CHAPTER 49 DEVELOPMENTAL DEFECTS AND PATHOPHYSIOLOGY
Developmental defects are among the most frequent causes of epilepsy, particularly of refractory epilepsy. They constitute a very broad range of pathological processes occurring during brain development. Each step of brain maturation, from neurogenesis to cortical organization, may be affected, which leads to several cortical malformations and/or molecular/cellular defects affecting synapses, neurotransmitter receptors, or ion channels. Advances in research, mainly in the fields of neuroimaging, electrophysiology, and genetics, have yielded a better understanding of the pathophysiology of these developmental epilepsies. This chapter focuses on malformations caused by abnormalities of cortical development (MCDs) responsible for epilepsy. Several aspects are covered: (1) definition and classification, (2) neurogenetics, (3) neuroimaging, and (4) electrophysiological-clinical data and relevance to epilepsy surgery. Each section highlights specific data of relevance to the pathophysiology of developmental defects associated with epilepsy.
DEFINITION AND CLASSIFICATION
MCDs have become more easily recognized in vivo since the 1980s because of technical improvements in magnetic resonance imaging (MRI). The term malformations caused by abnormalities of cortical development1 encompasses many forms of developmental defect resulting in architectural alteration of the cerebral cortex with or without abnormal cells (neuron and/or aberrant cells). MCDs are also referred to as disorders of cortical development,2 cortical dysplasias, and cortical dysgenesis. However, malformations caused by abnormalities of cortical development appears to us to be the most useful label.
MCDs comprise all architectural abnormalities occurring during the different processes involved in cortical formation that can be schematically divided into three overlapping steps (which are themselves not temporally separated): (1) cell proliferation, differentiation, and apoptosis; (2) neuronal migration; and (3) cortical organization. This scheme was used by Barkovich and colleagues to provide one of the most used classifications of MCD (Table 49-1).3 Other classifications based on either imaging aspects or etiology have also been proposed.4 However, the principal advantage of the Barkovich classification is that it incorporates several processes that combine to generate the complexity of clinical manifestations of MCDs, including embryological, genetic, anatomical, and neuropathological factors.
TABLE 49-1 Malformations Caused by Abnormalities of Cortical Development (MCDs): Classification Scheme
* Classically associated with epilepsy.
Adapted from Barkovich AJ, Kuzniecky RI, Jackson GD, et al: Classification system for malformations of cortical development: update 2001. Neurology 2001 57:2168-2178.
This chapter focuses on MCD as a cause of localization-related refractory epilepsy, excluding clinical manifestations with severe mental retardation. Therefore, only the following focal forms of MCD are discussed in this chapter: FCDs, cortical hamartomas of tuberous sclerosis, neoplastic MCD (i.e., dysembryoplastic neuroepithelial tumors [DNETs], ganglioglioma, and gangliocytoma), subcortical band heterotopia, periventricular (subependymal) and subcortical heterotopia, polymicrogyria and schizencephaly, and mild MCD (replacing the term microdysgenesis; see later discussion). Drug-resistant partial epilepsies associated with MCD represent a critical issue in pediatric and adult clinical neurology. In such cases, modern surgical approaches to epilepsy may represent a unique and curative solution for patients. A careful comprehensive presurgical assessment is required and must take into account all aspects of the epilepsy, from lesion to seizure phenomena. Recognition of MCD by magnetic resonance techniques has dramatically modified presurgical assessment. However, two critical issues remain: the precise localization of an epileptogenic zone responsible for seizure onset and the relationship between MRI-defined lesions and the epileptogenic zone, which may be complex.5 Moreover, up to 20% of MRI scans appear normal on visual inspection in such patients.6
No specific epidemiological studies on the prevalence of MCD are available in the literature; therefore, the only estimates of the proportion of each MCD subtype come from specialized tertiary centers.7,8 Nevertheless, it appears that FCDs are the principal cause of MCDs responsible for drug-resistant partial epilepsy that is potentially treatable by surgery. Therefore, since the first description of surgical specimens from epileptic patients by Taylor and colleagues in 1971,9 FCD has been of interest to physicians and researchers working with epilepsy surgery. Today, it is well recognized that FCD is not a homogeneous entity but exhibits different histopathological features with variable cytological components and degrees of architectural disruption. This observation implies a range of genetic and molecular mechanisms and variable alterations in cortical connectivity. This variability affects the visibility of FCD on MRI scans. However, from such diversity, it is possible to describe correlations between histopathological and MRI features and, to some extent, clinical electrophysiological features.10,11 Thus, Palmini and colleagues proposed a specific classification of FCDs that is clinically particularly useful (Table 49-2).12,13
TABLE 49-2 Focal Cortical Dysplasia (FCD): Classification Scheme
MCD, malformation caused by abnormalities of cortical development; MRI, magnetic resonance imaging.
