Chapter 21. Deformities in children
Crooked children are still referred to orthopaedic surgeons for treatment as they were in Andry’s time, but serious deformity is rare today and the majority of children referred to an orthopaedic children’s clinic have nothing more than a minor abnormality of the lower limbs requiring firm reassurance. Reassurance is not always easy; well-intentioned advice or a careless remark by a respected relative, usually a grandparent, can cause anxiety that is difficult to eradicate.
Before reassuring the parents that all is well, it is important to confirm that no serious abnormality is present and it is therefore essential to understand what is normal and what is not.
Normal milestones
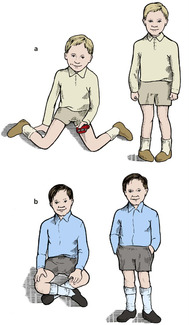
The milestones of normal development are very variable and accurate assessment may need a child development specialist. From an orthopaedic standpoint the most important milestones are those shown in Figure 21.1.
 |
| Fig. 21.1
Milestones of development: (a) holds the head up unsupported at 3 months; (b) sits up unaided at 6 months; (c) stands up unaided between 9 and 12 months; (d) walks between 12 and 18 months.
|
A paediatric opinion is needed if the child cannot do the following:
1. Sit by the age of 9 months.
2. Pull himself or herself upright by 12 months.
3. Walk by the age of 20 months.
Problems with walking
Not all children learn to walk at the same age and some are not very good at it when they do. Some children walk with an ungainly gait, as do some adults, and some learn to walk by using one leg as a prop at the front while the other pushes forwards from behind. This and other ‘trick’ gaits look bizarre but should correct spontaneously, although pathological and normal gaits can look very similar.
Remember DDH (p. 354)
Before reassuring a patient that a child’s gait is normal, always think of developmental dysplasia of the hip (DDH), particularly if the child is aged between 12 and 18 months (p. 354). If there are any of the following features, assume that the child has a dislocated hip until radiographs prove otherwise:
1. Limbs of unequal length.
2. An asymmetrical range of movement of the hips.
3. Asymmetrical skin creases.
4. A lurching gait.
5. A feeling of instability.
The consequences of missing DDH are very serious and radiographic examination of the pelvis is always justified if there is the slightest doubt about the stability of the hips in a child.
Bow legs and knock knees
Valgus or varus deformities in children are very common but seldom serious. Few children have rickets in this day and age but the herd memory of the disease is still active and bowed legs give very reasonable cause for alarm.
Many babies have varus tibiae at birth, and nappies, which hold the hips in abduction, make the bowing more obvious. The bowing also makes the toes turn slightly inward. If the bowing is very obvious the children are said to have outward-curved tibiae but the borderline between normal and abnormal is very vague.
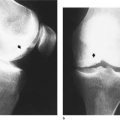
The varus will usually have corrected itself by the age of 3, when it is replaced by a slight valgus deformity at the knee (Fig. 21.2). The amount of valgus deformity is variable but it is permissible to have 10 cm between the ankles at the age of 4 (‘4 inches at 4 years’) when the deformity is at its greatest. A marked improvement can be expected soon after the child starts school at the age of 4 or 5 years.
 |
| Fig. 21.2
Normal valgus deformity of the leg. Ten centimetres between the medial malleoli at the age of 4 years will correct spontaneously.
|
Before reassuring the parents, it is important to exclude serious disease, of which the following are the most likely under the age of 5:
1. Vitamin D or C deficiency.
2. Blount’s disease, an abnormality of development at the upper end of the tibia which leads to progressive varus deformity.
3. Growth disorders such as epiphyseal injuries and epiphyseal dysplasia.
There are four factors to beware of in children with bow legs and knock knees:
1. More than 10 cm between the malleoli.
2. A family history of skeletal abnormality.
3. Asymmetry.
4. Abnormally short stature.
If one of these features is present there may be a serious developmental abnormality or a dislocated hip. If none then the child will probably develop normally.
In-toe gait
The commonest problem seen in a children’s orthopaedic clinic is an in-toe gait. An in-turned foot is very obvious, the child can fall and the gait attracts much comment.
There are three causes of an in-toe gait:
1. Anteversion of the femoral neck.
2. Metatarsus adductus.
3. Outward curved tibiae.
Anteversion of the femoral neck
The most common cause is abnormal rotation at the hip due to excessive anteversion of the neck of the femur relative to its shaft. This allows a greater range of internal rotation than external (Fig. 21.3). Abnormalities of rotation are much more obvious with the leg straight than with the hip and knee flexed.
 |
| Fig. 21.3
Anteversion and retroversion of the femoral neck: (a) children with anteversion of the femoral neck can sit with their feet out beside them and walk with the toes turned in; (b) children with retroversion can sit with the legs crossed and walk with the toes turned out.
|
In an adult, the ranges of internal and external rotation of the hip are roughly the same but in a child there may be 90° of internal rotation and only 30° of external rotation. Such a child will walk with the foot in the middle of the range of motion, i.e. in 30° of internal rotation, and will be able to squat with the legs turned outwards. It is not unusual for one of the parents to recall doing the same thing when young, which makes reassurance easy.
Treatment
No treatment, whether by splintage or operation, is helpful. If the patient has no external rotation at all, a rotational osteotomy of the femur may be required when growth is complete. Apart from this, nothing is needed except firm reassurance that the shape of the femur will gradually change so that the range of internal and external rotation approach each other. The position continues improving until 10 years of age.
Metatarsus adductus
Metatarsus adductus also causes an in-toe gait and is dealt with under foot deformities below.
