Chapter 7 Dementia
Neurologists and most other physicians continue to use the term dementia, which they see as a clinical condition or syndrome of a progressive decline in cognitive function that impairs daily activities. Neurologists require memory impairment plus one or more of the following: aphasia, apraxia, agnosia, or disturbance in executive function (see Chapter 8). Because their definition requires two domains, it excludes isolated amnesia (Greek, forgetfulness) or aphasia (Greek, speechlessness).
Disorders Related to Dementia
Congenital Cognitive Impairment
Physical manifestations of congenital cerebral injury, such as seizures and hemiparesis (“cerebral palsy,” see Chapter 13), frequently accompany Intellectual Developmental Disorder. In cases of genetic abnormalities, distinctive behavioral disturbances and anomalies of nonneurologic organs – the face, skin, ocular lenses, kidneys, and skeleton – often form syndromes.
Amnesia
Neurologists usually attribute amnesia to transient or permanent dysfunction of the hippocampus (Greek, sea horse) and other portions of the limbic system, which are based in the temporal and frontal lobes (see Fig. 16-5). Dementia, in contrast, usually results from extensive cerebral cortex dysfunction (see later).
Transient amnesia is an important, relatively common disturbance that usually consists of a suddenly occurring period of amnesia lasting only several minutes to several hours. It has several potential medical and neurologic explanations (Box 7-1). One of them, electroconvulsive therapy (ECT), routinely induces both anterograde and retrograde amnesia, with retrograde amnesia, especially for autobiographic information, tending to persist longer than anterograde amnesia. ECT-induced amnesia is more likely to occur or to be more pronounced following treatment with high electrical dosage, with bilateral rather than unilateral electrode placement, with use of alternating current, and three-times rather than two-times weekly administration. Without a pretreatment assessment of a patient’s memory and other aspects of cognitive function, clinicians may have problems separating ECT-induced amnesia from memory difficulties reflecting underlying depression, medications, and, especially in the elderly, pre-existing cognitive impairment.
Box 7-1
Commonly Cited Causes of Transient Amnesia
Various neuropsychologic and physical abnormalities usually accompany amnesia from neurologic conditions. For example, behavioral disturbances, depression, and headache are comorbid with posttraumatic amnesia. With severe traumatic brain injury (TBI, see Chapter 22), hemiparesis, ataxia, pseudobulbar palsy, or epilepsy accompanies posttraumatic amnesia. Similarly, in addition to its characteristic anterograde amnesia, the Wernicke–Korsakoff syndrome comprises ataxia and signs of a peripheral neuropathy (see later).
In another example, herpes simplex encephalitis causes amnesia accompanied by personality changes, complex partial seizures, and the Klüver–Bucy syndrome (see Chapters 12 and 16) because the virus typically enters the undersurface of the brain through the nasopharynx and attacks the frontal and temporal lobes. This condition occurs relatively frequently because herpes simplex is the most common cause of sporadically occurring (nonepidemic) viral encephalitis. (Human immunodeficiency virus [HIV] encephalitis, which does not cause this scenario, is epidemic.)
Conversely, apparent memory impairment may also appear as an aspect of several psychiatric disorders (see Chapter 11, dissociative amnesia). In general, individuals with amnesia from psychiatric illness or malingering (nonneurologic amnesia) lose memory for personal identity or emotionally laden events rather than recently acquired information. For example, a criminal deeply in debt may travel to another city and “forget” his debts, wife, and past associates, but he would retain his ability to recall people, events, and day-to-day transactions in his new life. Nonneurologic amnesia also characteristically produces inconsistent results on formal memory testing. In a simple example, a workman giving emphasis to the sequelae of a head injury may seem unable to recall three playing cards after 30 seconds, but half an hour after discussing neutral topics will recall three different cards after a 5-minute interval. Also, Amytal infusions may temporarily restore memories in individuals with nonneurologic amnesia, but not in those with brain damage.
Neuropsychologic Conditions
Confabulation is a neuropsychologic condition in which patients offer implausible explanations in a sincere, forthcoming, and typically jovial manner. Although individuals with confabulation disregard the truth, they do not intentionally deceive. Confabulation is a well-known aspect of Wernicke–Korsakoff syndrome, Anton syndrome (see cortical blindness, Chapter 12), and anosognosia (see Chapter 8). With these conditions referable to entirely different regions of the brain, confabulation lacks consistent anatomic correlations and physical features.
Discrete neuropsychologic disorders – aphasia, anosognosia, and apraxia – may occur alone, in various combinations, or as comorbidities of dementia (see Chapter 8). If one of them occurs with a comorbidity of dementia, it indicates that the dementia originates in “cortical” rather than “subcortical” dysfunction (see later). These disorders, unlike dementia, are attributable to discrete cerebral lesions. Sometimes only neuropsychologic testing can detect these disorders and tease them apart from dementia.
Normal Aging
Sleep
Among the elderly, times of falling asleep and awakening both phase-advance (occur earlier than usual), slow-wave sleep declines, and sleep fragments. In addition, restless legs syndrome and rapid eye movement (REM) behavioral disorder commonly disrupt their sleep (see Chapter 17). All these changes may occur whether or not the elderly have dementia.
Special Senses
Age-related deterioration of sensory organs impairs hearing and vision (see Chapter 12). Older individuals typically have small, less reactive pupils and some retinal degeneration. They require greater light, more contrast, and sharper focusing to be able to read and drive. Their hearing tends to be poorer, especially for speech discrimination. Their senses of taste and smell also deteriorate.
EEG and Imaging Changes
As individuals age, the electroencephalogram (EEG) background alpha activity slows to the lower end of the normal 8–12-Hz alpha range. Computed tomography (CT) and magnetic resonance imaging (MRI) often reveal decreased volume of the frontal and parietal lobes, atrophy of the cerebral cortex, expansion of the Sylvian fissure, and concomitantly increased volume of the lateral and third ventricles (see Figs 20-2, 20-3, and 20-18). In addition, MRI reveals white-matter hyperintensities (“white dots”). Although striking, these abnormalities are usually innocuous and, by themselves, do not reflect the onset of Alzheimer disease.
Dementia
Classifications and Causes
• Prevalence: Studies reporting on the epidemiology of dementia-producing illnesses vary by whether cases were clinical or postmortem, drawn from primary or tertiary care settings, or if the criteria were incidence or prevalence. By any measure, Alzheimer disease is the most prevalent cause of dementia, not only because its incidence is so high, but also, with its victims living for a relatively long time, its prevalence is also high. A typical breakdown of the prevalence of dementia-producing illnesses would list Alzheimer disease 70%, dementia with Lewy bodies (DLB) 15%, vascular cognitive impairment (VCI) or the preliminary DSM-5 term Vascular Neurocognitive Impairment 10%, frontotemporal dementia 5–10%, and all others, in total, 5–10%.
• Patient’s age at the onset of dementia: Similarly, beginning at age 65 years, those same illnesses cause almost all cases of dementia. However, individuals between 21 and 65 years are liable to succumb to different dementia-producing illnesses: HIV disease, substance abuse, severe TBI, end-stage multiple sclerosis (MS) (see Chapter 15), frontotemporal dementia, and VCI. In adolescence, the causes are different and more numerous (Box 7-2).
• Accompanying physical manifestations: Distinctive physical neurologic abnormalities that may accompany dementia and allow for a diagnosis by inspection include ocular motility impairments, gait apraxia (see later), myoclonus (see later and Chapter 18), peripheral neuropathy (see Box 5-1), chorea, other involuntary movement disorders (see Box 18-4), and lateralized signs, such as hemiparesis.
• Genetics: Of frequently occurring illnesses, Huntington disease, frontotemporal dementia, some prion illnesses, and, in certain families, Alzheimer disease follow an autosomal dominant pattern. Wilson disease follows an autosomal recessive pattern.
• Rapidity of onset: In patients with Alzheimer disease, dementia evolves over a period of many years to a decade, which serves as the reference point. In contrast, several diseases produce dementia within 6–12 months. These “rapidly progressive dementias” include HIV-associated dementia, frontotemporal dementia, dementia with Lewy bodies (see later), paraneoplastic limbic encephalitis (see Chapter 19), and, perhaps most notoriously, Creutzfeldt–Jakob disease and its variant.
