CHAPTER 70 DEMENTIA WITH LEWY BODIES
HISTORICAL PERSPECTIVE
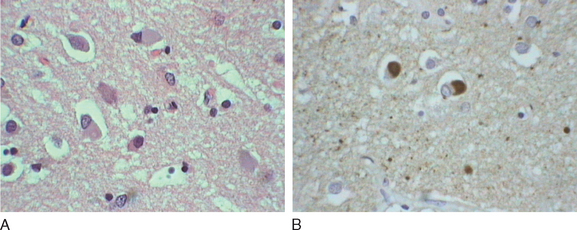
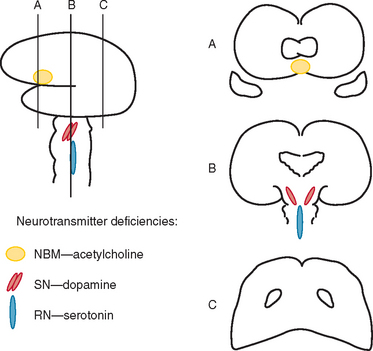
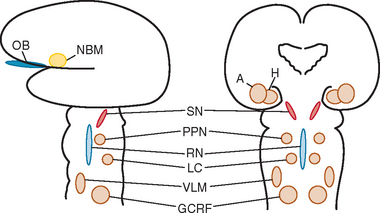
Okazaki and colleagues first described Lewy bodies in the cerebral cortex in patients with dementia in 1961.1 Few reports on Lewy body cortical pathology were published over the subsequent several years, perhaps partly because of the challenge of identifying Lewy bodies on standard hematoxylin- and eosin-stained cortical regions. In the 1980s, ubiquitin immunocytochemistry allowed easier recognition of Lewy bodies, but structures other than Lewy bodies are stained with ubiquitin.2,3 α-Synuclein immunocytochemistry was developed in the late 1990s, permitting easy identification of Lewy bodies and Lewy neurites (Fig. 70-1).4,5 Investigators from Japan, Europe, and the United States have pioneered much of the clinical and pathological characterization of Lewy body dementia/Lewy body disease since the mid-1980s.2–27 It is now clear that multiple neurotransmitter systems and structures in the brain are dysfunctional in dementia with Lewy bodies (DLB) (Figs. 70-2 and 70-3).
TERMINOLOGY
The terminology in the clinical and pathological characterization of Lewy body disease (LBD) has been confusing. Terms have included Lewy body disease, diffuse Lewy body disease, cortical Lewy body disease, Lewy body dementia, the Lewy body variant of Alzheimer’s disease, and senile dementia of the Lewy type. In 1995, the Consortium on Dementia with Lewy Bodies developed the consensus criteria for the clinical and neuropathological diagnoses. The report, published in 1996, suggested that the syndrome be termed dementia with Lewy bodies and that the neuropathological disorder be termed Lewy body disease.6 Therefore, most experts restrict use of the term dementia with Lewy bodies to the clinical syndrome; when characterizing autopsied material, the term Lewy body disease is used instead.
EPIDEMIOLOGY
The frequency of a DLB, based primarily on cases in hospital- and referral-based samples, has been approximately 15% to 25% of cases of irreversible dementia.6 More recently, in an autopsy-based study of dementia subjects in Olmsted County, Minnesota, the frequency of LBD was 10%.28 A population-based study in Finland suggested that among persons aged 75 years of age and older, the prevalences for dementia and DLB specifically were 22% and 5%, respectively.29 The few published data on frequency or prevalence suggest that DLB and LBD probably account for fewer than 25% of dementia cases and are closer to 5% to 15%. Prevalence may vary among populations, but data are inadequate for concluding whether there are significant differences on the basis of race or ancestral origin. No studies on incidence have been published to date. There does appear to be a male preponderance in DLB.6–8
CLINICAL FEATURES
A wide spectrum of symptoms and signs can occur in DLB (Table 70-1).6–8,24,27,30–34 Most symptoms can be categorized into one of five categories: cognitive impairment, neuropsychiatric features, motor dysfunction, sleep disorders, and autonomic dysfunction.
TABLE 70-1 Clinical Features Often Present in Dementia with Lewy Bodies
REM, rapid eye movement.
From Boeve B: Dementia with Lewy bodies. In Petersen R, ed: Continuum. Minneapolis: American Academy of Neurology, 2004, pp 81-112.
Cognitive Impairment
Executive and visuospatial functioning are the domains most consistently impaired in DLB.35 Symptoms of executive dysfunction include changes in problem solving, performance of sequential tasks, multitasking, and complex decision making. Difficulties with navigating in familial surroundings and problems sitting on a sofa or lying in the correct orientation on a bed are commonly voiced symptoms of visuospatial dysfunction. Memory impairment can vary from slight to very severe.35 Verbal blocking, in which a person tends to lose the train of thought in the middle of a sentence, is very common; this phenomenon can be mistaken for dysarthria or aphasia. Apathy and bradyphrenia are also common. Misidentification errors involving people can occur and are particularly upsetting when patients fail to recognize their own spouses or children. Reflections in mirrors may be mistaken for other individuals, and patients may speak to or argue with the perceived person. Most clinicians have regarded these cognitive symptoms as reflecting dysfunction of the frontosubcortical and parieto-occipital neural networks, as well as cholinergic depletion.
Fluctuations—periods of time when cognition and arousal are near normal and other periods of more marked confusion or decreased alertness—are considered a defining feature of DLB.6–8 The neural substrate responsible for fluctuations is not clear, but neurochemical alterations31 and sleep/wake dysregulation36 have been proposed.
Neuropsychiatric Features
Another defining feature of DLB is the presence of visual hallucinations.6,7,37 Often the hallucinations are first noted in the bedroom when the room is darkened at night; patients often visualize insects, animals, or people in the room, on the bed, or on the ceiling. These hallucinations are often vivid and fully formed, and patients often cannot be convinced that the stimuli are not truly present. In some, visual hallucinations can be frightening or can become the source of delusions (e.g., “That little girl has been stealing my money”). Visual illusions also are common and often coincide with the presence of visual hallucinations. Delusions are also frequent and typically have a paranoid quality, like the example just noted.37 Capgras’ syndrome—the belief that a relative or friend (usually spouse) has been replaced by an identical-appearing impostor—is also a feature of DLB.38 Depression occurs at some point in the illness in almost all patients with DLB, sometimes years before the onset of dementia.6,37 Anxiety is also common. Auditory, tactile, or olfactory hallucinations can also occur. Agitation and aggressive behavior are more variable; when present, they can be challenging to manage. Hypomania and overt bipolar disorder features can evolve, but they are atypical insofar as the onset usually occurs in patients in their 50s or 60s.
