419 |
Diabetes Mellitus: Complications |
Diabetes-related complications affect many organ systems and are responsible for the majority of morbidity and mortality associated with the disease. Strikingly, in the United States, diabetes is the leading cause of new blindness in adults, renal failure, and nontraumatic lower extremity amputation. Diabetes-related complications usually do not appear until the second decade of hyperglycemia. Because type 2 diabetes mellitus (DM) often has a long asymptomatic period of hyperglycemia before diagnosis, many individuals with type 2 DM have complications at the time of diagnosis. Fortunately, many of the diabetes-related complications can be prevented or delayed with early detection, aggressive glycemic control, and efforts to minimize the risks of complications.
Diabetes-related complications can be divided into vascular and nonvascular complications and are similar for type 1 and type 2 DM (Table 419-1). The vascular complications of DM are further subdivided into microvascular (retinopathy, neuropathy, nephropathy) and macrovascular complications (coronary heart disease [CHD], peripheral arterial disease [PAD], cerebrovascular disease). Microvascular complications are diabetes-specific, whereas macrovascular complications are similar to those in nondiabetics but occur at greater frequency in individuals with diabetes. Nonvascular complications include gastroparesis, infections, skin changes, and hearing loss. Whether type 2 DM increases the risk of dementia or impaired cognitive function is not clear.
|
DIABETES-RELATED COMPLICATIONS |
aThickened skin and reduced joint mobility.
GLYCEMIC CONTROL AND COMPLICATIONS
The microvascular complications of both type 1 and type 2 DM result from chronic hyperglycemia (Fig. 419-1). Evidence implicating a causative role for chronic hyperglycemia in the development of macrovascular complications is less conclusive. CHD events and mortality rate are two to four times greater in patients with type 2 DM and correlate with fasting and postprandial plasma glucose levels as well the hemoglobin A1c (HbA1c). Other factors such as dyslipidemia and hypertension also play important roles in macrovascular complications.
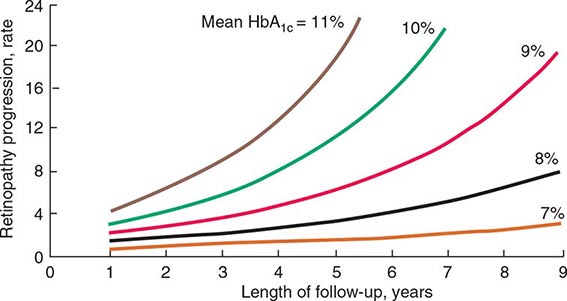
FIGURE 419-1 Relationship of glycemic control and diabetes duration to diabetic retinopathy. The progression of retinopathy in individuals in the Diabetes Control and Complications Trial is graphed as a function of the length of follow-up with different curves for different hemoglobin A1c (HbA1c) values. (Adapted from The Diabetes Control and Complications Trial Research Group: Diabetes 44:968, 1995.)
The Diabetes Control and Complications Trial (DCCT) provided definitive proof that reduction in chronic hyperglycemia can prevent many complications of type 1 DM (Fig. 419-1). This large multicenter clinical trial randomized more than 1400 individuals with type 1 DM to either intensive or conventional diabetes management and prospectively evaluated the development of diabetes-related complications during a mean follow-up of 6.5 years. Individuals in the intensive diabetes management group received multiple administrations of insulin each day (injection or pump) along with extensive educational, psychological, and medical support. Individuals in the conventional diabetes management group received twice-daily insulin injections and quarterly nutritional, educational, and clinical evaluation. The goal in the former group was normoglycemia; the goal in the latter group was prevention of symptoms of diabetes. Individuals in the intensive diabetes management group achieved a substantially lower HbA1c (7.3%) than individuals in the conventional diabetes management group (9.1%). After the DCCT results were reported in 1993, study participants continue to be followed in the Epidemiology of Diabetes Intervention and Complications (EDIC) trial, which recently completed 30 years of follow-up (DCCT + EDIC). At the end of the DCCT phase, study participants in both intensive and conventional arms were offered intensive therapy. However, during the subsequent follow-up of more than 18 years, the initial separation in glycemic control disappeared with both arms maintaining a mean HbA1c of 8.0%.
The DCCT phase demonstrated that improvement of glycemic control reduced nonproliferative and proliferative retinopathy (47% reduction), microalbuminuria (39% reduction), clinical nephropathy (54% reduction), and neuropathy (60% reduction). Improved glycemic control also slowed the progression of early diabetic complications. During the DCCT phase, weight gain (4.6 kg) and severe hypoglycemia (requiring assistance of another person to treat) were more common in the intensive therapy group. The benefits of an improvement in glycemic control occurred over the entire range of HbA1c values (Fig. 419-1), indicating that at any HbA1c level, an improvement in glycemic control is beneficial. The results of the DCCT predicted that individuals in the intensive diabetes management group would gain 7.7 additional years of vision, 5.8 additional years free from end-stage renal disease (ESRD), and 5.6 years free from lower extremity amputations. If all complications of DM were combined, individuals in the intensive diabetes management group would experience 15.3 more years of life without significant microvascular or neurologic complications of DM, compared to individuals who received standard therapy. This translates into an additional 5.1 years of life expectancy for individuals in the intensive diabetes management group. The 30-year follow-up data in the intensively treated group show a continued reduction in retinopathy, nephropathy, and cardiovascular disease. For example, individuals in the intensive therapy group had a 42–57% reduction in cardiovascular events (nonfatal myocardial infarction [MI], stroke, or death from a cardiovascular event) at a mean follow-up of 17 years, even though their subsequent glycemic control was the same as those in the conventional diabetes management group from years 6.5–17. During the EDIC phase, less than 1% of the cohort had become blind, lost a limb to amputation, or required dialysis.
The United Kingdom Prospective Diabetes Study (UKPDS) studied the course of >5000 individuals with type 2 DM for >10 years. This study used multiple treatment regimens and monitored the effect of intensive glycemic control and risk factor treatment on the development of diabetic complications. Newly diagnosed individuals with type 2 DM were randomized to (1) intensive management using various combinations of insulin, a sulfonylurea, or metformin or (2) conventional therapy using dietary modification and pharmacotherapy with the goal of symptom prevention. In addition, individuals were randomly assigned to different antihypertensive regimens. Individuals in the intensive treatment arm achieved an HbA1c of 7%, compared to a 7.9% HbA1c in the standard treatment group. The UKPDS demonstrated that each percentage point reduction in HbA1c was associated with a 35% reduction in microvascular complications. As in the DCCT, there was a continuous relationship between glycemic control and development of complications. Improved glycemic control also reduced the cardiovascular event rate in the follow-up period of >10 years.
One of the major findings of the UKPDS was that strict blood pressure control significantly reduced both macro- and microvascular complications. In fact, the beneficial effects of blood pressure control were greater than the beneficial effects of glycemic control. Lowering blood pressure to moderate goals (144/82 mmHg) reduced the risk of DM-related death, stroke, microvascular endpoints, retinopathy, and heart failure (risk reductions between 32 and 56%).
Similar reductions in the risks of retinopathy and nephropathy were also seen in a small trial of lean Japanese individuals with type 2 DM randomized to either intensive glycemic control or standard therapy with insulin (Kumamoto study). These results demonstrate the effectiveness of improved glycemic control in individuals of different ethnicity and, presumably, a different etiology of DM (i.e., phenotypically different from those in the DCCT and UKPDS). The Action to Control Cardiovascular Risk in Diabetes (ACCORD) and Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trials also found that improved glycemic control reduced microvascular complications.
Thus, these large clinical trials in type 1 and type 2 DM indicate that chronic hyperglycemia plays a causative role in the pathogenesis of diabetic microvascular complications. In both the DCCT and the UKPDS, cardiovascular events were reduced at follow-up of >10 years, even though the improved glycemic control was not maintained. The positive impact of a period of improved glycemic control on later disease has been termed a legacy effect or metabolic memory.
A summary of the features of diabetes-related complications includes the following. (1) Duration and degree of hyperglycemia correlate with complications. (2) Intensive glycemic control is beneficial in all forms of DM. (3) Blood pressure control is critical, especially in type 2 DM. (4) Survival in patients with type 1 DM is improving, and diabetes-related complications are declining. (5) Not all individuals with diabetes develop diabetes-related complications. Other incompletely defined factors appear to modulate the development of complications. For example, despite long-standing DM, some individuals never develop nephropathy or retinopathy. Many of these patients have glycemic control that is indistinguishable from those who develop microvascular complications, suggesting a genetic susceptibility for developing particular complications.
MECHANISMS OF COMPLICATIONS
Although chronic hyperglycemia is an important etiologic factor leading to complications of DM, the mechanism(s) by which it leads to such diverse cellular and organ dysfunction is unknown. An emerging hypothesis is that hyperglycemia leads to epigenetic changes (Chap. 82) that influence gene expression in affected cells. For example, this may explain the legacy effect or metabolic memory mentioned above.
Four theories, which are not mutually exclusive, on how hyperglycemia might lead to the chronic complications of DM include the following pathways. (1) Increased intracellular glucose leads to the formation of advanced glycosylation end products, which bind to a cell surface receptor, via the nonenzymatic glycosylation of intra- and extracellular proteins, leading to cross-linking of proteins, accelerated atherosclerosis, glomerular dysfunction, endothelial dysfunction, and altered extracellular matrix composition. (2) Hyperglycemia increases glucose metabolism via the sorbitol pathway related to the enzyme aldose reductase. However, testing of this theory in humans, using aldose reductase inhibitors, has not demonstrated beneficial effects. (3) Hyperglycemia increases the formation of diacylglycerol, leading to activation of protein kinase C, which alters the transcription of genes for fibronectin, type IV collagen, contractile proteins, and extracellular matrix proteins in endothelial cells and neurons. (4) Hyperglycemia increases the flux through the hexosamine pathway, which generates fructose-6-phosphate, a substrate for O-linked glycosylation and proteoglycan production, leading to altered function by glycosylation of proteins such as endothelial nitric oxide synthase or by changes in gene expression of transforming growth factor β (TGF-β) or plasminogen activator inhibitor-1.
Growth factors may play an important role in some diabetes-related complications, and their production is increased by most of these proposed pathways. Vascular endothelial growth factor A (VEGF-A) is increased locally in diabetic proliferative retinopathy and decreases after laser photocoagulation. TGF-β is increased in diabetic nephropathy and stimulates basement membrane production of collagen and fibronectin by mesangial cells. A possible unifying mechanism is that hyperglycemia leads to increased production of reactive oxygen species or superoxide in the mitochondria; these compounds may activate all four of the pathways described above. Although hyperglycemia serves as the initial trigger for complications of diabetes, it is still unknown whether the same pathophysiologic processes are operative in all complications or whether some pathways predominate in certain organs.
OPHTHALMOLOGIC COMPLICATIONS OF DIABETES MELLITUS
DM is the leading cause of blindness between the ages of 20 and 74 in the United States. The gravity of this problem is highlighted by the finding that individuals with DM are 25 times more likely to become legally blind than individuals without DM. Severe vision loss is primarily the result of progressive diabetic retinopathy and clinically significant macular edema. Diabetic retinopathy is classified into two stages: nonproliferative and proliferative. Nonproliferative diabetic retinopathy usually appears late in the first decade or early in the second decade of the disease and is marked by retinal vascular microaneurysms, blot hemorrhages, and cotton-wool spots (Fig. 419-2). Mild nonproliferative retinopathy may progress to more extensive disease, characterized by changes in venous vessel caliber, intraretinal microvascular abnormalities, and more numerous microaneurysms and hemorrhages. The pathophysiologic mechanisms invoked in nonproliferative retinopathy include loss of retinal pericytes, increased retinal vascular permeability, alterations in retinal blood flow, and abnormal retinal microvasculature, all of which can lead to retinal ischemia. A new concept is that the pathology involves inflammatory processes in the retinal neurovascular unit, which consists of neurons, glia, astrocytes, Muüller cells, and specialized vasculature.
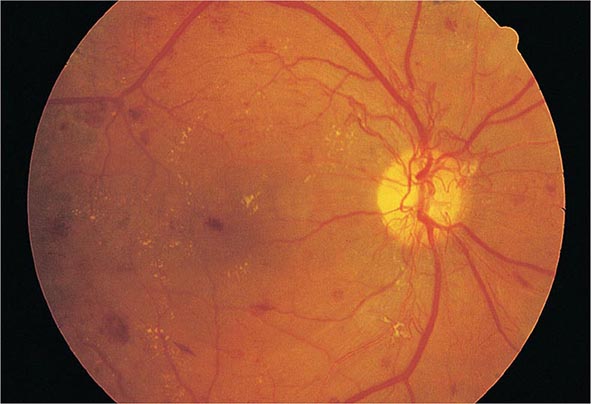
FIGURE 419-2 Diabetic retinopathy results in scattered hemorrhages, yellow exudates, and neovascularization. This patient has neovascular vessels proliferating from the optic disc, requiring urgent panretinal laser photocoagulation.
The appearance of neovascularization in response to retinal hypoxemia is the hallmark of proliferative diabetic retinopathy (Fig. 419-2). These newly formed vessels appear near the optic nerve and/or macula and rupture easily, leading to vitreous hemorrhage, fibrosis, and ultimately retinal detachment. Not all individuals with nonproliferative retinopathy go on to develop proliferative retinopathy, but the more severe the nonproliferative disease, the greater the chance of evolution to proliferative retinopathy within 5 years. This creates an important opportunity for early detection and treatment of diabetic retinopathy. Clinically significant macular edema can occur in the context of nonproliferative or proliferative retinopathy. Fluorescein angiography and optical coherence tomography are useful to detect macular edema, which is associated with a 25% chance of moderate visual loss over the next 3 years. Duration of DM and degree of glycemic control are the best predictors of the development of retinopathy; hypertension and nephropathy are also risk factors. Nonproliferative retinopathy is found in many individuals who have had DM for >20 years. Although there is genetic susceptibility for retinopathy, it confers less influence than either the duration of DM or the degree of glycemic control.
|
TREATMENT |
DIABETIC RETINOPATHY |
The most effective therapy for diabetic retinopathy is prevention. Intensive glycemic and blood pressure control will delay the development or slow the progression of retinopathy in individuals with either type 1 or type 2 DM. Paradoxically, during the first 6–12 months of improved glycemic control, established diabetic retinopathy may transiently worsen. Fortunately, this progression is temporary, and in the long term, improved glycemic control is associated with less diabetic retinopathy. Individuals with known retinopathy may be candidates for prophylactic laser photocoagulation when initiating intensive therapy. Once advanced retinopathy is present, improved glycemic control imparts less benefit, although adequate ophthalmologic care can prevent most blindness.
Regular, comprehensive eye examinations are essential for all individuals with DM (see Table 418-1). Most diabetic eye disease can be successfully treated if detected early. Routine, nondilated eye examinations by the primary care provider or diabetes specialist are inadequate to detect diabetic eye disease, which requires an ophthalmologist for optimal care of these disorders. Laser photocoagulation is very successful in preserving vision. Proliferative retinopathy is usually treated with panretinal laser photocoagulation, whereas macular edema is treated with focal laser photocoagulation and anti–vascular endothelial growth factor therapy (ocular injection). Aspirin therapy (650 mg/d) does not appear to influence the natural history of diabetic retinopathy.
RENAL COMPLICATIONS OF DIABETES MELLITUS
Diabetic nephropathy is the leading cause of chronic kidney disease (CKD), ESRD, and CKD requiring renal replacement therapy. Furthermore, the prognosis of diabetic patients on dialysis is poor, with survival comparable to many forms of cancer. Albuminuria in individuals with DM is associated with an increased risk of cardiovascular disease. Individuals with diabetic nephropathy commonly have diabetic retinopathy.
Like other microvascular complications, the pathogenesis of diabetic nephropathy is related to chronic hyperglycemia. The mechanisms by which chronic hyperglycemia leads to diabetic nephropathy, although incompletely defined, involve the effects of soluble factors (growth factors, angiotensin II, endothelin, advanced glycation end products [AGEs]), hemodynamic alterations in the renal microcirculation (glomerular hyperfiltration or hyperperfusion, increased glomerular capillary pressure), and structural changes in the glomerulus (increased extracellular matrix, basement membrane thickening, mesangial expansion, fibrosis). Some of these effects may be mediated through angiotensin II receptors. Smoking accelerates the decline in renal function. Because only 20–40% of patients with diabetes develop diabetic nephropathy, additional genetic or environmental susceptibility factors remain unidentified. Known risk factors include race and a family history of diabetic nephropathy. Diabetic nephropathy and ESRD secondary to DM develop more commonly in African Americans, Native Americans, and Hispanic individuals with diabetes.
The natural history of diabetic nephropathy is characterized by a fairly predictable sequence of events that was initially defined for individuals with type 1 DM but appears to be similar in type 2 DM (Fig. 419-3). Glomerular hyperperfusion and renal hypertrophy occur in the first years after the onset of DM and are associated with an increase of the glomerular filtration rate (GFR). During the first 5 years of DM, thickening of the glomerular basement membrane, glomerular hypertrophy, and mesangial volume expansion occur as the GFR returns to normal. After 5–10 years of type 1 DM, many individuals begin to excrete small amounts of albumin in the urine. The American Diabetes Association (ADA) recently suggested that the terms previously used to refer to increased urinary protein (microalbuminuria as defined as 30–299 mg/d in a 24-h collection or 30–299 μg/mg creatinine in a spot collection or macroalbuminuria as defined as >300 mg/24 h) be replaced by the phrases “persistent albuminuria (30–299 mg/24 h)” and “persistent albuminuria (≥300 mg/24 h)” to better reflect the continuous nature of albumin excretion in the urine as risk factor for nephropathy and cardiovascular disease (CVD). This chapter uses the terms microalbuminuria and macroalbuminuria. Although the appearance of microalbuminuria in type 1 DM is an important risk factor for progression to macroalbuminuria, only ~50% of individuals progress to macroalbuminuria over the next 10 years. In some individuals with type 1 diabetes and microalbuminuria of short duration, the microalbuminuria regresses. Microalbuminuria is also a risk factor for CVD. Once macroalbuminuria is present, there is a steady decline in GFR, and ~50% of individuals reach ESRD in 7–10 years. Once macroalbuminuria develops, blood pressure rises slightly and the pathologic changes are likely irreversible.

FIGURE 419-3 Time course of development of diabetic nephropathy. The relationship of time from onset of diabetes, the glomerular filtration rate (GFR), and the serum creatinine are shown. (Adapted from RA DeFranzo, in Therapy for Diabetes Mellitus and Related Disorders, 3rd ed. American Diabetes Association, Alexandria, VA, 1998.)
The nephropathy that develops in type 2 DM differs from that of type 1 DM in the following respects: (1) microalbuminuria or macroalbuminuria may be present when type 2 DM is diagnosed, reflecting its long asymptomatic period; (2) hypertension more commonly accompanies microalbuminuria or macroalbuminuria in type 2 DM; and (3) microalbuminuria may be less predictive of diabetic nephropathy and likelihood of progression to macroalbuminuria in type 2 DM, in large part due to increased CV mortality in this population. Finally, it should be noted that albuminuria in type 2 DM may be secondary to factors unrelated to DM, such as hypertension, congestive heart failure (CHF), prostate disease, or infection.
As part of comprehensive diabetes care (Chap. 418), albuminuria should be detected at an early stage when effective therapies can be instituted. Because some individuals with type 1 or type 2 DM have a decline in GFR in the absence of albuminuria, annual measurement of the serum creatinine to estimate GFR should also be performed. An annual microalbuminuria measurement (albumin-to-creatinine ratio in spot urine) is advised in individuals with type 1 or type 2 DM (Fig. 419-4). The urine protein measurement in a routine urinalysis does not detect these low levels of albumin excretion. Screening for albuminuria should commence 5 years after the onset of type 1 DM and at the time of diagnosis of type 2 DM.
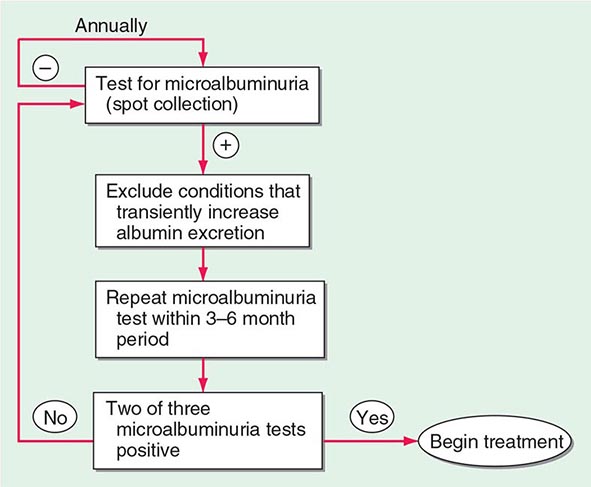
FIGURE 419-4 Screening for microalbuminuria should be performed in patients with type 1 diabetes for ≥5 years, in patients with type 2 diabetes, and during pregnancy. Non-diabetes-related conditions that might increase microalbuminuria are urinary tract infection, hematuria, heart failure, febrile illness, severe hyperglycemia, severe hypertension, and vigorous exercise. (Adapted from RA DeFranzo, in Therapy for Diabetes Mellitus and Related Disorders, 3rd ed. American Diabetes Association, Alexandria, VA, 1998.)
Type IV renal tubular acidosis (hyporeninemic hypoaldosteronism) may occur in type 1 or 2 DM. These individuals develop a propensity to hyperkalemia and acidemia, which may be exacerbated by medications (especially angiotensin-converting enzyme [ACE] inhibitors, angiotensin receptor blockers [ARBs], and spironolactone). Patients with DM are predisposed to radiocontrast-induced nephrotoxicity. Risk factors for radiocontrast-induced nephrotoxicity are preexisting nephropathy and volume depletion. Individuals with DM undergoing radiographic procedures with contrast dye should be well hydrated before and after dye exposure, and the serum creatinine should be monitored for 24–48 h following the procedure. Metformin should be held if indicated.
|
TREATMENT |
DIABETIC NEPHROPATHY |
The optimal therapy for diabetic nephropathy is prevention by control of glycemia (Chap. 418 outlines glycemic goals and approaches). Interventions effective in slowing progression of albuminuria include (1) improved glycemic control, (2) strict blood pressure control, and (3) administration of an ACE inhibitor or ARB. Dyslipidemia should also be treated.
Improved glycemic control reduces the rate at which microalbuminuria appears and progresses in type 1 and type 2 DM. However, once macroalbuminuria is present, it is unclear whether improved glycemic control will slow progression of renal disease. During the later phase of declining renal function, insulin requirements may fall as the kidney is a site of insulin degradation. As the GFR decreases with progressive nephropathy, the use and dose of glucose-lowering agents should be reevaluated (see Table 418-5). Some glucose-lowering medications (sulfonylureas and metformin) are contraindicated in advanced renal insufficiency.
Many individuals with type 1 or type 2 DM develop hypertension. Numerous studies in both type 1 and type 2 DM demonstrate the effectiveness of strict blood pressure control in reducing albumin excretion and slowing the decline in renal function. Blood pressure should be maintained at <140/90 mmHg in diabetic individuals.
Either ACE inhibitors or ARBs should be used to reduce the albuminuria and the associated decline in GFR that accompanies it in individuals with type 1 or type 2 DM (see “Hypertension,” below). Although direct comparisons of ACE inhibitors and ARBs are lacking, most experts believe that the two classes of drugs are equivalent in patient with diabetes. ARBs can be used as an alternative in patients who develop ACE inhibitor–associated cough or angioedema. After 2–3 months of therapy in patients with microalbuminuria, the drug dose is increased until the maximum tolerated dose is reached. Recent studies do not show benefit of intervention prior to onset of microalbuminuria. The combination of an ACE inhibitor and an ARB is not recommended and appears to be detrimental. If use of either ACE inhibitors or ARBs is not possible or the blood pressure is not controlled, then, diuretics, calcium channel blockers (nondihydropyridine class), or beta blockers should be used. These salutary effects are mediated by reducing intraglomerular pressure and inhibition of angiotensin-driven sclerosing pathways, in part through inhibition of TGF-β-mediated pathways.
The ADA does not suggest restriction of protein intake in diabetic individuals with albuminuria because studies have failed to show benefit.
Nephrology consultation should be considered when albuminuria appears and again when the estimated GFR is <60 mL/min per 1.743 m2. As compared with nondiabetic individuals, hemodialysis in patients with DM is associated with more frequent complications, such as hypotension (due to autonomic neuropathy or loss of reflex tachycardia), more difficult vascular access, and accelerated progression of retinopathy. Complications of atherosclerosis are the leading cause of death in diabetic individuals with nephropathy and hyperlipidemia should be treated aggressively. Renal transplantation from a living related donor is the preferred therapy but requires chronic immunosuppression. Combined pancreas-kidney transplant offers the promise of normoglycemia and freedom from dialysis.
NEUROPATHY AND DIABETES MELLITUS
Diabetic neuropathy occurs in ~50% of individuals with long-standing type 1 and type 2 DM. It may manifest as polyneuropathy, mononeuropathy, and/or autonomic neuropathy. As with other complications of DM, the development of neuropathy correlates with the duration of diabetes and glycemic control. Additional risk factors are body mass index (BMI) (the greater the BMI, the greater the risk of neuropathy) and smoking. The presence of CVD, elevated triglycerides, and hypertension is also associated with diabetic peripheral neuropathy. Both myelinated and unmyelinated nerve fibers are lost. Because the clinical features of diabetic neuropathy are similar to those of other neuropathies, the diagnosis of diabetic neuropathy should be made only after other possible etiologies are excluded (Chap. 459).
