder contraction in response to a standard meal, resulting in impaired gallbladder emptying. That these changes are related to pregnancy per se is supported by several studies that show reversal of these abnormalities quite rapidly after delivery. During pregnancy, gallbladder sludge develops in 20–30% of women and gallstones in 5–12%. Although biliary sludge is a common finding during pregnancy, it is usually asymptomatic and often resolves spontaneously after delivery. Gallstones, which are less common than sludge and frequently associated with biliary colic, may also disappear after delivery because of spontaneous dissolution related to bile becoming unsaturated with cholesterol postpartum.
Approximately 10–20% of persons with rapid weight reduction achieved through very-low-calorie dieting develop gallstones. In a study involving 600 patients who completed a 3-month, 520-kcal/d diet, UDCA in a dosage of 600 mg/d proved highly effective in preventing gallstone formation; gallstones developed in only 3% of UDCA recipients, compared to 28% of placebo-treated patients. In obese patients treated by gastric banding, 500 mg/d of UDCA reduced the risk of gallstone formation from 30% to 8% within a follow-up of 6 months.
To summarize, cholesterol gallstone disease occurs because of several defects, which include (1) bile supersaturation with cholesterol, (2) nucleation of cholesterol monohydrate with subsequent crystal retention and stone growth, and (3) abnormal gallbladder motor function with delayed emptying and stasis. Other important factors known to predispose to cholesterol-stone formation are summarized in Table 369-1.
|
PREDISPOSING FACTORS FOR CHOLESTEROL AND PIGMENT GALLSTONE FORMATION |
PIGMENT STONES Black pigment stones are composed of either pure calcium bilirubinate or polymer-like complexes with calcium and mucin glycoproteins. They are more common in patients who have chronic hemolytic states (with increased conjugated bilirubin in bile), liver cirrhosis, Gilbert’s syndrome, or cystic fibrosis. Gallbladder stones in patients with ileal diseases, ileal resection, or ileal bypass generally are also black pigment stones. Enterohepatic recycling of bilirubin in ileal disease states contributes to their pathogenesis. Brown pigment stones are composed of calcium salts of unconjugated bilirubin with varying amounts of cholesterol and protein. They are caused by the presence of increased amounts of unconjugated, insoluble bilirubin in bile that precipitates to form stones. Deconjugation of an excess of soluble bilirubin mono- and diglucuronides may be mediated by endogenous β-glucuronidase but may also occur by spontaneous hydrolysis. Sometimes, the enzyme is also produced when bile is chronically infected by bacteria, and such stones are brown. Pigment stone formation is frequent in Asia and is often associated with infections in the gallbladder and biliary tree (Table 369-1).
Diagnosis Procedures of potential use in the diagnosis of cholelithiasis and other diseases of the gallbladder are detailed in Table 369-2. Ultrasonography of the gallbladder is very accurate in the identification of cholelithiasis and has replaced oral cholecystography (Fig. 369-2A). Stones as small as 1.5 mm in diameter may be confidently identified provided that firm criteria are used (e.g., acoustic “shadowing” of opacities that are within the gallbladder lumen and that change with the patient’s position [by gravity]). In major medical centers, the false-negative and false-positive rates for ultrasound in gallstone patients are ~2–4%. Biliary sludge is material of low echogenic activity that typically forms a layer in the most dependent position of the gallbladder. This layer shifts with postural changes but fails to produce acoustic shadowing; these two characteristics distinguish sludges from gallstones. Ultrasound can also be used to assess the emptying function of the gallbladder.
|
DIAGNOSTIC EVALUATION OF THE GALLBLADDER |
FIGURE 369-2 Examples of ultrasound and radiologic studies of the biliary tract. A. An ultrasound study showing a distended gallbladder (GB) containing a single large stone (arrow), which casts an acoustic shadow. B. Endoscopic retrograde cholangiopancreatogram (ERCP) showing normal biliary tract anatomy. In addition to the endoscope and large vertical gallbladder filled with contrast dye, the common hepatic duct (CHD), common bile duct (CBD), and pancreatic duct (PD) are shown. The arrow points to the ampulla of Vater. C. Endoscopic retrograde cholangiogram (ERC) showing choledocholithiasis. The biliary tract is dilated and contains multiple radiolucent calculi. D. ERCP showing sclerosing cholangitis. The common bile duct shows areas that are strictured and narrowed.