The underlying cause of hallucinations, delusions, and agitation probably reflects dopaminergic dysfunction, and serotonin dysfunction is probably involved in depression, anxiety, and bipolar-type features. Rapid eye movement (REM) sleep/wakefulness dysregulation has also been proposed as a mechanism underlying visual hallucinations in Parkinson’s disease and psychosis, in which the dream imagery of REM sleep may invade wakefulness.39 The same mechanism has been proposed to underlie hallucinations associated with DLB.24,33,34 The fact that psychostimulants can sometimes ameliorate hallucinations and delusions, which is similar to what occurs in narcolepsy, supports this hypothesis.24,33
Motor Dysfunction
Parkinsonism unrelated to dopamine antagonist exposure is another defining characteristic of DLB.6–8 Signs and symptoms include masklike facies, stooped posture, shuffling gait, difficulty with fine motor skills, sialorrhea, tremor, and bradykinesia.40 Tremor tends to be more symmetrical and related to postural/action, rather than the unilateral and predominantly at-rest tremor that is typical of Parkinson’s disease. Myoclonus can also occur, and when the clinical course is rapid, differentiation from Creutzfeldt-Jakob disease can be difficult. Many of these symptoms and signs result from reduced dopaminergic activity.
Sleep Disorders
REM sleep behavior disorder (RBD) is common in DLB (as well as in Parkinson’s disease with or without dementia and in multiple-system atrophy).22–27,33,34,41,42 Patients appear to be acting out their dreams by screaming, swearing, punching, and kicking (Table 70-2).23,24,34 The theme of the dream is remarkably consistent across patients, almost always involving chasing or attacking, and the patient is usually protecting himself or herself against aggressors rather than being the attacker. When the patient is awakened and able to recall the dream content, the description of the dream tends to match the behaviors that were exhibited. Injuries such as pulled hair, bruises, lacerations, and broken bones have been described in patients and their bed partners. RBD often begins years or even decades before any cognitive or motor symptoms develop, and therefore RBD may be the first sign of an evolving neurodegenerative disorder in many individuals. Excessive daytime somnolence, in which patients struggle to stay awake through the day, is also common.43,44 Other sleep disorders in DLB include insomnia, obstructive sleep apnea, central sleep apnea, restless legs syndrome, and periodic limb movement during sleep.42 In one polysomnographic series of DLB patients, at least one sleep disorder was present in almost every case.45 These sleep disorders are important to recognize because treatments exist for each one, and clinical improvement can be dramatic in some instances when all sleep disorders are effectively treated.

TABLE 70-2 Typical Clinical Features of REM Sleep Behavior Disorder
Rights were not granted to include this table in electronic media. Please refer to the printed book.
From Boeve B, Silber M, Ferman T, et al: REM sleep behavior disorder in Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. In Bedard M, Agid Y, Chouinard S, et al, eds: Mental and Behavioral Dysfunction in Movement Disorders. Totowa, NJ: Humana Press, 2003, pp 383-397.
Dysfunction in brainstem neuronal networks are believed to underlie RBD, particularly the pedunculopontine nucleus, locus ceruleus, and gigantocellularis reticular formation (see Fig. 70-3), although the specific networks have not been fully defined.23,24 α-Synuclein–positive pathology is often present in the lateral hypothalamus, which could explain the hypersomnolence and narcolepsy-like features. Central sleep apnea syndrome is also probably caused by brainstem dysfunction. The specificity of these sleep disorders to LBD is still being studied, but at least for RBD, it occurs almost exclusively in the synucleinopathies and rarely in other neurodegenerative disorders.23,24
Autonomic Dysfunction
Orthostatic hypotension, impotence, urinary incontinence, and constipation are common in DLB,46–48 although the frequency of each feature in DLB has not been systematically studied. Lewy bodies have been found in the intermediolateral column of the spinal cord and in the sympathetic nerves in the thoracic and abdominal structures, which reflects the rather widespread nature of Lewy body pathology in the central and peripheral nervous systems.47,49
DIAGNOSTIC CRITERIA
The criteria for the clinical diagnosis of DLB as per the Consortium on Dementia with Lewy Bodies (“McKeith criteria”), originally published in 1996,6 were refined in 1999.7 However, clinicopathological analyses have shown that the accuracy of the clinical criteria has varied widely among groups of investigators50–55; the specificity for the Consortium on Dementia with Lewy Bodies criteria is adequate, but sensitivity is relatively low. Further refinements of the criteria have been suggested through 2004 after several consensus meetings. The 2004 proposed criteria for the clinical diagnosis of DLB, the Third Report of the DLB Consortium55a, are shown in Table 70-3. Attempts continue to be made to operationally define criteria for fluctuations and better characterize the cognitive aspects, visual hallucinations, parkinsonism, sleep aspects, and autonomic aspects of DLB.

TABLE 70-3 Revised Criteria for the Clinical Diagnosis of Dementia with Lewy Bodies (DLB)
Rights were not granted to include this table in electronic media. Please refer to the printed book.
From McKeith IG, Dickson DW, Lowe J, et al: Diagnosis and management of dementia with Lewy bodies: Third report of the DLB consortium. Neurology 2005; 65:1863-1872.
For the neuropathological criteria, the First and Second Reports of the DLB Consortium suggested that ubiquitin immunohistochemistry be used and required counting Lewy bodies to characterize LBD as being predominantly brainstem, limbic, or neocortical. The recommendations from the Third Report of the DLB Consortium are to use α-synuclein immunohistochemistry and a semiquantitative grading of lesion density (Table 70-4) rather than the counting methods previously proposed, inasmuch as it was agreed that the pattern of regional involvement is more important than total Lewy body count. The grading involves categorizing Lewy body densities as mild, moderate, severe, and very severe and then assessing the regional pattern of Lewy-related pathology by grading it on a template similar to that used in the Consortium to Establish a Registry of Alzheimer’s Disease for neuritic plaques. Finally, the probability that the neuropathological findings are associated with a DLB clinical syndrome will be determined, taking accounts of both Alzheimer’s and Lewy body–type pathologies (Table 70-5). This schema requires validation studies but clearly is a solid refinement of the originally proposed neuropathological characterization of DLB and LBD.

Rights were not granted to include this table in electronic media. Please refer to the printed book.
From McKeith IG, Dickson DW, Lowe J, et al: Dementia with Lewy bodies: diagnosis and management: Third report of the DLB Consortium. Neurology 2005; 65:1863-1872.

DIAGNOSTIC EVALUATION
The primary diagnostic considerations in any patient with cognitive-behavioral changes include mild cognitive impairment, Alzheimer’s disease, DLB, frontotemporal dementia, and vascular dementia (reviewed in detail by Knopman et al56). The American Academy of Neurology practice parameter on the evaluation of individuals with dementia suggests that laboratory testing, neuropsychological testing, and structural neuroimaging be performed.57 Other diagnostic procedures that can aid in the evaluation of patients with possible DLB, particularly for differentiating DLB from Alzheimer’s disease, include electroencephalography, single photon emission computed tomography, positron emission tomography, polysomnography, autonomic studies, and smell testing. These tests are reviewed in more detail in the following sections.
Blood/Urine
The role of laboratory testing is most helpful in identifying treatable causes of cognitive impairment.57 No specific findings on laboratory testing of blood or urine that are characteristic of DLB have been identified as yet.
