[level-membership-for-basic-science-category]
Dementia
General aspects
Clinical features
In most cases of dementia, such as Alzheimer’s disease, memory loss is a prominent and early component, but in certain types (e.g. frontotemporal dementia, discussed below) it is relatively spared. Loss of memory is usually accompanied by a marked decline in higher cognitive functions such as reasoning, visuospatial ability and language, together with changes in mood, behaviour and personality. The specific profile of higher cognitive deficits depends on the extent and distribution of pathology in the cerebral cortex (Fig. 12.1).
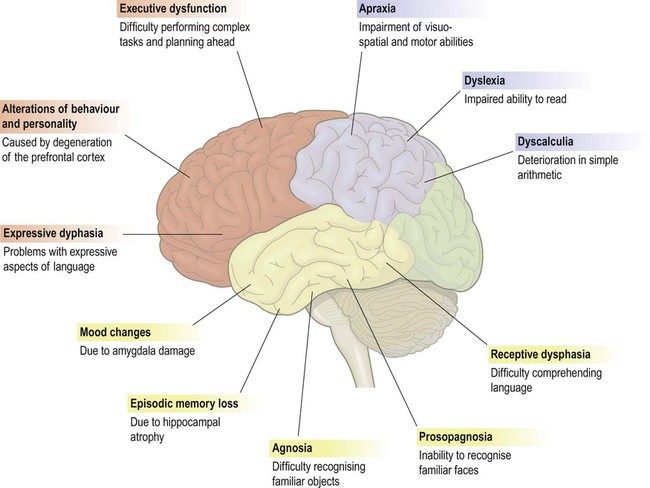
This figure shows some of the common features of dementia that are caused by degenerative changes in different parts of the cerebral cortex (frontal lobe = red, parietal lobe = blue, temporal lobe = yellow). The clinical picture in a particular person depends upon the distribution and severity of the changes.
Assessment and diagnosis
Psychometric testing
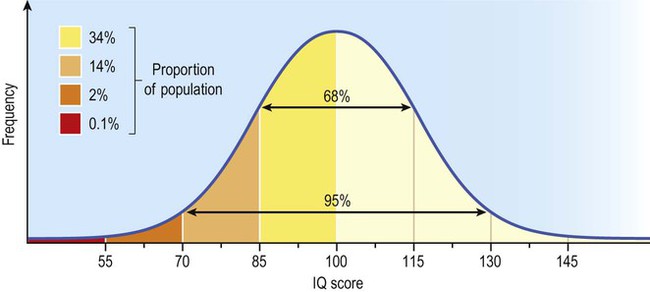
A widely used measure of cognitive ability is the intelligence quotient (IQ) which is a detailed assessment of reasoning, language and memory. Scores are standardized and age-corrected so that the average IQ is 100 and 95% of individuals score between 70 and 130 (Fig. 12.2). Repeated testing can be used to show changes in cognitive ability over time.
Types of dementia
The main types of dementia are discussed below. The most common cause is Alzheimer’s disease, accounting for around 65% of cases. Dementia with Lewy bodies (DLB) represents a further 20% and vascular dementia is responsible for 10% of cases. The remaining 5% include the frontotemporal dementias, which are an important cause of cognitive decline in people under the age of 60 (accounting for nearly half of cases in this age group, after Alzheimer’s disease). It should be noted that some patients have less severe cognitive impairment that falls short of the criteria for dementia (Clinical Box 12.1).
 Clinical Box 12.1:
Clinical Box 12.1:Reversible causes
Some forms of cognitive impairment are potentially treatable and should be excluded in the investigation of a patient with suspected dementia. These include nutritional deficiencies (e.g. vitamin B12, folate), endocrine disturbances (such as hypothyroidism), alcohol-related dementia (reversible with abstinence), syphilis (now rare in the developed world), depressive pseudodementia (treatable with antidepressant drugs) and normal pressure hydrocephalus (see Clinical Box 12.2). A ‘routine dementia screen’ therefore includes a range of blood tests and urinalysis, together with an MRI scan of the brain.
 Clinical Box 12.2: Normal pressure hydrocephalus
Clinical Box 12.2: Normal pressure hydrocephalus
Normal pressure hydrocephalus affects 1 in 200 people over the age of 65 and presents with a triad of dementia, gait disturbance and urinary incontinence. The cerebral ventricles are enlarged, but the CSF pressure is usually normal, although longer-term recording has revealed periodic pressure elevations in some people (hence the alternative term, intermittently raised pressure hydrocephalus). Up to 50% of patients show dramatic improvement after surgical implantation of a shunt to divert excess CSF to the abdomen (see Ch. 2, Clinical Box 2.2).





Alzheimer’s disease
Clinical aspects
The diagnosis of Alzheimer’s disease is predominantly clinical and post-mortem studies suggest that it is correct in at least 80% of cases. Two variants of Alzheimer’s disease that might cause diagnostic confusion are discussed in Clinical Box 12.3.
 Clinical Box 12.3:
Clinical Box 12.3:Visuospatial problems
Patients with Alzheimer’s disease often get lost in familiar places and may forget where they have left things. These features, together with impaired recall of daily events, reflect severe pathology in the medial temporal lobe. This involves structures such as the entorhinal cortex and hippocampus that are involved in spatial navigation and formation of episodic memories (see Ch. 3).
Neuroimaging
Functional brain imaging in early Alzheimer’s disease may show reduced blood flow and glucose metabolism in the posterior temporo-parietal regions and hippocampus (Fig. 12.3), before obvious structural changes are evident on MRI. Over time there is progressive brain atrophy, with ventricular dilatation, cortical thinning and hippocampal atrophy. Longitudinal studies show that people with Alzheimer’s disease lose brain tissue at a rate of 2% per year on average (up to 5% per year in the hippocampus), which is four times higher than in age-matched controls (Fig. 12.4).
