[level-membership-for-basic-science-category]
CHAPTER 59 Development of the thorax
THORACIC WALL AND DIAPHRAGM
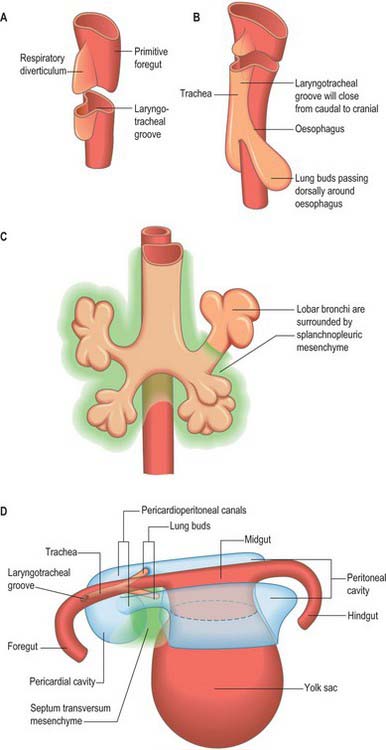
The thorax as an entity is not apparent in embryos until the end of the embryonic stage of development (stage 23). It develops around the early pericardial cavity and the associated pericardioperitoneal canals (Fig. 59.1A; see Fig. 59.3A). The pericardioperitoneal canals give rise to the pleural cavities surrounding the lungs and that part of the peritoneal cavity surrounding the lower end of the foregut, which becomes the oesophagus, stomach and duodenum within the thoracic cage. Later these portions of peritoneal cavity are sequestered into the abdominal cavity by the development of the diaphragm.
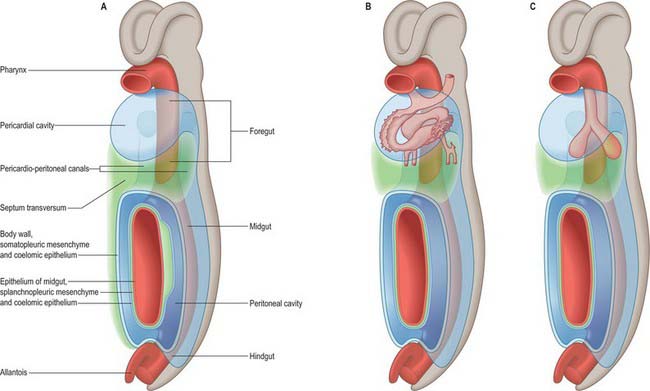
In stage 14 embryos, the heart is at the level of the upper cervical somites and above the upper limb buds. The thoracic somites are opposite the midgut. The putative thoracic region contains the pericardial cavity ventrally and the pericardioperitoneal canals posteriorly, on each side of the foregut. The future pleural cavities are as yet undefined regions of the pericardioperitoneal canals. Below the heart, septum transversum mesenchyme (see Fig. 73.5A) that has arisen from the caudal pericardial wall is being invaginated by endodermal epithelial cells from the foregut hepatic primordium.
The heart and pericardial cavity are relatively large in the early embryo (Fig. 59.1B; see Fig. 59.3A). Throughout development the lungs remain unexpanded and do not achieve their full size within the thorax, reflecting the fact that the placenta, and not the lungs, is the organ of fetal respiration. The lower partition of the thorax, the diaphragm, can be identified in stage 13 embryos; it migrates caudally in line with the craniocaudal progression of development and the elongation of the neck. The parietal pericardium remains attached to the diaphragm as it descends.
The lung buds are invested by splanchnopleuric mesenchyme derived from the medial walls of the pericardioperitoneal canals, whereas the lateral walls produce somatopleuric mesenchyme, which contributes to the body wall. This latter mesenchyme is penetrated by the developing ribs which arise from the thoracic sclerotomes. In the midline, the somatopleuric mesenchyme gives rise to the sternum and costal cartilages. The bony and cartilaginous cage provides insertions for the intercostal muscles, which arise from the ventrolateral edge of the epithelial plate of the somites. The somatopleuric coelomic epithelium, after its proliferative phase, gives rise to the mesothelium of the parietal layer of pleura.
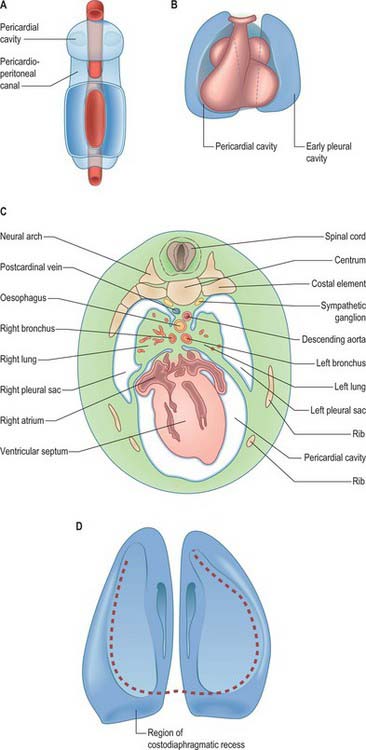
As the lung buds project into the pericardioperitoneal canals (Figs 59.1C, 59.2A), they subdivide them into primary pleural coeloms around the lung buds cranially, and paired peritoneal coeloms caudally, which are continuous with the wider peritoneal coelom around the mid- and hindguts. The communications with the pericardial and peritoneal coeloms become termed the pleuropericardial and pleuroperitoneal canals, respectively. When separation between these fluid-filled major coelomic regions is advancing towards completion, they are named the pericardial, pleural and peritoneal cavities. In early embryos, the cavities retain substantial volumes of fluid and their walls are separate: they provide the route for a primitive type of circulation until superseded by the blood vascular system. In later fetal and postnatal life, the cavity walls are coapted, so that a mere microscopic film of serous fluid intervenes between them.

A curved elevation of tissue, the pulmonary ridge, develops on the lateral wall of the pleural coelom and partly encircles the pleuropericardial canal. The ridge is continuous with the dorsolateral edge of the septum transversum. The developing lung bud abuts on the ridge, which as a result divides into two diverging membranes meeting at the septum transversum. One is cranially placed and termed the pleuropericardial membrane (Fig. 59.2A); embedded within it are the common cardinal vein and phrenic nerve, which reach the septum transversum by this route. The other membrane, caudally placed, is termed the pleuroperitoneal membrane (Fig. 59.2A,B). As the apical part of the lung forms, it invades and splits the body wall and extends cranially on the lateral aspect of the common cardinal vein, preceded by an extension from the primary pleural coelom to form part of the secondary, definitive, pleural sac. In this way the common cardinal vein and the phrenic nerve come to lie medially in the mediastinum. The pleuropericardial canal, which lies medial to the vein, is gradually narrowed to a slit, which is soon obliterated by the apposition and fusion of its margins. Closure occurs early and is mainly effected by the growth and expansion of the surrounding viscera (heart and great vessels, lungs, trachea and oesophagus), and not by active growth of the pleuropericardial membrane across the opening to the root of the lung.
In addition to its extension in a cranial direction, the lung and its associated visceral and parietal pleurae also enlarge ventromedially and caudodorsally. With the ventromedial extension, the lungs and pleurae therefore excavate and split the somatopleuric mesenchyme over the pericardium, separating the latter from the ventral and lateral thoracic walls (Fig. 59.3B–D). The ventrolateral fibrous pericardium, parietal serous pericardium and mediastinal parietal pleura, although topographically deep, are therefore somatopleuric in origin.
Congenital disorders of the chest wall
The dorsal portion of the thorax is derived from somites, the sclerotomal portions of which form the thoracic vertebrae and ribs (Ch. 44). Vertebral anomalies, including formation of hemivertebrae and block vertebrae (where cranial and caudal sclerotomal halves do not separate), may contribute to scoliosis. Five percent of scoliosis is congenital; it may be associated with multiple other anomalies, e.g. the VACTERL (Vertebral; Anorectal; Cardiac; Tracheal; Esophageal; Renal; Limb) association or with congenital syndromes such as Marfan’s and congenital neurofibromatosis. The diagnosis may be missed at birth. There are a number of syndromes in which the chest wall does not develop properly, and the lungs are consequently hypoplastic. These include Jeune’s syndrome, asphyxiating thoracic dystrophy (an autosomal recessive condition which may be associated with short limb dwarfism and polydactyly). Rib cage abnormalities also occur in thanatrophic dwarfism, achondroplasia, chondroectodermal dysplasia and giant exomphalos. Many of these conditions are fatal soon after birth; milder forms may improve with time, but affected individuals may need prolonged respiratory support.
DIAPHRAGM
The separation of the pleural and peritoneal cavities is effected by development of the diaphragm, which forms from a portion of the septum transversum mesenchyme above the developing liver (Fig. 59.2A,B). The septum transversum is a population of mesenchymal cells which arises from the coelomic wall of the caudal part of the pericardial cavity. As the population proliferates, it forms a condensation of mesenchyme, caudal to the pericardial cavity and extending from the ventral and lateral regions of the body wall to the foregut. Dorsal to it on each side are the relatively narrow pleuroperitoneal canals. The endodermal hepatic bud grows into the caudal part of the septum transversum, whereas the cranial portion will form the diaphragm.
The oesophagus and stomach are medial to the pleuroperitoneal canals. As development proceeds, the lower portion of the oesophagus inclines ventrally anterior to the descending thoracic aorta. Although the oesophagus has no true ventral or dorsal mesentery, in descriptions of diaphragmatic development the portion of mesenchyme between the oesophagus and aorta at the level of the forming diaphragm is often homologized with part of a dorsal meso-oesophagus. The pleuroperitoneal membranes, which remain small, are dorsolateral to the canals, and the mesonephric ridges, suprarenal glands and gonads are dorsal. Just as the enlargement of the pleural cavity cranially and ventrally is effected by a process of burrowing into the body wall, so is its caudodorsal enlargement. The expanding pleural cavities extend into the mesenchyme dorsal to the suprarenal glands, the gonads and (degenerating) mesonephric ridges. Thus somatopleuric mesenchyme is peeled off the dorsal body wall to form a substantial portion of the dorsolumbar part of the diaphragm (Fig. 59.3C). The pleuroperitoneal canal is closed by the fusion of its edges, which are carried together from posterolaterally to anteromedially by growth of the organs surrounding it, in particular by growth of the suprarenal gland. The right pleuroperitoneal canal closes earlier than the left, which presumably explains why an abnormal communication persisting between the pleural and peritoneal cavities is more frequently encountered on the left.
While these changes are occurring, the septum transversum undergoes a progressive alteration in relative position. The dorsal border of the septum transversum which initially lies opposite the second cervical segment, migrates caudally as the embryo grows and the heart enlarges. At first the ventral border moves more rapidly than the dorsal, but after the embryo has attained a length of 5 mm, the dorsal border migrates more rapidly. When the dorsal border of the septum transversum lies opposite the fourth cervical segment, the phrenic nerve (C3, 4 and 5) and portions of the corresponding myotomes, grow into it and accompany it in its later migrations. The dorsal border of the septum transversum does not come to lie opposite the last thoracic and first lumbar segments, the final position occupied by some of the dorsal attachments of the diaphragm, until the end of the second month. The main derivatives of the central part of the diaphragm lie at considerably more cranial levels.
Diaphragmatic herniae
Diaphragmatic herniae may result from failure of fusion of the component parts or from a primary defect. Posterolateral defects (Bochdalek’s hernia) are the most common (85–90%) and may be bilateral (5%) or unilateral. Of the unilateral defects, the left side is more commonly affected (80%). Although these hernias have been attributed to failure of fusion of the pleuroperitoneal membrane, there is increasing evidence that the primary abnormality is lung hypoplasia, and the herniation of the abdominal contents is secondary, which has important implications for treatment (Jesudason 2002). A non-muscular membranous sac, possibly derived from the pleuroperitoneal canal wall is present in 10–15% of cases, signifying the early occurrence of this lesion prior to closure of the pleuroperitoneal canal. Hernias between the costal and sternal origins (Morgagni hernia) are rare (1–2%). Midline defects in the central tendon arise from septum transversum defects. The incidence of congenital diaphragmatic hernias is about 1 : 3000 to 1 : 5000 in neonates, with a prenatal incidence of 1 : 2000.
OESOPHAGUS
The development of the oesophagus is described in Chapters 35 and 73. Failure of separation of the oesophagus and trachea is described on page 1036. Oesophageal atresia and tracheo-oesophageal fistula may present antenatally with polyhydramnios due to failure of fetal swallowing, and choking and inability to swallow saliva in the neonatal period.
Neonatal thorax and diaphragm
In expiration, there may be active braking of airflow, caused by inspiratory muscle activity and partial constriction of the larynx: this produces grunting on expiration especially if the lungs are stiff, for example in neonatal respiratory distress. A very preterm neonate is difficult to study, but respiration is likely to be even more compromised by the compliant chest wall and lack of reserve than it is in the term newborn (Mortola 2002).
At all ages, there is a reduction, if not loss, of tonic intercostal activity during rapid eye movement (REM) sleep. The mechanism is believed to be related to a descending spinal inhibition of the muscle spindle system. In addition, although during REM sleep the diaphragm descends further, this inspiratory effort is dissipated in sucking in the ribs and enlarging the abdomen, thus the rib cage and abdominal ventilatory movements become out of phase. The neonate is at particular risk in this respect, because the chest wall is flexible, and much of the infant sleep activity is of the REM type. Furthermore, the upper airway musculature may lose tone during REM sleep, so that the soft tissues of the pharynx are sucked in during inspiration, limiting the cross-sectional area of the upper airway, and further increasing the work of breathing.
HEART AND GREAT VESSELS
CELLS THAT GIVE RISE TO THE HEART
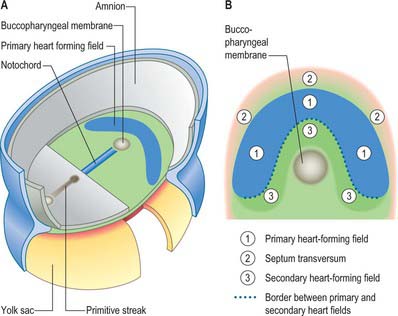
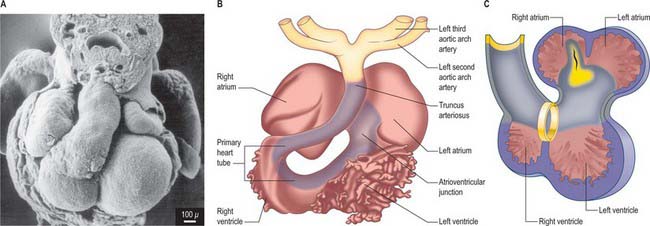
Primitive cardiac myocytes can first be seen in embryos at stage 9. During the onset of neurulation and somitogenesis, the intraembryonic coelom forms across the midline, initially above the endoderm, in a horse shoe-shaped area termed the cardiac crescent (Fig. 59.4). As the head fold emerges, the coelom undergoes a reversal, so that the future pericardial cavity comes to lie ventral to the endodermal foregut (Figs 59.1, 59.5). The splanchnopleuric wall of the pericardial coelom, subjacent to the endoderm, provides a germinal epithelium that produces early cardiac myocytes. It is characterized by the expression of myocardial specific markers, such as cardiac myosin heavy chain. This initial origin of myocardial cells is termed the primary heart-forming field, distinguishing it from later myocardial additions from mesenchyme localized central and peripheral to the cardiac crescent on the embryonic disc, termed the second heart-forming field (Fig. 59.4). It is not yet known whether these two sources of myocardium represent distinct myocardial lineages, or a single part which develops into separate components later on.