Cerebrospinal Fluid Analysis
On cerebrospinal fluid testing, low Aβ42 and normal tau levels58 have been reported in DLB, but this profile is also consistent with the diagnosis of Alzheimer’s disease. Hence, no specific findings on cerebrospinal fluid analysis that are diagnostic of DLB have been identified as yet.
Neuropsychological Testing
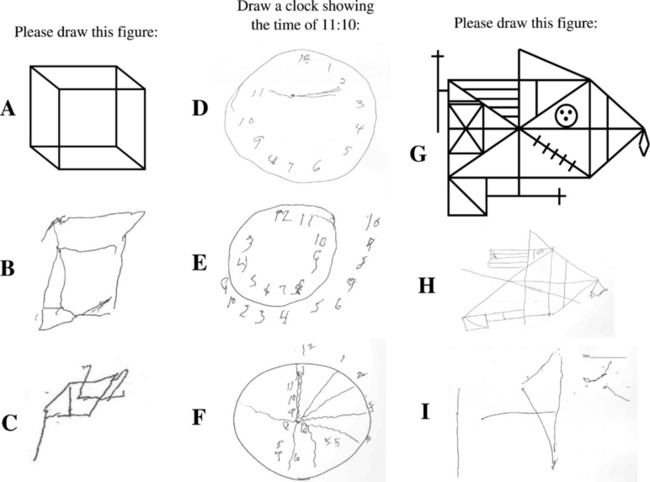
Neuropsychological testing typically reveals impairment on measures of attention/concentration and visuospatial functioning in DLB.13,35,59–61 Visuoconstructive abilities can be tested by having a patient draw a clock or copy the Necker cube, intersecting pentagons, and the Rey-Osterreith complex figure (Fig. 70-4). A similar pattern of deficits—impaired visual perceptual-organizational skills, constructional praxis, and verbal fluency—have been demonstrated in patients with dementia plus RBD.41 In a subsequent analysis in which the pattern of neuropsychological impairment was compared between one group of patients with RBD and dementia and another group of patients with autopsy-proved Alzheimer’s disease, a double dissociation was identified, in which the patients with RBD and dementia had worse impairment on measures of attention, visual perceptual-organizational skills, and letter fluency, whereas the patients with Alzheimer’s disease had had significantly worse performance on confrontation naming and verbal memory.25 The same pattern was then found in a group of patients who had dementia and RBD but did not have parkinsonism or visual hallucinations.26 These findings suggest that in the absence of visual hallucinations or parkinsonism, the presence of dementia and RBD may indicate underlying Lewy body disease. More recent neuropathological studies have corroborated this hypothesis.23,24 These studies strongly support the role of neuropsychological testing in the differential diagnosis in patients with dementia.
The most nebulous of the diagnostic features in DLB is fluctuations. Tools have been developed to differentiate fluctuations associated with DLB from those associated with other disorders,62–66 but these are currently being used by a small number of research investigators. An interview-based measure has been shown to reliably differentiate fluctuations by assessing the presence of four features: (1) drowsiness and lethargy, (2) hypersomnolence, (3) staring into space for long periods, and (4) disorganized speech in which the flow of ideas is unclear or not logical.27 The presence of three or four of these features occurred in 63% of patients with DLB, in comparison with 12% of patients with Alzheimer’s disease and only 0.5% of normal elderly persons. A score of 3 or 4 yielded a positive predictive value of 83% for the clinical diagnosis of DLB against an alternative diagnosis of Alzheimer’s disease. A score of less than 3 had a negative predictive value of 70% for the absence of a clinical diagnosis of DLB in favor of Alzheimer’s disease. Therefore, assessing these four features may aid in the diagnosis of DLB even in the office practice setting.
Electroencephalography
More background slowing on electroencephalography has been shown in patients with DLB than in those with Alzheimer’s disease,67 but no electroencephalographic findings are specific for DLB. Normal electroencephalographic findings may argue against DLB.
Structural Neuroimaging
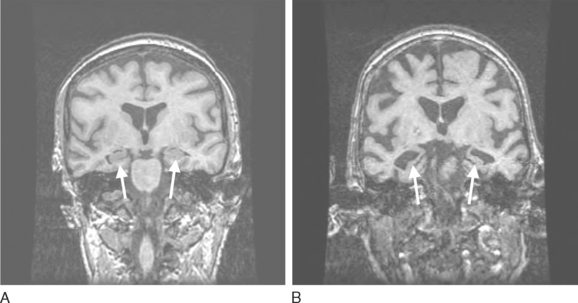
Structural neuroimaging with computed tomography or magnetic resonance imaging has classically been used to rule out a tumor, abscess, and hydrocephalus in evaluating patients with dementia. Attention is now turning to the presence and topography of atrophy. Hippocampal atrophy is well established in patients with mild cognitive impairment and Alzheimer’s disease,68–70 and patients with DLB appear to have less hippocampal atrophy on computed tomography and magnetic resonance imaging scans than do those with Alzheimer’s disease and vascular dementia.71–73 Hence, the finding of no significant hippocampal atrophy in a patient with mild to moderate dementia may suggest that the underlying histopathology is more likely to be LBD than Alzheimer’s disease (Fig. 70-5).