The plain abdominal film may detect gallstones containing sufficient calcium to be radiopaque (10–15% of cholesterol and ~50% of pigment stones). Plain radiography may also be of use in the diagnosis of emphysematous cholecystitis, porcelain gallbladder, limey bile, and gallstone ileus.
Oral cholecystography (OCG) has historically been a useful procedure for the diagnosis of gallstones but has been replaced by ultrasound and is regarded as obsolete. It may be used to assess the patency of the cystic duct and gallbladder emptying function. Further, OCG can also delineate the size and number of gallstones and determine whether they are calcified.
Radiopharmaceuticals such as 99mTc-labeled N-substituted iminodiacetic acids (HIDA, DIDA, DISIDA, etc.) are rapidly extracted from the blood and are excreted into the biliary tree in high concentration even in the presence of mild to moderate serum bilirubin elevations. Failure to image the gallbladder in the presence of biliary ductal visualization may indicate cystic duct obstruction, acute or chronic cholecystitis, or surgical absence of the organ. Such scans have some application in the diagnosis of acute cholecystitis.
Symptoms of Gallstone Disease Gallstones usually produce symptoms by causing inflammation or obstruction following their migration into the cystic duct or CBD. The most specific and characteristic symptom of gallstone disease is biliary colic that is a constant and often long-lasting pain (see below). Obstruction of the cystic duct or CBD by a stone produces increased intraluminal pressure and distention of the viscus that cannot be relieved by repetitive biliary contractions. The resultant visceral pain is characteristically a severe, steady ache or fullness in the epigastrium or right upper quadrant (RUQ) of the abdomen with frequent radiation to the interscapular area, right scapula, or shoulder.
Biliary colic begins quite suddenly and may persist with severe intensity for 30 min to 5 h, subsiding gradually or rapidly. It is steady rather than intermittent, as would be suggested by the word colic, which must be regarded as a misnomer, although it is in widespread use. An episode of biliary pain persisting beyond 5 h should raise the suspicion of acute cholecystitis (see below). Nausea and vomiting frequently accompany episodes of biliary pain. An elevated level of serum bilirubin and/or alkaline phosphatase suggests a common duct stone. Fever or chills (rigors) with biliary pain usually imply a complication, i.e., cholecystitis, pancreatitis, or cholangitis. Complaints of short-lasting, vague epigastric fullness, dyspepsia, eructation, or flatulence, especially following a fatty meal, should not be confused with biliary pain. Such symptoms are frequently elicited from patients with or without gallstone disease but are not specific for biliary calculi. Biliary colic may be precipitated by eating a fatty meal, by consumption of a large meal following a period of prolonged fasting, or by eating a normal meal; it is frequently nocturnal, occurring within a few hours of retiring.
Natural History Gallstone disease discovered in an asymptomatic patient or in a patient whose symptoms are not referable to cholelithiasis is a common clinical problem. Sixty to 80% of persons with asymptomatic gallstones remain asymptomatic over follow-up periods of up to 25 years. The probability of developing symptoms within 5 years after diagnosis is 2–4% per year and decreases in the years thereafter to 1–2%. The yearly incidence of complications is about 0.1–0.3%. Patients remaining asymptomatic for 15 years were found to be unlikely to develop symptoms during further follow-up, and most patients who did develop complications from their gallstones experienced prior warning symptoms. Similar conclusions apply to diabetic patients with silent gallstones. Decision analysis has suggested that (1) the cumulative risk of death due to gallstone disease while on expectant management is small, and (2) prophylactic cholecystectomy is not warranted.
Complications requiring cholecystectomy are much more common in gallstone patients who have developed symptoms of biliary pain. Patients found to have gallstones at a young age are more likely to develop symptoms from cholelithiasis than are patients >60 years at the time of initial diagnosis. Patients with diabetes mellitus and gallstones may be somewhat more susceptible to septic complications, but the magnitude of risk of septic biliary complications in diabetic patients is incompletely defined.