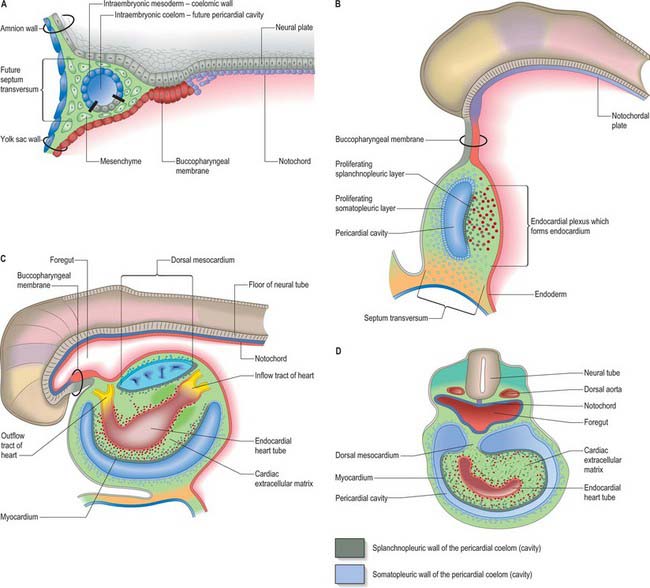
The endocardium also develops during stage 9 from the coelomic splanchnopleuric epithelium. Cells arise singly close to the ventro-lateral edges of the cranial intestinal portal and form an endocardial plexus between the splanchnopleuric coelomic epithelium and the foregut endoderm. These groups of cells are now termed angioblastic mesenchyme; they are amongst the earliest intraembryonic vascular precursors to appear and express markers for the endothelial cell lineage. The cells aggregate to form an epithelium, the endocardium, which encloses small cavities which coalesce in the vicinity of the developing foregut to establish bilateral, hollow endocardial tubes. The latter fuse across the midline progressively, commencing at the outflow tract, or arterial pole, and extending to the inflow tract, or venous pole (Fig. 59.5). By stage 10 a single endocardial tube is present and is almost completely surrounded by myocardial cells. This arrangement of an outer myocardial sleeve containing an inner endocardial tube constitutes the primary heart tube. The inner endocardial epithelium induces the myocardial cells to synthesize specific extracellular matrix proteins which form a fine extracellular reticulum that holds the endocardial tube apart from the developing myocardium. Close to the foregut endoderm, the myocardial cells at the reflections of the pericardial splanchnopleuric epithelium form the dorsal mesocardium, which may stabilize the developing endothelium and promote the fusion of the bilateral endocardial tubes. The dorsal mesocardium encompasses a mesenchymal population specifically referred to as mediastinal mesenchyme, and is contiguous with the splanchnopleuric mesenchyme surrounding the embryonic foregut.
The epicardium, sometimes included in descriptions of the myocardium as ‘epimyocardium’, is not present at the early stages of heart development (but see p. 1027).
Endocardial cushions
The extracellular matrix of the heart, historically termed cardiac jelly, promotes occlusion of the endocardial tubular lumen during myocardial contraction, thus providing mechanical assistance for the generation of the flow of blood. It also acts as a site for the deposition of inductive factors from the myocardial cells, which, in turn, modify the differentiation of specific endocardial cells. It has been called a gelatinoreticulum, a myoepicardial reticulum (Fig. 59.5C,D) and, more recently, the myocardial basement membrane. Here, the term cardiac extracellular matrix will be used. It is composed of, among other things, hyaluronic acid, hyaluronidase and fibronectin. Inductive signals originating from the myocardial cells cause a subset of endocardial cells lining the atrioventricular canal and the proximal outflow tract to transform into mesenchyme (cardiac mesenchyme); the endocardial cells in other regions of the heart tube, such as those in the ventricle, do not undergo such a transition. When activated by myocardial inductive factors, the endocardial cells lose their cell-to-cell associations, show decreased expression of neural cell adhesion molecule, and increased expression of substrate adhesion molecules such as chondroitin sulphate and fibronectin. They undergo rearrangement of their cytoskeleton necessary for migration, and they express type I procollagen. Uniquely, they retain their expression of endothelial markers. This epithelial to mesenchymal transition may, perhaps, be the only example of a mesenchymal population that is derived from an endothelial lineage.
Formation of cardiac mesenchymal cells at the atrioventricular canal and the proximal myocardial outflow tract is followed by their migration into the cardiac extracellular matrix. These cells proliferate between the endocardium and myocardium and, with local accumulation of extracellular matrix molecules, produce protrusions, termed endocardial or cardiac cushions, which bulge into the primary heart tube and initially provide the valvular mechanisms required in the atrioventricular canal and outflow tract. Their position corresponds to the future positions of the definitive cardiac valves. In the distal part of the outflow tract, which initially has myocardial walls, cells which are derived from the neural crest subsequently make significant contributions to the mesenchyme of the endocardial cushions. Although proper migration of these cells from the neural crest is crucial for normal development of the outflow tract and formation of the leaflets and sinuses of the arterial valves, their function is largely obscure. They are no longer found in the leaflets of the arterial valves in the formed heart, or in the muscular subpulmonary infundibulum, which is also derived from the outflow cushions. The endocardial cushions themselves ultimately fuse, forming a wedge of mesenchyme that serves to guide the union of the internal muscular septal structures (see p. 1024). At their time of fusion, the atrioventricular endocardial cushions are large relative to the size of the atrioventricular orifices; they will provide the scaffold for formation of the leaflets of the tricuspid and mitral valves.
Cardiac myocytes – contraction, conduction and automaticity
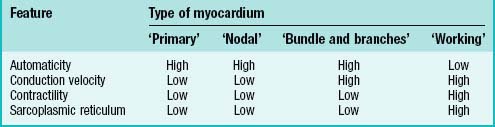
Cardiac myocytes share a number of characteristic features that distinguish them from other cells. All cardiac myocytes have sarcomeres and a sarcoplasmic reticulum and, in principle, share the capacity of producing an intrinsic cycle of electrical activity resulting in contraction. This phenomenon is called automaticity, or pacemaker activity. An absolute requirement for effective pacemaking is poor electrical coupling of the cells, which also implies slow conduction. It allows the cells to build up sufficient electrical charge which is then propagated through the surrounding myocardium. Thus ‘a small node can drive a large heart’ (see p. 1021).
Varying degrees of differentiation are seen in early populations of cardiac myocytes which can be categorized as forming working, nodal, conducting and primary myocardium (Table 59.1). Cells of the atrial and ventricular working myocardium display virtually no automaticity, but are well coupled and have well developed sarcomeres and sarcoplasmic reticular structures. The development of the synchronously (fast) contracting working myocardium requires fast conduction of the depolarizing impulse, and so the cells possess well-developed gap junctions. In marked contrast, the cells forming the nodes of the cardiac conduction system have the opposite phenotype, and resemble the myocytes which are found initially in the primary heart tube. The cells of the putative atrioventricular and peripheral ventricular conduction system have an ambiguous phenotype: the cells are well-coupled, thus allowing fast conduction of the depolarizing impulse, but otherwise retain an embryonic phenotype. The division does not imply that the cells belonging to one group are identical, but rather that they share distinguishing features developed to variable degrees.
HEART TUBE
During the process of embryonic folding, the midline splanchopleuric coelomic epithelium, which is derived from the primary heart field (cardiac crescent) becomes positioned ventral to the foregut. The splanchopleuric epithelium is proliferative and gives rise to mesenchymal populations and particularly early cardiac myocytes which arise from the coelomic wall adjacent to the endoderm of the foregut (Fig. 59.5). After folding, the second heart field contributes cells to both the arterial and venous poles of the heart. It has been suggested that the secondary heart field may contribute cells only to those cardiac components that are required for the pulmonary circulation, namely the right ventricle and outflow tract at the arterial pole, and the atrial septum and the dorsal atrial wall at the venous pole. Extending this suggestion, the original primary heart-forming field would give rise to those components that are required for the systemic circulation, namely the systemic venous sinus and its tributaries, the initial atrium, the left ventricle, and the arterial conus, as seen in the outflow tract of primitive fishes, although this latter structure has no homologue in mammalian hearts.
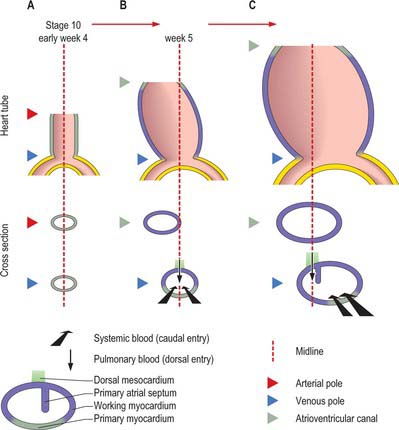
During the process of folding, the pericardial cavity and concomitantly the myocardium, gradually extend around the forming endocardial tube, leaving the dorsal mesocardium, a transient connection that is analogous to the mesentery of the intestines (Fig. 59.5C,D). The persisting stalk of the dorsal mesocardium connects the venous pole of the heart with the splanchnopleuric mesenchyme around the developing lung buds and with the septum transversum mesenchyme, which will give rise to the liver. The dorsal mesocardium is the site of early mediastinal mesenchyme production. It disappears as a mesenteric entity during the third week of development, when the embryo has from 4 to 12 somites; at the same time, the endocardial heart tube becomes entirely surrounded by the myocardium, and enclosed within the pericardial cavity. The breakdown of the dorsal mesocardium establishes a passage across the pericardial cavity, from side to side dorsal to the heart, which persists as the transverse sinus of the pericardium.
Looping of the heart tube
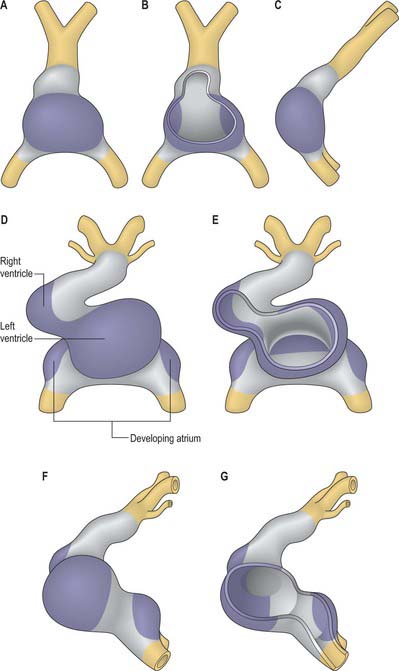
By the time the primary heart tube has formed its ventricular loop, it is possible to distinguish the atrial and ventricular components of the developing heart (Figs 59.6D-G, 59.7A,B) and to recognize an outflow tract connected with the aortic sac and developing pharyngeal arch arteries. The cells which, at this stage, make up the outflow tract, will, at later stages, be found within the right ventricle. At the proximal side of the outflow tract, cells are recruited to the developing right ventricle and, contemporaneously, new cells are recruited to the outflow tract from the second heart-forming field. The atrial and ventricular components are separated one from another by the atrioventricular canal, which at this stage has significant length. The systemic venous tributaries drain directly to the atrial cavity.
INFLOW TRACT
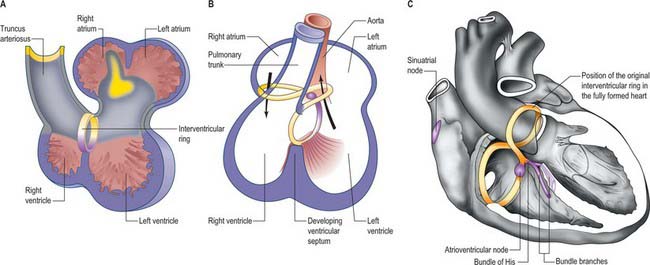
The working myocardium of the atrium differentiates locally at the dorsolateral sides of the heart tube (59.6D–G). The developing atrium then expands enormously, in dorsal, lateral and, most prominently, in a cranial direction. The cranial expansion is seen as pouches which become the left and right atrial appendages (Fig. 59.7A,B). The floor of the atrium, including the sinus venosus, and the atrioventricular canal, are made of primary myocardium. Less cellular proliferation occurs in these parts than in the expanding atrial chambers: the primary myocardium marks the inflow to, and the outflow from, the initial atrial chambers as ‘rings’ which are also associated with the formation of the conduction system (see p. 1021). The ring at the inflow defines the sinu-atrial junction, whilst the atrioventricular canal, which forms the atrial outlet during development, will eventually be incorporated into the definitive right and left atrial chambers as the atrial vestibules. Although it is possible to recognize the forming left and right atrial appendages at this stage, the right being more extensive than the left, the atrium itself has a single cavity and there is no evidence of septum formation. The myocardium of the sinus venosus and the newly forming mediastinal myocardium are smooth-walled, whereas the myocardium of the appendages shows ridges, the pectinate muscles, on the inner surface. The formation of the different appendages is under control of the Pitx2 signalling pathway. The myocardium of the appendages has a chamber phenotype, or is working myocardium; it expresses atrial natriuretic factor and connexin40 among other markers.
Right atrium
The further development of the right atrium is characterized by the incorporation of the sinus venosus into the right part of the primary atrium. This process is under control of the T-box transcription factor Tbx18. At about 4 weeks of development, the sinuatrial junction of the looping primary heart tube is positioned symmetrically in the midline (Fig. 59.8; see also Figs 13.1B, 73.8). The left and right common cardinal veins drain directly into the cavity of the primary atrium. The atrial myocardium extends to the margins of the pericardial cavity, and strictly speaking the sinus venosus is not yet formed, because the systemic venous tributaries are embedded within the mesenchyme of the septum transversum.
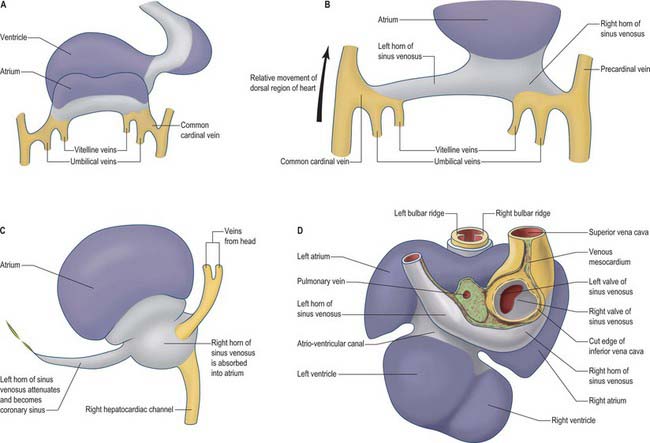
Fig. 59.8 The changes to the sinus venosus. A, Dorsal view of the early heart tube shown in Fig. 59.6D. B, Changes to the circulation brings the venous circulation to the right. This causes enlargement of the right horn of the sinus venosus and atrium and attenuation of the left horn of the sinus venosus. C, The right horn of the sinus venosus becomes absorbed into the atrium. The left horn of the sinus venosus becomes the coronary sinus. D, Dorsal view of the embryonic heart showing the relative changes to the sinus venosus.
During subsequent development, the pericardial cavity expands to enclose the terminal segments of the systemic venous tributaries, and at the same time their walls differentiate as myocardium. They can now be termed the left and right horns of the sinus venosus; each horn receives the union of the corresponding umbilical vein, vitelline radicles, and common cardinal vein (Fig. 59.8; see also Fig. 73.8). Concomitantly, the constriction between the left horn and the atrium becomes more pronounced. As the dorsal wall of the left atrium is formed from additions of mediastinal myocardium, the left horn becomes incorporated into the developing left atrioventricular junction, its orifice draining to the newly formed right atrium. At the same time, the left-sided venous tributaries diminish in size; the left common cardinal vein forms the oblique vein of the left atrium, and the left sinus horn forms the coronary sinus (Fig. 59.8), maintaining its own myocardial wall as it becomes incorporated into the atrioventricular junction.