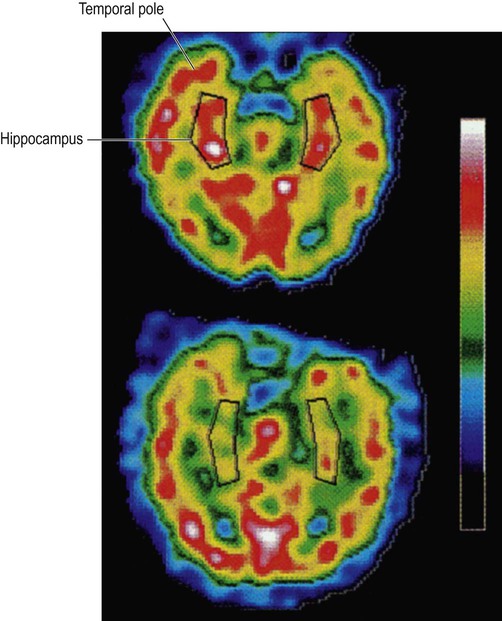
These images were obtained via single photon emission computer tomography (SPECT) scanning using a radioactive tracer and show regional cerebral blood flow. The upper image is a normal control subject and the lower image is from a patient with early Alzheimer’s disease. The sections are in the axial (horizontal) plane and pass through the temporal lobe. The black box shows the position of the hippocampus in the medial temporal lobe. From Rodriguez, G. et al.: Hippocampal perfusion in mild Alzheimer’s disease. Psychiatry Research: Neuroimaging (2000, pp. 65–74) with permission.
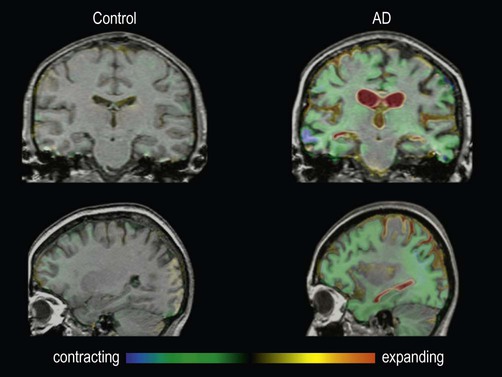
These are fluid-registered volumetric MRI scans from a 60-year-old patient with Alzheimer’s disease [right] and a normal age-matched control [left]. In each case, two MRI scans were acquired one year apart and have been registered together. Regions of brain loss are shown as blue-green; increases in CSF spaces are shown as red-yellow. In the patient with Alzheimer’s disease, there is marked volume loss in the hippocampi and temporal lobes, with widespread symmetrical atrophy in other parts of the cerebral hemispheres and dilation of the ventricles. The control shows normal age-related atrophy. Courtesy of Professor Nick Fox.



Risk factors
The most important risk factor for Alzheimer’s disease, apart from advancing age, is possession of a particular variant of the Apolipoprotein E gene (APOE) on chromosome 19 (discussed below). It is also more common in people with ischaemic heart disease, in those with limited educational attainments or lower socioeconomic status – and in association with previous head injury (Clinical Box 12.4).
 Clinical Box 12.4: Dementia pugilistica
Clinical Box 12.4: Dementia pugilistica
Dementia pugilistica or ‘punch drunk’ syndrome is a form of acquired cognitive impairment that occurs in up to a fifth of professional boxers (Latin: pugilator, boxer). It also occurs in other sports in which there may be mild head trauma, such as football and horse racing. The microscopic features are similar to Alzheimer’s disease, but the anatomical distribution is different. Pathology is often found in the substantia nigra of the midbrain and cognitive decline may be combined with features of Parkinson’s disease (in which nigral degeneration is a key pathological feature; see Ch. 13).




Pathological features
Plaques (Fig. 12.5)
A pathological hallmark of Alzheimer’s disease is the presence of plaques in the cerebral cortex, consisting of insoluble protein aggregates. Plaques are found in the extracellular space (between neurons) and are predominantly composed of amyloid beta peptide (Aβ). Like all forms of amyloid, the deposits take up the tissue stain Congo red and show apple-green birefringence under polarized light (see Ch. 8). Plaques can be identified using silver staining, but are best demonstrated using immunohistochemistry (antibody labelling).
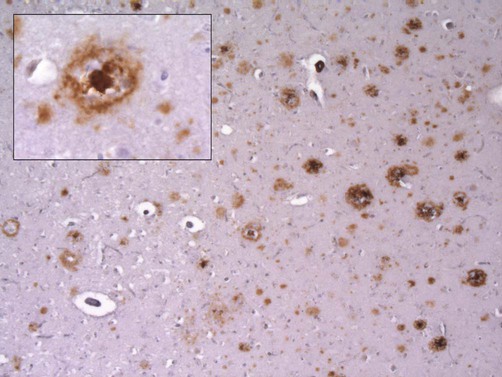
This micrograph demonstrates extensive amyloid beta deposits (amyloid plaques) in the cerebral cortex of a person who died of Alzheimer’s disease. Inset: a single ‘cored plaque’ with a dense central core of amyloid is shown at higher magnification [immunohistochemical preparation for Aβ peptide].
 Diffuse plaques are composed of amyloid beta peptide in a non-fibrillary (non-amyloid) form and may be numerous in older people who do not have dementia.
Diffuse plaques are composed of amyloid beta peptide in a non-fibrillary (non-amyloid) form and may be numerous in older people who do not have dementia.
 Neuritic plaques are surrounded by abnormal dystrophic neurites (thickened, tortuous neuronal processes) and sometimes have a dense, central core of amyloid (‘cored plaques’; see Fig. 12.5).
Neuritic plaques are surrounded by abnormal dystrophic neurites (thickened, tortuous neuronal processes) and sometimes have a dense, central core of amyloid (‘cored plaques’; see Fig. 12.5).
Unlike diffuse plaques, neuritic plaques are strongly associated with cognitive decline. Aβ is also deposited in the walls of blood vessels, leading to cerebral amyloid angiopathy (Clinical Box 12.5).
 Clinical Box 12.5: Cerebral amyloid angiopathy
Clinical Box 12.5: Cerebral amyloid angiopathy
Aβ is deposited in cortical blood vessels in 90% of patients with Alzheimer’s disease, which is termed cerebral amyloid angiopathy (CAA) (see Ch. 8, Fig. 8.14). Amyloid deposition leads to degeneration of vascular smooth muscle, weakening the vessel wall and predisposing to microinfarctions and intracerebral haemorrhage (see Ch. 10, Fig. 10.2B). CAA can occur in the absence of dementia and accounts for a quarter of intracerebral haemorrhages in the elderly. The bleeding is superficial or ‘cortical’ rather than deep (in contrast to hypertension-related haemorrhages, which frequently occur in the region of the basal ganglia).
