[level-membership-for-internal-medicine-category]
Chapter 20 Diabetes mellitus and other disorders of metabolism
Diabetes mellitus
Hyperglycaemia, insulin and insulin action
Insulin structure and secretion
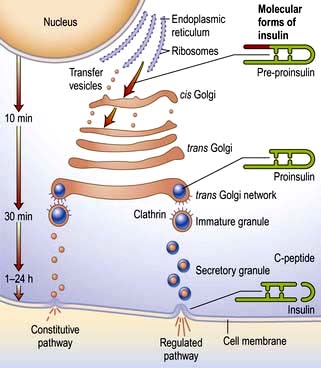
Insulin is the key hormone involved in the storage and controlled release within the body of the chemical energy available from food. It is coded for on chromosome 11 and synthesized in the beta cells of the pancreatic islets (Fig. 20.1). The synthesis, intracellular processing and secretion of insulin by the beta cell is typical of the way that the body produces and manipulates many peptide hormones. Figure 20.2 illustrates the cellular events triggering the release of insulin-containing granules. After secretion, insulin enters the portal circulation and is carried to the liver, its prime target organ. About 50% of secreted insulin is extracted and degraded in the liver; the residue is broken down by the kidneys. C-peptide is only partially extracted by the liver (and hence provides a useful index of the rate of insulin secretion) but is mainly degraded by the kidneys.
An outline of glucose metabolism
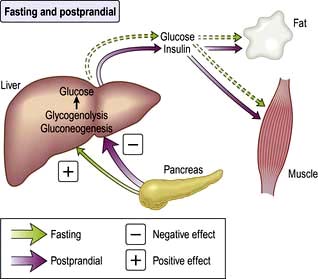
Insulin is a major regulator of intermediary metabolism, although its actions are modified in many respects by other hormones. Its actions in the fasting and postprandial states differ (Fig. 20.3). In the fasting state, its main action is to regulate glucose release by the liver, and in the postprandial state, it additionally promotes glucose uptake by fat and muscle. The effect of counter-regulatory hormones (glucagon, epinephrine (adrenaline), cortisol and growth hormone) is to cause greater production of glucose from the liver and less utilization of glucose in fat and muscle for a given level of insulin.
 GLUT-1 – enables basal non-insulin-stimulated glucose uptake into many cells (see Fig. 6.29).
GLUT-1 – enables basal non-insulin-stimulated glucose uptake into many cells (see Fig. 6.29).

 GLUT-3 – enables non-insulin-mediated glucose uptake into brain neurones and placenta.
GLUT-3 – enables non-insulin-mediated glucose uptake into brain neurones and placenta.
 GLUT-4 – enables much of the peripheral action of insulin. It is the channel through which glucose is taken up into muscle and adipose tissue cells following stimulation of the insulin receptor (Fig. 20.4).
GLUT-4 – enables much of the peripheral action of insulin. It is the channel through which glucose is taken up into muscle and adipose tissue cells following stimulation of the insulin receptor (Fig. 20.4).
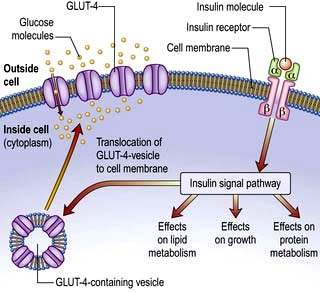
This is a glycoprotein (400 kDa), coded for on the short arm of chromosome 19, which straddles the cell membrane of many cells (Fig. 20.4). It consists of a dimer with two α-subunits, which include the binding sites for insulin, and two β-subunits, which traverse the cell membrane. When insulin binds to the α-subunits it induces a conformational change in the β-subunits, resulting in activation of tyrosine kinase and initiation of a cascade response involving a host of other intracellular substrates. One consequence of this is migration of the GLUT-4 glucose transporter to the cell surface and increased transport of glucose into the cell. The insulin-receptor complex is then internalized by the cell, insulin is degraded, and the receptor is recycled to the cell surface.
Classification of diabetes
Diabetes may be primary (idiopathic) or secondary (Table 20.1). Primary diabetes is classified into:


Table 20.1 Aetiological classification of diabetes mellitus, based on classification by the American Diabetes Association (ADA)
Note: Patients with any form of diabetes may require insulin treatment at some stage of their disease. Such use of insulin does not, of itself, classify the patient.
(Adapted from ADA. Diagnosis and classification of diabetes mellitus. Diabetes Care 2008; 31(Suppl 1):S55–S60.)
The key clinical features of the two main forms of diabetes are listed in Table 20.2. Type 1 and type 2 diabetes represent two distinct diseases from the epidemiological point of view, but clinical distinction can sometimes be difficult. The two diseases should from a clinical point of view be seen as a spectrum, distinct at the two ends but overlapping to some extent in the middle. Hybrid forms are increasingly recognized, and patients with immune-mediated diabetes (type 1) may, for example, also be overweight and insulin resistant. This is sometimes referred to as ‘double diabetes’. It is more relevant to give the patient the right treatment on clinical grounds than to worry about how to label their diabetes. The classification of primary diabetes continues to evolve. Monogenic forms have been identified (see p. 1007), in some cases with significant therapeutic implications. Although secondary diabetes accounts for barely 1–2% of all new cases at presentation, it should not be missed because the cause can sometimes be treated. All forms of diabetes derive from inadequate insulin secretion relative to the needs of the body, and progressive insulin secretory failure is characteristic of both common forms of diabetes. Thus, some patients with immune-mediated diabetes type 1 may not at first require insulin, whereas many with type 2 diabetes will eventually do so.
Table 20.2 The spectrum of diabetes: a comparison of type 1 and type 2 diabetes mellitus
| Type 1 | Type 2 | |
|---|---|---|
|
Age |
Younger (usually <30) |
Older (usually >30) |
|
Weight |
Lean |
Overweight |
|
Symptom duration |
Weeks |
Months/years |
|
Higher risk ethnicity |
Northern European |
Asian, African, Polynesian and American-Indian |
|
Seasonal onset |
Yes |
No |
|
Heredity |
HLA-DR3 or DR4 in >90% |
No HLA links |
|
Pathogenesis |
Autoimmune disease |
No immune disturbance |
|
Ketonuria |
Yes |
No |
|
Clinical |
Insulin deficiency |
Partial insulin deficiency initially |
|
|
± ketoacidosis |
± hyperosmolar state |
|
|
Always need insulin |
Need insulin when beta cells fail over time |
|
Biochemical |
C-peptide disappears |
C-peptide persists |
Type 1 diabetes mellitus
Epidemiology
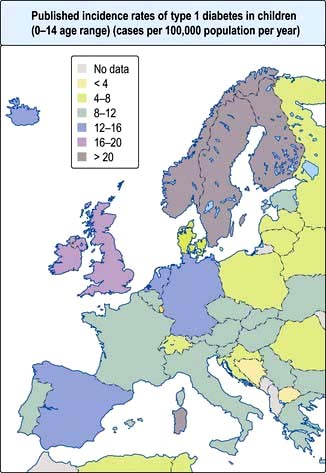
Type 1 diabetes is a disease of insulin deficiency. In western countries almost all patients have the immune-mediated form of the disease, otherwise known as type 1A. Type 1 diabetes is a disease of childhood, reaching a peak incidence around the time of puberty, but can present at any age. A ‘slow-burning’ variant with slower progression to insulin deficiency occurs in later life and is sometimes called latent autoimmune diabetes in adults (LADA). LADA may be difficult to distinguish from type 2 diabetes. Clinical clues are: leaner build, rapid progression to insulin therapy following an initial response to other therapies, and the presence of circulating islet autoantibodies. The highest rates of type 1 diabetes in the world are seen in Finland and other Northern European countries, and on the island of Sardinia, which for unknown reasons, has the second highest rate in the world (Fig. 20.5). The incidence of type 1 diabetes appears to be increasing in most populations. In Europe, the annual increase is of the order of 2–3%, and is most marked in children under the age of 5 years. WHO estimated in 1995 that there were 19.4 million people with type 1 diabetes and that the number will rise to 57.2 million by 2025.
Causes
Autoimmunity and type 1 diabetes
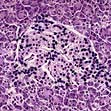
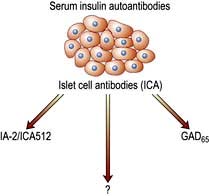
Type 1 diabetes is associated with other organ-specific autoimmune diseases including autoimmune thyroid disease, coeliac disease, Addison’s disease and pernicious anaemia. Autopsies of patients who died following diagnosis of type 1 diabetes show infiltration of the pancreatic islets by mononuclear cells. This appearance, known as insulitis, resembles that in other autoimmune diseases such as thyroiditis. Several islet antigens have been characterized, and these include insulin itself, the enzyme glutamic acid decarboxylase (GAD), protein tyrosine phosphatase (IA-2) (Fig. 20.6) and the cation transporter ZnT8. Recent studies have shown that GAD immunotherapy has no benefit. The observation that treatment with immunosuppressive agents such as ciclosporin prolongs beta-cell survival in newly diagnosed patients has confirmed that the disease is immune-mediated.
Environmental factors
The incidence of childhood type 1 diabetes is rising across Europe at the rate of 2–3% each year, suggesting that environmental factor(s) are involved in its pathogenesis. Islet autoantibodies (see above) appear in the first few years of life, indicating prenatal or early postnatal interactions with the environment. Exposures to dietary constituents, enteroviruses such as Coxsackie B4 and relative deficiency of vitamin D are possible candidates, but their role in the causation of the disease has yet to be confirmed. A cleaner environment with less early stimulation of the immune system in childhood may increase susceptibility for type 1 diabetes, as for atopic/allergic conditions (the hygiene hypothesis) (see p. 824), and more rapid weight gain in childhood and adolescence leading to increased insulin resistance might accelerate clinical onset (the accelerator hypothesis).
Type 2 diabetes mellitus
Epidemiology
Type 2 diabetes is associated with central obesity, hypertension, hypertriglyceridaemia, a decreased HDL-cholesterol, disturbed haemostatic variables and modest increases in a number of pro-inflammatory markers. Insulin resistance is strongly associated with many of these variables, as is increased cardiovascular risk. This group of conditions is referred to as the metabolic syndrome (see p. 223). The International Diabetes Federation has proposed criteria based on increased waist circumference (or BMI >30) plus two of the following: diabetes (or fasting glucose >6.0 mmol/L), hypertension, raised triglycerides or low HDL cholesterol. On this definition, about one-third of the adult population has features of the syndrome, not necessarily associated with diabetes. Critics would argue that the metabolic syndrome is not a distinct entity, but one end of a continuum in the relationship between exercise, lifestyle and bodyweight on the one hand, and genetic make-up on the other, and that diagnosis adds little to standard clinical practice in terms of diagnosis, prognosis or therapy.
Causes
Abnormalities of insulin secretion and action
The relative role of secretory failure versus insulin resistance in the pathogenesis of type 2 diabetes has been much debated, but even massively obese individuals with a fully functioning beta-cell mass do not necessarily develop diabetes, which implies that some degree of beta-cell dysfunction is necessary. Insulin binds normally to its receptor on the surface of cells in type 2 diabetes, and the mechanisms of ‘insulin resistance’ are still poorly understood. Insulin resistance is, however, associated with central obesity and accumulation of intracellular triglyceride in muscle and liver in type 2 diabetes, and a high proportion of patients have non-alcoholic fatty liver disease (NAFLD), see page 303. It has long been stated that patients with type 2 diabetes retain up to 50% of their beta-cell mass at the time of diagnosis, as compared with healthy controls, but the shortfall is greater than this when they are matched with healthy individuals who are equally obese. In addition, patients with type 2 diabetes almost all show islet amyloid deposition at autopsy, derived from a peptide known as amylin or islet amyloid polypeptide (IAPP), which is co-secreted with insulin. It is not known if this is a cause or consequence of beta-cell secretory failure.
Monogenic diabetes mellitus
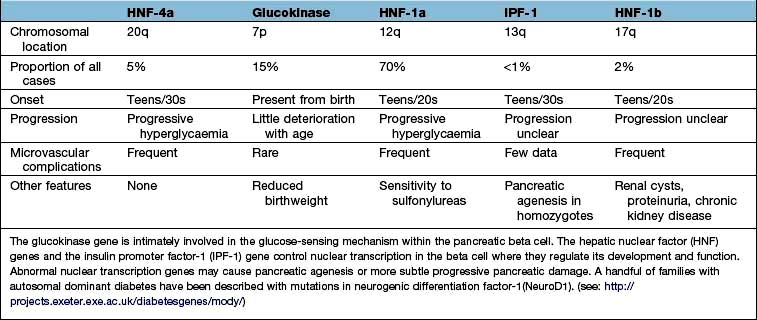
The genetic causes of some rare forms of diabetes are shown in Table 20.3. Considerable progress has been made in understanding these rare variants of diabetes. Genetic defects of beta-cell function (previously called ‘maturity-onset diabetes of the young’, MODY) are dominantly inherited, and several variants have been described, each associated with different clinical phenotypes (Table 20.4). These should be considered in people presenting with early-onset diabetes in association with an affected parent and early-onset diabetes in ~50% of relatives. They can often be treated with a sulfonylurea.
| Disorder | Features |
|---|---|
|
Insulin receptor mutations |
Obesity, marked insulin resistance, hyperandrogenism in women, acanthosis nigricans (areas of hyperpigmented skin) |
|
Maternally inherited diabetes and deafness (MIDD) |
Mutation in mitochondrial DNA. Diabetes onset before age 40. Variable deafness, neuromuscular and cardiac problems, pigmented retinopathy |
|
Wolfram’s syndrome (DIDMOAD – diabetes insipidus, diabetes mellitus, optic atrophy and deafness) |
Recessively inherited. Mutation in the transmembrane gene, WFS1. Insulin-requiring diabetes and optic atrophy in the first decade. Diabetes insipidus and sensorineural deafness in the second decade progressing to multiple neurological problems. Few live beyond middle age |
|
Severe obesity and diabetes |
Alström’s, Bardet–Biedl and Prader–Willi syndromes. Retinitis pigmentosa, mental insufficiency and neurological disorders |
|
Disorders of intracellular insulin signalling. All with severe insulin resistance |
Leprechaunism, Rabson–Mendenhall syndrome, pseudoacromegaly, partial lipodystrophy: lamin A/C gene mutation |
|
Genetic defects of beta-cell function |
See Table 20.4 |
FURTHER READING
Chan JC, Malik V, Jia W et al. Diabetes in Asia: epidemiology, risk factors and pathophysiology. JAMA 2009; 301:2129–2140.
Ramachandran A, Ma RC, Snehalatha C. Diabetes in Asia. Lancet 2010; 375:408–418.
Sturnvoll M, Goldstein BJ, van Haefken TW. Type 2 diabetes; pathogenesis and treatment. Lancet 2008; 371:2153–2156.
Clinical presentation of diabetes
Presentation may be acute, subacute or asymptomatic.
Acute presentation
Young people often present with a 2–6-week history and report the classic triad of symptoms:








Asymptomatic diabetes
Physical examination at diagnosis
Evidence of weight loss and dehydration may be present, and the breath may smell of ketones. Older patients may present with established complications, and the presence of the characteristic retinopathy is diagnostic of diabetes. In occasional patients, there will be physical signs of an illness causing secondary diabetes (see Table 20.1). Patients with severe insulin resistance may have acanthosis nigricans, which is characterized by blackish pigmentation at the nape of the neck and in the axillae (p. 1217).
Diagnosis and investigation of diabetes
Diabetes is easy to diagnose when overt symptoms are present, and a glucose tolerance test is hardly ever necessary for clinical purposes. The oral glucose tolerance test has, however, allowed more detailed epidemiological characterization based on the existence of separate glucose thresholds for macrovascular and microvascular disease. These correspond with the levels for the diagnosis of impaired glucose tolerance (IGT) and diabetes as specified by the WHO criteria set out in Box 20.1. Epidemiological studies show that for every person with known diabetes, there is another undiagnosed in the population. A much larger proportion fall into the intermediate category of impaired glucose tolerance.
 Box 20.1
Box 20.1
WHO diagnostic criteria
WHO criteria for the diagnosis of diabetes are:




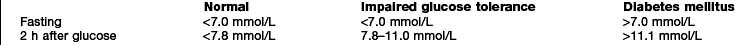




Haemoglobin A1c (HbA1c)
Other investigations
No further tests are needed to diagnose diabetes. Other routine investigations include urine testing for protein, a full blood count, urea and electrolytes, liver biochemistry and random lipids. The latter test is useful to exclude an associated hyperlipidaemia and, if elevated, should be repeated fasting after diabetes has been brought under control. Diabetes may be secondary to other conditions (see Table 20.1), may be precipitated by underlying illness and be associated with autoimmune disease or hyperlipidaemia. Hypertension is present in 50% of patients with type 2 diabetes and a higher proportion of African and Caribbean patients.
Treatment of diabetes
Diet
The diet for people with diabetes is no different from that considered healthy for everyone. Table 20.5 lists recommendations on the ideal composition of this diet. To achieve this, food for people with diabetes should be:
 low in sugar (though not sugar free)
low in sugar (though not sugar free)
 high in starchy carbohydrate (especially foods with a low glycaemic index), i.e. slower absorption
high in starchy carbohydrate (especially foods with a low glycaemic index), i.e. slower absorption


Table 20.5 Recommended composition of the diet for people with diabetes, with comments on how this may be achieved
| Component of diet | Comment |
|---|---|
|
Protein |
1 g/kg ideal bodyweight (approx.) |
|
Total fat |
<35% of energy intake. Limit: fat/oil in cooking, fried foods, processed meats (burgers, salami, sausages), high-fat snacks (crisps, cake, nuts, chocolate, biscuits, pastry). Encourage: lower-fat dairy products (skimmed milk, reduced-fat cheese, low-fat yoghurt), lean meat |
|
Saturated and trans-unsaturated fat |
<10% of total energy intake |
|
n-6 polyunsaturated fat |
<10% of total energy intake |
|
n-3 polyunsaturated fat |
No absolute quantity recommended. Eat fish, especially oily fish, once or twice weekly. Fish oil supplements not recommended |
|
Cis-monounsaturated fat |
10–20% of total energy intake (olive oil, avocado) |
|
Total carbohydrate |
40–60% of total energy intake Encourage: artificial (intense) sweeteners instead of sugar (sugar-free fizzy drinks, squashes and cordials). Limit: fruit juices, confectionery, cake, biscuits |
|
Sucrose |
Up to 10% of total energy intake, provided this is eaten in the context of a healthy diet (examples: fibre-rich breakfast cereals, baked beans) |
|
Fibre |
No absolute quantity recommended. Soluble fibre has beneficial effects on glycaemic and lipid metabolism. Insoluble fibre has no direct effects on glycaemic metabolism, but benefits satiety and gastrointestinal health |
|
Vitamins and antioxidants |
Best taken as fruit and vegetables (five portions per day) in a mixed diet. There is no evidence for the use of supplements |
|
Alcohol |
Not forbidden. Its energy content should be taken into account, as should its tendency to cause delayed hypoglycaemia in those treated with insulin |
|
Salt |
<6 g/day (lower in hypertension) |
Tablet treatment for type 2 diabetes
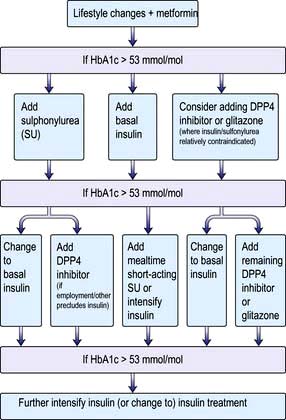
Diet and lifestyle changes are the key to successful treatment of type 2 diabetes, and no amount of medication will succeed where these have failed. The concept is that controlling diabetes is not just a matter of swallowing tablets, and these should in general never be prescribed until lifestyle changes have been implemented. Tablets will however be needed if satisfactory metabolic control (see ‘Measuring control’ below) is not established within 4–6 weeks. A consensus treatment pathway is shown in Figure 20.9 (p. 1013). The three main options are metformin, a sulfonylurea or a thiazolidinedione.
Sulfonylureas (Table 20.6)
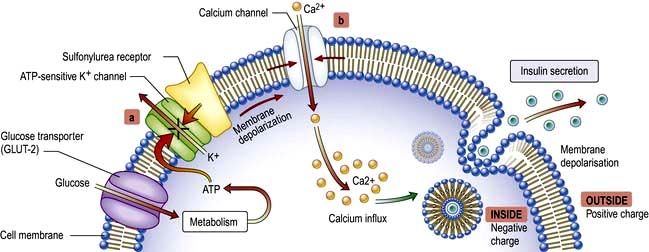
These act upon the beta cell to promote insulin secretion in response to glucose and other secretagogues. They are ineffective in patients without a functional beta-cell mass, and they are usually avoided in pregnancy. Their action is to bind to the sulfonylurea receptor on the cell membrane, which closes ATP-sensitive potassium channels and blocks potassium efflux. The resulting depolarization promotes influx of calcium, a signal for insulin release (Fig. 20.2). Sulfonylureas are cheap and more effective than the other agents in achieving short-term (1–3 years) glucose control, but their effect wears off as the beta-cell mass declines. There are theoretical concerns that they might hasten beta-cell apoptosis and they promote weight gain, and are best avoided in the overweight. They can also cause hypoglycaemia and although the episodes are generally mild, fatal hypoglycaemia may occur. Severe cases should always be admitted to hospital, monitored carefully, and treated with a continuous glucose infusion since some sulfonlyureas have long half-lives. Sulfonylureas should be used with care in patients with liver disease. Patients with renal impairment should only be given those primarily excreted by the liver. Tolbutamide is the safest drug in the very elderly because of its short duration of action.
Table 20.6 Properties of the most commonly used sulfonylureas
| Drug | Features |
|---|---|
|
Tolbutamide |
Lower maximal efficacy than other sulfonylureas |
|
Short half-life – preferable in elderly |
|
|
Largely metabolized by liver – can use in renal impairment |
|
|
Glibenclamide |
Long biological half-life |
|
Severe hypoglycaemia |
|
|
Do not use in the elderly |
|
|
Glipizide and Glimepiride |
Active metabolites |
|
Renal excretion – avoid in renal impairment |
|
|
Gliclazide |
Intermediate biological half-life |
|
Largely metabolized by liver – can use in renal impairment |
|
|
More costly |
|
|
Chlorpropamide |
Very long biological half-life |
|
Renal excretion – avoid in renal impairment |
|
|
1–2% develop inappropriate ADH-like syndrome |
|
|
Facial flush with alcohol |
|
|
Very inexpensive – major issue for developing countries |
|
|
Can produce fatal hypoglycaemia |
|
|
Not recommended in the elderly |
Meglitinides
Meglitinides, e.g. repaglinide and nateglinide, are insulin secretagogues. Meglitinides are the non-sulfonylurea moiety of glibenclamide. As with the sulfonylureas, they act via closure of the K+-ATP channel in the beta cells (see Fig. 20.2). They are short-acting agents that promote insulin secretion in response to meals. Their effects are similar to that of the short-acting sulfonylurea tolbutamide, but they are much more costly.
Dipeptidyl peptidase-4 (DPP4) inhibitors
These enhance the incretin effect (Box 20.2). The enzyme dipeptidyl peptidase 4 (DPP4) rapidly inactivates GLP-1 as this is released into the circulation. Inhibition of this enzyme thus potentiates the effect of endogenous GLP-1 secretion. Four agents are currently available (linagliptin, saxagliptin, sitagliptin and vildagliptin) with more likely to be available in the future. They have a moderate effect in lowering blood glucose and are weight neutral. They are most effective in the early stages of type 2 diabetes when insulin secretion is relatively preserved, and are currently recommended for second-line use in combination with metformin or a sulfonylurea. Adverse events are uncommon: the main side-effect is nausea, and there have been occasional reports of acute pancreatitis. Their place in the management of type 2 diabetes has yet to be fully established. Although the short-term safety record is good, DPP4 is widely distributed in the body, and the long-term consequences of inhibition of this enzyme in other tissues are unknown.
 Box 20.2
Box 20.2