• Reversibility: The most common conditions that neurologists usually list as “reversible causes of dementia” are depression, over-medication, hypothyroidism, B12 deficiency, other metabolic abnormalities, subdural hematomas, and normal-pressure hydrocephalus (NPH). Although reversible dementias are rightfully sought, the results of treatment are discouraging. Only about 9% of dementia cases are potentially reversible and physicians actually partially or fully reverse less than 1%.
• Cortical and subcortical dementias: According to a cortical/subcortical distinction, cortical dementias consist of illnesses in which neuropsychologic signs of cortical injury – typically aphasia, agnosia, and apraxia – accompany dementia. Because the brain’s subcortical areas are relatively untouched, patients remain alert, attentive, and ambulatory. Alzheimer disease serves as the prime example of a cortical dementia.
Mental Status Testing
Mini-Mental State Examination (MMSE) (Fig. 7-1)

FIGURE 7-1 Mini-Mental State Examination (MMSE). Points are assigned for correct answers. Scores of 20 points or less indicate dementia, delirium, schizophrenia, or affective disorders – alone or in combination. However, such low scores are not found in normal elderly people or in those with neuroses or personality disorders. MMSE scores fall in proportion to dementia-induced impairments in daily living as well as cognitive decline. For example, scores of 20 correlate with inability to keep appointments or use a telephone. Scores of 15 correlate with inability to dress or groom.
(Adapted from Folstein MF, Folstein SE, McHugh PR. “Mini-Mental state:” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. © 1975, 1998 MiniMental LLC.)
Alzheimer Disease Assessment Scale (ADAS)
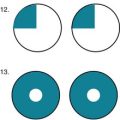
The ADAS consists of cognitive and noncognitive sections. Its cognitive section (ADAS-Cog) (Fig. 7-2) includes not only standard tests of language, comprehension, memory, and orientation, but also tests of visuospatial ability, such as drawing geometric figures, and physical tasks that reflect ideational praxis, such as folding a paper into an envelope. Patients obtain scores of 0–70 points in proportion to worsening performance.
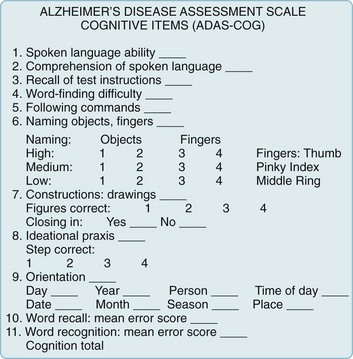
FIGURE 7-2 The cognitive section of the Alzheimer Disease Assessment Scale (ADAS-Cog), the standard test for assessing pharmacologic intervention, has been designed to measure many important aspects of cognitive function that are apt to deteriorate in Alzheimer disease. The noncognitive section measures mood, attention, delusions, and motor activity, such as pacing and tremors. As dementia begins or worsens, patients’ responses change from no impairment (where the patient receives 0 points) to severe cognitive impairment (5 points). In other words, as dementia worsens, patients accumulate points. Total ADAS-Cog scores for patients with mild to moderate Alzheimer disease range from 15 to 25 and, as cognitive function declines, scores increase by 6–12 points yearly.
(From Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984;141:1356–1364. Reprinted with permission from the American Journal of Psychiatry, Copyright 1984. American Psychiatric Association.)
Montreal Cognitive Assessment (MoCA) (Fig. 7-3)
The MoCA, which has greater sensitivity than the MMSE, readily detects mild impairment. It is useful when a patient’s only symptom is memory impairment or physicians suspect cognitive impairment in individuals who score 25 points or better on the MMSE. Neurologists also administer the MoCA to patients with any neurodegenerative illness – a category that includes Alzheimer, Parkinson, and dementia with Lewy bodies diseases, and frontotemporal dementia. In Parkinson disease, the MoCA compared to the MMSE is more sensitive in detecting early cognitive impairment (see Chapter 18).

FIGURE 7-3 The Montreal Cognitive Assessment (MoCA). This assessment includes eight cognitive domains. As with the MMSE, the total score reaches 30 and values lower than 26 indicate cognitive impairment. However, some authors found that a cut-off of 23 gave a sensitivity and specificity of at least 95%.
(Copyright Z. Nasreddine MD. Reproduced with permission. The test and instruction may be accessed at www.mocast.org.)
Further Testing
• For very intelligent, well-educated, or highly functional individuals with symptoms compatible with dementia whose screening tests fail to show a cognitive impairment.
• For individuals whose ability to execute critical occupational or personal decisions, including legal competency, must be assured.
• For assessing individuals with confounding deficits, such as intellectual development disorder, learning disabilities, minimal education, deafness, or aphasia.
• In distinguishing dementia from depression, other psychiatric disturbances, and malingering.
• In distinguishing Alzheimer disease from frontotemporal dementia.
Laboratory Evaluation in Dementia
Depending on the clinical evaluation, neurologists generally request a series of laboratory tests (Box 7-3). Although testing is expensive (see Appendix 2), it may allow a firm diagnosis or even detect a potentially correctable cause of dementia. In addition, certain tests may reveal the illness before individuals manifest cognitive impairment, i.e., in their preclinical or presymptomatic state. Neurologists modify the testing protocol if dementia has developed in an adolescent (see Box 7-2) or has progressed rapidly.
Physicians should reserve certain other tests for particular indications. For instance, if adolescents or young adults develop dementia, an evaluation might include serum ceruloplasmin determination and slit-lamp examination for Wilson disease; urine toxicology screens for drug abuse; and, depending on the circumstances, urine analysis for metachromatic granules and arylsulfatase-A activity for metachromatic leukodystrophy (see Chapter 5). Likewise, physicians should judiciously request serologic tests for autoimmune or inflammatory disease, serum Lyme disease titer determinations, and other tests for systemic illnesses.
Alzheimer Disease
Dementia
In the dementia stage, patients may remain superficially conversant, sociable, able to perform routine tasks, and physically intact. Nevertheless, the spouse or caregiver will report that they suffer from incapacitating memory impairment and inability to cope with new situations. Patients’ language impairment typically includes a decrease in spontaneous verbal output, an inability to find words (anomia), use of incorrect words (paraphasic errors), and a tendency to circumvent forgotten words – elements of aphasia (see Chapter 8).
Neuropsychiatric Manifestations
Wandering constitutes a particularly troublesome, although not unique, manifestation of Alzheimer disease. It may originate in any combination of numerous disturbances, including memory impairment, visuospatial perceptual difficulties, delusions and hallucinations, akathisia (see Chapter 18), and mundane activities, such as looking for food.
Alzheimer patients also lose their normal circadian sleep–wake pattern to a greater degree than cognitively intact elderly people (see Chapter 17). Their sleep becomes fragmented throughout the day and night. The disruption of their sleep parallels the severity of their dementia.
Physical Signs
Patients with Alzheimer disease characteristically have no physical impairment, except for one small but intriguing finding – anosmia (see Chapter 4). Even before the onset of dementia, Alzheimer disease patients require high concentrations of a volatile substance to detect and identify it. Neurologists also find anosmia in patients with Parkinson disease and dementia with Lewy bodies disease, but not in those with VCI or depression. Patients with TBI with or without dementia also have anosmia because they have had shearing of the olfactory nerve fibers at the cribriform plate (see Chapter 22).
Until Alzheimer disease advances to unequivocal dementia, patients usually remain ambulatory and able to feed themselves. The common sight of an Alzheimer disease patient walking steadily but aimlessly through a neighborhood characterizes the disparity between intellectual and motor deficits. When patients reach advanced stages of the illness, physicians can elicit frontal release signs (Fig. 7-4), increased jaw jerk reflex (see Fig. 4-13), and Babinski signs. Unlike patients with VCI, those with Alzheimer disease do not have lateralized signs. They eventually become mute, fail to respond to verbal requests, remain confined to bed, and assume a decorticate (fetal) posture. They frequently slip into a persistent vegetative state (see Chapter 11).