ACUTE AND CHRONIC CHOLECYSTITIS
Acute Cholecystitis Acute inflammation of the gallbladder wall usually follows obstruction of the cystic duct by a stone. Inflammatory response can be evoked by three factors: (1) mechanical inflammation produced by increased intraluminal pressure and distention with resulting ischemia of the gallbladder mucosa and wall, (2) chemical inflammation caused by the release of lysolecithin (due to the action of phospholipase on lecithin in bile) and other local tissue factors, and (3) bacterial inflammation, which may play a role in 50–85% of patients with acute cholecystitis. The organisms most frequently isolated by culture of gallbladder bile in these patients include Escherichia coli, Klebsiella spp., Streptococcus spp., and Clostridium spp.
Acute cholecystitis often begins as an attack of biliary pain that progressively worsens. Approximately 60–70% of patients report having experienced prior attacks that resolved spontaneously. As the episode progresses, however, the pain of acute cholecystitis becomes more generalized in the right upper abdomen. As with biliary colic, the pain of cholecystitis may radiate to the interscapular area, right scapula, or shoulder. Peritoneal signs of inflammation such as increased pain with jarring or on deep respiration may be apparent. The patient is anorectic and often nauseated. Vomiting is relatively common and may produce symptoms and signs of vascular and extracellular volume depletion. Jaundice is unusual early in the course of acute cholecystitis but may occur when edematous inflammatory changes involve the bile ducts and surrounding lymph nodes.
A low-grade fever is characteristically present, but shaking chills or rigors are not uncommon. The RUQ of the abdomen is almost invariably tender to palpation. An enlarged, tense gallbladder is palpable in 25–50% of patients. Deep inspiration or cough during subcostal palpation of the RUQ usually produces increased pain and inspiratory arrest (Murphy’s sign). Localized rebound tenderness in the RUQ is common, as are abdominal distention and hypoactive bowel sounds from paralytic ileus, but generalized peritoneal signs and abdominal rigidity are usually lacking, in the absence of perforation.
The diagnosis of acute cholecystitis is usually made on the basis of a characteristic history and physical examination. The triad of sudden onset of RUQ tenderness, fever, and leukocytosis is highly suggestive. Typically, leukocytosis in the range of 10,000–15,000 cells per microliter with a left shift on differential count is found. The serum bilirubin is mildly elevated (<85.5 μmol/L [5 mg/dL]) in fewer than half of patients, whereas about one-fourth have modest elevations in serum aminotransferases (usually less than a fivefold elevation). Ultrasound will demonstrate calculi in 90–95% of cases and is useful for detection of signs of gallbladder inflammation including thickening of the wall, pericholecystic fluid, and dilatation of the bile duct. The radionuclide (e.g., HIDA) biliary scan may be confirmatory if bile duct imaging is seen without visualization of the gallbladder.
Approximately 75% of patients treated medically have remission of acute symptoms within 2–7 days following hospitalization. In 25%, however, a complication of acute cholecystitis will occur despite conservative treatment (see below). In this setting, prompt surgical intervention is required. Of the 75% of patients with acute cholecystitis who undergo remission of symptoms, ~25% will experience a recurrence of cholecystitis within 1 year, and 60% will have at least one recurrent bout within 6 years. In view of the natural history of the disease, acute cholecystitis is best treated by early surgery whenever possible.
Mirizzi’s syndrome is a rare complication in which a gallstone becomes impacted in the cystic duct or neck of the gallbladder causing compression of the CBD, resulting in CBD obstruction and jaundice. Ultrasound shows gallstone(s) lying outside the hepatic duct. Endoscopic retrograde cholangiopancreatography (ERCP) (Fig. 369-2B), percutaneous transhepatic cholangiography (PTC), or magnetic resonance cholangiopancreatography (MRCP) will usually demonstrate the characteristic extrinsic compression of the CBD. Surgery consists of removing the cystic duct, diseased gallbladder, and the impacted stone. The preoperative diagnosis of Mirizzi’s syndrome is important to avoid CBD injury.