Polyneuropathy/Mononeuropathy The most common form of diabetic neuropathy is distal symmetric polyneuropathy. It most frequently presents with distal sensory loss and pain, but up to 50% of patients do not have symptoms of neuropathy. Hyperesthesia, paresthesia, and dysesthesia also may occur. Any combination of these symptoms may develop as neuropathy progresses. Symptoms may include a sensation of numbness, tingling, sharpness, or burning that begins in the feet and spreads proximally. Neuropathic pain develops in some of these individuals, occasionally preceded by improvement in their glycemic control. Pain typically involves the lower extremities, is usually present at rest, and worsens at night. Both an acute (lasting <12 months) and a chronic form of painful diabetic neuropathy have been described. The acute form is sometimes treatment-related, occurring in the context of improved glycemic control. As diabetic neuropathy progresses, the pain subsides and eventually disappears, but a sensory deficit in the lower extremities persists. Physical examination reveals sensory loss, loss of ankle deep-tendon reflexes, and abnormal position sense.
Diabetic polyradiculopathy is a syndrome characterized by severe disabling pain in the distribution of one or more nerve roots. It may be accompanied by motor weakness. Intercostal or truncal radiculopathy causes pain over the thorax or abdomen. Involvement of the lumbar plexus or femoral nerve may cause severe pain in the thigh or hip and may be associated with muscle weakness in the hip flexors or extensors (diabetic amyotrophy). Fortunately, diabetic polyradiculopathies are usually self-limited and resolve over 6–12 months.
Mononeuropathy (dysfunction of isolated cranial or peripheral nerves) is less common than polyneuropathy in DM and presents with pain and motor weakness in the distribution of a single nerve. Mononeuropathies can occur at entrapment sites such as carpal tunnel or be noncompressive. A vascular etiology for noncompressive mononeuropathies has been suggested, but the pathogenesis is unknown. Involvement of the third cranial nerve is most common and is heralded by diplopia. Physical examination reveals ptosis and ophthalmoplegia with normal pupillary constriction to light. Sometimes other cranial nerves, such as IV, VI, or VII (Bell’s palsy), are affected. Peripheral mononeuropathies or simultaneous involvement of more than one nerve (mononeuropathy multiplex) may also occur.
Autonomic Neuropathy Individuals with long-standing type 1 or 2 DM may develop signs of autonomic dysfunction involving the cholinergic, noradrenergic, and peptidergic (peptides such as pancreatic polypeptide, substance P, etc.) systems. DM-related autonomic neuropathy can involve multiple systems, including the cardiovascular, gastrointestinal, genitourinary, sudomotor, and metabolic systems. Autonomic neuropathies affecting the cardiovascular system cause a resting tachycardia and orthostatic hypotension. Reports of sudden death have also been attributed to autonomic neuropathy. Gastroparesis and bladder-emptying abnormalities are often caused by the autonomic neuropathy seen in DM (discussed below). Hyperhidrosis of the upper extremities and anhidrosis of the lower extremities result from sympathetic nervous system dysfunction. Anhidrosis of the feet can promote dry skin with cracking, which increases the risk of foot ulcers. Autonomic neuropathy may reduce counterregulatory hormone release (especially catecholamines), leading to an inability to sense hypoglycemia appropriately (hypoglycemia unawareness; Chap. 420), thereby subjecting the patient to the risk of severe hypoglycemia and complicating efforts to improve glycemic control.
|
TREATMENT |
DIABETIC NEUROPATHY |
Treatment of diabetic neuropathy is less than satisfactory. Improved glycemic control should be aggressively pursued and will improve nerve conduction velocity, but symptoms of diabetic neuropathy may not necessarily improve. Efforts to improve glycemic control in long-standing diabetes may be confounded by autonomic neuropathy and hypoglycemia unawareness. Risk factors for neuropathy such as hypertension and hypertriglyceridemia should be treated. Avoidance of neurotoxins (alcohol) and smoking, supplementation with vitamins for possible deficiencies (B12, folate; Chap. 96e), and symptomatic treatment are the mainstays of therapy. Loss of sensation in the foot places the patient at risk for ulceration and its sequelae; consequently, prevention of such problems is of paramount importance. Patients with symptoms or signs of neuropathy should check their feet daily and take precautions (footwear) aimed at preventing calluses or ulcerations. If foot deformities are present, a podiatrist should be involved.
Chronic, painful diabetic neuropathy is difficult to treat but may respond to duloxetine, amitriptyline, gabapentin, valproate, pregabalin, or opioids. Two agents, duloxetine and pregabalin, have been approved by the U.S. Food and Drug Administration (FDA) for pain associated with diabetic neuropathy, but no treatments are satisfactory. No direct comparisons of agents are available, and it is reasonable to switch agents if there is no response or if side effects develop. Referral to a pain management center may be necessary. Because the pain of acute diabetic neuropathy may resolve over time, medications may be discontinued as progressive neuronal damage from DM occurs.
Therapy of orthostatic hypotension secondary to autonomic neuropathy is also challenging. A variety of agents have limited success (fludrocortisone, midodrine, clonidine, octreotide, and yohimbine), but each has significant side effects. Nonpharmacologic maneuvers (adequate salt intake, avoidance of dehydration and diuretics, and lower extremity support hose) may offer some benefit.
GASTROINTESTINAL/GENITOURINARY DYSFUNCTION
Long-standing type 1 and 2 DM may affect the motility and function of the gastrointestinal (GI) and genitourinary systems. The most prominent GI symptoms are delayed gastric emptying (gastroparesis) and altered small- and large-bowel motility (constipation or diarrhea). Gastroparesis may present with symptoms of anorexia, nausea, vomiting, early satiety, and abdominal bloating. Microvascular complications (retinopathy and neuropathy) are usually present. Nuclear medicine scintigraphy after ingestion of a radiolabeled meal may document delayed gastric emptying, but may not correlate well with the patient’s symptoms. Noninvasive “breath tests” following ingestion of a radiolabeled meal have been developed, but are not yet validated. Although parasympathetic dysfunction secondary to chronic hyperglycemia is important in the development of gastroparesis, hyperglycemia itself also impairs gastric emptying. Nocturnal diarrhea, alternating with constipation, is a feature of DM-related GI autonomic neuropathy. In type 1 DM, these symptoms should also prompt evaluation for celiac sprue because of its increased frequency. Esophageal dysfunction in long-standing DM may occur but is usually asymptomatic.
Diabetic autonomic neuropathy may lead to genitourinary dysfunction including cystopathy and female sexual dysfunction (reduced sexual desire, dyspareunia, reduced vaginal lubrication). Symptoms of diabetic cystopathy begin with an inability to sense a full bladder and a failure to void completely. As bladder contractility worsens, bladder capacity and the postvoid residual increase, leading to symptoms of urinary hesitancy, decreased voiding frequency, incontinence, and recurrent urinary tract infections. Diagnostic evaluation includes cystometry and urodynamic studies.
Erectile dysfunction and retrograde ejaculation are very common in DM and may be one of the earliest signs of diabetic neuropathy (Chap. 67). Erectile dysfunction, which increases in frequency with the age of the patient and the duration of diabetes, may occur in the absence of other signs of diabetic autonomic neuropathy.
|
TREATMENT |
GASTROINTESTINAL/GENITOURINARY DYSFUNCTION |
Current treatments for these complications of DM are inadequate. Improved glycemic control should be a primary goal, because some aspects (neuropathy, gastric function) may improve. Smaller, more frequent meals that are easier to digest (liquid) and low in fat and fiber may minimize symptoms of gastroparesis. Metoclopramide has been used but is now restricted in both the United States and Europe and not advised for long-term use. Gastric electrical stimulatory devices are available but not approved. Diabetic diarrhea in the absence of bacterial overgrowth is treated symptomatically (Chap. 349).
Diabetic cystopathy should be treated with scheduled voiding or self-catheterization. Drugs that inhibit type 5 phosphodiesterase are effective for erectile dysfunction, but their efficacy in individuals with DM is slightly lower than in the nondiabetic population (Chap. 67). Sexual dysfunction in women may be improved with use of vaginal lubricants, treatment of vaginal infections, and systemic or local estrogen replacement.
CARDIOVASCULAR MORBIDITY AND MORTALITY
CVD is increased in individuals with type 1 or type 2 DM. The Framingham Heart Study revealed a marked increase in PAD, coronary artery disease, MI, and CHF (risk increase from one- to fivefold) in DM. In addition, the prognosis for individuals with diabetes who have coronary artery disease or MI is worse than for nondiabetics. CHD is more likely to involve multiple vessels in individuals with DM. In addition to CHD, cerebrovascular disease is increased in individuals with DM (threefold increase in stroke). Thus, after controlling for all known cardiovascular risk factors, type 2 DM increases the cardiovascular death rate twofold in men and fourfold in women.
The American Heart Association has designated DM as a “CHD risk equivalent,” and type 2 DM patients without a prior MI have a similar risk for coronary artery–related events as nondiabetic individuals who have had a prior MI. However, the cardiovascular risk assessment in type 2 DM should encompass a more nuanced approach. Cardiovascular risk is lower and not equivalent in a younger individual with a brief duration of type 2 DM compared to an older individual with long-standing type 2DM. Because of the extremely high prevalence of underlying CVD in individuals with diabetes (especially in type 2 DM), evidence of atherosclerotic vascular disease (e.g., cardiac stress test) should be sought in an individual with diabetes who has symptoms suggestive of cardiac ischemia or peripheral or carotid arterial disease. The screening of asymptomatic individuals with diabetes for CHD, even with a risk-factor scale, is not recommended because recent studies have not shown a clinical benefit. The absence of chest pain (“silent ischemia”) is common in individuals with diabetes, and a thorough cardiac evaluation should be considered prior to major surgical procedures.
The increase in cardiovascular morbidity and mortality rates in diabetes appears to relate to the synergism of hyperglycemia with other cardiovascular risk factors. Risk factors for macrovascular disease in diabetic individuals include dyslipidemia, hypertension, obesity, reduced physical activity, and cigarette smoking. Additional risk factors more prevalent in the diabetic population include microalbuminuria, macroalbuminuria, an elevation of serum creatinine, abnormal platelet function and endothelial dysfunction The possibility of atherogenic potential of insulin is suggested by the data in nondiabetic individuals showing higher serum insulin levels (indicative of insulin resistance) in association with greater risk of cardiovascular morbidity and mortality. However, treatment with insulin and the sulfonylureas did not increase the risk of CVD in individuals with type 2 DM.
|
TREATMENT |
CARDIOVASCULAR DISEASE |
In general, the treatment of coronary disease is not different in the diabetic individual (Chap. 293). Revascularization procedures for CHD, including percutaneous coronary interventions (PCI) and coronary artery bypass grafting (CABG), may be less efficacious in the diabetic individual. Initial success rates of PCI in diabetic individuals are similar to those in the nondiabetic population, but diabetic patients have higher rates of restenosis and lower long-term patency and survival rates in older studies.
Aggressive cardiovascular risk modification in all individuals with DM and glycemic control should be individualized, as discussed in Chap. 418. In patients with known CHD and type 2 DM, an ACE inhibitor (or ARB), a statin, and acetylsalicylic acid (ASA; aspirin) should be considered. Past trepidation about using beta blockers in individuals who have diabetes should not prevent use of these agents because they clearly benefit diabetic patients after MI. In patients with CHF, thiazolidinediones should not be used (Chap. 418). However, metformin can be used in patients with stable CHF if the renal function is normal.
Antiplatelet therapy reduces cardiovascular events in individuals with DM who have CHD and is recommended. Current recommendations by the ADA include the use of aspirin for primary prevention of coronary events in diabetic individuals with an increased 10-year cardiovascular risk >10% (at least one risk factor such as hypertension, smoking, family history, albuminuria, or dyslipidemia in men >50 years or women >60 years of age). ASA is not recommended for primary prevention in those with a 10-year cardiovascular risk <10%. The aspirin dose is the same as in nondiabetic individuals.
Cardiovascular Risk Factors • DYSLIPIDEMIA Individuals with DM may have several forms of dyslipidemia (Chap. 421). Because of the additive cardiovascular risk of hyperglycemia and hyperlipidemia, lipid abnormalities should be assessed aggressively and treated as part of comprehensive diabetes care (Chap. 418). The most common pattern of dyslipidemia is hypertriglyceridemia and reduced high-density lipoprotein (HDL) cholesterol levels. DM itself does not increase levels of low-density lipoprotein (LDL), but the small dense LDL particles found in type 2 DM are more atherogenic because they are more easily glycated and susceptible to oxidation.
Almost all treatment studies of diabetic dyslipidemia have been performed in individuals with type 2 DM because of the greater frequency of dyslipidemia in this form of diabetes. Interventional studies have shown that the beneficial effects of LDL reduction with statins are similar in the diabetic and nondiabetic populations. Large prospective trials of primary and secondary intervention for CHD have included some individuals with type 2 DM, and subset analyses have consistently found that reductions in LDL reduce cardiovascular events and morbidity in individuals with DM. No prospective studies have addressed similar questions in individuals with type 1 DM. Because the frequency of CVD is low in children and young adults with diabetes, assessment of cardiovascular risk should be incorporated into the guidelines discussed below.
Based on the guidelines provided by the ADA, priorities in the treatment of dyslipidemia are as follows: (1) lower the LDL cholesterol, (2) raise the HDL cholesterol, and (3) decrease the triglycerides. A treatment strategy depends on the pattern of lipoprotein abnormalities. Initial therapy for all forms of dyslipidemia should include dietary changes, as well as the same lifestyle modifications recommended in the nondiabetic population (smoking cessation, blood pressure control, weight loss, increased physical activity). The dietary recommendations for individuals with DM include increased monounsaturated fat and carbohydrates and reduced saturated fats and cholesterol (Chap. 421). According to guidelines of the ADA, the target lipid values in diabetic individuals (age >40 years) without CVD should be as follows: LDL <2.6 mmol/L (100 mg/dL); HDL >1 mmol/L (40 mg/dL) in men and >13 mmol/L (50 mg/dL) in women; and triglycerides <1.7 mmol/L (150 mg/dL). In patients >40 years, the ADA recommends addition of a statin, regardless of the LDL level, in patients with CHD and those without CHD who have CHD risk factors. Recently released guidelines by the American College of Cardiology (ACC) and American Heart Association (AHA) differ slightly and recommend that diabetic individuals aged 40–75 without CHD and a LDL of 70–189 mg/dl receive “moderate” intensity statin therapy (Chap. 291e). Improvement in glycemic control will lower triglycerides and have a modest beneficial effect by raising HDL.
If the patient is known to have CHD, the ADA recommends an LDL goal of <18 mmol/L (70 mg/dL) as an “option” (in keeping with evidence that such a goal is beneficial in nondiabetic individuals with CHD [Chap. 421]). The ACC/AHA guidelines do not advocate a specific LDL for statin therapy. HMG-CoA reductase inhibitors are the agents of choice for lowering LDL. Combination therapy with an HMG-CoA reductase inhibitor and a fibrate or another lipid-lowering agent (ezetimibe, niacin) may be considered but increases the possibility of side effects such as myositis and has not been shown to be beneficial. Nicotinic acid effectively raises HDL and can be used in patients with diabetes, but may worsen glycemic control and increase insulin resistance and has not been shown to provide additional benefit beyond statin therapy alone. Bile acid–binding resins should not be used if hypertriglyceridemia is present. In large clinical trials, statin usage is associated with a mild increase in the risk of developing type 2 DM. This risk is greatest in individuals with other risk factors for type 2 DM (Chap. 417). However, the cardiovascular benefits of statin use outweigh the mildly increased risk of diabetes.
HYPERTENSION Hypertension can accelerate other complications of DM, particularly CVD, nephropathy, and retinopathy. In targeting a goal of blood pressure of <140/80 mmHg, therapy should first emphasize lifestyle modifications such as weight loss, exercise, stress management, and sodium restriction. The BP goal should be individualized. In some younger individuals, the provider may target a blood pressure of <130/80 mmHg. Realizing that more than one agent is usually required to reach the blood pressure goal, the ADA recommends that all patients with diabetes and hypertension be treated with an ACE inhibitor or an ARB. Subsequently, agents that reduce cardiovascular risk (beta blockers, thiazide diuretics, and calcium channel blockers) should be incorporated into the regimen. ACE inhibitors and ARBs are likely equivalent in most patients with diabetes and renal disease. Serum potassium and renal function should be monitored.
Because of the high prevalence of atherosclerotic disease in individuals with type 2 DM, the possibility of renovascular hypertension should be considered when the blood pressure is not readily controlled.
LOWER EXTREMITY COMPLICATIONS
DM is the leading cause of nontraumatic lower extremity amputation in the United States. Foot ulcers and infections are also a major source of morbidity in individuals with DM. The reasons for the increased incidence of these disorders in DM involve the interaction of several pathogenic factors: neuropathy, abnormal foot biomechanics, PAD, and poor wound healing. The peripheral sensory neuropathy interferes with normal protective mechanisms and allows the patient to sustain major or repeated minor trauma to the foot, often without knowledge of the injury. Disordered proprioception causes abnormal weight bearing while walking and subsequent formation of callus or ulceration. Motor and sensory neuropathy lead to abnormal foot muscle mechanics and to structural changes in the foot (hammer toe, claw toe deformity, prominent metatarsal heads, Charcot joint). Autonomic neuropathy results in anhidrosis and altered superficial blood flow in the foot, which promote drying of the skin and fissure formation. PAD and poor wound healing impede resolution of minor breaks in the skin, allowing them to enlarge and to become infected.
Many individuals with type 2 DM develop a foot ulcer (great toe or metatarsophalangeal areas are most common), and a significant subset who develop an ulceration will ultimately undergo amputation (14–24% risk with that ulcer or subsequent ulceration). Risk factors for foot ulcers or amputation include male sex, diabetes for >10 years, peripheral neuropathy, abnormal structure of foot (bony abnormalities, callus, thickened nails), PAD, smoking, history of previous ulcer or amputation, visual impairment, and poor glycemic control. Large calluses are often precursors to or overlie ulcerations.
|
TREATMENT |
LOWER EXTREMITY COMPLICATIONS |
The optimal therapy for foot ulcers and amputations is prevention through identification of high-risk patients, education of the patient, and institution of measures to prevent ulceration. High-risk patients should be identified during the routine, annual foot examination performed on all patients with DM (see “Ongoing Aspects of Comprehensive Diabetes Care” in Chap. 418). If the monofilament test or one of the other tests is abnormal, the patient is diagnosed with loss of protective sensation (LOPS; Chap. 417). Providers should consider screening for asymptomatic PAD in individuals >50 years of age who have diabetes and other risk factors using ankle-brachial index testing in high-risk individuals (Chap. 302). Patient education should emphasize (1) careful selection of footwear, (2) daily inspection of the feet to detect early signs of poor-fitting footwear or minor trauma, (3) daily foot hygiene to keep the skin clean and moist, (4) avoidance of self-treatment of foot abnormalities and high-risk behavior (e.g., walking barefoot), and (5) prompt consultation with a health care provider if an abnormality arises. Patients at high risk for ulceration or amputation may benefit from evaluation by a foot care specialist. Calluses and nail deformities should be treated by a podiatrist. Interventions directed at risk factor modification include orthotic shoes and devices, callus management, nail care, and prophylactic measures to reduce increased skin pressure from abnormal bony architecture. Attention to other risk factors for vascular disease (smoking, dyslipidemia, hypertension) and improved glycemic control are also important.
Despite preventive measures, foot ulceration and infection are common and represent a serious problem. Due to the multifactorial pathogenesis of lower extremity ulcers, management of these lesions is multidisciplinary and often demands expertise in orthopedics, vascular surgery, endocrinology, podiatry, and infectious diseases. The plantar surface of the foot is the most common site of ulceration. Ulcers may be primarily neuropathic (no accompanying infection) or may have surrounding cellulitis or osteomyelitis. Cellulitis without ulceration is also frequent and should be treated with antibiotics that provide broad-spectrum coverage, including anaerobes (see below).
An infected ulcer is a clinical diagnosis, because superficial culture of any ulceration will likely find multiple possible bacterial species. The infection surrounding the foot ulcer is often the result of multiple organisms, with aerobic gram-positive cocci (staphylococci including MRSA, Group A and B streptococci) being most common and with aerobic gram-negative bacilli and/or obligate anaerobes as co-pathogens.
Gas gangrene may develop in the absence of clostridial infection. Cultures taken from the surface of the ulcer are not helpful; a culture from the debrided ulcer base or from purulent drainage or aspiration of the wound is the most helpful. Wound depth should be determined by inspection and probing with a blunt-tipped sterile instrument. Plain radiographs of the foot should be performed to assess the possibility of osteomyelitis in chronic ulcers that have not responded to therapy. Magnetic resonance imaging (MRI) is the most specific modality, with nuclear medicine scans and labeled white cell studies as alternatives. Surgical debridement is often necessary.
Osteomyelitis is best treated by a combination of prolonged antibiotics (IV, then oral) and/or possibly debridement of infected bone. The possible contribution of vascular insufficiency should be considered in all patients. Peripheral arterial bypass procedures are often effective in promoting wound healing and in decreasing the need for amputation of the ischemic limb (Chap. 302).
A consensus statement from the ADA identified six interventions with demonstrated efficacy in diabetic foot wounds: (1) off-loading, (2) debridement, (3) wound dressings, (4) appropriate use of antibiotics, (5) revascularization, and (6) limited amputation. Off-loading is the complete avoidance of weight bearing on the ulcer, which removes the mechanical trauma that retards wound healing. Bed rest and a variety of orthotic devices or contact casting limit weight bearing on wounds or pressure points. Surgical debridement is important and effective, but clear efficacy of other modalities for wound cleaning (enzymes, soaking, whirlpools) is lacking. Dressings such as hydrocolloid dressings promote wound healing by creating a moist environment and protecting the wound. Antiseptic agents should be avoided. Topical antibiotics are of limited value. Referral for physical therapy, orthotic evaluation, and rehabilitation should occur once the infection is controlled.
Mild or non-limb-threatening infections can be treated with oral antibiotics directed predominantly at methicillin-susceptible staphylococci and streptococci (e.g., dicloxacillin, cephalosporin, amoxicillin/clavulanate). However the increasing prevalence of MRSA often requires the use of clindamycin, doxycycline, or trimethoprim-sulfamethoxazole. Trimethoprim-sulfamethoxazole exhibits less reliable coverage of streptococci than the β-lactams, and diabetic patients may develop adverse effects including acute kidney injury and hyperkalemia. Surgical debridement of necrotic tissue, local wound care (avoidance of weight bearing over the ulcer), and close surveillance for progression of infection are crucial. More severe infections require IV antibiotics as well as bed rest and local wound care. Urgent surgical debridement may be required. Optimization of glycemic control should be a goal. IV antibiotics should provide broad-spectrum coverage directed toward Staphylococcus aureus, including MRSA, streptococci, gram-negative aerobes, and anaerobic bacteria. Initial antimicrobial regimens include vancomycin plus a β-lactam/β-lactamase inhibitor or carbapenem or vancomycin plus a combination of quinolone plus metronidazole. Daptomycin, ceftaroline, or linezolid may be substituted for vancomycin. If the infection surrounding the ulcer is not improving with IV antibiotics, reassessment of antibiotic coverage and reconsideration of the need for surgical debridement or revascularization are indicated. With clinical improvement, oral antibiotics and local wound care can be continued on an outpatient basis with close follow-up.
INFECTIONS
Individuals with DM have a greater frequency and severity of infection. The reasons for this include incompletely defined abnormalities in cell-mediated immunity and phagocyte function associated with hyperglycemia, as well as diminished vascularization. Hyperglycemia aids the colonization and growth of a variety of organisms (Candida and other fungal species). Many common infections are more frequent and severe in the diabetic population, whereas several rare infections are seen almost exclusively in the diabetic population. Examples of this latter category include rhinocerebral mucormycosis, emphysematous infections of the gallbladder and urinary tract, and “malignant” or invasive otitis externa. Invasive otitis externa is usually secondary to P. aeruginosa infection in the soft tissue surrounding the external auditory canal, usually begins with pain and discharge, and may rapidly progress to osteomyelitis and meningitis. These infections should be sought, in particular, in patients presenting with severe hyperglycemia (Chap. 418).
Pneumonia, urinary tract infections, and skin and soft tissue infections are all more common in the diabetic population. In general, the organisms that cause pulmonary infections are similar to those found in the nondiabetic population; however, gram-negative organisms, S. aureus, and Mycobacterium tuberculosis are more frequent pathogens. Urinary tract infections (either lower tract or pyelonephritis) are the result of common bacterial agents such as Escherichia coli, although several yeast species (Candida and Torulopsis glabrata) are commonly observed. Complications of urinary tract infections include emphysematous pyelonephritis and emphysematous cystitis. Bacteriuria occurs frequently in individuals with diabetic cystopathy. Susceptibility to furunculosis, superficial candidal infections, and vulvovaginitis are increased. Poor glycemic control is a common denominator in individuals with these infections. Diabetic individuals have an increased rate of colonization of S. aureus in the skinfolds and nares. Diabetic patients also have a greater risk of postoperative wound infections.