Outward curved tibiae
Outward curved tibiae can also cause an in-toe gait. No treatment is required.
Out-toe gait
Retroversion of the femoral neck
A smaller number of children have a greater range of external rotation than internal and walk with their feet turned out ‘like Charlie Chaplin’. These children can also sit with their legs crossed, impossible for children with excessive internal rotation. External rotation of the leg is also seen in DDH and this must be excluded before reassuring the parents. As always, check the rest of the child for deformities.
Treatment
As with internal rotation of the hip, firm reassurance is all that is required.
Toe-walkers
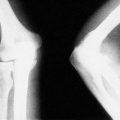
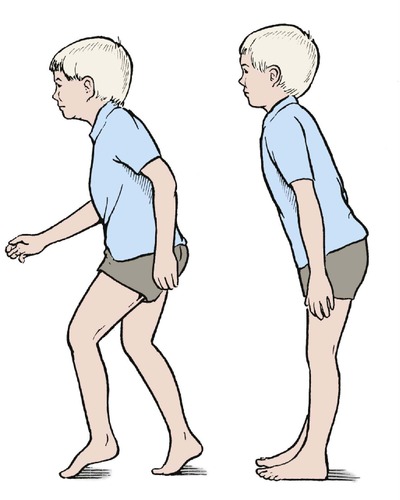
In some children, the Achilles tendon is tight and restricts dorsiflexion of the ankle so that the child walks on tiptoe and cannot stand with the heels flat (Fig. 21.4).
 |
| Fig. 21.4
Toe-walkers walk on tiptoe and lean slightly forward when standing.
|
The feet and ankles appear normal at first glance and the parents may already have been reassured that ‘there is nothing wrong’ by the time they reach an orthopaedic clinic. On careful examination, dorsiflexion of the ankle is restricted or absent, the child cannot squat with the heels to the ground and stands with a characteristic forward stoop. If they attempt to stand upright, the children fall over backwards. The signs are worse when the child is barefoot.
It must be remembered that hyperactive children and patients with mild cerebral palsy may also walk on their toes. Be sure to exclude neurological abnormalities in toe-walkers!
Treatment
Serial plasters are sometimes effective but lengthening of the Achilles tendon is often required.
Tight hamstrings
If the child has tight hamstrings which prevent forward flexion, suspect spondylolisthesis (p. 457). Radiographs of the lumbosacral spine are essential in any child with tight hamstrings or calf muscles.
Foot deformities
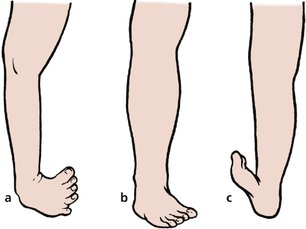
Foot deformities (Fig. 21.5) can be divided into two groups:
1. Forefoot deformities, more common and usually trivial.
2. Hindfoot deformities, less common and more serious.
 |
| Fig. 21.5
Foot deformities: (a) talipes equinovarus with a wasted calf; (b) metatarsus adductus; (c) talipes calcaneovalgus.
|
Metatarsus adductus
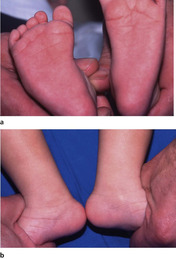
The commonest forefoot deformity is metatarsus adductus or hooked forefoot, which is usually noticed at about the age of 6 months when children begin to pull themselves upright. The deformity occurs at the midtarsal joint (Fig. 21.6a) and the hindfoot is completely normal. This can be confirmed by covering the forefoot while examining the hindfoot. When this is done, the foot looks entirely normal (Fig. 21.6b).
 |
| Fig. 21.6
Metatarsus adductus. The deformity arises in the middle of the foot (a). If the forefoot is covered over (b), the foot cannot be distinguished from normal.
|
The cause is not known but an attractive suggestion is that the forefoot is pushed inwards by the child while sleeping face down with their bottom in the air, a position only stable if the feet are pointing towards each other. It is certainly true that the condition begins to correct when the child is too ‘bottom-heavy’ to sleep in this position, usually at the age of about 18 months.
Treatment
Treatment begins by reassuring the parents that the child does not have a club foot and that the deformity corrects itself spontaneously in over 90% of patients.
Before the age of 3 years, no active treatment is required apart from gentle stretching of the medial side of the forefoot by holding the heel in one hand and the forefoot in the other. This should be done twice daily when dressing or bathing the child.
After the age of 3, treatment depends on the mobility of the foot. If gentle pressure corrects the deformity completely, observation can be continued, but if it cannot be corrected, serial casts are needed. A below-knee cast is applied in the position of maximum correction and changed at 2 week intervals to achieve a gradual correction. If serial casts are unsuccessful and the deformity is still marked by the age of 6, surgical correction may be necessary.
Congenital talipes equinovarus
The most common type of club foot, congenital talipes equinovarus (CTEV), is a deformity of the whole foot, which is pulled downwards (equinus) and inwards (varus) (Fig. 21.7).
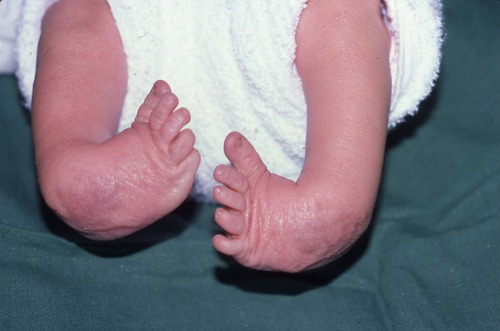 |
| Fig. 21.7
Talipes equinovarus in a neonate.
|
The condition has been recognized for centuries but remains an enigma. The demi-god Vulcan, who injured his foot when cast from heaven, is depicted with a club foot. The poet Byron also had a club foot.