Adapted from Palmini A, Najm I, Avanzini G, et al: Terminology and classification of the cortical dysplasias. Neurology 2004 62(6, Suppl 3):S2-S8.
In the same report, the result of a panel discussion between epileptologists, neuroradiologists, and neuropathologists specializing in the field, Palmini and colleagues12 also brought clarity to the concept of microdysgenesis. This category of MCD is important because it represents another frequent cause of drug-resistant partial epilepsy in which MRI scans are normal.14 The term microdysgenesis has been used to describe microscopic changes that constitute cortical laminar disorganization; abnormal cortical myelinated fibers; neuronal clustering; and heterotopic or excessively numerous neurons in white matter, subcortical areas, or cortical layer I. Today, the term mild MCD is preferred to describe such microscopic histopathological changes. Palmini and colleagues classified mild MCD into type I (with ectopically placed neurons in or adjacent to layer I) and type II (with microscopic neuronal heterotopia outside layer I).
NEUROGENETICS
Since the first reports of familial cases of lissencephaly and subcortical heterotopia2 in the early 1990s, the genetic basis of MCD has been increasingly recognized.15,16 Like many developmental neurological disorders, MCD is the result of a combination of genetic defect and gestational environmental insult.17 Thus, although genetic discoveries have brought new insights and a better understanding of the causes of MCD, they have also revealed more complexity as concerns an understanding of pathophysiology. Indeed, the same mutation may lead to different types of MCD, and conversely, the same phenotype may be linked to different mutations.7
Lissencephaly and subcortical band heterotopia are the first MCDs for which a genetic basis was found. However, these MCDs are not commonly associated with epilepsy. They represent a spectrum of abnormalities, and both can be encountered in the same families. Two genes have been discovered: LIS118 on chromosome 17 and DCX19 on chromosome X. LIS1 mutations can lead to isolated lissencephaly or a more severe phenotype (Miller-Dieker syndrome) in the case of heterozygous deletions. DCX mutations can lead to lissencephaly in boys, subcortical band heterotopia in girls, or mixed phenotypes.
Several familial forms of polymicrogyria have been linked to a number of genes, mainly on chromosome X.20,21 Familial schizencephaly linked to a mutation within the EMX2 gene has also been reported.21
Finally, one report linked a mutation of the TSC1 gene to molecular defects associated with FCD.22
NEUROIMAGING
Conventional Magnetic Resonance Imaging
Focal Cortical Dysplasia
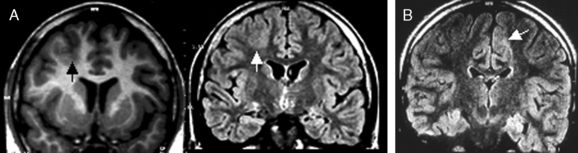
Not all the subtypes of FCD (Fig. 49-1) can be identified on MRI. Type I FCD is almost always invisible on MRI and is usually discovered on neuropathological examination after surgery. The most commonly identified lesions on MRI are those associated with type II (Taylor-type) FCD. Several features can be observed: focal cortical thickening, blurring of the junction between gray and white matter, increased signal intensity on T2-weighted, proton density, or fluid-attenuated inversion recovery imaging (FLAIR), classically linked to the balloon cell content of the FCD (type IIB) and extension of this hypersignal from the cortex to the ventricle (also called transmantle dysplasia in the literature).
Hamartomas of the Tuberous Sclerosis Complex
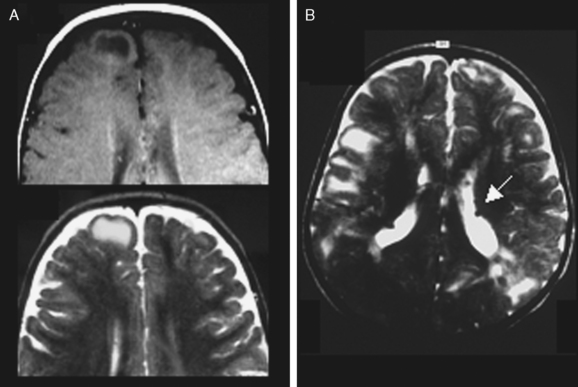
Typical cortical hamartomas (Fig. 49-2) potentially responsible for refractory epilepsy are usually called tubers and may mimic some of the features of FCD. However, they do not show regular cortical thickening or transmantle hypersignal spreading to the ventricle. The more typical findings are cortical or subcortical hyperintensity on T2-weighted images. Tubers are generally associated with other characteristic hamartomous lesions such as subependymal nodules and subependymal giant cell astrocytomas. They may also be calcified, and signal appearances may change with age.