DERMATOLOGIC MANIFESTATIONS
The most common skin manifestations of DM are xerosis and pruritus and are usually relieved by skin moisturizers. Protracted wound healing and skin ulcerations are also frequent complications. Diabetic dermopathy, sometimes termed pigmented pretibial papules, or “diabetic skin spots,” begins as an erythematous macule or papule that evolves into an area of circular hyperpigmentation. These lesions result from minor mechanical trauma in the pretibial region and are more common in elderly men with DM. Bullous diseases, such as bullosa diabeticorum (shallow ulcerations or erosions in the pretibial region), are also seen. Necrobiosis lipoidica diabeticorum is an uncommon disorder, accompanying diabetes in predominantly young women. This usually begins in the pretibial region as an erythematous plaque or papules that gradually enlarge, darken, and develop irregular margins, with atrophic centers and central ulceration. They are often painful. Vitiligo occurs at increased frequency in individuals with type 1 DM. Acanthosis nigricans (hyperpigmented velvety plaques seen on the neck, axilla, or extensor surfaces) is sometimes a feature of severe insulin resistance and accompanying diabetes. Generalized or localized granuloma annulare (erythematous plaques on the extremities or trunk) and scleredema (areas of skin thickening on the back or neck at the site of previous superficial infections) are more common in the diabetic population. Lipoatrophy and lipohypertrophy can occur at insulin injection sites but are now unusual with the use of human insulin.
420 |
Hypoglycemia |
Hypoglycemia is most commonly caused by drugs used to treat diabetes mellitus or by exposure to other drugs, including alcohol. However, a number of other disorders, including critical organ failure, sepsis and inanition, hormone deficiencies, non-β-cell tumors, insulinoma, and prior gastric surgery, can cause hypoglycemia (Table 420-1). Hypoglycemia is most convincingly documented by Whipple’s triad: (1) symptoms consistent with hypoglycemia, (2) a low plasma glucose concentration measured with a precise method (not a glucose monitor), and (3) relief of symptoms after the plasma glucose level is raised. The lower limit of the fasting plasma glucose concentration is normally ∼70 mg/dL (∼3.9 mmol/L), but lower venous glucose levels occur normally, late after a meal, during pregnancy, and during prolonged fasting (>24 h). Hypoglycemia can cause serious morbidity; if severe and prolonged, it can be fatal. It should be considered in any patient with episodes of confusion, an altered level of consciousness, or a seizure.
|
CAUSES OF HYPOGLYCEMIA IN ADULTS |
Source: From PE Cryer et al: J Clin Endocrinol Metab 94:709, 2009. ©The Endocrine Society, 2009.
SYSTEMIC GLUCOSE BALANCE AND GLUCOSE COUNTERREGULATION
Glucose is an obligate metabolic fuel for the brain under physiologic conditions. The brain cannot synthesize glucose or store more than a few minutes’ supply as glycogen and therefore requires a continuous supply of glucose from the arterial circulation. As the arterial plasma glucose concentration falls below the physiologic range, blood-to-brain glucose transport becomes insufficient to support brain energy metabolism and function. However, redundant glucose counterregulatory mechanisms normally prevent or rapidly correct hypoglycemia.
Plasma glucose concentrations are normally maintained within a relatively narrow range—roughly 70–110 mg/dL (3.9–6.1 mmol/L) in the fasting state, with transient higher excursions after a meal—despite wide variations in exogenous glucose delivery from meals and in endogenous glucose utilization by, for example, exercising muscle. Between meals and during fasting, plasma glucose levels are maintained by endogenous glucose production, hepatic glycogenolysis, and hepatic (and renal) gluconeogenesis (Fig. 420-1). Although hepatic glycogen stores are usually sufficient to maintain plasma glucose levels for ∼8 h, this period can be shorter if glucose demand is increased by exercise or if glycogen stores are depleted by illness or starvation.
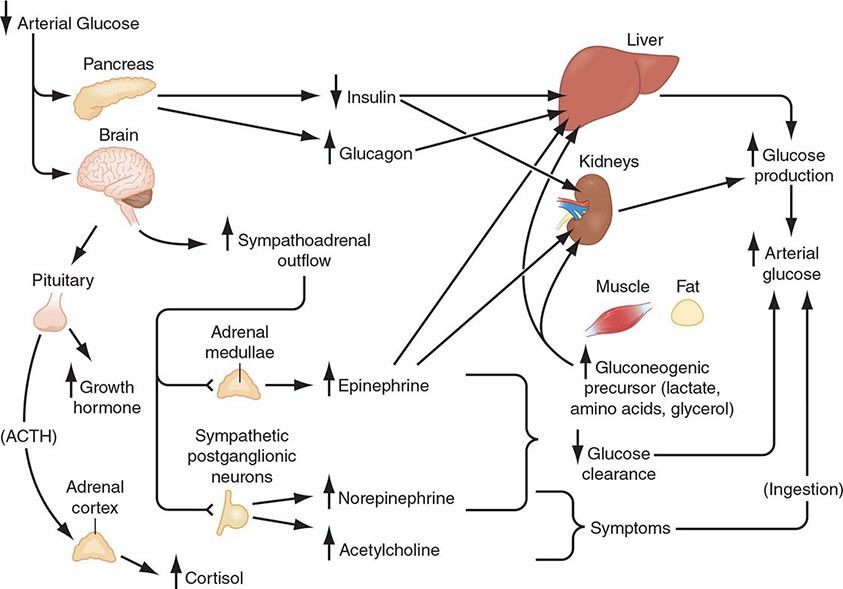
FIGURE 420-1 Physiology of glucose counterregulation: mechanisms that normally prevent or rapidly correct hypoglycemia. In insulin-deficient diabetes, the key counterregulatory responses—suppression of insulin and increases in glucagon—are lost, and stimulation of sympathoadrenal outflow is attenuated. ACTH, adrenocorticotropic hormone.
Gluconeogenesis normally requires low insulin levels and the presence of anti-insulin (counterregulatory) hormones together with a coordinated supply of precursors from muscle and adipose tissue to the liver (and kidneys). Muscle provides lactate, pyruvate, alanine, glutamine, and other amino acids. Triglycerides in adipose tissue are broken down into fatty acids and glycerol, which is a gluconeogenic precursor. Fatty acids provide an alternative oxidative fuel to tissues other than the brain (which requires glucose).
Systemic glucose balance—maintenance of the normal plasma glucose concentration—is accomplished by a network of hormones, neural signals, and substrate effects that regulate endogenous glucose production and glucose utilization by tissues other than the brain (Chap. 417). Among the regulatory factors, insulin plays a dominant role (Table 420-2; Fig. 420-1). As plasma glucose levels decline within the physiologic range in the fasting state, pancreatic β-cell insulin secretion decreases, thereby increasing hepatic glycogenolysis and hepatic (and renal) gluconeogenesis. Low insulin levels also reduce glucose utilization in peripheral tissues, inducing lipolysis and proteolysis and consequently releasing gluconeogenic precursors. Thus, a decrease in insulin secretion is the first defense against hypoglycemia.
|
PHYSIOLOGIC RESPONSES TO DECREASING PLASMA GLUCOSE CONCENTRATIONS |
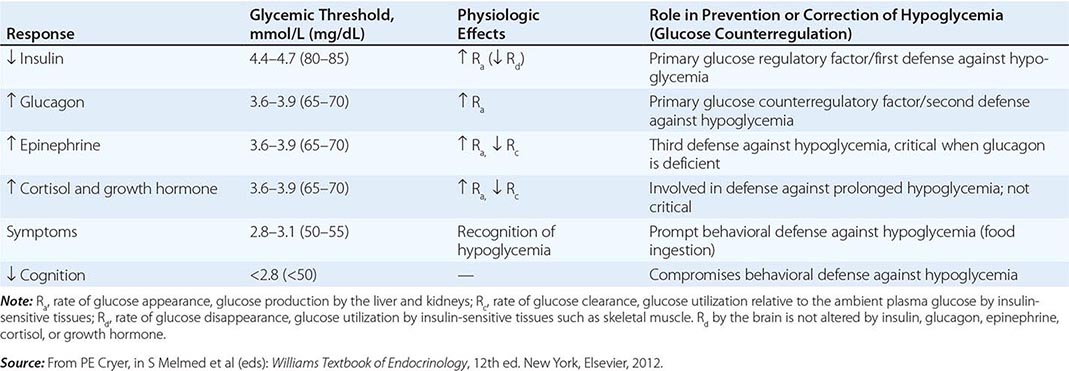
As plasma glucose levels decline just below the physiologic range, glucose counterregulatory (plasma glucose–raising) hormones are released (Table 420-2; Fig. 420-1). Among these, pancreatic α-cell glucagon, which stimulates hepatic glycogenolysis, plays a primary role. Glucagon is the second defense against hypoglycemia. Adrenomedullary epinephrine, which stimulates hepatic glycogenolysis and gluconeogenesis (and renal gluconeogenesis), is not normally critical. However, it becomes critical when glucagon is deficient. Epinephrine is the third defense against hypoglycemia. When hypoglycemia is prolonged beyond ∼4 h, cortisol and growth hormone also support glucose production and restrict glucose utilization to a limited amount (∼20% compared to epinephrine). Thus cortisol and growth hormone play no role in defense against acute hypoglycemia.
As plasma glucose levels fall further, symptoms prompt behavioral defense against hypoglycemia, including the ingestion of food (Table 420-2; Fig. 420-1). The normal glycemic thresholds for these responses to decreasing plasma glucose concentrations are shown in Table 420-2. However, these thresholds are dynamic. They shift to higher-than-normal glucose levels in people with poorly controlled diabetes, who can experience symptoms of hypoglycemia when their glucose levels decline toward the normal range (pseudohypoglycemia). On the other hand, thresholds shift to lower-than-normal glucose levels in people with recurrent hypoglycemia; e.g., patients with aggressively treated diabetes or an insulinoma have symptoms at glucose levels lower than those that cause symptoms in healthy individuals.
Clinical Manifestations Neuroglycopenic manifestations of hypoglycemia are the direct result of central nervous system glucose deprivation. These features include behavioral changes, confusion, fatigue, seizure, loss of consciousness, and, if hypoglycemia is severe and prolonged, death. Neurogenic (or autonomic) manifestations of hypoglycemia result from the perception of physiologic changes caused by the central nervous system–mediated sympathoadrenal discharge that is triggered by hypoglycemia. They include adrenergic symptoms (mediated largely by norepinephrine released from sympathetic postganglionic neurons but perhaps also by epinephrine released from the adrenal medullae), such as palpitations, tremor, and anxiety, as well as cholinergic symptoms (mediated by acetylcholine released from sympathetic postganglionic neurons), such as sweating, hunger, and paresthesias. Clearly, these are nonspecific symptoms. Their attribution to hypoglycemia requires that the corresponding plasma glucose concentration be low and that the symptoms resolve after the glucose level is raised (as delineated by Whipple’s triad).
Common signs of hypoglycemia include diaphoresis and pallor. Heart rate and systolic blood pressure are typically increased but may not be raised in an individual who has experienced repeated, recent episodes of hypoglycemia. Neuroglycopenic manifestations are often observable. Transient focal neurologic deficits occur occasionally. Permanent neurologic deficits are rare.
Etiology and Pathophysiology Hypoglycemia is most commonly a result of the treatment of diabetes. This topic is therefore addressed before other causes of hypoglycemia are considered.
HYPOGLYCEMIA IN DIABETES
Impact and Frequency Hypoglycemia is the limiting factor in the glycemic management of diabetes mellitus. First, it causes recurrent morbidity in most people with type 1 diabetes (T1DM) and in many with advanced type 2 diabetes (T2DM), and it is sometimes fatal. Second, it precludes maintenance of euglycemia over a lifetime of diabetes and thus full realization of the well-established microvascular benefits of glycemic control. Third, it causes a vicious cycle of recurrent hypoglycemia by producing hypoglycemia-associated autonomic failure—i.e., the clinical syndromes of defective glucose counterregulation and of hypoglycemia unawareness (see later).
Hypoglycemia is a fact of life for people with T1DM. They suffer an average of two episodes of symptomatic hypoglycemia per week and at least one episode of severe, at least temporarily disabling hypoglycemia each year. An estimated 6–10% of people with T1DM die as a result of hypoglycemia. The incidence of hypoglycemia is lower in T2DM than in T1DM. However, its prevalence in insulin-requiring T2DM is surprisingly high. Recent studies investigating insulin-pump or multiple-injection therapies have revealed a hypoglycemia prevalence approaching 70%. In fact, as patients with T2DM outnumber those with T1DM by ten- to twentyfold, the prevalence of hypoglycemia is now greater in T2DM. Insulin, a sulfonylurea, or a glinide can cause hypoglycemia in T2DM. Metformin, thiazolidinediones, α-glucosidase inhibitors, glucagon-like peptide 1 (GLP-1) receptor agonists, and dipeptidyl peptidase IV (DPP-IV) inhibitors should not cause hypoglycemia. However, they increase the risk when combined with one of the sulfonylureas or glinides, or with insulin. Notably, the frequency of hypoglycemia approaches that in T1DM as persons with T2DM develop absolute insulin deficiency and require more complex treatment with insulin.
Conventional Risk Factors The conventional risk factors for hypoglycemia in diabetes are identified on the basis of the premise that relative or absolute insulin excess is the sole determinant of risk. Relative or absolute insulin excess occurs when (1) insulin (or insulin secretagogue) doses are excessive, ill-timed, or of the wrong type; (2) the influx of exogenous glucose is reduced (e.g., during an overnight fast or after missed meals or snacks); (3) insulin-independent glucose utilization is increased (e.g., during exercise); (4) sensitivity to insulin is increased (e.g., with improved glycemic control, in the middle of the night, late after exercise, or with increased fitness or weight loss); (5) endogenous glucose production is reduced (e.g., after alcohol ingestion); and (6) insulin clearance is reduced (e.g., in renal failure). However, these conventional risk factors alone explain a minority of episodes; other factors are typically involved.
Hypoglycemia-Associated Autonomic Failure (HAAF) While marked insulin excess alone can cause hypoglycemia, iatrogenic hypoglycemia in diabetes is typically the result of the interplay of relative or absolute therapeutic insulin excess and compromised physiologic and behavioral defenses against falling plasma glucose concentrations (Table 420-2; Fig. 420-2). Defective glucose counterregulation compromises physiologic defense (particularly decrements in insulin and increments in glucagon and epinephrine), and hypoglycemia unawareness compromises behavioral defense (ingestion of carbohydrate).
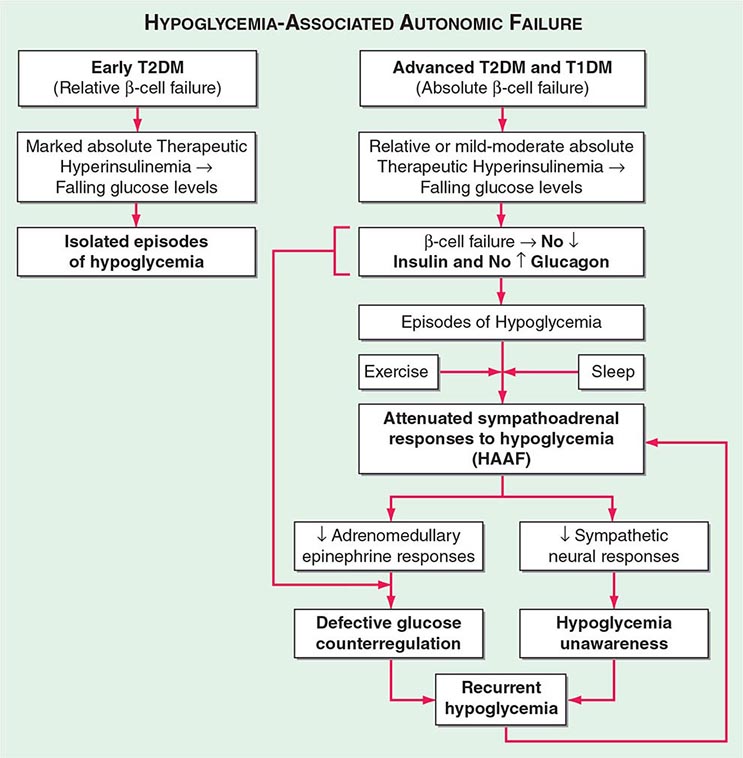
FIGURE 420-2 Hypoglycemia-associated autonomic failure (HAAF) in insulin-deficient diabetes. T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus. (Modified from PE Cryer: Hypoglycemia in Diabetes. Pathophysiology, Prevalence, and Prevention, 2nd ed. © American Diabetes Association, 2012.)
DEFECTIVE GLUCOSE COUNTERREGULATION In the setting of absolute endogenous insulin deficiency, insulin levels do not decrease as plasma glucose levels fall; the first defense against hypoglycemia is lost. Furthermore, probably because the decrement in intraislet insulin is normally a signal to stimulate glucagon secretion, glucagon levels do not increase as plasma glucose levels fall further; a second defense against hypoglycemia is lost. Finally, the increase in epinephrine levels, a third defense against hypoglycemia, in response to a given level of hypoglycemia is typically attenuated. The glycemic threshold for the sympathoadrenal (adrenomedullary epinephrine and sympathetic neural norepinephrine) response is shifted to lower plasma glucose concentrations. That shift is typically the result of recent antecedent iatrogenic hypoglycemia. In the setting of absent decrements in insulin and of absent increments in glucagon, the attenuated increment in epinephrine causes the clinical syndrome of defective glucose counterregulation. Affected patients are at ≥25-fold greater risk of severe iatrogenic hypoglycemia during aggressive glycemic therapy for their diabetes than are patients with normal epinephrine responses. This functional—and potentially reversible—disorder is distinct from classic diabetic autonomic neuropathy—a structural and irreversible disorder.
HYPOGLYCEMIA UNAWARENESS The attenuated sympathoadrenal response (largely the reduced sympathetic neural response) to hypoglycemia causes the clinical syndrome of hypoglycemia unawareness—i.e., loss of the warning adrenergic and cholinergic symptoms that previously allowed the patient to recognize developing hypoglycemia and therefore to abort the episode by ingesting carbohydrates. Affected patients are at a sixfold increased risk of severe iatrogenic hypoglycemia during aggressive glycemic therapy of their diabetes.
HAAF IN DIABETES The concept of HAAF in diabetes posits that recent antecedent iatrogenic hypoglycemia (or sleep or prior exercise) causes both defective glucose counterregulation (by reducing the epinephrine response to a given level of subsequent hypoglycemia in the setting of absent insulin and glucagon responses) and hypoglycemia unawareness (by reducing the sympathoadrenal response to a given level of subsequent hypoglycemia). These impaired responses create a vicious cycle of recurrent iatrogenic hypoglycemia (Fig. 420-2). Hypoglycemia unawareness and, to some extent, the reduced epinephrine component of defective glucose counterregulation are reversible by as little as 2–3 weeks of scrupulous avoidance of hypoglycemia in most affected patients.
On the basis of this pathophysiology, additional risk factors for hypoglycemia in diabetes include (1) absolute insulin deficiency, indicating that insulin levels will not decrease and glucagon levels will not increase as plasma glucose levels fall; (2) a history of severe hypoglycemia or of hypoglycemia unawareness, implying recent antecedent hypoglycemia, as well as prior exercise or sleep, indicating that the sympathoadrenal response will be attenuated; and (3) lower hemoglobin A1c (HbA1c) levels or lower glycemic goals that, all other factors being equal, increase the probability of recent antecedent hypoglycemia.
Hypoglycemia Risk Factor Reduction Several recent multicenter, randomized, controlled trials investigating the potential benefits of tight glucose control in either inpatient or outpatient settings have reported a high prevalence of severe hypoglycemia. In the NICE-SUGAR study, attempts to control in-hospital plasma glucose values towards physiologic levels resulted in increased mortality risk. The ADVANCE and ACCORD studies and the Veterans Affairs Diabetes Trial (VADT) also found a significant incidence of severe hypoglycemia among T2DM patients. Severe hypoglycemia with accompanying serious cardiovascular morbidity and mortality also occurred in the standard (e.g., not receiving intensified treatment) control group in both the ACCORD study and the VADT. Thus, severe hypoglycemia can and does occur at HbA1c values of 8–9% in both T1DM and T2DM. Somewhat surprisingly, all three studies found little or no benefit of intensive glucose control to reduce macrovascular events in T2DM. In fact, the ACCORD study was ended early because of the increased mortality rate in the intensive glucose control arm. Whether iatrogenic hypoglycemia was the cause of the increased mortality risk is not known. In light of these findings, some new recommendations and paradigms have been formulated. Whereas there is little debate regarding the need to reduce hyperglycemia in the hospital, the glycemic maintenance goals have been modified to lie between 140 and 180 mg/dL. Accordingly, the benefits of insulin therapy and reduced hyperglycemia can be obtained while the prevalence of hypoglycemia is reduced.
Similarly, evidence exists that intensive glucose control can reduce the prevalence of microvascular disease in both T1DM and T2DM. These benefits need to be weighed against the increased prevalence of hypoglycemia. Certainly, the level of glucose control (i.e., the HbA1c level) should be evaluated for each patient. Multicenter trials have demonstrated that individuals with recently diagnosed T1DM or T2DM can have better glycemic control with less hypoglycemia. In addition, there is still long-term benefit in reducing HbA1c values from higher to lower, albeit still above recommended levels. Perhaps a reasonable therapeutic goal is the lowest HbA1c level that does not cause severe hypoglycemia and that preserves awareness of hypoglycemia.
Pancreatic transplantation (both whole-organ and islet-cell) has been used in part as a treatment for severe hypoglycemia. Generally, rates of hypoglycemia are reduced after transplantation. This decrease appears to be due to increased physiologic insulin and glucagon responses during hypoglycemia.
The use of continuous glucose monitors offers some promise as a method of reducing hypoglycemia while improving HbA1c. Other interventions to stimulate counterregulatory responses, such as selective serotonin-reuptake inhibitors, β-adrenergic receptor antagonists, opiate receptor antagonists, and fructose, remain experimental and have not been assessed in large-scale clinical trials.
Thus, intensive glycemic therapy (Chap. 418) needs to be applied along with the patient’s education and empowerment, frequent self-monitoring of blood glucose, flexible insulin (and other drug) regimens (including the use of insulin analogues, both short- and longer-acting), individualized glycemic goals, and ongoing professional guidance, support, and consideration of both the conventional risk factors and those indicative of compromised glucose counterregulation. Given a history of hypoglycemia unawareness, a 2- to 3-week period of scrupulous avoidance of hypoglycemia is indicated.
HYPOGLYCEMIA WITHOUT DIABETES
There are many causes of hypoglycemia (Table 420-1). Because hypoglycemia is common in insulin- or insulin secretagogue–treated diabetes, it is often reasonable to assume that a clinically suspicious episode is the result of hypoglycemia. On the other hand, because hypoglycemia is rare in the absence of relevant drug-treated diabetes, it is reasonable to conclude that a hypoglycemic disorder is present only in patients in whom Whipple’s triad can be demonstrated.
Particularly when patients are ill or medicated, the initial diagnostic focus should be on the possibility of drug involvement and then on critical illnesses, hormone deficiency, or non–islet cell tumor hypoglycemia. In the absence of any of these etiologic factors and in a seemingly well individual, the focus should shift to possible endogenous hyperinsulinism or accidental, surreptitious, or even malicious hypoglycemia.
Drugs Insulin and insulin secretagogues suppress glucose production and stimulate glucose utilization. Ethanol blocks gluconeogenesis but not glycogenolysis. Thus, alcohol-induced hypoglycemia typically occurs after a several-day ethanol binge during which the person eats little food, with consequent glycogen depletion. Ethanol is usually measurable in blood at the time of presentation, but its levels correlate poorly with plasma glucose concentrations. Because gluconeogenesis becomes the predominant route of glucose production during prolonged hypoglycemia, alcohol can contribute to the progression of hypoglycemia in patients with insulin-treated diabetes.
Many other drugs have been associated with hypoglycemia. These include commonly used drugs such as angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists, β-adrenergic receptor antagonists, quinolone antibiotics, indomethacin, quinine, and sulfonamides.
Critical Illness Among hospitalized patients, serious illnesses such as renal, hepatic, or cardiac failure; sepsis; and inanition are second only to drugs as causes of hypoglycemia.
Rapid and extensive hepatic destruction (e.g., toxic hepatitis) causes fasting hypoglycemia because the liver is the major site of endogenous glucose production. The mechanism of hypoglycemia in patients with cardiac failure is unknown. Hepatic congestion and hypoxia may be involved. Although the kidneys are a source of glucose production, hypoglycemia in patients with renal failure is also caused by the reduced clearance of insulin and the reduced mobilization of gluconeogenic precursors in renal failure.
Sepsis is a relatively common cause of hypoglycemia. Increased glucose utilization is induced by cytokine production in macrophage-rich tissues such as the liver, spleen, and lung. Hypoglycemia develops if glucose production fails to keep pace. Cytokine-induced inhibition of gluconeogenesis in the setting of nutritional glycogen depletion, in combination with hepatic and renal hypoperfusion, may also contribute to hypoglycemia.
Hypoglycemia can be seen with starvation, perhaps because of loss of whole-body fat stores and subsequent depletion of gluconeogenic precursors (e.g., amino acids), necessitating increased glucose utilization.
Hormone Deficiencies Neither cortisol nor growth hormone is critical to the prevention of hypoglycemia, at least in adults. Nonetheless, hypoglycemia can occur with prolonged fasting in patients with primary adrenocortical failure (Addison’s disease) or hypopituitarism. Anorexia and weight loss are typical features of chronic cortisol deficiency and likely result in glycogen depletion. Cortisol deficiency is associated with impaired gluconeogenesis and low levels of gluconeogenic precursors; these associations suggest that substrate-limited gluconeogenesis, in the setting of glycogen depletion, is the cause of hypoglycemia. Growth hormone deficiency can cause hypoglycemia in young children. In addition to extended fasting, high rates of glucose utilization (e.g., during exercise or in pregnancy) or low rates of glucose production (e.g., after alcohol ingestion) can precipitate hypoglycemia in adults with previously unrecognized hypopituitarism.