Functional Neuroimaging
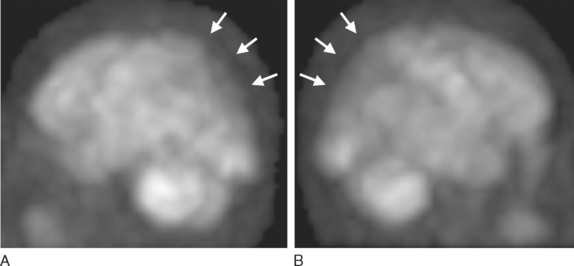
Parietal and particularly occipital hypoperfusion on single photon emission computed tomography and hypometabolism positron emission tomography have been common in patients with DLB (Fig. 70-6). In two studies comparing the occipital hypoperfusion/hypometabolism of DLB to findings in Alzheimer’s disease, the sensitivity was approximately 64% to 90%, whereas the specificity was approximately 80% to 86%.74,75 Therefore, the presence of occipital abnormalities on functional imaging studies in patients with dementia is suggestive of DLB, although this is not specific to DLB.76
Polysomnography
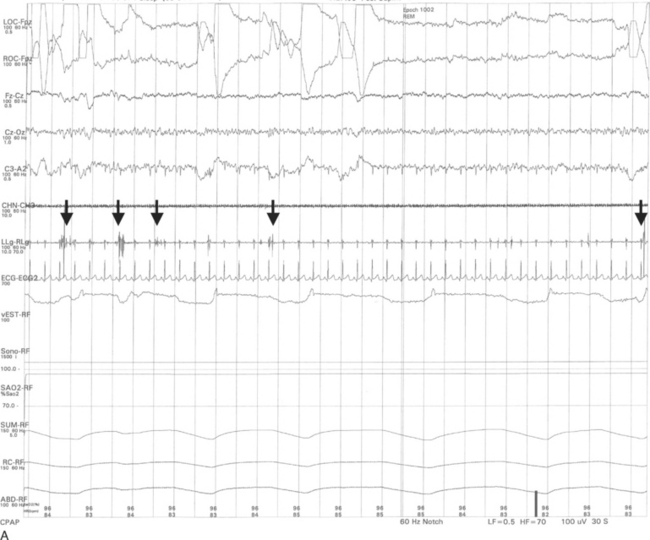
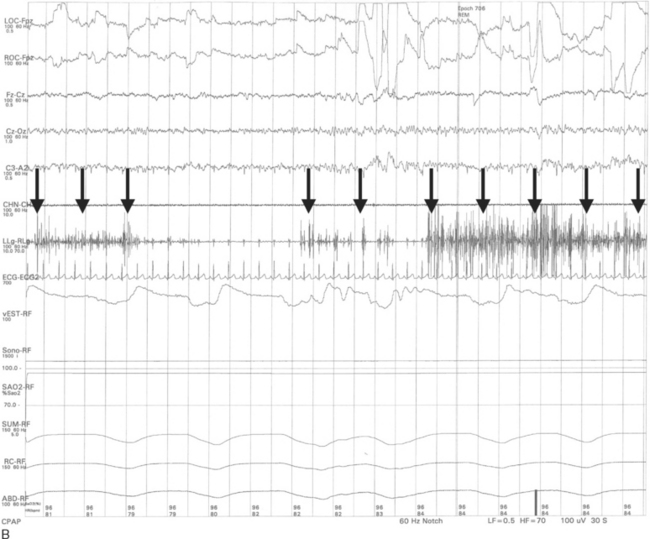
The important polysomnographic finding in RBD involves the loss of the normal electromyographic atonia during REM sleep (Fig. 70-7), also known as REM sleep without atonia (RSWA). RSWA therefore represents the electrophysiological substrate for RBD, but the diagnosis of RBD requires (1) RSWA plus (2) either a history of dream enactment behavior or dream enactment behavior during REM sleep captured on the polysomnography.77 Complicating matters is the fact that some individuals have the polysomnographic finding of RSWA but do not exhibit abnormal behaviors during polysomnography and do not have a history of nightmares and dream enactment behavior. Although the presence of RSWA may be a predictor for subsequent development of RBD, this issue has never been formally studied. Therefore, it is difficult to justify performing polysomnography if the only question is whether the patient has the electrophysiological finding of RSWA. If a patient is exhibiting dream enactment behavior that is potentially injuri ous to himself or herself and the bed partner, polysomnography can be easily justified, because clonazepam, the drug of choice for RBD, would not be desirable for a cognitively impaired patient unless the clinician is certain about the presence of RBD. Also, if a patient has features suggestive of another primary sleep disorder (e.g., loud disruptive snoring and apneic pauses, suggestive of obstructive sleep apnea; hypersomnia associated with nocturnal leg jerks, suggestive of symptomatic periodic limb movement during sleep), then a polysomnogram would be reasonable. If there is sufficient REM sleep on the polysomnogram, the clinician can then scrutinize the record, thereby providing a means of determining whether RSWA and RBD are present.
Autonomic Testing
It is now well established that autonomic dysfunction occurs in many patients with DLB (Fig. 70-8).46,47,78 In a comparative analysis involving subjects with DLB, Parkinson’s disease, and multiple-system atrophy, the degree of autonomic dysfunction for DLB was more severe than in Parkinson’s disease but less than in multiple-system atrophy.48 Furthermore, marked degeneration of the ventrolateral medulla—a structure important in the central control of autonomic functioning—was identified in pathologically proved multiple-system atrophy, in comparison with much milder degeneration in the ventrolateral medulla in pathologically proved LBD; this suggests that dysfunction in the peripheral autonomic system but not in the ventrolateral medulla is inherent in LBD.49 In patients with dementia in whom diagnosis is challenging, the presence of significant abnormalities on thermoregulatory sweat testing and autonomic reflex screening may favor underlying LBD rather than either Alzheimer’s disease or a disorder within the frontotemporal lobar degeneration spectrum.

Smell Testing
Anosmia has been studied extensively in Parkinson’s disease,79,80 as well as in Alzheimer’s disease.81 Reports have suggested that anosmia in the setting of mild cognitive impairment may be predictive of subsequent conversion to Alzheimer’s disease,82 and anosmia in the setting of RBD may be predictive of an underlying synucleinopathy such as LBD.83 According to Braak and associates’ neuropathological staging system of Parkinson’s disease,9 α-synuclein–positive pathology in the olfactory structures, as well as in the medulla, occurs very early (stage 1) in Parkinson’s disease (see Fig. 70-3); such pathology gradually ascends up the brainstem and eventually to limbic and neocortical structures. Hence, at least in Parkinson’s disease, the progression of anosmia to RBD to parkinsonism to dementia fits well into Braak and associates’ paradigm. Whether the quantitative and qualitative features of anosmia in patients with dementia have predictive value in determining the underlying neurodegenerative disorder remains to be tested.
MANAGEMENT
No therapy that significantly alters synuclein pathophysiology, which is presumed to be central to Lewy body disease, has yet been identified. Management must therefore be directed toward target symptoms (Table 70-6). Although only a few double-blind, placebocontrolled clinical trials have been carried out specifically in DLB patients, there is sufficient experience to provide some suggestions for therapy. The tenet “start low and go slow” is particularly important in DLB patients.
TABLE 70-6 Symptoms, Behaviors, and Disorders in Dementia with Lewy Bodies: Select Medications with Suggested Dosing Schedules*
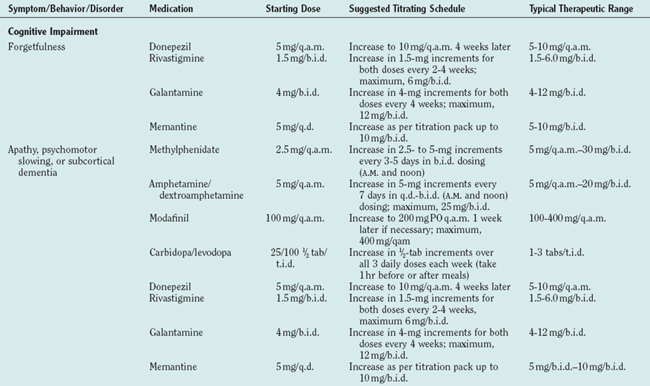
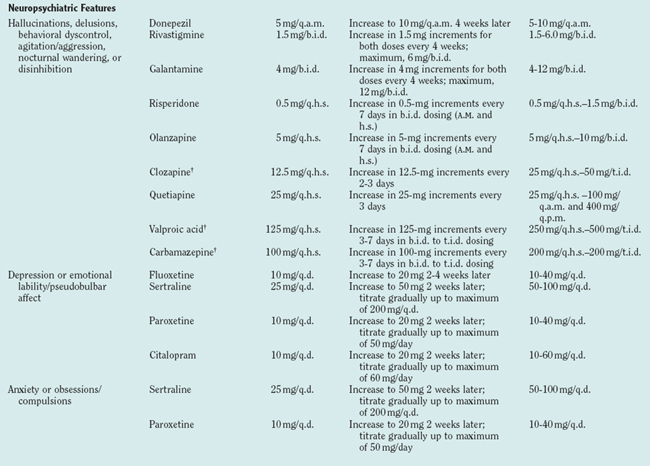



Although limbic and neocortical neuronal loss clearly occurs in DLB, the severity appears to be less than that in Alzheimer’s disease and other dementias (as exemplified in Fig. 70-5).4,5,11,84 Furthermore, depletion of the cholinergic basal forebrain, classically viewed as part of Alzheimer’s disease, is often even more profound in LBD.4,5 Decreases in dopamine and serotonin are also inherent in LBD. Thus, the marked neurotransmitter deficiencies but better preservation of viable neurons suggests that medical therapy may be as effective in DLB as in other dementing disorders or more so. The following approach addresses strategies for the five categories of symptoms described previously.