Neurofibrillary tangles (Fig. 12.6)
The second major pathological finding in Alzheimer’s disease is the neurofibrillary tangle (NFT). This is a filamentous inclusion composed of the microtubule-associated protein tau, which is normally present in axons (see Ch.5).
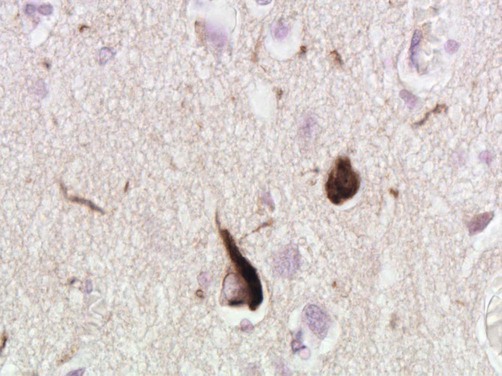
Micrograph showing two cortical neurons containing neurofibrillary tangles. The small, rounded inclusion (right) is a ‘globose’ tangle whilst the larger, more pyramidal-shaped inclusion (left) is a ‘flame’ tangle. The different appearances of the pathological inclusions reflect the shape of the neuronal cell body, which is filled with abnormal tau protein. A few fine ‘neuropil threads’ can also be seen, representing neuronal processes filled with tau [immunohistochemistry for hyperphosphorylated tau protein].
Tau is a phosphoprotein with 79 serine and threonine phosphorylation sites, less than half of which are normally phosphorylated. Neurofibrillary tangles contain hyperphosphorylated tau in the form of paired helical filaments (PHFs) that resemble twisted ribbons (Fig. 12.7).

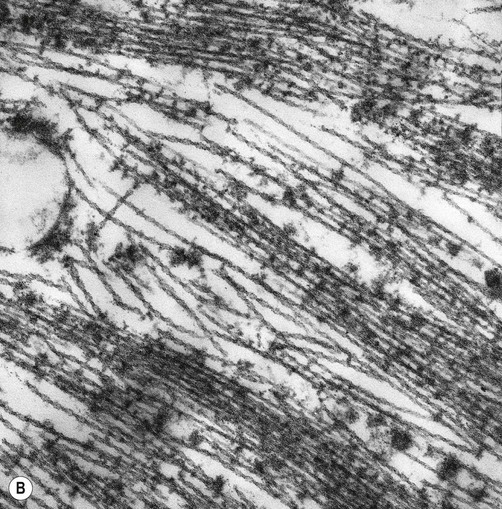
(A) Illustration showing the twisted structure of PHF-tau, which has a diameter of 22 nm and a periodicity of 80 nm; (B) An electron micrograph showing paired helical filament tau. From Ellison and Love: Neuropathology 2e (Mosby 2003) with permission.
Neuronal and synaptic loss
Quantitative post-mortem studies in patients with Alzheimer’s disease have shown significant neuronal loss in the hippocampus and medial temporal lobe. The entorhinal cortex is particularly affected, with more than 90% neuronal loss in advanced Alzheimer’s disease. This area gives rise to the perforant path (a major afferent projection to the hippocampus, see Ch. 3), meaning that the hippocampus is effectively ‘disconnected’ from the neocortex in Alzheimer’s disease.
More widespread loss of nerve cells and synaptic connections is found throughout the cerebral cortex and there is a reduction in the number of dendritic spines (see Ch. 5). Cell death also occurs in the brain stem, including the noradrenergic loci coerulei (singular: locus coeruleus) in the pons, which are normally pigmented but appear pale in Alzheimer’s disease.
Profound neuronal loss occurs in Meynert’s nucleus, a large cholinergic nucleus in the base of each cerebral hemisphere which contributes to the diffuse neurochemical system for acetylcholine (see Ch. 1). Degeneration of this projection (which diffusely innervates the neocortex and hippocampus) has a negative impact on memory, attention and cognition. For this reason, agents that potentiate cholinergic neurotransmission may improve symptoms in Alzheimer’s disease (see below).





Pathogenesis of Alzheimer’s disease
Accumulation of amyloid beta is widely believed to be a key initiating event in the development of Alzheimer’s disease. This appears to trigger a cascade of pathological events in the neuron, leading to tau hyperphosphorylation and tangle formation: the amyloid cascade hypothesis. Neuronal injury is thought to be mediated by a combination of cytopathic mechanisms including oxidative stress, excitotoxicity and inflammation, culminating in programmed cell death (see Ch. 8).
Formation of amyloid beta
Amyloid beta is a soluble peptide of 40 or 42 amino acids (Aβ40, Aβ42). It is produced by proteolytic cleavage of amyloid precursor protein (APP) (Fig. 12.8A). This is a large transmembrane protein that appears to be involved in synaptic plasticity and learning. There are six isoforms of APP in the human brain, composed of 695–770 amino acids (depending on alternative splicing). It has a single intramembranous portion, a large extracellular domain and a short cytoplasmic tail. The Aβ sequence is partly contained in the intramembranous portion.
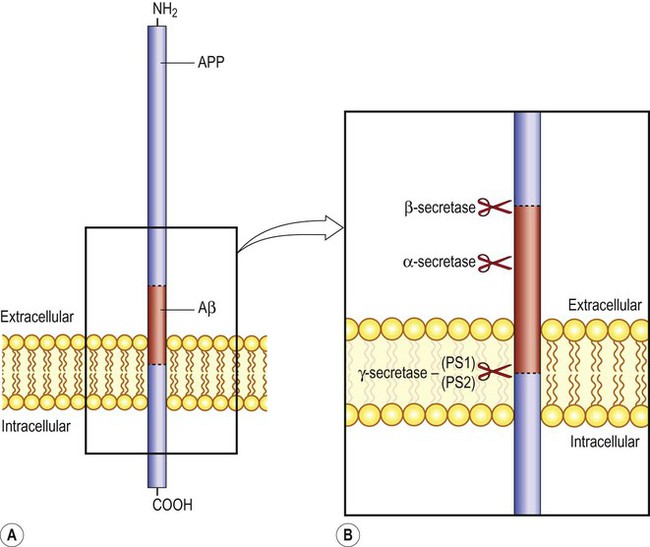
(A) The sequence corresponding to the amyloid beta peptide (Aβ) is shown in red, partially embedded in the neuronal plasma membrane; (B) A closer view, showing the sites of action of the main APP processing enzymes. Gamma-secretase is a multi-subunit integral membrane protein that includes the enzymes presenilin-1 and presenilin-2 (PS1/PS2).