Hypoglycemia is not a feature of the epinephrine-deficient state that results from bilateral adrenalectomy when glucocorticoid replacement is adequate, nor does it occur during pharmacologic adrenergic blockade when other glucoregulatory systems are intact. Combined deficiencies of glucagon and epinephrine play a key role in the pathogenesis of iatrogenic hypoglycemia in people with insulin-deficient diabetes, as discussed earlier. Otherwise, deficiencies of these hormones are not usually considered in the differential diagnosis of a hypoglycemic disorder.
Non-β-Cell Tumors Fasting hypoglycemia, often termed non–islet cell tumor hypoglycemia, occurs occasionally in patients with large mesenchymal or epithelial tumors (e.g., hepatomas, adrenocortical carcinomas, carcinoids). The glucose kinetic patterns resemble those of hyperinsulinism (see next), but insulin secretion is suppressed appropriately during hypoglycemia. In most instances, hypoglycemia is due to overproduction of an incompletely processed form of insulin-like growth factor II (“big IGF-II”) that does not complex normally with circulating binding proteins and thus more readily gains access to target tissues. The tumors are usually apparent clinically, plasma ratios of IGF-II to IGF-I are high, and free IGF-II levels (and levels of pro-IGF-II [1–21]) are elevated. Curative surgery is seldom possible, but reduction of tumor bulk may ameliorate hypoglycemia. Therapy with a glucocorticoid, a growth hormone, or both has also been reported to alleviate hypoglycemia. Hypoglycemia attributed to ectopic IGF-I production has been reported but is rare.
Endogenous Hyperinsulinism Hypoglycemia due to endogenous hyperinsulinism can be caused by (1) a primary β-cell disorder—typically a β-cell tumor (insulinoma), sometimes multiple insulinomas, or a functional β-cell disorder with β-cell hypertrophy or hyperplasia; (2) an antibody to insulin or to the insulin receptor; (3) a β-cell secretagogue such as a sulfonylurea; or perhaps (4) ectopic insulin secretion, among other very rare mechanisms. None of these causes is common.
The fundamental pathophysiologic feature of endogenous hyperinsulinism caused by a primary β-cell disorder or an insulin secretagogue is the failure of insulin secretion to fall to very low levels during hypoglycemia. This feature is assessed by measurement of plasma insulin, C-peptide (the connecting peptide that is cleaved from proinsulin to produce insulin), proinsulin, and glucose concentrations during hypoglycemia. Insulin, C-peptide, and proinsulin levels need not be high relative to normal, euglycemic values; rather, they are inappropriately high in the setting of a low plasma glucose concentration. Critical diagnostic findings are a plasma insulin concentration ≥3 μU/mL (≥18 pmol/L), a plasma C-peptide concentration ≥0.6 ng/mL (≥0.2 nmol/L), and a plasma proinsulin concentration ≥5.0 pmol/L when the plasma glucose concentration is <55 mg/dL (<3.0 mmol/L) with symptoms of hypoglycemia. A low plasma β-hydroxybutyrate concentration (≤2.7 mmol/L) and an increment in plasma glucose level of >25 mg/dL (>1.4 mmol/L) after IV administration of glucagon (1.0 mg) indicate increased insulin (or IGF) actions.
The diagnostic strategy is (1) to measure plasma glucose, insulin, C-peptide, proinsulin, and β-hydroxybutyrate concentrations and to screen for circulating oral hypoglycemic agents during an episode of hypoglycemia and (2) to assess symptoms during the episode and seek their resolution following correction of hypoglycemia by IV injection of glucagon (i.e., to document Whipple’s triad). This is straightforward if the patient is hypoglycemic when seen. Since endogenous hyperinsulinemic disorders usually, but not invariably, cause fasting hypoglycemia, a diagnostic episode may develop after a relatively short outpatient fast. Serial sampling during an inpatient diagnostic fast of up to 72 h or after a mixed meal is more problematic. An alternative is to give patients a detailed list of the required measurements and ask them to present to an emergency room, with the list, during a symptomatic episode. Obviously, a normal plasma glucose concentration during a symptomatic episode indicates that the symptoms are not the result of hypoglycemia.
An insulinoma—an insulin-secreting pancreatic islet β-cell tumor—is the prototypical cause of endogenous hyperinsulinism and therefore should be sought in patients with a compatible clinical syndrome. However, insulinoma is not the only cause of endogenous hyperinsulinism. Some patients with fasting endogenous hyperinsulinemic hypoglycemia have diffuse islet involvement with β-cell hypertrophy and sometimes hyperplasia. This pattern is commonly referred to as nesidioblastosis, although β-cells budding from ducts are not invariably found. Other patients have a similar islet pattern but with postprandial hypoglycemia, a disorder termed noninsulinoma pancreatogenous hypoglycemia. Postgastric bypass postprandial hypoglycemia, which most often follows Roux-en-Y gastric bypass, is also characterized by diffuse islet involvement and endogenous hyperinsulinism. Some have suggested that exaggerated GLP-1 responses to meals cause hyperinsulinemia and hypoglycemia, but the relevant pathogenesis has not been clearly established. If medical treatments with agents such as an α-glucosidase inhibitor, diazoxide, or octreotide fail, partial pancreatectomy may be required. Autoimmune hypoglycemias include those caused by an antibody to insulin that binds post-meal insulin and then gradually disassociates, with consequent late postprandial hypoglycemia. Alternatively, an insulin receptor antibody can function as an agonist. The presence of an insulin secretagogue, such as a sulfonylurea or a glinide, results in a clinical and biochemical pattern similar to that of an insulinoma but can be distinguished by the presence of the circulating secretagogue. Finally, there are reports of very rare phenomena such as ectopic insulin secretion, a gain-of-function insulin receptor mutation, and exercise-induced hyperinsulinemia.
Insulinomas are uncommon, with an estimated yearly incidence of 1 in 250, 000. Because more than 90% of insulinomas are benign, they are a treatable cause of potentially fatal hypoglycemia. The median age at presentation is 50 years in sporadic cases, but the tumor usually presents in the third decade when it is a component of multiple endocrine neoplasia type 1 (Chap. 408). More than 99% of insulinomas are within the substance of the pancreas, and the tumors are usually small (<2.0 cm in diameter in 90% of cases). Therefore, they come to clinical attention because of hypoglycemia rather than mass effects. CT or MRI detects ∼70–80% of insulinomas. These methods detect metastases in the roughly 10% of patients with a malignant insulinoma. Transabdominal ultrasound often identifies insulinomas, and endoscopic ultrasound has a sensitivity of ∼90%. Somatostatin receptor scintigraphy is thought to detect insulinomas in about half of patients. Selective pancreatic arterial calcium injections, with the endpoint of a sharp increase in hepatic venous insulin levels, regionalize insulinomas with high sensitivity, but this invasive procedure is seldom necessary except to confirm endogenous hyperinsulinism in the diffuse islet disorders. Intraoperative pancreatic ultrasonography almost invariably localizes insulinomas that are not readily palpable by the surgeon. Surgical resection of a solitary insulinoma is generally curative. Diazoxide, which inhibits insulin secretion, or the somatostatin analogue octreotide can be used to treat hypoglycemia in patients with unresectable tumors; everolimus, an mTOR (mammalian target of rapamycin) inhibitor, is promising.
ACCIDENTAL, SURREPTITIOUS, OR MALICIOUS HYPOGLYCEMIA
Accidental ingestion of an insulin secretagogue (e.g., as the result of a pharmacy or other medical error) or even accidental administration of insulin can occur. Factitious hypoglycemia, caused by surreptitious or even malicious administration of insulin or an insulin secretagogue, shares many clinical and laboratory features with insulinoma. It is most common among health care workers, patients with diabetes or their relatives, and people with a history of other factitious illnesses. However, it should be considered in all patients being evaluated for hypoglycemia of obscure cause. Ingestion of an insulin secretagogue causes hypoglycemia with increased C-peptide levels, whereas exogenous insulin causes hypoglycemia with low C-peptide levels reflecting suppression of insulin secretion.
Analytical error in the measurement of plasma glucose concentrations is rare. On the other hand, glucose monitors used to guide treatment of diabetes are not quantitative instruments, particularly at low glucose levels, and should not be used for the definitive diagnosis of hypoglycemia. Even with a quantitative method, low measured glucose concentrations can be artifactual—e.g., the result of continued glucose metabolism by the formed elements of the blood ex vivo, particularly in the presence of leukocytosis, erythrocytosis, or thrombocytosis or with delayed separation of the serum from the formed elements (pseudohypoglycemia).
INBORN ERRORS OF METABOLISM CAUSING HYPOGLYCEMIA
Nondiabetic hypoglycemia also results from inborn errors of metabolism. Such hypoglycemia most commonly occurs in infancy but can also occur in adulthood. Cases in adults can be classified into those resulting in fasting hypoglycemia, postprandial hypoglycemia, and exercise-induced hypoglycemia.
Fasting Hypoglycemia Although rare, disorders of glycogenolysis can result in fasting hypoglycemia. These disorders include glycogen storage disease (GSD) of types 0, I, III, and IV and Fanconi-Bickel syndrome (Chap. 433e). Patients with GSD types I and III characteristically have high blood lactate levels before and after meals, respectively. Both groups have hypertriglyceridemia, but ketones are high in GSD type III. Defects in fatty acid oxidation also result in fasting hypoglycemia. These defects can include (1) defects in the carnitine cycle; (2) fatty-acid β-oxidation disorders; (3) electron transfer disturbances; and (4) ketogenesis disorders. Finally, defects in gluconeogenesis (fructose-1, 6-biphosphatase) have been reported to result in recurrent hypoglycemia and lactic acidosis.
Postprandial Hypoglycemia Inborn errors of metabolism resulting in postprandial hypoglycemia are also rare. These errors include (1) glucokinase, SUR1, and Kir6.2 potassium channel mutations; (2) congenital disorders of glycosylation; and (3) inherited fructose intolerance.
Exercise-Induced Hypoglycemia Exercise-induced hypoglycemia, by definition, follows exercise. It results in hyperinsulinemia caused by increased activity of monocarboxylate transporter 1 in β cells.
421 |
Disorders of Lipoprotein Metabolism |
Lipoproteins are complexes of lipids and proteins that are essential for transport of cholesterol, triglycerides, and fat-soluble vitamins. Previously, lipoprotein disorders were the purview of specialized lipidologists, but the demonstration that lipid-lowering therapy significantly reduces the clinical complications of atherosclerotic cardiovascular disease (ASCVD) has brought the diagnosis and treatment of these disorders into the domain of the internist. The number of individuals who are candidates for lipid-lowering therapy continues to increase. Therefore, the appropriate diagnosis and management of lipoprotein disorders is of critical importance in the practice of medicine. This chapter reviews normal lipoprotein physiology, the pathophysiology of disorders of lipoprotein metabolism, the effects of diet and other environmental factors that influence lipoprotein metabolism, and the practical approaches to the diagnosis and management of lipoprotein disorders.
LIPOPROTEIN METABOLISM
LIPOPROTEIN CLASSIFICATION AND COMPOSITION
Lipoproteins are large macromolecular complexes composed of lipids and proteins that transport poorly soluble lipids (primarily triglycerides, cholesterol, and fat-soluble vitamins) through body fluids (plasma, interstitial fluid, and lymph) to and from tissues. Lipoproteins play an essential role in the absorption of dietary cholesterol, long-chain fatty acids, and fat-soluble vitamins; the transport of triglycerides, cholesterol, and fat-soluble vitamins from the liver to peripheral tissues; and the transport of cholesterol from peripheral tissues to the liver and intestine.
Lipoproteins contain a core of hydrophobic lipids (triglycerides and cholesteryl esters) surrounded by a shell of hydrophilic lipids (phospholipids, unesterified cholesterol) and proteins (called apolipoproteins) that interact with body fluids. The plasma lipoproteins are divided into five major classes based on their relative density (Fig. 421-1 and Table 421-1): chylomicrons, very-low-density lipoproteins (VLDLs), intermediate-density lipoproteins (IDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs). Each lipoprotein class comprises a family of particles that vary in density, size, and protein composition. Because lipid is less dense than water, the density of a lipoprotein particle is primarily determined by the amount of lipid per particle. Chylomicrons are the most lipid-rich and therefore least dense lipoprotein particles, whereas HDLs have the least lipid and are therefore the most dense lipoproteins. In addition to their density, lipoprotein particles can be classified according to their size, determined either by nondenaturing gel electrophoresis or by nuclear magnetic resonance profiling. There is a strong inverse relationship between density and size, with the largest particles being the most buoyant (chylomicrons) and the smallest particles being the most dense (HDL).
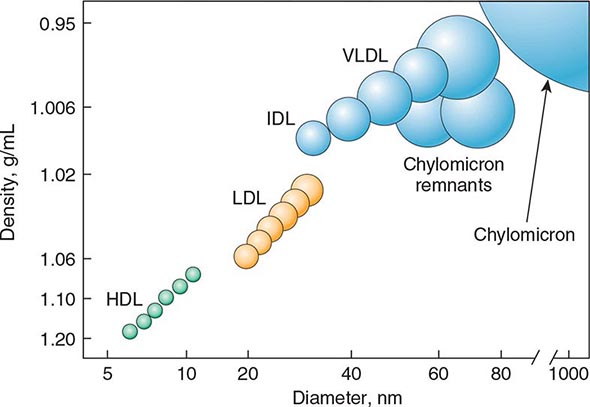
FIGURE 421-1 The density and size distribution of the major classes of lipoprotein particles. Lipoproteins are classified by density and size, which are inversely related. HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein.
|
MAJOR LIPOPROTEIN CLASSES |
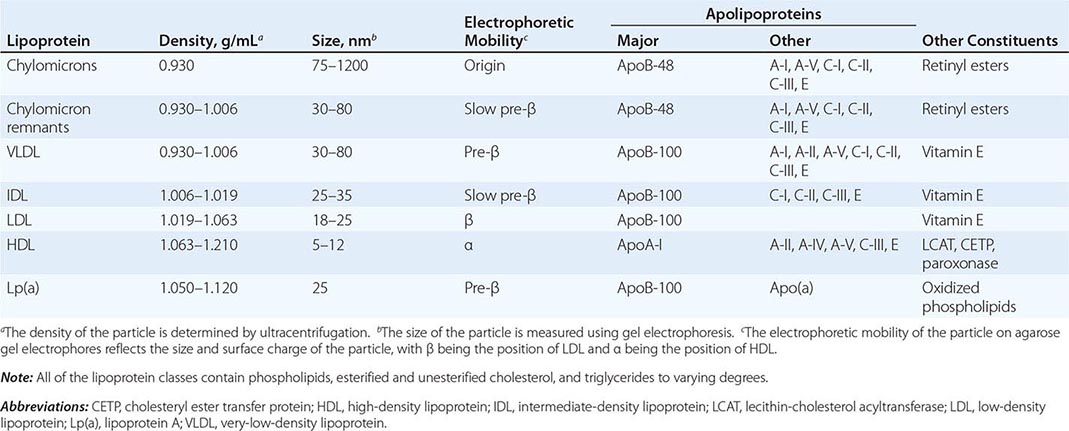
The proteins associated with lipoproteins, called apolipoproteins (Table 421-2), are required for the assembly, structure, function, and metabolism of lipoproteins. Apolipoproteins activate enzymes important in lipoprotein metabolism and act as ligands for cell surface receptors. ApoB is a very large protein and is the major structural protein of chylomicrons, VLDLs, IDLs, and LDLs; one molecule of apoB, either apoB-48 (chylomicron) or apoB-100 (VLDL, IDL, or LDL), is present on each lipoprotein particle. The human liver synthesizes apoB-100, and the intestine makes apoB-48, which is derived from the same gene by mRNA editing. HDLs have different apolipoproteins that define this lipoprotein class, most importantly apoA-I, which is synthesized in the liver and intestine and is found on virtually all HDL particles. ApoA-II is the second most abundant HDL apolipoprotein and is on approximately two-thirds of the HDL particles. ApoC-I, apoC-II, and apoC-III participate in the metabolism of triglyceride-rich lipoproteins. ApoE also plays a critical role in the metabolism and clearance of triglyceride-rich particles. Most apolipoproteins, other than apoB, exchange actively among lipoprotein particles in the blood. Apolipoprotein(a) [apo(a)] is a distinctive apolipoprotein and is discussed more below.
|
MAJOR APOLIPOPROTEINS |
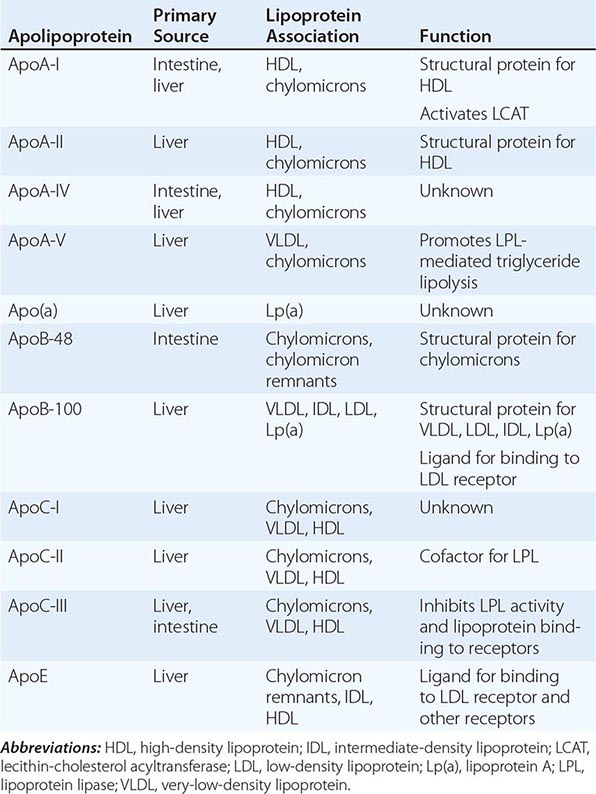
TRANSPORT OF INTESTINALLY DERIVED DIETARY LIPIDS BY CHYLOMICRONS
One critical role of lipoproteins is the efficient transport of dietary lipids from the intestine to tissues that require fatty acids for energy or store and metabolize lipids (Fig. 421-2). Dietary triglycerides are hydrolyzed by lipases within the intestinal lumen and emulsified with bile acids to form micelles. Dietary cholesterol, fatty acids, and fat-soluble vitamins are absorbed in the proximal small intestine. Cholesterol and retinol are esterified (by the addition of a fatty acid) in the enterocyte to form cholesteryl esters and retinyl esters, respectively. Longer-chain fatty acids (>12 carbons) are incorporated into triglycerides and packaged with apoB-48, cholesteryl esters, retinyl esters, phospholipids, and cholesterol to form chylomicrons. Nascent chylomicrons are secreted into the intestinal lymph and delivered via the thoracic duct directly to the systemic circulation, where they are extensively processed by peripheral tissues before reaching the liver. The particles encounter lipoprotein lipase (LPL), which is anchored to a glycosylphosphatidylinositol-anchored protein, GPIHBP1, that is attached to the endothelial surfaces of capillaries in adipose tissue, heart, and skeletal muscle (Fig. 421-2). The triglycerides of chylomicrons are hydrolyzed by LPL, and free fatty acids are released. ApoC-II, which is transferred to circulating chylomicrons from HDL, acts as a required cofactor for LPL in this reaction. The released free fatty acids are taken up by adjacent myocytes or adipocytes and either oxidized to generate energy or reesterified and stored as triglyceride. Some of the released free fatty acids bind albumin before entering cells and are transported to other tissues, especially the liver. The chylomicron particle progressively shrinks in size as the hydrophobic core is hydrolyzed and the hydrophilic lipids (cholesterol and phospholipids) and apolipoproteins on the particle surface are transferred to HDL, creating chylomicron remnants.
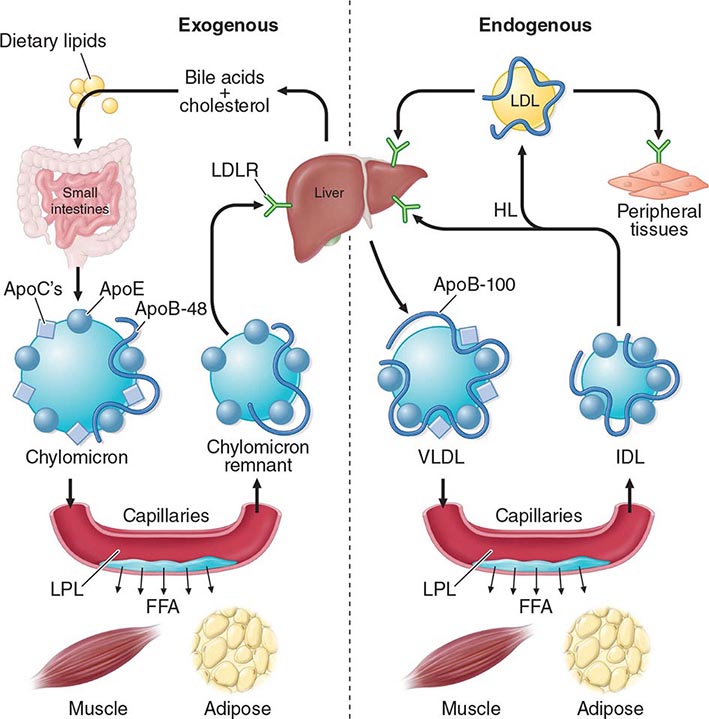
FIGURE 421-2 The exogenous and endogenous lipoprotein metabolic pathways. The exogenous pathway transports dietary lipids to the periphery and the liver. The endogenous pathway transports hepatic lipids to the periphery. FFA, free fatty acid; HL, hepatic lipase; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; LPL, lipoprotein lipase; VLDL, very-low-density lipoprotein.
Chylomicron remnants are rapidly removed from the circulation by the liver through a process that requires apoE as a ligand for receptors in the liver. Consequently, few, if any, chylomicrons or chylomicron remnants are generally present in the blood after a 12-h fast, except in patients with certain disorders of lipoprotein metabolism.
TRANSPORT OF HEPATICALLY DERIVED LIPIDS BY VLDL AND LDL
Another key role of lipoproteins is the transport of hepatic lipids from the liver to the periphery (Fig. 421-2). VLDL particles resemble chylomicrons in protein composition but contain apoB-100 rather than apoB-48 and have a higher ratio of cholesterol to triglyceride (~1 mg of cholesterol for every 5 mg of triglyceride). The triglycerides of VLDL are derived predominantly from the esterification of long-chain fatty acids in the liver. The packaging of hepatic triglycerides with the other major components of the nascent VLDL particle (apoB-100, cholesteryl esters, phospholipids, and vitamin E) requires the action of the enzyme microsomal triglyceride transfer protein (MTP). After secretion into the plasma, VLDL acquires multiple copies of apoE and apolipoproteins of the C series by transfer from HDL. As with chylomicrons, the triglycerides of VLDL are hydrolyzed by LPL, especially in muscle, heart, and adipose tissue. After the VLDL remnants dissociate from LPL, they are referred to as IDLs, which contain roughly similar amounts of cholesterol and triglyceride. The liver removes approximately 40–60% of IDL by LDL receptor–mediated endocytosis via binding to apoE. The remainder of IDL is remodeled by hepatic lipase (HL) to form LDL. During this process, phospholipids and triglyceride in the particle are hydrolyzed, and all apolipoproteins except apoB-100 are transferred to other lipoproteins. Approximately 70% of LDL is removed from the circulation by the liver in a similar manner as IDL; however, in this case, apoB, rather than apoE, binds the LDL receptor.
Lp(a) is a lipoprotein similar to LDL in lipid and protein composition, but it contains an additional protein called apolipoprotein(a) [apo(a)]. Apo(a) is synthesized in the liver and attached to apoB-100 by a disulfide linkage. The major site of clearance of Lp(a) is the liver, but the uptake pathway is not known.
HDL METABOLISM AND REVERSE CHOLESTEROL TRANSPORT
All nucleated cells synthesize cholesterol, but only hepatocytes and enterocytes can effectively excrete cholesterol from the body, into either the bile or the gut lumen. In the liver, cholesterol is secreted into the bile, either directly or after conversion to bile acids. Cholesterol in peripheral cells is transported from the plasma membranes of peripheral cells to the liver and intestine by a process termed “reverse cholesterol transport” that is facilitated by HDL (Fig. 421-3).
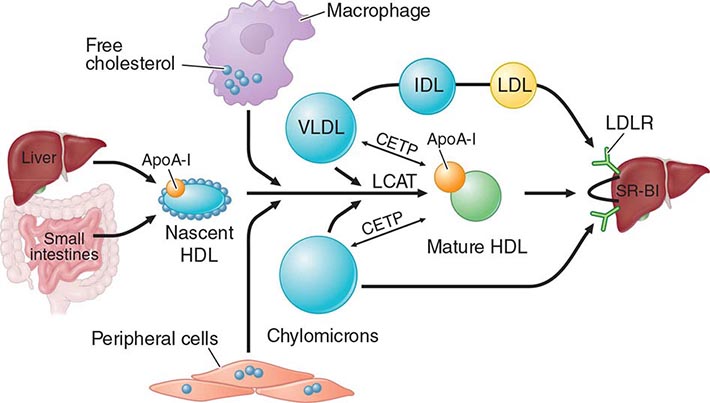
FIGURE 421-3 High-density lipoprotein (HDL) metabolism and reverse cholesterol transport. This pathway transports excess cholesterol from the periphery back to the liver for excretion in the bile. The liver and the intestine produce nascent HDLs. Free cholesterol is acquired from macrophages and other peripheral cells and esterified by lecithin-cholesterol acyltransferase (LCAT), forming mature HDLs. HDL cholesterol can be selectively taken up by the liver via SR-BI (scavenger receptor class BI). Alternatively, HDL cholesteryl ester can be transferred by cholesteryl ester transfer protein (CETP) from HDLs to very-low-density lipoproteins (VLDLs) and chylomicrons, which can then be taken up by the liver. IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor.