Cognitive Impairment
The cholinergic deficit in DLB is now well established, and the cholinesterase inhibitors have been shown in open-label and double-blind, placebocontrolled studies to modestly improve cognition and functional abilities (reviewed in detail by Simard and van Reekum85). The currently available cholinesterase inhibitors include tacrine, donepezil, rivastigmine, and galantamine. Because frequent dosing and laboratory monitoring are necessary with tacrine, this agent is rarely used. Despite the concern that cholinergic stimulation might worsen parkinsonism, this has not been a serious problem in clinical experience and controlled studies.86 Thus, clinicians should consider prescribing one of these agents for patients with DLB in whom there is no contraindication to its use.
Studies with relatively few patients have shown improvement with memantine in parkinsonism in patients with Parkinson’s disease,87 but there are no published data on this drug in DLB patients. Clinical experience has shown that some patients with DLB do benefit from memantine, but visual hallucinations, gait impairment, and worsened cognition can occur; these typically resolve on discontinuation of the drug.
Psychostimulants, carbidopa/levodopa, and the dopamine agonists can theoretically improve cognition, apathy, and psychomotor slowing, but no controlled studies demonstrating efficacy of these agents have been published to date.
Neuropsychiatric Features
Visual hallucination is one of the most frequent and pervasive features of DLB, but drug therapy is not required if the hallucinations are not frightening. When they are frightening, or when paranoia develops in concert with hallucinations, drug therapy is often necessary. Conventional neuroleptic agents, which have been classically used to manage hallucinations, can cause striking and irreversible parkinsonism (a phenomenon termed neuroleptic sensitivity), and use of these agents is now strongly discouraged in the management of DLB,88 which underscores the importance of accurate diagnosis in this condition. The cholinesterase inhibitors have been shown to ameliorate hallucinations, as well as apathy.86,89–91 There are reports of neuroleptic sensitivity even among the newer atypical neuroleptic agents, and some of these have been minimally effective for psychotic features.63 The following agents have been reported to improve hallucinations, delusions, or agitation: clozapine,92–94 risperidone,95,96 olanzapine,97,98 quetiapine,94,99 and the cholinesterase inhibitors. Therefore, if problematic hallucinations, delusions, or agitation occurs in patients with DLB who do not respond to the cholinesterase inhibitors, clinicians should consider quetiapine, clozapine, or olanzapine. There is insufficient evidence on the efficacy of ziprasidone and aripiprazole. Orthostatism can occur with any of the atypical neuroleptic agents. Valproic acid and carbamazepine have mood-stabilizing properties and may be appropriate for some patients.
The raphe nucleus is compromised in DLB, which is the likely substrate for depression. The selective serotonin reuptake inhibitors are usually effective and well tolerated. Tricyclic antidepressants, because of their anticholinergic properties, should generally be avoided in DLB. Paroxetine also has anticholinergic effects and should be used cautiously in DLB. Electroconvulsive therapy can be effective in some patients without significantly worsening cognition.100 The selective serotonin reuptake inhibitors and buspirone can improve anxiety.
Motor Dysfunction
When significant parkinsonism is present in patients with DLB, the challenge for the clinician is to ameliorate this feature without exacerbating psychotic symptoms, hypersomnolence, and orthostatism. Experience has shown that many of the parkinsonian signs and symptoms of DLB can respond to carbidopa/levodopa and the dopamine agonists, but these agents must be used cautiously. In one open-label study, levodopa was generally well tolerated, but only one third of the patients experienced significant improvement in motor functioning.101 In those with refractory depression warranting electroconvulsive therapy, parkinsonism can be ameliorated through unknown mechanisms. Invasive strategies for managing parkinsonism, such as pallidotomy and deep brain stimulation, are not appropriate for patients with DLB.
Autonomic Dysfunction
Orthostatic hypotension can occur in DLB; it is probably a result of degenerative changes in the intermediolateral cell column of the spinal cord and peripheral autonomic system.48,49 Liberal amounts of salt in the diet, salt tablets, thigh-high compression stockings, fludrocortisone, and midodrine are additional considerations. Although certainly not a first-line agent for the management of orthostatic hypotension, the cholinesterase inhibitors have been shown to ameliorate orthostatic hypotension102; whether this occurs consistently in patients with DLB remains to be seen.
Sleep Disorders
REM Sleep Behavior Disorder
RBD is manifested as violent dreams or nightmares and potentially injurious dream reenactment behavior, and the diagnosis is usually not difficult if the features listed in Table 70-2 are present. However, patients with moderate to severe obstructive sleep apnea can have features identical to those of RBD; the nightmares and behaviors are typically eliminated with nasal continuous positive airway pressure. Hence, patients should be considered for polysomnography with or without a trial of nasal continuous positive airway pressure if there is a history suggestive of RBD and/or obstructive sleep apnea.