The right sinus horn increases rapidly in size with growth of the liver (see Figs 73.8, 73.9). The vitello-umbilical blood flow enters the right horn through a wide but short hepatocardiac channel, which becomes the cranial end of the inferior vena cava. The right horn also receives the right common cardinal vein, draining the blood from the right side of the body (Fig. 59.8B,C). Later, when transverse connections are established between the cardinal veins, the blood from the left side of the body also reaches the heart via the veins draining the right side (see Fig. 13.4). As these changes take place, the right sinus horn, including the proximal parts of the superior and inferior cardinal veins, becomes incorporated into the right atrium, forming the smooth-walled systemic venous sinus, also known as the sinus venarum.
The right sinus horn opens into the right atrium through its dorsal and caudal walls (Fig. 59.8; see Fig. 59.12). The sinuatrial orifice becomes elongated and slit-like, guarded by two muscular folds, the left and right sinuatrial (venous) valves. These two valves meet cranially and become continuous with a fold that projects from the atrial roof, the septum spurium. The valves also meet caudally, and merge with the inferior atrioventricular cushion. With ongoing development, the cranial part of the right sinu-atrial valve loses its fold-like form, but its position is indicated in the adult heart by the site of the crista terminalis of the right atrium. Its caudal part forms the valve of the coronary sinus, also known as the Thebesian valve, and most of the valve of the inferior vena cava (Eustachian valve). The union of the two valvular remnants then passes through the tissue which separates the orifice of the coronary sinus from the fossa ovalis. This area is known as the ‘sinus septum’, but in reality this ‘septum’ is no more than a muscular fold in the dorsal wall of the right atrium. The continuation of the venous valves persists as the tendon of Todaro, an important landmark to the location of the atrioventricular node in the definitive heart. The left venous valve blends with the right side of the atrial septum; there is usually no trace of it in the postnatal heart.
Left atrium
Following the formation of the primary atrium and the left atrial appendage, the left atrium takes shape by the formation and incorporation of mediastinal myocardium. While the tributaries of the sinus venosus approach the atrium caudally, the differentiating pulmonary veins gain their entrance to the atrial cavity through the dorsal mesocardium (Fig. 59.5). The topographical relationships seen in the postnatal heart are thus established as soon as differentiation of the pulmonary venous portal occurs.
Early in the development of the atrium, the pulmonary vein develops as a solitary channel from angiogenic cells derived from the dorsal mesocardium, and establishes continuity with the vascular plexus formed in the mediastinal mesenchyme around the developing lung buds. The solitary pulmonary vein opens into the caudo-dorsal wall of the atrium adjacent to the developing atrioventricular junction, its atrial orifice being flanked by two prominent ridges. The primary atrial septum develops from the right ridge, following incorporation of the left sinus horn into the right side of the primary atrium, thus confining the pulmonary venous orifice to the developing left atrium (Fig. 59.12). The pulmonary vein initially branches within the dorsal mediastinal mesenchyme, its tributaries draining blood from the developing lung. With continuing development, the walls of the venous channels become surrounded by myocardium, this process occurring to the level of the second bifurcation. The veins then expand, and are incorporated into the roof of the left atrium, eventually forming the greater part of its cavity. However, all four pulmonary veins do not achieve their separate opening into the atrial roof until well after the completion of atrial septation, and probably not until the 10th or 11th week of development. Variations in the precise pattern of pulmonary venous drainage are quite common. With these changes, the left half of the primary atrium becomes progressively restricted to the mature appendage. The myocardial sleeves surrounding the pulmonary orifices taper off, and become intermingled with fibrous tissue. In later life, it is likely that this intermingling of myocardial and fibrous tissues forms the substrate for some forms of atrial fibrillation. The opening of the solitary pulmonary vein to the left of the right pulmonary ridge is an essential pre-requisite for atrial septation.
VENTRICLES
The ventricles develop at the ventral side of the looping primary heart tube during the fourth week of development (Figs 59.6, 59.7). The left ventricle develops from the stem of the Y-shaped heart, the right ventricle develops later, downstream relative to the left ventricle, when more myocardium has been added to the cardiac tube. As a result of the looping of the heart tube, the right ventricle is positioned at the right of the left ventricle, which is a prerequisite for the appropriate connection with the expanding atrial component of the heart. Unlike the atrial chambers, the morphological differences between the right and left ventricles are not part of the general asymmetry between the right- and left-sided organs of the body, but rather are under control of the signalling pathways which determine caudo-cranial differentiation. Retinoic acid, and its downstream transcription factor Tbx5, play a crucial role in this process.
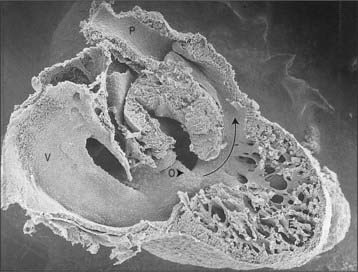
The myocardium at the inner curvature, the original dorsal side of the cardiac tube, remains smooth-walled and maintains its molecular phenotype, whereas the myocardium at the outer curvature of the myocardial tube displays trabeculations in the fifth week of development (Fig. 59.7B). By stage 17, the trabeculations have achieved a typical spatial orientation, giving a sponge-like appearance to the internal aspect of both ventricles (Fig. 59.9). The definitive trabeculations, coarse in the right ventricle but much finer in the left, are first observed about the 40th day of gestation: they appear initially in the walls of both ventricles at the level of the atrioventricular junction and develop towards the apex of the heart. By the time the fetus is 10 weeks old, the trabeculations are much sparser, and are confined to the apical regions. This process of remodeling is accomplished without the intervention of macrophages or inflammatory cells in the immediate interstitium. The ventricular myocardium, encompassing the trabeculations and exterior wall, possesses a chamber phenotype, the myocytes expressing, among other proteins, the gap-junctional protein Cx40 and atrial natriuretic peptide. This myocardium stops proliferating and differentiates into the fast-conducting peripheral ventricular conduction system, whereas the outer layer becomes highly proliferative and forms the compact layer of the ventricular wall.
OUTFLOW TRACT
Given that different authors use different terminologies, it is not surprising that formation of the outflow tract remains one of the most confusing areas of cardiac development. The recent finding that the stem of the heart tube contains precursor cells exclusively for the left ventricle makes it essential to adopt descriptions which follow dynamic events, rather than continuing to use static names. Thus, as soon as the primary heart tube bends to produce its ventral ventricular expansion, by definition the developing ventricle achieves inlet and outlet components, even though these structures are not the definitive ventricular inlets and outlets. Cells that, initially, are found in the ventricular outflow tract subsequently become cells of the right ventricle. Furthermore, working myocytes can easily be distinguished from the primary myocytes of the straight heart tube. The primary myocardium is smooth-walled, whereas the developing myocardium of the ventricles is trabeculated, and expresses specific markers such as atrial natriuretic peptide and connexin40. Thus, that part of the primary heart tube downstream of the developing ventricular loop is termed the outflow tract, irrespective of the fact that eventually it will largely become incorporated within the definitive right ventricle (Figs 59.6, 59.7). Moreover, within the inner curvature of the ventricular loop, the walls of the outflow tract and atrioventricular canal fade into one another without a clear boundary. Indeed, ventricular working myocardium never develops at the inner curvature. It is within this ventricular part of the primary heart tube that the cushions of the atrioventricular canal and the outflow tract must achieve appropriate connections with the muscular ventricular septum in order to divide this part of the tube into left and right compartments. The cavity of the tube within which these events take place has been called ‘interventricular foramen’. This is incorrect, since it is rather the region between adjacent parts of the primary heart tube. It is, nonetheless, the remodelling of this inner curvature that sets the scene for the completion of cardiac septation.
CARDIAC FUNCTION AND THE CONDUCTION SYSTEM
Although only mammalian hearts have a well-defined conduction system, an essentially similar electrocardiogram can be recorded from animals as diverse as fish and man, indicating that the electrical connections between the cardiac components have been conserved during vertebrate evolution. Indeed, the development of the conduction system of the heart is inextricably associated with the development of the basic building plan for the heart. The conduction system is best defined as the system that initiates and conducts the sinus impulse. In mammals, it encompasses the sinu-atrial node, the atrioventricular node, the atrioventricular bundle, the bundle branches and their terminal ramifications. The nodes and the atrioventricular bundle can be considered the central conduction system, while the bundle branches and their ramifications represent the peripheral ventricular conduction system. The sinus impulse is generated in the sinus node, whence it is rapidly propagated through the atrial myocardium towards the atrioventricular node, where propagation is delayed. After an essential period of atrioventricular delay, the impulse travels rapidly through the atrioventricular conduction axis and the peripheral ramifications to reach the ventricular myocardium, which is then rapidly depolarized. Following repolarization, the sequence recurs in endless fashion throughout the life of the individual. The term ‘conduction system’, as opposed to the working myocardium of the chambers, may suggest that the working myocardium does not conduct, while the conduction system conducts rapidly. In order to produce powerful synchronous contractions, the working myocardium of the chambers must also, of necessity, conduct rapidly, whereas to act as a pacemaker, the cells of the nodes need to be poorly coupled, and hence display slow conduction.
As development proceeds, the ventricular chambers develop in the cranial part of the heart tube, by ventral expansion, and the atrial chambers in the caudal part by dorso-lateral expansion (Fig. 59.6). An adult type of electrocardiogram can be recorded from such hearts, showing rapid atrial depolarization, a period of atrioventricular delay, and rapid ventricular depolarization. The electrocardiographic tracings reflect the development of fast-conducting components within a slowly conducting heart tube, i.e. the development of a morphologic conduction system. The newly developed working myocardium expresses atrial natriuretic factor, along with the gap junctional proteins connexin40 and 43, which permit fast conduction.
Like the early peristaltic heart, flow through the developing chambered heart remains directional because the dominant pacemaking activity is still at the inlet of the heart. Peristaltic hearts do not need valves, whereas chambered hearts require the presence of one-way valves at the inlets to, and the outlets from, the chambers. The slowly conducting atrioventricular canal, interposed between the atrium and the ventricle, and the outflow tract positioned between the ventricle and the great arteries, are the parts which contain the endocardial cushions. These are able to function as sphincteric valves, as a result of their prolonged duration of contraction. These components retain their function until the definitive leaflets of the one-way valves have been sculpted from the cushions. The primary myocardium of the outflow tract does not regress until the arterial valves have been formed, and does not disappear in its entirety until around the twelfth week of human development. The primary myocardium of the atrioventricular canal is eventually incorporated into the atrial vestibules at the same time as the formation of the atrioventricular valves, becoming sequestrated on the atrial side of the atrioventricular junctions between 6 and 12 weeks of development (Fig. 59.10). An important part of the canal persists dorsally, where it differentiates into the slowly conducting atrioventricular node.
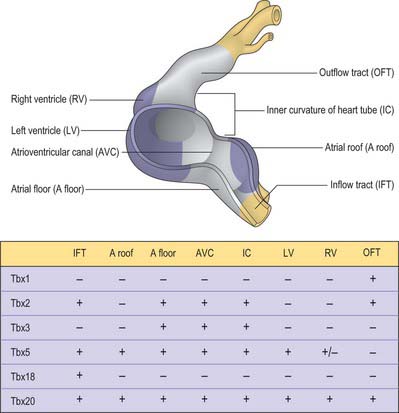
Considerable progress has been made over recent years in deciphering the pathways involved in establishing the building plan of the heart (Fig. 59.11). The combined action of the transcription factors Tbx5 and Nkx2–5 is required for the formation of the myocardium of the atrial and ventricular chambers. Tbx5 is expressed in a gradient over the heart tube, decreasing in concentration from caudal to cranial and may impose positional information. However, its pattern cannot explain the localized formation of the chambers and the conduction system. Localized expression of the transcriptional repressors Tbx2 and Tbx3 in the inflow tract, the floor of the atrium, the atrioventricular canal, the inner curvature of the ventricular region, and the outflow tract prevents the differentiation of primary into working myocardium. Tbx2 and Tbx3 effectively compete for binding to the promoters of the chamberspecific genes, such as atrial natriuretic factor and connexin40. The remaining primary myocardium of these regions initially induces the endocardium to undergo epithelial-to-mesenchymal transition, by which process the cardiac cushions become filled with mesenchyme. The myocardium also participates in the alignment of the atrial and ventricular chambers (which is essential for proper cardiac septation, and which is guided by the fusing cardiac cushions), and it forms the conduction system, including the sinus and atrioventricular nodes, the atrioventricular bundle, the floor of the developing right atrium (the internodal region), and the entirety of the atrioventricular canal. In the early chamber-forming heart of the mouse, Tbx3 is expressed in the atrioventricular canal, the floor of the atrium and around the orifices of the systemic venous tributaries, but not in the dorsal mesocardium surrounding the entrance of the pulmonary vein. During subsequent development, the domain of expression of Tbx3 expands from the atrioventricular canal to form a crescent on the crest of the ventricular septum. The dorsal aspect of this crescent develops into the atrioventricular bundle, while the atrioventricular node is formed at the dorsal junction with the atrioventricular canal. The ventral parts subsequently disappear during development.
SEPTATION OF THE EMBRYONIC CARDIAC COMPARTMENTS
The flow of blood on the right and left sides does not intermingle even in the developing heart, because flow is laminar, and the pressures are similar at the right and left sides. It is only after birth that the pressures in the ‘left’ systemic circulation become higher than those in the ‘right’ pulmonary circulation, and therefore the different flows have to be separated physically in order to prevent admixture of oxygenated and deoxygenated blood. Prior to birth the lungs are not functioning and blood flow to them is small. Even so, normal development of the heart requires that all cardiac compartments receive an adequate amount of blood. To this end, even before birth, the systemic blood of the right atrium is guided to the left atrium so that the left half of the heart and the brain is provided with a normal flow of blood. The right ventricle, in contrast, drains to the dorsal aorta via the ductus arteriosus (see Fig. 35.8). The walls that separate the right and left sides of the heart are largely formed in the fourth and fifth week of development. Some time later, not only the components of the primary heart tube, specifically the atrioventricular canal and the outflow tract, but also the atrial and ventricular chambers, must be separated physically into right and left halves. However, each compartment is separated in markedly different fashion.
Septation of the atrial chambers
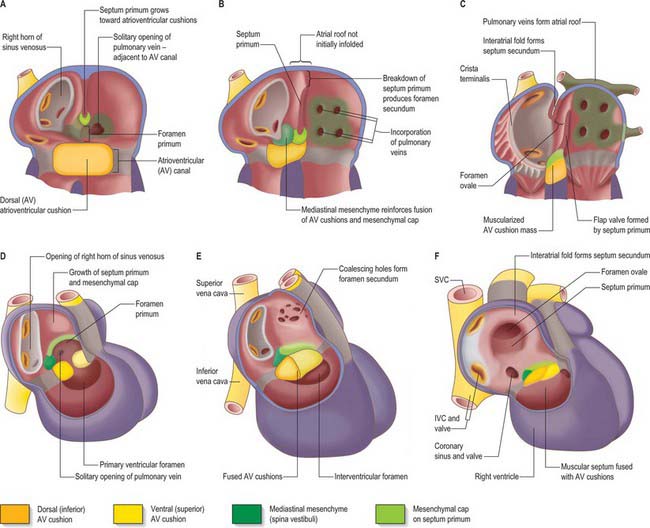
As a prelude to atrial septation, the right horn of the sinus venosus is incorporated into the right part of the atrium and the pulmonary vein arises as a midline structure within the dorsal mesocardium. Internal separation into right and left atria is mainly effected by growth of two septa coupled with remodelling of the venous compartments. At the end of the fourth week of development, a crescentic fold, the primary atrial septum or septum primum, grows by active proliferation from the dorso-cranial atrial roof into the direction of the atrioventricular canal, extending downwards just to the right of the entrance of the pulmonary vein (Fig. 59.12A,D). It is separated from the left sinu-atrial valve by the interseptovalvular space. The leading edge of the septum primum is covered by a mesenchymal cap in continuity dorsally with extracardiac mesenchyme (mediastinal mesenchyme) derived from the dorsal mesocardium which forms the vestibular spine, or spina vestibuli. The ventral horn of the septum primum reaches the ventral (cranial) atrioventricular cushion, while the dorsal horn reaches the dorsal (caudal) cushion. Thus the primary atrial foramen, the foramen or ostium primum, is entirely surrounded by mesenchyme derived from endocardium. Ventral and caudal to the advancing edge of the septum primum, the developing atrial chambers communicate through the foramen primum (Fig. 59.12A,D).