Nascent HDL particles are synthesized by the intestine and the liver. Newly secreted apoA-I rapidly acquires phospholipids and unesterified cholesterol from its site of synthesis (intestine or liver) via efflux promoted by the membrane protein ATP-binding cassette protein A1 (ABCA1). This process results in the formation of discoidal HDL particles, which then recruit additional unesterified cholesterol from cells or circulating lipoproteins. Within the HDL particle, the cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT), a plasma enzyme associated with HDL, and the more hydrophobic cholesteryl ester moves to the core of the HDL particle. As HDL acquires more cholesteryl ester, it becomes spherical, and additional apolipoproteins and lipids are transferred to the particles from the surfaces of chylomicrons and VLDLs during lipolysis.
HDL cholesterol is transported to hepatocytes by both an indirect and a direct pathway. HDL cholesteryl esters can be transferred to apoB-containing lipoproteins in exchange for triglyceride by the cholesteryl ester transfer protein (CETP). The cholesteryl esters are then removed from the circulation by LDL receptor–mediated endocytosis. HDL cholesterol can also be taken up directly by hepatocytes via the scavenger receptor class B1 (SR-B1), a cell surface receptor that mediates the selective transfer of lipids to cells.
HDL particles undergo extensive remodeling within the plasma compartment by a variety of lipid transfer proteins and lipases. The phospholipid transfer protein (PLTP) transfers phospholipids from other lipoproteins to HDL or among different classes of HDL particles. After CETP- and PLTP-mediated lipid exchange, the triglyceride-enriched HDL becomes a much better substrate for HL, which hydrolyzes the triglycerides and phospholipids to generate smaller HDL particles. A related enzyme called endothelial lipase hydrolyzes HDL phospholipids, generating smaller HDL particles that are catabolized faster. Remodeling of HDL influences the metabolism, function, and plasma concentrations of HDL.
DISORDERS OF ELEVATED CHOLESTEROL AND TRIGLYCERIDES
Disorders of lipoprotein metabolism are collectively referred to as “dyslipidemias.” Dyslipidemias are generally characterized clinically by increased plasma levels of cholesterol, triglycerides, or both, variably accompanied by reduced levels of HDL cholesterol. Because plasma lipids are commonly screened (see below), dyslipidemia is frequently seen in clinical practice. The majority of patients with dyslipidemia have some combination of genetic predisposition (often polygenic) and environmental contribution (lifestyle, medical condition, or drug). Many, but not all, patients with dyslipidemia are at increased risk for ASCVD, the primary reason for making the diagnosis, as intervention may reduce this risk. In addition, patients with substantially elevated levels of triglycerides may be at risk for acute pancreatitis and require intervention to reduce this risk.
Although literally hundreds of proteins influence lipoprotein metabolism and may interact to produce dyslipidemia in an individual patient, there are a limited number of discrete “nodes” that regulate lipoprotein metabolism. These include: (1) assembly and secretion of triglyceride-rich VLDLs by the liver; (2) lipolysis of triglyceride-rich lipoproteins by LPL; (3) receptor-mediated uptake of apoB-containing lipoproteins by the liver; (4) cellular cholesterol metabolism in the hepatocyte and the enterocyte; and (5) neutral lipid transfer and phospholipid hydrolysis in the plasma. The following discussion will focus on these regulatory nodes, recognizing that in many cases these nodes interact with and influence each other.
DYSLIPIDEMIA CAUSED BY EXCESSIVE HEPATIC SECRETION OF VLDL
Excessive production of VLDL by the liver is one of the most common causes of dyslipidemia. Individuals with excessive hepatic VLDL production usually have elevated fasting triglycerides and low levels of HDL cholesterol (HDL-C), with variable elevations in LDL cholesterol (LDL-C) but usually elevated plasma levels of apoB. A cluster of other metabolic risk factors are often found in association with VLDL overproduction, including obesity, glucose intolerance, insulin resistance, and hypertension (the so-called metabolic syndrome, Chap. 422). Some of the major factors that drive hepatic VLDL secretion include obesity, insulin resistance, a high-carbohydrate diet, alcohol use, exogenous estrogens, and genetic predisposition.
Secondary Causes of VLDL Overproduction • HIGH-CARBOHYDRATE DIET Dietary carbohydrates are converted to fatty acids in the liver. Some of the newly synthesized fatty acids are esterified forming triglycerides (TGs) and secreted as constituents of VLDL. Thus, excessive intake of calories as carbohydrates, which is frequent in Western societies, leads to increased hepatic VLDL-TG secretion.
ALCOHOL Regular alcohol consumption inhibits hepatic oxidation of free fatty acids, thus promoting hepatic TG synthesis and VLDL secretion. Regular alcohol use also raises plasma levels of HDL-C and should be considered in patients with the unusual combination of elevated TGs and elevated HDL-C.
OBESITY AND INSULIN RESISTANCE (See also Chaps. 416 and 417) Obesity and insulin resistance are frequently accompanied by dyslipidemia characterized by elevated plasma levels of TG, low HDL-C, variable levels of LDL-C, and increased levels of small dense LDL. The increase in adipocyte mass and accompanying decreased insulin sensitivity associated with obesity have multiple effects on lipid metabolism, with one of the major effects being excessive hepatic VLDL production. More free fatty acids are delivered from the expanded and insulin-resistant adipose tissue to the liver, where they are reesterified in hepatocytes to form TGs, which are packaged into VLDLs for secretion into the circulation. In addition, the increased insulin levels promote increased fatty acid synthesis in the liver. In insulin-resistant patients who progress to type 2 diabetes mellitus, dyslipidemia remains common, even when the patient is under relatively good glycemic control. In addition to increased VLDL production, insulin resistance can also result in decreased LPL activity, resulting in reduced catabolism of chylomicrons and VLDLs and more severe hypertriglyceridemia (see below).
NEPHROTIC SYNDROME (See also Chap. 335) Nephrotic syndrome is a classic cause of excessive VLDL production. The molecular mechanism of VLDL overproduction remains poorly understood but has been attributed to the effects of hypoalbuminemia leading to increased hepatic protein synthesis. Effective treatment of the underlying renal disease often normalizes the lipid profile, but most patients with chronic nephrotic syndrome require lipid-lowering drug therapy.
CUSHING’S SYNDROME (See also Chap. 406) Endogenous or exogenous glucocorticoid excess is associated with increased VLDL synthesis and secretion and hypertriglyceridemia. Patients with Cushing’s syndrome frequently have dyslipidemia especially characterized by hypertriglyceridemia and low HDL-C, although elevations in plasma levels of LDL-C can also be seen.
Primary (Genetic) Causes of VLDL Overproduction Genetic variation influences hepatic VLDL production. A number of genes have been identified in which common and low-frequency variants likely contribute to increased VLDL production, likely involving interactions with diet and other environmental factors. The best recognized inherited condition associated with VLDL overproduction is familial combined hyperlipidemia.
FAMILIAL COMBINED HYPERLIPIDEMIA (FCHL) FCHL is generally characterized by elevations in plasma levels of TGs (VLDL) and LDL-C (including small dense LDL) and reduced plasma levels of HDL-C. It is estimated to occur in approximately 1 in 100–200 individuals and is an important cause of premature coronary heart disease (CHD); approximately 20% of patients who develop CHD under age 60 have FCHL. FCHL can manifest in childhood but is usually not fully expressed until adulthood. The disease clusters in families, with affected family members typically have one of three possible phenotypes: (1) elevated plasma levels of LDL-C, (2) elevated plasma levels of TGs due to elevation in VLDL, or (3) elevated plasma levels of both LDL-C and TG. The lipoprotein profile can switch among these three phenotypes in the same individual over time and may depend on factors such as diet, exercise, weight, and insulin sensitivity. Patients with FCHL almost always have significantly elevated plasma levels of apoB. The levels of apoB are disproportionately high relative to the plasma LDL-C concentration, indicating the presence of small, dense LDL particles, which are characteristic of this syndrome.
Individuals with this phenotype generally share the same metabolic defect, namely overproduction of VLDL by the liver. The molecular etiology of this condition remains poorly understood, and no single gene has been identified in which mutations cause this disorder. It is likely that defects in a combination of genes can cause the condition, suggesting that a more appropriate term for the disorder might be polygenic combined hyperlipidemia.
The presence of a mixed dyslipidemia (plasma TG levels between 200 and 600 mg/dL and total cholesterol levels between 200 and 400 mg/dL, usually with HDL-C levels <40 mg/dL in men and <50 mg/dL in women) and a family history of mixed dyslipidemia and/or premature CHD strongly suggests the diagnosis. Individuals with this phenotype should be treated aggressively due to significantly increased risk of premature CHD. Decreased dietary intake of simple carbohydrates, aerobic exercise, and weight loss can all have beneficial effects on the lipid profile. Patients with diabetes should be aggressively treated to maintain good glucose control. Most patients with FCHL require lipid-lowering drug therapy, starting with statins, to reduce lipoprotein levels and lower the risk of cardiovascular disease.
LIPODYSTROPHY Lipodystrophy is a condition in which the generation of adipose tissue generally or in certain fat depots is impaired. Lipodystrophies are often associated with insulin resistance and elevated plasma levels of VLDL and chylomicrons due to increased fatty acid synthesis and VLDL production, as well as reduced clearance of TG-rich particles. This disorder can be especially difficult to control. Patients with congenital generalized lipodystrophy are very rare and have nearly complete absence of subcutaneous fat, accompanied by profound insulin resistance and leptin deficiency, and accumulation of TGs in multiple tissues including the liver. Some patients with generalized lipodystrophy have been treated successfully with leptin administration. Partial lipodystrophy is somewhat more common and can be caused by mutations in several different genes, most notably lamin A. Partial lipodystrophy is usually characterized by increased truncal fat accompanied by markedly reduced or absent subcutaneous fat in the extremities and buttocks. These patients generally have insulin resistance, often quite severe, accompanied by type 2 diabetes, hepatosteatosis, and dyslipidemia. The dyslipidemia is usually characterized by elevated TGs and cholesterol and can be difficult to manage clinically. Patients with partial lipodystrophy are at substantially increased risk of atherosclerotic vascular disease and should therefore be treated aggressively for their dyslipidemia with statins and, if necessary, additional lipid-lowering therapies.
DYSLIPIDEMIA CAUSED BY IMPAIRED LIPOLYSIS OF TRIGLYCERIDE-RICH LIPOPROTEINS
Impaired lipolysis of the TGs in TG-rich lipoproteins (TRLs) also commonly contributes to dyslipidemia. As noted above, LPL is the key enzyme responsible for hydrolyzing the TGs in chylomicrons and VLDL. LPL is synthesized and secreted into the extracellular space from adipocytes, myocytes, and cardiomyocytes. It is then transported from the subendothelial to the vascular endothelial surfaces by GPIHPB1. LPL is also synthesized in macrophages. Individuals with impaired LPL activity, whether secondary or due to a primary genetic disorder, have elevated fasting TGs and low levels of HDL-C, usually without elevation in LDL-C or apoB. Insulin resistance, in addition to causing excessive VLDL production, can also cause impaired LPL activity and lipolysis. A number of common and low-frequency genetic variants have been described that influence LPL activity, and single-gene Mendelian disorders that reduce LPL activity have also been described (Table 421-3).
|
PRIMARY HYPERLIPOPROTEINEMIAS CAUSED BY KNOWN SINGLE-GENE MUTATIONS |
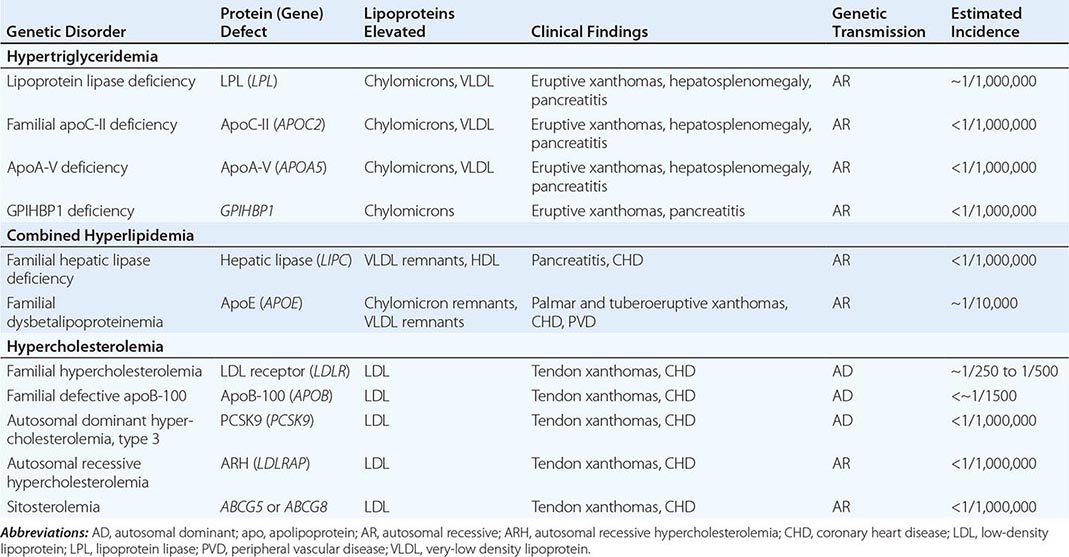
Secondary Causes of Impaired Lipolysis of TRLs • OBESITY AND INSULIN RESISTANCE (See also Chaps. 415e, 416, and 417) In addition to hepatic overproduction of VLDL, as discussed above, obesity, insulin resistance, and type 2 diabetes have been reported to be associated with variably reduced LPL activity. This may be due in part to the effects of tissue insulin resistance leading to reduced transcription of LPL in skeletal muscle and adipose, as well as to increased production of the LPL inhibitor apoC-III by the liver. This reduction in LPL activity often contributes to the dyslipidemia seen in these patients.
Primary (Genetic) Causes and Genetic Predisposition to Impaired Lipolysis of TRLs • FAMILIAL CHYLOMICRONEMIA SYNDROME As noted above, LPL is required for the hydrolysis of TGs in chylomicrons and VLDLs, and apoC-II is a cofactor for LPL. Genetic deficiency or inactivity of either protein results in impaired lipolysis and profound elevations in plasma chylomicrons. These patients can also have elevated plasma levels of VLDL, but chylomicronemia predominates. The fasting plasma is turbid, and if left at 4°C (39.2°F) for a few hours, the chylomicrons float to the top and form a creamy supernatant. In these disorders, collectively called the familial chylomicronemia syndrome, fasting TG levels are almost invariably >1000 mg/dL. Fasting cholesterol levels are also elevated but to a lesser degree.
LPL deficiency has autosomal recessive inheritance and has a frequency of approximately 1 in 1 million in the population. ApoC-II deficiency is also recessive in inheritance pattern and is even less common than LPL deficiency. Multiple different mutations in the LPL and APOC2 genes cause these diseases. Obligate LPL heterozygotes often have mild-to-moderate elevations in plasma TG levels, whereas individuals heterozygous for mutation in apoC-II do not have hypertriglyceridemia.
Both LPL and apoC-II deficiency usually present in childhood with recurrent episodes of severe abdominal pain due to acute pancreatitis. On funduscopic examination, the retinal blood vessels are opalescent (lipemia retinalis). Eruptive xanthomas, which are small, yellowish-white papules, often appear in clusters on the back, buttocks, and extensor surfaces of the arms and legs. These typically painless skin lesions may become pruritic. Hepatosplenomegaly results from the uptake of circulating chylomicrons by reticuloendothelial cells in the liver and spleen. For unknown reasons, some patients with persistent and pronounced chylomicronemia never develop pancreatitis, eruptive xanthomas, or hepatosplenomegaly. Premature CHD is not generally a feature of familial chylomicronemia syndromes.
The diagnoses of LPL and apoC-II deficiency are established enzymatically in specialized laboratories by assaying TG lipolytic activity in postheparin plasma. Blood is sampled after an IV heparin injection to release the endothelial-bound LPL. LPL activity is profoundly reduced in both LPL and apoC-II deficiency; in patients with apoC-II deficiency, it normalizes after the addition of normal plasma (providing a source of apoC-II). Molecular sequencing of the genes can be used to confirm the diagnosis.
The major therapeutic intervention in familial chylomicronemia syndrome is dietary fat restriction (to as little as 15 g/d) with fat-soluble vitamin supplementation. Consultation with a registered dietician familiar with this disorder is essential. Caloric supplementation with medium-chain TGs, which are absorbed directly into the portal circulation, can be useful, but there is uncertainty about their hepatic safety with prolonged use. If dietary fat restriction alone is not successful in resolving the chylomicronemia, fish oils have been effective in some patients. In patients with apoC-II deficiency, apoC-II can be provided by infusing fresh-frozen plasma to resolve the chylomicronemia in the acute setting. Management of patients with familial chylomicronemia syndrome is particularly challenging during pregnancy when VLDL production is increased. A gene therapy approach, called alipogene tiparvovec, is approved for LPL deficiency in Europe; it involves multiple intramuscular injections of an adeno-associated viral vector encoding a gain-of-function LPL variant, leading to skeletal myocyte expression of LPL.
APOA-V DEFICIENCY Another apolipoprotein, ApoA-V, facilitates the association of VLDL and chylomicrons with LPL and promotes their hydrolysis. Individuals harboring loss-of-function mutations in both APOA5 alleles develop hyperchylomicronemia. Heterozygosity for variants in APOA5 that reduce its function contributes to the polygenic basis of hypertriglyceridemia.
GPIHBP1 DEFICIENCY Homozygosity for mutations that interfere with GPIHBP1 synthesis or folding cause severe hypertriglyceridemia by compromising the transport of LPL to the vascular endothelium. The frequency of chylomicronemia due to mutations in GHIHBP1 has not been established but appears to be very rare.
FAMILIAL HYPERTRIGLYCERIDEMIA (FHTG) FHTG is characterized by elevated fasting TGs without a clear secondary cause, average to below average LDL-C levels, low HDL-C levels, and a family history of hypertriglyceridemia. Plasma LDL-C levels are often reduced due to defective conversion of TG-rich particles to LDL. In contrast to FCHL, apoB levels are not elevated. The identification of other first-degree relatives with hypertriglyceridemia is useful in making the diagnosis. Unlike in FCHL, this condition is not generally associated with a significantly increased risk of CHD. However, if the hypertriglyceridemia is exacerbated by environmental factors, medical conditions, or drugs, the TGs can rise to a level at which acute pancreatitis is a risk. Indeed, management of patients with this condition is mostly geared toward reduction of TGs to prevent pancreatitis.
Individuals with this phenotype generally have reduced lipolysis of TRLs, although overproduction of VLDL by the liver can also contribute. No single gene has been identified in which mutations cause this disorder, whereas combinations of gene variants have been shown to cause this phenotype. A more appropriate term for this condition might be polygenic hypertriglyceridemia.
It is important to consider and rule out secondary causes of the hypertriglyceridemia as discussed above. Increased intake of simple carbohydrates, obesity, insulin resistance, alcohol use, estrogen treatment, and certain medications can exacerbate this phenotype. Patients who are at high risk for CHD due to other risk factors should be treated with statin therapy. In patients who are otherwise not at high risk for CHD, lipid-lowering drug therapy can frequently be avoided with appropriate dietary and lifestyle changes. Patients with plasma TG levels >500 mg/dL after a trial of diet and exercise should be considered for drug therapy with a fibrate or fish oil to reduce TGs in order to prevent pancreatitis.
DYSLIPIDEMIA CAUSED BY IMPAIRED HEPATIC UPTAKE OF APOB-CONTAINING LIPOPROTEINS
Impaired uptake of LDL and remnant lipoproteins by the liver is another common cause of dyslipidemia. As discussed above, the LDL receptor is the major receptor responsible for uptake of LDL and remnant particles by the liver. Downregulation of LDL receptor activity or genetic variation that reduces the activity of the LDL receptor pathway leads to elevations in LDL-C. One major factor that reduces LDL receptor activity is a diet high in saturated and trans fats. Other medical conditions that reduce LDL receptor activity include hypothyroidism and estrogen deficiency. In addition, genetic variation in a number of genes influences LDL clearance, and mutations in some of these genes cause several discrete Mendelian disorders of elevated LDL-C (Table 421-3).
Secondary Causes of Impaired Hepatic Uptake of Lipoproteins • HYPOTHYROIDISM (See also Chap. 405) Hypothyroidism is associated with elevated plasma LDL-C levels due primarily to a reduction in hepatic LDL receptor function and delayed clearance of LDL. Thyroid hormone increases hepatic expression of the LDL receptor. Hypothyroid patients also frequently have increased levels of circulating IDL, and some patients with hypothyroidism also have mild hypertriglyceridemia. Because hypothyroidism is often subtle and therefore easily overlooked, all patients presenting with elevated plasma levels of LDL-C, especially if there has been an unexplained increase in LDL-C, should be screened for hypothyroidism. Thyroid replacement therapy usually ameliorates the hypercholesterolemia; if not, the patient probably has a primary lipoprotein disorder and may require lipid-lowering drug therapy with a statin.
CHRONIC KIDNEY DISEASE (See also Chap. 335) Chronic kidney disease (CKD) is often associated with mild hypertriglyceridemia (<300 mg/dL) due to the accumulation of VLDLs and remnant lipoproteins in the circulation. TG lipolysis and remnant clearance are both reduced in patients with renal failure. Because the risk of ASCVD is increased in end-stage renal disease, subjects with hyperlipidemia, they should usually be aggressively treated with lipid-lowering agents, even though there is inadequate data at present to indicate that this population benefits from LDL-lowering therapy.
Patients with solid organ transplants often have increased lipid levels due to the effect of the drugs required for immunosuppression. These patients can present a difficult clinical management problem, since statins should be used cautiously in these patients due to untoward muscle-related side effects.
Primary (Genetic) Causes of Impaired Hepatic Uptake of Lipoproteins Genetic variation contributes substantially to elevated LDL-C levels in the general population. It has been estimated that at least 50% of variation in LDL-C is genetically determined. Many patients with elevated LDL-C have polygenic hypercholesterolemia characterized by hypercholesterolemia in the absence of secondary causes of hypercholesterolemia (other than dietary factors) or a primary Mendelian disorder. In patients who are genetically predisposed to higher LDL-C levels, diet plays a key role; indeed increased saturated and trans fats in the diet shifts the entire distribution of LDL levels in the population to the right. Inheritance of several variants that together elevate LDL-C, coupled with diet, is generally the cause of this condition; <10% of first-degree relatives themselves have hypercholesterolemia. However, single-gene (Mendelian) causes of elevated LDL-C are relatively common and should be considered in the differential diagnosis of elevated LDL-C.
FAMILIAL HYPERCHOLESTEROLEMIA (FH) FH, also known as autosomal dominant hypercholesterolemia (ADH) type 1, is an autosomal co-dominant disorder characterized by elevated plasma levels of LDL-C in the absence of hypertriglyceridemia. FH is caused by loss-of-function mutations in the gene encoding the LDL receptor. The reduction in LDL receptor activity in the liver results in a reduced rate of clearance of LDL from the circulation. The plasma level of LDL increases to a level such that the rate of LDL production equals the rate of LDL clearance by residual LDL receptor as well as non-LDL receptor mechanisms. More than 1600 different mutations have been reported in association with FH. The elevated levels of LDL-C in FH are primarily due to delayed removal of LDL from the blood; in addition, because the removal of IDL is also delayed, the production of LDL from IDL is also increased. Individuals with two mutated LDL receptor alleles (FH homozygotes, or compound heterozygotes) have much higher LDL-C levels than those with one mutant allele (FH heterozygotes).
 Heterozygous FH is caused by the inheritance of one mutant LDL receptor allele. The population frequency of heterozygous FH due to LDL receptor mutations was originally estimated to be 1 in 500 individuals, but recent data suggest it may be as high as approximately 1 in 250 individuals, making it one of the most common single-gene disorders in humans. FH has a higher prevalence in certain founder populations, such as South African Afrikaners, Christian Lebanese, and French Canadians. Heterozygous FH is characterized by elevated plasma levels of LDL-C (usually 200–400 mg/dL) and normal levels of TGs. Patients with heterozygous FH have hypercholesterolemia from birth, and disease recognition is usually based on detection of hypercholesterolemia on routine screening, the appearance of tendon xanthomas, or the development of symptomatic cardiovascular disease. Inheritance is dominant, meaning that the condition was inherited from one parent and ~50% of the patient’s siblings can be expected to have hypercholesterolemia. The family history is frequently positive for premature CHD on the side of the family from which the mutation was inherited. Physical findings in many, but not all, patients with heterozygous FH include corneal arcus and tendon xanthomas particularly involving the dorsum of the hands and the Achilles tendons. Untreated heterozygous FH is associated with a markedly increased risk of cardiovascular disease. Untreated men with heterozygous FH have an ~50% chance of having a myocardial infarction before age 60 years, and women with heterozygous FH are at substantially increased risk as well. The age of onset of cardiovascular disease is highly variable and depends on the specific molecular defect, the level of LDL-C, and coexisting cardiovascular risk factors. FH heterozygotes with elevated plasma levels of Lp(a) (see below) appear to be at greater risk for cardiovascular disease.