Neoplastic MCDs
Dysembryoplastic neuroepithelial tumors
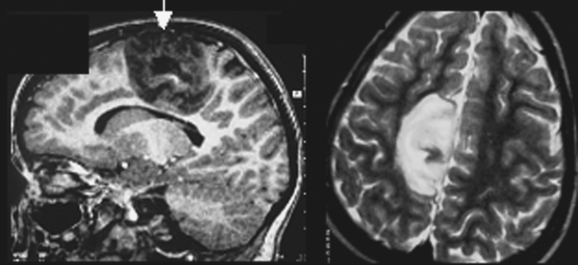
DNETs (Fig. 49-3) are nonmalignant neoplastic malformations in cortical “locations.” DNETs can share MRI features with FCD (cortical thickening, T2 hyperintensity) but are found more often in the temporal lobes. They are, as in the case of tubers, usually well circumscribed, without blurring of the junction between the gray and white matter, although they may involve the white matter. A typical feature is molding of the overlying skull. Calcification, best seen on computed tomographic scan, and cystic components may be observed. Enhancement with contrast material is rare. DNETs must be differentiated from low-grade gliomas.
Ganglioglioma
Gangliogliomas (Fig. 49-4) are difficult to differentiate from DNETs, although greater enhancement with contrast material is generally seen in gangliogliomas. The presence of T2 hyperintensity, cysts, and high-contrast enhancement and the lack of perifocal edema may suggest a low-grade or pilocytic astrocytoma. Superficial enhancement extending to the leptomeninges may likewise suggest the diagnosis of a pleomorphic xanthoastrocytoma.
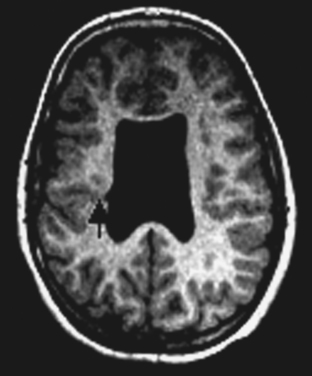
Subcortical Band Heterotopia
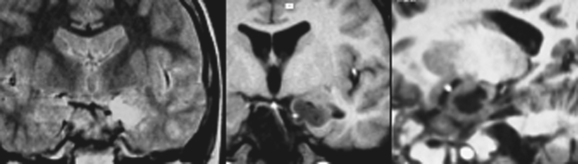
Subcortical band heterotopia (Fig. 49-5) manifests radiologically as a band of cortical tissue underlying the cortical surface from which it is separated by a thin strip of white matter. The band comprises normal appearing neurons with a signal similar to that of cortical gray matter. Band thickness may vary, as may position along the fronto-occipital axis, leading to a spectrum of type and severity of cognitive impairment.
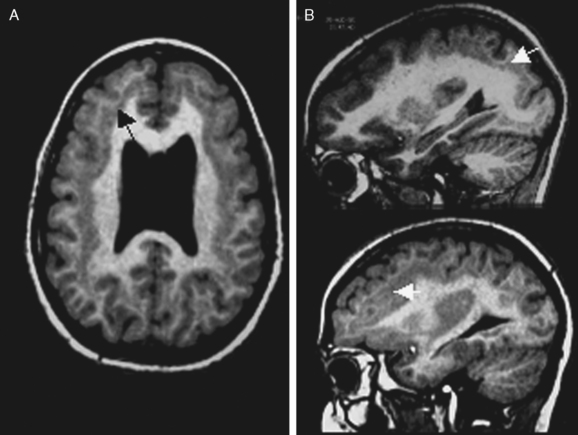
Polymicrogyria
Polymicrogyria (Fig. 49-6) is a gyration defect resulting in a cortex composed of microgyri with excessive folding. However, in thick MRI slices, the appearances of polymicrogyria may be confused with pachygyria, which resembles thick and smooth cortex. Thin slices reveal the excessively folded and thin cortex. This pattern is often bilateral (either symmetrical or asymmetrical) but may be unilateral and diffuse or well localized. Several morphoclinical forms have been described: bilateral perisylvian, frontal, and parieto-occipital.
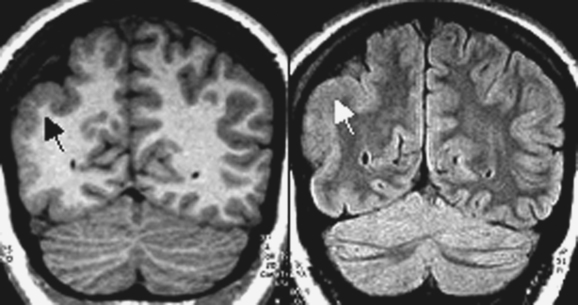
Schizencephaly
Schizencephaly (Fig. 49-7) manifests as a transcerebral cleft, with edges lined by cortical tissue, often associated with polymicrogyria along the cleft borders. Bilateral forms occur, and there is an association with other brain malformations of the septum, optic nerve, corpus callosum, cingulate cortex, or hippocampus.