ACALCULOUS CHOLECYSTITIS In 5–10% of patients with acute cholecystitis, calculi obstructing the cystic duct are not found at surgery. In >50% of such cases, an underlying explanation for acalculous inflammation is not found. An increased risk for the development of acalculous cholecystitis is especially associated with serious trauma or burns, with the postpartum period following prolonged labor, and with orthopedic and other nonbiliary major surgical operations in the postoperative period. It may possibly complicate periods of prolonged parenteral hyperalimentation. For some of these cases, biliary sludge in the cystic duct may be responsible. Other precipitating factors include vasculitis, obstructing adenocarcinoma of the gallbladder, diabetes mellitus, torsion of the gallbladder, “unusual” bacterial infections of the gallbladder (e.g., Leptospira, Streptococcus, Salmonella, or Vibrio cholerae), and parasitic infestation of the gallbladder. Acalculous cholecystitis may also be seen with a variety of other systemic disease processes (e.g., sarcoidosis, cardiovascular disease, tuberculosis, syphilis, actinomycosis).
Although the clinical manifestations of acalculous cholecystitis are indistinguishable from those of calculous cholecystitis, the setting of acute gallbladder inflammation complicating severe underlying illness is characteristic of acalculous disease. Ultrasound or computed tomography (CT) examinations demonstrating a large, tense, static gallbladder without stones and with evidence of poor emptying over a prolonged period may be diagnostically useful in some cases. The complication rate for acalculous cholecystitis exceeds that for calculous cholecystitis. Successful management of acute acalculous cholecystitis appears to depend primarily on early diagnosis and surgical intervention, with meticulous attention to postoperative care.
ACALCULOUS CHOLECYSTOPATHY Disordered motility of the gallbladder can produce recurrent biliary pain in patients without gallstones. Infusion of an octapeptide of CCK can be used to measure the gallbladder ejection fraction during cholescintigraphy. The surgical findings have included abnormalities such as chronic cholecystitis, gallbladder muscle hypertrophy, and/or a markedly narrowed cystic duct. Some of these patients may well have had antecedent gallbladder disease. The following criteria can be used to identify patients with acalculous cholecystopathy: (1) recurrent episodes of typical RUQ pain characteristic of biliary tract pain, (2) abnormal CCK cholescintigraphy demonstrating a gallbladder ejection fraction of <40%, and (3) infusion of CCK reproducing the patient’s pain. An additional clue would be the identification of a large gallbladder on ultrasound examination. Finally, it should be noted that sphincter of Oddi dysfunction can also give rise to recurrent RUQ pain and CCK-scintigraphic abnormalities.
EMPHYSEMATOUS CHOLECYSTITIS So-called emphysematous cholecystitis is thought to begin with acute cholecystitis (calculous or acalculous) followed by ischemia or gangrene of the gallbladder wall and infection by gas-producing organisms. Bacteria most frequently cultured in this setting include anaerobes, such as Clostridium welchii or Clostridium perfringens, and aerobes, such as E. coli. This condition occurs most frequently in elderly men and in patients with diabetes mellitus. The clinical manifestations are essentially indistinguishable from those of nongaseous cholecystitis. The diagnosis is usually made on plain abdominal film by finding gas within the gallbladder lumen, dissecting within the gallbladder wall to form a gaseous ring, or in the pericholecystic tissues. The morbidity and mortality rates with emphysematous cholecystitis are considerable. Prompt surgical intervention coupled with appropriate antibiotics is mandatory.
Chronic Cholecystitis Chronic inflammation of the gallbladder wall is almost always associated with the presence of gallstones and is thought to result from repeated bouts of subacute or acute cholecystitis or from persistent mechanical irritation of the gallbladder wall by gallstones. The presence of bacteria in the bile occurs in >25% of patients with chronic cholecystitis. The presence of infected bile in a patient with chronic cholecystitis undergoing elective cholecystectomy probably adds little to the operative risk. Chronic cholecystitis may be asymptomatic for years, may progress to symptomatic gallbladder disease or to acute cholecystitis, or may present with complications (see below).