As far as we know, the condition is caused by failure of growth in the posteromedial muscles of the calf, particularly tibialis posterior, the toe flexors, gastrocnemius and soleus. The muscles are entirely normal in all other respects but are simply too small for the patient. The bones of the forefoot, and sometimes the tibia and fibula, may be shorter than those of the opposite side. The condition is more common if a relative is affected or there is any other genetic abnormality.
Because the muscles on the medial side of the foot and calf are too short, the foot is pulled downwards and inwards with the ankle in flexion. As growth proceeds, the talonavicular joint is distorted, the navicular is pulled off the talus medially and a bony deformity quickly develops as the soft bones of childhood mould themselves to surrounding tissues.
Treatment
No treatment will make the foot and calf completely normal and this should be explained to parents as gently as possible before treatment is begun. It is unkind to lead the parents to believe that a complete cure is possible.
The aims of treatment are twofold:
1. To prevent bony deformity developing.
2. To keep the foot plantigrade; i.e. in such a position that it can be placed flat on the ground.
The Ponseti method of treatment is gaining popularity. This involves sequential gentle manipulation and splinting of the foot with serial casts for a considerable period of time (Fig. 21.8). The treatment is started as soon after birth as possible and continued until the foot is held in the desired position. A tendo Achilles lengthening (percutaneous tenotomy) is often required to achieve a plantigrade foot. Subsequent night braces may need to be worn up to 4 years of age.
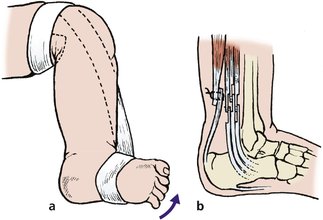 |
| Fig. 21.8
Treatment of CTEV: (a) strapping is applied to pull the foot upwards and outwards; (b) if this fails, the posteromedial structures must be lengthened and the posterior capsule of the ankle divided.
|
If the deformity is still present or is unable to be corrected then an operation is needed to lengthen the structures on the medial side of the foot and calf as well as release of the posterior capsule of the ankle joint. This will produce a good appearance but as growth proceeds the short structures may need to be lengthened again.
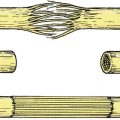
If the position is still unsatisfactory when growth is complete (Fig. 21.9), a triple arthrodesis will be required to fuse the following three joints (see Fig. 25.3):
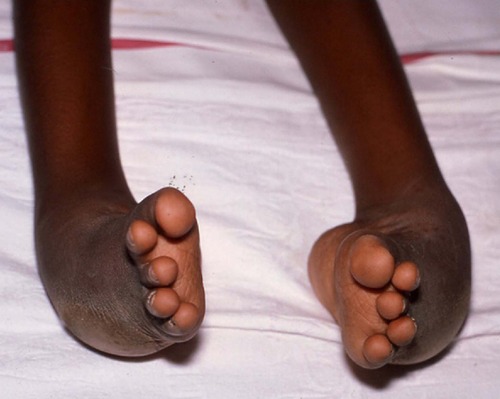 |
| Fig. 21.9
End result of untreated CTEV.
|
1. Calcaneocuboid.
2. Subtalar.
3. Talonavicular.
Congenital vertical talus
A congenital vertical talus produces a spectacularly flat foot, usually obvious at birth. Radiographs are difficult to interpret in early life but the vertical position of the talus is usually obvious (Fig. 21.10).
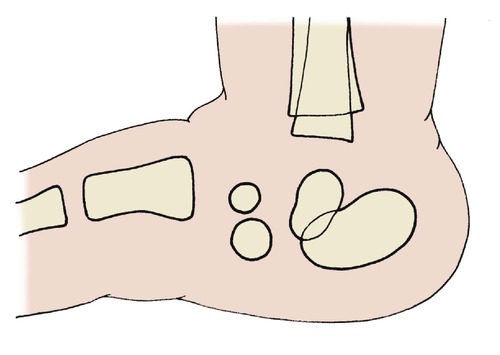 |
| Fig. 21.10
The position of the bones in a child with congenital vertical talus.
|
Treatment. Although rare, the condition must be recognized and treated promptly by open reduction. This is one type of flat foot for which reassurance is not appropriate.
Talipes calcaneovalgus
A calcaneovalgus foot points upwards and the heel points downwards. The deformity is usually postural and is commonest in large babies delivered of small mothers. The deformity corrects itself within the first 12 months of life without treatment, but firm reassurance that all is well cannot be given until more serious conditions have been excluded, particularly DDH.
Calcaneovalgus deformities are also seen in patients with meningomyelocoele and abnormalities of the cauda equina. A thorough neurological examination of the lower limbs is therefore important.
Flat foot
Flat feet in adults is dealt with on page 442. Two types of flat foot are seen in children:
1. Mobile flat foot
2. Rigid flat foot.
Mobile flat foot
By far the more common type is mobile (or flexible) flat foot. The deformity is only apparent when the child stands with the feet flat to the ground; it disappears when the child is standing on tiptoe or lying relaxed on a couch.
Treatment. Most patients with this condition have generalized ligamentous laxity and require no treatment because the feet become normal as growth proceeds and ligaments become tighter. Special shoes are only needed if the wear on one part of the shoe is excessive, yet shoes with a 3–5 mm inside wedge or firm arch support are often recommended. There is no evidence that they hasten the natural resolution of the deformity but prescribing special shoes may be comforting to parents who then feel that ‘something is being done’.
