Developmental and Positional Anomalies of The Kidneys
Renal Embryology
The pronephros, which has no adult function, induces the mesonephros to differentiate into the mesonephric duct during the fourth to eighth week of fetal life. The mesonephric duct is the basis of the Wolffian system, which develops into the seminal vesicles, vas deferens, epididymis, and efferent ductules of the testis in boys, and the epoophoron and paraophoron (vestigial remnants between the fallopian tube and ovary) in girls. Between weeks 9 and 12, the ureteric bud branches off the mesonephric duct, contacts the metanephric blastema bud, and induces the entire collecting system of ureter, renal pelvis, calyx, and collecting tubules. The kidney develops via induction of the metanephric blastema by the ureteric bud into Bowman’s capsule, the convoluted tubules, and the loop of Henle.1 Figure 53-1 illustrates the progression of development from pronephros, to mesonephros, to metanephros.
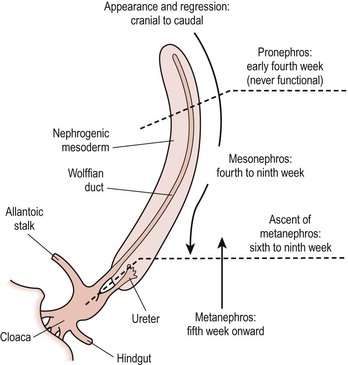
FIGURE 53-1 Development of the kidney. (Redrawn from Gray SW, Skandalakis JE. Embryology for Surgeons. Philadelphia: WB Saunders; 1972. p. 444.)
The kidneys begin at the upper sacral level with the renal pelvis facing anteriorly. The kidneys ascend either because the lumbar and sacral regions grow faster than the cervical and thoracic regions between 4 to 8 weeks, or because there is active migration. As the kidneys ascend, the renal pelvis rotates medially by 90°, leading to the normal configuration of the renal pelvis lying medial to the parenchyma. During this time, the blood supply shifts from inferior branches of the aorta to more cephalad branches, with the final renal artery being located at about L2. Failure of normal ascent leads to the persistence of a low-lying blood supply.1
Renal Dysplasia and Hypoplasia
Since the development of the kidney depends on proper interaction between the ureteric bud and the metanephric blastema, it should not be surprising that an abnormality in the location of the ureteral orifice is associated with abnormally induced renal tissue.2 Examination of the thickness of the renal parenchyma and number of glomeruli associated with normal and ectopic ureters in fetal specimens suggests that it is the initial interaction between bud and blastema, rather than subsequent obstruction or VUR, that determines if normal renal tissue will develop.2 Figure 53-2 shows how a ureter which arises in the proper trigonal location (A, E, F) is associated with normal renal parenchyma whereas a ureter arising from a more cranial location (B, C, D) or caudal location (G, H) is associated with progressively less normal renal parenchyma.
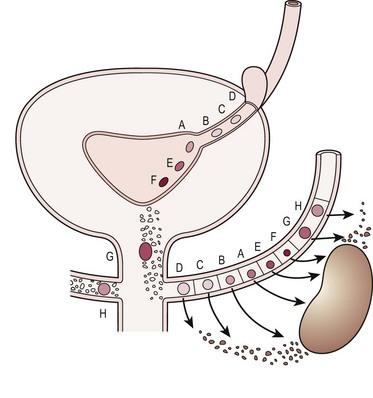
FIGURE 53-2 Relation of ureteral orifice location and associated metanephric tissue. (Redrawn from Mackie GG, Stephens FD. Duplex kidneys: A correlation of renal dysplasia with position of the ureteral orifice. J Urol 1975;114:274–80.)