Complications of Cholecystitis • EMPYEMA AND HYDROPS Empyema of the gallbladder usually results from progression of acute cholecystitis with persistent cystic duct obstruction to superinfection of the stagnant bile with a pus-forming bacterial organism. The clinical picture resembles that of cholangitis with high fever; severe RUQ pain; marked leukocytosis; and often, prostration. Empyema of the gallbladder carries a high risk of gram-negative sepsis and/or perforation. Emergency surgical intervention with proper antibiotic coverage is required as soon as the diagnosis is suspected.
Hydrops or mucocele of the gallbladder may also result from prolonged obstruction of the cystic duct, usually by a large solitary calculus. In this instance, the obstructed gallbladder lumen is progressively distended, over a period of time, by mucus (mucocele) or by a clear transudate (hydrops) produced by mucosal epithelial cells. A visible, easily palpable, nontender mass sometimes extending from the RUQ into the right iliac fossa may be found on physical examination. The patient with hydrops of the gallbladder frequently remains asymptomatic, although chronic RUQ pain may also occur. Cholecystectomy is indicated, because empyema, perforation, or gangrene may complicate the condition.
GANGRENE AND PERFORATION Gangrene of the gallbladder results from ischemia of the wall and patchy or complete tissue necrosis. Underlying conditions often include marked distention of the gallbladder, vasculitis, diabetes mellitus, empyema, or torsion resulting in arterial occlusion. Gangrene usually predisposes to perforation of the gallbladder, but perforation may also occur in chronic cholecystitis without premonitory warning symptoms. Localized perforations are usually contained by the omentum or by adhesions produced by recurrent inflammation of the gallbladder. Bacterial superinfection of the walled-off gallbladder contents results in abscess formation. Most patients are best treated with cholecystectomy, but some seriously ill patients may be managed with cholecystostomy and drainage of the abscess. Free perforation is less common but is associated with a mortality rate of ~30%. Such patients may experience a sudden transient relief of RUQ pain as the distended gallbladder decompresses; this is followed by signs of generalized peritonitis.
FISTULA FORMATION AND GALLSTONE ILEUS Fistula formation into an adjacent organ adherent to the gallbladder wall may result from inflammation and adhesion formation. Fistulas into the duodenum are most common, followed in frequency by those involving the hepatic flexure of the colon, stomach or jejunum, abdominal wall, and renal pelvis. Clinically “silent” biliary-enteric fistulas occurring as a complication of acute cholecystitis have been found in up to 5% of patients undergoing cholecystectomy. Asymptomatic cholecystoenteric fistulas may sometimes be diagnosed by finding gas in the biliary tree on plain abdominal films. Barium contrast studies or endoscopy of the upper gastrointestinal tract or colon may demonstrate the fistula. Treatment in the symptomatic patient usually consists of cholecystectomy, CBD exploration, and closure of the fistulous tract.
Gallstone ileus refers to mechanical intestinal obstruction resulting from the passage of a large gallstone into the bowel lumen. The stone customarily enters the duodenum through a cholecystoenteric fistula at that level. The site of obstruction by the impacted gallstone is usually at the ileocecal valve, provided that the more proximal small bowel is of normal caliber. The majority of patients do not give a history of either prior biliary tract symptoms or complaints suggestive of acute cholecystitis or fistula formation. Large stones, >2.5 cm in diameter, are thought to predispose to fistula formation by gradual erosion through the gallbladder fundus. Diagnostic confirmation may occasionally be found on the plain abdominal film (e.g., small-intestinal obstruction with gas in the biliary tree and a calcified, ectopic gallstone) or following an upper gastrointestinal series (cholecystoduodenal fistula with small-bowel obstruction at the ileocecal valve). Laparotomy with stone extraction (or propulsion into the colon) remains the procedure of choice to relieve obstruction. Evacuation of large stones within the gallbladder should also be performed. In general, the gallbladder and its attachment to the intestines should be left alone.