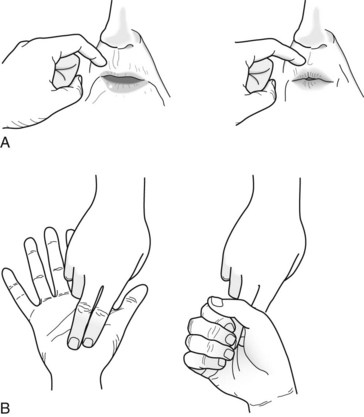
FIGURE 7-4 The snout and grasp reflexes – frontal lobe release reflexes – are frequently present in individuals with severe dementia. A, Physicians elicit the snout reflex by tapping the patient’s upper lip with a finger or a percussion hammer. This reflex causes the patient’s lips to purse and lower facial muscles to pout. B, Physicians elicit the grasp reflex by stroking the patient’s palm crosswise or the fingers lengthwise. The reflex causes the patient to grasp the physicians’ fingers and fail to release despite requests.
Laboratory Tests
In about one-half of Alzheimer patients, PET for glucose metabolism shows areas of decreased metabolism in the bilateral parietal and temporal association cortex (see Fig. 20-29). These hypometabolic areas are vague at first, but as the disease progresses, hypometabolism spreads to the frontal lobe cortex and becomes more distinct. PET remains unsuitable for routine testing, mostly because of its low sensitivity and specificity, as well as its cost. The changes are more specific and more cost-effective for frontotemporal dementia (see later). PET for amyloid accumulation is useful in the diagnosis of the preclinical stage of Alzheimer disease, but it is probably unnecessary in the symptomatic stage. Similarly, CSF analysis for depressed concentrations of amyloid and elevated concentrations of tau protein may be helpful in the preclinical stage, but superfluous in the symptomatic stage. Neurologists have not yet defined the role of PET and advanced CSF analysis in MCI.
Neurologists almost never request cerebral cortex biopsies for diagnostic purposes – mostly because routine evaluation is approximately 90% specific and sensitive.* In addition, histologic findings of Alzheimer disease differ mostly quantitatively, rather than qualitatively, from age-related changes. As a last resort, cerebral cortex biopsies can help establish a diagnosis of herpes simplex encephalitis, familial Alzheimer disease, and Creutzfeldt–Jakob disease or its variant.
Pathology
Cerebral atrophy naturally leads to compensatory dilation of the lateral and third ventricles. Of these, the temporal horns of the lateral ventricles expand the most. Nevertheless, in Alzheimer disease and most other illnesses that cause dementia, the anterior horns of the lateral ventricles maintain their concave (bowed inward) shape because of the indentation on their lateral border by the head of the preserved caudate nucleus (see Figs 20-2 and 20-18). The exception is Huntington disease, where atrophy of the head of the caudate nucleus permits the ventricle to expand laterally and assume a convex (bowed outward) shape (see Fig. 20-5).
Another histologic feature of Alzheimer disease is the loss of neurons in the frontal and temporal lobes. Neuron loss in the nucleus basalis of Meynert (also known as the substantia innominata), which is a group of large neurons located near the septal region beneath the globus pallidus (see Fig. 21-4), constitutes the most distinctive change. Their loss depletes cerebral acetylcholine (ACh: see later). Of all these histologic features, loss of neuron synapses correlates most closely with dementia.
Amyloid Deposits
Returning to the formation of Aβ, enzymes encoded on chromosome 21 cleave amyloid precursor protein (APP) to precipitate Aβ in the plaques (Fig. 7-5). Aβ differs from amyloid deposited in viscera as part of various systemic illnesses, such as multiple myeloma and amyloidosis. In Alzheimer disease, Aβ accumulates early and permanently in the cerebral cortex and vital subcortical regions.
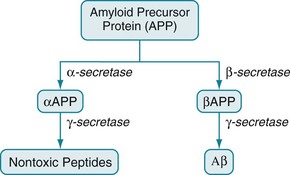
FIGURE 7-5 The amyloid precursor protein (APP) consists of 770 amino acids. Three secretase enzymes (α-, β-, and γ-) cleave it into polypeptide fragments. In one pathway, α-secretase cleaves APP to form the polypeptide αAPP. Then γ-secretase cleaves αAPP into soluble, nontoxic polypeptides. In the other pathway, β-secretase cleaves APP into a different polypeptide, βAPP. Then γ-secretase cleaves βAPP to form the insoluble, toxic, 42-amino-acid polypeptide, Aβ.
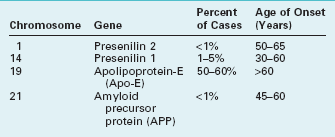
• All known genetic mutations associated with Alzheimer disease increase Aβ production (chromosomes 1, 14, 21) or Aβ aggregation (chromosome 19).
• The most powerful established genetic risk factor, which involves apolipoprotein-E (Apo-E), promotes Aβ deposition.
• Aβ is toxic in vitro probably because it binds to cerebral ACh receptors and poisons cerebral mitochondria.
• Accumulation of Aβ is a requirement for the development of Alzheimer disease.
• At least in some nonhuman studies, antibodies to Aβ reduce clinical and pathologic aspects of Alzheimer disease.
Biochemical Abnormalities
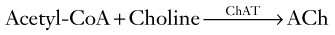
The cholinergic hypothesis, drawn from these biochemical observations, postulates that reduced cholinergic activity causes the dementia of Alzheimer disease. In addition to the histologic and biochemical data, supporting evidence includes the finding that, even in normal individuals, blocking cerebral ACh receptors causes profound memory impairments. For example, an injection of scopolamine, which has central anticholinergic activity, induces a several-minute episode of Alzheimer-like cognitive impairment that can be reversed with physostigmine (Fig. 7-6). The cholinergic hypothesis led to the introduction of donepezil (Aricept) and other cholinesterase inhibitors in an attempt at preserving ACh activity by inactivating its metabolic enzyme, cholinesterase (see Fig. 7-6 and later).
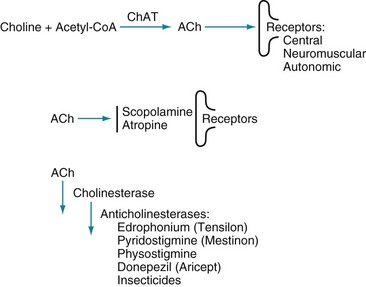
FIGURE 7-6 Top, The enzyme choline acetyltransferase (ChAT) catalyzes the reaction of choline and acetyl-coenzyme A (ACoA) to form acetylcholine (ACh) in neurons of the central (CNS), peripheral (PNS), and autonomic nervous system. When released from its presynaptic neurons, ACh interacts with postsynaptic ACh receptors. Middle, However, various substances block ACh from interacting with its receptors. For example, scopolamine, which readily crosses the blood–brain barrier, blocks ACh–receptor interaction in the CNS. Likewise, atropine blocks ACh receptors, but predominantly those in the autonomic nervous system. (Unless its concentration is great, atropine does not cross the blood–brain barrier.) Bottom, Cholinesterase metabolizes ACh. Thus, enzyme degradation rather than reuptake terminates ACh activity. (In contrast, reuptake terminates most dopamine and serotonin activity.) Anticholinesterases or cholinesterase inhibitors block cholinesterase and preserve ACh concentrations. The most widely known application of this strategy is using donepezil (Aricept) and other cholinesterase inhibitors to preserve ACh activity in Alzheimer disease. Similarly, physostigmine, a powerful anticholinesterase medicine that can cross the blood–brain barrier, purportedly briefly corrects ACh deficits in tardive dyskinesia as well as Alzheimer disease (see Chapter 18). Physostigmine might also counteract excessive anticholinergic activity in tricyclic antidepressant overdose or reverse the effects of scopolamine or atropine. Using cholinesterase inhibitors is also applicable to PNS disorders. For example, edrophonium (Tensilon) and pyridostigmine (Mestinon) are anticholinesterases that restore neuromuscular junction ACh activity in myasthenia gravis (see Fig. 6-2). In the opposite situation, insecticides contain powerful anticholinesterases that create excessive ACh activity at neuromuscular junctions, which paralyzes insects by overstimulating muscle contractions.