Injection therapies for type 2 diabetes
GLP-1 agonists
Exenatide and liraglutide are injectable long-acting analogues of GLP-1, which enhance the incretin effect (Box 20.2). They promote insulin release, inhibit glucagon release, reduce appetite and delay gastric emptying, thus blunting the postprandial rise in plasma glucose and promoting weight loss. Their main clinical disadvantage is the need for subcutaneous injection (twice daily for exenatide and once daily for liraglutide), and their major advantage is improving glucose control whilst inducing useful weight reduction. They work well in 70% but have limited benefit in 30% of those treated. Side-effects include nausea, acute pancreatitis and acute kidney injury. At present they are used as an alternative to insulin, particularly in the overweight. A once weekly version of exenatide has been developed.
Other therapies
 Intestinal enzyme inhibitors include acarbose, a sham sugar that competitively inhibits α-glucosidase enzymes situated in the brush border of the intestine, reducing absorption of dietary carbohydrate. Undigested starch may then enter the large intestine where it will be broken down by fermentation. Abdominal discomfort, flatulence and diarrhoea can result, and dosage needs careful adjustment to avoid these side-effects.
Intestinal enzyme inhibitors include acarbose, a sham sugar that competitively inhibits α-glucosidase enzymes situated in the brush border of the intestine, reducing absorption of dietary carbohydrate. Undigested starch may then enter the large intestine where it will be broken down by fermentation. Abdominal discomfort, flatulence and diarrhoea can result, and dosage needs careful adjustment to avoid these side-effects.

 Gastric banding and gastric bypass surgery (see p. 220) have been used in those with marked obesity unresponsive to 6 months’ intensive attempts at dieting and graded exercise. NICE recommends consideration of surgery in those with a BMI >40, or in those with BMI >35 and co-morbidities such as diabetes or hypertension which will be alleviated by weight loss. In the USA, the FDA-recommended BMI thresholds are lower. The risks of surgery are not insignificant, and long-term specialist care and follow-up are needed, including psychological support and nutritional supplements for those with bowel resection, but these concerns should be balanced against the risk of patients staying as they are. About one-third of patients become non-diabetic after gastric bypass, but the condition may recur.
Gastric banding and gastric bypass surgery (see p. 220) have been used in those with marked obesity unresponsive to 6 months’ intensive attempts at dieting and graded exercise. NICE recommends consideration of surgery in those with a BMI >40, or in those with BMI >35 and co-morbidities such as diabetes or hypertension which will be alleviated by weight loss. In the USA, the FDA-recommended BMI thresholds are lower. The risks of surgery are not insignificant, and long-term specialist care and follow-up are needed, including psychological support and nutritional supplements for those with bowel resection, but these concerns should be balanced against the risk of patients staying as they are. About one-third of patients become non-diabetic after gastric bypass, but the condition may recur.
Insulin treatment
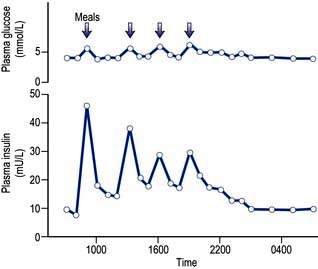
Insulin is found in every vertebrate, and the key parts of the molecule show few species differences. Small differences in the amino acid sequence may alter the antigenicity of the molecule. The glucose and insulin profiles in normal subjects are shown in Figure 20.7.
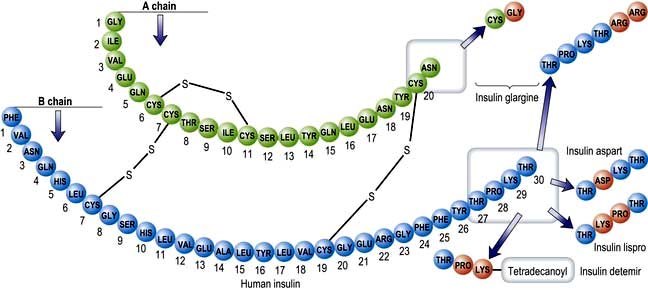
Insulins derived from beef or pig pancreas have been replaced in most countries by biosynthetic human insulin. This is produced by adding a DNA sequence coding for insulin or proinsulin into cultured yeast or bacterial cells. Short-acting insulins are used for pre-meal injection in multiple dose regimens, for continuous intravenous infusion in labour or during medical emergencies, and in patients using insulin pumps. Human insulin is absorbed slowly, reaching a peak 60–90 min after subcutaneous injection, and its action tends to persist after meals, predisposing to hypoglycaemia. Absorption is delayed because soluble insulin is in the form of stable hexamers (six insulin molecules around a zinc core) and needs to dissociate to monomers or dimers before it can enter the circulation. Short-acting insulin analogues have been engineered to dissociate more rapidly following injection without altering the biological effect. Insulin analogues (Fig. 20.8) such as the rapid-acting insulins (insulin lispro, insulin aspart and insulin glulisine) enter the circulation more rapidly than human soluble insulin, and also disappear more rapidly. Although widely used, the short-acting analogues have little effect upon overall glucose control in most patients, mainly because improved postprandial glucose is balanced by higher levels before the next meal. A Cochrane review has concluded that there is little evidence as to their benefit in type 2 diabetes.
Practical management of diabetes
All patients with diabetes require advice about diet and lifestyle. Lifestyle changes, i.e. controlling weight, stopping smoking and taking regular exercise, can prevent or delay the onset of type 2 diabetes in people with glucose intolerance. Good glycaemic control is unlikely to be achieved with insulin or oral therapy when diet is neglected, especially when the patient is also overweight. Regular exercise helps to control weight and reduces cardiovascular risk. Blood pressure control is vital using an angiotensin converting enzyme (ACE) inhibitor or angiotensin II receptor (AIIR) antagonist (see p. 782); Most patients will also benefit from a statin and low-dose aspirin (see p. 1022).
FURTHER READING
Inzucchi SE, Bergenstal RM, Buse JB et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach. Position Statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care, published online 19 April 2012.
Rejeski W et al. Lifestyle change and mobility in obese adults with type 2 diabetes. N Engl J Med 2012; 366:1209–1217.
The great majority of patients presenting over the age of 40 will have type 2 diabetes, but do not miss the occasional type 1 patient presenting late. An approach to their management is illustrated in Figure 20.9. Goals of treatment are described on page 1016. Type 2 diabetes is characterized by progressive beta-cell failure, and glucose control deteriorates over time, requiring a progressive and pre-emptive escalation of diabetes therapy. Regular review is essential for this to be achieved. Most patients on tablets will eventually require insulin, and it is helpful to explain this from the outset. The most widespread error in management at this stage is procrastination; the patient whose control is inadequate on oral therapy should start insulin without undue delay. Targets for glucose control are discussed later (p. 1016).
There is little consensus regarding the optimal insulin regimen in type 2 diabetes, but an intermediate insulin given at night with metformin during the day is initially as effective as multidose insulin regimens in controlling glucose levels, and is less likely to promote weight gain, which is a common complication of insulin therapy. Metformin is a useful adjunct to insulin in those able to tolerate it. Addition of a morning dose of insulin may become necessary to control postprandial hyperglycaemia. Twice-daily injections of pre-mixed soluble and isophane insulins (i.e. biphasic isophane insulin) are widely used and reasonably effective (Fig. 20.10a). More aggressive treatment, with multiple injections or continuous infusion pumps, is increasingly used in younger patients with type 2 diabetes.
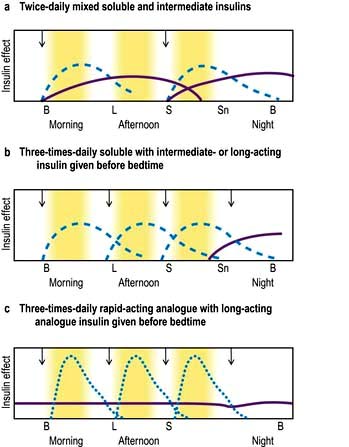
Principles of insulin treatment
Insulin administration
In healthy individuals a sharp increase in insulin occurs after meals; this is superimposed on a constant background of secretion (Fig. 20.7). Insulin therapy attempts to reproduce this pattern, but ideal control is difficult to achieve for four reasons:
 In normal subjects, insulin is secreted directly into the portal circulation and reaches the liver in high concentration; about 50% of the insulin produced by the pancreas is cleared by the liver. By contrast, insulin injected subcutaneously passes into the systemic circulation before passage to the liver. Insulin-treated patients therefore have lower portal levels of insulin and higher systemic levels relative to the physiological situation.
In normal subjects, insulin is secreted directly into the portal circulation and reaches the liver in high concentration; about 50% of the insulin produced by the pancreas is cleared by the liver. By contrast, insulin injected subcutaneously passes into the systemic circulation before passage to the liver. Insulin-treated patients therefore have lower portal levels of insulin and higher systemic levels relative to the physiological situation.