Renal dysplasia and hypoplasia can be considered errors in renal induction. Figure 53-3 shows varying changes from agenesis to dysplasia and hypoplasia of the kidney. Although dysplasia is technically a histologic term, it refers to kidneys which contain primitive tubules either focally or diffusely. These ducts are lined by epithelium and surrounded by swirls of primitive collagen. No treatment is necessary for the dysplastic kidney, but there is an increased risk of reflux in the contralateral kidney.3 Hypoplastic kidneys are small, normal kidneys with a decreased number of nephrons. Dysplasia can also occur in hypoplastic kidneys. While secondary hypoplasia can occur due to infection or obstruction, two types of hypoplastic kidneys are clinically important: the oligomeganephronic type, and the Ask–Upmark kidney. In oligomeganephronia, there is a decrease in the number of nephrons with an associated hypertrophy of the ones which are present. Patients present with polyuria and failure to concentrate their urine, but no hypertension. Imaging with ultrasound (US) reveals small kidneys. Medical management with protein restriction, and high fluid and salt intake is initiated. Once the glomerular filtration rate drops significantly, dialysis is required.4 The Ask–Upmark kidney was initially felt to be a developmental problem, but is now believed to represent reflux nephropathy. The key finding is a small kidney with segmental hypoplasia, probably secondary to ascending pyelonephritis. VUR and hypertension are usually present. Most patients are over 10 years of age with a 2 : 1 female : male ratio. If the disease is unilateral, nephrectomy may cure the hypertension. Bilateral disease is managed medically.5
Renal Agenesis
Unilateral renal agenesis occurs in 1 : 1,000 live births with a 2 : 1 male predominance.6,7 Unilateral renal agenesis can result in compensatory hypertrophy of the contralateral kidney. The left kidney is more likely to be affected in unilateral renal agenesis.8 Since unilateral renal agenesis is asymptomatic and eventual renal function is normal, the diagnosis is usually made on prenatal ultrasound, or it is incidentally found during imaging for other abdominal symptoms. Sometimes it can be suspected on plain abdominal films if the colon is medially deviated at the splenic or hepatic flexures.9 These patients should consider obtaining a medical alert bracelet so that in case of traumatic injury, the solitary kidney is not inadvertently removed.
In a newborn with the prenatal diagnosis of unilateral renal agenesis, physical examination at the time of birth should be focused on detecting the anomalies present in the VACTERL association (Box 53-1).10 A voiding cystourethrogram (VCUG) should also be obtained since approximately 30% of VACTERL patients with unilateral renal agenesis will have VUR in the contralateral kidney.10
Females with unilateral renal agenesis should have their genital anatomy evaluated since up to 30% will have an abnormality of the Müllerian duct due to the Mayer–Rokitansky syndrome (Müllerian, uterine, upper vaginal duplications with or without obstruction, or vaginal agenesis).11,12 The abnormal induction of the mesonephric duct is believed to cause partial or complete nonunion of the paired Müllerian ducts.13 Conversely, 40% of patients with abnormalities of the Müllerian organs will have unilateral renal agenesis or ectopia.14 In patients with duplicated vaginas and unilateral vaginal agenesis, the side without a vagina is also the side without a kidney.13
If the diagnosis of Mayer–Rokitansky is not made prenatally, the patients can present either as infants with hydrocolpos, or as adolescents with lower abdominal pain after the onset of menses due to an obstructed vagina or uterus (with or without duplication). Magnetic resonance imaging (MRI) is useful in delineating the pelvic anatomy in these cases. In vaginal agenesis, the vagina is only present as a shallow pouch. There is a wide variety of abnormalities of the vagina, uterus, and fallopian tubes (Fig. 53-4), but the ovaries are embryologically normal.
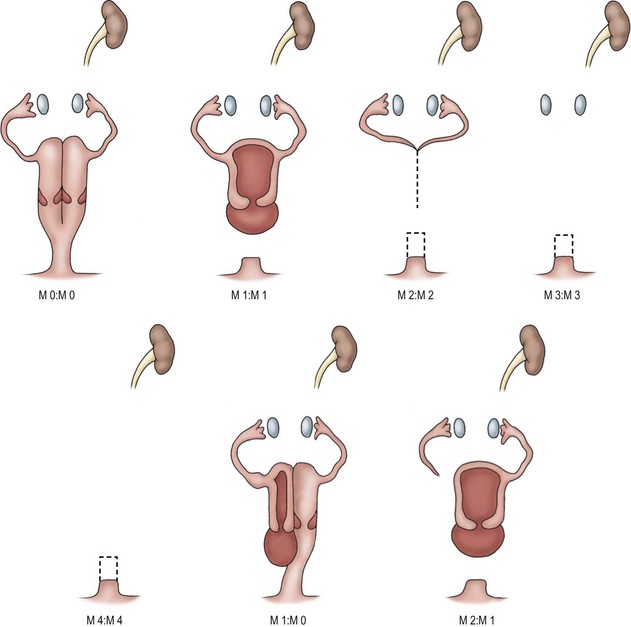
FIGURE 53-4 Variations in Müllerian anatomy in Mayer–Rokitansky syndrome. M0, Right or left vagina and uterus, or duplex vagina and uterus with partial or complex septum. M1, Partial or complete absence of vagina. M2, Absence of vagina and uterus. M3, Absence of vagina, uterus, and fallopian tube. M4, Absence of vagina, uterus, fallopian tube, and ovary. (Redrawn from Tarry WR, Duckett JW, Stephens FD. The Mayer-Rokitansky syndrome: Pathogenesis, classification and management. J Urol 1989;136:648–52.)