Rigid flat foot
Rigid (or ‘spastic’) flat foot is present if the deformity does not disappear when the child stands on tiptoe or lies flat. The rigidity is due partly to joint abnormalities and partly to spasm of the surrounding muscles. Further investigation is needed to exclude abnormal tarsal coalitions (p. 434) and other structural deformities.
Treatment depends on the underlying pathology. Special shoes or operation may be needed.
Children’s shoes
Much nonsense is talked about children’s shoes as a cause of foot deformity. Two points are worth considering:
1. Deformed feet occur in developing countries where shoes are not worn at all.
2. Attempts to correct foot deformity by diligent external splintage are generally unsuccessful.
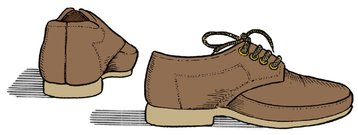
It is therefore difficult to understand how a comfortable shoe worn for perhaps 12 h a day can lead to a permanent alteration of growth. Nevertheless, it is important that children should wear good shoes with the following features (Fig. 21.11):
 |
| Fig. 21.11
A good child’s shoe. The heel is flat on the ground, support for the heel is firm and the inner side of the shoe is well supported. There is ample room for growth.
|
1. A firm heel to prevent the hindfoot rolling into inversion or eversion.
2. A firm medial edge to support the longitudinal arch.
3. A firm flat sole.
Such a shoe will support the foot and give stable contact with the ground.
Developmental dysplasia of the hip (DDH)
Previously known as congenital dislocation of the hip, this is a serious condition if not diagnosed and treated within the first months of life. The incidence varies from race to race but is approximately 1.5 per 1000 live births in European nations. Girls are affected eight times more often than boys, the left hip is affected more often than the right, and the incidence is higher if a relative is affected. One-third of dislocated hips have an abnormality of the opposite hip as well.
Diagnosis
The diagnosis is made by clinical examination at the routine postnatal examination, assisted by ultrasound examination. Radiographs are unhelpful because the femoral head does not start to calcify until the age of 10 weeks, often later. Several tests are described (Barlow’s, Ortolani’s, von Rosen’s), all very similar.
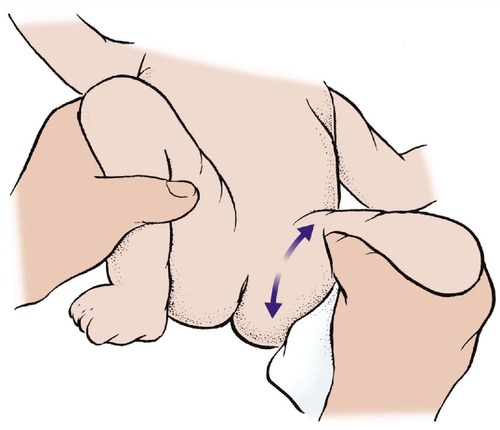
Clinical examination. The child is laid supine, without nappies, and the femur held between the thumb and forefinger (Fig. 21.12). The hip is abducted and the femoral head moved backwards and forwards relative to the acetabulum; i.e. up and down relative to the couch. If the hip is unstable, an unmistakable thud or jolt is felt as the femur moves in and out of the acetabulum.
 |
| Fig. 21.12
A test for DDH in the neonate. Backward and forward pressure on the femur in full flexion and abduction can move the head of the femur in and out of the acetabulum.
|
Correctly performed, this test will identify almost every unstable hip, but there are two exceptions. The first is a very small group in which the femoral head slips slowly out of the acetabulum during the first year of life, and the second is the very unusual hip that is irreducible at birth.
Abnormal clicks are found in about 20 per 1000 live births at the immediate postnatal examination, but fall to about 6 per 1000 after 2 weeks.
Ultrasound examination allows the hips to be placed in one of four categories. In most centres the examination is advised in ‘high risk’ hips; i.e. babies with a click on clinical examination, a family history of DDH, or other predisposing factor.
If ultrasound shows the hip to be dislocated or the acetabular roof to be sloping, a splint or harness is applied and retained until ultrasound shows the hip is reduced.
It is better to treat 100 patients needlessly in this way than miss one patient with DDH, even though incorrectly applied splintage which holds the hip in excess abduction can cause aseptic necrosis of the femoral head.
Treatment
Treatment depends on the time of diagnosis – the sooner the diagnosis, the easier the treatment.
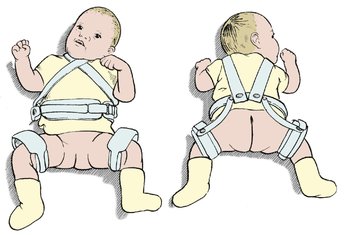
At birth. If DDH is diagnosed within the first week of life, it can be corrected by the child wearing double nappies or using a harness which holds the hips abducted and flexed (Fig. 21.13). One study showed that the condition was far commoner in North American Indians, who wrapped their babies in a papoose, than in racially identical Eskimos who carried their babies on their backs with the hips abducted and flexed.
 |
| Fig. 21.13
Cambridge splint for DDH.