Risk Factors and Genetic Causes
Genetic Causes
Unlike the gene on chromosome 19, which codes for Apo-E alleles, mutations on at least three other chromosomes – 1, 14, and 21 – unequivocally cause Alzheimer disease (Table 7-1). In other words, while the Apo-E gene confers susceptibility, these mutations – “obligate” genes – actually cause the illness. All of them usually transmit Alzheimer disease in an autosomal dominant pattern, promote Aβ production, and lead to its deposition in plaques. Although mutations account for only 5–10% of all cases, they determine the fate of many individuals, provide a window into the pathogenesis of Alzheimer disease, and probably account for the vast majority of cases of “early-onset Alzheimer disease” (symptoms appearing before 60 years of age).
Treatment of Dementia
A complementary strategy, similar to maintaining ACh neuromuscular junction activity in myasthenia (see Chapter 6), attempts to preserve or increase cerebral ACh concentration by reducing ACh metabolism by cholinesterases. In Alzheimer disease treatment, several commercially available cholinesterase inhibitors that penetrate the blood–brain barrier – donepezil, rivastigmine, and galantamine – produce modest, temporary (approximately 9–12-month) improvement in cognitive tests, “global evaluations,” and measurements of quality of life. They may either reduce certain troublesome symptoms, such as depression, psychosis, and anxiety, or make them amenable to psychotropics. The cholinesterase inhibitor, donepezil, reduces the risk of progression of dementia for as long as 1 year; however, by 36 months it has little or no effect. Cholinesterase inhibitors may also slow the progression of dementia in Parkinson disease and DLB, which are also characterized by an ACh deficit (see later), but not in the other neurodegenerative illnesses, MCI, pure VCI, TBI, or delirium.
A completely different approach consisted of immunizations against amyloid. The expectation was that antibodies would attack and destroy amyloid plaques. Researchers first administered an antiamyloid vaccine to mice with an inherited Alzheimer-like illness and then to patients with Alzheimer disease and presymptomatic individuals destined to develop the disease because they were carrying mutations that doomed them to the illness. Although the vaccination program was successful in the mice, human volunteers derived no benefit or suffered inflammatory encephalitis. Looking toward a different method of reducing the amyloid burden, researchers attempted to interrupt the synthesis of Aβ by administering inhibitors of β–secretase or γ–secretase (see Fig. 7-5). However, a large clinical trial of a γ–secretase inhibitor exacerbated rather than helped the problem.
Related Disorders
Dementia with Lewy Bodies
Neurologists named DLB for its histologic signature: cortical spherical intracytoplasmic inclusions, Lewy bodies, composed of a circular, eosinophilic dense proteinaceous core surrounded by loose fibrils. The core contains mostly aggregates of α-synuclein – a 140-amino-acid protein encoded on chromosome 4 – and a less specific substance, ubiquitin. The concentration of the Lewy bodies correlates with the severity of dementia. (Lewy bodies are widely known as a marker for Parkinson disease, where they populate the basal ganglia rather than the cerebral cortex [see Chapter 18].)
Another common symptom, which ultimately occurs in about 50% of DLB patients and often complicates Parkinson disease patients, is REM sleep behavior disorder (see Chapter 17). Normal individuals during REM sleep are rendered quadriparetic except for respiratory and ocular movement. In contrast, individuals with REM sleep behavior disorder, whether from DLB, Parkinson disease, or no apparent reason, resist the usual REM-induced paralysis and act as though they are participating in their dreams. For example, they make running, punching, and similar movements while dreaming. Long-acting benzodiazepines, such as clonazepam, suppress the REM sleep behavior disturbance in DLB disease as well as in its other causes.
Frontal Lobe Disorders
Injuries
Neurologists find several general manifestations of frontal lobe damage that vary and occur in different combinations. Patients are characteristically apathetic and indifferent to their surroundings, ongoing events, and underlying illness. They also have comparable cognitive slowness. Their impairments prevent them from making transitions, changing sets, and adopting alternate strategies. Patients also have slowed thinking (bradyphrenia), lack of emotions, and a paucity of speech that can range from reticence to silence (abulia). In addition, if an illness or injury damages their dominant frontal lobe’s language center, patients will have impaired verbal output and other signs of aphasia (see Chapter 8).
Commonly cited causes of bilateral frontal lobe damage include TBI, glioblastoma multiforme, metastatic tumors, metachromatic leukodystrophy, MS, ruptured anterior cerebral artery aneurysm, and infarction of both anterior cerebral arteries. In the abandoned frontal lobotomy, which physicians had introduced to control psychotic thought and behavior long before the first-generation antipsychotic agents, neurosurgeons injected sclerosing agents into the frontal lobe or severed its underlying large white-matter tracts (see Fig. 20-23). Patients who underwent the procedure showed less agitation, but usually at the expense of developing apathy, restricted spontaneous verbal output, indifference to social conventions, and impaired abstract reasoning.
In contrast to bilateral frontal lobe injury producing neuropsychologic impairments, modern-day surgical removal of the anterior, nondominant frontal lobe causes little, if any, impairment. For example, neurosurgeons routinely remove this “silent area” of the brain in cases of tumor, arteriovenous malformation, or seizure focus refractory to antiepileptic drugs (AEDs) (see Chapter 10).
Frontotemporal Dementia
Major varieties of frontotemporal dementia include ones characterized by behavioral disturbances, nonfluent aphasia (see Chapter 8), and motor neuron signs (see Chapter 5). The behavioral variant, which is most important, reflects degeneration primarily of the frontal rather than the temporal lobes. As with other disorders resulting from frontal lobe degeneration, patients with the behavioral variant show a broad decline in affect and behavior. In fact, physicians may reasonably initially misdiagnose frontotemporal dementia as major depression, bipolar disorder, obsessive-compulsive disorder, or other psychiatric illnesses. Neurologic criteria for a “possible” diagnosis specifically require that patients show three of six disturbances:
4. Perseverative or compulsive behaviors
5. Hyperorality (excessive – sometime compulsive – talking, eating, or cigarette smoking)
Notably, these criteria do not require the presence of dementia. From a practical viewpoint, patients have memory problems, but overall their cognitive impairments are neither specific nor pronounced. Routine cognitive assessment testing does not readily separate Alzheimer disease from frontotemporal dementia. However, astute clinicians will not confuse these two illnesses (Table 7-2).
TABLE 7-2 Features Distinguishing Alzheimer Disease and Frontotemporal Dementia
| Feature | Alzheimer Disease | Frontotemporal Dementia |
|---|---|---|
| Age at onset (years) | >65 | 53 (mean) |
| Memory impairments | Early, pronounced | Subtle, at least initially, with preserved visuospatial ability |
| Behavior abnormalities | None until middle or late stage | Early and prominent perseverative and compulsive behavior; hyperorality; impaired executive ability |
| Language impairment | Except for anomias, none until late stage | Paraphasias, anomias, and decreased fluency |
| CT/MRI appearance | General atrophy, but especially parietal and temporal lobes | Frontal and temporal lobe atrophy |
| Histologic marker | Aβ-amyloid accumulation | Tau accumulation |
CT, computed tomography; MRI, magnetic resonance imaging.
Progressive Supranuclear Palsy
Just as DLB represents a hybrid of Alzheimer and Parkinson diseases, PSP represents a hybrid of a dementia-producing illness and distinctive conjugate eye movement disorder (see Chapter 12). Preliminary DSM-5 nomenclature would include PSP as a Neurocognitive Disorder due to Another Medical Condition.
PSP patients present with parkinsonism, except that they rarely have a tremor. They have predominantly axial rigidity that forces their head, neck, and entire spine to remain overly upright and unnaturally straight (Fig. 7-7). Their posture contrasts with the typical Parkinson disease patients’ flexed head and neck and kyphosis of the spine (see Fig. 18-9, A). In addition, their postural instability occurs at the onset of PSP and accounts for their falling, which is sometimes incapacitating or even fatal.