 The absorption of subcutaneous insulin into the circulation is variable.
The absorption of subcutaneous insulin into the circulation is variable.

A multiple injection regimen with short-acting insulin and a longer-acting insulin at night is appropriate for most younger patients (Fig. 20.10b). The advantages of multiple injection regimens are that the insulin and the food go in at roughly the same time so that meal times and sizes can vary, without greatly disturbing metabolic control. The flexibility of multiple injection regimens is of great value to patients with busy jobs, shift workers and those who travel regularly. Some recovery of endogenous insulin secretion may occur over the first few months (the ‘honeymoon period’) in type 1 patients and the insulin dose may need to be reduced or even stopped for a period. Requirements rise thereafter. Strict glucose control from diagnosis in type 1 diabetes prolongs beta-cell function, resulting in better glucose levels and less hypoglycaemia. Target blood glucose values should normally be 4–7 mmol/L before meals and 4–10 mmol/L after meals, assuming that this can be achieved without troublesome hypoglycaemia.
All patients need careful training for a life with insulin. This is best achieved outside hospital, provided that adequate facilities exist for outpatient diabetes education. A scheme for adjusting insulin regimens is given in Table 20.7. DAFNE is described on page 1010.
Table 20.7 Guide to adjusting insulin dosage according to blood glucose test results
| Blood glucose persistently too high | Blood glucose persistently too low | |
|---|---|---|
|
Before breakfast |
Increase evening long-acting insulin |
Reduce evening long-acting insulin |
|
Before lunch |
Increase morning short-acting insulin |
Reduce morning short-acting insulin or increase mid-morning snack |
|
Before evening meal |
Increase morning long-acting insulin or lunch short-acting insulin |
Reduce morning long-acting insulin or lunch short-acting insulin or increase mid-afternoon snack |
|
Before bed |
Increase evening short-acting insulin |
Reduce evening short-acting insulin |
When to use insulin analogues
Hypoglycaemia between meals and particularly at night is the limiting factor for many patients on multiple injection regimens. The more expensive rapid-acting insulin analogues (Fig. 20.10c) are a useful substitute for soluble insulin in some patients. They reduce the frequency of nocturnal hypoglycaemia due to reduced carry-over effect from the day-time. They are often used on grounds of convenience, since patients can inject shortly before meals but standard insulins injected at the same time give equivalent overall control. High or erratic morning blood sugar readings can prove a problem for about a quarter of all patients on conventional multiple injection regimens, because the bedtime intermediate-acting insulin falls and the absorption is variable. The long-acting insulin analogues insulin glargine and insulin detemir may help to overcome these problems and reduce the risk of nocturnal hypoglycaemia.
Complications of insulin therapy
The most common cause of mild insulin resistance is obesity. Occasional unstable patients require massive insulin doses, sometimes with a fluctuating requirement. Rare syndromes of insulin resistance may be present, but most cases are unexplained. Insulin resistance associated with antibodies directed against the insulin receptor has been reported in patients with acanthosis nigricans (Table 20.3).
Hypoglycaemia during insulin treatment
 Checking that a bedtime snack is taken regularly
Checking that a bedtime snack is taken regularly


 Changing to a rapid-acting insulin analogue, with a long-lasting insulin analogue at night.
Changing to a rapid-acting insulin analogue, with a long-lasting insulin analogue at night.