The goals of therapy for RBD are to minimize the nightmares and abnormal behavior, and because injuries to patients and their bed partners can occur, treatment should be commenced in patients with the potential for injury.24,42 Simple measures to minimize the potential for injury involve counseling patients and their bed partners to move lamps and furniture away from the bed and to place a mattress or cushion on the floor beside the bed. Clonazepam, the drug of choice for most patients who have RBD without dementia, is usually effective at 0.25 to 0.5 mg per night; dosages higher than 1 mg are sometimes necessary.24,42–44 Clonazepam is not an ideal agent in patients with dementia, but experience has shown that most patients tolerate the drug well at low dosages.24,42 Melatonin can also be effective at 3 to 12 mg per night either as monotherapy or in conjunction with clonazepam.103 Experience suggests that quetiapine can also be effective in managing RBD.24,42
Insomnia
Insomnia can be caused by one or more primary sleep disorders (e.g., restless legs syndrome, periodic limb movement during sleep, obstructive sleep apnea, or central sleep apnea syndrome), by depression, and by medications.42 Frequent arousals for no apparent reason may be an intrinsic part of the disease, presumably caused by degenerative changes in the sleep/wake circuits in the brainstem and hypothalamus. A carefully documented sleep history can lead to the correct diagnoses, but polysomnography is often necessary because the cardinal features of obstructive sleep apnea, central sleep apnea, and periodic limb movement during sleep may not be apparent to the patient or bed partner. Contrary to popular belief among clinicians and the lay public, nasal continuous positive airway pressure therapy or bilevel positive airway pressure is tolerated by many patients with dementia.42 Although certainly not universal among DLB patients with one or more sleep disorders, marked clinical improvements in alertness and cognition can occur with effective management of the sleep issues.42 Insomnia can also be caused by medications, particularly the cholinesterase inhibitors, and patients can minimize insomnia by taking donepezil in the morning or taking rivastigmine or galantamine no later than the evening meal. Other drugs effective for primary insomnia include trazodone, chloral hydrate, zolpidem, zaleplon, and the atypical neuroleptic agents (e.g., quetiapine, olanzapine, clozapine, or risperidone). Melatonin was evaluated as part of the Melatonin for Sleep Disturbance in Alzheimer’s Disease clinical trial, but there were no group differences between those treated with low-dose melatonin, high-dose melatonin, and placebo.104 However, as pointed out in the report by the investigators, melatonin may be effective in rare instances. Fluoxetine, venlafaxine, and bupropion may precipitate or aggravate insomnia, whereas mirtazapine may ameliorate insomnia. Numerous nonpharmacological strategies are also available for managing insomnia.105
Excessive Daytime Somnolence
Excessive daytime somnolence can be caused by primary sleep disorders, depression, and medications as noted previously; adequate evaluation may necessitate polysomnography with or without multiple sleep latency tests.42 Interestingly, there is evidence that some patients with Parkinson’s disease and psychosis have narcolepsy-like features,39 and the same may be true of patients with DLB.24,34 Although psychostimulants would be expected to exacerbate hallucinations and delusions in patients with DLB, experience has shown that excessive daytime somnolence, as well as hallucinations and delusions, can be well managed with agents such as modafinil and methylphenidate.24 This is a controversial aspect in the management of DLB, and clearly controlled trials are necessary to justify use of psychostimulants in this population.
CONCLUSIONS AND FUTURE DIRECTIONS
Parkinson’s disease/Lewy body disease has classically been considered a dopamine deficiency disorder. Technical developments and considerable attention directed to the study of dementia and parkinsonism since the mid-1980s have revealed that the syndrome of DLB and the disorder of LBD are clearly far more complex, with motor, cognitive, neuropsychiatric, sleep, and autonomic manifestations, as well as multiple neurotransmitter systems involved. As a more comprehensive approach evolves in the characterization and management of DLB patients, the quality of life for patients and their families can be optimized. Further research on α-synuclein and other protein aggregation abnormalities in LBD offer hope that disease-altering and preventive therapies may some day be developed.
Barber R, McKeith I, Ballard C, et al. A comparison of medial and lateral temporal lobe atrophy in dementia with Lewy bodies and Alzheimer’s disease: magnetic resonance imaging volumetric study. Dementia Geriatr Cogn Disord. 2001;12:198-205.
Boeve B. Dementia with Lewy bodies. In: Petersen R, editor. Continuum. Minneapolis: American Academy of Neurology; 2004:81-112.
Ferman et al Ferman T, Smith G, Boeve B, et al: Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol (in press).
McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebocontrolled international study. Lancet. 2000;356:2031-2036.
McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113-1124.
McKeith I, Mintzer J, Aarsland D, et al. Dementia with Lewy bodies. Lancet Neurology. 2004;3:19-28.
Thaisetthawatkul P, Boeve B, Benarroch E, et al. Autonomic dysfunction in dementia with Lewy bodies. Neurology. 2004;62:1804-1809.
1 Okazaki H, Lipkin L, Aronson S. Diffuse intracytoplasmic ganglionic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J Neuropathol Exp Neurol. 1961;20:237-244.
2 Kosaka K. Dementia and neuropathology in Lewy body disease. Adv Neurol. 1993;60:456-463.
3 Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(Suppl):S73-S78.
4 Dickson DW. Tau and synuclein and their role in neuropathology. Brain Pathol. 1999;9:657-661.
5 Dickson D. Dementia with Lewy bodies: neuropathology. J Geriatr Psychiatr Neurol. 2002;15:210-216.
6 McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113-1124.
7 McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53:902-905.
8 McKeith I, Mintzer J, Aarsland D, et al. Dementia with Lewy bodies. Lancet Neurology. 2004;3:19-28.
9 Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197-211.
10 Braak H, Ghebremedhin E, Rub U, et al. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121-134.
11 Dickson DW, Crystal H, Mattiace LA, et al. Diffuse Lewy body disease: light and electron microscopic immunocytochemistry of senile plaques. Acta Neuropathol. 1989;78:572-584.
12 Dickson DW, Ruan D, Crystal H, et al. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s disease: light and electron microscopic immunocytochemistry of CA2–3 neurites specific to DLBD. Neurology. 1991;41:1402-1409.
13 Salmon D, Galasko D, Hansen L, et al. Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn. 1996;31:148-164.
14 Hansen L, Salmon D, Galasko D, et al. The Lewy body variant of Alzheimer’s disease: a clinical and pathologic entity. Neurology. 1990;40:1-8.
15 Hansen L, Samuel W. Criteria for Alzheimer’s disease and the nosology of dementia with Lewy bodies. Neurology. 1997;48:126-132.
16 Lippa CF, Pulaski-Salo D, Dickson DW, et al. Alzheimer’s disease, Lewy body disease and aging: a comparative study of the perforant pathway. J Neurol Sci. 1997;147:161-166.
17 Lippa C, Smith T, Perry E. Dementia with Lewy bodies: choline acetyltransferase parallels nucleus basalis pathology. J Neural Transm. 1999;105:525-535.
18 Lippa CF, McKeith I. Dementia with Lewy bodies: improving diagnostic criteria [Comment]. Neurology. 2003;60:1571-1572.
19 Duda J, Lee V, Trojanowski J. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121-127.
20 Trojanowski J, Lee V. Parkinson’s disease and related neurodegenerative synucleinopathies linked to progressive accumulations of synuclein aggregates in brain. Parkinsonism Relat Disord. 2001;7:247-251.
21 Duda J, Giasson B, Mabon M, et al. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol. 2002;52:205-210.
22 Boeve B, Silber M, Ferman T, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16:622-630.
23 Boeve B, Silber M, Parisi J, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology. 2003;61:40-45.
24 Boeve B, Silber M, Ferman T. REM sleep behavior disorder in Parkinson’s disease and dementia with Lewy bodies. J Geriatr Psychiatry Neurol. 2004;17:146-157.
25 Ferman TJ, Boeve B, Smith GE, et al. REM sleep behavior disorder and dementia: cognitive differences when compared with AD. Neurology. 1999;52:951-957.