|
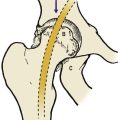
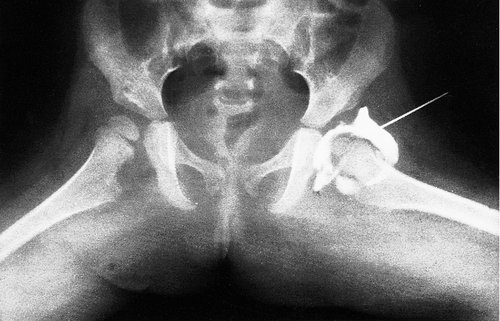
An appliance such as the Cambridge splint, Pavlik harness or von Rosen splint holds the hips in the same position and will achieve a stable hip if worn correctly for 12 weeks. The pelvis is then examined radiologically or with ultrasound to confirm that the hip is reduced. If the plain radiograph is not conclusive, an arthrogram is performed (Fig. 21.14). If the hip is satisfactory at 12 weeks, the joint is likely to be virtually normal but the child should be reviewed regularly until skeletal maturity. The patient can then be discharged but should be urged to check her or his own children’s and grandchildren’s hips for dislocation.
 |
| Fig. 21.14
Arthrogram of a congenitally dislocated hip after reduction. Note that the acetabulum is well formed but not as well calcified as the healthy side.
|
At 2 months. If the condition is not diagnosed at birth it may be identified at routine examination 8 weeks later, when it should be apparent from the restriction of hip abduction. DDH diagnosed at this age can be managed conservatively with a brace traction or plaster immobilization.
At 12–18 months. If the dislocation is not found at the 8 week examination the diagnosis will probably be missed until the age of 12–18 months, when the child begins to walk with a limp and a rolling gait. The diagnosis can be confirmed on clinical examination because there will be shortening of the limb, the foot will be externally rotated, the skin creases asymmetrical and the Trendelenburg sign positive (Fig. 21.15).
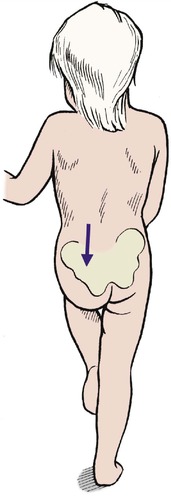 |
| Fig. 21.15
Positive Trendelenburg test at the age of 15 months in a child with a dislocated left hip.
|
At this stage the hip cannot be made perfect but a series of operations will usually achieve a stable joint, although at the price of great physical and emotional upset for the child and the family.
Because the prognosis is so much better if the diagnosis is made early, special care should be taken with the neonatal examination. A missed DDH is a disaster. The only patients who can lead a moderately normal life with a dislocated hip are those with both hips dislocated, because their problem is then symmetrical (Fig. 21.16).
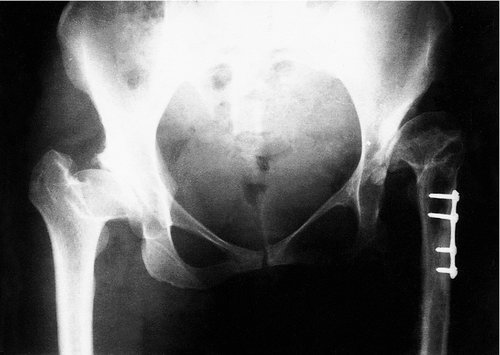 |
| Fig. 21.16
Bilateral DDH. The left hip remains dislocated despite an osteotomy to attempt reduction. Both femoral heads have dysplasia from damage due to splinting in extreme abduction. This damage can be avoided.
|
Neurological disorders
Spina bifida and meningomyelocoele
Meningomyelocoele is a congenital anomaly of the spinal cord in which there is defective tubulation of the neural plate, which remains open (Fig. 21.17). Neural tissues, including the roots and long tracts, lie visible on the child’s back and do not function. The lesion is often surrounded by hair (see Fig. 21.17). The extent of the lesion is variable and it may be associated with the Arnold–Chiari malformation and hydrocephalus.
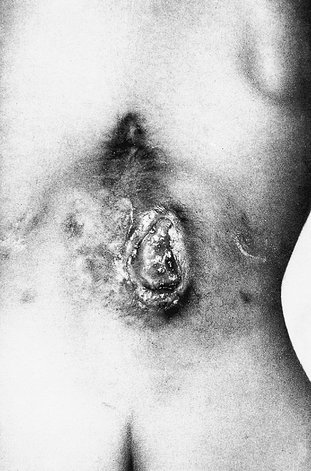 |
| Fig. 21.17
A meningomyelocoele.
|
Treatment
The problems of meningomyelocoele are enormous. Paediatricians, urologists and neurosurgeons are all as much involved as orthopaedic surgeons.
The management of the orthopaedic problems is complex and is determined as much by the intellectual potential of the child as by the deformity. If the child has mental impairment and is unlikely to manage anything more than a wheelchair existence, it is unkind to inflict operation, calipers and intensive physiotherapy. These cause just as much emotional strain as they would to a normal child or an adult and it may be wiser to help the child come to terms with a wheelchair existence.
The appropriate treatment for the individual child can be selected during the first 2 years of life, and decisions made then will determine future policy. In general, the best approach is a realistic assessment of the child’s potential and minimal surgery to prevent deformity.
Diastematomyelia
Diastematomyelia is dealt with on page 460.
Cerebral palsy
Cerebral palsy is caused by a lesion of the immature brain, often sustained at birth. It leads to a variety of neurological disorders, including spastic diplegia and choreoathetosis.
Clinical features
The principal feature is loss of voluntary control of the muscles. The commonest pattern is increased tone in the flexor muscle groups on one side producing a spastic hemiparesis, or spasticity of both lower limbs. Spinal deformities are also seen. In spastic hemiparesis the lower limb is usually more seriously affected than the upper (Fig. 21.18). Mental retardation, athetosis and other abnormalities may also be present.