Functional Imaging and Advanced Magnetic Resonance Imaging Techniques
Functional Imaging
Several imaging studies have investigated the potential functions of dysplastic cortex. This issue is of particular interest in relation to the importance of individual MCD-associated pathophysiology for surgical planning. Functional MRI studies with motor, visual, or cognitive tasks have shown that hemimegalencephaly, subcortical band heterotopia, periventricular heterotopia, FCD, and polymicrogyria may all be functionally active.23–25 These findings confirm those of previous positron emission tomographic studies, which demonstrated participation of MCD in functionally reorganized cortex.26 These findings suggest that careful mapping of cerebral functions before surgery is vital in cases of MCD located in functionally eloquent cortex.27
Diffusion Tensor Imaging and Tractography
Diffusion tensor imaging is a magnetic resonance technique in which the three-dimensional measurement of tissue water diffusion is used to infer both the integrity (quantification of diffusivity and anisotropy) and orientation of white matter axonal fibers in vivo (tractography). Parametric quantification of diffusivity and anisotropy has revealed structural abnormalities beyond visually defined structural lesions on conventional MRI, which indicates more widespread morphological changes than those visible with conventional scanning.28 Tractography has been used in a preliminary manner to study the architecture of white matter in MCD. It has been used to show normal-appearing connectivity of band heterotopia to underlying cortex—a result that concords with functionality demonstrated by functional MRI.29 Conversely, tractography has also demonstrated diffuse impairment of white matter connectivity patterns.30 Further research is needed to better define reorganization of the brain in terms of neural connectivity. This may lead to a better understanding of the pathophysiology of MCD, especially the anatomical substrate of neural networks implicated in epileptogenic zones, which may themselves be more widespread than conventional MRI visible lesions.
Morphometry
Several analytical techniques can quantify structural changes from T1-weighted images, including the respective amounts of white and gray matter present regionally. Such analyses have demonstrated both increased and decreased amounts of gray matter within and beyond visible lesions, which, again, shows that anatomical defects are more widespread than they appear on conventional structural scanning.31,32
Metabolic Imaging
Metabolic imaging by means of magnetic resonance spectroscopic imaging (MRSI) or positron emission tomography has also contributed to a better description of biochemical disturbances associated with MCD. MRSI shows differential patterns of metabolite changes (mainly affecting N-acetyl-aspartate, choline-containing compounds, and creatine-phosphocreatine ratios) according to type of MCD, and may be used to differentiate FCD and DNET from low-grade gliomas.33,34 Positron emission tomography with positron-labeled amino acids can differentiate DNET from other benign tumors associated with epilepsy, or differentiate active epileptogenic tubers from nonactive ones in tuberous sclerosis.35,36 MRSI and positron emission tomography (with one of the positron-labeled tracers flumazenil, serotonin, and glucose) are promising, and such techniques may provide a description of metabolic abnormalities linked to epileptiform discharges rather than lesions per se.37–39
ELECTROPHYSIOLOGICAL-CLINICAL DATA AND RELEVANCE TO EPILEPSY SURGERY
Electrophysiology
Animal Model Studies
Most animal models of MCD have been produced through genetic modification or environmental insults applied in utero to rodent embryos.40,41 Three models of focal cortical malformation are available: (1) a pharmacological model with methylazoxymethanol, (2) an irradiation model, and (3) a cold lesion model. These interventions usually result in heterotopia or microgyria. Thus, the models result in MCD with abnormal connectivity through aberrant circuitry.40 It is still a matter of debate whether heterotopic tissue secondarily triggers seizures that propagate to the cortex. It is more likely that more complex heterotopic-cortical neuronal networks may be involved in the generation of seizures.
Human In Vitro Studies
In vitro studies have provided fundamental research data on the pathophysiology of MCD in humans, with new insights particularly into type II FCD (which probably represents the most frequent cause of refractory partial epilepsy considered for surgery). Intrinsic epileptogenicity has now been well demonstrated by studies of FCD surgical specimens.2 Data support the idea that cytomegalic neurons rather than balloon cells are the generators of epileptogenic activity.42 However, on MRI scans, T2 hyperintensity probably results from the presence of balloon cells.13 Thus, there is a potential discrepancy between MRI-visible abnormalities and electrically discharging lesions. Surrounding normal tissue may also demonstrate hyperexcitability.