Amyloid processing
There are two APP processing pathways (Fig. 12.9) mediated by a family of proteolytic enzymes called secretases (see also Fig. 12.8B). The pathogenic Aβ fragment is released from APP by sequential beta and gamma secretase cleavage. The C-terminal fragment is released into the cytoplasm and is involved in nuclear signalling. APP is encoded on chromosome 21, which accounts for the high incidence of Alzheimer-type changes in people with Down’s syndrome (trisomy 21) who have an extra copy of the chromosome. This leads to increased production of Aβ, associated with a greater than 50% chance that the brain will contain plaques and tangles by early middle age.
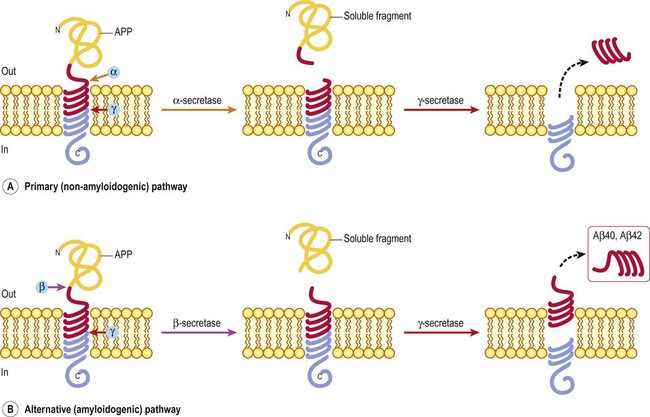
(A) The primary (alpha) pathway involves sequential cleavage by alpha and gamma secretases, which destroys the pathogenic (Aβ) fragment and is therefore non-amyloidogenic; (B) The alternative (beta) pathway involves sequential cleavage by beta and gamma secretases, which releases the pathogenic Aβ species. The alternative pathway is therefore potentially amyloidogenic.
Primary (alpha) pathway
The primary pathway for APP processing involves sequential activity of alpha and gamma secretase (Fig. 12.9A). Alpha secretase releases a large soluble APP fragment into the extracellular fluid. This is followed by gamma secretase cleavage, which acts on the intramembranous portion of APP. The cleavage point of alpha secretase falls in the middle of the Aβ sequence, thereby destroying the pathogenic peptide. The primary pathway is therefore non-amyloidogenic and since it is dominant, very little Aβ is normally produced in the brain.
Alternative (beta) pathway
This pathway involves the sequential action of beta and gamma secretases. It liberates amyloid beta from APP and is therefore potentially amyloidogenic (Fig. 12.9B). Beta-secretase cleaves APP at the distal end of the Aβ sequence and gamma secretase acts at the proximal end (within the plasma membrane). The alternative pathway releases either a 40 or 42 amino acid species of amyloid beta.
Ratio of Aβ40 to Aβ42
This is determined by the multi-protein gamma-secretase complex that includes the enzymes presenilin-1 and presenilin-2 (PS-1 and PS-2) and affects the pattern and distribution of the pathological changes. Aβ42 tends to form plaques in the cerebral cortex and is more associated with dementia. The more soluble Aβ40 fragment is frequently deposited in blood vessels, causing cerebral amyloid angiopathy. Several inherited forms of Alzheimer’s disease involve mutations in genes concerned with APP processing that increase the amount of Aβ (or Aβ42) (Clinical Box 12.6).
 Clinical Box 12.6: Familial Alzheimer’s disease
Clinical Box 12.6: Familial Alzheimer’s disease







Formation of neurofibrillary tangles
The next step in the ‘amyloid cascade’ is abnormal phosphorylation of the microtubule-associated protein tau. This normally binds to (and stabilizes) axonal microtubules, but in its hyperphosphorylated form, tau dissociates from microtubules and tends to self-aggregate (Fig. 12.10). This may reflect excessive activity in tau kinases (which add phosphate groups to proteins) such as glycogen synthase kinase 3-beta (GSK-3β) and cyclin-dependent kinase 5 (CDK5) but the mechanism is not certain.
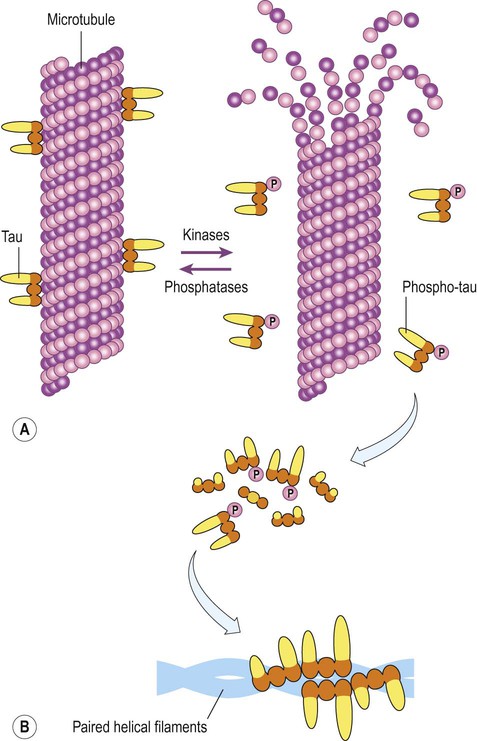
(A) In its unphosphorylated state, tau binds to microtubules and helps to stabilize them. Phosphorylation is regulated by the activity of kinases (which add phosphate groups) and phosphatases (which remove them); (B) Hyperphosphorylated tau monomers dissociate from microtubules and, at sufficient concentrations, tend to self-aggregate to form paired helical filament tau (the main constituent of neurofibrillary tangles).
Once dissociated from microtubules, monomers of phosphorylated tau aggregate into protofilaments (oligomers) and then filaments, eventually forming mature neurofibrillary tangles. The spread of tangle pathology through the brain follows six Braak stages, which can be divided into early, intermediate and late phases (Fig. 12.11).