As the foramen primum diminishes in size, the upper margin of the septum primum perforates by apoptosis, and thus right to left shunting of blood continues through a secondary foramen, the foramen or ostium secundum. The latter is formed before the end of the fifth week of development. Initially it lacks an upper rim, since its upper border is the atrial roof. The foramen primum is finally occluded in the median plane, as the edge of the septum primum merges with the fusing atrioventricular cushions (Fig. 59.12B,E). The foramen secundum then enlarges to permit free passage of blood from the right to the left atrium.
The solitary opening of the pulmonary vein into the roof of the left atrium, which is originally adjacent to the atrioventricular junction, becomes incorporated into the atrial wall, ultimately forming four separate orifices at the corners of the atrial roof. As this development continues, the muscular atrial roof itself invaginates on the right side of the primary atrial septum (Fig. 59.12B,E), the invaginated fold constituting the septum secundum. The free edge of the septum secundum overlaps the foramen secundum. When left atrial pressure exceeds right atrial pressure in postnatal life, the septum primum closes against the septum secundum as a flap-valve (Fig. 59.12C,F). The passage taken by blood as it passes from the right atrium, under the crescentic edge of the septum secundum, then obliquely towards and through the foramen secundum to the left atrium, persists throughout intrauterine life as the foramen ovale. At first, the foramen ovale is sited cranio-dorsally relative to the atrial septum, but with remoulding of the venous components, it achieves a cranio-ventral position.
Septation and appropriate positioning of the atrioventricular canal
Extracellular matrix accumulates between the endocardium and myocardium of the primary heart tube. However, it mostly disappears in the regions of ballooning of the chamber myocardium of the developing atria and ventricles. The matrix becomes filled with mesenchyme in the persisting regions of the primary heart tube. In conventional accounts of the process, these regions are called the atrioventricular canal and the outflow tract, or conotruncus. The dorsal or inferior atrioventricular cushion continues into the floor of the atrium, which is made of primary myocardium. As the cushion extends dorsally, it meets the mediastinal mesenchyme in the region initially termed the spina vestibuli, where it continues as a mesenchymal cap running along the leading edge of the septum primum until it meets the ventral, or cranial, atrioventricular cushion (Fig. 59.12 and see Fig. 59.14). The dorsal cushion also has a significant ventricular extension in the inner curvature of the heart tube, which comes to lie on top of the developing muscular ventricular septum. The two atrioventricular cushions fuse in the sixth week of development, dividing the atrioventricular canal into its right and left components. The cushions are very large relative to the canal, leaving narrow right and left slits, which increase markedly in size during further development.
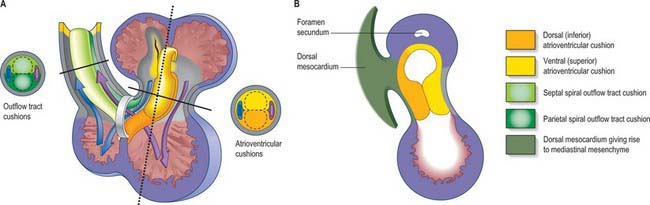
Fig. 59.14 Septation of ventricles and outflow tract. A, Frontal view of heart at stage 13 with the outflow tract reflected to the right. Lines show the level of transverse sections of the atrioventricular cushions and outflow tract. Note that blood from both atria pass through the embryonic interventricular foramen at this stage. B, Sagittal section of heart along the dotted line in A showing the atrioventicular junctional region seen from the right side. The atrioventricular cushions extend onto the leading edge of the septum primum (although the mesenchymal cap on the atrial septum, see Fig. 59.12, probably has a different developmental origin).
At the start of stage 10, the venous pole, the atrioventricular canal, the developing left ventricle and the outflow tract are all positioned symmetrically around the midline (Fig. 59.13A). As a result of the subsequent rapid growth of the cardiac tube, the atrioventricular canal moves in its entirety to the left (Fig. 59.13B), as the ventricular part of the heart tube loops to the right, thus placing the developing left ventricle on the left, and the forming right ventricle on the right (Fig. 59.7). This ventricular loop is conspicuous throughout the fourth and fifth weeks of development, and it is at this stage that a deep interventricular groove appears externally. Over this period the atrial floor, including the developing systemic venous sinus (sinus venosus) and the atrioventricular canal, move to the right (Fig. 59.13C). The developing pulmonary vein remains anchored in the midline and, subsequent to these manoeuvres, the atrioventricular canal once more becomes positioned in the midline: this process facilitates the appropriate connections of the developing muscular septum with the atrioventricular and outflow cushions. The orifice of the sinus venosus is by now positioned to the right. From the outset, the muscular ventricular septum develops in line with the right side of the dorsal (inferior) atrioventricular cushion (Fig. 59.14), so that the separating right atrium always has direct access to the developing right ventricle. The process also ensures that space remains ventrally for incorporation of the subaortic component of the outflow tract into the left ventricle.
Septation of the ventricles
Separation between the right and left ventricles is initially heralded by the appearance of a caudal crescentic ridge within the ventricular loop. The trabeculated parts of the ventricles contain less extracellular matrix than the walls of the primary heart tube, and it is these parts of the chambers that expand on each side of the ridge that remains between them. The crest of the developing septum, therefore, is its oldest portion. The more apical parts are added concomitant with enlargement of the chambers, as if they were expanding like balloons. The impression can be gained that the dorsal and ventral horns of the ventricular septum grow along the ventricular walls, meeting and fusing with the right extremities of the dorsal and ventral cushions of the atrioventricular canal. In reality, the crest of the septum marks the position of the original primary heart tube, and becomes the atrioventricular bundle. The septum has a free sickle-shaped margin that, together with the fused caudal surface of the endocardial cushions, bounds an ovoid foramen. Previously this foramen has erroneously been termed the interventricular, or bulboventricular foramen: it is no more than a locus within the cavity of the primary heart tube. The apical trabecular components of the ventricles balloon from the ventral side of the primary heart tube. At the dorsal side, or the inner curvature, there is no chamber myocardium, only the smooth walls of the primary heart tube. Therefore, from the outset of the process, the forming apical parts of the ventricles are separated by a muscular septum. The foramen marked caudally by the crest of the ventricular septum provides the initial inlet to the developing right ventricle, and the outlet for the developing left ventricle. Completion of ventricular septation requires division of this primary foramen, rather than its closure. It also demands appropriate connection between the muscular septum and the divided atrioventricular canal, including its cushions, and with the divided outflow tract, along with its cushions (Fig 59.14).
Septation and appropriate positioning of the outflow tract
Key to separation of the outflow tract is the appearance of the two outflow ridges, or cushions, which spiral one round another as they run from the distal end of the right ventricle, where they are positioned septally and parietally, to the aortic sac (Figs 59.14, 59.15). Within the aortic sac, a transverse wedge of tissue, termed the aorto-pulmonary septum, separates the origins of the arteries traversing the fourth and sixth pharyngeal arteries. The septum is formed from dorsal mediastinal mesenchyme. Myocardial precursor cells and non-myocardial cells are added to the outflow tract, the latter forming the intra-pericardial components of the arterial trunks. Neural crest cells migrate from the pharyngeal mediastinal myocardium into the outflow cushions. They do not appear to populate the septum itself, but form the larger parts of the walls of the intrapericardial portions of the truncus arteriosus. The precise relationship between the outflow cushions and the newly formed aortic arch arteries, which never possess a septum between them, has still to be assessed.

The spiral outflow ridges start to fuse from the sixth week onward, initially in the distal part of the outflow tract, and fusions continue gradually in the direction of the ventricular septum (Figs 59.15, 59.16A). The most proximal parts of the cushions remain unfused when the aorta and pulmonary trunk have gained their separate existence within the pericardial cavity. At this time, the proximal part of the outflow tract, upstream to the intrapericardial arterial trunks, remains encased in a myocardial sleeve. The arterial valves and sinuses then develop within the more distal part of this myocardial outflow tract; the outer myocardial wall does not disappear completely until well after the completion of septation. The most proximal parts of the cushions are the last ones to fuse, and they then muscularize and join the muscular ventricular septum, thereby joining the aorta into the left ventricle. The dorsal atrioventricular cushion maintains its mesenchymal character, becoming the membranous part of the interventricular septum in the formed heart. Formation of its interventricular and atrioventricular components does not become evident until the septal leaflet of the tricuspid valve delaminates from the surface of the ventricular septum.
NON-MYOCARDIAL COMPONENTS OF THE HEART
Atrioventricular valves
The atrioventricular valves develop as shelf-like projections from the margins of the atrioventricular orifices. They are directed as almost complete conical sheets towards the ventricles, their advancing edges continuing, initially as trabecular ridges, deep into the ventricular cavity. With continued differential growth and excavation on their ventricular aspects, each sheet develops major indentations forming the mitral and tricuspid orifices and defining their principal leaflets. Minor marginal indentations subdivide some leaflets into scallops. Each leaflet develops functionally significant regional variations in surface texture and its core condenses as a collagenous fibrous lamina, supported either by the fibro-fatty tissue of the atrioventricular groove, or by a fibrous anulus. The anterior leaflet of the tricuspid valve and the aortic and mural leaflets of the mitral valve all appear at about the time of fusion of the atrioventricular and outflow cushions. Delamination of the septal leaflet of the tricuspid valve occurs after the completion of ventricular septation, producing the interventricular and atrioventricular components of the membranous septum, and continuing until at least week 11 (Fig. 59.16B).
Aortic and pulmonary valves
The aortic and pulmonary valves form from endocardial cushions within the distal end of the myocardial outflow tract and become populated by neural crest cells, in addition to endocardial and pharyngeal derived mesenchyme. The fusing outflow cushions divide, providing two rudiments each for the developing aortic and pulmonary valves. The remaining leaflet for each valve is then produced from the newly appearing intercalated cushions (Fig. 59.15). Each cushion-derived intrusion grows, and is also excavated on its mural aspect to form a semilunar valvular leaflet. Similar events affect the adjacent wall. In this way, the pouches between the valves and the walls of the cushions gradually enlarge, the mural aspect of each cushion forming the valvular sinuses. The core of each leaflet forms a collagenous fibrous lamina, delicate and thin in each crescentic lunule, thick and compact in the central nodule, with marginal radiate and basal bands. The dynamic nature of cardiac development makes it difficult to describe the positions of the leaflets precisely in space and therefore it is best to describe the right, left, and non-coronary sinuses and leaflets of the aortic valve, and the adjacent and non-adjacent leaflets of the pulmonary valve.
FETAL CIRCULATION
As the cardiovascular system develops, the embryonic circulation (see Ch. 13) is modified into a fetal circulation which maintains a placental circulation and is also able to establish the pulmonary circulation at birth. Extensive remodeling of the early aortic arch arteries and (initially bilateral) dorsal aortae occurs (see Ch. 35). The dorsal aortae persist on the cranial side of the third aortic arches as continuations of the internal carotid arteries (see Fig. 35.8). The dorsal aorta between the third and fourth aortic arches, the carotid duct, diminishes and finally disappears. From the fourth arch to the origin of the seventh intersegmental artery, the right dorsal aorta becomes part of the right subclavian artery, and caudal to the seventh intersegmental artery, the right dorsal aorta disappears as far as the locus of fusion of the thoracic aortae. After disappearance of the left carotid duct, the remainder persists to form the descending part of the arch of the aorta. The fused right and left embryonic dorsal aortae persist as the definitive descending thoracic and abdominal aorta. A constriction, the aortic isthmus, is sometimes present in the aorta between the final site of origin of the left subclavian artery and the orifice of the arterial duct.
Concomitant changes also occur in the early venous system. The precardinal veins enlarge as the head and brain develop. They are further augmented by the subclavian veins from the upper limb buds, which become the chief tributaries of the common cardinal veins, gradually assuming an almost vertical position as the heart descends into the thorax (see Fig. 13.4). That part of the original precardinal vein rostral to the subclavian vein is now the internal jugular vein, and their confluence is the brachiocephalic vein of each side. The right and left common cardinal veins are originally of the same diameter: by the development of a large oblique transverse connection, the left brachiocephalic vein carries blood across from the left to the right (see Fig. 13.4). The part of the original right precardinal vein between the junction of the two brachiocephalic veins and the azygos veins forms the upper part of the superior vena cava. The caudal part of this vessel, below the entrance of the azygos vein, is formed by the right common cardinal vein. The left precardinal and left common cardinal veins caudal to the transverse branching of the left brachiocephalic vein largely atrophy: the precardinal constitutes the terminal part of the left superior intercostal vein, while the common cardinal is represented by the ligament of the left superior vena cava and the oblique vein of the left atrium (see Fig. 13.4). The remainder of the left superior intercostal vein is developed from the cranial end of the postcardinal vein and drains the second, third and, on occasion, the fourth, intercostal veins. The oblique vein passes downwards across the back of the left atrium to open into the coronary sinus, which represents the persistent left horn of the systemic venous sinus.
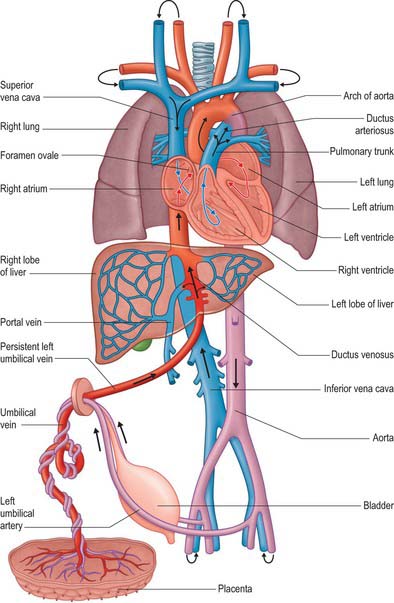
The fetal circulation contains a number of relatively large vessels which permit the majority of the blood flow to bypass the liver and lungs. The placenta serves as the organ for fetal nutrition and excretion, receiving deoxygenated fetal blood and returning it oxygenated and detoxified. Fetal blood reaches the placenta via two umbilical arteries and in early fetal life returns by two umbilical veins (Fig. 59.17). The right umbilical vein later disappears, whereas the persisting left umbilical vein enters the abdomen at the umbilicus, traverses the edge of the falciform ligament to reach the hepatic surface, and then joins the left branch of the portal vein at the hepatic portal. Opposite the junction, a large vessel, the ductus venosus, arises and ascends posterior to the liver to join the left hepatic vein near its termination in the inferior vena cava. (For a detailed developmental account of the circumhepatic veins, see Ch. 73, Figs 73.8–73.10.) The portal vein is small in the fetus compared to the size of the umbilical vein. Parts of the left branch of the umbilical vein, proximal and distal to their junctions, function as branches of the portal vein, carrying oxygenated blood to the right and left parts of the liver. Blood in the left umbilical vein therefore reaches the inferior vena cava by three routes: some enters the liver directly and reaches the vena cava via the hepatic veins; a considerable quantity circulates through the liver with portal venous blood before also entering by the hepatic veins; the remainder is bypassed into the inferior vena cava by the ductus venosus.