26 Ferman T, Boeve B, Smith G, et al. Dementia with Lewy bodies may present as dementia with REM sleep behavior disorder without parkinsonism or hallucinations. J Int Neuropsychol Soc. 2002;8:907-914.
27 Ferman T, Smith G, Boeve B. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62:181-187.
28 Knopman D, Parisi J, Boeve B, et al. Vascular dementia in a population-based autopsy study. Arch Neurol. 2003;60:569-575.
29 Rahkonen T, Eloniemi-Sulkava U, Rissanen S, et al. Dementia with Lewy bodies according to the consensus criteria in a general population aged 75 years or older. J Neurol Neurosurg Psychiatry. 2003;74:720-724.
30 Ballard C, Holmes C, McKeith I, et al. Psychiatric morbidity in dementia with Lewy bodies: a prospective clinical and neuropathological comparative study with Alzheimer’s disease. Am J Psychiatry. 1999;156:1039-1045.
31 Ballard C, O’Brien J, Gray A, et al. Attention and fluctuating attention in patients with dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2001;58:977-982.
32 Galasko D. Lewy bodies and dementia. Curr Neurol Neurosci Rep. 2001;1:435-441.
33 Boeve B, Silber M, Ferman T, et al. REM sleep behavior disorder in Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. In: Bedard M, Agid Y, Chouinard S, et al, editors. Mental and Behavioral Dysfunction in Movement Disorders. Totowa, NJ: Humana Press; 2003:383-397.
34 Boeve B. Dementia with Lewy bodies. In: Petersen R, editor. Continuum. Minneapolis: American Academy of Neurology; 2004:81-112.
35 Simard M, van Reekum R, Cohen T. A review of the cognitive and behavioral symptoms in dementia with Lewy bodies. J Neuropsychiatr Clin Neurosci. 2000;12:425-450.
36 Ferman T, Boeve B, Silber M, et al. Is fluctuating cognition in dementia with Lewy bodies attributable to an underlying sleep disorder? Sleep. 2001;24:A374.
37 Aarsland D, Ballard C, Larsen J, et al. A comparative study of psychiatric symptoms in dementia with Lewy bodies and Parkinson’s disease with and without dementia. Int J Geriatr Psychiatry. 2001;16:528-536.
38 Marantz A, Verghese J. Capgras’ syndrome in dementia with Lewy bodies. J Geriatr Psychiatr Neurol. 2002;15:239-241.
39 Arnulf I, Bonnet AM, Damier P, et al. Hallucinations, REM sleep, and Parkinson’s disease: a medical hypothesis. Neurology. 2000;55:281-288.
40 Burn DJ, Rowan EN, Minett T, et al. Extrapyramidal features in Parkinson’s disease with and without dementia and dementia with Lewy bodies: a cross-sectional comparative study. Mov Disord. 2003;18:884-889.
41 Boeve BF, Silber MH, Ferman TJ, et al. REM sleep behavior disorder and degenerative dementia: an association likely reflecting Lewy body disease. Neurology. 1998;51:363-370.
42 Boeve B, Silber M, Ferman T. Current management of sleep disturbances in dementia. Curr Neurol Neurosci Rep. 2001;2:169-177.
43 Olson E, Boeve B, Silber M. Rapid eye movement sleep behavior disorder: demographic, clinical, and laboratory findings in 93 cases. Brain. 2000;123:331-339.
44 Schenck C, Mahowald M. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep. 2002;25:120-138.
45 Boeve B, Ferman T, Silber M, et al. Sleep disturbances in dementia with Lewy bodies involve more than REM sleep behavior disorder. Neurology. 2003;60:A79.
46 Ballard C, Shaw F, McKeith I, et al. High prevalence of neurovascular instability in neurodegenerative dementias. Neurology. 1998;51:1760-1762.
47 Hishikawa N, Hashizume Y, Yoshida M, et al. Clinical and neuropathological correlates of Lewy body disease. Acta Neuropathol (Berl). 2003;105:341-350.
48 Thaisetthawatkul P, Boeve B, Benarroch E, et al. Autonomic dysfunction in dementia with Lewy bodies. Neurology. 2004;62:1804-1809.
49 Benarroch E, Schmeichel A, Low P, et al. Involvement of medullary regions controlling sympathetic output in Lewy body disease. Brain. 2005;128:338-344.
50 Hohl U, Tiraboschi P, Hansen L, et al. Diagnostic accuracy of dementia with Lewy bodies. Arch Neurol. 2000;57:347-351.
51 Lopez O, Litvan I, Catt K, et al. Accuracy of four clinical diagnostic criteria for the diagnosis of neurodegenerative dementias. Neurology. 1999;53:1292-1299.
52 Lopez O, Hamilton R, Becker J, et al. Severity of cognitive impairment and the clinical diagnosis of AD with Lewy bodies. Neurology. 2000;54:1780-1787.
53 McKeith IG, Ballard CG, Perry RH, et al. Prospective validation of consensus criteria for the diagnosis of dementia with Lewy bodies. Neurology. 2000;54:1050-1058.
54 Mega M, Masterman D, Benson D, et al. Dementia with Lewy bodies: reliability and validity of clinical and pathologic criteria. Neurology. 1996;47:1403-1409.
55 Verghese J, Crystal HA, Dickson DW, et al. Validity of clinical criteria for the diagnosis of dementia with Lewy bodies. Neurology. 1999;53:1974-1982.
55a McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB consortium. Neurology. 2005;65:1863-1872.
56 Knopman D, Boeve B, Petersen R. Essentials of the proper diagnosis of mild cognitive impairment, dementia, and major subtypes of dementia. Mayo Clin Proc. 2003;78:1290-1308.
57 Knopman D, DeKosky S, Cummings J, et al. Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56:1143-1153.
58 Kanemaru K, Kameda N, Yamanouchi H. Decreased CSF amyloid β42 and normal tau levels in dementia with Lewy bodies. Neurology. 2000;54:1875-1876.
59 Mori E, Shimomura T, Fujimori M, et al. Visuoperceptual impairment in dementia with Lewy bodies. Arch Neurol. 2000;57:489-493.
60 Simard M, van Reekum R, Myran D. Visuospatial impairment in dementia with Lewy bodies and Alzheimer’s disease: a process analysis approach. Int J Geriatr Psychiatry. 2003;18:387-391.
61 Ferman T, Smith G, Boeve B, et al: Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol (in press).
62 Doubleday E, Snowden J, Varma A, et al. Qualitative performance characteristics differentiate dementia with Lewy bodies and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2002;72:602-607.
63 Walker Z, Grace J, Overshot R, et al. Olanzapine in dementia with Lewy bodies: a clinical study. Int J Geriatr Psychiatry. 1999;14:459-466.
64 Walker M, Ayre G, Cummings J, et al. The Clinician Assessment of Fluctuation and the One Day Fluctuation Assessment Scale. Two methods to assess fluctuating confusion in dementia. Br J Psychiatry. 2000;177:252-256.