Several potential mechanisms of hyperexcitability have been demonstrated. They are mediated by cellular changes (loss of inhibitory GABAergic interneurones, increase of abnormally oriented pyramidal neurons exhibiting excitatory receptors) and molecular factors (N-methyl-D-aspartate–mediated excitatory mechanisms and GABAA receptor–mediated synchronicity) leading to epileptogenicity.43 With regard to the different forms of MCD, FCD is much more epileptogenic than nodular heterotopia, polymicrogyria, or schizencephaly.2
Human In Vivo Studies
Surface recordings
In all types of MCD classically associated with epilepsy, Raymond and colleagues found in a study of 100 patients no interictal electroencephalographic (EEG) abnormality in only 15% of cases.8 Generalized EEG features (synchronous and bilateral) were observed in 19%. Most of these patients had bilateral or diffuse MCD consisting of heterotopias, polymicrogyria, or a tuberous sclerosis complex. However, in 63 of 68 patients with focal or unilateral MCD, the EEG features were either focal or lateralized.
There are particular features described in FCD. Thus, focal rhythmic interictal discharges on electroencephalography are considered typical of FCD. They correspond to continuous epileptiform discharges recorded with electrocorticography.44 This feature is very significant for localization of the epileptogenic zone. Magnetoencephalography and electroencephalography with source localization of interictal spikes may also help localize the origin of epileptogenic processes. Using both techniques together in cases of polymicrogyria and FCD, several investigators have demonstrated spike source localizations concordant with the location of MRI visible lesions.45 Thus, interictal event localization may help define the epileptogenicity of a given case of MCD.
Intracranial recordings
Numerous researchers have investigated the intracranial electrophysiological features of MCD, using either electrocorticography46–48 or stereoelectroencephalography (SEEG).10,49–52 They have focused on specific MCDs: FCD,10,46,48,49 DNET,47 nodular heterotopia,52 or band heterotopia.51 Intrinsic epileptogenicity has been well demonstrated in FCD, but seizures may also be initiated in surrounding cortex. As already mentioned, ictal onset involves dysmorphic neurons and never balloon cells; therefore, T2 hyperintensity seen on MRI are not correlated perfectly with the epileptogenic zone. The cortex surrounding a DNET is much more epileptogenic than the tumor itself. In nodular heterotopia, specific interictal patterns are recorded from nodules, but seizures are not initiated solely in them; they are also initiated from overlying cortex or simultaneously in both. Polymicrogyria and band heterotopia are characterized by a broad epileptogenic zone involving complex networks comprising both normal- and abnormal-appearing cortex.51
Epilepsy Surgery for MCD
Surgical Outcomes: Prognostic Factors
Numerous studies have investigated the potential benefit of MCD surgery. Surgery can render about 40% of MCD patients seizure free for at least 2 years for all types and locations.5,8 The remaining 60% of patients experience improvement or no change. The outcome is more variable in view of the different types and locations of MCD. Presurgical evaluation, type of resection, and specific prognostic factors may affect the eventual outcome in individual cases.
MCD subtype
The pathological type and grade of MCD have an effect on surgical outcome. DNET and FCD surgery lead to the best results, rendering seizure free 40% to 65% of patients with FCD and up to 80% with DNET.11,53,54 FCD subtype I has a better prognosis.11 The heterotopias are usually considered less amenable to surgery. However, working with SEEG, Tassi and colleagues52 reported seven patients with unilateral nodular heterotopia rendered seizure free after surgery. None of their bilaterally heterotopic patients became seizure free, but they experienced a decrease in seizure frequency. Surgery for band heterotopia is classically associated with a poor outcome.55,56 However, it seems that SEEG-guided surgery may lead to freedom from seizures in some cases.50 Poor outcome is also associated with polymicrogyria and schizencephaly, but, again, there are reports of freedom from seizures after intracranial recording–guided surgery.5
Complete lesion resection
Complete resection of the lesion is considered a major prognostic factor for good postsurgical outcome, especially in FCD and neoplastic dysplasia (DNET and ganglioglioma).46
Complete resection of epileptogenic zone
It is now well demonstrated (after lengthy debate) that resection of both lesional and nonlesional epileptogenic areas is necessary for surgery to be successful. This is easy and obvious in cases in which lesion and epileptogenic zone overlap but less so when the epileptogenic zone involves a distributed epileptogenic network that is more widespread than the visible lesion.5,49,52
MCD and epileptogenic zone location
Outcome may also depend on the lobe affected; a high rate of freedom from seizures postoperatively is associated particularly with temporal lobe lesions.46 Different locations in the brain imply potential involvement of different types of neural system. This is consistent with the postulate that not only the type of lesion but also the type of cortex (i.e., primary or association) and its connectivity play a part in determining the character of the epileptogenic zone (possibly via kindling mechanisms).