LIMEY (MILK OF CALCIUM) BILE AND PORCELAIN GALLBLADDER Calcium salts in the lumen of the gallbladder in sufficient concentration may produce calcium precipitation and diffuse, hazy opacification of bile or a layering effect on plain abdominal roentgenography. This so-called limey bile, or milk of calcium bile, is usually clinically innocuous, but cholecystectomy is recommended, especially when it occurs in a hydropic gallbladder. In the entity called porcelain gallbladder, calcium salt deposition within the wall of a chronically inflamed gallbladder may be detected on the plain abdominal film. Cholecystectomy is advised in all patients with porcelain gallbladder because in a high percentage of cases this finding appears to be associated with the development of carcinoma of the gallbladder.
Postcholecystectomy Complications Early complications following cholecystectomy include atelectasis and other pulmonary disorders, abscess formation (often subphrenic), external or internal hemorrhage, biliary-enteric fistula, and bile leaks. Jaundice may indicate absorption of bile from an intraabdominal collection following a biliary leak or mechanical obstruction of the CBD by retained calculi, intraductal blood clots, or extrinsic compression.
Overall, cholecystectomy is a very successful operation that provides total or near-total relief of preoperative symptoms in 75–90% of patients. The most common cause of persistent postcholecystectomy symptoms is an overlooked symptomatic nonbiliary disorder (e.g., reflux esophagitis, peptic ulceration, pancreatitis, or—most often—irritable bowel syndrome). In a small percentage of patients, however, a disorder of the extrahepatic bile ducts may result in persistent symptomatology. These so-called postcholecystectomy syndromes may be due to (1) biliary strictures, (2) retained biliary calculi, (3) cystic duct stump syndrome, (4) stenosis or dyskinesia of the sphincter of Oddi, or (5) bile salt–induced diarrhea or gastritis.
CYSTIC DUCT STUMP SYNDROME In the absence of cholangiographically demonstrable retained stones, symptoms resembling biliary pain or cholecystitis in the postcholecystectomy patient have frequently been attributed to disease in a long (>1 cm) cystic duct remnant (cystic duct stump syndrome). Careful analysis, however, reveals that postcholecystectomy complaints are attributable to other causes in almost all patients in whom the symptom complex was originally thought to result from the existence of a long cystic duct stump. Accordingly, considerable care should be taken to investigate the possible role of other factors in the production of postcholecystectomy symptoms before attributing them to cystic duct stump syndrome.
PAPILLARY DYSFUNCTION, PAPILLARY STENOSIS, SPASM OF THE SPHINCTER OF ODDI, AND BILIARY DYSKINESIA Symptoms of biliary colic accompanied by signs of recurrent, intermittent biliary obstruction may be produced by acalculous cholecystopathy, papillary stenosis, papillary dysfunction, spasm of the sphincter of Oddi, and biliary dyskinesia. Papillary stenosis is thought to result from acute or chronic inflammation of the papilla of Vater or from glandular hyperplasia of the papillary segment. Five criteria have been used to define papillary stenosis: (1) upper abdominal pain, usually RUQ or epigastric; (2) abnormal liver tests; (3) dilatation of the CBD upon ERCP examination; (4) delayed (>45 min) drainage of contrast material from the duct; and (5) increased basal pressure of the sphincter of Oddi, a finding that may be of only minor significance. An alternative to ERCP is magnetic resonance cholangiography (MRC) if ERCP and/or biliary manometry are either unavailable or not feasible. After exclusion of acalculous cholecystopathy, treatment consists of endoscopic or surgical sphincteroplasty to ensure wide patency of the distal portions of both the bile and pancreatic ducts. The greater the number of the preceding criteria present, the greater is the likelihood that a patient does have a degree of papillary stenosis sufficient to justify correction. The factors usually considered as indications for sphincterotomy include (1) prolonged duration of symptoms, (2) lack of response to symptomatic treatment, (3) presence of severe disability, and (4) the patient’s choice of sphincterotomy over surgery (given a clear understanding on his or her part of the risks involved in both procedures).