CHANGES IN THE FETAL CIRCULATION AND OCCLUSION OF FETAL VESSELS AFTER BIRTH
Ductus arteriosus
The ductus arteriosus shunts blood from the pulmonary trunk to the arch of the aorta, bypassing the fetal lungs (Figs 59.17, 59.18). It arises as a direct continuation of the pulmonary trunk at the point where it divides into right and left pulmonary arteries. It is 8–12 mm long, and joins the aorta at an angle of 30–35° on the left side, anterolaterally, below the origin of the left subclavian artery. The opening of the ductus arteriosus into the aorta is greatly elongated. Its diameter at its origin from the pulmonary trunk, when distended with blood, is 4–5 mm, which is nearly equal to the diameter of the adjacent ascending aorta (5–6 mm). Both arteries taper to a smaller diameter as they pass inferiorly and the aorta remains slightly larger (4 mm) (Fig. 59.19). In the neonate, the ductus arteriosus is closely related to the left primary bronchus inferiorly and the thymus gland anteriorly.
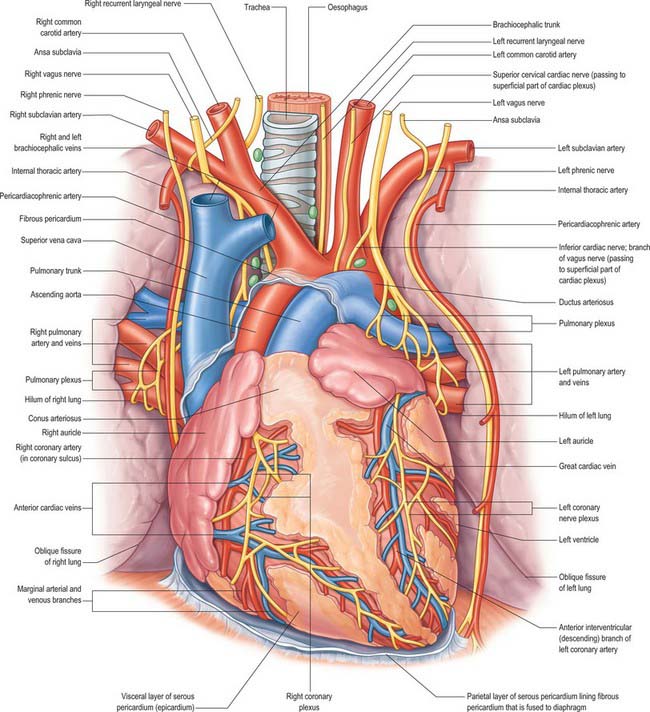
It is essential that the ductus arteriosus remains patent during intrauterine life. Prostaglandins appear to have a role in maintaining this patency. Fetal and neonatal ductal tissue can produce prostaglandins E2, I2 and F2a, which inhibit the ability of the ductus to contract in response to oxygen.
Closure of the ductus arteriosus
The first stage of ductal closure is completed within 10–15 hours and the second stage takes 2–3 weeks. The first stage consists of contraction of the smooth muscle cells and development of subendothelial oedema. Destruction of the endothelium and proliferation of the intima subsequently occurs, and leads to permanent closure. Diverse factors which may promote ductal closure have been identified. They include: increased oxygen tension; increased plasma catecholamine concentrations; suppression of prostaglandin I2 production; switching off prostaglandin E receptors; a synergistic role of prostaglandin F2a and oxygen concentrations; a decrease in plasma adenosine concentration. After birth, these interrelated events result in the closure of the ductus arteriosus. It has been proposed that the high oxygen tension of the reversed blood flow through the ductus initiates the synthesis of a hydroperoxy fatty acid which suppresses prostacyclin production, thus exposing the ductus to the contractile effects of prostaglandin endoperoxide. After closure, the duct becomes the ligamentum arteriosum, which connects the left pulmonary artery (near its origin) with the aortic arch.
FETAL AND NEONATAL HEART
At full term, the heart is situated midway between the crown of the head and the lower level of the buttocks (see Fig. 14.4). The anterior surface is formed mainly by the right atrium and right ventricle, as it is in the adult. This surface is usually covered by the thymus, which may extend over the base of the right ventricle. The heart is relatively large at birth, weighing around 20 grams. The cardiac output is around 550 ml per minute, and the blood pressure is 80/46 mmHg. The heart rate during fetal life, as term approaches, is around 150 beats per minute. It increases at birth to around 180 beats, decreases over the first 10 minutes after birth to 170 beats, and reaches 120–140 beats per minute from 15 minutes to 1 hour after birth. Any signs of fetal distress will increase this general basic level. The heart rate decreases further with increasing age: it is normally 113–127 beats per minute from 6 months to 1 year, and settles to around 100 beats per minute by the end of the first year.
Considered relative to the thoracic landmarks, the foramen ovale lies at the level of the third intercostal space, with its long axis in the median plane. It is 4–6 mm in vertical length and 3–4 mm wide (see Fig. 14.4). It is almost exactly in the coronal plane of the body, so that blood passes from the anterior, or ventral, right atrium posteriorly and upwards to reach the upper and posterior part of the left atrium. After birth, the intra-atrial pressures are equalized, and the free edge of the flap valve formed by the primary atrial septum is kept in contact with the left side of the rims of the fossa, promoting subsequent anatomic fusion, albeit that the foramen remains probe-patent in up to one-third of all individuals. The initially free crescentic margin of the infolded superior interatrial fold forms the border of the fossa after fusion; the flap valve formed by the primary septum accounts for its floor in the adult heart.
CONGENITAL HEART DEFECTS
Atrial septal defects
A persistent communication between the atrial chambers within the fossa ovale is common, and results from the failure of the flap valve of the primary atrial septum to fuse with the infolded muscular rims of the fossa. When the flap valve is still able to overlap the rims, the communication is of no functional significance as long as left atrial pressure is greater than right, which is usually the case. However, when the flap valve is smaller than the fossa ovale, or when it is perforate, there is a true atrial septal defect (Fig. 59.20A).
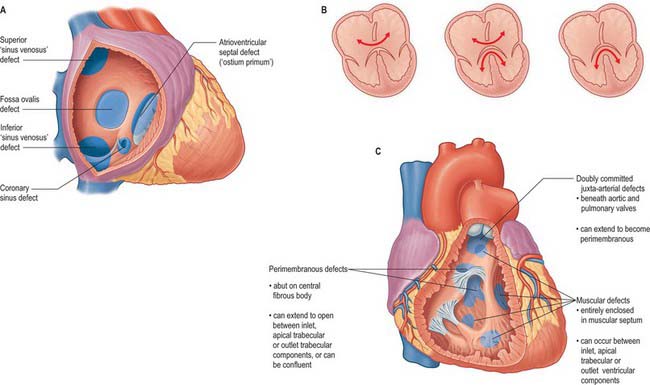
In normal development, the free leading edge of the septum primum merges with the atrioventricular endocardial cushions, permitting subsequent formation of the atrioventricular septum. When this does not happen, there is a common atrioventricular junction and an atrioventricular septal defect. This can occur when the leaflets of the atrioventricular valves are fused to the crest of the ventricular septum (Fig. 59.20A), producing an interatrial communication at the expected site of the atrioventricular septum. This is the so-called ostium primum defect, and in reality is an atrioventricular septal defect permitting exclusive atrial shunting. Other interatrial communications can be formed in the mouths of the vena cavae, most frequently the superior vena cava, and are usually associated with drainage of the right pulmonary veins into the cavo-atrial junction. Known as sinus venosus defects (Fig. 59.20A), their essential feature is a bi-atrial connection of the involved caval vein. An interatrial communication can also occur through the mouth of the coronary sinus when there is a deficiency or absence of the walls that usually separate the sinus from the left atrium.
Atrioventricular septal defects result from failure of fusion of the endocardial atrioventricular cushions, producing a common atrioventricular orifice and deficiencies of the adjacent septal structures (Fig. 59.20B). The common orifice is guarded by a basically common valve, with superior and inferior leaflets bridging the scooped-out ventricular septum and tethered in both right and left ventricles. Although the left component of the valve thus formed is often interpreted as a ‘cleft mitral valve’, in reality it bears no resemblance to the normally structured mitral valve, because it has three leaflets; the ‘cleft’ forms the zone of apposition between the left ventricular components of the bridging leaflets. Two major subgroups are identified. The more frequent pattern has a common atrioventricular orifice, and the potential for shunting through the septal defect at both atrial and ventricular levels (Fig. 59.20B). The less common form has separate right and left atrioventricular orifices, and shunting occurs only at atrial level. Occasionally, when the bridging leaflets are attached to the underside of the atrial septum, shunting is confined at ventricular level, and is typically small.
Ventricular septal defects
The most common defect of the ventricular septum occurs around the expected site of the membranous septum in the right wall of the aortic vestibule, below the zone of apposition between the non-coronary and right coronary leaflets of the aortic valve (Fig. 59.20C). The defect is closely related to the septal leaflet of the tricuspid valve, but can extend to open into the ventricular outlet beneath the supraventricular crest. It occurs because the ventricular septum is incompletely closed by its membranous component, and it is often associated with overriding of the crest of the muscular septum by the aortic orifice, together with pulmonary stenosis or atresia and hypertrophy of the right ventricle (this latter combination is known as Fallot’s tetralogy). Rarely, the pulmonary trunk can be normal or even dilated with this combination of abnormalities (Eisenmenger complex). Peri-membranous defects, which retain the remnant of the membranous septum as part of their perimeter, can also occur with abnormal ventriculo–arterial connections, e.g. the pulmonary trunk may override the muscular septum, (the Taussig–Bing anomaly). In perimembranous ventricular septal defects, the atrioventricular bundle and its right and left branches are always found along the postero-inferior margin of the defect.
ABNORMAL CONNECTIONS OF THE GREAT ARTERIES AND VEINS
A number of outflow anomalies may occur should the outflow cushions either fail to develop, or fuse in inappropriate fashion. When the spiral septum cushions fail to fuse, the result is a common arterial trunk, represented by an undivided arterial channel, guarded by a common arterial valve, positioned above and astride the free margin of the muscular ventricular septum (Fig. 59.21). There is, therefore, a coexisting juxta-arterial deficiency of the ventricular septum. The right and left pulmonary arteries usually arise via a confluent segment, but can take independent origin from the common arterial trunk, which continues as the ascending aorta. The common valve usually has three leaflets, but may have two, four or more. The lesion is almost certainly linked to abnormal migration of cells into the heart from the neural crest.
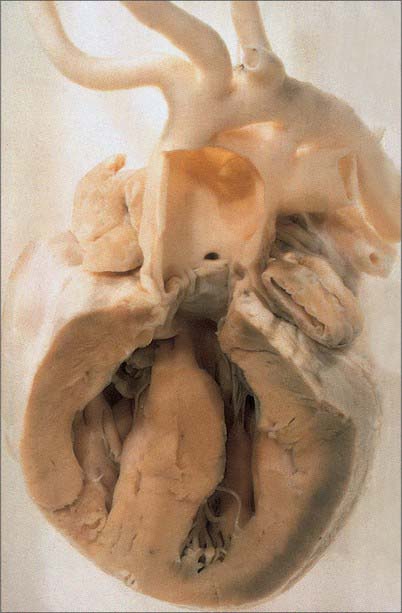
Double outlet ventricle exists when the greater parts of both arterial valves are attached within the same ventricle, almost always the right. For the circulation to continue, it is then necessary for the ventricular septum to be deficient, although the septal defect can rarely close as a secondary event. The position of the septal defect serves for subclassification. It is usually beneath the aorta or the pulmonary trunk, but can be doubly committed or even non-committed.
A right aortic arch is found most frequently with Fallot’s tetralogy or with a common arterial trunk. It can also exist, together with a left arch, in various combinations known as arterial rings, which compress the oesophagus, giving so-called dysphagia lusoria. Persistent patency of the ductus arteriosus must be distinguished from delayed closure. The persistently patent ductus can be an obligatory part of the circulation when associated with aortic or pulmonary atresia. Coarctation of the aorta can be found as an isolated lesion when the ductus arteriosus is closed, or with an open duct, when it is more likely to be associated with additional lesions within the heart (see p. 984).
NEONATAL ARTERIAL AND VENOUS VESSELS
Central venous catheterization
Small-bore catheters can be fed into large central veins or into the right atrium via needles or catheters inserted in the peripheral veins. Typically, the median cubital or basilic veins are used in the upper limb and the long saphenous vein at the medial malleolus in the lower limb. The tip of the catheter is sited at the entrance to the right atrium. The required catheter length is assessed from direct measurement of the distance between the point of surface entry in the limb to the right atrium, estimated at midsternal level. For details of umbilical catheterization, see Chapter 73.
RESPIRATORY TREE
The development of the respiratory diverticulum can first be seen at stage 12 (approximately 26 days), when there is a sharp onset of epithelial proliferation within the foregut at regions of the endoderm tube destined to become the lungs, stomach, liver and dorsal pancreas. The specialist respiratory epithelium forms from the endoderm; the other elements of the airway wall are of mesenchymal origin. The future respiratory epithelium bulges ventrally into the investing splanchnopleuric mesenchyme, then grows caudally as a bulb-shaped tube (Figs 59.1, 59.22; see Fig. 73.9). By stage 13, the caudal end of the tube has divided asymmetrically to form the future primary bronchi: with growth the right primary bronchus becomes orientated more caudally whereas the left extends more transversely. The trachea is clearly recognizable at stage 14. From this time the origin of the trachea remains close to its site of evagination from the future oesophagus, however, longitudinal growth of the trachea causes the region of the future carina to descend. Failure of complete separation between trachea and oesophagus will result in the baby being born with a tracheo-oesophageal fistula (see below). Lung development is controlled by groups of homeobox genes, transcription factors and growth factors (summarized in detail by Roth-Kleiner & Post 2005). The formation of the lung bud is dependent on the transcription factors hepatocyte nuclear factor-3β (HNF-3 β), thyroid transcription factor (TTF-1) and sonic hedgehog (SHH). Gli proteins and retinoic acid receptors are implicated in the mesenchymal-epithelial cell interactions that are essential for lung branching morphogenesis.
There is a sexual dimorphism in lung development. Androgens delay fetal lung maturation while stimulating fetal lung growth: in males, type II cells are less mature than in females as they develop during the cannalicular phase, perhaps because androgens block the effects of cortisol on fibroblast–pneumocyte factor concentrations. It is possible that in male fetuses surfactant production is delayed. The clinical counterpart is the observation that preterm males may have worse respiratory distress than females after control for other risk factors such as degree of prematurity.
TRACHEA
The trachea starts to develop at stage 12, as a ventral outgrowth from the endodermal foregut into the mesenchyme surrounding the sinus venosus and inflow tract of the heart (Fig. 59.1B,C; see Figs 73.3, 73.5). The point at which the original respiratory diverticulum buds from the foregut, the laryngotracheal groove, remains at a constant level during the embryonic period, and the trachea lengthens distally as the bifurcation point descends. The respiratory diverticulum generally becomes surrounded by angiogenic mesenchyme which connects to the developing sixth aortic arch artery and which is essential for airway branching. By stage 17, the mesenchyme around the trachea is beginning to condense to form cartilage.
Initially, the tracheal mesenchyme is continuous with that surrounding the ventral wall of the oesophagus (Fig. 59.3). Progressive lengthening and continued division of the tracheal bud, together with deviation of the lung buds dorsally, isolates the oesophagus and trachea within tissue-specific mesenchyme and facilitates regional differentiation, not only between trachea and lungs, but also within the lungs themselves, i.e. the number of lobes, or the degree of growth and maturity of a particular lung. Each lung develops by a process of dichotomous branching. For branching to occur, a cleft must develop in the tip (or side) of the epithelial tube. The epithelium then evaginates each side of the cleft, forming new branches which lengthen, and the process is then repeated. At the tips of the developing epithelial buds, the mesenchyme is flattened and densely packed, whereas it forms an ordered row of cuboidal cells along the side of the bud and in the clefts. Cells in both arrangements send processes towards the epithelial basal lamina, which is thicker in the clefts, but so attenuated as to be almost indistinguishable on the tips of the buds where the epithelium and mesenchymal cells form intimate contacts. Tenascin, an extracellular matrix molecule, is present in the budding and distal tip regions, but absent in the clefts. Conversely, fibronectin, an extracellular matrix molecule found commonly in basal laminae, is found in the clefts and along the sides of the developing bronchi, but not on the budding and distal tips.