Perspectives for Improving Surgical Outcome: The Concepts of Hidden Lesion and Distributed Epileptogenic Network
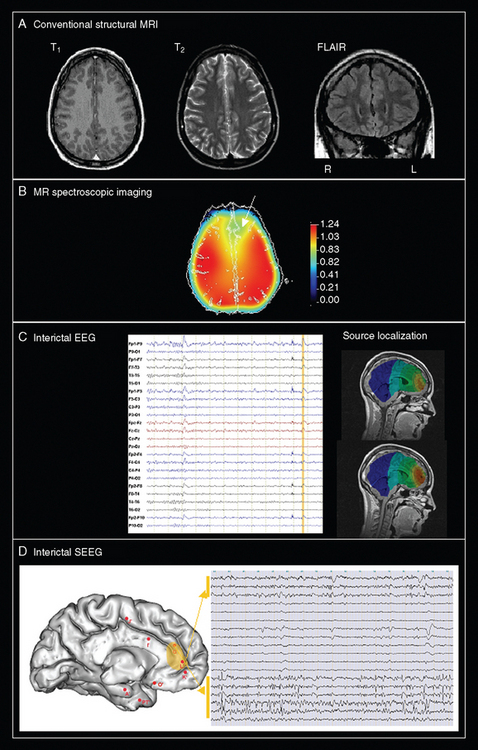
A critical issue is why surgical failures happen. In other words, what is the exact pathophysiology of MCD-related epilepsy? Each type of MCD should be approached specifically, but there are two classical theories that provide possible explanations. The first theoretical assumption is to consider that the dysplastic tissue seen on MRI represents only an obvious problem and that other histological abnormalities (not only surrounding but also potentially remote from the visible lesion) may initiate seizures despite removal of the obvious lesion. This argument is supported by the finding of remote abnormalities through advanced MRI techniques. The second theory is to consider that epilepsy associated with MCD may be initiated in an epileptogenic network more or less distributed, depending on the type of MCD and the neural system involved. This second explanation could account for the clinical observation that complete, histologically proven lesion resection in some cases fails to ameliorate the epilepsy, whereas in other cases, incomplete lesion resection may lead to freedom from seizures. The second hypothesis implies that presurgical evaluation should rely more on electrophysiological and functional imaging than structural imaging alone. Both theories must be taken into consideration when efforts are made to improve the outcome of epilepsy surgery for MCD. Only a comprehensive approach comprising clinical, electrophysiological, and multimodal imaging assessments will lead to a better understanding of the exact pathophysiology of MCD in individual patients seen in clinical practice (Fig. 49-8).
Barkovich AJ, Kuzniecky RI, Jackson GD, et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57:2168-2178.
Guerrini R, Andermann F, Canapicchi R, et al, editors. Dysplasias of Cerebral Cortex and Epilepsy. New York: Lippincott-Raven, 1996.
Palmini A. Disorders of cortical development. Curr Opin Neurol. 2000;13:183-192.
Sarnat HB. Cerebral Dysgenesis: Embryology and Clinical Expression. New York: Oxford University Press, 1992.
Sisodiya SM. Malformations of cortical development: burdens and insights from important causes of human epilepsy. Lancet Neurol. 2004;3:29-38.
1 Barkovich AJ, Kuzniecky RI, Dobyns WB, et al. A classification scheme for malformations of cortical development. Neuropediatrics. 1996;27:59-63.
2 Palmini A. Disorders of cortical development. Curr Opin Neurol. 2000;13:183-192.
3 Barkovich AJ, Kuzniecky RI, Jackson GD, et al. Classification system for malformations of cortical development: update 2001. Neurology. 2001;57:2168-2178.
4 Sarnat HB, Flores-Sarnat L. A new classification of malformations of the nervous system: an integration of morphological and molecular genetic criteria as patterns of genetic expression. Eur J Paediatr Neurol. 2001;5:57-64.
5 Sisodiya SM. Surgery for malformations of cortical development causing epilepsy. Brain. 2000;123(Pt 6):1075-1091.
6 Duncan JS. Imaging and epilepsy. Brain. 1997;120(Pt 2):339-377.
7 Sisodiya SM. Malformations of cortical development: burdens and insights from important causes of human epilepsy. Lancet Neurol. 2004;3:29-38.
8 Raymond AA, Fish DR, Sisodiya SM, et al. Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumour and dysgenesis of the archicortex in epilepsy. Clinical, EEG and neuroimaging features in 100 adult patients. Brain. 1995;118(Pt 3):629-660.