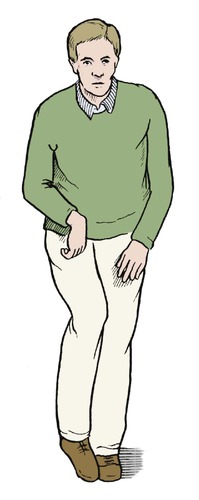 |
| Fig. 21.18
The deformity of cerebral palsy in an adult. The flexor groups of the elbow, wrist, fingers, hip and ankle all have increased tone, which is not balanced by the opposing extensor groups.
|
Examination
Because the flexor muscles are tight the limbs are pulled into characteristic positions. The foot is held in equinus, the knees are flexed and the hips adducted and flexed. The arm is held across the body, with the wrist and elbow flexed.
If the patient is relaxed the spasm in the flexors can easily be overcome by gentle manipulation but returns as soon as the limb is released or the child becomes agitated.
Treatment
Conservative treatment is important and the parents need much care and support. Physiotherapy helps to relieve muscle spasm and makes the best use of normal muscles. Calipers support the limbs and control unwanted movements.
If conservative treatment fails, operation is required to relieve flexor spasm by the following means:
1. Denervating muscles.
2. Lengthening tendons.
3. Moving muscle attachments.
In older patients, joints can be stabilized and bony deformities corrected by osteotomy or arthrodesis.
Selecting treatment is difficult and the whole patient must be considered. The equinus deformity of the foot, for example, is easily corrected by lengthening the Achilles tendon but the child may depend on that equinus deformity to compensate for the associated flexion deformities at the hip and the knee. In short, the deformity does not need to be corrected simply because it is there, but only if correction will improve the function of the limb as a whole.
Adductor spasm can lead to progressive subluxation of the hip and make it difficult to clean the perineum. The spasm can be relieved by dividing the adductor tendons and the obturator nerve. There may be a place for botulinum toxin injections to eradicate the spasm in some cases.
Poliomyelitis
Poliomyelitis, or infantile paralysis, causes deformities in children and should be a piece of history. Immunization is very effective if properly done, yet the disease is still seen.
The condition differs from cerebral palsy in two ways:
1. The paralysis is flaccid, not spastic.
2. Any muscle can be involved and the condition does not affect flexor muscles more than extensors. Individual nerve roots are involved.
Treatment
The aim of treatment is to produce a balanced limb with stable joints.
Conservative. Physiotherapy is important to overcome contractures and rehabilitate muscles; calipers or braces will often stabilize a flail limb but they are not strong enough to control active and unopposed muscles.
Congenital deformities
A host of minor congenital deformities exist but not all require treatment and it is usually advisable to defer operation until growth is complete (Fig. 21.19 and Fig. 21.20). In deciding whether or not to operate, remember that function is more important than appearance; to turn a painless digit that works well but looks odd into a useless one that hurts but looks perfect is a poor exchange.
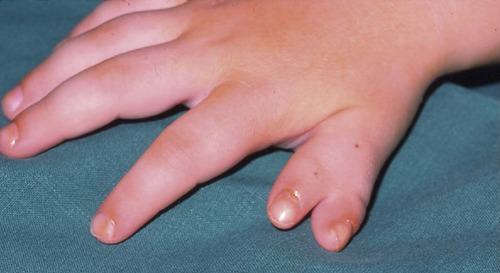 |
| Fig. 21.19
Reduplicated thumb.
|
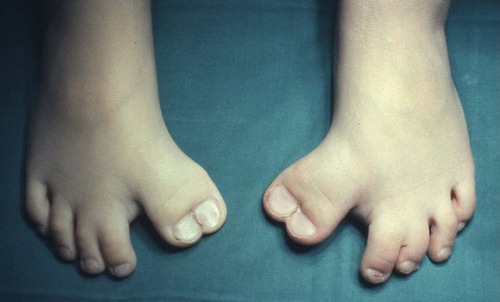 |
| Fig. 21.20
Reduplicated great toes.
|
Fortunately, it is seldom necessary to correct a deformity until growth is complete, by which time the patient will be able to make her or his own decision.
Hammer toe (or mallet toe)
Fixed flexion deformity of the distal interphalangeal joint of the second toe is a familial disorder that can cause pressure problems at the tip of the toe (Fig. 21.21a).
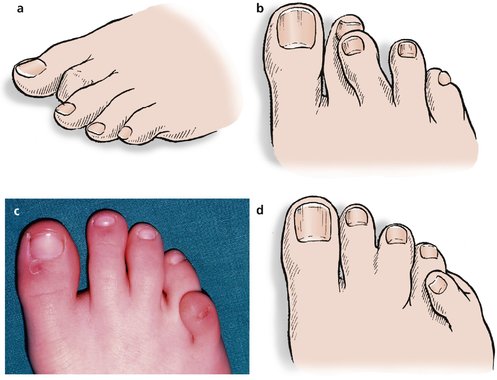 |
| Fig. 21.21
(a) Mallet toe deformity; (b) crossed second and third toes; (c) overriding fifth toe; (d) overriding fifth toe and webbed second and third toes.
|
Treatment
If the symptoms warrant it, the toe can be straightened or the tip amputated when growth is finished. Before skeletal maturity, simple flexor tenotomy will relieve the deformity.
Crossed second and third toes
The second and third toes sometimes cross over each other and cause difficulty with shoe wear (Fig. 21.21b).
Treatment
No disability is likely from this deformity and correction is seldom needed.