Bilateral renal agenesis occurs in 1 : 4,800 live births, and has a 3 : 1 male predominance.15 Infants affected with bilateral renal agenesis present with oligohydramnios, pulmonary hypoplasia, Potter’s facies (low-set ears, broad flat nose, a prominent skin fold beginning over the eye and running to the cheek), and the great majority die soon after birth from their pulmonary hypoplasia. The renal arteries and ureters are usually absent, and the bladder is underdeveloped. The vas is usually present, but female genital structures are usually abnormal.16,17 The adrenals are usually present but appear round, instead of flattened, due to the lack of compression by the kidneys.15 Prenatal diagnosis is useful in determining that heroic efforts at extracorporeal membrane oxygenation or hemodialysis are not indicated after delivery.
Supernumerary Kidney
This is a rare condition in which a completely separate kidney is found in addition to two normally positioned kidneys. The additional kidney has its own blood supply and parenchyma, and usually is found caudal to the normal kidney. It is usually smaller than the normally positioned kidney. This additional kidney represents abnormal induction of metanephric blastema by an abnormally directed ureteric bud, either as a separate ureteral bud from the mesonephric duct, or as part of a ‘Y’ duplication. If the supernumerary kidney is located cranial to the normal kidney, the ureter is usually completely separate and may enter the bladder ectopically. Presumably this is a result of a completely separate ureteral bud inducing the metanephric blastema and migrating very low on the mesonephric duct, separate from the normally positioned kidney.18,19 If the ureter ends ectopically, it may present as incontinence in a girl, or as infection in a poorly functioning renal unit. The diagnosis can be difficult.20 Stone disease and hydronephrosis can be found in up to 50% of patients. Treatment should be reserved for these problems as the presence of a supernumerary kidney itself is not worrisome.19 Like other ectopic kidneys, these kidneys may be more subject to trauma, so a medical alert bracelet may be helpful.
Renal Ectopia
Failure of rotation, while not strictly ectopia, usually results in a kidney in which the renal pelvis is anteriorly directed. In the unusual situation in which hyper-rotation occurs, the renal pelvis can actually point posteriorly. The renal vessels are normally positioned. The renal pelvis and calyces will often appear abnormal on an intravenous urogram due to their unusual orientation. With oblique views the anatomy can be established, and does not usually require repair, even in poorly functioning units. One method to localize even poorly functioning ectopic renal tissue is with a nuclear medicine study. There are two technical factors to be considered in interpreting renal scans in ectopic kidneys. First, the radionuclide in the bladder can overlap a pelvic kidney so a catheter may need to be inserted for the study. Second, the pelvic kidney is located further anterior than orthotopic kidneys, and the function may be artificially lowered by the distance of the kidney from the camera. Placing the patient prone may result in a more accurate assessment. Magnetic resonance urography (MRU) is another emerging technique for localizing ectopic renal units.
Simple ectopia results in a kidney which is located anywhere from the pelvis to the diaphragm. The incidence is 1 : 1,000 live births with a 3 : 2 male predominance.21 The contralateral kidney often also has a rotational abnormality or ectopia. The development of the ipsilateral adrenal gland is unaffected. A ‘thoracic kidney’ is actually subdiaphragmatic, although it may lie in the chest through a focal eventration of the diaphragm. It is not associated with a true congenital diaphragmatic hernia.22 An ectopic ‘abdominal kidney’ is above the iliac crest, the ‘lumbar kidney’ is anterior to the iliac vessels at the sacral promontory, and the ‘pelvic kidney’ is below the aortic bifurcation and opposite the sacrum. All of these ectopic kidneys are more susceptible to trauma since they are not as well protected by the lower rib cage and are anterior in position. It may be advisable for these patients to avoid contact sports in which there is a risk of abdominal trauma.
Most ectopic kidneys are asymptomatic and are detected either on prenatal ultrasound or incidentally on other imaging studies. Ectopic kidneys are at higher risk for UPJ obstruction, VUR, and stone formation. The anatomy can include an extrarenal pelvis and infundibulum, and a high insertion of the ureter into the pelvis. This anatomical arrangement can mimic a UPJ obstruction so careful evaluation is necessary to avoid unnecessary surgery.23 More than half will have a dilated renal pelvis. Of these, half are due to obstruction, 25% are due to reflux, and 25% are merely dilated without UPJ obstruction.24 For repair of UPJ obstruction with a high insertion of the ureter, a side to side ureteropyelostomy or ureterocalycostomy to a dilated lower pole calyx is sometimes required to obtain dependent drainage.