In the normal neonate, the trachea is relatively small in relation to the larynx (Fig. 14.4). The walls of the trachea are relatively thick and the tracheal cartilages are relatively closer together than in the adult. The trachea begins at the upper border of the sixth cervical vertebra, a relationship that is conserved with growth, and it bifurcates at the level of the third or fourth thoracic vertebra.
LUNGS
Branching of the conducting airways is generally complete by week 16. Airways subsequently increase in size, but not in number. Alveolar development is largely (but not completely) a postnatal phenomenon. The development of pre-acinar blood vessels follows the conducting airway development, whereas development of intra-acinar blood vessels follows the development of the alveolar ducts and alveolar sacs. Lung development is described histologically as progressing through embryonic, pseudoglandular, canalicular, saccular and alveolar phases. (For further details see Rosenthal & Bush 2002; Bush 2005.)
Embryonic phase
The lung buds grow dorsally, passing each side of the relatively smaller oesophagus and bulge into the medial walls of the laterally situated pericardioperitoneal canals (Fig. 59.22; see Fig. 73.5A). The investing splanchnopleuric mesenchyme surrounding the lung buds contains a mixed population of cells. Further mesenchymal cells will differentiate into the smooth muscle cells that surround both the respiratory tubes and the blood vessels. In stage 13 embryos, proliferation of the adjacent splanchnopleuric coelomic epithelium (of the primary pleural cavities) is especially evident. The proliferative activity decreases in stage 14, and the mesenchyme becomes arranged in zones around the developing endoderm. Key molecules in airway branching include the fibroblast growth factors (FGF) especially FGF-10, and their four receptors. There is almost certainly redundancy in the system, since mice null for either FGF-1 or FGF-7 exhibit normal lung development, despite the potent in vitro effects of these mediators. The key receptor appears to be FGFR2IIIb, and those ligands that bind it. SHH is another gene important in lung development: it is tightly regulated and over-expression leads to the formation of excessive lung mesenchyme. The three members of the gli family of transcription factors are implicated in SHH signalling. SHH produced in distal epithelial cells appears to act on mesenchymal cells to suppress FGF-10 to limit budding. Other signals interacting with this process include bone morphogenetic protein 4 and TGF-β1.
At stage 15, angiogenetic mesenchyme is apparent around the primary bronchi. It forms an extensive capillary network around each lung bud, receiving blood from the developing sixth aortic arch artery and draining it into an anastomosis connected to the dorsal surface of the left atrium in the mediastinal mesenchyme. The pulmonary arteries arise from the sixth aortic arch arteries and the pulmonary veins develop from a solitary channel derived from the dorsal mesocardium which establishes continuity with the vascular plexus formed in the mediastinal mesenchyme. The pulmonary veins become surrounded by myocardium to the level of the second bifurcation; the veins themselves expand and are incorporated into the roof of the left atrium: cardiac muscle is therefore found in the central branches of the pulmonary venous tree (Hislop 2005).
The lung buds on each side of the oesophagus project dorsally into the pericardioperitoneal canals at stage 15. After this stage, the coelomic epithelium at the perimeter of the lung surface follows a differentiation pathway to form the visceral pleura. Lobar or secondary bronchi can be seen at stage 16, and the bronchopulmonary segments are present at stage 17 (see Fig. 73.3). Later stages of respiratory development involve the repeated division of the bronchial tree to form the subsegmental bronchi.
Alveolar phase
The formation of millions of alveoli is accomplished by a complex process of folding and division. Existing walls of distal airspaces become thinner as pneumocyte epithelial cells flatten, and some cells undergo apoptosis. Ridges subsequently grow out from the sides of the saccule walls, forming primitive alveoli. The secondary septa contain a double capillary layer, and further new alveoli form by the infolding of one of these layers, further subdividing the air spaces. The double capillary networks then undergo remodeling to form the familiar single capillary sheet around each alveolus. Conducting airways also undergo profound postnatal changes, and exhibit increased smooth muscle and bronchoconstrictor responsiveness. Although the precise molecular signalling for these processes is unknown, it is acknowledged to be tightly developmentally regulated, thus dexamethasone treatment of rats during the alveolarization period permanently abrogates alveolar development. This finding implies that iatrogenic damage to the developing alveoli when ventilating a preterm baby with oxygen-enriched mixtures is probably irreversible.
Development of the intrapulmonary vasculature and lymphatics
As the pulmonary veins enlarge they become separated from the airways by lymphatic vessels which lie within connective tissue. At around week 12, the peripheral veins develop a single layer of smooth muscle cells in their walls. This layer is derived only from the splanchnopleuric mesenchyme and not from bronchial smooth muscle (as occurs in the arteries). The lumen of each vein is relatively large and the wall is relatively thin at all levels. Arteries and veins continue to develop in the canalicular phase, probably by angiogenesis; dividing cells are seen in the peripheral capillaries. The epithelium of the most peripheral conducting airways flattens when the developing capillaries come to lie immediately subjacent to it during this phase. (For a review of the development of the pulmonary circulation see Hislop 2005.)
Lymphatic capillaries and vessels run within bronchovascular bundles. Lymphatic channels develop from outgrowths of vascular endothelial cells. The molecular trigger for lymphatic sprouting is VEGF-C and D, acting through the VEGF receptor 3 (Janér et al 2006).
Intra-uterine maturation of the lungs
During development, the mucous glands of the trachea and bronchi secrete a chloride-rich fluid, which usually passes up the respiratory tract to mix with the amniotic fluid. The relationship between lung fluid and amniotic fluid is far more complex than was previously believed. Pulmonary hypoplasia at birth may be associated with severe congenital urinary obstruction and oligohydramnios (Potter’s syndrome). In renal agenesis, reduced bronchial branching occurs as early as 12–14 weeks of gestation (i.e. at a time before amniotic fluid is produced by the kidneys), which suggests that a direct renal factor supports lung development. Later, the presence of amniotic fluid is necessary for normal fetal lung development. The fetal lung is a net fluid secretor, the output of fluid reaching as high as 5 ml/kg shortly before birth. Most of the fluid produced within the lungs remains there because of the mechanical effect exerted by amniotic fluid pressure, and normally only a small amount of this fluid contributes to the amniotic fluid. The normal functioning of the kidneys regulates the volume and pressure of the lung airway fluid and may in turn provide the pressure needed for expansion and enlargement of the bronchial and pulmonary systems. Interestingly, obstruction to the fetal airway causes accelerated maturation of alveoli; this approach has been used therapeutically in congenital diaphragmatic hernia, where intermittent inflation and deflation of an intra-tracheal balloon in a lamb model resulted in better lung growth and maturation (Nelson et al 2005).
FUNCTIONAL CHANGES IN THE LUNG AROUND THE TIME OF BIRTH
Just prior to birth, the placenta is the organ of respiration. Virtually all the venous return to the right heart is shunted away from the lungs through the foramen ovale and the ductus arteriosus. Pulmonary vascular resistance (PVR) is maintained at a very high level by the muscular precapillary vessels. At birth, the umbilical cord is tied and cut, and within seconds the lungs must take over all respiratory function in order to maintain life. They also must convert from secreting fluid to absorbing it. The mechanisms that mediate these dramatic changes are obscure. However, it is clear that the first breath results in vasodilatation by at least two mechanisms. First, the mechanical effect of traction on the vasculature: as the chest wall expands, it pulls open the pulmonary vessels. Second, oxygen entering the lungs for the first time produces pulmonary vasodilatation. The experimental demonstration that PVR falls even if the fetus is delivered into an atmosphere of pure nitrogen, implies that other mediators, including cyclo-oxygenase metabolites, are undoubtedly important. The gene for cyclo-oxygenase 1 (but not 2) in endothelium and vascular smooth muscle shows enhanced expression in late fetal and early post-natal life. Endothelin receptor expression increases around the time of birth, implying a role for this system in postnatal adaptation. The role of nitric oxide in postnatal adaptation is controversial, and there are likely to be important species differences. Nitric oxide synthase (NOS) is more abundant in young compared with mature animals, and smooth muscle sensitivity to nitric oxide may be greater at birth than in older animals. The effects of mediators may differ ante- and postnatally, e.g. endothelin (ET)-1 causes vasodilatation in fetal sheep, but vasoconstriction postnatally.
POSTNATAL LUNG DEVELOPMENT
Lung volume increases most rapidly during the first year of life and also increases more rapidly than airway calibre during this time, a finding which is consistent with the concept of dysanaptic lung growth, i.e. a dissociation between the growth of the lung parenchyma and the airways. Most of the alveoli have been formed by two years of age, and subsequent lung growth primarily results from an increase in the size of individual alveoli. Forced vital capacity is reported to be the same in male and female infants and very young children (Jones et al 2000), which is consistent with post mortem morphometric measurements. By the second year of life the rate of increase in forced vital capacity is similar to that reported in older children.
During childhood, lung volumes and flow rates increase linearly with height, with a greater intercept and more positive slope in boys compared to girls (Rosenthal et al 1993a & b). The linearity of these changes is interrupted by puberty, when important shape changes occur in the thorax, particularly in boys, and are mirrored by changes in the size of the underlying lung (and in particular the alveoli) as manifest by an abrupt increase in vital capacity, forced expired volume in one second, and total lung capacity. Thereafter, the change in lung size again proceeds in a linear fashion. High resolution computed tomographic scanning has been used to measure airway dimensions, although this technique should be used with caution because of the radiation exposure. Alveolar size can now be measured using hyperpolarized helium, using magnetic resonance imaging.
Normal postnatal pulmonary arterial development
lasts from birth to about postnatal day 4, and concerns the immediate adaptation to extrauterine life. At birth, the endothelial cells of the precapillary arteries are squat and have narrow bases on the subendothelium, a low surface : volume ratio and many surface projections. Five minutes after birth, the endothelial cells are thinner and gradually show less cell overlap, the surface : volume ratio increases, few cell projections are seen, the vessel wall becomes thinner and the lumen diameter increases (Fig. 59.23). The smooth muscle cells show a significant reduction in diameter during this time.
CONGENITAL MALFORMATIONS OF THE TRACHEA, BRONCHI AND LUNGS
This is an enormous topic and only a small number of abnormalities can be mentioned here (for further information, see Bush 2001; Abel et al 2006). The concept of the lung as six ‘trees’ has been mentioned earlier: all but the systemic venous tree may contribute to a congenital thoracic malformation (CTM). In addition, congenital abnormalities of the heart and great vessels, and of the chest and abdominal walls, including neuromuscular disease, may impact on lung development. It is somewhat artificial to describe airway malformations in isolation, and the possibility of associated vascular abnormalities must always be considered. Moreover, descriptions of what is seen clinically should be kept separate from speculations about the embryological origins of the abnormality.
Disorders of the proximal airways
Tracheomalacia and bronchomalacia
Abnormalities in cartilage development lead to the affected airways being ‘floppy’, and collapsing during inspiration or expiration, depending on whether they are outside or inside the bony thorax, respectively: these abnormalities are termed tracheomalacia when the trachea is affected, and bronchomalacia when the bronchi are involved. The abnormality may involve a small, localized area, or may be more generalized (e.g. Williams–Campbell syndrome – congenital deficiency of the airway cartilage – in which there is diffuse bronchomalacia from the second to the seventh generation of bronchi). Tracheobronchomalacia usually presents in early infancy with cough, tachypnoea, stridor and wheeze; it may also be associated with cardiac or respiratory abnormalities, such as absent pulmonary valve syndrome, or tracheo-oesophageal fistula, respectively. Patients with significant obstruction and apnoea may be treated by the insertion of airway stents if the disease is localized; generalized disease may mandate respiratory support, either with non-invasive mask ventilation during sleep, or via a tracheostomy, depending on severity. Surgical treatment by aortopexy is controversial.
Tracheo-oesophageal fistulae
Tracheo-oesophageal fistulae are the most common abnormalities of the lower respiratory tract, and occur in about 1 in 3000 births. They usually present in the newborn with recurrent respiratory distress and choking spells, although late presentation in adult life has been described. The anomaly may be associated with oesophageal atresia. Normally, the oesophagus lengthens with the rapid elongation of the embryo up to 7 weeks. The rapid proliferation of the oesophageal lumen partially obliterates the mucosa and recanalization is not complete until 8 weeks. The cellular processes that lead to separation of the trachea and oesophagus occasionally produce oesophageal atresia. Tracheo-oesophageal atresia is rare, with an incidence of 1 in 3000 to 1 in 4500 births. Five types of tracheo-oesophageal fistulae may be recognized (Fig. 59.24). In almost all cases, the oesophagus ends blindly and the stomach is connected to the lower end of the trachea. Because of this connection, the abdomen becomes rapidly distended with air once the baby is delivered and starts breathing. Prenatally, polyhydramnios may be a clinical feature, but may not be apparent until the third trimester. In two of the five types of tracheo-oesophageal fistulae there is no communication between the stomach and the upper gut. Such cases are identifiable on ultrasound, because the stomach should always be visible at a 20-week examination, and its absence on ultrasound should prompt further evaluation.
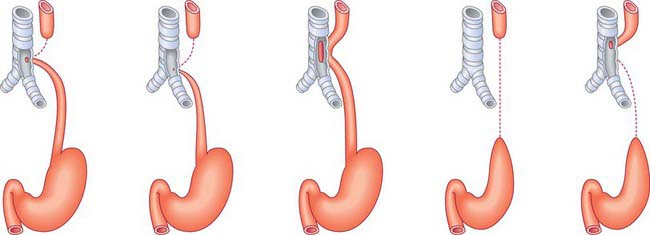
Abel R, Bush A, Chitty L, Harcourt J, Nicholson A. Congenital lung disease. In: Chernick V, Boat T, Wilmott R, Bush A. Kendig’s Disorders of the Respiratory Tract in Children. 7th edn. Edinburgh: Elsevier; 2006:280-316.
Allen J, Gripp KW. Haddad GG, Abman SH, Chernick V. Development of the thoracic cage, Basic Mechanisms of Pediatric Respiratory Disease. 2nd edn.. Hamilton, Ont: BC Decker; 2002:124-138.
Anderson RH, Webb S, Brown NA, Lamers WH, Moorman AFM. Development of the heart. 2. Septation of the atriums and ventricles. Heart. 2003;89:949-958.
Anderson RH, Webb S, Brown NA, Lamers WH, Moorman AFM. Development of the Heart. 3. Formation of the ventricular outflow tracts, arterial valves, and intrapericardial arterial trunks. Heart. 2003;89:1110-1118.
Anderson RH, Christoffels VM, Moorman AFM. Controversies concerning the anatomical definition of the conduction tissues. Anat Record B New Anat. 2004;280:8-14.
Anderson RH, Brown NA, Moorman AFM. Development and structures of the venous pole of the heart. Dev Dyn. 2006;235:2-9.
Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826-837.
Bush A. Congenital lung disease: a plea for clear thinking and clear nomenclature. Pediatr Pulmonol. 2001;32:328-337.
A new classification of congenital lung malformations..
Bush A. Asthma research: The real action is in children. Paediatr Respir Rev. 2005;6:101-110.
A review of how early problems with lung growth and development impact on later lung disease..
Christoffels VM, Burch JBE, Moorman AFM. Architectural plan for the heart: Early patterning and delineation of the chambers and the nodes. Trends in Cardiovasc Med. 2004;14:301-307.
de Lange FJ, Moorman AFM, Anderson RH, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645-654.
Hislop A. Developmental biology of the pulmonary circulation. Paediatr Respir Rev. 2005;6:35-43.
Janér J, Lassus P, Haglund C, Paavonen K, Alitalo K, Andersson S. Pulmonary vascular endothelial growth factor-C in development and lung injury in preterm infants. Am J Respir Crit Care Med. 2006;174:326-330.