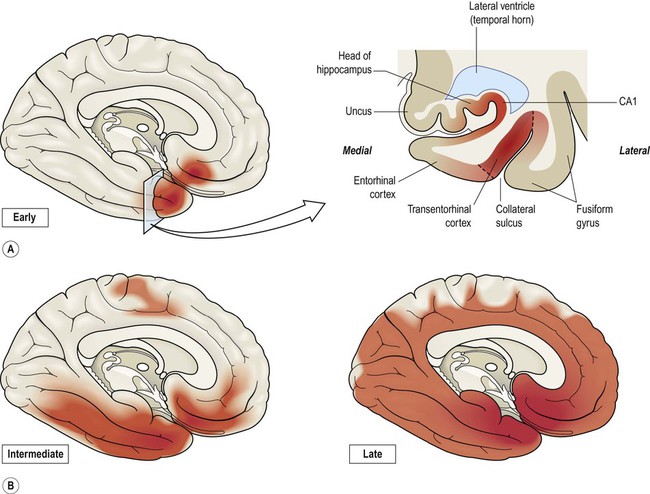
(A) Coronal section through the temporal lobe showing the distribution of tangle pathology in early (Braak I–II) Alzheimer’s disease. The earliest and most severely affected area (between the dashed lines) is the transentorhinal cortex; (B) Progression of tangle pathology in the intermediate (Braak III–IV) and late (Braak V–VI) stages.




Pathological ageing and cognitive reserve
Post-mortem studies have shown that the typical pathological changes of Alzheimer’s disease are present to a greater or lesser extent in many elderly people with no history of dementia. This is referred to as pathological ageing. In some cases the pathology is advanced, despite no evidence of cognitive decline during life. This is thought to reflect greater cognitive reserve capacity in some people, due to higher levels of premorbid intelligence or education (Fig. 12.12).
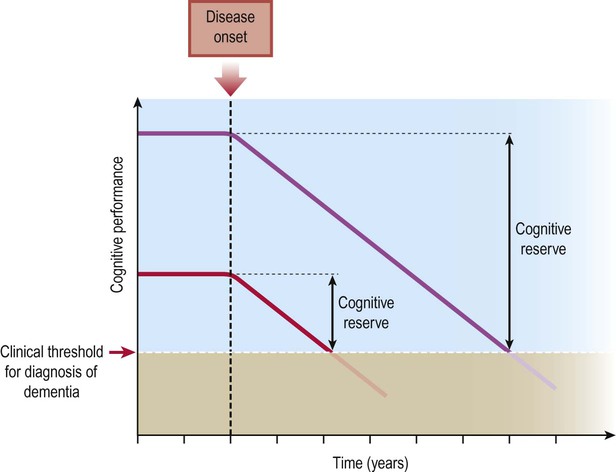
Individuals of average intelligence (red line) may show clinical signs of dementia much earlier after disease onset than people with a higher degree of premorbid intelligence or education (purple line), assuming that the rate of progression is similar.





Treatment of Alzheimer’s disease
Pharmacological agents
A second agent used in patients with Alzheimer’s disease is memantine, an NMDA receptor antagonist that inhibits glutamatergic neurotransmission (see Ch. 7). The mechanism is uncertain, but may include protection against neuronal calcium overload. Clinical trials have shown some benefit in patients with moderate dementia, but not in those with mild cognitive decline.
Disease-modifying agents






One of the most promising approaches to date has been Aβ vaccination, which has achieved almost complete plaque clearance in mouse models. Follow-up post-mortem studies in patients with Alzheimer’s disease have confirmed some degree of plaque clearance, but: (i) only 20% of patients generate anti-Aβ antibodies; (ii) the improvement in cognitive state is minimal; and (iii) severe meningoencephalitis (inflammation of the brain and its coverings) occurs in 1 in 20 people. Theoretical concerns have also been raised that disrupting plaques may release harmful oligomeric species.



Dementia with Lewy bodies
Clinical features
The three core elements of dementia with Lewy bodies (in addition to cognitive decline) are: (i) visual hallucinations; (ii) fluctuation in cognitive performance; and (iii) features of parkinsonism, such as tremor, rigidity and bradykinesia (see Ch. 13). Additional findings in support of the diagnosis include reduced dopamine levels in the basal ganglia or a sleep disorder (Clinical Box 12.7).
 Clinical Box 12.7:
Clinical Box 12.7:
Psychological testing and neuroimaging
Functional brain imaging often shows reduced metabolism and cerebral blood flow in the occipital and posterior temporoparietal regions (Fig. 12.13). This is in contrast to other forms of dementia in which the posterior hemisphere tends to be spared and may account for the prominent visual hallucinations in DLB.
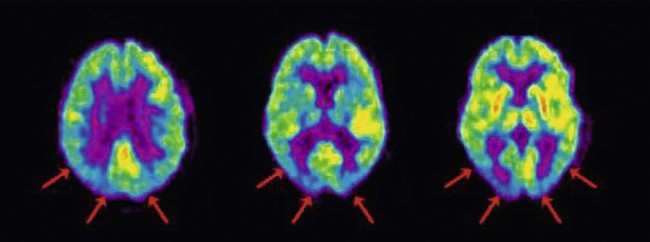
Functional brain imaging in patients with DLB may show reduced glucose consumption (metabolism) in the occipital and posterior temporoparietal regions, as in this case. [The image was obtained by positron-emission tomography (PET) scanning, using 18F-FDG, a positron-emitting isotope of glucose]. From Silverman, D et al: Positron emission tomography scans obtained for the evaluation of cognitive dysfunction. Seminars in Nuclear Medicine. Neuronuclear Imaging (2008, pp. 251–261) with permission.



Pathological features
Macroscopic examination of the brain is often unremarkable but there may be generalized atrophy and dilation of the ventricles, with pallor of the substantia nigra (see Ch. 13, Fig. 13.5) and locus coeruleus (see Ch. 1). The microscopic features are indistinguishable from Parkinson’s disease with dementia (PDD) and the distinction between PDD and DLB is purely clinical (see Ch. 13, Clinical Box 13.1).
Lewy bodies
Microscopic examination of the brain shows pathological inclusions (abnormal protein aggregates) in the cytoplasm of surviving neurons. These rounded intraneuronal structures, called Lewy bodies, appear bright pink on standard histological preparations (since they take up the red tissue dye eosin) and are often surrounded by a pale halo (Fig. 12.14A). Lewy bodies are best demonstrated by immunohistochemistry for alpha-synuclein protein, which is the major constituent (Fig. 12.14B). Cortical Lewy bodies are similar to those found in the brain stem, but lack a halo and are best seen on immunohistochemistry (Fig. 12.14C). Many patients also have some degree of Alzheimer’s disease pathology (Fig. 12.14D) and there may be a synergistic interaction between alpha-synuclein and Aβ.