Heterozygous FH is caused by the inheritance of one mutant LDL receptor allele. The population frequency of heterozygous FH due to LDL receptor mutations was originally estimated to be 1 in 500 individuals, but recent data suggest it may be as high as approximately 1 in 250 individuals, making it one of the most common single-gene disorders in humans. FH has a higher prevalence in certain founder populations, such as South African Afrikaners, Christian Lebanese, and French Canadians. Heterozygous FH is characterized by elevated plasma levels of LDL-C (usually 200–400 mg/dL) and normal levels of TGs. Patients with heterozygous FH have hypercholesterolemia from birth, and disease recognition is usually based on detection of hypercholesterolemia on routine screening, the appearance of tendon xanthomas, or the development of symptomatic cardiovascular disease. Inheritance is dominant, meaning that the condition was inherited from one parent and ~50% of the patient’s siblings can be expected to have hypercholesterolemia. The family history is frequently positive for premature CHD on the side of the family from which the mutation was inherited. Physical findings in many, but not all, patients with heterozygous FH include corneal arcus and tendon xanthomas particularly involving the dorsum of the hands and the Achilles tendons. Untreated heterozygous FH is associated with a markedly increased risk of cardiovascular disease. Untreated men with heterozygous FH have an ~50% chance of having a myocardial infarction before age 60 years, and women with heterozygous FH are at substantially increased risk as well. The age of onset of cardiovascular disease is highly variable and depends on the specific molecular defect, the level of LDL-C, and coexisting cardiovascular risk factors. FH heterozygotes with elevated plasma levels of Lp(a) (see below) appear to be at greater risk for cardiovascular disease.
No definitive diagnostic test for heterozygous FH is available, except in certain founder populations where selected mutations predominate. Most LDL receptor mutations are private and require sequencing of the LDL receptor gene for identification. Sequencing for clinical diagnosis is available but not standard of care and is rarely performed in the United States, because the clinical utility of identifying the specific mutation has not been demonstrated. A family history of hypercholesterolemia and/or premature coronary disease is supportive of the diagnosis. Secondary causes of significant hypercholesterolemia such as hypothyroidism, nephrotic syndrome, and obstructive liver disease should be excluded.
Heterozygous FH patients should be aggressively treated to lower plasma levels of LDL-C, starting in childhood. Initiation of a diet low in saturated and trans fats is recommended, but heterozygous FH patients virtually always require lipid-lowering drug therapy for effective control of their LDL-C levels. Statins are effective in heterozygous FH and are clearly the drug class of choice, and usually a more potent member of the class. However, some heterozygous FH patients cannot achieve adequate control of their LDL-C levels even with high-dose statin therapy and require additional drugs; a cholesterol absorption inhibitor and/or a bile acid sequestrant are the next-line classes of drugs. Currently, heterozygous FH patients whose LDL-C levels remain markedly elevated (>200 mg/dL with cardiovascular disease [CVD] or >300 mg/dL without CVD) on maximally tolerated drug therapy are candidates for LDL apheresis, a physical method of purging the blood of LDL in which the LDL particles are selectively removed from the circulation; LDL apheresis is usually performed every 2 weeks. A new class of drugs known as PCSK9 inhibitors is under clinical development and has the potential to effectively control LDL-C levels in the vast majority of patients with heterozygous FH who are inadequately controlled on a statin alone or who are statin intolerant.
Homozygous FH is caused by mutations in both alleles of the LDL receptor and therefore much rarer than heterozygous FH. Patients with homozygous FH have been classified into those patients with virtually no detectable LDL receptor activity (receptor negative) and those patients with markedly reduced but detectable LDL receptor activity (receptor defective). LDL-C levels in patients with homozygous FH range from about 400 to >1000 mg/dL, with receptor-defective patients at the lower end and receptor-negative patients at the higher end of the range. TGs are usually normal. Many patients with homozygous FH, particularly receptor-negative patients, present in childhood with cutaneous xanthomas on the hands, wrists, elbows, knees, heels, or buttocks. The devastating consequence of homozygous FH is accelerated ASCVD, which often presents in childhood or early adulthood. Atherosclerosis often develops first in the aortic root, where it can cause aortic valvular or supravalvular stenosis, and typically extends into the coronary ostia, which become stenotic. Symptoms can be atypical, and sudden death is not uncommon. Untreated, receptor-negative patients with homozygous FH rarely survive beyond the second decade; patients with receptor-defective LDL receptor defects have a better prognosis but almost invariably develop clinically apparent atherosclerotic vascular disease by age 30, and often much sooner. Carotid and femoral disease develops later in life and is usually not clinically significant.
Homozygous FH should be suspected in a child or young adult with LDL >400 mg/dL without secondary cause. Cutaneous xanthomas, evidence of CVD, and hypercholesterolemia in both parents all are supportive of the diagnosis. Although the specific mutations in the LDL receptor can usually be identified by DNA sequencing, this is not generally performed, and the diagnosis is usually made on clinical grounds.
Patients with homozygous FH must be treated aggressively to delay the onset and progression of CVD. Receptor defective patients sometimes respond to statins and other LDL-lowering drug classes such as a cholesterol absorption inhibitor or a bile acid sequestrant, which upregulate the LDL receptor activity. Two drugs that reduce the hepatic production of VLDL and thus LDL, a small-molecule inhibitor of the microsomal TG transfer protein (MTP) and an antisense oligonucleotide to apoB, are approved in the United States for the treatment of adults with homozygous FH and can be considered. PCSK9 inhibitors, which work through increasing LDL receptor availability, appear to have some benefit in receptor-defective patients and are under clinical development. LDL apheresis is used to lower plasma LDL levels in these patients and can promote regression of xanthomas as well as slow the progression of atherosclerosis. Because the liver is quantitatively the most important tissue for removing circulating LDLs via the LDL receptor, liver transplantation is effective in decreasing plasma LDL-C levels in this disorder but is infrequently used because of the associated problems with immunosuppression.
FAMILIAL DEFECTIVE APOB-100 (FDB) FDB, also known as autosomal dominant hypercholesterolemia (ADH) type 2, is a dominantly inherited disorder that clinically resembles heterozygous FH with elevated LDL-C levels and normal TGs. FDB is caused by mutations in the gene encoding apoB-100, specifically in LDL receptor–binding domain of apoB-100. Several different mutations have been identified, but a single mutation predominates: substitution of glutamine for arginine at position 3500. The mutation results in a reduction in the affinity of LDL binding to the LDL receptor, so LDL is removed from the circulation at a reduced rate. FDB is less common than FH but is more prevalent in individuals of central European descent; the Lancaster County (United States) Amish are a founder population in which the prevalence of FDB is as high as 1 in 10 individuals. FDB is characterized by elevated plasma LDL-C levels with normal TGs; tendon xanthomas can be seen, although not as frequently as in FH, and there is an associated increase in risk of CHD. Patients with FDB cannot be clinically distinguished from patients with heterozygous FH, although patients with FDB tend to have somewhat lower plasma levels of LDL-C than FH heterozygotes, presumably due to the fact that IDL clearance is not impaired in this disorder. Homozygotes for FDB mutations have higher LDL-C levels than FDB heterozygotes but are not as severely affected as homozygous FH patients. The apoB-100 gene mutations can be detected directly through sequencing of the receptor-binding region of the apoB gene or genotyping for the most common mutation, but genetic diagnosis is not generally performed because there is no direct implication for clinical management. As with FH, patients are treated with statins first and, if necessary, with additional classes of LDL-lowering drugs.
AUTOSOMAL DOMINANT HYPERCHOLESTEROLEMIA DUE TO MUTATIONS IN PCSK9 (ADH-PCSK9 OR ADH3) ADH-PCSK9, also known as autosomal dominant hypercholesterolemia (ADH) type 3, is a very rare autosomal dominant disorder caused by gain-of-function mutations in proprotein convertase subtilisin/kexin type 9 (PCSK9). PCSK9 is a secreted protein that binds to the LDL receptor, targeting it for degradation. Normally, after LDL binds to the LDL receptor, it is internalized along with the receptor, and in the low pH of the endosome, the LDL receptor dissociates from the LDL and recycles to the cell surface. When PCSK9 binds the receptor, the complex is internalized and the receptor is directed to the lysosome, rather than to the cell surface. The missense mutations in PCSK9 that cause hypercholesterolemia enhance the activity of PCSK9. As a consequence, the number of hepatic LDL receptors is reduced. Patients with ADH-PCSK9 are similar clinically to patients with FH. They may be particularly responsive to PCSK9 inhibitors in clinical development. Loss-of-function mutations in PCSK9 cause low LDL-C levels (see below).
AUTOSOMAL RECESSIVE HYPERCHOLESTEROLEMIA (ARH) ARH is a very rare disorder that is mostly seen in individuals of Sardinian descent. The disease is caused by mutations in a protein, ARH (also called LDLR adaptor protein, LDLRAP), which is required for LDL receptor–mediated endocytosis in the liver. ARH binds to the cytoplasmic domain of the LDL receptor and links the receptor to the endocytic machinery. In the absence of LDLRAP, LDL binds to the extracellular domain of the LDL receptor, but the lipoprotein-receptor complex fails to be internalized. ARH, like homozygous FH, is characterized by hypercholesterolemia, tendon xanthomas, and premature coronary artery disease (CAD). The levels of plasma LDL-C tend to be intermediate between the levels present in FH homozygotes and FH heterozygotes, and CAD is not usually symptomatic until the third decade. LDL receptor function in cultured fibroblasts is normal or only modestly reduced in ARH, whereas LDL receptor function in lymphocytes and the liver is negligible. Unlike FH homozygotes, the hyperlipidemia responds to treatment with statins, but these patients usually require additional therapy to lower plasma LDL-C to acceptable levels.
SITOSTEROLEMIA Sitosterolemia is a rare autosomal recessive disease that can result in severe hypercholesterolemia, tendon xanthomas, and premature ASCVD. Sitosterolemia is caused by loss-of-function mutations in either of two members of the ATP-binding cassette (ABC) half transporter family, ABCG5 and ABCG8. These genes are expressed in enterocytes and hepatocytes. The proteins heterodimerize to form a functional complex that transports plant sterols such as sitosterol and campesterol, and animal sterols, predominantly cholesterol, across the biliary membrane of hepatocytes into the bile and across the intestinal luminal surface of enterocytes into the gut lumen. In normal individuals, <5% of dietary plant sterols are absorbed by the proximal small intestine. The small amounts of plant sterols that enter the circulation are preferentially excreted into the bile. Thus, levels of plant sterols are kept very low in tissues. In sitosterolemia, the intestinal absorption of sterols is increased and biliary and fecal excretion of the sterols is reduced, resulting in increased plasma and tissue levels of both plant sterols and cholesterol. The increase in hepatic sterol levels results in transcriptional suppression of the expression of the LDL receptor, resulting in reduced uptake of LDL and substantially increased LDL-C levels. In addition to the usual clinical picture of hypercholesterolemia (i.e., tendon xanthomas and premature ASCVD), these patients also have anisocytosis and poikilocytosis of erythrocytes and megathrombocytes due to the incorporation of plant sterols into cell membranes. Episodes of hemolysis and splenomegaly are a distinctive clinical feature of this disease compared to other genetic forms of hypercholesterolemia and can be a clue to the diagnosis.
Sitosterolemia should be suspected in a patient with severe hypercholesterolemia without a family history of such or who responds dramatically to dietary therapy and/or ezetimibe but not statins. Sitosterolemia can be diagnosed by a laboratory finding of a substantial increase in the plasma level of sitosterol and/or other plant sterols. It is important to make the diagnosis, because bile acid sequestrants and cholesterol-absorption inhibitors are the most effective agents to reduce LDL-C and plasma plant sterol levels in these patients.
CHOLESTERYL ESTER STORAGE DISEASE (CESD) CESD, also known as lysosomal acid lipase deficiency, is an autosomal recessive disorder characterized by elevated LDL-C, usually in association with low HDL-C, together with progressive fatty liver ultimately leading to hepatic fibrosis. Plasma TG levels can also be mild to moderately increased in this disorder. The most severe form of this disorder, Wolman’s disease, presents in infancy and is rapidly fatal. Both Wolman’s disease and CESD are caused by loss-of-function variants in both alleles of the gene encoding lysosomal acid lipase (LAL; gene name LIPA). LAL is responsible for hydrolyzing neutral lipids, particularly TGs and cholesteryl esters, after delivery to the lysosome by cell-surface receptors such as the LDL receptor. It is particularly important in the liver, which clears large amounts of lipoproteins from the circulation. Genetic deficiency of LAL results in accumulation of neutral lipid in the hepatocytes, leading to hepatosplenomegaly, microvesicular steatosis, and ultimately fibrosis and end-stage liver disease. The etiology of the elevated LDL-C levels is uncertain; one study suggested that VLDL production is increased, but impaired LDL receptor–mediated clearance of LDL is also likely.
CESD should be particularly suspected in nonobese patients with elevated LDL-C, low HDL-C, and evidence of fatty liver in the absence of overt insulin resistance. The diagnosis can be made with a dried blood spot assay of LAL activity and confirmed by DNA genotyping for the most common mutation, followed if necessary by sequencing of the gene to find the second mutation. Liver biopsy is required to assess the degree of inflammation and fibrosis. It is important to make the diagnosis because it has implications for liver monitoring and potentially for therapeutic approaches under development.
FAMILIAL DYSBETALIPOPROTEINEMIA (FDBL) FDBL (also known as type III hyperlipoproteinemia) is usually a recessive disorder characterized by a mixed hyperlipidemia (elevated cholesterol and TGs) due to the accumulation of remnant lipoprotein particles (chylomicron remnants and VLDL remnants, or IDL). ApoE is present in multiple copies on chylomicron remnants and IDL, and mediates their removal via hepatic lipoprotein receptors (Fig. 421-2). FDBL is due to genetic variants of apoE, most commonly apoE2, that result in an apoE protein with reduced ability to bind lipoprotein receptors. The APOE gene is polymorphic in sequence, resulting in the expression of three common isoforms: apoE3, which is the most common; and apoE2 and apoE4, which both differ from apoE3 by a single amino acid. Although associated with slightly higher LDL-C levels and increased CHD risk, the apoE4 allele is not associated with FDBL. Individuals who carry one or two apoE4 alleles have an increased risk of Alzheimer’s disease. ApoE2 has a lower affinity for the LDL receptor; therefore, chylomicron remnants and IDL containing apoE2 are removed from plasma at a slower rate. Individuals who are homozygous for the E2 allele (the E2/E2 genotype) comprise the most common subset of patients with FDBL.
Approximately 0.5% of the general population are apoE2/E2 homozygotes, but only a small minority of these individuals actually develop hyperlipidemia characteristic of FDBL. In most cases, an additional, sometimes identifiable, factor precipitates the development of hyperlipoproteinemia. The most common precipitating factors are a high-fat diet, diabetes mellitus, obesity, hypothyroidism, renal disease, HIV infection, estrogen deficiency, alcohol use, or certain drugs. The disease seldom presents in women before menopause. Other mutations in apoE can cause a dominant form of FDBL where the hyperlipidemia is fully manifest in the heterozygous state, but these mutations are very rare.
Patients with FDBL usually present in adulthood with hyperlipidemia, xanthomas, or premature coronary or peripheral vascular disease. In FDBL, in contrast to other disorders of elevated TGs, the plasma levels of cholesterol and TG are often elevated to a similar degree, and the level of HDL-C is usually normal or reduced. Two distinctive types of xanthomas, tuberoeruptive and palmar, are seen in FDBL patients. Tuberoeruptive xanthomas begin as clusters of small papules on the elbows, knees, or buttocks and can grow to the size of small grapes. Palmar xanthomas (alternatively called xanthomata striata palmaris) are orange-yellow discolorations of the creases in the palms and wrists. Both of these xanthoma types are virtually pathognomonic for FDBL. Subjects with FDBL have premature ASCVD and tend to have more peripheral vascular disease than is typically seen in FH.
The definitive diagnosis of FDBL can be made either by the documentation of very high levels of remnant lipoproteins or by identification of the apoE2/E2 genotype. A variety of methods are used to identify remnant lipoproteins in the plasma, including “β-quantification” by ultracentrifugation (ratio of directly measured VLDL-C to total plasma TG >0.30), lipoprotein electrophoresis (broad β band), or nuclear magnetic resonance lipoprotein profiling. The Friedewald formula for calculation of LDL-C is not valid in FDBL because the VLDL particles are depleted in TG and enriched in cholesterol. The plasma levels of LDL-C are actually low in this disorder due to defective metabolism of VLDL to LDL. DNA-based methods (apoE genotyping) can be performed to confirm homozygosity for apoE2. However, absence of the apoE2/E2 genotype does not strictly rule out the diagnosis of FDBL, because other mutations in apoE can (rarely) cause this condition.
Because FDBL is associated with increased risk of premature ASCVD, it should be treated aggressively. Other metabolic conditions that can worsen the hyperlipidemia (see above) should be managed. Patients with FDBL are typically diet-responsive and can respond favorably to weight reduction and to low-cholesterol, low-fat diets. Alcohol intake should be curtailed. Pharmacologic therapy is often required, and statins are the first line in management. In the event of statin intolerance or insufficient control of hyperlipidemia, cholesterol absorption inhibitors, fibrates, and niacin are also effective in the treatment of FDBL.
HEPATIC LIPASE DEFICIENCY Hepatic lipase (HL; gene name LIPC) is a member of the same gene family as LPL and hydrolyzes TGs and phospholipids in remnant lipoproteins and HDL. Hydrolysis of lipids in remnant particles by HL contributes to their hepatic uptake via an apoE-mediated process. HL deficiency is a very rare autosomal recessive disorder characterized by elevated plasma levels of cholesterol and TGs (mixed hyperlipidemia) due to the accumulation of lipoprotein remnants, accompanied by elevated plasma level of HDL-C. The diagnosis is confirmed by measuring HL activity in postheparin plasma and/or confirmation of loss-of-function mutations in both alleles of HL/LIPC. Due to the small number of patients with HL deficiency, the association of this genetic defect with ASCVD is not entirely clear, although anecdotally patients with HL deficiency who have premature CVD have been described. As with FDBL, statin therapy is recommended to reduce remnant lipoproteins and cardiovascular risk.
Additional Secondary Causes of Dyslipidemia Many of the secondary causes of dyslipidemia (Table 421-4) have been described above. Additional considerations are discussed here.
|
SECONDARY CAUSES OF DYSLIPIDEMIA |
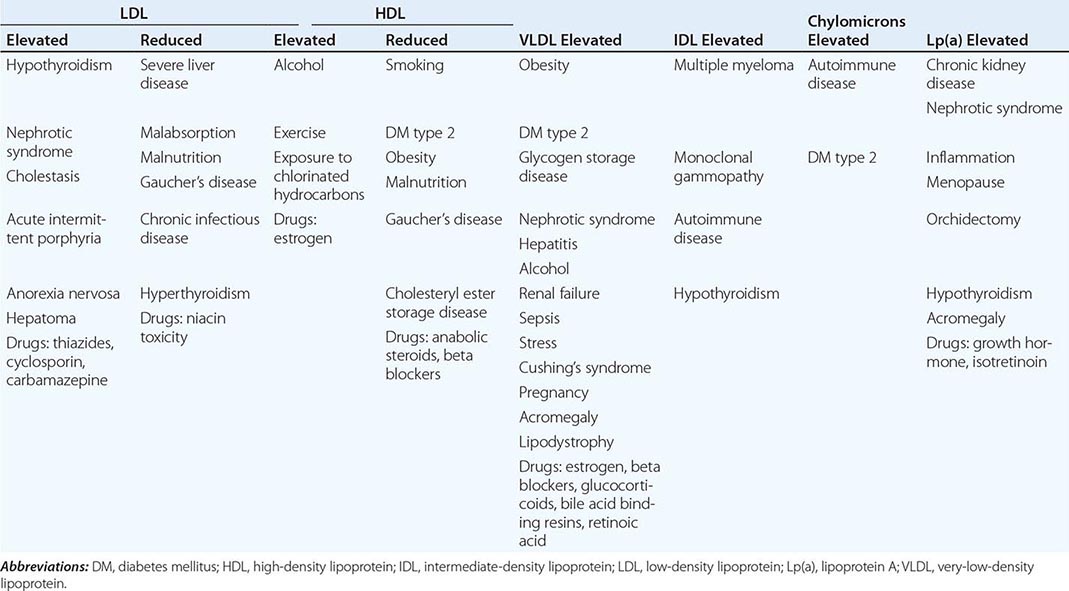
LIVER DISORDERS (See also Chap. 357) Because the liver is the principal site of formation and clearance of lipoproteins, liver disorders can affect plasma lipid levels in a variety of ways. Hepatitis due to infection, drugs, or alcohol is often associated with increased VLDL synthesis and mild to moderate hypertriglyceridemia. Severe hepatitis and liver failure are associated with dramatic reductions in plasma cholesterol and TGs due to reduced lipoprotein biosynthetic capacity.
Cholestasis is associated with hypercholesterolemia, which can be very severe. A major pathway by which cholesterol is excreted from the body is via secretion into bile, either directly or after conversion to bile acids, and cholestasis blocks this critical excretory pathway. In cholestasis, free cholesterol, coupled with phospholipids, is secreted into the plasma as a constituent of a lamellar particle called LP-X. The particles can deposit in skinfolds, producing lesions resembling those seen in patients with FDBL (xanthomata strata palmaris). Planar and eruptive xanthomas can also be seen in patients with cholestasis.
DRUGS Many drugs have an impact on lipid metabolism and can result in significant alterations in the lipoprotein profile (Table 421-4). Estrogen administration is associated with increased VLDL and HDL synthesis, resulting in elevated plasma levels of both TGs and HDL-C. This lipoprotein pattern is distinctive because the levels of plasma TG and HDL-C are typically inversely related. Plasma TG levels should be monitored when birth control pills or postmenopausal estrogen therapy is initiated to ensure that the increase in VLDL production does not lead to severe hypertriglyceridemia. Use of low-dose preparations of estrogen or the estrogen patch can minimize the effect of exogenous estrogen on lipids.
INHERITED CAUSES OF LOW LEVELS OF APOB-CONTAINING LIPOPROTEINS
Plasma concentrations of LDL-C <60 mg/dL are unusual. Although in some cases LDL-C levels in this range may be reflective of malnutrition or serious chronic illness, LDL-C <60 mg/dL in an otherwise healthy individual suggests an inherited condition. The major inherited causes of low LDL-C are reviewed here.
Abetalipoproteinemia The synthesis and secretion of apoB-containing lipoproteins in the enterocytes of the proximal small bowel and in the hepatocytes of the liver involve a complex series of events that coordinate the coupling of various lipids with apoB-48 and apoB-100, respectively. Abetalipoproteinemia is a rare autosomal recessive disease caused by loss-of-function mutations in the gene encoding microsomal TG transfer protein (MTP; gene name MTTP), a protein that transfers lipids to nascent chylomicrons and VLDLs in the intestine and liver, respectively. Plasma levels of cholesterol and TG are extremely low in this disorder, and chylomicrons, VLDLs, LDLs, and apoB are undetectable in plasma. The parents of patients with abetalipoproteinemia (obligate heterozygotes) have normal plasma lipid and apoB levels. Abetalipoproteinemia usually presents in early childhood with diarrhea and failure to thrive due to fat malabsorption. The initial neurologic manifestations are loss of deep tendon reflexes, followed by decreased distal lower extremity vibratory and proprioceptive sense, dysmetria, ataxia, and the development of a spastic gait, often by the third or fourth decade. Patients with abetalipoproteinemia also develop a progressive pigmented retinopathy presenting with decreased night and color vision, followed by reductions in daytime visual acuity and ultimately progressing to near-blindness. The presence of spinocerebellar degeneration and pigmented retinopathy in this disease has resulted in some patients with abetalipoproteinemia being misdiagnosed as having Friedreich’s ataxia.
Most of the clinical manifestations of abetalipoproteinemia result from defects in the absorption and transport of fat-soluble vitamins. Vitamin E and retinyl esters are normally transported from enterocytes to the liver by chylomicrons, and vitamin E is dependent on VLDL for transport out of the liver and into the circulation. As a consequence of the inability of these patients to secrete apoB-containing particles, patients with abetalipoproteinemia are markedly deficient in vitamin E and are also mildly to moderately deficient in vitamins A and K. Patients with abetalipoproteinemia should be referred to specialized centers for confirmation of the diagnosis and appropriate therapy. Treatment consists of a low-fat, high-caloric, vitamin-enriched diet accompanied by large supplemental doses of vitamin E. It is imperative that treatment be initiated as soon as possible to prevent development of neurologic sequelae, which can progress even with appropriate therapy. New therapies for this serious disease are needed.
Familial Hypobetalipoproteinemia (FHBL) FHBL generally refers to a condition of low total cholesterol, LDL-C, and apoB due to mutations in apoB. Most of the mutations causing FHBL result in a truncated apoB protein, resulting in impaired assembly and secretion of chylomicrons from enterocytes and VLDL from the liver. Mutations that result in VLDL particles containing a truncated apoB protein are cleared from the circulation at an accelerated rate, which also contributes to patients with this disorder having low levels of LDL-C and apoB. Individuals heterozygous for these mutations usually have LDL-C levels <60–80 mg/dL and also tend to have lower levels of plasma TG. Many FHBL patients have elevated levels of hepatic fat (due to reduced VLDL export) and sometimes have increased levels of liver transaminases, although it appears that these patients infrequently develop associated inflammation and fibrosis.
Mutations in both apoB alleles cause homozygous FHBL, an extremely rare disorder resembling abetalipoproteinemia with nearly undetectable LDL-C and apoB. The neurologic defects in this form of hypobetalipoproteinemia tend to be less severe than is typically seen in abetalipoproteinemia. Homozygous hypobetalipoproteinemia can be distinguished from abetalipoproteinemia by examining the inheritance pattern of the plasma LDL-C level. The levels of LDL-C and apoB are normal in the parents of patients with abetalipoproteinemia and low in those of patients with homozygous hypobetalipoproteinemia.