The role of VEGF and its receptors in the development of the lymphatic system..
Jesudason EC. Challenging embryological theories on congenital diaphragmatic hernia: Future therapeutic implications for paediatric surgery. Ann R Coll Surg Engl. 2002;84:252-259.
Jones M, Castile R, Davis S, et al. Forced expiratory flows and volumes in infants: Normative data and lung growth. Am J Respir Crit Care Med. 2000;161:353-359.
Lamers WH, Moorman AFM. Cardiac septation: A late contribution of the embryonic primary myocardium to heart morphogenesis. Circ Res. 2002;91:93-103.
Mommersteeg MTM, Soufan AT, de Lange FJ, et al. Two distinct pools of mesenchyme contribute to the development of the atrial septum. Circ Res. 2006;99:351-353.
Moorman AFM, Christoffels VM. Cardiac chamber formation: Development, genes and evolution. Physiol Rev. 2003;83:1223-1267.
Moorman AFM, Webb S, Brown NA, Lamers WH, Anderson RH. Development of the heart: I. Formation of the cardiac chambers and arterial trunks. Heart. 2003;89:806-814.
Moorman AFM, Christoffels VM. Development of the cardiac conduction system: A matter of chamber development. Novartis Found Symp. 2003;250:25-34.
Mortola JP. Comparative aspects of neonatal respiratory mechanics. In: Haddad GG, Abman SH, Chernick V. Basic Mechanisms of Pediatric Respiratory Disease. 2nd edn. Hamilton, Ont: BC Decker; 2002:171-178.
Nelson SM, Hajivassiliou CA, Haddock G, et al. Rescue of the hypoplastic lung by prenatal cyclical strain. Am J Respir Crit Care Med. 2005;171:1395-1402.
How tracheal occlusion can be used in an animal model to improve lung growth and maturation..
Rosenthal M, Bain SH, Cramer D, Helms P, Denison D, Bush A, Warner JO. Lung function in white children aged 4–19 years: I. Spirometry. Thorax. 1993;48:794-802.
Rosenthal M, Cramer D, Bain SH, Denison D, Bush A, Warner JO. Lung function in white children aged 4–19 years: II. Single breath analysis and plethysmography. Thorax. 1993;48:803-808.
Rosenthal M, Bush A. The growing lung: Normal development, and the long-term effects of pre- and post-natal insults. Eur Respir Monogr. 2002;7:1-24. In: Bush A, Zach M, Carlsen K-H (eds) Growing up with lung disease: The lung in transition to adult life.
Roth-Kleiner M, Post M. Similarities and dissimilarities of branching and septation during lung development. Pediatr Pulmonol. 2005;40:113-134.
An account of the molecular mechanisms underlying normal lung development..
Soufan AT, van den Hoff MJB, Ruijter JM. Reconstruction of the patterns of gene expression in the developing mouse heart reveals an architectural arrangement that facilitates the understanding of atrial malformations and arrhythmias. Circ Res. 2004;95:1207-1215.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 59 Development of the thorax
THORACIC WALL AND DIAPHRAGM
The thorax as an entity is not apparent in embryos until the end of the embryonic stage of development (stage 23). It develops around the early pericardial cavity and the associated pericardioperitoneal canals (Fig. 59.1A; see Fig. 59.3A). The pericardioperitoneal canals give rise to the pleural cavities surrounding the lungs and that part of the peritoneal cavity surrounding the lower end of the foregut, which becomes the oesophagus, stomach and duodenum within the thoracic cage. Later these portions of peritoneal cavity are sequestered into the abdominal cavity by the development of the diaphragm.
In stage 14 embryos, the heart is at the level of the upper cervical somites and above the upper limb buds. The thoracic somites are opposite the midgut. The putative thoracic region contains the pericardial cavity ventrally and the pericardioperitoneal canals posteriorly, on each side of the foregut. The future pleural cavities are as yet undefined regions of the pericardioperitoneal canals. Below the heart, septum transversum mesenchyme (see Fig. 73.5A) that has arisen from the caudal pericardial wall is being invaginated by endodermal epithelial cells from the foregut hepatic primordium.
The heart and pericardial cavity are relatively large in the early embryo (Fig. 59.1B; see Fig. 59.3A). Throughout development the lungs remain unexpanded and do not achieve their full size within the thorax, reflecting the fact that the placenta, and not the lungs, is the organ of fetal respiration. The lower partition of the thorax, the diaphragm, can be identified in stage 13 embryos; it migrates caudally in line with the craniocaudal progression of development and the elongation of the neck. The parietal pericardium remains attached to the diaphragm as it descends.
The lung buds are invested by splanchnopleuric mesenchyme derived from the medial walls of the pericardioperitoneal canals, whereas the lateral walls produce somatopleuric mesenchyme, which contributes to the body wall. This latter mesenchyme is penetrated by the developing ribs which arise from the thoracic sclerotomes. In the midline, the somatopleuric mesenchyme gives rise to the sternum and costal cartilages. The bony and cartilaginous cage provides insertions for the intercostal muscles, which arise from the ventrolateral edge of the epithelial plate of the somites. The somatopleuric coelomic epithelium, after its proliferative phase, gives rise to the mesothelium of the parietal layer of pleura.
As the lung buds project into the pericardioperitoneal canals (Figs 59.1C, 59.2A), they subdivide them into primary pleural coeloms around the lung buds cranially, and paired peritoneal coeloms caudally, which are continuous with the wider peritoneal coelom around the mid- and hindguts. The communications with the pericardial and peritoneal coeloms become termed the pleuropericardial and pleuroperitoneal canals, respectively. When separation between these fluid-filled major coelomic regions is advancing towards completion, they are named the pericardial, pleural and peritoneal cavities. In early embryos, the cavities retain substantial volumes of fluid and their walls are separate: they provide the route for a primitive type of circulation until superseded by the blood vascular system. In later fetal and postnatal life, the cavity walls are coapted, so that a mere microscopic film of serous fluid intervenes between them.
A curved elevation of tissue, the pulmonary ridge, develops on the lateral wall of the pleural coelom and partly encircles the pleuropericardial canal. The ridge is continuous with the dorsolateral edge of the septum transversum. The developing lung bud abuts on the ridge, which as a result divides into two diverging membranes meeting at the septum transversum. One is cranially placed and termed the pleuropericardial membrane (Fig. 59.2A); embedded within it are the common cardinal vein and phrenic nerve, which reach the septum transversum by this route. The other membrane, caudally placed, is termed the pleuroperitoneal membrane (Fig. 59.2A,B). As the apical part of the lung forms, it invades and splits the body wall and extends cranially on the lateral aspect of the common cardinal vein, preceded by an extension from the primary pleural coelom to form part of the secondary, definitive, pleural sac. In this way the common cardinal vein and the phrenic nerve come to lie medially in the mediastinum. The pleuropericardial canal, which lies medial to the vein, is gradually narrowed to a slit, which is soon obliterated by the apposition and fusion of its margins. Closure occurs early and is mainly effected by the growth and expansion of the surrounding viscera (heart and great vessels, lungs, trachea and oesophagus), and not by active growth of the pleuropericardial membrane across the opening to the root of the lung.
In addition to its extension in a cranial direction, the lung and its associated visceral and parietal pleurae also enlarge ventromedially and caudodorsally. With the ventromedial extension, the lungs and pleurae therefore excavate and split the somatopleuric mesenchyme over the pericardium, separating the latter from the ventral and lateral thoracic walls (Fig. 59.3B–D). The ventrolateral fibrous pericardium, parietal serous pericardium and mediastinal parietal pleura, although topographically deep, are therefore somatopleuric in origin.
Congenital disorders of the chest wall
The dorsal portion of the thorax is derived from somites, the sclerotomal portions of which form the thoracic vertebrae and ribs (Ch. 44). Vertebral anomalies, including formation of hemivertebrae and block vertebrae (where cranial and caudal sclerotomal halves do not separate), may contribute to scoliosis. Five percent of scoliosis is congenital; it may be associated with multiple other anomalies, e.g. the VACTERL (Vertebral; Anorectal; Cardiac; Tracheal; Esophageal; Renal; Limb) association or with congenital syndromes such as Marfan’s and congenital neurofibromatosis. The diagnosis may be missed at birth. There are a number of syndromes in which the chest wall does not develop properly, and the lungs are consequently hypoplastic. These include Jeune’s syndrome, asphyxiating thoracic dystrophy (an autosomal recessive condition which may be associated with short limb dwarfism and polydactyly). Rib cage abnormalities also occur in thanatrophic dwarfism, achondroplasia, chondroectodermal dysplasia and giant exomphalos. Many of these conditions are fatal soon after birth; milder forms may improve with time, but affected individuals may need prolonged respiratory support.
DIAPHRAGM
The separation of the pleural and peritoneal cavities is effected by development of the diaphragm, which forms from a portion of the septum transversum mesenchyme above the developing liver (Fig. 59.2A,B). The septum transversum is a population of mesenchymal cells which arises from the coelomic wall of the caudal part of the pericardial cavity. As the population proliferates, it forms a condensation of mesenchyme, caudal to the pericardial cavity and extending from the ventral and lateral regions of the body wall to the foregut. Dorsal to it on each side are the relatively narrow pleuroperitoneal canals. The endodermal hepatic bud grows into the caudal part of the septum transversum, whereas the cranial portion will form the diaphragm.
The oesophagus and stomach are medial to the pleuroperitoneal canals. As development proceeds, the lower portion of the oesophagus inclines ventrally anterior to the descending thoracic aorta. Although the oesophagus has no true ventral or dorsal mesentery, in descriptions of diaphragmatic development the portion of mesenchyme between the oesophagus and aorta at the level of the forming diaphragm is often homologized with part of a dorsal meso-oesophagus. The pleuroperitoneal membranes, which remain small, are dorsolateral to the canals, and the mesonephric ridges, suprarenal glands and gonads are dorsal. Just as the enlargement of the pleural cavity cranially and ventrally is effected by a process of burrowing into the body wall, so is its caudodorsal enlargement. The expanding pleural cavities extend into the mesenchyme dorsal to the suprarenal glands, the gonads and (degenerating) mesonephric ridges. Thus somatopleuric mesenchyme is peeled off the dorsal body wall to form a substantial portion of the dorsolumbar part of the diaphragm (Fig. 59.3C). The pleuroperitoneal canal is closed by the fusion of its edges, which are carried together from posterolaterally to anteromedially by growth of the organs surrounding it, in particular by growth of the suprarenal gland. The right pleuroperitoneal canal closes earlier than the left, which presumably explains why an abnormal communication persisting between the pleural and peritoneal cavities is more frequently encountered on the left.
While these changes are occurring, the septum transversum undergoes a progressive alteration in relative position. The dorsal border of the septum transversum which initially lies opposite the second cervical segment, migrates caudally as the embryo grows and the heart enlarges. At first the ventral border moves more rapidly than the dorsal, but after the embryo has attained a length of 5 mm, the dorsal border migrates more rapidly. When the dorsal border of the septum transversum lies opposite the fourth cervical segment, the phrenic nerve (C3, 4 and 5) and portions of the corresponding myotomes, grow into it and accompany it in its later migrations. The dorsal border of the septum transversum does not come to lie opposite the last thoracic and first lumbar segments, the final position occupied by some of the dorsal attachments of the diaphragm, until the end of the second month. The main derivatives of the central part of the diaphragm lie at considerably more cranial levels.
Diaphragmatic herniae
Diaphragmatic herniae may result from failure of fusion of the component parts or from a primary defect. Posterolateral defects (Bochdalek’s hernia) are the most common (85–90%) and may be bilateral (5%) or unilateral. Of the unilateral defects, the left side is more commonly affected (80%). Although these hernias have been attributed to failure of fusion of the pleuroperitoneal membrane, there is increasing evidence that the primary abnormality is lung hypoplasia, and the herniation of the abdominal contents is secondary, which has important implications for treatment (Jesudason 2002). A non-muscular membranous sac, possibly derived from the pleuroperitoneal canal wall is present in 10–15% of cases, signifying the early occurrence of this lesion prior to closure of the pleuroperitoneal canal. Hernias between the costal and sternal origins (Morgagni hernia) are rare (1–2%). Midline defects in the central tendon arise from septum transversum defects. The incidence of congenital diaphragmatic hernias is about 1 : 3000 to 1 : 5000 in neonates, with a prenatal incidence of 1 : 2000.
OESOPHAGUS
The development of the oesophagus is described in Chapters 35 and 73. Failure of separation of the oesophagus and trachea is described on page 1036. Oesophageal atresia and tracheo-oesophageal fistula may present antenatally with polyhydramnios due to failure of fetal swallowing, and choking and inability to swallow saliva in the neonatal period.
Neonatal thorax and diaphragm
In expiration, there may be active braking of airflow, caused by inspiratory muscle activity and partial constriction of the larynx: this produces grunting on expiration especially if the lungs are stiff, for example in neonatal respiratory distress. A very preterm neonate is difficult to study, but respiration is likely to be even more compromised by the compliant chest wall and lack of reserve than it is in the term newborn (Mortola 2002).
At all ages, there is a reduction, if not loss, of tonic intercostal activity during rapid eye movement (REM) sleep. The mechanism is believed to be related to a descending spinal inhibition of the muscle spindle system. In addition, although during REM sleep the diaphragm descends further, this inspiratory effort is dissipated in sucking in the ribs and enlarging the abdomen, thus the rib cage and abdominal ventilatory movements become out of phase. The neonate is at particular risk in this respect, because the chest wall is flexible, and much of the infant sleep activity is of the REM type. Furthermore, the upper airway musculature may lose tone during REM sleep, so that the soft tissues of the pharynx are sucked in during inspiration, limiting the cross-sectional area of the upper airway, and further increasing the work of breathing.
HEART AND GREAT VESSELS
CELLS THAT GIVE RISE TO THE HEART
Primitive cardiac myocytes can first be seen in embryos at stage 9. During the onset of neurulation and somitogenesis, the intraembryonic coelom forms across the midline, initially above the endoderm, in a horse shoe-shaped area termed the cardiac crescent (Fig. 59.4). As the head fold emerges, the coelom undergoes a reversal, so that the future pericardial cavity comes to lie ventral to the endodermal foregut (Figs 59.1, 59.5). The splanchnopleuric wall of the pericardial coelom, subjacent to the endoderm, provides a germinal epithelium that produces early cardiac myocytes. It is characterized by the expression of myocardial specific markers, such as cardiac myosin heavy chain. This initial origin of myocardial cells is termed the primary heart-forming field, distinguishing it from later myocardial additions from mesenchyme localized central and peripheral to the cardiac crescent on the embryonic disc, termed the second heart-forming field (Fig. 59.4). It is not yet known whether these two sources of myocardium represent distinct myocardial lineages, or a single part which develops into separate components later on.
The endocardium also develops during stage 9 from the coelomic splanchnopleuric epithelium. Cells arise singly close to the ventro-lateral edges of the cranial intestinal portal and form an endocardial plexus between the splanchnopleuric coelomic epithelium and the foregut endoderm. These groups of cells are now termed angioblastic mesenchyme; they are amongst the earliest intraembryonic vascular precursors to appear and express markers for the endothelial cell lineage. The cells aggregate to form an epithelium, the endocardium, which encloses small cavities which coalesce in the vicinity of the developing foregut to establish bilateral, hollow endocardial tubes. The latter fuse across the midline progressively, commencing at the outflow tract, or arterial pole, and extending to the inflow tract, or venous pole (Fig. 59.5). By stage 10 a single endocardial tube is present and is almost completely surrounded by myocardial cells. This arrangement of an outer myocardial sleeve containing an inner endocardial tube constitutes the primary heart tube. The inner endocardial epithelium induces the myocardial cells to synthesize specific extracellular matrix proteins which form a fine extracellular reticulum that holds the endocardial tube apart from the developing myocardium. Close to the foregut endoderm, the myocardial cells at the reflections of the pericardial splanchnopleuric epithelium form the dorsal mesocardium, which may stabilize the developing endothelium and promote the fusion of the bilateral endocardial tubes. The dorsal mesocardium encompasses a mesenchymal population specifically referred to as mediastinal mesenchyme, and is contiguous with the splanchnopleuric mesenchyme surrounding the embryonic foregut.