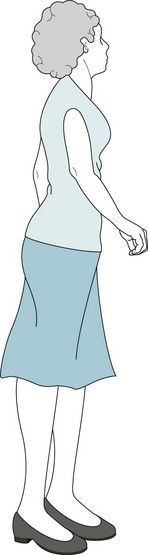
FIGURE 7-7 Axial rigidity and bradykinesia force progressive supranuclear palsy patients to hold themselves straight and walk slowly. They resemble Parkinson disease patients, but their erect posture differs from the flexed posture of Parkinson disease (see Fig. 18-9A).
Its pathognomonic feature, which may not appear until 3 years after the onset of parkinsonism, consists of patients’ losing their ability to look voluntarily in vertical directions and then laterally. After being unable to look upward or downward, they eventually lose lateral eye movement, and then their eyes stay fixed in a straight-ahead position. To circumvent the lack of supranuclear (cortical) input, neurologists can recruit the labyrinthine system and brainstem systems: They typically rock the patients’ head and neck up and down to elicit “doll’s eye” or oculocephalic vertical reflex movements. In PSP, this maneuver forces the eyes to move appropriately (Fig. 7-8). The limited voluntary vertical eye movement, but with the ability of the examiner to overcome it, constitutes the best clinical diagnostic test for PSP and separates it from other dementia-producing illnesses.

FIGURE 7-8 Left, Progressive supranuclear palsy patients typically have a scowl because of pseudobulbar-like continuous contractions of their facial muscles. Middle, This patient, who has characteristically lost his ability to look first in vertical then lateral directions, cannot comply with the examiner’s request to look downward. Right, However, when the examiner rocks the patient’s head back and forth (performs an oculocephalic maneuver), his eyes dip well below the meridian.
Other Dementias
Vascular Cognitive Impairment
Also known among neurologists as “multi-infarct dementia” or “vascular dementia,” and psychiatrists as Vascular Neurocognitive Disorders according to the preliminary version of DSM-5, VCI is essentially the dementia that results from cerebral infarctions (strokes) or any other cerebral vascular disease (see Chapter 11). The disorder has consistent neuropsychologic characteristics. However, unlike Alzheimer disease, it rarely causes an isolated amnesia. In addition to having cognitive impairment, VCI patients may have aphasia, dyscalculia, anosognosia, or other neuropsychologic disorder depending on the location of underlying strokes. VCI’s primary distinction is that the underlying strokes also cause abnormal physical findings, such as hemiparesis, hemianopsia, dysarthria, ataxia, and pseudobulbar palsy. Gait impairment so regularly occurs that it places most cases of VCI into the category of subcortical dementia. In VCI – unlike in Alzheimer disease, DLB, and frontotemporal dementia – physical signs overshadow cognitive impairments. Finally, unlike those other dementia-producing illnesses, VCI typically follows a step-wise deterioration that presumably reflects a progressive accumulation of strokes.
Among risk factors for VCI, pre-existing cognitive impairment – especially from Alzheimer disease – exerts the greatest influence. Risk factors for stroke are naturally risk factors for VCI (see Chapter 11). Even vascular disease in other organs, such as coronary artery disease, increases the risk for VCI. However, despite its importance in other circumstances, Apo-E4 is not a risk factor for VCI.
Wernicke–Korsakoff Syndrome
In its acute stages, Wernicke–Korsakoff syndrome includes combinations of ataxia and ocular motility abnormalities, including conjugate gaze paresis, abducens nerve paresis, and nystagmus. However, only a minority of patients display all these abnormalities. With alcoholism, with or without Wernicke–Korsakoff syndrome, patients develop a peripheral neuropathy and cerebellar atrophy. Because the cerebellar atrophy particularly affects the vermis (see Chapter 2), alcoholism leads to an ataxic gait (see Fig. 2-13).
CT and MRI may be normal or show cerebral and cerebellar atrophy. In acute Wernicke–Korsakoff syndrome, distinctive and potentially fatal petechial hemorrhages develop in the mamillary bodies and periaqueductal gray matter (see Fig. 18-2). The damage to these structures, which are elements of the limbic system (see Fig. 16-5), explains the amnesia. Also, because the periaqueductal gray matter is immediately adjacent to the nuclei of third and sixth cranial nerves and the medial longitudinal fasciculus, damage to this region explains the ocular motility abnormalities (see Chapters 4 and 12).
Other Causes of Dementia in Alcoholics
Rarely, but interestingly, alcoholics can develop degeneration of the corpus callosum that causes a “split-brain syndrome” (see Marchiafava–Bignami syndrome, Chapter 8). They are also susceptible to seizures from either excessive alcohol use or alcohol withdrawal. In either case, because the underlying problem is a metabolic aberration, the seizures are more likely to be generalized, tonic-clonic rather than complex partial. Infants of severely alcoholic mothers are often born with the fetal alcohol syndrome, which includes facial anomalies, low birth weight, microcephaly, and tremors. Most importantly, fetal alcohol syndrome usually includes an Intellectual Developmental Disorder.
Normal-Pressure Hydrocephalus
NPH is a quintessential correctable cause of dementia. Most cases of NPH are idiopathic, but meningitis or subarachnoid hemorrhage often precedes it. In those cases, inflammatory material or blood probably clogs the arachnoid villi overlying the brain and impairs reabsorption of CSF. As CSF production continues despite inadequate reabsorption, excessive CSF accumulates in the ventricles and expands them to the point of producing hydrocephalus (Fig. 7-9).
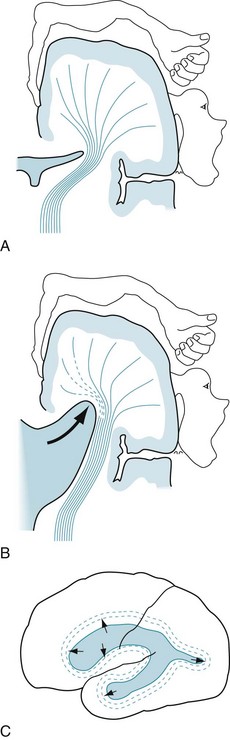
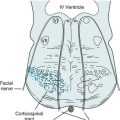
FIGURE 7-9 A and B, Ventricular expansion, as in normal-pressure hydrocephalus, results in compression of brain parenchyma and stretching of the corticospinal and other tracts of the internal capsule (see Fig. 18-1). Gait impairment (apraxia) and urinary incontinence are prominent normal-pressure hydrocephalus symptoms because the ventricular expansion stretches and most disrupts the tracts that supply the legs and the voluntary muscles of the bladder. C, Also, internal pressure on the frontal lobes leads to cognitive impairment and psychomotor retardation.
NPH is essentially a syndrome of three elements: dementia, urinary incontinence, and gait apraxia. The dementia conforms to the subcortical classification because it entails slowing of thought and gait, but spares language skills. Although the dementia may bring the patient to a psychiatrist’s attention, gait apraxia is generally the initial, most consistent, and most prominent feature of NPH (Fig. 7-10). It is also the first to improve with treatment (see later). Urinary incontinence consists of urgency and frequency that progress to incontinence, and it also improves with treatment. The physical features of NPH easily separate it from Alzheimer disease and other dementia-producing illnesses.
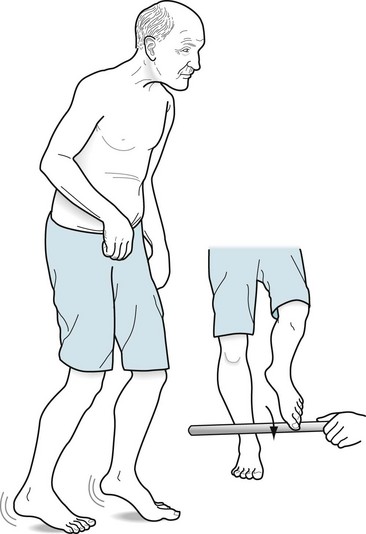
FIGURE 7-10 Gait apraxia, the hallmark of normal-pressure hydrocephalus, appears in several aspects of gait testing. Patients with gait apraxia fail to alternate their leg movements and do not shift their weight to their forward foot. They tend to pick up the same leg twice and attempt to elevate their weight-bearing foot. When their weight remains on the foot that they attempt to raise, that foot has a tremor and appears to be stuck or “magnetized” to the floor. Gait apraxia is particularly evident when patients have a magnetic gait when they start a turn and then require multiple steps to complete 180°. Despite being unable to walk, they can typically step over a stick because their stepping reflex is relatively preserved.