Measuring the metabolic control of diabetes
Urine tests



Home blood glucose testing
Blood tests offer invaluable feedback to everyone affected by diabetes, but their routine use varies according to need. Patients soon learn to provide their own profiles using finger-prick blood samples and reagent strips which can be read with the aid of a meter. Blood is taken from the side of a finger tip (not from the tip, which is densely innervated) using a special lancet usually fitted to a spring-loaded device, a range of which are available. The fasting blood glucose concentration is reproducible in diet or tablet-treated type 2 diabetes, and is therefore a useful guide to therapy, supplemented by occasional tests after meals. Those on insulin treatment require more frequent testing in order to adjust their therapy and avoid hypoglycaemia. Regular profiles (e.g. four daily samples on at least two days each week) are needed in those on intensified therapy, and should be recorded electronically or in a record book. Patients on insulin are encouraged to adjust their insulin dose as appropriate (Table 20.7) and should be able to obtain advice over the telephone when needed. Blood glucose monitoring does not in itself result in better control, but is essential to those wishing to achieve it.
Targets for glucose control
Data from the UK Prospective Diabetic Study (UKPDS) and the Diabetes Control and Complications Trial (DCCT) suggest that patients with both type 1 diabetes and type 2 diabetes should ideally aim to run their glycosylated haemoglobin readings below 7.0% (53 mmol/mol) in order to reduce the risk of long-term microvascular complications (Table 20.8). Hypoglycaemia, patterns of eating and lifestyle, weight problems and problems accepting and coping with diabetes limit what can be achieved (see p. 1017). Some will, but most will not, be able to reach these target values, particularly as their duration of diabetes increases. Realistic goals should be set for each patient, taking into account what is likely to be achievable, and this applies in particular to elderly patients and those with a limited prognosis.
Table 20.8 Target goals of risk factors for diabetic patients
| Parameter | Ideal | Reasonable but not ideal |
|---|---|---|
|
HbA1c |
<7% (53 mmol/mol) |
<8% (64 mmol/mol) |
|
Blood pressure (mmHg) |
<130/80 |
<140/80 |
|
Total cholesterol (mmol/L) |
<4.0 |
<5.0 |
|
LDL |
<2.0 |
<3.0 |
|
HDL* |
>1.1 |
>0.8 |
|
Triglycerides |
<1.7 |
<2.0 |
Standards from American Diabetes Association (2003).
Regular checks for patients with diabetes
Box 20.3 is modified from the guidelines set out in The European Patients’ Charter published by the St Vincent Declaration Steering Committee of the WHO. The charter sets out goals for both the healthcare team and the patient.
 Box 20.3
Box 20.3
Regular checks for patients with diabetes




Checked at least once a year
 Biochemical assessment of metabolic control (e.g. glycosylated Hb test)
Biochemical assessment of metabolic control (e.g. glycosylated Hb test)


 Measure plasma lipids (except in extreme old age)
Measure plasma lipids (except in extreme old age)

 Examine state of retina (ophthalmoscope or retinal photo)
Examine state of retina (ophthalmoscope or retinal photo)
 Test urine for proteinuria/microalbuminuria
Test urine for proteinuria/microalbuminuria
 Test blood for renal function (creatinine, eGFR)
Test blood for renal function (creatinine, eGFR)
 Check condition of feet, pulses and neurology
Check condition of feet, pulses and neurology
 Review cardiovascular risk factors
Review cardiovascular risk factors


FURTHER READING
DCCT/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive insulin therapy. N Engl J Med 2000; 342:381–389.
Holman RR, Paul SK, Bethel MA et al. Long-term follow-up after tight control of blood pressure in type 2 diabetes. N Engl J Med 2008; 359:1565–1576.
Holman RR, Paul SK, Bethel MA et al. 10 year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetics. Lancet 2008; 371:1800–1809.
The ACCORD Study Group. Long-term effects of intensive glucose lowering on cardiovascular outcomes. N Engl J Med 2011; 364:818–828.
Psychosocial implications of diabetes
 You cannot take a ‘holiday’ from diabetes – yet the human psyche is poorly developed to cope with unremitting adversity.
You cannot take a ‘holiday’ from diabetes – yet the human psyche is poorly developed to cope with unremitting adversity.











Diabetic metabolic emergencies
|
Ketonuria |
Detectable ketone levels in the urine; it should be appreciated that ketonuria occurs in fasted non-diabetics and may be found in relatively well-controlled patients with insulin-dependent diabetes mellitus |
|
Ketosis |
Elevated plasma ketone levels in the absence of acidosis |
|
Diabetic ketoacidosis |
A metabolic emergency in which hyperglycaemia is associated with a metabolic acidosis due to greatly raised (>5 mmol/L) ketone levels |
|
Hyperosmolar hyperglycaemic state |
A metabolic emergency in which uncontrolled hyperglycaemia induces a hyperosmolar state in the absence of significant ketosis |
|
Lactic acidosis |
A metabolic emergency in which elevated lactic acid levels induce a metabolic acidosis. In diabetic patients it is rare and associated with biguanide therapy |
Diabetic ketoacidosis



Pathogenesis
Ketoacidosis is a state of uncontrolled catabolism associated with insulin deficiency. Insulin deficiency is a necessary precondition since only a modest elevation in insulin levels is sufficient to inhibit hepatic ketogenesis, and stable patients do not readily develop ketoacidosis when insulin is withdrawn. Other factors include counter-regulatory hormone excess and fluid depletion. The combination of insulin deficiency with excess of its hormonal antagonists leads to the parallel processes shown in Figure 20.11. In the absence of insulin, hepatic glucose production accelerates, and peripheral uptake by tissues such as muscle is reduced. Rising glucose levels lead to an osmotic diuresis, loss of fluid and electrolytes, and dehydration. Plasma osmolality rises and renal perfusion falls. In parallel, rapid lipolysis occurs, leading to elevated circulating free fatty-acid levels. The free fatty acids are broken down to fatty acyl-CoA within the liver cells, and this in turn is converted to ketone bodies within the mitochondria (Fig. 20.12). Accumulation of ketone bodies produces a metabolic acidosis. Vomiting leads to further loss of fluid and electrolytes. The excess ketones are excreted in the urine but also appear in the breath, producing a distinctive smell similar to that of acetone. Respiratory compensation for the acidosis leads to hyperventilation, graphically described as ‘air hunger’. Progressive dehydration impairs renal excretion of hydrogen ions and ketones, aggravating the acidosis. As the pH falls below 7.0 ([H+] >100 nmol/L), pH-dependent enzyme systems in many cells function less effectively. Untreated, severe ketoacidosis is invariably fatal.
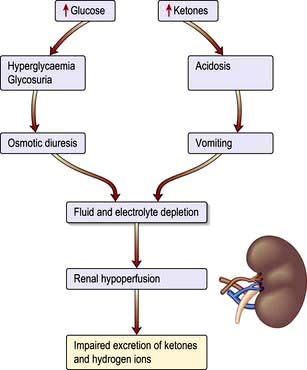
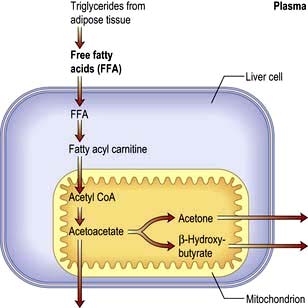
Diagnosis
The severity of DKA can be assessed as follows. One or more suggest severe DKA.








Management (pathophysiology)
The principles of management are as follows (Emergency Box 20.1); these should be carried out in a high dependency area.
 Replace the fluid losses with 0.9% saline. Average loss of water is 5–7 litres with a sodium loss of 500 mmol.
Replace the fluid losses with 0.9% saline. Average loss of water is 5–7 litres with a sodium loss of 500 mmol.





 Emergency Box 20.1
Emergency Box 20.1
Guidelines for the diagnosis and management of diabetic ketoacidosis










Phase 1





















Problems of management
 Hypotension. This may lead to renal shutdown. Plasma expanders (or whole blood) are therefore given if the systolic blood pressure is below 80 mmHg. A central venous pressure line is useful in this situation. A bladder catheter is inserted if no urine is produced within 2 h, but routine catheterization is not necessary.
Hypotension. This may lead to renal shutdown. Plasma expanders (or whole blood) are therefore given if the systolic blood pressure is below 80 mmHg. A central venous pressure line is useful in this situation. A bladder catheter is inserted if no urine is produced within 2 h, but routine catheterization is not necessary.
 Coma. The usual principles apply (see p. 1093). It is essential to pass a nasogastric tube to prevent aspiration, since gastric stasis is common and carries the risk of aspiration pneumonia if a drowsy patient vomits.
Coma. The usual principles apply (see p. 1093). It is essential to pass a nasogastric tube to prevent aspiration, since gastric stasis is common and carries the risk of aspiration pneumonia if a drowsy patient vomits.