While endoscopic techniques for treatment of UPJ obstruction have been used in children,25 the presence of anomalous vessels suggests that either an open or laparoscopic approach would be safer than endoscopic incision of a UPJ obstruction in an ectopic kidney. The advent of computed tomography (CT) angiography and MRU has made the assessment of anomalous vessels in ectopic or horseshoe kidneys easier and less invasive.
Fusion Defects
Horseshoe Kidney
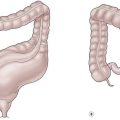
A horseshoe kidney is found in 1 : 400 live births, and has a 2 : 1 male predominance.26 The kidney is usually lower than normal, since the lower poles fuse in the midline and drape anteriorly over the spine. The isthmus can be fibrotic or contain parenchyma. This anomaly is believed to occur between 4 to 6 weeks of life, since the orientation of the renal pelvis is anterior. It is proposed that as the kidneys ‘hurdle’ the iliac vessels during ascent, they come into contact at the lower pole and fuse (Fig. 53-5). Other variations of upper pole and mid-pole contact are possible, but much less common than the usual lower pole fusion. The kidney is usually low due to its inability to ascend past the inferior mesenteric artery. Each renal moiety retains its ureter, which is draped over the isthmus. The renal pelvis is usually anterior. The arterial supply varies from the normal single vessel to each moiety to vessels arising from any conceivable nearby blood supply. Horseshoe kidneys are more commonly found in patients with sacral agenesis, high cloacas, and Turner syndrome (45,XO gonadal dysgenesis).27 They are associated with a higher risk of renal cell carcinoma and Wilms tumor.28–30 The presence of a Wilms tumor in a horseshoe kidney was not suspected preoperatively in 13/41 patients despite imaging studies.30
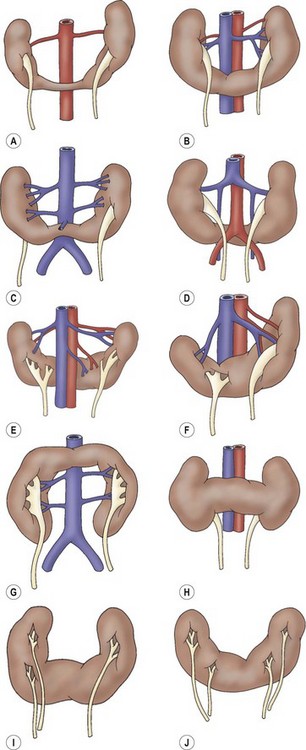
FIGURE 53-5 Variations of horseshoe kidney. (Redrawn from Banjamin JA, Schullian DM. Observations of kidneys with horseshoe configuration: The contribution of Leonardo Botallo. J Hist Med Allied Sci 1950;5:315, after Gutierrez, 1931.)
One-third of patients with horseshoe kidney have no symptoms. The patients with symptoms often complain of vague abdominal or back pain. 10% have ureteral duplication, 50% have VUR, and 33% have UPJ obstruction.27,31,32 Repair of UPJ obstruction in a horseshoe kidney requires placement of the anastomosis to avoid a secondary kinking at the UPJ. Division of the isthmus is not required. Treatment of kidney stones in horseshoe kidneys can be accomplished by extracorporeal shock wave lithotripsy, ureteroscopy, or percutaneous nephrolithotomy. Percutaneous approaches are sometimes difficult as the kidneys do not reside next to the body wall, making access to the collecting system difficult. However, percutaneous approaches result in a higher stone-free rate than ureteroscopy or shock wave lithotripsy.33,34 Although there is no increase in the rate of metabolic abnormalities in patients with horseshoe kidneys and kidney stones, suggesting that stasis in an extrarenal pelvis contributes to the formation of kidney stones,35 patients with a horseshoe kidney and kidney stones are more likely to have hypocitraturia than other patients with kidney stones.36
Cystic Renal Disease and Cystic Tumors
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
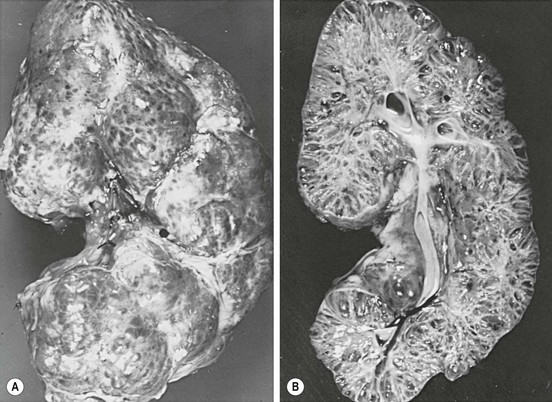
This disease was formerly called infantile polycystic kidney disease, which is inaccurate since it can present in older patients. While it occurs in 1 : 40,000 live births, many patients die soon afterwards. The kidneys are bilaterally enlarged, with very small cysts radially oriented throughout parenchyma (Fig. 53-6). The cysts represent dilated collecting tubules. Periportal hepatic fibrosis also occurs in varying degrees, and can lead to portal hypertension. The hepatic involvement appears to be inversely proportional to the renal involvement. The disease has been classified into four forms.37 The severe perinatal form (>90% renal involvement) leads to death by six weeks from pulmonary hypoplasia. The neonatal form (60% renal involvement) is usually lethal by one year. The infantile form (25% renal involvement) results in hepatosplenomegaly, with survival up to 10 years. The juvenile form (<10% renal involvement) has severe periportal fibrosis. Some patients survive up to 15 years, but the development of portal hypertension is usually lethal. Since this is an autosomal recessive disease, family screening should be undertaken to determine which siblings are carriers.