In NPH, CT and MRI show ventricular dilation, particularly of the temporal horns (see Figs 20-7 and 20-19), and sometimes signs of CSF reabsorption across ventricular surfaces. They show minimal or no cerebral atrophy. Nevertheless, diagnosing NPH exclusively by CT and MRI is unreliable because the findings are nonspecific, particularly because they resemble cerebral atrophy with resultant hydrocephalus (hydrocephalus ex vacuo, see Fig. 20-3). The CSF pressure and its protein and glucose concentrations are normal.
Infections
Neurosyphilis
Both neurologists and psychiatrists encounter patients with syphilis if the infection has spread to the substance of the CNS. Neuropathologists call an infection of either brain or spinal cord tissue “parenchymatous syphilis,” an infection of the brain “general paresis,” and the spinal cord “tabes dorsalis” (see Chapter 2).
General paresis, the brain infection, initially causes the insidious onset of nonspecific cognitive impairments and personality changes. If the infection remains untreated, patients develop dementia, hallucinations, and disordered thinking. However, delusions of grandeur, despite their notoriety, rarely arise. The cognitive impairments are often accompanied by any of a wide variety of physical abnormalities, including dysarthria, tremors, Argyll Robertson pupils (see Chapter 12), or loss of hearing or vision.
Subacute Sclerosing Panencephalitis
Symptoms and signs gradually arise. Early manifestations include nonspecific poor school work, behavioral disturbances, restlessness, and personality changes. Over weeks, cognition deteriorates into dementia and a weakness in one limb evolves into complete bodily paralysis. Patients may have partial or generalized seizures, but the hallmark of SSPE is myoclonus (see Chapter 18).
Clinicians can confirm a diagnosis of SSPE by finding an elevated CSF measles antibody titer. An EEG is also diagnostically helpful: It shows periodic sharp-wave complexes or a burst suppression pattern (see Fig. 10-6). (The EEG patterns in Creutzfeldt–Jakob disease and SSPE are similar, but their demography, clinical picture, and histology are entirely different.) Histologic examinations in SSPE reveal intranuclear eosinophilic inclusions (Cowdry bodies). By way of contrast, Lewy bodies are intracytoplasmic eosinophilic inclusions.
Creutzfeldt–Jakob and Related Diseases
Prions
Testing for Prion Infection.
During the course of Creutzfeldt–Jakob disease, the EEG usually shows periodic sharp-wave complexes or a burst suppression pattern that can confirm a clinical impression (see Fig. 10-6). As another marker, the CSF in almost 90% of cases contains “14-3-3” protein. Although sensitive, this CSF protein falls short of being pathognomonic of Creutzfeldt–Jakob disease because it is also present in other illnesses characterized by acute neuron death, such as encephalitis, hypoxia, and tumors. In addition, the CSF in Creutzfeldt–Jakob disease, as in Alzheimer disease, usually contains an elevated concentration of tau protein. MRI findings may indicate Creutzfeldt–Jakob disease as well as exclude mass lesions from consideration. Some studies have tentatively diagnosed Creutzfeldt–Jakob disease by locating PrPSc deposits in olfactory nerves and even in extraneural organs, such as the spleen and muscle.
Variant Creutzfeldt–Jakob Disease
Physicians should note two aspects of vCJD. It was one of the causes of dementia in adolescents and young adults (see Box 7-2). Its initial symptoms were largely psychiatric and unaccompanied by objective physical neurologic correlates.
Several conditions mimic Creutzfeldt–Jakob disease and vCJD. Lithium or bismuth intoxication or thyroid (Hashimoto) encephalopathy may produce myoclonus with dementia; however, these conditions usually cause more of a delirium than dementia, and all are readily diagnosable with specific blood tests. Paraneoplastic syndromes might also produce wasting, myoclonus, ataxia, and dementia (see Chapter 19).
Lyme Disease
Studies have shown that many patients with chronic Lyme disease do not have a persistently positive serum Lyme titer. Even if their titer were persistently positive, the persistence would be analogous to lifelong abnormal serologies after successful syphilis treatment. Moreover, when Lyme tests are positive, the titers do not correlate with memory impairments. Confusing matters somewhat, because the infectious agent, Borrelia burgdorferi, is a spirochete as in syphilis, serum FTA-ABS and VDRL tests may turn positive in Lyme disease. Chronic Lyme disease patients often report that continual, multiple antibiotic or other nontraditional treatments keep their symptoms at bay, but large-scale studies do not support such claims. Nevertheless, the source of chronic Lyme symptoms remains enigmatic. Some symptoms fall into the realm of depression or chronic fatigue syndrome (see Chapter 6).
Human Immunodeficiency Virus-Associated Dementia
Manifestations
Patients eventually decline into a persistent vegetative state (see Chapter 11). Myoclonus, extrapyramidal signs, and slowed ocular saccades and pursuits (see Chapter 12) – signs of basal ganglia and brainstem impairment – may also complicate this phase.
Treatment
Combination ART, highly active ART, or simply ART, which are essentially the same treatment, improve cognitive function in HAD patients as well as reducing the systemic complications of HIV infection. Instituting ART as early as possible may reduce the risk of developing HAND. However, ART is not a panacea or even easy to take. Patients must strictly adhere to a demanding multidrug schedule. Many patients, especially those with denial, depression, or dementia, have difficulty adhering to it. Some components do not improve cognitive function because they cannot cross the blood–brain barrier. Several, such as efavirenz and zidovudine, can cause cognitive impairment or even psychosis. Components that rely on the cytochrome P450 enzyme system for metabolism potentially create adverse drug–drug interactions. Furthermore, some produce neuropathy, interfere with methadone and precipitate opiate withdrawal, or, because they interfere with mitochondria metabolism, cause myopathy (see Chapters 5 and 6).
AIDS-Induced Cerebral Lesions
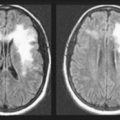
Second to HIV itself, Toxoplasma gondii causes the most common AIDS-related CNS infection, cerebral toxoplasmosis. This protozoon typically produces multiple ring-shaped enhancing lesions that are readily detectable by CT or MRI (see Fig. 20-11). Neurologists usually institute antibiotics once they have reached a clinical diagnosis of toxoplasmosis because this infection is so common in the HIV patient population, gives a distinctive appearance on imaging studies, and readily responds to antibiotics. They recommend a cerebral biopsy for patients who do not improve after several weeks of treatment.
Through damage to oligodendroglia, the infection leads to demyelination in the white (Greek, leuko) matter of the brain and spinal cord (see Fig. 15-10). As its name implies, PML produces multiple, widespread, and often confluent white-matter lesions. PML is the only virus-induced disorder that causes cerebral demyelination in humans.
Other AIDS-Related Conditions
The spinal cord is subject to HIV infection as well as lymphoma. HIV infection of the spinal cord (myelitis or vacuolar myelopathy) produces paraparesis, the Romberg sign, and other indications of spinal cord injury. The damage is located predominantly in the posterior columns (the fasciculi gracilis and cuneatus [see Chapter 2]). Although this pattern resembles combined system disease, vitamin B12 treatment does not alleviate the problem.
Delirium/Toxic-Metabolic Encephalopathy
Characteristics
Symptoms arise over several hours to several days and then fluctuate hourly to daily. They usually resolve with successful treatment of the underlying medical problem. However, improvement occasionally lags (Fig. 7-11).