9 Taylor DC, Falconer MA, Bruton CJ, et al. Focal dysplasia of the cerebral cortex in epilepsy. J Neurol Neurosurg Psychiatry. 1971;34:369-387.
10 Tassi L, Colombo N, Garbelli R, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain. 2002;125(Pt 8):1719-1732.
11 Fauser S, Schulze-Bonhage A, Honegger J, et al. Focal cortical dysplasias: surgical outcome in 67 patients in relation to histological subtypes and dual pathology. Brain. 2004;127(Pt 11):2406-2418.
12 Palmini A, Najm I, Avanzini G, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62(6, Suppl 3):S2-S8.
13 Widdess-Walsh P, Kellinghaus C, Jeha L, et al. Electroclinical and imaging characteristics of focal cortical dysplasia: correlation with pathological subtypes. Epilepsy Res. 2005;67(1–2):25-33.
14 Eriksson SH, Malmgren K, Nordborg C. Microdysgenesis in epilepsy. Acta Neurol Scand. 2005;111:279-290.
15 Walsh CA. Genetic malformations of the human cerebral cortex. Neuron. 1999;23:19-29.
16 Guerrini R, Carrozzo R. Epilepsy and genetic malformations of the cerebral cortex. Am J Med Genet. 2001;106:160-173.
17 Palmini A, Andermann E, Andermann F. Prenatal events and genetic factors in epileptic patients with neuronal migration disorders. Epilepsia. 1994;35:965-973.
18 Reiner O, Carrozzo R, Shen Y, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit–like repeats. Nature. 1993;364:717-721.
19 des Portes V, Francis F, Pinard JM, et al. doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH). Hum Mol Genet. 1998;7:1063-1070.
20 Villard L, Nguyen K, Cardoso C, et al. A locus for bilateral perisylvian polymicrogyria maps to Xq28. Am J Hum Genet. 2002;70:1003-1008.
21 Granata T, Farina L, Faiella A, et al. Familial schizencephaly associated with EMX2 mutation. Neurology. 1997;48:1403-1406.
22 Becker AJ, Urbach H, Scheffler B, et al. Focal cortical dysplasia of Taylor’s balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to tuberous sclerosis. Ann Neurol. 2002;52:29-37.
23 Spreer J, Martin P, Greenlee MW, et al. Functional MRI in patients with band heterotopia. Neuroimage. 2001;14:357-365.
24 Innocenti GM, Maeder P, Knyazeva MG, et al. Functional activation of microgyric visual cortex in a human. Ann Neurol. 2001;50:672-676.
25 Janszky J, Ebner A, Kruse B, et al. Functional organization of the brain with malformations of cortical development. Ann Neurol. 2003;53:759-767.
26 Richardson MP, Koepp MJ, Brooks DJ, et al. Cerebral activation in malformations of cortical development. Brain. 1998;121(Pt 7):1295-1304.
27 Chang BS, Walsh CA. Mapping form and function in the human brain: the emerging field of functional neuroimaging in cortical malformations. Epilepsy Behav. 2003;4:618-625.
28 Eriksson SH, Rugg-Gunn FJ, Symms MR, et al. Diffusion tensor imaging in patients with epilepsy and malformations of cortical development. Brain. 2001;124(Pt 3):617-626.
29 Eriksson SH, Symms MR, Rugg-Gunn FJ, et al. Exploring white matter tracts in band heterotopia using diffusion tractography. Ann Neurol. 2002;52:327-334.
30 Lim CC, Yin H, Loh NK, et al. Malformations of cortical development: high-resolution MR and diffusion tensor imaging of fiber tracts at 3T. AJNR Am J Neuroradiol. 2005;26:61-64.
31 Sisodiya SM, Free SL, Stevens JM, et al. Widespread cerebral structural changes in patients with cortical dysgenesis and epilepsy. Brain. 1995;118(Pt 4):1039-1050.
32 Colliot O, Bernasconi N, Khalili N, et al. Individual voxel-based analysis of gray matter in focal cortical dysplasia. Neuroimage. 2006;29:162-171.
33 Li LM, Cendes F, Bastos AC, et al. Neuronal metabolic dysfunction in patients with cortical developmental malformations: a proton magnetic resonance spectroscopic imaging study. Neurology. 1998;50:755-759.
34 Vuori K, Kankaanranta L, Hakkinen AM, et al. Low-grade gliomas and focal cortical developmental malformations: differentiation with proton MR spectroscopy. Radiology. 2004;230:703-708.
35 Maehara T, Nariai T, Arai N, et al. Usefulness of [11C]methionine PET in the diagnosis of dysembryoplastic neuroepithelial tumor with temporal lobe epilepsy. Epilepsia. 2004;45:41-45.