The epicardium, sometimes included in descriptions of the myocardium as ‘epimyocardium’, is not present at the early stages of heart development (but see p. 1027).
Endocardial cushions
The extracellular matrix of the heart, historically termed cardiac jelly, promotes occlusion of the endocardial tubular lumen during myocardial contraction, thus providing mechanical assistance for the generation of the flow of blood. It also acts as a site for the deposition of inductive factors from the myocardial cells, which, in turn, modify the differentiation of specific endocardial cells. It has been called a gelatinoreticulum, a myoepicardial reticulum (Fig. 59.5C,D) and, more recently, the myocardial basement membrane. Here, the term cardiac extracellular matrix will be used. It is composed of, among other things, hyaluronic acid, hyaluronidase and fibronectin. Inductive signals originating from the myocardial cells cause a subset of endocardial cells lining the atrioventricular canal and the proximal outflow tract to transform into mesenchyme (cardiac mesenchyme); the endocardial cells in other regions of the heart tube, such as those in the ventricle, do not undergo such a transition. When activated by myocardial inductive factors, the endocardial cells lose their cell-to-cell associations, show decreased expression of neural cell adhesion molecule, and increased expression of substrate adhesion molecules such as chondroitin sulphate and fibronectin. They undergo rearrangement of their cytoskeleton necessary for migration, and they express type I procollagen. Uniquely, they retain their expression of endothelial markers. This epithelial to mesenchymal transition may, perhaps, be the only example of a mesenchymal population that is derived from an endothelial lineage.
Formation of cardiac mesenchymal cells at the atrioventricular canal and the proximal myocardial outflow tract is followed by their migration into the cardiac extracellular matrix. These cells proliferate between the endocardium and myocardium and, with local accumulation of extracellular matrix molecules, produce protrusions, termed endocardial or cardiac cushions, which bulge into the primary heart tube and initially provide the valvular mechanisms required in the atrioventricular canal and outflow tract. Their position corresponds to the future positions of the definitive cardiac valves. In the distal part of the outflow tract, which initially has myocardial walls, cells which are derived from the neural crest subsequently make significant contributions to the mesenchyme of the endocardial cushions. Although proper migration of these cells from the neural crest is crucial for normal development of the outflow tract and formation of the leaflets and sinuses of the arterial valves, their function is largely obscure. They are no longer found in the leaflets of the arterial valves in the formed heart, or in the muscular subpulmonary infundibulum, which is also derived from the outflow cushions. The endocardial cushions themselves ultimately fuse, forming a wedge of mesenchyme that serves to guide the union of the internal muscular septal structures (see p. 1024). At their time of fusion, the atrioventricular endocardial cushions are large relative to the size of the atrioventricular orifices; they will provide the scaffold for formation of the leaflets of the tricuspid and mitral valves.
Cardiac myocytes – contraction, conduction and automaticity
Cardiac myocytes share a number of characteristic features that distinguish them from other cells. All cardiac myocytes have sarcomeres and a sarcoplasmic reticulum and, in principle, share the capacity of producing an intrinsic cycle of electrical activity resulting in contraction. This phenomenon is called automaticity, or pacemaker activity. An absolute requirement for effective pacemaking is poor electrical coupling of the cells, which also implies slow conduction. It allows the cells to build up sufficient electrical charge which is then propagated through the surrounding myocardium. Thus ‘a small node can drive a large heart’ (see p. 1021).
Varying degrees of differentiation are seen in early populations of cardiac myocytes which can be categorized as forming working, nodal, conducting and primary myocardium (Table 59.1). Cells of the atrial and ventricular working myocardium display virtually no automaticity, but are well coupled and have well developed sarcomeres and sarcoplasmic reticular structures. The development of the synchronously (fast) contracting working myocardium requires fast conduction of the depolarizing impulse, and so the cells possess well-developed gap junctions. In marked contrast, the cells forming the nodes of the cardiac conduction system have the opposite phenotype, and resemble the myocytes which are found initially in the primary heart tube. The cells of the putative atrioventricular and peripheral ventricular conduction system have an ambiguous phenotype: the cells are well-coupled, thus allowing fast conduction of the depolarizing impulse, but otherwise retain an embryonic phenotype. The division does not imply that the cells belonging to one group are identical, but rather that they share distinguishing features developed to variable degrees.
HEART TUBE
During the process of embryonic folding, the midline splanchopleuric coelomic epithelium, which is derived from the primary heart field (cardiac crescent) becomes positioned ventral to the foregut. The splanchopleuric epithelium is proliferative and gives rise to mesenchymal populations and particularly early cardiac myocytes which arise from the coelomic wall adjacent to the endoderm of the foregut (Fig. 59.5). After folding, the second heart field contributes cells to both the arterial and venous poles of the heart. It has been suggested that the secondary heart field may contribute cells only to those cardiac components that are required for the pulmonary circulation, namely the right ventricle and outflow tract at the arterial pole, and the atrial septum and the dorsal atrial wall at the venous pole. Extending this suggestion, the original primary heart-forming field would give rise to those components that are required for the systemic circulation, namely the systemic venous sinus and its tributaries, the initial atrium, the left ventricle, and the arterial conus, as seen in the outflow tract of primitive fishes, although this latter structure has no homologue in mammalian hearts.
During the process of folding, the pericardial cavity and concomitantly the myocardium, gradually extend around the forming endocardial tube, leaving the dorsal mesocardium, a transient connection that is analogous to the mesentery of the intestines (Fig. 59.5C,D). The persisting stalk of the dorsal mesocardium connects the venous pole of the heart with the splanchnopleuric mesenchyme around the developing lung buds and with the septum transversum mesenchyme, which will give rise to the liver. The dorsal mesocardium is the site of early mediastinal mesenchyme production. It disappears as a mesenteric entity during the third week of development, when the embryo has from 4 to 12 somites; at the same time, the endocardial heart tube becomes entirely surrounded by the myocardium, and enclosed within the pericardial cavity. The breakdown of the dorsal mesocardium establishes a passage across the pericardial cavity, from side to side dorsal to the heart, which persists as the transverse sinus of the pericardium.
Looping of the heart tube
By the time the primary heart tube has formed its ventricular loop, it is possible to distinguish the atrial and ventricular components of the developing heart (Figs 59.6D-G, 59.7A,B) and to recognize an outflow tract connected with the aortic sac and developing pharyngeal arch arteries. The cells which, at this stage, make up the outflow tract, will, at later stages, be found within the right ventricle. At the proximal side of the outflow tract, cells are recruited to the developing right ventricle and, contemporaneously, new cells are recruited to the outflow tract from the second heart-forming field. The atrial and ventricular components are separated one from another by the atrioventricular canal, which at this stage has significant length. The systemic venous tributaries drain directly to the atrial cavity.
INFLOW TRACT
The working myocardium of the atrium differentiates locally at the dorsolateral sides of the heart tube (59.6D–G). The developing atrium then expands enormously, in dorsal, lateral and, most prominently, in a cranial direction. The cranial expansion is seen as pouches which become the left and right atrial appendages (Fig. 59.7A,B). The floor of the atrium, including the sinus venosus, and the atrioventricular canal, are made of primary myocardium. Less cellular proliferation occurs in these parts than in the expanding atrial chambers: the primary myocardium marks the inflow to, and the outflow from, the initial atrial chambers as ‘rings’ which are also associated with the formation of the conduction system (see p. 1021). The ring at the inflow defines the sinu-atrial junction, whilst the atrioventricular canal, which forms the atrial outlet during development, will eventually be incorporated into the definitive right and left atrial chambers as the atrial vestibules. Although it is possible to recognize the forming left and right atrial appendages at this stage, the right being more extensive than the left, the atrium itself has a single cavity and there is no evidence of septum formation. The myocardium of the sinus venosus and the newly forming mediastinal myocardium are smooth-walled, whereas the myocardium of the appendages shows ridges, the pectinate muscles, on the inner surface. The formation of the different appendages is under control of the Pitx2 signalling pathway. The myocardium of the appendages has a chamber phenotype, or is working myocardium; it expresses atrial natriuretic factor and connexin40 among other markers.
Right atrium
The further development of the right atrium is characterized by the incorporation of the sinus venosus into the right part of the primary atrium. This process is under control of the T-box transcription factor Tbx18. At about 4 weeks of development, the sinuatrial junction of the looping primary heart tube is positioned symmetrically in the midline (Fig. 59.8; see also Figs 13.1B, 73.8). The left and right common cardinal veins drain directly into the cavity of the primary atrium. The atrial myocardium extends to the margins of the pericardial cavity, and strictly speaking the sinus venosus is not yet formed, because the systemic venous tributaries are embedded within the mesenchyme of the septum transversum.
Fig. 59.8 The changes to the sinus venosus. A, Dorsal view of the early heart tube shown in Fig. 59.6D. B, Changes to the circulation brings the venous circulation to the right. This causes enlargement of the right horn of the sinus venosus and atrium and attenuation of the left horn of the sinus venosus. C, The right horn of the sinus venosus becomes absorbed into the atrium. The left horn of the sinus venosus becomes the coronary sinus. D, Dorsal view of the embryonic heart showing the relative changes to the sinus venosus.
During subsequent development, the pericardial cavity expands to enclose the terminal segments of the systemic venous tributaries, and at the same time their walls differentiate as myocardium. They can now be termed the left and right horns of the sinus venosus; each horn receives the union of the corresponding umbilical vein, vitelline radicles, and common cardinal vein (Fig. 59.8; see also Fig. 73.8). Concomitantly, the constriction between the left horn and the atrium becomes more pronounced. As the dorsal wall of the left atrium is formed from additions of mediastinal myocardium, the left horn becomes incorporated into the developing left atrioventricular junction, its orifice draining to the newly formed right atrium. At the same time, the left-sided venous tributaries diminish in size; the left common cardinal vein forms the oblique vein of the left atrium, and the left sinus horn forms the coronary sinus (Fig. 59.8), maintaining its own myocardial wall as it becomes incorporated into the atrioventricular junction.
The right sinus horn increases rapidly in size with growth of the liver (see Figs 73.8, 73.9). The vitello-umbilical blood flow enters the right horn through a wide but short hepatocardiac channel, which becomes the cranial end of the inferior vena cava. The right horn also receives the right common cardinal vein, draining the blood from the right side of the body (Fig. 59.8B,C). Later, when transverse connections are established between the cardinal veins, the blood from the left side of the body also reaches the heart via the veins draining the right side (see Fig. 13.4). As these changes take place, the right sinus horn, including the proximal parts of the superior and inferior cardinal veins, becomes incorporated into the right atrium, forming the smooth-walled systemic venous sinus, also known as the sinus venarum.
The right sinus horn opens into the right atrium through its dorsal and caudal walls (Fig. 59.8; see Fig. 59.12). The sinuatrial orifice becomes elongated and slit-like, guarded by two muscular folds, the left and right sinuatrial (venous) valves. These two valves meet cranially and become continuous with a fold that projects from the atrial roof, the septum spurium. The valves also meet caudally, and merge with the inferior atrioventricular cushion. With ongoing development, the cranial part of the right sinu-atrial valve loses its fold-like form, but its position is indicated in the adult heart by the site of the crista terminalis of the right atrium. Its caudal part forms the valve of the coronary sinus, also known as the Thebesian valve, and most of the valve of the inferior vena cava (Eustachian valve). The union of the two valvular remnants then passes through the tissue which separates the orifice of the coronary sinus from the fossa ovalis. This area is known as the ‘sinus septum’, but in reality this ‘septum’ is no more than a muscular fold in the dorsal wall of the right atrium. The continuation of the venous valves persists as the tendon of Todaro, an important landmark to the location of the atrioventricular node in the definitive heart. The left venous valve blends with the right side of the atrial septum; there is usually no trace of it in the postnatal heart.
Left atrium
Following the formation of the primary atrium and the left atrial appendage, the left atrium takes shape by the formation and incorporation of mediastinal myocardium. While the tributaries of the sinus venosus approach the atrium caudally, the differentiating pulmonary veins gain their entrance to the atrial cavity through the dorsal mesocardium (Fig. 59.5). The topographical relationships seen in the postnatal heart are thus established as soon as differentiation of the pulmonary venous portal occurs.
Early in the development of the atrium, the pulmonary vein develops as a solitary channel from angiogenic cells derived from the dorsal mesocardium, and establishes continuity with the vascular plexus formed in the mediastinal mesenchyme around the developing lung buds. The solitary pulmonary vein opens into the caudo-dorsal wall of the atrium adjacent to the developing atrioventricular junction, its atrial orifice being flanked by two prominent ridges. The primary atrial septum develops from the right ridge, following incorporation of the left sinus horn into the right side of the primary atrium, thus confining the pulmonary venous orifice to the developing left atrium (Fig. 59.12). The pulmonary vein initially branches within the dorsal mediastinal mesenchyme, its tributaries draining blood from the developing lung. With continuing development, the walls of the venous channels become surrounded by myocardium, this process occurring to the level of the second bifurcation. The veins then expand, and are incorporated into the roof of the left atrium, eventually forming the greater part of its cavity. However, all four pulmonary veins do not achieve their separate opening into the atrial roof until well after the completion of atrial septation, and probably not until the 10th or 11th week of development. Variations in the precise pattern of pulmonary venous drainage are quite common. With these changes, the left half of the primary atrium becomes progressively restricted to the mature appendage. The myocardial sleeves surrounding the pulmonary orifices taper off, and become intermingled with fibrous tissue. In later life, it is likely that this intermingling of myocardial and fibrous tissues forms the substrate for some forms of atrial fibrillation. The opening of the solitary pulmonary vein to the left of the right pulmonary ridge is an essential pre-requisite for atrial septation.
VENTRICLES
The ventricles develop at the ventral side of the looping primary heart tube during the fourth week of development (Figs 59.6, 59.7). The left ventricle develops from the stem of the Y-shaped heart, the right ventricle develops later, downstream relative to the left ventricle, when more myocardium has been added to the cardiac tube. As a result of the looping of the heart tube, the right ventricle is positioned at the right of the left ventricle, which is a prerequisite for the appropriate connection with the expanding atrial component of the heart. Unlike the atrial chambers, the morphological differences between the right and left ventricles are not part of the general asymmetry between the right- and left-sided organs of the body, but rather are under control of the signalling pathways which determine caudo-cranial differentiation. Retinoic acid, and its downstream transcription factor Tbx5, play a crucial role in this process.
The myocardium at the inner curvature, the original dorsal side of the cardiac tube, remains smooth-walled and maintains its molecular phenotype, whereas the myocardium at the outer curvature of the myocardial tube displays trabeculations in the fifth week of development (Fig. 59.7B). By stage 17, the trabeculations have achieved a typical spatial orientation, giving a sponge-like appearance to the internal aspect of both ventricles (Fig. 59.9). The definitive trabeculations, coarse in the right ventricle but much finer in the left, are first observed about the 40th day of gestation: they appear initially in the walls of both ventricles at the level of the atrioventricular junction and develop towards the apex of the heart. By the time the fetus is 10 weeks old, the trabeculations are much sparser, and are confined to the apical regions. This process of remodeling is accomplished without the intervention of macrophages or inflammatory cells in the immediate interstitium. The ventricular myocardium, encompassing the trabeculations and exterior wall, possesses a chamber phenotype, the myocytes expressing, among other proteins, the gap-junctional protein Cx40 and atrial natriuretic peptide. This myocardium stops proliferating and differentiates into the fast-conducting peripheral ventricular conduction system, whereas the outer layer becomes highly proliferative and forms the compact layer of the ventricular wall.