PCSK9 Deficiency Another inherited cause of low LDL-C results from loss-of-function mutations in PCSK9. PCSK9 is a secreted protein that binds to the extracellular domain of the LDL receptor in the liver and promotes the degradation of the receptor. Heterozygosity for nonsense mutations in PCSK9 that interfere with the synthesis of the protein are associated with increased hepatic LDL receptor activity and reduced plasma levels of LDL-C. Such mutations are particularly frequent in individuals of African descent. Individuals who are heterozygous for a loss-of-function mutation in PCSK9 have an ~30–40% reduction in plasma levels of LDL-C and have a substantial protection from CHD relative to those without a PCSK9 mutation, presumably due to having lower plasma cholesterol levels since birth. This observation led to the development of PCSK9 inhibitors as a new strategy for reducing LDL-C levels and cardiovascular risk. Homozygotes for these nonsense mutations have been reported and have extremely low LDL-C levels (<20 mg/dL) but appear otherwise healthy. A sequence variation of somewhat higher frequency (R46L) is found predominantly in individuals of European descent. This mutation impairs, but does not completely destroy, PCSK9 function. As a consequence, the plasma levels of LDL-C in individuals carrying this mutation are more modestly reduced (~15–20%); individuals with these mutations have a 45% reduction in ASCVD risk.
DISORDERS OF REDUCED HDL CHOLESTEROL
Low levels of HDL-C are very commonly encountered in clinical practice. Low HDL-C is an important independent predictor of increased cardiovascular risk and has been used regularly in standardized risk calculators, including the most recent one from the American Heart Association (AHA)/American College of Cardiology (ACC). However, it remains very uncertain whether low HDL-C is directly causal for the development of ASCVD. HDL metabolism is strongly influenced by TRLs, insulin resistance, and inflammation, among other environmental and medical factors. Thus the HDL-C measurement integrates a number of cardiovascular risk factors, potentially explaining its strong inverse association with ASCVD.
The majority of patients with low HDL-C have some combination of genetic predisposition and secondary factors. Variants in dozens of genes have been shown to influence HDL-C levels. Even more important quantitatively, obesity and insulin resistance have strong suppressive effects on HDL-C, and low HDL-C in these conditions is widely observed. Furthermore, the vast majority of patients with elevated TGs have reduced levels of HDL-C. Most patients with low HDL-C who have been studied in detail have accelerated catabolism of HDL and its associated apoA-I as the physiologic basis for the low HDL-C. Importantly, although HDL-C remains an important biomarker for assessing cardiovascular risk, it is not currently a direct target of intervention for raising the level in order to reduce cardiovascular risk. Certain therapeutic approaches in clinical development, such as inhibitors of CETP (see below), have the potential to change this paradigm.
INHERITED CAUSES OF VERY LOW LEVELS OF HDL-C
Mutations in genes encoding proteins that play critical roles in HDL synthesis and catabolism can result in reductions in plasma levels of HDL-C. Unlike the genetic forms of hypercholesterolemia, which are invariably associated with premature coronary atherosclerosis, genetic forms of hypoalphalipoproteinemia (low HDL-C) are often not associated with clearly increased risk of ASCVD.
Gene Deletions in the APOA5-A1-C3-A4 Locus and Coding Mutations in APOA1 Complete genetic deficiency of apoA-I due to a complete deletion of the APOA1 gene results in the virtual absence of circulating HDL and appears to increase the risk of premature ASCVD. The genes encoding APOA5, APOA1, APOC3, and APOA4 are clustered together on chromosome 11. Some patients with no apoA-I have genomic deletions that include other genes in the cluster. ApoA-I is required for LCAT activity. In the absence of LCAT, free cholesterol levels increase in both plasma (not HDL) and in tissues. The free cholesterol can form deposits in the cornea and in the skin, resulting in corneal opacities and planar xanthomas. Premature CHD is associated with apoA-I deficiency.
Missense and nonsense mutations in the apoA-I gene are present in some patients with low plasma levels of HDL-C (usually 15–30 mg/dL), but are a rare cause of low plasma HDL-C levels. Most individuals with low plasma HDL-C levels due to missense mutations in apoA-I do not appear to have premature CHD. Patients who are heterozygous for an Arg173Cys substitution in apoA-I (so-called apoA-IMilano) have very low plasma levels of HDL-C due to impaired LCAT activation and accelerated clearance of the HDL particles containing the abnormal apoA-I. Despite having very low plasma levels of HDL-C, these individuals do not have an increased risk of premature CHD.
A few selected missense mutations in apoA-I and apoA-II promote the formation of amyloid fibrils, which can cause systemic amyloidosis.
Tangier Disease (ABCA1 Deficiency) Tangier disease is a rare autosomal co-dominant form of extremely low plasma HDL-C levels that is caused by mutations in the gene encoding ABCA1, a cellular transporter that facilitates efflux of unesterified cholesterol and phospholipids from cells to apoA-I (Fig. 421-3). ABCA1 in the liver and intestine rapidly lipidates the apoA-I secreted from the basolateral membranes of these tissues. In the absence of ABCA1, the nascent, poorly lipidated apoA-I is immediately cleared from the circulation. Thus, patients with Tangier disease have extremely low circulating plasma levels of HDL-C (<5 mg/dL) and apoA-I (<5 mg/dL). Cholesterol accumulates in the reticuloendothelial system of these patients, resulting in hepatosplenomegaly and pathognomonic enlarged, grayish yellow or orange tonsils. An intermittent peripheral neuropathy (mononeuritis multiplex) or a sphingomyelia-like neurologic disorder can also be seen in this disorder. Tangier disease is probably associated with some increased risk of premature atherosclerotic disease, although the association is not as robust as might be anticipated, given the very low levels of HDL-C and apoA-I in these patients. Patients with Tangier disease also have low plasma levels of LDL-C, which may attenuate the atherosclerotic risk. Obligate heterozygotes for ABCA1 mutations have moderately reduced plasma HDL-C levels (15–30 mg/dL), and their risk of premature CHD remains uncertain.
Familial LCAT Deficiency This rare autosomal recessive disorder is caused by mutations in LCAT, an enzyme synthesized in the liver and secreted into the plasma, where it circulates associated with lipoproteins (Fig. 421-3). As reviewed above, the enzyme is activated by apoA-I and mediates the esterification of cholesterol to form cholesteryl esters. Consequently, in familial LCAT deficiency, the proportion of free cholesterol in circulating lipoproteins is greatly increased (from ~25% to >70% of total plasma cholesterol). Deficiency in this enzyme interferes with the maturation of HDL particles and results in rapid catabolism of circulating apoA-I.
Two genetic forms of familial LCAT deficiency have been described in humans: complete deficiency (also called classic LCAT deficiency) and partial deficiency (also called fish-eye disease). Progressive corneal opacification due to the deposition of free cholesterol in the cornea, very low plasma levels of HDL-C (usually <10 mg/dL), and variable hypertriglyceridemia are characteristic of both disorders. In partial LCAT deficiency, there are no other known clinical sequelae. In contrast, patients with complete LCAT deficiency have hemolytic anemia and progressive renal insufficiency that eventually leads to end-stage renal disease. Remarkably, despite the extremely low plasma levels of HDL-C and apoA-I, premature ASCVD is not a consistent feature of either LCAT deficiency or fish eye disease. The diagnosis can be confirmed in a specialized laboratory by assaying plasma LCAT activity or by sequencing the LCAT gene.
Primary Hypoalphalipoproteinemia The condition of low plasma levels of HDL-C (the “alpha lipoprotein”) is referred to as hypoalphalipoproteinemia. Primary hypoalphalipoproteinemia is defined as a plasma HDL-C level below the tenth percentile in the setting of relatively normal cholesterol and TG levels, no apparent secondary causes of low plasma HDL-C, and no clinical signs of LCAT deficiency or Tangier disease. This syndrome is often referred to as isolated low HDL. A family history of low HDL-C facilitates the diagnosis of an inherited condition, which may follow an autosomal dominant pattern. The metabolic etiology of this disease appears to be primarily accelerated catabolism of HDL and its apolipoproteins. Some of these patients may have ABCA1 mutations and therefore technically have heterozygous Tangier disease. Several kindreds with primary hypoalphalipoproteinemia and an increased incidence of premature CHD have been described, although it is not clear if the low HDL-C level is the cause of the accelerated atherosclerosis in these families. Association of hypoalphalipoproteinemia with premature CHD may depend on the specific nature of the gene defect or the underlying metabolic defect that either directly or indirectly causes the low plasma HDL-C level.
INHERITED CAUSES OF VERY HIGH LEVELS OF HDL-C
CETP Deficiency Loss-of-function mutations in both alleles of the gene encoding CETP cause substantially elevated HDL-C levels (usually >150 mg/dL). As noted above, CETP transfers cholesteryl esters from HDL to apoB-containing lipoproteins (Fig. 421-3). Absence of this transfer activity results in an increase in the cholesteryl ester content of HDL and a reduction in plasma levels of LDL-C. The large, cholesterol-rich HDL particles circulating in these patients are cleared at a reduced rate. CETP deficiency was first diagnosed in Japanese persons and is rare outside of Japan. The relationship of CETP deficiency to ASCVD remains unresolved. Heterozygotes for CETP deficiency have only modestly elevated HDL-C levels. Based on the phenotype of high HDL-C in CETP deficiency, pharmacologic inhibition of CETP is under development as a new therapeutic approach to both raise HDL-C levels and lower LDL-C levels, but whether it will reduce risk of ASCVD remains to be determined.
SCREENING, DIAGNOSIS, AND MANAGEMENT OF DISORDERS OF LIPOPROTEIN METABOLISM
SCREENING
Plasma lipid and lipoprotein levels should be measured in all adults, preferably after a 12-h overnight fast. In most clinical laboratories, the total cholesterol and TGs in the plasma are measured enzymatically, and then the cholesterol in the supernatant is measured after precipitation of apoB-containing lipoproteins to determine the HDL-C. The LDL-C is then estimated using the following equation:
LDL-C = total cholesterol – (TG/5) – HDL-C
(The VLDL cholesterol content is estimated by dividing the plasma TG by 5, reflecting the ratio of TG to cholesterol in VLDL particles.) This formula (the Friedewald formula) is reasonably accurate if test results are obtained on fasting plasma and if the TG level does not exceed ~200 mg/dL; by convention it cannot be used if the TG level is >400 mg/dL. LDL-C can be directly measured by a number of methods. Further evaluation and treatment are based primarily on the clinical assessment of absolute cardiovascular risk using risk calculators such as the AHA/ACC risk calculator based on a large amount of observational data.
DIAGNOSIS
A critical first step in managing a lipoprotein disorder is to attempt to determine the class or classes of lipoproteins that are increased or decreased in the patient. Once the hyperlipidemia is accurately classified, efforts should be directed to rule out any possible secondary causes of the hyperlipidemia (Table 421-4). Although many patients with hyperlipidemia have a primary (i.e., genetic) cause of their lipid disorder, secondary factors frequently contribute to the hyperlipidemia. A careful social, medical, and family history should be obtained. A fasting glucose should be obtained in the initial workup of all subjects with an elevated TG level. Nephrotic syndrome and chronic renal insufficiency should be excluded by obtaining urine protein and serum creatinine. Liver function tests should be performed to rule out hepatitis and cholestasis. Hypothyroidism should be ruled out by measuring serum thyroid-stimulating hormone.
Once secondary causes have been ruled out, attempts should be made to diagnose the primary lipid disorder because the underlying genetic defect can provide important prognostic information regarding the risk of developing CHD, the response to drug therapy, and the management of other family members. Obtaining the correct diagnosis often requires a detailed family medical history, lipid analyses in family members, and sometimes specialized testing.
Severe Hypertriglyceridemia If the fasting plasma TG level is >1000 mg/dL, the patient has chylomicronemia. If the cholesterol-to-TG ratio is >10, familial chylomicronemia syndrome must be considered, and LPL activity measured in postheparin plasma can help with making that diagnosis. Most adults with chylomicronemia also have elevated VLDL levels. These individuals usually do not have a Mendelian disorder but instead are genetically predisposed and have secondary factors (diet, obesity, glucose intolerance, alcohol ingestion, estrogen therapy) that contribute to the hyperlipidemia. Such patients are a risk of acute pancreatitis and should be treated to reduce their TG levels and thus their risk of pancreatitis.
Severe Hypercholesterolemia If the levels of LDL-C are very high (greater than a ninety-fifth percentile for age and sex), it is likely that the patient has a genetic cause of hypercholesterolemia. At present, there is no compelling reason to perform molecular studies to further refine the molecular diagnosis because the clinical management is not affected. Recessive forms of severe hypercholesterolemia are rare, but if a patient with severe hypercholesterolemia has parents with normal cholesterol levels, ARH, sitosterolemia, and CESD should be considered. Patients with more moderate hypercholesterolemia that does not segregate in families as a monogenic trait are likely to have polygenic hypercholesterolemia.
Combined Hyperlipidemia The most common errors in the diagnosis of lipid disorders involve patients with combined hyperlipidemia. Elevations in the plasma levels of both cholesterol and TGs are seen in patients with increased plasma levels of VLDL and LDL or of remnant lipoproteins. A β-quantification to determine the VLDL cholesterol/TG ratio in plasma (see discussion of FDBL) or a direct measurement of the plasma LDL-C should be performed at least once prior to initiation of lipid-lowering therapy to determine if the hyperlipidemia is due to the accumulation of remnants or to an increase in both LDL and VLDL. Measurement of plasma apoB levels can help identify patients with FCHL who may require more aggressive treatment.
APPROACH TO THE PATIENT:
Lipoprotein Disorders
The major goals in the clinical management of lipoprotein disorders are: (1) prevention of acute pancreatitis in patients with severe hypertriglyceridemia; and (2) prevention of CVD and related cardiovascular events.
MANAGEMENT OF SEVERE HYPERTRIGLYCERIDEMIA TO PREVENT PANCREATITIS
Although the observational relationship between severe hypertriglyceridemia, particularly chylomicronemia, and acute pancreatitis is well-established, there has never been a clinical trial designed or powered to prove that intervention to reduce TGs reduces the risk of pancreatitis. Nevertheless, it is generally considered appropriate medical practice to intervene in patients with TGs >500 mg/dL in order to reduce the risk of pancreatitis. It remains controversial whether individuals with severe hypertriglyceridemia are at increased risk for ASCVD.
Lifestyle Modifying the lifestyle of the patient with severe hypertriglyceridemia often is associated with a significant reduction in plasma TG level. Patients who drink alcohol should be encouraged to decrease or preferably eliminate their intake. Patients with severe hypertriglyceridemia often benefit from a formal dietary consultation with a dietician intimately familiar with counseling patients on the dietary management of high TGs. Dietary fat intake should be restricted to reduce the formation of chylomicrons in the intestine. The excessive intake of simple carbohydrates should be discouraged because insulin drives TG production in the liver. Aerobic exercise and even increase in regular physical activity can have a positive effect in reducing TG levels and should be strongly encouraged. For patients who are overweight, weight loss can help to reduce TG levels. In extreme cases, bariatric surgery has been shown to not only produce effective weight loss but also substantially reduce plasma TG levels.
Pharmacologic Therapy for Severe Hypertriglyceridemia Despite the above interventions, however, many patients with severe hypertriglyceridemia require pharmacologic therapy (Table 421-5). Patients who persist in having fasting TG >500 mg/dL despite active lifestyle management are candidates for pharmacologic therapy. There are three classes of drugs that are used for management of these patients: fibrates, omega-3 fatty acids (fish oils), and niacin. In addition, statins can reduce plasma TG levels and also reduce ASCVD risk.
|
SUMMARY OF THE MAJOR APPROVED DRUGS USED FOR THE TREATMENT OF DYSLIPIDEMIA |
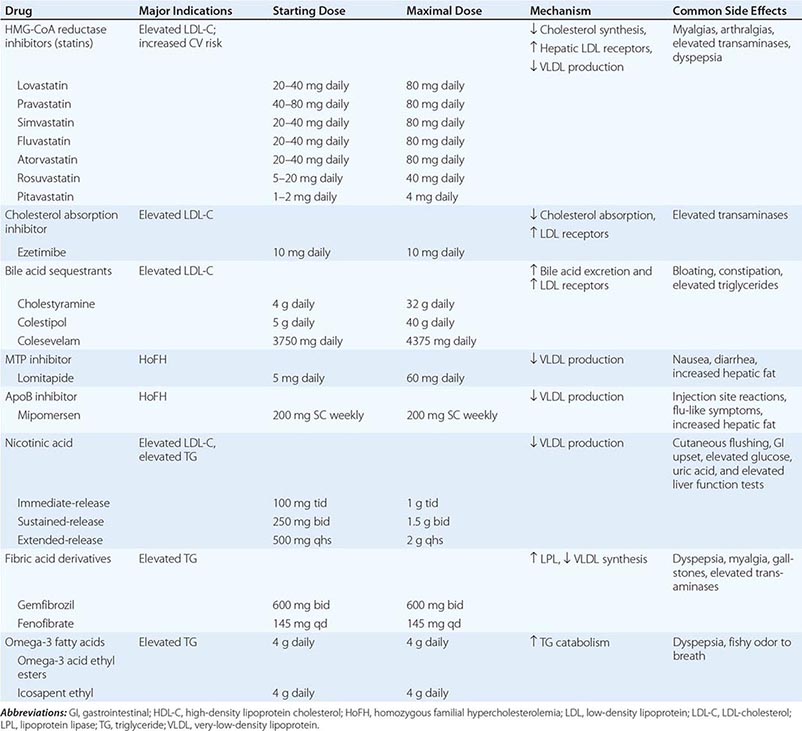
FIBRATES Fibric acid derivatives, or fibrates, are agonists of PPARα, a nuclear receptor involved in the regulation of lipid metabolism. Fibrates stimulate LPL activity (enhancing TG hydrolysis), reduce apoC-III synthesis (enhancing lipoprotein remnant clearance), promote β-oxidation of fatty acids, and may reduce VLDL TG production. Fibrates are a first-line therapy for severe hypertriglyceridemia (>500 mg/dL). This class of therapeutic agents sometimes lowers but more often raises the plasma level of LDL-C in individuals with severe hypertriglyceridemia. Fibrates are generally well tolerated, but are associated with an increase in the incidence of gallstones. Fibrates can cause myopathy, especially when combined with other lipid-lowering therapy (statins, niacin), and can raise creatinine. Fibrates should be used with caution in patients with CKD. Importantly, fibrates can potentiate the effect of warfarin and certain oral hypoglycemic agents, so the anticoagulation status and plasma glucose levels should be closely monitored in patients on these agents.
OMEGA 3 FATTY ACIDS (FISH OILS) Omega-3 fatty acids, or omega-3 polyunsaturated fatty acids (n-3 PUFAs), commonly known as fish oils, are present in high concentration in fish and in flaxseed. The most widely used n-3 PUFAs for the treatment of hyperlipidemias are the two active molecules in fish oil: eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). n-3 PUFAs have been concentrated into tablets and in doses of 3–4 g/d are effective at lowering fasting TG levels. Fish oils are a reasonable consideration for first-line therapy in patients with severe hypertriglyceridemia (>500 mg/dL) to prevent pancreatitis. Fish oils can cause an increase in plasma LDL-C levels in some patients. In general, fish oils are well tolerated, with the major side effect being dyspepsia. They appear to be safe, at least at doses up to 3–4 g, but can be associated with a prolongation in the bleeding time.
NICOTINIC ACID Nicotinic acid, or niacin, is a B-complex vitamin that has been used as a lipid-modifying agent for more than five decades. Niacin suppresses lipolysis in the adipocyte through its effect on the niacin receptor GPR109A and has other effects on hepatic lipid metabolism that are poorly understood. Niacin reduces plasma TG and LDL-C levels and also raises the plasma concentration of HDL-C. Because it has a number of side effects and can be difficult to use, it is at best a third-line agent for the management of severe hypertriglyceridemia. Niacin therapy is generally started at lower doses and gradually titrated up to higher doses. The most frequent side effect of niacin is cutaneous flushing, which is mediated by activating GPR109A in the skin. Niacin can cause dyspepsia and can exacerbate esophageal reflux and peptic ulcer disease. Mild elevations in transaminases occur in up to 15% of patients treated with any form of niacin. Niacin can raise plasma levels of uric acid and precipitate gouty attacks in susceptible patients. Acanthosis nigricans, a dark-colored coarse skin lesion, and maculopathy are infrequent side effects of niacin.
MANAGEMENT OF CHOLESTEROL TO PREVENT CARDIOVASCULAR DISEASE
In contrast to hypertriglyceridemia and pancreatitis, there are abundant and compelling data that intervention to reduce LDL-C substantially reduces the risk of CVD, including myocardial infarction and stroke, as well as total mortality. Thus, it is imperative that patients with hypercholesterolemia be assessed for cardiovascular risk and for the need for intervention. It is also worth noting that patients at high risk for CVD who have plasma LDL-C levels in the “normal” or average range also benefit from intervention to reduce LDL-C levels.
Lifestyle The first approach to a patient with hypercholesterolemia and high cardiovascular risk is to make any necessary lifestyle changes. In obese patients, efforts should be made to reduce body weight to the ideal level. Patients should receive dietary counseling to reduce the content of saturated fats, trans fats, and cholesterol in the diet. Regular aerobic exercise has relatively little impact on reducing plasma LDL-C levels, although it has cardiovascular benefits independent of LDL lowering.
Pharmacologic Therapy for Hypercholesterolemia The decision to use LDL-lowering drug therapy (Table 421-5)—with a statin being first-line therapy—depends on the level of LDL-C as well as the level of cardiovascular risk. In general, patients with a Mendelian disorder of elevated LDL-C such as FH must be treated to reduce the very high lifetime risk of CVD, and treatment should be initiated as early as possible in adulthood or, in some cases, during childhood.
Otherwise, the decision to initiate LDL-lowering drug therapy is generally determined by the level of cardiovascular risk. In patients with established CVD, statin therapy is well supported by clinical trial data and should be used regardless of the LDL-C level. For patients >40 years old without clinical CVD, the AHA/ACC risk calculator (http://my.americanheart.org/professional/StatementsGuidelines/PreventionGuidelines/Prevention-Guidelines_UCM_457698_SubHomePage.jsp) can be used to determine the 10-year absolute risk for CVD, and current guidelines suggest that a 10-year risk >7.5% merits consideration of statin therapy regardless of plasma LDL-C level. For younger patients, the assessment of lifetime risk of CVD may help inform the decision to start a statin.
HMG-CoA REDUCTASE INHIBITORS (STATINS) Statins inhibit HMG-CoA reductase, a key enzyme in cholesterol biosynthesis. By inhibiting cholesterol biosynthesis, statins lead to increased hepatic LDL receptor activity and accelerated clearance of circulating LDL, resulting in a dose-dependent reduction in plasma levels of LDL-C. The magnitude of LDL lowering associated with statin treatment varies widely among individuals, but once a patient is on a statin, the doubling of the statin dose produces an ~6% further reduction in the level of plasma LDL-C. The statins currently available differ in their LDL-C–reducing potency (Table 421-5). Currently, there is no convincing evidence that any of the different statins confer an advantage that is independent of the effect on LDL-C. Statins also reduce plasma TGs in a dose-dependent fashion, which is roughly proportional to their LDL-C–lowering effects (if the TGs are <400 mg/dL). Statins have a modest HDL-raising effect (5–10%) that is not generally dose-dependent.
Statins are well tolerated and can be taken in tablet form once a day. Potential side effects include dyspepsia, headaches, fatigue, and muscle or joint pains. Severe myopathy and even rhabdomyolysis occur rarely with statin treatment. The risk of statin-associated myopathy is increased by the presence of older age, frailty, renal insufficiency, and coadministration of drugs that interfere with the metabolism of statins, such as erythromycin and related antibiotics, antifungal agents, immunosuppressive drugs, and fibric acid derivatives (particularly gemfibrozil). Severe myopathy can usually be avoided by careful patient selection, avoidance of interacting drugs, and instructing the patient to contact the physician immediately in the event of unexplained muscle pain. In the event of muscle symptoms, the plasma creatine kinase (CK) level should be obtained to differentiate myopathy from myalgia. Serum CK levels need not be monitored on a routine basis in patients taking statins, because an elevated CK in the absence of symptoms does not predict the development of myopathy and does not necessarily suggest the need for discontinuing the drug.
Another consequence of statin therapy can be elevation in liver transaminases (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]). They should be checked before starting therapy, at 2–3 months, and then annually. Substantial (greater than three times the upper limit of normal) elevation in transaminases is relatively rare, and mild-to-moderate (one to three times normal) elevation in transaminases in the absence of symptoms need not mandate discontinuing the medication. Severe clinical hepatitis associated with statins is exceedingly rare, and the trend is toward less frequent monitoring of transaminases in patients taking statins. The statin-associated elevation in liver enzymes resolves upon discontinuation of the medication.
Statins appear to be remarkably safe. Meta-analyses of large randomized controlled clinical trials with statins do not suggest an increase in any major noncardiac diseases except type 2 diabetes. A small excess percentage of those taking statins will develop diabetes but the benefits associated with the reduction in cardiovascular events outweigh the increase in incidence of diabetes. Statins are the drug class of choice for LDL-C reduction and are by far the most widely used class of lipid-lowering drugs.
CHOLESTEROL ABSORPTION INHIBITORS Cholesterol within the lumen of the small intestine is derived from the diet (about one-third) and the bile (about two-thirds) and is actively absorbed by the enterocyte through a process that involves the protein NPC1L1. Ezetimibe (Table 421-5) is a cholesterol absorption inhibitor that binds directly to and inhibits NPC1L1 and blocks the intestinal absorption of cholesterol. Ezetimibe (10 mg) inhibits cholesterol absorption by almost 60%, resulting in a reduction in delivery of dietary sterols in the liver and an increase in hepatic LDL receptor expression. The mean reduction in plasma LDL-C on ezetimibe (10 mg) is 18%, and the effect is additive when used in combination with a statin. Effects on TG and HDL-C levels are negligible. When used in combination with a statin, monitoring of liver transaminases is recommended. The only roles for ezetimibe in monotherapy are in patients who do not tolerate statins and in sitosterolemia.
