[level-membership-for-dermatology-category]
Chapter 16 Deposition disorders
Touart DM, Sau P: Cutaneous deposition diseases. Part I, J Am Acad Dermatol 39:149–171, 1998.
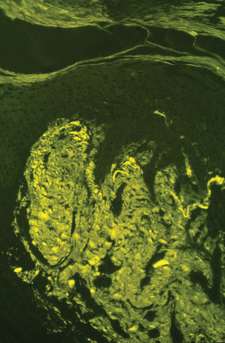
| CLINICAL DISORDER | AMYLOID PROTEIN PRECURSOR | AMYLOID PROTEIN |
|---|---|---|
| Primary systemic amyloidosis | Immunoglobulin light chain | AL |
| Myeloma-associated amyloidosis | Immunoglobulin light chain | AL |
| Secondary systemic amyloidosis | Serum amyloid A lipoprotein | AA |
| Primary localized cutaneous amyloidosis | ||
| Macular amyloidosis | Keratinocyte tonofilaments | − |
| Lichen amyloidosis | Keratinocyte tonofilaments | − |
| Nodular amyloidosis | Immunoglobulin light chain (produced locally by plasma cells) | AL |
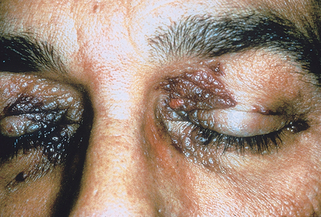
Lecha M, Puy H, Deybach JC: Erythropoietic protoporphyria, Orphanet J Rare Dis 4:19, 2009.
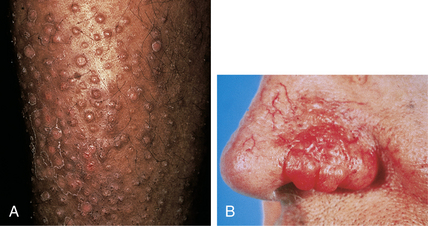
Jackson EM, English JC 3rd: Diffuse cutaneous mucinoses, Dermatol Clin 20:493–501, 2002.
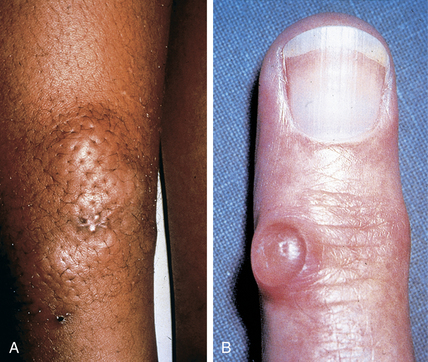
Boin F, Hummers LK: Scleroderma-like fibrosing disorders, Rheum Dis Clin North Am 34:199–220, 2008.
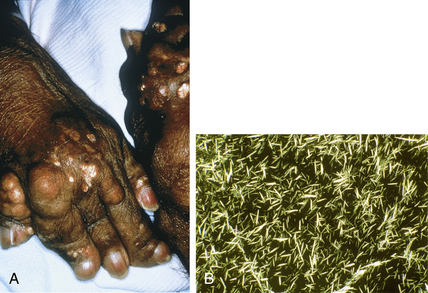
Eggebeen AT: Gout: an update, Am Fam Physician 76:801–808, 2007.
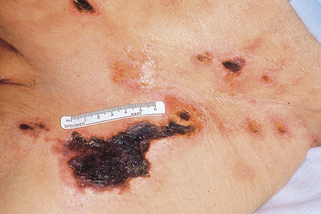
Daudén E, Oñate MJ: Calciphylaxis, Dermatol Clin 26:557–568, 2008.
Touart DM, Sau P: Cutaneous deposition diseases. Part II, J Am Acad Dermatol 39:527–544, 1998.
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Chapter 16 Deposition disorders
Touart DM, Sau P: Cutaneous deposition diseases. Part I, J Am Acad Dermatol 39:149–171, 1998.
| CLINICAL DISORDER | AMYLOID PROTEIN PRECURSOR | AMYLOID PROTEIN |
|---|---|---|
| Primary systemic amyloidosis | Immunoglobulin light chain | AL |
| Myeloma-associated amyloidosis | Immunoglobulin light chain | AL |
| Secondary systemic amyloidosis | Serum amyloid A lipoprotein | AA |
| Primary localized cutaneous amyloidosis | ||
| Macular amyloidosis | Keratinocyte tonofilaments | − |
| Lichen amyloidosis | Keratinocyte tonofilaments | − |
| Nodular amyloidosis | Immunoglobulin light chain (produced locally by plasma cells) | AL |