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
While ADPKD tends to clinically present in the third to fifth decade, it has been diagnosed in the ultrasound era in asymptomatic children as well. The cysts in ADPKD are different in configuration, being few and scattered in distinction to those seen in ARPKD. This condition occurs in 1 : 500 patients.38 Patients usually present with flank pain, hematuria, hypertension, and possibly renal failure, if there are extensive bilateral cysts. Neonates can present with renal enlargement, although children from affected families who are screened usually only have a few cysts. Failure to see cysts on screening ultrasound in a child at risk for ARPKD does not exclude the disease since the cysts can develop later in life. Linkage analysis of the loci on chromosome 4 and 16 is more sensitive.39 The cysts are located throughout the cortex and medulla, although the fetal form seems to affect the glomeruli predominantly. Hepatic involvement is limited to biliary cysts. Associated findings include cysts in the spleen, pancreas, and lungs, mitral valve prolapse, colon diverticuli, and berry aneurysms of the circle of Willis.
Hypertension is commonly found in these children, and may be part of the presentation. Renal failure in childhood is very rare. Periodic evaluation of blood pressure and proteinuria during childhood is recommended.40,41 Unlike ARPKD, there is no increased risk of renal cell carcinoma. Renal transplant candidates can obtain organs from family members who have been screened for the disease.
Multicystic Dysplastic Kidney (MCDK)
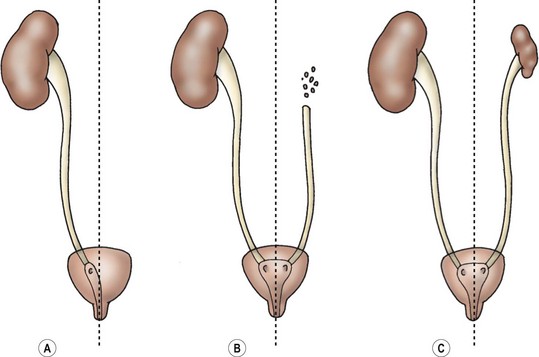
The multicystic dysplastic kidney is believed to be caused by severe early ureteral obstruction or a failure in ureteric bud-metanephric blastema induction.42,43 The main differential diagnosis is severe hydronephrosis due to UPJ obstruction. Radiographically, this occurs when the peripheral cysts surround a dominant central cyst mimicking the renal pelvis (‘hydronephrotic’ form of MCDK). The classic ultrasound appearance shows cysts randomly distributed throughout the kidney without a dominant medial cyst or evidence of communication between cysts. The parenchyma, if present, has abnormal echogenicity and is seen between the cysts, instead of being arranged on their periphery. A renal scan will show no function in a MCDK. The affected area may be the upper pole of a duplicated collecting system, or one-half of a horseshoe kidney.