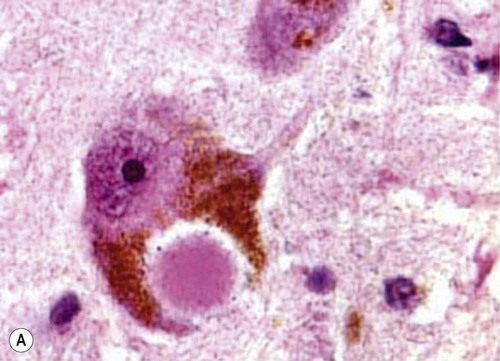
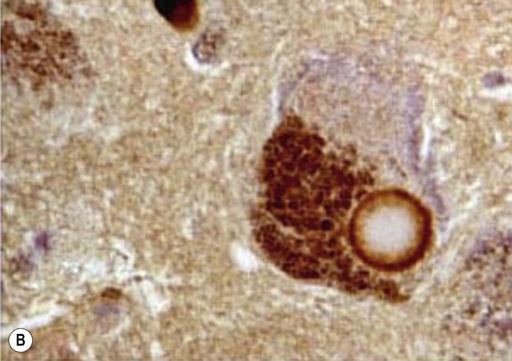
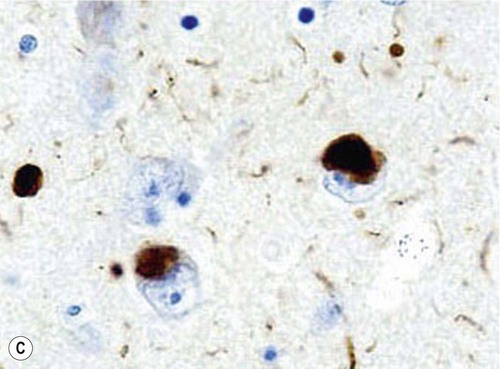
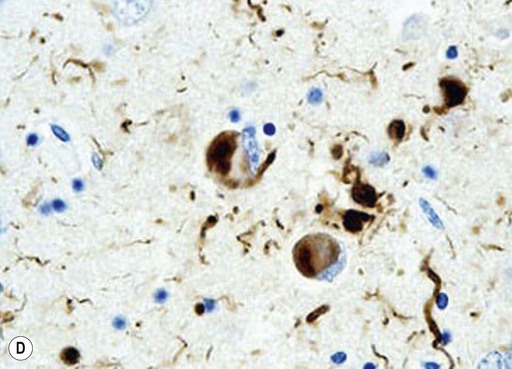
(A) This is a routine haematoxylin and eosin (H&E)-stained section from the substantia nigra of a patient who had dementia with Lewy bodies (DLB). It shows a bright pink Lewy body in the neuronal cytoplasm, surrounded by brown neuromelanin pigment; (B) Lewy bodies can be demonstrated using immunohistochemistry (antibody labelling) for alpha-synuclein, which is the main protein constituent; (C) Cortical Lewy bodies are best detected by immunostaining for alpha-synuclein, in this case in the temporal lobe of a patient with DLB; (D) Immunohistochemistry for alpha-synuclein in a patient with Alzheimer’s disease showing co-existing Lewy bodies and Lewy neurites in the temporal lobe. From Popescu, A and Lippa, CF: Parkinsonian syndromes: Parkinson’s disease dementia, dementia with Lewy bodies and progressive supranuclear palsy. Clinical Neuroscience Research, Non-Alzheimer’s Disease Dementias (2004, pp. 461–468) with permission.
Progression of pathology
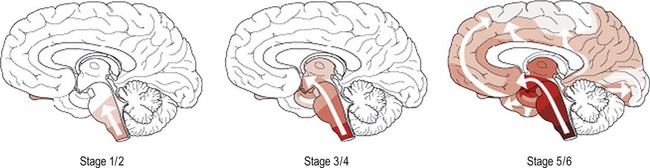
The pathological changes spread through the brain in a predictable fashion, analogous to the orderly progression of tangles in Alzheimer’s disease (Fig. 12.15). Six Braak stages are described, which are the same as those in idiopathic Parkinson’s disease (see also Ch. 13, Fig. 13.7). Synuclein-positive inclusions appear first in the olfactory bulb and medulla (stage I), before progressing to the pons (stage II) and midbrain (stage III). The first three stages are thus largely confined to the brain stem. Inclusions are next encountered in the limbic lobe, including the entorhinal cortex and amygdala (stage IV). The last two stages are neocortical, with inclusions in the higher-order association cortices (stage V) and finally in the primary sensory and motor areas (stage VI). The pathology and pathogenesis of the synuclein-related disorders (or synucleinopathies) is discussed further in Chapter 13.



Vascular dementia
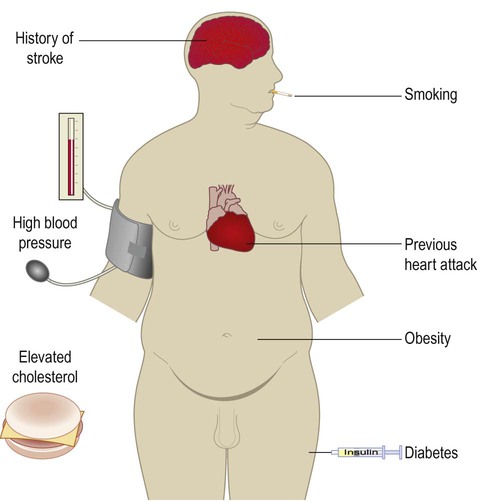
Vascular dementia is the third most common cause of acquired cognitive decline in older people, accounting for approximately 10% of cases. The risk factors are the same as those for ischaemic heart disease and stroke (Fig. 12.16). The term vascular cognitive impairment is used to describe a non-progressive decline in intellectual ability with a vascular aetiology.
Clinical features
People with vascular dementia may show a step-wise loss of cognitive ability consistent with a series of strokes, referred to as multi-infarct dementia (Fig. 12.17). In others, the decline is more gradual and is dominated by generalized slowing of cognition with disturbed frontal executive function (i.e. bradyphrenia or ‘subcortical-type’ dementia). This is often due to diffuse white matter disease, sometimes referred to as Binswanger’s encephalopathy (pronounced: BINSE-vangers).