Overriding fifth toe (digiti quinti varus)
In this condition the little toe lies transversely across the top of the fourth toe, producing a curious appearance and making shoe wear difficult (Fig. 21.21c, d).
Treatment
The deformity may need correction in early adolescence if the symptoms justify it. The operation, Butler’s procedure, is radical and involves dissecting the toe free until it is held only by vessels and nerves. The toe is then ‘reimplanted’ in the correct position. Less radical procedures are ineffective.
Lobster claw hand
Lobster claw hand (Fig. 21.22) is inherited as an autosomal dominant and involves dysplasia of the middle rays of the hand and foot, producing a bizarre appearance that rarely interferes with function.
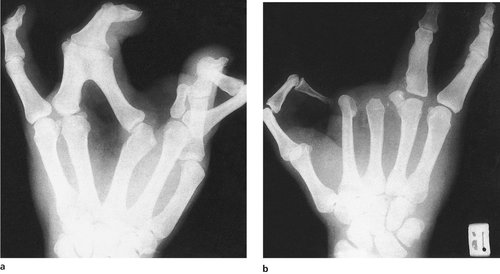 |
| Fig. 21.22
(a), (b) Lobster claw hand.
|
Absence of parts (Fig. 21.23)
The absence of a limb is known as amelia and is extremely rare. Absence of part of a limb is more common and is known as phocomelia. This term has nothing to do with ‘focal’ and means that the limbs look like the flippers of a seal. Phocomelia can arise from many causes but the best known in recent years was the drug thalidomide.
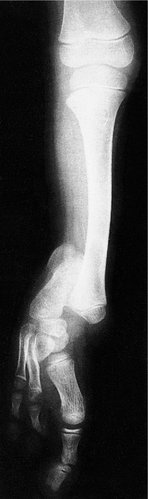 |
| Fig. 21.23
Congenital absence of the fibula.
|
Any limb can be involved to any degree, but proximal focal femoral deficiency creates special difficulties with weight-bearing.
Treatment is by braces and appliances.
Scoliosis
Very few patients have scoliosis requiring treatment but the condition attracts much attention. There are several types.
Types of scoliosis
1. Non-structural (postural) curves due, for example, to limb inequality. There is no rotation of the vertebrae with these curves.
2. Structural curves, which have rotation and sometimes wedging of the vertebrae.
Structural curves can be subdivided into four groups:
1. Idiopathic scoliosis.
2. Congenital and infantile.
3. Neuromuscular.
4. Miscellaneous.
Idiopathic scoliosis
Idiopathic scoliosis may be infantile (0–3 years), juvenile (3–10 years), adolescent (10 years to maturity), or adult (after maturity), according to the time of onset. Of these, adolescent idiopathic scoliosis is the commonest type (Fig. 21.24). It develops during the adolescent growth spurt, is commoner in girls than boys, and thoracic curves are usually convex to the right. The cause is unknown.
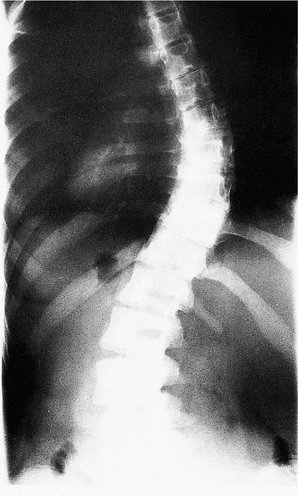 |
| Fig. 21.24
Idiopathic thoracolumbar scoliosis.
|
In its mildest form there is a slight curve of the spine, which may pass completely unnoticed or may be part of a more obvious asymmetry. Over 10% of adolescents have some degree of chest asymmetry. In more severe forms the spine can become grossly deformed if not treated, producing the classic hunchback of history. The hunchback of Nôtre Dame is said to have had a facial deformity and this makes neurofibromatosis the most likely diagnosis.
The thoracic spine is most often involved and has the most serious consequences, but deformities of the more flexible lumbar, thoracolumbar and cervical spines are also seen.
Natural history
The deformity appears at the start of the adolescent growth spurt and increases rapidly over the next 2 or 3 years. Progression continues until growth ceases. Some progress to moderate, mild or severe deformity, but some do not progress at all.
Untreated, the deformity may produce no obvious locomotor disability but it may cause cosmetic problems, cardiopulmonary compromise, pain from secondary degenerative changes in the spinal joints and loss of balance when sitting.
Clinical features
There are five important clinical features:
1. The spine is curved. The curve is best seen by asking the child to bend forward and inspecting the contour of the spine from behind (see Fig. 2.6). Structural curves in the spine will be exaggerated and postural curves reduced.
2. The shoulders are not level.
3. The waist is asymmetrical.
4. There is asymmetry of the chest or loin on forward bending.
5. There may be features of an underlying disorder, e.g. café-au-lait patches in neurofibromatosis or hairy patches in spina bifida.
Treatment
Not all deformities become worse but it is wise to review the patients regularly so that those which do progress can be treated promptly. There are three forms of treatment:
1. Corrective casts may stop infantile idiopathic curves progressing if applied early.
2. Braces may stop progression in some curves, but this is an area of controversy. Some evidence suggests that conservative treatment, including braces, has no effect at all and that the patients whose deformity was ‘controlled’ by devices such as braces were the ones who would not have progressed anyway.
3. Operation may be needed in severe and increasing deformity. The curve can be corrected by instrumentation to the back or front of the spine and held with internal fixation devices or it can be fused permanently in the corrected position. Both are major surgical undertakings and sometimes followed by serious complications, including paraplegia.