The MCDK is the most common renal cystic mass in the newborn. Currently, most are detected on prenatal ultrasound. Bilateral forms are not compatible with life. Postnatal evaluation consists of a VCUG to look for VUR in the contralateral kidney, which is found 30% of the time.44 If there is significant hydronephrosis (caliectasis) in the contralateral kidney (this occurs 12% of the time), then a diuretic renal scan may be necessary. Contralateral UPJ obstruction or VUR is more likely with a smaller MCDK or a lower ureteral atresia ipsilateral to the MCDK.45,46 There are reports of malignancy arising from a MCDK, although it is unclear whether the affected kidneys were truly MCDK.47 Hypertension has also been reported in association with MCDK, although resection is not always curative, and the rate does not appear to be any higher than the general population.48
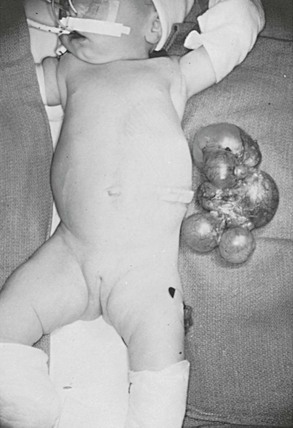
MCDK usually involute, but they can occasionally grow.48 The follow-up is repeat imaging with ultrasound every six months for the first two years of life. It is not usually feasible to monitor a patient indefinitely for a MCDK. We have taken an operative approach at 18 to 24 months of life if the MCDK is not involuting, or if parenchyma remains visible on the ultrasound. Although the indications are controversial, the kidney can be removed at that age via laparoscopy or a small incision as an outpatient procedure (Fig. 53-7). Occasionally, the MCDK can involute prenatally, leaving a ureter with a small nubbin of tissue in the renal fossa. These were previously called ‘aplastic kidneys,’ but are now felt to represent the remnants of a MCDK.
Cystic Nephroma
Formerly called a ‘multilocular cyst,’ this is a well demarcated tumor of cysts with an overall round configuration, lined with epithelium and septae which contain tubules.49 It is considered to be the benign end of a spectrum progressing from cystic Wilms tumor, cystic partially differentiated nephroblastoma, to cystic nephroma. It usually is found in boys under age 4 (male : female ratio 2 : 1) or women over 30 (female : male ratio 8 : 1). It is rarely bilateral and is cured by partial nephrectomy, shelling out the tumor by following the plane of the pseudocapsule. There is a risk of sarcomatous degeneration in adults if it is not removed.50
Cystic Partially Differentiated Nephroblastoma
This lesion was formerly called a multilocular cystic nephroma. It is radiologically identical to the cystic nephroma, and can only be diagnosed pathologically. The majority of patients are boys less than 2 years old, or women in their third to fourth decade. A classic (but not diagnostic) radiologic finding is herniation of a parenchymal mass into the renal pelvis.22 The tumor is usually well circumscribed. Hemorrhage and calcification are usually absent. Pathologically, it differs from the cystic nephroma in that there is blastema found in the septations.
Patients usually present with an asymptomatic flank mass, and occasionally hematuria. Operative treatment consists of partial nephrectomy as for cystic nephroma, since the tumors rarely recur and are not multifocal. No chemotherapy is required for stage I (limited to capsule, fully resected) tumors. Although experience is limited, stage II (outside renal capsule but fully resected) are usually treated with vincristine, dactinomycin, and doxorubicin. Four year survival for both stages is 100%.51,52
Simple Cysts and Calyceal Diverticuli
The simple renal cyst on ultrasound has the following characteristics: distinct wall, no internal echoes, and posterior enhancement. If these criteria are not met, a CT scan is obtained to confirm that the fluid does not enhance. The differential diagnosis is a calyceal diverticulum or hydrocalyx, both of which communicate with the collecting system, and in which the fluid should enhance on either IVP or CT. Ultrasound is able to detect milk of calcium layering within a diverticulum. Calyceal diverticuli require treatment when they harbor stones or infection. In the IVP era, 40% of calyceal diverticuli were felt to be symptomatic.53 In the ultrasound era, with its greater number of incidental findings, it is not clear how often calyceal diverticuli require treatment. Minimally invasive approaches such as percutaneous, laparoscopic, and ureteroscopic ablation appear to be equally successful.54
Simple cysts reside in the cortex and are lined by simple columnar epithelium. They can grow, resorb, or remain the same size. They are usually asymptomatic and are found incidentally. Once they are found, the authors usually follow with ultrasound at 3 to 6 month intervals to determine if the cyst is growing. The underlying concern is whether or not this cyst is the first sign of ADPKD. A family history of renal cystic disease, renal failure, or death in the neonatal period from unknown causes should be sought. Biopsy to rule out tumor, followed by drainage, or unroofing should only be undertaken if the cyst characteristics are other than those listed for a simple cyst, or if the cyst becomes symptomatic due to obstruction of an infundibulum or the UPJ. Minimally invasive approaches such as percutaneous puncture with instillation of sclerosing agents (absolute alcohol, bismuth, povidone-iodine55) or laparoscopic decortication56 may shift the threshold for treatment of large asymptomatic simple cysts.
References
1. Gray, SW, Skandalakis, JE. The kidney and ureter. In: Embryology for Surgeons. Philadelphia: WB Saunders; 1972:443–518.
2. Mackie, GG, Stephens, FD. Duplex kidneys: A correlation of renal dysplasia with position of the ureteral orifice. J Urol. 1975; 114:274–280.