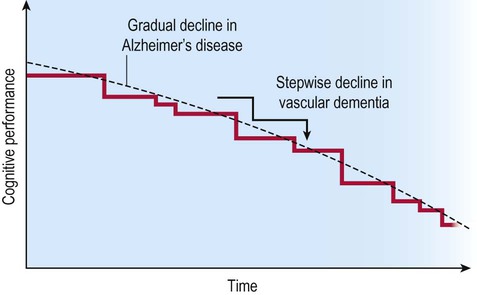
Progression is classically said to occur in a step-wise fashion consistent with a series of small strokes, in contrast to the gradual decline seen in Alzheimer’s disease.
Distinction from Alzheimer’s disease
Associated damage to the basal ganglia sometimes leads to a pseudo-parkinsonian gait with short, shuffling steps: the marche à petit pas (French: walking with small steps) and there may be focal neurological signs consistent with one or more previous strokes. The typical imaging appearances are shown in Figure 12.18.
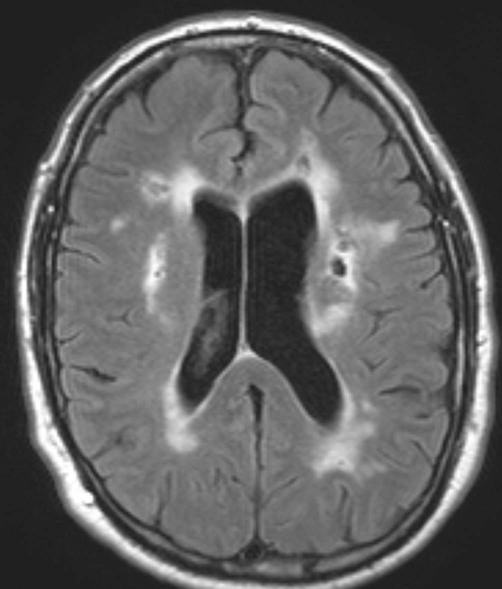
There is confluent hyperintensity in the periventricular white matter and patches of hyperintensity in the deep and subcortical white matter. Additionally, there are several small focal cavities (with the same signal intensity as CSF) which are typical of ‘old’ lacunar infarcts [T2-weighted MRI with CSF suppression: fluid-attenuation inversion recovery or ‘FLAIR’ sequence]. Courtesy of Dr Andrew MacKinnon.
Pathological features
Vascular dementia may be caused by: (i) small vessel disease; (ii) large vessel disease; or (iii) a combination of the two. The term mixed dementia is used if there are co-existent features of Alzheimer’s disease or another dementia. A rare inherited form of vascular dementia is discussed in Clinical Box 12.8.
 Clinical Box 12.8:
Clinical Box 12.8:Small vessel disease
This is the most common finding in patients with vascular dementia. It is frequently associated with arterial hypertension, diabetes and other vascular risk factors. High blood pressure damages the walls of small and medium-sized blood vessels, leading to replacement of smooth muscle by collagen. This gives an amorphous or ‘hyaline’ (Greek: glassy) appearance under the microscope, termed hyaline arteriosclerosis (Fig. 12.19A). Some vessels show more striking wall destruction with replacement of smooth muscle by lipid-laden foam cells, termed lipohyalinosis.
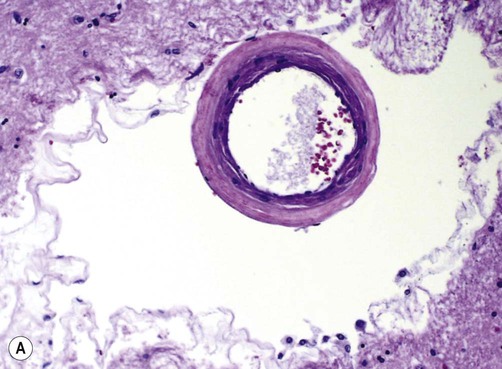
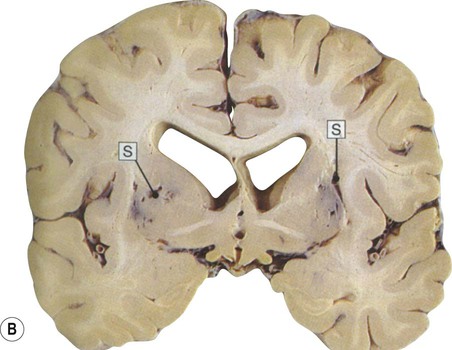
(A) Micrograph of a deep hemispheric blood vessel showing replacement of vascular smooth muscle by (pink) hyaline material. There is also marked enlargement of the perivascular space. From Prayson, R: Neuropathology 1e (Churchill Livingstone 2005) with permission; (B) Photograph of a coronal brain section showing multiple lacunar infarcts in the basal ganglia as a result of small vessel disease (including prominent slit-like spaces, labelled “S”). From Stevens and Lowe: Pathology 2e (Mosby 2000) with permission.
Both types of small vessel pathology contribute to ‘hardening’ of the arteries and arterioles, referred to as arteriosclerosis and arteriolar sclerosis respectively (Greek: sklerōs, hard). Normally distensible blood vessels are thus converted into rigid pipes with limited ability to dilate in response to fluctuations in blood pressure. This leads to small strokes (less than 1 cm in diameter) called lacunar infarcts (Latin: lacūna, hole or gap) (Fig. 12.19B). There may also be a more general attenuation of the subcortical white matter with expansion of perivascular spaces, creating a ‘moth-eaten’ appearance.






Frontotemporal dementia
Frontotemporal dementia (FTD) is a clinical term for a group of conditions in which there is selective degeneration of the frontal and temporal lobes (Fig. 12.20). The lifetime prevalence is 1 in 6000 and it is equally common in males and females. FTD accounts for less than 5% of dementia overall, but is the second most common cause in people under 60 (after Alzheimer’s disease). Onset is typically in middle age, with an average survival of around 6–8 years. A family history is present in 40% of cases.
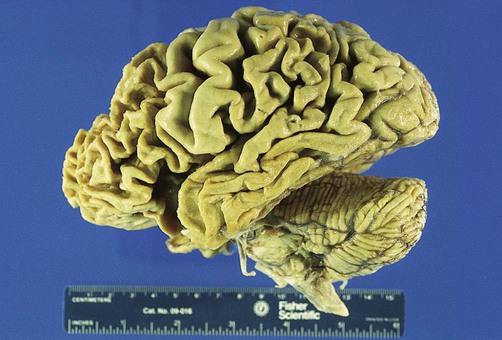
Macroscopic photograph showing selective degeneration of the frontal and temporal lobes. The primary sensory and motor areas are spared. From Prayson, R: Neuropathology 1e (Churchill Livingstone 2005) with permission.