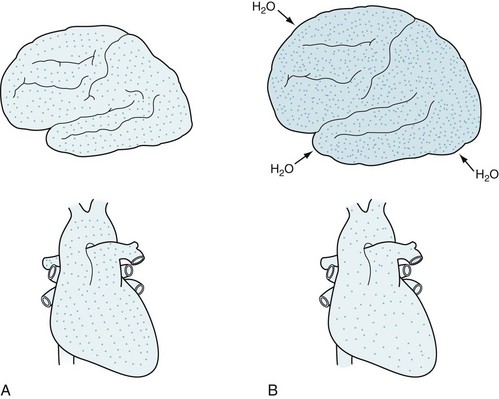
FIGURE 7-11 A distressing clinical situation occurs when patients deteriorate – become confused, lethargic, and agitated – after apparent correction of certain metabolic abnormalities. A, In cases of uremia or hyperglycemia, the brain and serum contain an approximately equal concentration of solute (dots). B, Overly vigorous dialysis or insulin administration clears solute more rapidly from the serum than the brain, leaving solute concentration in the brain much greater than in the serum. The concentration gradient causes free water to move into the brain, which produces cerebral edema.
Some signs allow a bedside diagnosis of a specific underlying illness. For example, patients with Wernicke–Korsakoff syndrome typically have oculomotor palsies, nystagmus, ataxia, and polyneuropathy. Those with hepatic or uremic encephalopathy have asterixis (Fig. 7-12). Those with uremia, penicillin intoxication, meperidine (Demerol) treatment, and several other metabolic encephalopathies have myoclonus. Narcotic or barbiturate intoxication causes constricted pupils, but amphetamine, atropine, or sympathomimetic drug intoxication causes dilated pupils.
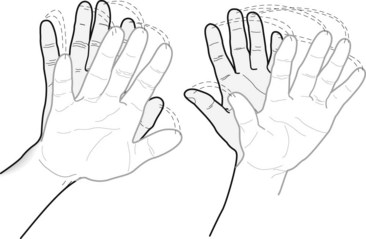
FIGURE 7-12 Neurologists elicit asterixis by having patients extend their arms and hands, as though they were stopping traffic. Their hands intermittently quickly fall forward and slowly return, as though waving goodbye.
EEG typically shows background slowing and disorganization (see Chapter 10); however, numerous conditions cause the same alterations. Some EEG findings suggest a specific etiology. For example, triphasic waves often indicate hepatic or uremic encephalopathy (see Fig. 10-5). Neurologists often attribute the EEG changes, as well as the patient’s depressed level of consciousness, to dysfunction of the reticular activating system. CT and MRI are usually normal, but they frequently show cerebral atrophy because of age-related atrophy or comorbid dementia-producing illness. Neurologists routinely order a CT or MRI to exclude a structural lesion as either a cause or complication of delirium. For example, they might order a CT to exclude an acute subdural hematoma in an alcoholic patient.
Causes
Innumerable disorders, alone or in combination, may cause delirium; however, only a few account for the majority of cases in acute care hospitals and nursing homes (Box 7-4). Medicines are a frequently occurring, inapparent cause of delirium. Medicines administered in a standard, small dose, but given to a previously unexposed patient may have this unintended effect. Medicine-related problems also include the build-up of toxic concentrations because of inadequate hepatic metabolism or renal clearance, drug–drug interactions, or absorption of ocular or topical medicines. Ironically, medicines for psychiatric and neurologic disorders – anticholinergics, AEDs, antiparkinson agents, opioids, and serotonin agents – readily cross the blood–brain barrier and produce a delirium. In a surprising and paradoxical example, intramuscular injections of olanzapine administered for schizophrenia may produce confusion, agitation, anxiety, sedation, and more severely depressed levels of consciousness rather than calming the patient. Researchers have labeled this reaction the post-injection delirium/sedation syndrome and attribute it to the medicine seeping into the vasculature.
Precautions in Diagnosing Alzheimer Disease
Even though this and other chapters have presented numerous causes of dementia and delirium, Alzheimer disease occupies the default position in the diagnosis of dementia. Physicians should maintain vigilance and mind the “red flags” that warn against its diagnosis (Box 7-5). Although they certainly do not preclude a diagnosis of Alzheimer disease, these red flags will help prevent a misdiagnosis in cases of toxic-metabolic encephalopathy, psychiatric disorders, dementia in young adults (see Box 7-2), and neurodegenerative illnesses that strike at more than the brain’s cognitive centers.
Box 7-5
Red Flags for an Alzheimer Disease Diagnosis
• Age younger than 65 years: see Box 7-2
• Fluctuating level of consciousness: see toxic-metabolic encephalopathy, Lewy bodies dementia
• Behavior, emotional, or personality disturbances overshadowing cognitive impairment: see frontotemporal dementia and HIV dementia
• Rapidly – 6–12-month – progressive development, see Creutzfeldt–Jakob disease, vCJD, paraneoplastic limbic encephalitis (Chapter 19), HAD, frontotemporal dementia
Clarfield AM. The decreasing prevalence of reversible dementias. Arch Intern Med. 2003;163:2219–2229.
Folstein MR, Folstein SE, McHugh PR. “Mini-Mental State:” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198.
Iverson DJ, Groseth GS, Reger MA, et al. Practice parameter: Evaluation and management of driving risk in dementia. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2010;74:1316–1324.
Luis CA, Keegan AP, Mullan M. Cross validation of the Montreal Cognitive Assessment in community dwelling older adults residing in the Southeastern US. Int J Geriatr Psychiatry. 2009;24:197–201.
Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53:695–699.
Thurman DJ, Stevens JA, Rao JK. Practice parameter: Assessing patients in neurologic practice for risk of falls. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2008;70:473–479.
Allan LM, Ballard CG, Burn DJ, et al. Prevalence and severity of gait disorders in Alzheimer’s and non-Alzheimer’s dementias. J Am Geriatr Soc. 2005;53:1681–1687.
Ballard C, Hanney ML, Theodoulou M, et al. The dementia antipsychotic withdrawal trial (DART-AD): Long-term follow-up of a randomized placebo-controlled trial. Lancet Neurol. 2009;8:151–157.
Green RC, Roberts JS, Cupples LA, et al. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med. 2009;361:245–254.
Larner AJ. Cholinesterase inhibitors. Expert Rev Neurother. 2010;10:1699–1705.
Mayeux R. Early Alzheimer’s disease. N Engl J Med. 2010;362:2194–2201.
Meyer GD, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67:949–956.
Ott BR, Heindel WC, Papandonatos GD. A survey of voter participation by cognitively impaired elderly patients. Neurology. 2003;60:1546–1548.
Petersen RC. Mild cognitive impairment. N Engl J Med. 2011;364:2227–2234.
Querfurth HW, LaFeria FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344.
Rossetti HC, Lacritz LH, Weiner MF. Normative data for the Montreal Cognitive Assessment (MoCA) in a population-based sample. Neurology. 2011;77:1272–1275.
Schneider LS, Tariot PN, Dagerman KS, et al. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355:1525–1538.
Sultzer DL, David SM, Tariot PN, et al. Clinical symptom responses to atypical antipsychotic medications in Alzheimer disease. Am J Psychiatry. 2008;165:844–854.
Yaffe K, Weston A, Graff-Radford NR, et al. Association of plasma beta-amyloid and cognitive reserve with subsequent cognitive decline. JAMA. 2011;305:261–266.
Cohen RA, Gongvatana A. The persistence of HIV-associated neurocognitive dysfunction and the effects of comorbidities. Neurology. 2010;75:2052–2053.
Heaton RK, Clifford DB, Franklin DR, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–2096.
Rackstraw S. HIV-related neurocognitive impairment – a review. Psychol Health Med. 2011;16:548–563.
Thompson A, Silverman B, Treisman G. Psychotropic medications and HIV. HIV/AIDS. 2006;42:1305–1310.
Devinsky O. The neurology of Capgras syndrome. Rev Neurol Dis. 2008;5:97–100.
Geser F, Wenning GK, Poewe W, et al. How to diagnose dementia with Lewy bodies: State of the art. Mov Disord. 2005;12(Suppl 12):S11–S20.