3. Atiyeh, B, Husmann, D, Baum, M. Contralateral renal abnormalities in multicystic-dysplastic kidney disease. J Pediatr. 1992; 121:65–67.
4. Royer, P, Habib, R, Broyer, M, et al. L’Hypoplasie renale bilaterale congenitale avec reduction du nombre et hypertrophie des nephrons chez l’enfant. Ann Pediatr (Paris). 1962; 38:133–146.
5. Arant, BS, Jr., Sotelo-Avila, C, Bernstein, J. Segmental “hypoplasia” of the kidney (Ask-Upmark). J Pediatr. 1979; 95:931–939.
6. Doroshow, LW, Abeshouse, BS. Congenital unilateral solitary kidney: Report of 37 cases and a review of the literature. Urol Surv. 1961; 11:219–229.
7. Sheih, CP, Hung, CS, Wei, CF, et al. Cystic dilatations within the pelvis in patients with ipsilateral renal agenesis or dysplasia. J Urol. 1990; 144:324–327.
8. Kohn, G, Borns, PF. The association of bilateral and unilateral renal aplasia in the same family. J Pediatr. 1973; 83:95–97.
9. Mascatello, V, Lebowitz, RL. Malposition of the colon in left renal agenesis and ectopia. Radiology. 1976; 120:371–376.
10. Kolon, TF, Gray, CL, Sutherland, RW, et al. Upper urinary tract manifestations of the VACTERL association. J Urol. 2000; 163:1949–1951.
11. Downs, RA, Lane, JW, Burns, E. Solitary pelvic kidney. Its clinical implications. Urology. 1973; 1:51–56.
12. Thompson, DP, Lynn, HB. Genital anomalies associated with solitary kidney. Mayo Clin Proc. 1966; 41:538–548.
13. Tarry, WF, Duckett, JW, Stephens, FD. The Mayer-Rokitansky syndrome: Pathogenesis, classification, and management. J Urol. 1986; 136:648–652.
14. Griffin, JE, Edwards, C, Madden, JD, et al. Congenital absence of the vagina. The Mayer-Rokitansky-Kuster-Hauser syndrome. Ann Internal Med. 1976; 85:224–236.
15. Potter, EL. Bilateral absence of ureters and kidneys: A report of 50 cases. Obstet Gynecol. 1965; 25:3–12.
16. Ashley, DJ, Mostofi, FK. Renal agenesis and dysgenesis. J Urol. 1960; 83:211–230.
17. Carpentier, PJ, Potter, EL. Nuclear sex and genital malformation in 48 cases of renal agenesis, with especial reference to nonspecific female pseudoheramphroditism. Am J Obstet Gynecol. 1959; 78:235–258.
18. Geisinger, JG. Supernumerary kidney. J Urol. 1937; 38:331.
19. N’Guessan, G, Stephens, FD. Supernumerary kidney. J Urol. 1983; 130:649–653.
20. Weiss, JP, Duckett, JW, Snyder, HM. Single unilateral vaginal ectopic ureter: Is it really a rarity? J Urol. 1984; 132:1177–1179.
21. Malek, RS, Kelalis, PP, Burke, EC. Ectopic kidney in children and frequency of association with other malformations. Mayo Clinic Proc. 1971; 46:461–467.
22. Zagoria, RL, Tung, GA. The kidney and retroperitoneum: Anatomy and congenital anomalies. In: Zagoria RL, ed. Genitourinary Radiology: The Requisites. St. Louis: Mosby; 1997:51–79.
23. Dretler, SP, Pfister, R, Hendren, WH. Extrarenal calyces in the ectopic kidney. J Urol. 1970; 103:406–410.
24. Gleason, PE, Kelalis, PP, Husmann, DA, et al. Hydronephrosis in renal ectopia: Incidence, etiology, and significance. J Urol. 1994; 151:1660–1661.
25. Jabbour, ME, Goldfischer, ER, Stravodimos, KG, et al. Endopyelotomy for horseshoe and ectopic kidneys. J Urol. 1998; 160:694–697.
26. Dees, J. Clinical importance of congenital anomalies of upper urinary tract. J Urol. 1941; 46:659.
27. Boatman, DL, Kolln, CP, Flocks, RH. Congenital anomalies associated with horseshoe kidneys. J Urol. 1972; 107:205–207.
28. Buntley, D. Malignancy associated with horseshoe kidney. Urology. 1976; 8:146–148.
29. Hohenfellner, M, Schultz-Lampel, D, Lempel, A, et al. Tumor in the horseshoe kidney: Clinical implications and review of embryogenesis. J Urol. 1992; 147:1098–1102.