BILE ACID SEQUESTRANTS (RESINS) Bile acid sequestrants bind bile acids in the intestine and promote their excretion rather than reabsorption in the ileum. To maintain the bile acid pool size, the liver diverts cholesterol to bile acid synthesis. The decreased hepatic intracellular cholesterol content results in upregulation of the LDL receptor and enhanced LDL clearance from the plasma. Bile acid sequestrants, including cholestyramine, colestipol, and colesevelam (Table 421-5), primarily reduce plasma LDL-C levels but can cause an increase in plasma TGs. Therefore, patients with hypertriglyceridemia generally should not be treated with bile acid–binding resins. Cholestyramine and colestipol are insoluble resins that must be suspended in liquids. Colesevelam is available as tablets but generally requires up to six to seven tablets per day for effective LDL-C lowering. Most side effects of resins are limited to the gastrointestinal tract and include bloating and constipation. Because bile acid sequestrants are not systemically absorbed, they are very safe and the cholesterol-lowering drug of choice in children and in women of childbearing age who are lactating, pregnant, or could become pregnant. They are effective in combination with statins and in combination with ezetimibe and are particularly useful with one or both of these drugs for patients with severe hypercholesterolemia or those with statin intolerance.
SPECIALIZED DRUGS FOR HOMOZYGOUS FH Two “orphan” drugs are approved specifically for the management of homozygous FH. They include a small-molecule inhibitor of MTP, called lomitapide, and an antisense oligonucleotide against apoB, called mipomersen. These drugs reduce VLDL production and LDL-C levels in homozygous FH patients. Due to their mechanism of action, each drug causes an increase in hepatic fat, the long-term consequences of which are unknown. In addition, lomitapide is associated with gastrointestinal-related side effects, and mipomersen is associated with skin reactions and flu-like symptoms.
LDL APHERESIS Patients who remain severely hypercholesterolemic despite optimally tolerated drug therapy are candidates for LDL apheresis. In this process, the patient’s plasma is passed over a column that selectively removes the LDL, and the LDL-depleted plasma is returned to the patient. Patients on maximally tolerated combination drug therapy who have CHD and a plasma LDL-C level >200 mg/dL or no CHD and a plasma LDL-C level >300 mg/dL are candidates for every-other-week LDL apheresis and should be referred to a specialized lipid center.
422 |
The Metabolic Syndrome |
The metabolic syndrome (syndrome X, insulin resistance syndrome) consists of a constellation of metabolic abnormalities that confer increased risk of cardiovascular disease (CVD) and diabetes mellitus. Evolution of the criteria for the metabolic syndrome since the original definition by the World Health Organization in 1998 reflects growing clinical evidence and analysis by a variety of consensus conferences and professional organizations. The major features of the metabolic syndrome include central obesity, hypertriglyceridemia, low levels of high-density lipoprotein (HDL) cholesterol, hyperglycemia, and hypertension (Table 422-1).
|
NCEP:ATPIIIa 2001 AND HARMONIZING DEFINITION CRITERIA FOR THE METABOLIC SYNDROME |
EPIDEMIOLOGY
 The most challenging feature of the metabolic syndrome to define is waist circumference. Intraabdominal circumference (visceral adipose tissue) is considered most strongly related to insulin resistance and risk of diabetes and CVD, and for any given waist circumference the distribution of adipose tissue between SC and visceral depots varies substantially. Thus, within and between populations, there is a lesser vs. greater risk at the same waist circumference. These differences in populations are reflected in the range of waist circumferences considered to confer risk in different geographic locations (Table 422-1).
The most challenging feature of the metabolic syndrome to define is waist circumference. Intraabdominal circumference (visceral adipose tissue) is considered most strongly related to insulin resistance and risk of diabetes and CVD, and for any given waist circumference the distribution of adipose tissue between SC and visceral depots varies substantially. Thus, within and between populations, there is a lesser vs. greater risk at the same waist circumference. These differences in populations are reflected in the range of waist circumferences considered to confer risk in different geographic locations (Table 422-1).
The prevalence of the metabolic syndrome varies around the world, in part reflecting the age and ethnicity of the populations studied and the diagnostic criteria applied. In general, the prevalence of the metabolic syndrome increases with age. The highest recorded prevalence worldwide is among Native Americans, with nearly 60% of women ages 45–49 and 45% of men ages 45–49 meeting the criteria of the National Cholesterol Education Program and Adult Treatment Panel III (NCEP:ATPIII). In the United States, the metabolic syndrome is less common among African-American men and more common among Mexican-American women. Based on data from the National Health and Nutrition Examination Survey (NHANES) 2003–2006, the age-adjusted prevalence of the metabolic syndrome in U.S. adults without diabetes is 28% for men and 30% for women. In France, studies of a cohort of 30- to 60-year-olds have shown a <10% prevalence for each sex, although 17.5% of people 60–64 years of age are affected. Greater global industrialization is associated with rising rates of obesity, which are expected to increase the prevalence of the metabolic syndrome dramatically, especially as the population ages. Moreover, the rising prevalence and severity of obesity among children is reflected in features of the metabolic syndrome in a younger population.
The frequency distribution of the five components of the syndrome for the U.S. population (NHANES III) is summarized in Fig. 422-1. Increases in waist circumference predominate among women, whereas increases in fasting plasma triglyceride levels (i.e., to >150 mg/dL), reductions in HDL cholesterol levels, and hyperglycemia are more likely in men.
FIGURE 422-1 Prevalence of the metabolic syndrome components, from NHANES 2003–2006. NHANES, National Health and Nutrition Examination Survey; TG, triglyceride; HDL-C, high-density lipoprotein cholesterol; BP, blood pressure. The prevalence of elevated glucose includes individuals with known diabetes mellitus. (Created from data in ES Ford et al: J Diabetes 2:1753, 2010.)
RISK FACTORS
Overweight/Obesity Although the metabolic syndrome was first described in the early twentieth century, the worldwide overweight/obesity epidemic has recently been the force driving its increasing recognition. Central adiposity is a key feature of the syndrome, and the syndrome’s prevalence reflects the strong relationship between waist circumference and increasing adiposity. However, despite the importance of obesity, patients who are of normal weight may also be insulin resistant and may have the metabolic syndrome.
Sedentary Lifestyle Physical inactivity is a predictor of CVD events and the related risk of death. Many components of the metabolic syndrome are associated with a sedentary lifestyle, including increased adipose tissue (predominantly central), reduced HDL cholesterol, and increased triglycerides, blood pressure, and glucose in genetically susceptible persons. Compared with individuals who watch television or videos or use the computer <1 h daily, those who do so for >4 h daily have a twofold increased risk of the metabolic syndrome.
Aging The metabolic syndrome affects nearly 50% of the U.S. population older than age 50, and at >60 years of age women are more often affected than men. The age dependency of the syndrome’s prevalence is seen in most populations around the world.
Diabetes Mellitus Diabetes mellitus is included in both the NCEP and the harmonizing definitions of the metabolic syndrome. It is estimated that the great majority (~75%) of patients with type 2 diabetes or impaired glucose tolerance have the metabolic syndrome. The presence of the metabolic syndrome in these populations relates to a higher prevalence of CVD than in patients who have type 2 diabetes or impaired glucose tolerance but do not have this syndrome.
Cardiovascular Disease Individuals with the metabolic syndrome are twice as likely to die of cardiovascular disease as those who do not, and their risk of an acute myocardial infarction or stroke is threefold higher. The approximate prevalence of the metabolic syndrome among patients with coronary heart disease (CHD) is 50%, with a prevalence of ~35% among patients with premature coronary artery disease (before or at age 45) and a particularly high prevalence among women. With appropriate cardiac rehabilitation and changes in lifestyle (e.g., nutrition, physical activity, weight reduction, and—in some cases—pharmacologic therapy), the prevalence of the syndrome can be reduced.
Lipodystrophy Lipodystrophic disorders in general are associated with the metabolic syndrome. Both genetic lipodystrophy (e.g., Berardinelli-Seip congenital lipodystrophy, Dunnigan familial partial lipodystrophy) and acquired lipodystrophy (e.g., HIV-related lipodystrophy in patients receiving antiretroviral therapy) may give rise to severe insulin resistance and many of the components of the metabolic syndrome.
ETIOLOGY
Insulin Resistance The most accepted and unifying hypothesis to describe the pathophysiology of the metabolic syndrome is insulin resistance, which is caused by an incompletely understood defect in insulin action (Chap. 417). The onset of insulin resistance is heralded by postprandial hyperinsulinemia, which is followed by fasting hyperinsulinemia and ultimately by hyperglycemia.
An early major contributor to the development of insulin resistance is an overabundance of circulating fatty acids (Fig. 422-2). Plasma albumin-bound free fatty acids are derived predominantly from adipose-tissue triglyceride stores released by intracellular lipolytic enzymes. Fatty acids are also derived from the lipolysis of triglyceride-rich lipoproteins in tissues by lipoprotein lipase. Insulin mediates both antilipolysis and the stimulation of lipoprotein lipase in adipose tissue. Of note, the inhibition of lipolysis in adipose tissue is the most sensitive pathway of insulin action. Thus, when insulin resistance develops, increased lipolysis produces more fatty acids, which further decrease the antilipolytic effect of insulin. Excessive fatty acids enhance substrate availability and create insulin resistance by modifying downstream signaling. Fatty acids impair insulin-mediated glucose uptake and accumulate as triglycerides in both skeletal and cardiac muscle, whereas increased glucose production and triglyceride accumulation take place in the liver.
FIGURE 422-2 Pathophysiology of the metabolic syndrome. Free fatty acids (FFAs) are released in abundance from an expanded adipose tissue mass. In the liver, FFAs result in increased production of glucose and triglycerides and secretion of very low density lipoproteins (VLDLs). Associated lipid/lipoprotein abnormalities include reductions in high-density lipoprotein (HDL) cholesterol and an increased low-density lipoprotein (LDL) particle number (no.). FFAs also reduce insulin sensitivity in muscle by inhibiting insulin-mediated glucose uptake. Associated defects include a reduction in glucose partitioning to glycogen and increased lipid accumulation in triglyceride (TG). The increase in circulating glucose, and to some extent FFAs, increases pancreatic insulin secretion, resulting in hyperinsulinemia. Hyperinsulinemia may result in enhanced sodium reabsorption and increased sympathetic nervous system (SNS) activity and contribute to hypertension, as might higher levels of circulating FFAs. The proinflammatory state is superimposed and contributory to the insulin resistance produced by excessive FFAs. The enhanced secretion of interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) produced by adipocytes and monocyte-derived macrophages results in more insulin resistance and lipolysis of adipose tissue triglyceride stores to circulating FFAs. IL-6 and other cytokines also enhance hepatic glucose production, VLDL production by the liver, hypertension and insulin resistance in muscle. Cytokines and FFAs also increase hepatic production of fibrinogen and adipocyte production of plasminogen activator inhibitor 1 (PAI-1), resulting in a prothrombotic state. Higher levels of circulating cytokines stimulate hepatic production of C-reactive protein (CRP). Reduced production of the anti-inflammatory and insulin-sensitizing cytokine adiponectin is also associated with the metabolic syndrome. (Modified from RH Eckel et al: Lancet 365:1415, 2005.)
Leptin resistance has also been raised as a possible pathophysiologic mechanism to explain the metabolic syndrome. Physiologically, leptin reduces appetite, promotes energy expenditure, and enhances insulin sensitivity. In addition, leptin may regulate cardiac and vascular function through a nitric oxide–dependent mechanism. However, when obesity develops, hyperleptinemia ensues, with evidence of leptin resistance in the brain and other tissues resulting in inflammation, insulin resistance, hyperlipidemia, and a plethora of cardiovascular disorders, such as hypertension, atherosclerosis, CHD, and heart failure.
The oxidative stress hypothesis provides a unifying theory for aging and the predisposition to the metabolic syndrome. In studies of insulin-resistant individuals with obesity or type 2 diabetes, the offspring of patients with type 2 diabetes, and the elderly, a defect in mitochondrial oxidative phosphorylation that leads to the accumulation of triglycerides and related lipid molecules in muscle has been identified.
Recently, the gut microbiome has emerged as an important contributor to the development of obesity and related metabolic disorders, including the metabolic syndrome. Although the mechanism remains uncertain, interaction among genetic predisposition, diet, and the intestinal flora is important.
Increased Waist Circumference Waist circumference is an important component of the most recent and frequently applied diagnostic criteria for the metabolic syndrome. However, measuring waist circumference does not reliably distinguish increases in SC adipose tissue from those in visceral fat; this distinction requires CT or MRI. With increases in visceral adipose tissue, adipose tissue–derived free fatty acids are directed to the liver. In contrast, increases in abdominal SC fat release lipolysis products into the systemic circulation and avert more direct effects on hepatic metabolism. Relative increases in visceral versus SC adipose tissue with increasing waist circumference in Asians and Asian Indians may explain the greater prevalence of the syndrome in those populations than in African-American men, in whom SC fat predominates. It is also possible that visceral fat is a marker for—but not the source of—excess postprandial free fatty acids in obesity.
Dyslipidemia (See also Chap. 421) In general, free fatty acid flux to the liver is associated with increased production of ApoB-containing, triglyceride-rich, very low-density lipoproteins (VLDLs). The effect of insulin on this process is complex, but hypertriglyceridemia is an excellent marker of the insulin-resistant condition. Not only is hypertriglyceridemia a feature of the metabolic syndrome, but patients with the metabolic syndrome have elevated levels of ApoCIII carried on VLDLs and other lipoproteins. This increase in ApoCIII is inhibitory to lipoprotein lipase, further contributing to hypertriglyceridemia and also associated with more atherosclerotic cardiovascular disease.
The other major lipoprotein disturbance in the metabolic syndrome is a reduction in HDL cholesterol. This reduction is a consequence of changes in HDL composition and metabolism. In the presence of hypertriglyceridemia, a decrease in the cholesterol content of HDL is a consequence of reduced cholesteryl ester content of the lipoprotein core in combination with cholesteryl ester transfer protein–mediated alterations in triglyceride that make the particle small and dense. This change in lipoprotein composition also results in increased clearance of HDL from the circulation. These changes in HDL have a relationship to insulin resistance that is probably indirect, occurring in concert with the changes in triglyceride-rich lipoprotein metabolism.
In addition to HDLs, low-density lipoproteins (LDLs) are modified in composition in the metabolic syndrome. With fasting serum triglycerides at >2.0 mM (~180 mg/dL), there is almost always a predominance of small, dense LDLs, which are thought to be more atherogenic although their association with hypertriglyceridemia and low HDLs make their independent contribution to CVD events difficult to assess. Individuals with hypertriglyceridemia often have increases in cholesterol content of both VLDL1 and VLDL2 subfractions and in LDL particle number. Both of these lipoprotein changes may contribute to atherogenic risk in patients with the metabolic syndrome.
Glucose Intolerance (See also Chap. 417) Defects in insulin action in the metabolic syndrome lead to impaired suppression of glucose production by the liver and kidney and reduced glucose uptake and metabolism in insulin-sensitive tissues—i.e., muscle and adipose tissue. The relationship between impaired fasting glucose or impaired glucose tolerance and insulin resistance is well supported by studies of humans, nonhuman primates, and rodents. To compensate for defects in insulin action, insulin secretion and/or clearance must be modified so that euglycemia is sustained. Ultimately, this compensatory mechanism fails, usually because of defects in insulin secretion, resulting in progression from impaired fasting glucose and/or impaired glucose tolerance to diabetes mellitus.
 Hypertension The relationship between insulin resistance and hypertension is well established. Paradoxically, under normal physiologic conditions, insulin is a vasodilator with secondary effects on sodium reabsorption in the kidney. However, in the setting of insulin resistance, the vasodilatory effect of insulin is lost but the renal effect on sodium reabsorption is preserved. Sodium reabsorption is increased in whites with the metabolic syndrome but not in Africans or Asians. Insulin also increases the activity of the sympathetic nervous system, an effect that may be preserved in the setting of insulin resistance. Insulin resistance is characterized by pathway-specific impairment in phosphatidylinositol-3-kinase signaling. In the endothelium, this impairment may cause an imbalance between the production of nitric oxide and the secretion of endothelin 1, with a consequent decrease in blood flow. Although these mechanisms are provocative, evaluation of insulin action by measurement of fasting insulin levels or by homeostasis model assessment shows that insulin resistance contributes only partially to the increased prevalence of hypertension in the metabolic syndrome.
Hypertension The relationship between insulin resistance and hypertension is well established. Paradoxically, under normal physiologic conditions, insulin is a vasodilator with secondary effects on sodium reabsorption in the kidney. However, in the setting of insulin resistance, the vasodilatory effect of insulin is lost but the renal effect on sodium reabsorption is preserved. Sodium reabsorption is increased in whites with the metabolic syndrome but not in Africans or Asians. Insulin also increases the activity of the sympathetic nervous system, an effect that may be preserved in the setting of insulin resistance. Insulin resistance is characterized by pathway-specific impairment in phosphatidylinositol-3-kinase signaling. In the endothelium, this impairment may cause an imbalance between the production of nitric oxide and the secretion of endothelin 1, with a consequent decrease in blood flow. Although these mechanisms are provocative, evaluation of insulin action by measurement of fasting insulin levels or by homeostasis model assessment shows that insulin resistance contributes only partially to the increased prevalence of hypertension in the metabolic syndrome.
Another possible mechanism underlying hypertension in the metabolic syndrome is the vasoactive role of perivascular adipose tissue. Reactive oxygen species released by NADPH oxidase impair endothelial function and result in local vasoconstriction. Other paracrine effects could be mediated by leptin or other proinflammatory cytokines released from adipose tissue, such as tumor necrosis factor α.
Hyperuricemia is another consequence of insulin resistance and is commonly observed in the metabolic syndrome. There is growing evidence not only that uric acid is associated with hypertension but also that reduction of uric acid normalizes blood pressure in hyperuricemic adolescents with hypertension. The mechanism appears to be related to an adverse effect of uric acid on nitric acid synthase in the macula densa of the kidney and stimulation of the renin-angiotensin aldosterone system.
Proinflammatory Cytokines The increases in proinflammatory cytokines—including interleukins 1, 6, and 18; resistin; tumor necrosis factor α; and the systemic biomarker C-reactive protein—reflect overproduction by the expanded adipose tissue mass (Fig. 422-2). Adipose tissue–derived macrophages may be the primary source of proinflammatory cytokines locally and in the systemic circulation. It remains unclear, however, how much of the insulin resistance is caused by the paracrine effects of these cytokines and how much by the endocrine effects.
Adiponectin Adiponectin is an anti-inflammatory cytokine produced exclusively by adipocytes. Adiponectin enhances insulin sensitivity and inhibits many steps in the inflammatory process. In the liver, adiponectin inhibits the expression of gluconeogenic enzymes and the rate of glucose production. In muscle, adiponectin increases glucose transport and enhances fatty acid oxidation, partially through the activation of AMP kinase. Adiponectin levels are reduced in the metabolic syndrome. The relative contributions of adiponectin deficiency and overabundance of the proinflammatory cytokines are unclear.
CLINICAL FEATURES
Symptoms and Signs The metabolic syndrome typically is not associated with symptoms. On physical examination, waist circumference may be expanded and blood pressure elevated. The presence of either or both of these signs should prompt the clinician to search for other biochemical abnormalities that may be associated with the metabolic syndrome. Less frequently, lipoatrophy or acanthosis nigricans is found on examination. Because these physical findings characteristically are associated with severe insulin resistance, other components of the metabolic syndrome should be expected.
Associated Diseases • CARDIOVASCULAR DISEASE The relative risk for new-onset CVD in patients with the metabolic syndrome who do not have diabetes averages 1.5–3 fold. However, an 8-year follow-up of middle-aged participants in the Framingham Offspring Study documented that the population-attributable CVD risk in the metabolic syndrome was 34% among men and only 16% among women. In the same study, both the metabolic syndrome and diabetes predicted ischemic stroke, with greater risk among patients with the metabolic syndrome than among those with diabetes alone (19% vs. 7%) and a particularly large difference among women (27% vs. 5%). Patients with the metabolic syndrome are also at increased risk for peripheral vascular disease.
TYPE 2 DIABETES Overall, the risk for type 2 diabetes among patients with the metabolic syndrome is increased three- to fivefold. In the Framingham Offspring Study’s 8-year follow-up of middle-aged participants, the population-attributable risk for developing type 2 diabetes was 62% among men and 47% among women.
Other Associated Conditions In addition to the features specifically associated with the metabolic syndrome, other metabolic alterations accompany insulin resistance. Those alterations include increases in ApoB and ApoCIII, uric acid, prothrombotic factors (fibrinogen, plasminogen activator inhibitor 1), serum viscosity, asymmetric dimethylarginine, homocysteine, white blood cell count, proinflammatory cytokines, C-reactive protein, microalbuminuria, nonalcoholic fatty liver disease and/or nonalcoholic steatohepatitis, polycystic ovary syndrome, and obstructive sleep apnea.
NONALCOHOLIC FATTY LIVER DISEASE (SEE ALSO CHAP. 367e) Fatty liver is a relatively common condition, affecting 25–45% of the U.S. population. However, in nonalcoholic steatohepatitis, triglyceride accumulation and inflammation coexist. Nonalcoholic steatohepatitis is now present in 3–12% of the population of the United States and other Western countries. Of patients with the metabolic syndrome, ~25–60% have nonalcoholic fatty liver disease and up to 35% have nonalcoholic steatohepatitis. As the prevalence of overweight/obesity and the metabolic syndrome increases, nonalcoholic steatohepatitis may become one of the more common causes of end-stage liver disease and hepatocellular carcinoma.
HYPERURICEMIA (SEE ALSO CHAP. 431e) Hyperuricemia reflects defects in insulin action on the renal tubular reabsorption of uric acid and may contribute to hypertension through its effect on the endothelium. An increase in asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, also relates to endothelial dysfunction. In addition, microalbuminuria may be caused by altered endothelial pathophysiology in the insulin-resistant state.
POLYCYSTIC OVARY SYNDROME (SEE ALSO CHAP. 412) Polycystic ovary syndrome is highly associated with insulin resistance (50–80%) and the metabolic syndrome, with a prevalence of the syndrome between 40% and 50%. Women with polycystic ovary syndrome are two to four times more likely to have the metabolic syndrome than are women without polycystic ovary syndrome.
OBSTRUCTIVE SLEEP APNEA (SEE ALSO CHAP. 38) Obstructive sleep apnea is commonly associated with obesity, hypertension, increased circulating cytokines, impaired glucose tolerance, and insulin resistance. With these associations, it is not surprising that individuals with obstructive sleep apnea frequently have the metabolic syndrome. Moreover, when biomarkers of insulin resistance are compared between patients with obstructive sleep apnea and weight-matched controls, insulin resistance is found to be more severe in those with apnea. Continuous positive airway pressure treatment improves insulin sensitivity in patients with obstructive sleep apnea.
DIAGNOSIS
The diagnosis of the metabolic syndrome relies on fulfillment of the criteria listed in Table 422-1, as assessed using tools at the bedside and in the laboratory. The medical history should include evaluation of symptoms for obstructive sleep apnea in all patients and polycystic ovary syndrome in premenopausal women. Family history will help determine risk for CVD and diabetes mellitus. Blood pressure and waist circumference measurements provide information necessary for the diagnosis.
Laboratory Tests Measurement of fasting lipids and glucose is needed in determining whether the metabolic syndrome is present. The measurement of additional biomarkers associated with insulin resistance can be individualized. Such tests might include those for ApoB, high-sensitivity C-reactive protein, fibrinogen, uric acid, urinary microalbumin, and liver function. A sleep study should be performed if symptoms of obstructive sleep apnea are present. If polycystic ovary syndrome is suspected on the basis of clinical features and anovulation, testosterone, luteinizing hormone, and follicle-stimulating hormone should be measured.
|
TREATMENT |
THE METABOLIC SYNDROME |
LIFESTYLE (SEE ALSO CHAP. 416)
Obesity is the driving force behind the metabolic syndrome. Thus, weight reduction is the primary approach to the disorder. With weight reduction, improvement in insulin sensitivity is often accompanied by favorable modifications in many components of the metabolic syndrome. In general, recommendations for weight loss include a combination of caloric restriction, increased physical activity, and behavior modification. Caloric restriction is the most important component, whereas increases in physical activity are important for maintenance of weight loss. Some but not all evidence suggests that the addition of exercise to caloric restriction may promote greater weight loss from the visceral depot. The tendency for weight regain after successful weight reduction underscores the need for long-lasting behavioral changes.
Diet Before prescribing a weight-loss diet, it is important to emphasize that it has taken the patient a long time to develop an expanded fat mass; thus, the correction need not occur quickly. Given that ~3500 kcal = 1 lb of fat, ~500-kcal restriction daily equates to weight reduction of 1 lb per week. Diets restricted in carbohydrate typically provide a rapid initial weight loss. However, after 1 year, the amount of weight reduction is minimally reduced or no different from that with caloric restriction alone. Thus, adherence to the diet is more important than which diet is chosen. Moreover, there is concern about low-carbohydrate diets enriched in saturated fat, particularly for patients at risk for CVD. Therefore, a high-quality dietary pattern—i.e., a diet enriched in fruits, vegetables, whole grains, lean poultry, and fish—should be encouraged to maximize overall health benefit.