 Late complications. These include pneumonia and deep-vein thrombosis (DVT prophylaxis, see p. 429 is essential) and occur especially in the comatose or elderly patient.
Late complications. These include pneumonia and deep-vein thrombosis (DVT prophylaxis, see p. 429 is essential) and occur especially in the comatose or elderly patient.

Hyperosmolar hyperglycaemic state
This condition, in which severe hyperglycaemia develops without significant ketosis, is the metabolic emergency characteristic of uncontrolled type 2 diabetes. Patients present in middle or later life, often with previously undiagnosed diabetes. Common precipitating factors include consumption of glucose-rich fluids, concurrent medication such as thiazide diuretics or steroids, and intercurrent illness. The hyperosmolar hyperglycaemic state and ketoacidosis represent two ends of a spectrum rather than two distinct disorders (Box 20.4). The biochemical differences may partly be explained as follows:
 Age. The extreme dehydration characteristic of hyperosmolar hyperglycaemic state may be related to age. Old people experience thirst less acutely, and more readily become dehydrated. In addition, the mild renal impairment associated with age results in increased urinary losses of fluid and electrolytes.
Age. The extreme dehydration characteristic of hyperosmolar hyperglycaemic state may be related to age. Old people experience thirst less acutely, and more readily become dehydrated. In addition, the mild renal impairment associated with age results in increased urinary losses of fluid and electrolytes.