Criteria for diagnosing dyskinesia of the sphincter of Oddi are even more controversial than those for papillary stenosis. Proposed mechanisms include spasm of the sphincter, denervation sensitivity resulting in hypertonicity, and abnormalities of the sequencing or frequency rates of sphincteric-contraction waves. When thorough evaluation has failed to demonstrate another cause for the pain, and when cholangiographic and manometric criteria suggest a diagnosis of biliary dyskinesia, medical treatment with nitrites or anticholinergics to attempt pharmacologic relaxation of the sphincter has been proposed. Endoscopic biliary sphincterotomy (EBS) or surgical sphincteroplasty may be indicated in patients who fail to respond to a 2- to 3-month trial of medical therapy, especially if basal sphincter of Oddi pressures are elevated. EBS has become the procedure of choice for removing bile duct stones and for other biliary and pancreatic problems.
BILE SALT–INDUCED DIARRHEA AND GASTRITIS Postcholecystectomy patients may develop symptoms of dyspepsia, which have been attributed to duodenogastric reflux of bile. However, firm data linking these symptoms to bile gastritis after surgical removal of the gallbladder are lacking. Cholecystectomy induces persistent changes in gut transit, and these changes effect a noticeable modification of bowel habits. Cholecystectomy shortens gut transit time by accelerating passage of the fecal bolus through the colon with marked acceleration in the right colon, thus causing an increase in colonic bile acid output and a shift in bile acid composition toward the more diarrheagenic secondary bile acids, i.e. deoxycholic acid. Diarrhea that is severe enough, i.e., three or more watery movements per day, can be classified as postcholecystectomy diarrhea, and this occurs in 5–10% of patients undergoing elective cholecystectomy. Treatment with bile acid–sequestering agents such as cholestyramine or colestipol is often effective in ameliorating troublesome diarrhea.
THE HYPERPLASTIC CHOLECYSTOSES
The term hyperplastic cholecystoses is used to denote a group of disorders of the gallbladder characterized by excessive proliferation of normal tissue components.
Adenomyomatosis is characterized by a benign proliferation of gallbladder surface epithelium with glandlike formations, extramural sinuses, transverse strictures, and/or fundal nodule (“adenoma” or “adenomyoma”) formation.
Cholesterolosis is characterized by abnormal deposition of lipid, especially cholesteryl esters, within macrophages in the lamina propria of the gallbladder wall. In its diffuse form (“strawberry gallbladder”), the gallbladder mucosa is brick red and speckled with bright yellow flecks of lipid. The localized form shows solitary or multiple “cholesterol polyps” studding the gallbladder wall. Cholesterol stones of the gallbladder are found in nearly half the cases. Cholecystectomy is indicated in both adenomyomatosis and cholesterolosis when symptomatic or when cholelithiasis is present.
The prevalence of gallbladder polyps in the adult population is ~5%, with a marked male predominance. Few significant changes have been found over a 5-year period in asymptomatic patients with gallbladder polyps <10 mm in diameter. Cholecystectomy is recommended in symptomatic patients, as well as in asymptomatic patients >50 years of age, or in those whose polyps are >10 mm in diameter or associated with gallstones or polyp growth on serial ultrasonography.
DISEASES OF THE BILE DUCTS
CONGENITAL ANOMALIES
Biliary Atresia and Hypoplasia Atretic and hypoplastic lesions of the extrahepatic and large intrahepatic bile ducts are the most common biliary anomalies of clinical relevance encountered in infancy. The clinical picture is one of severe obstructive jaundice during the first month of life, with pale stools. When biliary atresia is suspected on the basis of clinical, laboratory, and imaging findings, the diagnosis is confirmed by surgical exploration and operative cholangiography. Approximately 10% of cases of biliary atresia are treatable with Roux-en-Y choledochojejunostomy, with the Kasai procedure (hepatic portoenterostomy) being attempted in the remainder in an effort to restore some bile flow. Most patients, even those having successful biliary-enteric anastomoses, eventually develop chronic cholangitis, extensive hepatic fibrosis, and portal hypertension.