65 Walker M, Ayre G, Cummings J, et al. Quantifying fluctuation in dementia with Lewy bodies, Alzheimer’s disease, and vascular dementia. Neurology. 2000;54:1616-1625.
66 Walker M, Ayre G, Perry E, et al. Quantification and characterization of fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease. Dementia Geriatr Cogn Disord. 2000;11:327-335.
67 Briel RC, McKeith IG, Barker WA, et al. EEG findings in dementia with Lewy bodies and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1999;66:401-403.
68 Jack CRJr, Petersen RC, Xu YC, et al. Hippocampal atrophy and apolipoprotein E genotype are independently associated with Alzheimer’s disease. Ann Neurol. 1998;43:303-310.
69 Jack CRJr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55:484-489.
70 Jack CRJr, Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52:1397-1403.
71 Barber R, McKeith I, Ballard C, et al. A comparison of medial and lateral temporal lobe atrophy in dementia with Lewy bodies and Alzheimer’s disease: magnetic resonance imaging volumetric study. Dementia Geriatr Cogn Disord. 2001;12:198-205.
72 Barber R, Gholkar A, Scheltens P, et al. Medial temporal lobe atrophy on MRI in dementia with Lewy bodies. Neurology. 1999;52:1153-1158.
73 Barber R, Ballard C, McKeith IG, et al. MRI volumetric study of dementia with Lewy bodies: a comparison with AD and vascular dementia. Neurology. 2000;54:1304-1309.
74 Lobotesis K, Fenwick J, Phipps A, et al. Occipital hypoperfusion on SPECT in dementia with Lewy bodies but not AD. Neurology. 2001;56:643-649.
75 Minoshima S, Foster N, Sima A, et al. Alzheimer’s disease versus dementia with Lewy bodies: cerebral metabolic distinction with autopsy confirmation. Ann Neurol. 2001;50:358-365.
76 Tang-Wai D, Graff-Radford N, Boeve B, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004;63:1168-1174.
77 Mahowald M, Schenck C. REM sleep behavior disorder. In: Kryger M, Roth T, Dement W, editors. Principles and Practice of Sleep Medicine. 3rd ed. Philadelphia: WB Saunders; 2000:724-741.
78 Pakiam AS, Bergeron C, Lang AE. Diffuse Lewy body disease presenting as multiple system atrophy. Can J Neurol Sci. 1999;26:127-131.
79 Tissingh G, Berendse H, Bergmans P, et al. Loss of olfaction in de novo and treated Parkinson’s disease: possible implications for early diagnosis. Mov Disord. 2001;16:41-46.
80 Ponsen M, Stoffers D, Booij J, et al. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol. 2004;56:173-181.
81 Peters J, Hummel T, Kratzsch T, et al. Olfactory function in mild cognitive impairment and Alzheimer’s disease: an investigation using psychophysical and electrophysiological techniques. Am J Psychiatry. 2003;160:1995-2002.
82 Wang Q, Tian L, Huang Y, et al. Olfactory identification and apolipoprotein E epsilon 4 allele in mild cognitive impairment. Brain Res. 2002;951:77-81.
83 Stiasny-Kolster K, Doerr Y, Möller J, et al. Combination of “idiopathic” REM sleep behaviour disorder and olfactory dysfunction as possible indicator for α-synucleinopathy demonstrated by dopamine transporter FP-CIT-SPECT. Brain. 2005;128:126-137.
84 Dickson DW, Davies P, Mayeux R, et al. Diffuse Lewy body disease. Neuropathological and biochemical studies of six patients. Acta Neuropathol. 1987;75:8-15.
85 Simard M, van Reekum R. The acetylcholinesterase inhibitors for treatment of cognitive and behavioral symptoms in dementia with Lewy bodies. J Neuropsychiatry Clin Neurosci. 2004;16:409-425.
86 McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebocontrolled international study. Lancet. 2000;356:2031-2036.
87 Merello M, Nouzeilles M, Cammarota A, et al. Effect of memantine (NMDA antagonist) on Parkinson’s disease: a double-blind crossover randomized study. Clin Neuropharmacol. 1999;22:273-276.
88 McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305:673-678.
89 Shea C, MacKnight C, Rockwood K. Donepezil for treatment of dementia with Lewy bodies: a case series of nine patients. Int Psychogeriatr. 1998;10:229-238.
90 Fergusson E, Howard R. Donepezil for the treatment of psychosis in dementia with Lewy bodies. Int J Geriatr Psychiatry. 2000;15:280-281.
91 Lanctot KL, Herrmann N. Donepezil for behavioural disorders associated with Lewy bodies: a case series. Int J Geriatr Psychiatry. 2000;15:338-345.
92 Chacko RC, Hurley RA, Harper RG, et al. Clozapine for acute and maintenance treatment of psychosis in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 1995;7:471-475.
93 Valldeoriola F, Nobbe FA, Tolosa E. Treatment of behavioural disturbances in Parkinson’s disease. J Neural Transm Suppl. 1997;51:175-204.
94 Dewey RJ, O’Suilleabhain P. Treatment of drug-induced psychosis with quetiapine and clozapine in Parkinson’s disease. Neurology. 2000;55:1753-1754.
95 Workman RHJr, Orengo CA, Bakey AA, et al. The use of risperidone for psychosis and agitation in demented patients with Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 1997;9:594-597.
96 Leopold NA. Risperidone treatment of drug-related psychosis in patients with parkinsonism. Mov Disord. 2000;15:301-304.
97 Aarsland D, Larsen JP, Lim NG, et al. Olanzapine for psychosis in patients with Parkinson’s disease with and without dementia. J Neuropsychiatry Clin Neurosci. 1999;11:392-394.
98 Cummings J, Street J, Masterman D, et al. Efficacy of olanzapine in the treatment of psychosis in dementia with Lewy bodies. Dementia Geriatr Cogn Disord. 2002;13:67-73.
99 Takahashi H, Yoshida K, Sugita T, et al. Quetiapine treatment of psychotic symptoms and aggressive behavior in patients with dementia with Lewy bodies: a case series. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:549-553.
100 Rasmussen KJr, Russell J, Kung S, et al. Electroconvulsive therapy for patients with major depression and probable Lewy body dementia. J ECT. 2003;19:103-109.
101 Molloy S, McKeith I, O’Brien J, et al. The role of levodopa in the management of dementia with Lewy bodies. J Neurol Neurosurg Psychiatry. 2005;76:1200-1203.
102 Singer W, Opfer-Gehrking T, McPhee B, et al. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry. 2003;74:1294-1298.
103 Boeve B, Silber M, Ferman T. Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: results in 14 patients. Sleep Med. 2003;4:281-284.
104 Singer C, Tractenberg R, Kaye J, et al. A multicenter, placebocontrolled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep. 2003;26:893-901.
105 Hauri P, Linde S. No More Sleepless Nights, 2nd ed. New York: John Wiley & Sons, 1996.