30. Neville, H, Ritchey, ML, Shamberger, RC, et al. The occurrence of Wilms’ tumor in horseshoe kidneys: A report from the National Wilms’ Tumor Study Group (NWTSG). J Pediatr Surg. 2002; 37:1134–1137.
31. Segura, JW, Kelalis, PP, Burke, EC. Horseshoe kidney in children. J Urol. 1972; 108:333–336.
32. Whitehouse, GH. Some urographic aspects of horseshoe kidney anomaly – a review of 59 cases. Clin Radiol. 1975; 26:107–114.
33. Yohannes, P, Smith, AD. The endourological management of complications associated with horseshoe kidney. J Urol. 2002; 168:5–8.
34. Miller, NL, Matlaga, BR, Handa, SE, et al. The presence of horseshoe kidney does not affect the outcome of percutaneous nephrolithotomy. J Endourol. 2008; 22:1219–1225.
35. Evans, WP, Resnick, MI. Horseshoe kidney and urolithiasis. J Urol. 1981; 125:620–621.
36. Raj, GV, Auge, BK, Assimos, D, et al. Metabolic abnormalities associated with renal calculi in patients with horseshoe kidney. J Endourol. 2004; 12:157–161.
37. Blyth, H, Ockenden, BG. Polycystic disease of kidney and liver presenting in childhood. J Med Genet. 1971; 8:257–284.
38. Gabow, PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993; 329:332–342.
39. Gabow, PA, Kimberling, WJ, Strain, JD, et al. Utility of ultrasonography in the diagnosis of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol. 1997; 8:105–110.
40. Ravine, D, Walker, RG, Gibson, RN, et al. Treatable complications in undiagnosed cases of autosomal dominant polycystic kidney disease. Lancet. 1991; 337:127–129.
41. Zerres, K, Rudnik-Schoneborn, S, Deget, F. Routine examination of children at risk of autosomal dominant polycystic kidney disease. Lancet. 1992; 339:1356–1357.
42. Beck, AD. The effect of intra-uterine urinary obstruction upon the development of the fetal kidney. J Urol. 1971; 105:784–789.
43. Osathanondh, V, Potter, EL. Pathogenesis of polycystic kidneys: Historical survey. Arch Pathol. 1964; 77:459–465.
44. Flack, CE, Bellinger, MF. The multicystic dysplastic kidney and contralateral vesicoureteral reflux: protection of the solitary kidney. J Urol. 1993; 150:1873–1874.
45. Cendron, J, Gubler, JP, Valayer, J, et al. Dysplasie multikystique du rein chez enfant. A propos de 45 observations. J Urol Nephrol (Paris). 1973; 79:773–799.
46. Cendron, J, Kiriakos, S. Rein multikystique. J Urol Nephrol (Paris). 1976; 82(S2):322–333.
47. Beckwith, JB. Comment, Wilms’ tumor and multicystic dysplastic kidney disease. J Urol. 1997; 158:2259–2260.
48. Wacksman, J, Phipps, L. Report of the multicystic kidney registry: Preliminary findings. J Urol. 1993; 150:1870–1872.
49. Joshi, VV, Beckwith, JB. Multilocular cyst of the kidney (cystic nephroma) and cystic, partially differentiated nephroblastoma. Terminology and criteria for diagnosis. Cancer. 1989; 64:466–479.
50. Castillo, OA, Boyle, ET, Jr., Kramer, SA. Multilocular cysts of the kidney. A study of 29 patients and review of literature. Urology. 1991; 37:156–162.
51. Blakely, ML, Shamberger, RC, Norkool, P, et al. Outcome of children with cystic partially differentiated nephroblastoma treated with and without chemotherapy. J Pediatr Surg. 2003; 38:897–900.
52. Luithle, T, Szavay, P, Furtwangler, R, et al. Treatment of cystic nephroma and cystic partially differentiated nephroblastoma – A report from SIOP/GPOH study group. J Urol. 2007; 177:294–296.
53. Timmons, JW, Jr., Malek, RS, Hattery, RR, et al. Caliceal diverticulum. J Urol. 1975; 114:6–9.
54. Canales, B, Monga, M. Surgical management of the calyceal diverticulum. Curr Opin Urol. 2003; 13:255–260.
55. Phelan, M, Zajko, A, Hrebinko, RL. Preliminary results of percutaneous treatment of renal cysts with povidone-iodine sclerosis. Urology. 1999; 53:816–817.
56. Lifson, BJ, Teichman, JM, Hulbert, JC. Role and long-term results of laparoscopic decortication in solitary cystic and autosomal dominant polycystic kidney disease. J Urol. 1998; 159:702–706.