Choledochal Cysts Cystic dilatation may involve the free portion of the CBD, i.e., choledochal cyst, or may present as diverticulum formation in the intraduodenal segment. In the latter situation, chronic reflux of pancreatic juice into the biliary tree can produce inflammation and stenosis of the extrahepatic bile ducts leading to cholangitis or biliary obstruction. Because the process may be gradual, ~50% of patients present with onset of symptoms after age 10. The diagnosis may be made by ultrasound, abdominal CT, MRC, or cholangiography. Only one-third of patients show the classic triad of abdominal pain, jaundice, and an abdominal mass. Ultrasonographic detection of a cyst separate from the gallbladder should suggest the diagnosis of choledochal cyst, which can be confirmed by demonstrating the entrance of extrahepatic bile ducts into the cyst. Surgical treatment involves excision of the “cyst” and biliary-enteric anastomosis. Patients with choledochal cysts are at increased risk for the subsequent development of cholangiocarcinoma.
Congenital Biliary Ectasia Dilatation of intrahepatic bile ducts may involve either the major intrahepatic radicles (Caroli’s disease), the inter- and intralobular ducts (congenital hepatic fibrosis), or both. In Caroli’s disease, clinical manifestations include recurrent cholangitis, abscess formation in and around the affected ducts, and, often, brown pigment gallstone formation within portions of ectatic intrahepatic biliary radicles. Ultrasound, MRC, and CT are of great diagnostic value in demonstrating cystic dilatation of the intrahepatic bile ducts. Treatment with ongoing antibiotic therapy is usually undertaken in an effort to limit the frequency and severity of recurrent bouts of cholangitis. Progression to secondary biliary cirrhosis with portal hypertension, extrahepatic biliary obstruction, cholangiocarcinoma, or recurrent episodes of sepsis with hepatic abscess formation is common.
CHOLEDOCHOLITHIASIS
Pathophysiology and Clinical Manifestations Passage of gallstones into the CBD occurs in ~10–15% of patients with cholelithiasis. The incidence of common duct stones increases with increasing age of the patient, so that up to 25% of elderly patients may have calculi in the common duct at the time of cholecystectomy. Undetected duct stones are left behind in ~1–5% of cholecystectomy patients. The overwhelming majority of bile duct stones are cholesterol stones formed in the gallbladder, which then migrate into the extrahepatic biliary tree through the cystic duct. Primary calculi arising de novo in the ducts are usually brown pigment stones developing in patients with (1) hepatobiliary parasitism or chronic, recurrent cholangitis; (2) congenital anomalies of the bile ducts (especially Caroli’s disease); (3) dilated, sclerosed, or strictured ducts; or (4) an MDR3 (ABCB4) gene defect leading to impaired biliary phospholipids secretion (low phospholipid–associated cholesterol cholelithiasis). Common duct stones may remain asymptomatic for years, may pass spontaneously into the duodenum, or (most often) may present with biliary colic or a complication.
Complications • CHOLANGITIS Cholangitis may be acute or chronic, and symptoms result from inflammation, which usually is caused by at least partial obstruction to the flow of bile. Bacteria are present on bile culture in ~75% of patients with acute cholangitis early in the symptomatic course. The characteristic presentation of acute cholangitis involves biliary pain, jaundice, and spiking fevers with chills (Charcot’s triad). Blood cultures are frequently positive, and leukocytosis is typical. Nonsuppurative acute cholangitis is most common and may respond relatively rapidly to supportive measures and to treatment with antibiotics. In suppurative acute cholangitis, however, the presence of pus under pressure in a completely obstructed ductal system leads to symptoms of severe toxicity—mental confusion, bacteremia, and septic shock. Response to antibiotics alone in this setting is relatively poor, multiple hepatic abscesses are often present, and the mortality rate approaches 100% unless prompt endoscopic or surgical relief of the obstruction and drainage of infected bile are carried out. Endoscopic management of bacterial cholangitis is as effective as surgical intervention. ERCP with endoscopic sphincterotomy is safe and the preferred initial procedure for both establishing a definitive diagnosis and providing effective therapy.
[/not-level-membership-for-internal-medicine-category]