Clinical features
Selective degeneration of the frontal and temporal lobes leads to a progressive deterioration in behaviour, personality and language. In difficult cases, specialized neuroimaging techniques may be helpful in distinguishing between FTD and Alzheimer’s disease (Clinical Box 12.9).
 Clinical Box 12.9: Neuroimaging in frontotemporal dementia
Clinical Box 12.9: Neuroimaging in frontotemporal dementia
A soluble amyloid-binding molecule has been developed that is able to cross the blood–brain barrier, called Pittsburgh interesting compound B. This can be radioactively labelled and used to demonstrate deposition of amyloid beta peptide in the brains of living people (called PIB scanning). A potential clinical application of this technique is in the differentiation between patients with frontotemporal dementia and Alzheimer’s disease with prominent involvement of the frontal and temporal lobes (Fig. 12.21).
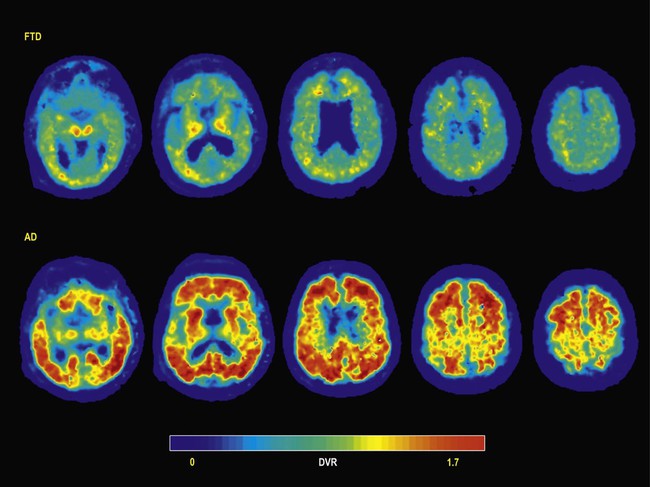
PIB-PET imaging in the patient with Alzheimer’s disease reveals diffuse cortical binding of amyloid beta, in contrast to non-specific white matter labelling in the patient with FTD. In both cases, pathological confirmation of the diagnosis was subsequently obtained at post-mortem examination. Axial distribution volume ratio (DVR) images provided courtesy of Dr Gil Rabinovici.
Frontotemporal dementia subtypes
Behavioural variant of FTD
The behavioural variant of frontotemporal dementia accounts for 50% of cases. It is characterized by striking personality change with relative preservation of memory and language. Common features include disinhibition (e.g. inappropriate comments, reduced tact, self-centred behaviour) and emotional blunting (apathy, emotional coldness or loss of empathy). Many patients have elements of obsessive-compulsive disorder (OCD) (see Ch. 3, Clinical Box 3.10) and there may be altered eating habits with weight gain and a craving for sweet foods. Neuroimaging typically shows bilateral frontal (especially orbitofrontal) atrophy which is often worse on the non-dominant side (Fig. 12.22A). Many of these cases were formally referred to as ‘Pick’s disease’ (Clinical Box 12.10).
 Clinical Box 12.10:
Clinical Box 12.10: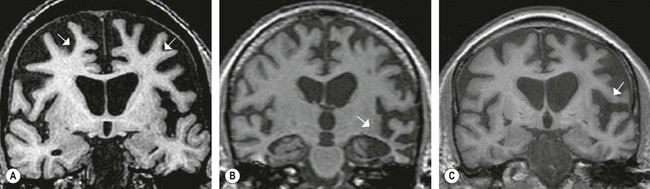
(A) In the behavioural variant of FTD there is usually bilateral frontal lobe atrophy which is often worse on the non-dominant side; (B) In semantic dementia severe bilateral temporal lobe atrophy is typical; (C) Progressive non-fluent aphasia is characterized by dominant (usually left) hemisphere atrophy in the inferior frontal gyrus and perisylvian region, corresponding to the left-hemisphere language areas. [Coronal T1-weighted MRI scans. The arrows indicate the most severely affected areas.] From Schott, JM and Fox, NC: Structural imaging in the dementias. J Psychiat (Old Age Psychiatr) (2007, pp. 503–507) with permission.
Semantic dementia
In semantic dementia the ability to understand the meaning of words and concepts is gradually lost. Inability to name objects (anomia) is an early feature and there is gradual erosion of semantic categories used to classify people and objects. In the early stages this may lead to naming errors (e.g. referring to a bus as a car). Later, more general categories are used to describe particular objects (e.g. apples, oranges and bananas are all described as ‘fruit’). In advanced stages, semantic categories are lost altogether (e.g. living versus non-living, plant versus animal) and everyday objects may appear strange or frightening. Neuroimaging shows severe bilateral temporal lobe atrophy (Fig. 12.22B).
Progressive non-fluent aphasia
Individuals with progressive non-fluent aphasia (PNFA) have marked expressive language difficulties (aphasia) with word substitutions and word-finding problems, together with errors in grammar, syntax and pronunciation. This tends to affect verbs, prepositions and function words more than nouns (cf. Broca’s dysphasia; see Ch. 3). In some cases there is progressive apraxia of speech, which has more to do with motor control of the speech articulators. Neuroimaging in PNFA typically shows asymmetric brain atrophy which is most severe in the left inferior frontal lobe and ‘perisylvian region’, meaning the area surrounding the lateral sulcus, corresponding to the left-hemisphere language areas (Fig. 12.22C).
FTD and motor neuron disease
Up to 15% of patients with frontotemporal dementia also have features of motor neuron disease (MND). This is characterized by progressive muscle weakness and wasting, together with increased muscle tone and reflexes, due to loss of motor neurons in the cerebral cortex and spinal cord (see Ch. 4, Clinical Box 4.9). In these cases a diagnosis of FTD-MND is made. Conversely, at least 30% of patients with motor neuron disease develop cognitive decline which may be severe enough to meet the criteria for frontotemporal dementia. This reflects an overlap between the molecular pathology of FTD and MND (discussed below).









[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]<
[/not-level-membership-for-basic-science-category]