Congenital and infantile scoliosis
Congenital deformities such as hemivertebra give rise to a deformity present at birth. Correction is difficult or impossible.
Infantile idiopathic scoliosis develops during the first 3 years of life, is commoner in boys than in girls, and the thoracic curve is usually convex to the left, the opposite of adolescent idiopathic scoliosis. Ninety per cent of thoracic curves resolve spontaneously but the rest need treatment with braces or internal fixation if severe. Even if no treatment is required, it is sensible to obtain an opinion on the value of a brace in the individual case.
Neuromuscular scoliosis
Neurological imbalance of the spinal muscles from poliomyelitis, spina bifida, neurofibromato-sis or other neurological disorders produces a severe scoliosis which cannot always be treated successfully. Braces or internal fixation may be required.
Other conditions
Limb inequality
To have limbs of equal length is rare; there is usually a difference of up to 1 cm. Because the lower limb is about 1 metre long, this represents a ‘tolerance’ of 1%.
Management of limb inequality
< 2 cm at maturity – no treatment.
2–5 cm – raise to shoe.
> 5 cm – operation sometimes.
Treatment
Shortening of less than 2 cm is scarcely noticeable, and up to 5 cm can be treated by a raise to the shoe. Shortening greater than 5cm can be treated either by interfering with the epiphysis to arrest growth in the longer limb or shortening the longer limb when growth is complete.
Alternatively, the shorter limb can be elongated by distraction apparatus applied either across a transverse cut in the diaphysis or the epiphyseal plate. Operation is not easy and vessels, nerves and tendons may not stretch as easily as the divided bone (Fig. 21.25).
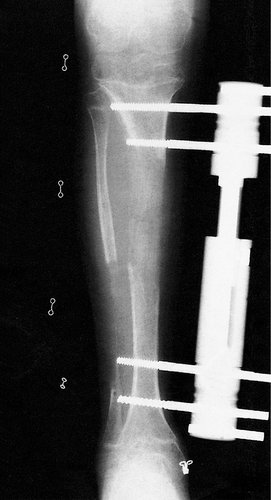 |
| Fig. 21.25
Radiograph of leg lengthening.
|
To determine how much the limb must be lengthened, or when growth should be arrested, requires an accurate assessment of the likely difference in limb length when growth is complete. This information is derived from tables and graphs showing the rate of growth of the individual epiphyses at different ages.
Arthrogryposis multiplex congenita
Arthrogryposis is a rare condition characterized by the replacement of striated muscle with fibrous tissue and by soft tissue contractures. The result is a disabling loss of movement in many joints accompanied by a severe talipes and often dislocation of the hip. The degree of involvement is variable.
The elbows are usually fixed in extension and the wrists in flexion. The muscles of respiration are also involved and a few patients die of respiratory insufficiency. The patients are of normal intelligence and usually very determined.
Treatment
Treatment consists of soft tissue release and osteotomy to bring the limbs into the most useful position.
Radial club hand
Congenital anomalies of the hand are less common than the foot but radial club hand is seen and is usually associated with an absent or deficient radius, which allows the hand to ‘fall off’ the arm.
Treatment
Treatment consists of stabilizing the hand on the ulna, either with a brace or by operation.
Torticollis
Torticollis, or wry-neck, is now seldom seen but once occupied much orthopaedic time. The condition is the result of excessive stretching of the sternomastoid during delivery; improved obstetric care has reduced its incidence dramatically.
The area of damaged muscle contracts to form a firm fibrous mass in the muscle known as a sternomastoid ‘tumour’. With growth, the head is pulled progressively over to one side by the tight contracture of sternomastoid (Fig. 21.26).
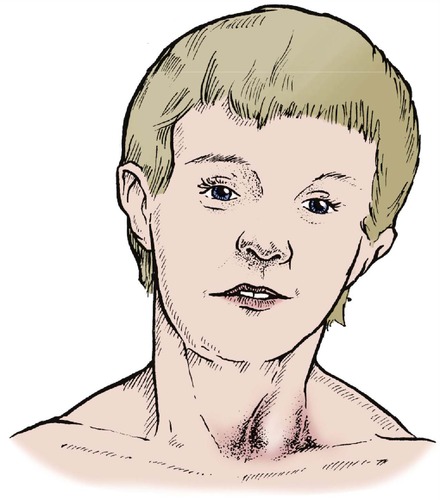 |
| Fig. 21.26
Torticollis due to sternomastoid ‘tumour’. Note the asymmetrical eyes and tilted face.
|
Treatment
Physiotherapy is only effective in the first year of life. If the deformity persists the sternomastoid must be released from the clavicle and the head held in the corrected position until the patient no longer tilts it. The operation should be done early for two reasons:
1. Growth changes produce a very odd asymmetrical face.
Pseudarthrosis of the tibia
Pseudarthrosis of the tibia is a rare condition and should be differentiated from neurofibromatosis. In essence, the middle third of the tibia is deficient and either breaks with trivial injury or is completely absent. The appearances are very similar to an atrophic non-union in an adult and union is just as difficult to achieve.
Children’s fractures usually unite rapidly and it is not known why the tibia sometimes behaves in this pernicious manner.
Treatment is by internal fixation and bone grafting. Amputation has been required in the past, but with solid fixation and bone grafting this should be avoided.
Congenital dislocation of the knee
This is a very rare condition consisting of hyperextension of the knee so that the knees appear to be mounted backwards. The condition is not analagous to DDH.
Treatment
Conservative treatment may be effective but open correction is often needed. Gentle manipulation early on is recommended. Open release of the contracted tissue is often needed and disorders of patella–femoral movement are common.