36 Kagawa K, Chugani DC, Asano E, et al. Epilepsy surgery outcome in children with tuberous sclerosis complex evaluated with alpha-[11C]methyl-L-tryptophan positron emission tomography (PET). J Child Neurol. 2005;20:429-438.
37 Hammers A, Koepp MJ, Brooks DJ, et al. Periventricular white matter flumazenil binding and postoperative outcome in hippocampal sclerosis. Epilepsia. 2005;46:944-948.
38 Merlet I, Ryvlin P, Costes N, et al. Statistical parametric mapping of 5-HT1A receptor binding in temporal lobe epilepsy with hippocampal ictal onset on intracranial EEG. Neuroimage. 2004;22:886-896.
39 Guye M, Ranjeva JP, Le Fur Y, et al. 1H-MRS imaging in intractable frontal lobe epilepsies characterized by depth electrode recording. Neuroimage. 2005;26:1174-1183.
40 Chevassus-Au-Louis N, Congar P, Represa A, et al. Neuronal migration disorders: heterotopic neocortical neurons in CA1 provide a bridge between the hippocampus and the neocortex. Proc Natl Acad Sci U S A. 1998;95:10263-10268.
41 Chevassus-au-Louis N, Baraban SC, Gaiarsa JL, et al. Cortical malformations and epilepsy: new insights from animal models. Epilepsia. 1999;40:811-821.
42 Cepeda C, Andre VM, Vinters HV, et al. Are cytomegalic neurons and balloon cells generators of epileptic activity in pediatric cortical dysplasia? Epilepsia. 2005;46(Suppl 5):82-88.
43 Avoli M, Louvel J, Mattia D, et al. Epileptiform synchronization in the human dysplastic cortex. Epilept Disord. 2003;5(Suppl 2):S45-S50.
44 Gambardella A, Palmini A, Andermann F, et al. Usefulness of focal rhythmic discharges on scalp EEG of patients with focal cortical dysplasia and intractable epilepsy. Electroencephalogr Clin Neurophysiol. 1996;98:243-249.
45 Bast T, Oezkan O, Rona S, et al. EEG and MEG source analysis of single and averaged interictal spikes reveals intrinsic epileptogenicity in focal cortical dysplasia. Epilepsia. 2004;45:621-631.
46 Palmini A, Gambardella A, Andermann F, et al. Intrinsic epileptogenicity of human dysplastic cortex as suggested by corticography and surgical results. Ann Neurol. 1995;37:476-487.
47 Seo DW, Hong SB. Epileptogenic foci on subdural recording in intractable epilepsy patients with temporal dysembryoplastic neuroepithelial tumor. J Korean Med Sci. 2003;18:559-565.
48 Boonyapisit K, Najm I, Klem G, et al. Epileptogenicity of focal malformations due to abnormal cortical development: direct electrocorticographic-histopathologic correlations. Epilepsia. 2003;44:69-76.
49 Chassoux F, Devaux B, Landre E, et al. Stereoelectroencephalography in focal cortical dysplasia: a 3D approach to delineating the dysplastic cortex. Brain. 2000;123(Pt 8):1733-1751.
50 Francione S, Kahane P, Tassi L, et al. Stereo-EEG of interictal and ictal electrical activity of a histologically proved heterotopic gray matter associated with partial epilepsy. Electroencephalogr Clin Neurophysiol. 1994;90:284-290.
51 Mai R, Tassi L, Cossu M, et al. A neuropathological, stereo-EEG, and MRI study of subcortical band heterotopia. Neurology. 2003;60:1834-1838.
52 Tassi L, Colombo N, Cossu M, et al. Electroclinical, MRI and neuropathological study of 10 patients with nodular heterotopia, with surgical outcomes. Brain. 2005;128(Pt 2):321-337.
53 Nolan MA, Sakuta R, Chuang N, et al. Dysembryoplastic neuroepithelial tumors in childhood: long-term outcome and prognostic features. Neurology. 2004;62:2270-2276.
54 Bingaman WE. Surgery for focal cortical dysplasia. Neurology. 2004;62(6, Suppl 3):S30-S34.
55 Dubeau F, Palmini A, Fish D, et al. The significance of electrocorticographic findings in focal cortical dysplasia: a review of their clinical, electrophysiological and neurochemical characteristics. Electroencephalogr Clin Neurophysiol Suppl. 1998;48:77-96.
56 Bernasconi A, Martinez V, Rosa-Neto P, et al. Surgical resection for intractable epilepsy in “double cortex” syndrome yields inadequate results. Epilepsia. 2001;42:1124-1129.