Goldman JG, Goetz CG, Brandabur M, et al. Effects of dopaminergic medications on psychosis and motor function in dementia with Lewy bodies. Mov Disord. 2008;23:2248–2250.
McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB consortium. Neurology. 2005;65:1863–1872.
Nagahma Y, Okina T, Suzuki N, et al. Neural correlates of psychiatric symptoms in dementia with Lewy bodies. Brain. 2010;133:557–567.
Dotosn VM, Beydoun MA, Zonderman AB. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology. 2010;75:27–34.
Goodman WK. Electroconvulsive therapy in the spotlight. N Engl J Med. 2011;364:1785–1789.
Saczyski JS, Beiser A, Seshadri S, et al. Depressive symptoms and risk of dementia: The Framingham Heart Study. Neurology. 2010;75:35–41.
Dubois B, Slachevsky A, Litvan I, et al. The FAB: A Frontal Assessment Battery at bedside. Neurology. 2000;55:1621–1626.
Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia. Ann Neurol. 2006;59:952–962.
Kertesz A, Blair M, McMonagle P, et al. The diagnosis and course of frontotemporal dementia. Alzheimer Dis Assoc Disord. 2007;21:155–163.
Kucharski A. History of frontal lobotomy in the United States, 1935–1955. Neurosurgery. 1984;14:762–772.
Rascovsky K, Hodges JR, Knopman D. Sensitivity of revised diagnostic criteria for the behavioral variant of frontotemporal dementia. Brain. 2011;134:2456–2477.
Feder HM, Johnson BJB, O’Connell S, et al. A critical appraisal of “chronic Lyme disease”. N Engl J Med. 2007;357:1422–1430.
Halperin JJ. Neurologic manifestations of Lyme disease. Curr Infect Dis Rep. 2011;13:360–366.
CreutzfeldtJakob Disease and Related Illnesses
Binelli S, Agazzi P, Canafoglia L, et al. Myoclonus in Creutzfeldt–Jakob disease. Mov Dis. 2010;25:2818–2827.
Heath CA, Cooper SA, Murray K, et al. Validation of diagnostic criteria for variant Creutzfeldt–Jakob disease. Ann Neurol. 2010;67:761–770.
Heath CA, Cooper SA, Murray K, et al. Diagnosing variant Creutzfeldt–Jakob disease: A retrospective analysis of the first 150 cases in the UK. J Neurol Neurosurg Psychiatry. 2011;82:646–651.
Prusiner SB. Neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526.
Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt–Jakob disease. Neurology. 2006;66:286–287.
Schott JM, Reiniger L, Thom M, et al. Brain biopsy in dementia: Clinical indications and diagnostic approach. Acta Neuropathol. 2010;120:327–341.
Williams ES, Miller MW. Chronic wasting disease in deer and elk. Rev Sci Tech Off. 2002;1:305–316.
Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt–Jakob disease. Brain. 2009;132:2659–2668.
Delirium (Toxic-Metabolic Encephalopathy)
Detke HC, McDonnell DP, Brunner E, et al. Post-injection delirium/sedation syndrome in patients with schizophrenia treated with olanzapine long-acting injection: Analysis of cases. BMC Psychiatry. 2010;10:43.
Grover S, Malhotra S, Bharadwaj R, et al. Delirium in children and adolescents. Int J Psychiatry Med. 2009;39:179–187.
Grover S, Mattoo SK, Gupta N. Usefulness of atypical antipsychotics and choline esterase inhibitors in delirium: A review. Pharmacopsychiatry. 2011;44:43–54.
Inouye SK. Delirium in older persons. N Engl J Med. 2006;354:1157–1165.
Lonergan E, Luxenberg J, Areosa-Sastre A, et al. Benzodiazepines for delirium. Cochrane Database Syst Rev. 1, 2009. D006397
Meagher DJ, Leonard M, Donnelly S, et al. A comparison of neuropsychiatric and cognitive profiles in delirium, dementia, comorbid delirium-dementia and cognitively intact controls. J Neurol Neurosurg Psychiatry. 2010;81:876–881.
Ozbolt LB, Paniagua MA, Kaiser RM. Atypical antipsychotics for the treatment of delirious elders. J Am Med Dir Assoc. 2008;9:18–28.
Wong CL, Holyroyd-Leduc J, Simel DL, et al. Does this patient have delirium? Value of bedside instruments. JAMA. 2010;304:779–786.
Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713.
Malouf R, Birks J. Donepezil for vascular cognitive impairment. Cochrane Database Syst. Rev 1, 2004. CD004395
Sachdev PS, Brodaty H, Valenzuela MJ, et al. The neuropsychological profile of vascular cognitive impairment in stroke and TIA patients. Neurology. 2004;62:912–919.
Solfrizzi V, Scafato E, Capurso C, et al. Metabolic syndrome and the risk of vascular dementia: The Italian Longitudinal Study on Ageing. J Neurol Neurosurg Psychiatry. 2010;81:433–440.
Chahine LM, Khoriaty RN, Tomford WJ, et al. The changing face of neurosyphilis. Int J Stroke. 2011;6:136–143.
Ghanem KG. Neurosyphilis: A historical perspective and review. CNS Neurosci Ther. 2010;16:e157–e168.
Gutierrez J, Issacson RS, Koppel BS. Subacute sclerosing panencephalitis: An update. Dev Med Child Neurol. 2010;52:901–907.
Klassen BT, Ahlskog JE. Normal pressure hydrocephalus: How often does the diagnosis hold water? Neurology. 2011;77:1119–1125.
Lair L, Naidech AM. Modern neuropsychiatric presentation of neurosyphilis. Neurology. 2004;63:1331–1333.
Sonia M, Lalit D, Shobha B, et al. Subacute sclerosing panencephalitis in a tertiary care centre in post measles vaccination era. J Commun Dis. 2009;41:161–167.
Yatabe Y, Hashimoto M, Kaneda K, et al. Neuropsychiatric symptoms of progressive supranuclear palsy in a dementia clinic. Psychogeriatrics. 2011;11:54–59.
Chapter 7 Questions and Answers
7. A 75-year-old retired football player stated that during the past 3–5 years he has become generally weaker and has lost muscle bulk, particularly in his hands. Because of imbalance, he has also mostly lost the ability to ride a bicycle. Nevertheless, he plays golf, walks a mile, and performs his activities of daily living. He scored 28 on the Mini-Mental State Examination (MMSE). A routine medical evaluation reveals no particular abnormality. Which is the most likely explanation for his symptoms?
10. A malnourished man with a history of chronic alcohol abuse was admitted with seizures and postictal delirium. His physicians had treated him with antiepileptic drugs, thiamine, niacin, glucose, and, because his serum sodium was 119 mEq/L, a rapid infusion of hypertonic saline. When the delirium persisted for several days, his physicians solicited a neurology consultant who found nystagmus, dysarthria, quadriparesis, and ataxia, as well as confusion and disorientation. Computed tomography (CT) of the head and routine blood tests showed no significant abnormalities. Which is the most likely explanation of his physical findings?
19–22. What is the effect of the following substances on cerebral ACh activity?
19-a. Centrally acting cholinesterase inhibitors, such as donepezil (Aricept), which are often prescribed as treatment for Alzheimer disease, increase cerebral ACh activity. In contrast, peripherally acting anticholinesterases, such as pyridostigmine (Mestinon), correct the neuromuscular junction ACh activity deficit in myasthenia, but because they do not penetrate the blood–brain barrier, they have no effect on cognition or other cerebral function.
20-b. Scopolamine, like atropine, is a centrally acting anticholinergic medication. In routine pharmacologic doses, neither scopolamine nor atropine crosses the blood–brain barrier; however, in high doses both penetrate the blood–brain barrier and may cause a toxic psychosis.
21-a. Organic phosphate insecticides usually contain anticholinesterases. They produce an abundance of ACh that overwhelms the neuromuscular junction and the autonomic nervous system.
22-a. Physostigmine is a centrally acting anticholinesterase that can penetrate the blood–brain barrier, but it has a short half-life.