 Box 20.4
Box 20.4
Electrolyte changes in diabetic ketoacidosis and the hyperosmolar hyperglycaemic state
Examples of blood values
| Severe ketoacidosis | Hyperosmolar hyperglycaemic state | |
|---|---|---|
|
Na+ (mmol/L) |
140 |
155 |
|
K+ (mmol/L) |
5 |
5 |
|
Cl− (mmol/L) |
100 |
110 |
|
HCO3− (mmol/L) |
5 |
25 |
|
Urea (mmol/L) |
8 |
15 |
|
Glucose (mmol/L) |
30 |
50 |
|
Arterial pH |
7.0 |
7.35 |
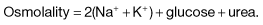
For instance, in the example of severe ketoacidosis given above:
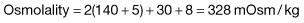
and in the example of the hyperosmolar hyperglycaemic state:
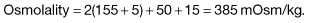
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 20 Diabetes mellitus and other disorders of metabolism
Diabetes mellitus
Hyperglycaemia, insulin and insulin action
Insulin structure and secretion
Insulin is the key hormone involved in the storage and controlled release within the body of the chemical energy available from food. It is coded for on chromosome 11 and synthesized in the beta cells of the pancreatic islets (Fig. 20.1). The synthesis, intracellular processing and secretion of insulin by the beta cell is typical of the way that the body produces and manipulates many peptide hormones. Figure 20.2 illustrates the cellular events triggering the release of insulin-containing granules. After secretion, insulin enters the portal circulation and is carried to the liver, its prime target organ. About 50% of secreted insulin is extracted and degraded in the liver; the residue is broken down by the kidneys. C-peptide is only partially extracted by the liver (and hence provides a useful index of the rate of insulin secretion) but is mainly degraded by the kidneys.
An outline of glucose metabolism
Insulin is a major regulator of intermediary metabolism, although its actions are modified in many respects by other hormones. Its actions in the fasting and postprandial states differ (Fig. 20.3). In the fasting state, its main action is to regulate glucose release by the liver, and in the postprandial state, it additionally promotes glucose uptake by fat and muscle. The effect of counter-regulatory hormones (glucagon, epinephrine (adrenaline), cortisol and growth hormone) is to cause greater production of glucose from the liver and less utilization of glucose in fat and muscle for a given level of insulin.
GLUT-1 – enables basal non-insulin-stimulated glucose uptake into many cells (see Fig. 6.29).
GLUT-3 – enables non-insulin-mediated glucose uptake into brain neurones and placenta.
GLUT-4 – enables much of the peripheral action of insulin. It is the channel through which glucose is taken up into muscle and adipose tissue cells following stimulation of the insulin receptor (Fig. 20.4).
This is a glycoprotein (400 kDa), coded for on the short arm of chromosome 19, which straddles the cell membrane of many cells (Fig. 20.4). It consists of a dimer with two α-subunits, which include the binding sites for insulin, and two β-subunits, which traverse the cell membrane. When insulin binds to the α-subunits it induces a conformational change in the β-subunits, resulting in activation of tyrosine kinase and initiation of a cascade response involving a host of other intracellular substrates. One consequence of this is migration of the GLUT-4 glucose transporter to the cell surface and increased transport of glucose into the cell. The insulin-receptor complex is then internalized by the cell, insulin is degraded, and the receptor is recycled to the cell surface.
Classification of diabetes
Diabetes may be primary (idiopathic) or secondary (Table 20.1). Primary diabetes is classified into:
Table 20.1 Aetiological classification of diabetes mellitus, based on classification by the American Diabetes Association (ADA)
Note: Patients with any form of diabetes may require insulin treatment at some stage of their disease. Such use of insulin does not, of itself, classify the patient.
(Adapted from ADA. Diagnosis and classification of diabetes mellitus. Diabetes Care 2008; 31(Suppl 1):S55–S60.)
The key clinical features of the two main forms of diabetes are listed in Table 20.2. Type 1 and type 2 diabetes represent two distinct diseases from the epidemiological point of view, but clinical distinction can sometimes be difficult. The two diseases should from a clinical point of view be seen as a spectrum, distinct at the two ends but overlapping to some extent in the middle. Hybrid forms are increasingly recognized, and patients with immune-mediated diabetes (type 1) may, for example, also be overweight and insulin resistant. This is sometimes referred to as ‘double diabetes’. It is more relevant to give the patient the right treatment on clinical grounds than to worry about how to label their diabetes. The classification of primary diabetes continues to evolve. Monogenic forms have been identified (see p. 1007), in some cases with significant therapeutic implications. Although secondary diabetes accounts for barely 1–2% of all new cases at presentation, it should not be missed because the cause can sometimes be treated. All forms of diabetes derive from inadequate insulin secretion relative to the needs of the body, and progressive insulin secretory failure is characteristic of both common forms of diabetes. Thus, some patients with immune-mediated diabetes type 1 may not at first require insulin, whereas many with type 2 diabetes will eventually do so.
Table 20.2 The spectrum of diabetes: a comparison of type 1 and type 2 diabetes mellitus
| Type 1 | Type 2 | |
|---|---|---|
|
Age |
Younger (usually <30) |
Older (usually >30) |
|
Weight |
Lean |
Overweight |
|
Symptom duration |
Weeks |
Months/years |
|
Higher risk ethnicity |
Northern European |
Asian, African, Polynesian and American-Indian |
|
Seasonal onset |
Yes |
No |
|
Heredity |
HLA-DR3 or DR4 in >90% |
No HLA links |
|
Pathogenesis |
Autoimmune disease |
No immune disturbance |
|
Ketonuria |
Yes |
No |
|
Clinical |
Insulin deficiency |
Partial insulin deficiency initially |
|
|
± ketoacidosis |
± hyperosmolar state |
|
|
Always need insulin |
Need insulin when beta cells fail over time |
|
Biochemical |
C-peptide disappears |
C-peptide persists |
Type 1 diabetes mellitus
Epidemiology
Type 1 diabetes is a disease of insulin deficiency. In western countries almost all patients have the immune-mediated form of the disease, otherwise known as type 1A. Type 1 diabetes is a disease of childhood, reaching a peak incidence around the time of puberty, but can present at any age. A ‘slow-burning’ variant with slower progression to insulin deficiency occurs in later life and is sometimes called latent autoimmune diabetes in adults (LADA). LADA may be difficult to distinguish from type 2 diabetes. Clinical clues are: leaner build, rapid progression to insulin therapy following an initial response to other therapies, and the presence of circulating islet autoantibodies. The highest rates of type 1 diabetes in the world are seen in Finland and other Northern European countries, and on the island of Sardinia, which for unknown reasons, has the second highest rate in the world (Fig. 20.5). The incidence of type 1 diabetes appears to be increasing in most populations. In Europe, the annual increase is of the order of 2–3%, and is most marked in children under the age of 5 years. WHO estimated in 1995 that there were 19.4 million people with type 1 diabetes and that the number will rise to 57.2 million by 2025.
Causes
Autoimmunity and type 1 diabetes
Type 1 diabetes is associated with other organ-specific autoimmune diseases including autoimmune thyroid disease, coeliac disease, Addison’s disease and pernicious anaemia. Autopsies of patients who died following diagnosis of type 1 diabetes show infiltration of the pancreatic islets by mononuclear cells. This appearance, known as insulitis, resembles that in other autoimmune diseases such as thyroiditis. Several islet antigens have been characterized, and these include insulin itself, the enzyme glutamic acid decarboxylase (GAD), protein tyrosine phosphatase (IA-2) (Fig. 20.6) and the cation transporter ZnT8. Recent studies have shown that GAD immunotherapy has no benefit. The observation that treatment with immunosuppressive agents such as ciclosporin prolongs beta-cell survival in newly diagnosed patients has confirmed that the disease is immune-mediated.
Environmental factors
The incidence of childhood type 1 diabetes is rising across Europe at the rate of 2–3% each year, suggesting that environmental factor(s) are involved in its pathogenesis. Islet autoantibodies (see above) appear in the first few years of life, indicating prenatal or early postnatal interactions with the environment. Exposures to dietary constituents, enteroviruses such as Coxsackie B4 and relative deficiency of vitamin D are possible candidates, but their role in the causation of the disease has yet to be confirmed. A cleaner environment with less early stimulation of the immune system in childhood may increase susceptibility for type 1 diabetes, as for atopic/allergic conditions (the hygiene hypothesis) (see p. 824), and more rapid weight gain in childhood and adolescence leading to increased insulin resistance might accelerate clinical onset (the accelerator hypothesis).
Type 2 diabetes mellitus
Epidemiology
Type 2 diabetes is associated with central obesity, hypertension, hypertriglyceridaemia, a decreased HDL-cholesterol, disturbed haemostatic variables and modest increases in a number of pro-inflammatory markers. Insulin resistance is strongly associated with many of these variables, as is increased cardiovascular risk. This group of conditions is referred to as the metabolic syndrome (see p. 223). The International Diabetes Federation has proposed criteria based on increased waist circumference (or BMI >30) plus two of the following: diabetes (or fasting glucose >6.0 mmol/L), hypertension, raised triglycerides or low HDL cholesterol. On this definition, about one-third of the adult population has features of the syndrome, not necessarily associated with diabetes. Critics would argue that the metabolic syndrome is not a distinct entity, but one end of a continuum in the relationship between exercise, lifestyle and bodyweight on the one hand, and genetic make-up on the other, and that diagnosis adds little to standard clinical practice in terms of diagnosis, prognosis or therapy.
Causes
Abnormalities of insulin secretion and action
The relative role of secretory failure versus insulin resistance in the pathogenesis of type 2 diabetes has been much debated, but even massively obese individuals with a fully functioning beta-cell mass do not necessarily develop diabetes, which implies that some degree of beta-cell dysfunction is necessary. Insulin binds normally to its receptor on the surface of cells in type 2 diabetes, and the mechanisms of ‘insulin resistance’ are still poorly understood. Insulin resistance is, however, associated with central obesity and accumulation of intracellular triglyceride in muscle and liver in type 2 diabetes, and a high proportion of patients have non-alcoholic fatty liver disease (NAFLD), see page 303. It has long been stated that patients with type 2 diabetes retain up to 50% of their beta-cell mass at the time of diagnosis, as compared with healthy controls, but the shortfall is greater than this when they are matched with healthy individuals who are equally obese. In addition, patients with type 2 diabetes almost all show islet amyloid deposition at autopsy, derived from a peptide known as amylin or islet amyloid polypeptide (IAPP), which is co-secreted with insulin. It is not known if this is a cause or consequence of beta-cell secretory failure.
Monogenic diabetes mellitus
The genetic causes of some rare forms of diabetes are shown in Table 20.3. Considerable progress has been made in understanding these rare variants of diabetes. Genetic defects of beta-cell function (previously called ‘maturity-onset diabetes of the young’, MODY) are dominantly inherited, and several variants have been described, each associated with different clinical phenotypes (Table 20.4). These should be considered in people presenting with early-onset diabetes in association with an affected parent and early-onset diabetes in ~50% of relatives. They can often be treated with a sulfonylurea.
| Disorder | Features |
|---|---|
|
Insulin receptor mutations |
Obesity, marked insulin resistance, hyperandrogenism in women, acanthosis nigricans (areas of hyperpigmented skin) |
|
Maternally inherited diabetes and deafness (MIDD) |
Mutation in mitochondrial DNA. Diabetes onset before age 40. Variable deafness, neuromuscular and cardiac problems, pigmented retinopathy |
|
Wolfram’s syndrome (DIDMOAD – diabetes insipidus, diabetes mellitus, optic atrophy and deafness) |
Recessively inherited. Mutation in the transmembrane gene, WFS1. Insulin-requiring diabetes and optic atrophy in the first decade. Diabetes insipidus and sensorineural deafness in the second decade progressing to multiple neurological problems. Few live beyond middle age |
|
Severe obesity and diabetes |
Alström’s, Bardet–Biedl and Prader–Willi syndromes. Retinitis pigmentosa, mental insufficiency and neurological disorders |
|
Disorders of intracellular insulin signalling. All with severe insulin resistance |
Leprechaunism, Rabson–Mendenhall syndrome, pseudoacromegaly, partial lipodystrophy: lamin A/C gene mutation |
|
Genetic defects of beta-cell function |
See Table 20.4 |
FURTHER READING
Chan JC, Malik V, Jia W et al. Diabetes in Asia: epidemiology, risk factors and pathophysiology. JAMA 2009; 301:2129–2140.
Ramachandran A, Ma RC, Snehalatha C. Diabetes in Asia. Lancet 2010; 375:408–418.
Sturnvoll M, Goldstein BJ, van Haefken TW. Type 2 diabetes; pathogenesis and treatment. Lancet 2008; 371:2153–2156.
Clinical presentation of diabetes
Presentation may be acute, subacute or asymptomatic.
Acute presentation
Young people often present with a 2–6-week history and report the classic triad of symptoms:
Asymptomatic diabetes
Physical examination at diagnosis
Evidence of weight loss and dehydration may be present, and the breath may smell of ketones. Older patients may present with established complications, and the presence of the characteristic retinopathy is diagnostic of diabetes. In occasional patients, there will be physical signs of an illness causing secondary diabetes (see Table 20.1). Patients with severe insulin resistance may have acanthosis nigricans, which is characterized by blackish pigmentation at the nape of the neck and in the axillae (p. 1217).
