CHAPTER 83 Deep Brain Stimulation
Mechanisms of Action
Key Developments
The practical application of DBS has benefited from several important neurosurgical innovations and discoveries since the late 1800s that were motivated by the need to improve the efficacy of subcortical ablative procedures for treating patients with neurological disorders (Fig. 83-1). Perhaps the most noteworthy of these advances was reported by Spiegel and associates1 in 1947, when they described the use of a human stereotactic frame derived from and similar in concept to that reported by Horsley and Clarke2 for animal work. The stereotactic frame was developed to allow subcortical lesions to be made with more precision and accuracy. Using a variation of this apparatus, Albe-Fessard and colleagues3 demonstrated the practical application of a neurosurgical targeting technique known as intraoperative microelectrode recording. This approach involved sampling neuronal spike activity from a microelectrode inserted in the brain as a means of identifying regions with pathologic activity and then using that information to refine anatomic maps before ablative procedures. Neurosurgical targeting also progressed with the advent of computed tomography (CT) and magnetic resonance imaging (MRI), which allow one to determine the coordinates for a target based on the specific anatomy of the patient’s brain.
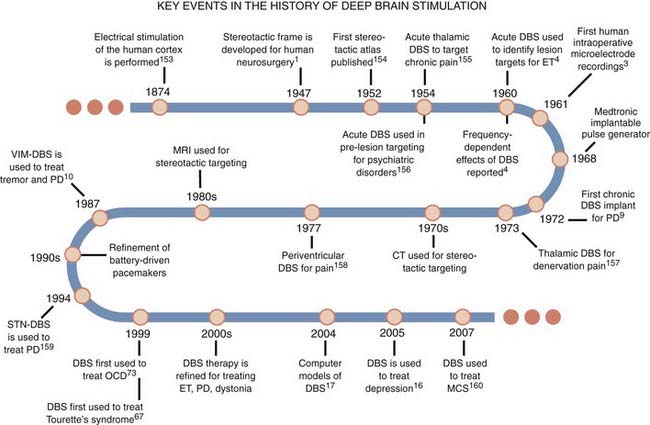
The seminal study of the potential application of DBS in humans was performed by Hassler and colleagues4 in 1960. They described a series of parkinsonian patients who received electrical stimulation via electrodes introduced into the globus pallidus for the purpose of creating therapeutic ablations. They observed that low-frequency stimulation (<25 Hz) in the globus pallidus could exacerbate contralateral tremor, whereas high-frequency stimulation (25 to 100 Hz) through the same electrode could alleviate or abolish tremor entirely. They suggested that electrical stimulation could be used to pinpoint regions of the brain where lesions would reduce the motor symptoms of movement disorders. Following the reported success of intraoperative stimulation for mitigating pain and reducing the motor signs of movement disorders,5–8 the next step was to deliver this therapy continuously through an implanted system. Bechtereva and colleagues9 were the first to describe a semichronic DBS system, but not until the maturation of battery-powered implantable pulse generators (IPGs) in the 1980s and the work of Benabid and colleagues10 were chronic DBS systems fully realized.11 Since then, DBS systems have been implanted in tens of thousands of patients with medication-refractory neurological and psychiatric disorders. For many of these disorders, electrical stimulation has now supplanted ablation as the surgical treatment of choice because DBS is reversible and can be titrated to maximize therapeutic benefit.
General Principles
Implantation Surgery
Once the final implantation coordinates are determined physiologically, the mapping electrode is removed, and the DBS lead is implanted. The Medtronic Model 3387 and 3389 DBS leads (Fridley, MN) are the only leads currently approved by the U.S. Food and Drug Administration. They consist of four cylindrical electrode contacts (1.5 mm high, 1.27 mm in diameter) spaced either 1.5 mm (Model 3387) or 0.5 mm (Model 3389) apart from one another (Fig. 83-2A). During insertion of the DBS lead, fluoroscopy is often used to confirm that the lead has reached the correct depth and that it has not moved during the process of securing it at the cranium. Stimulation through the DBS lead is then performed using a handheld screener, and symptoms and side effects are evaluated in real time to verify that the threshold intensities for generating adverse side effects are sufficiently greater than those necessary to generate therapeutic effects.
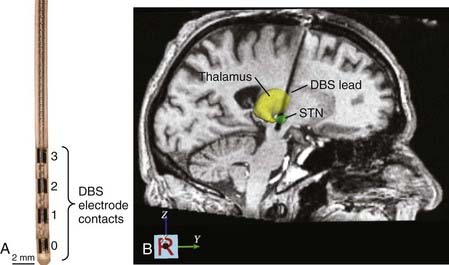
Once implanted, the DBS lead is secured by means of a bur-hole cap, and any extra length of lead wire is carefully placed beneath the galea. Postoperative CT and MRI scans are generally obtained to verify lead location and to confirm that hemorrhage has not occurred (Fig. 83-2B). The remainder of the DBS system, which consists of an IPG placed within a subcutaneous infraclavicular pocket and an extension cable that is tunneled percutaneously along the neck to connect the distal end of the DBS lead to the IPG, is implanted during a second procedure performed with the patient under general anesthesia.
Stimulation Parameters
After the patient has recuperated from surgery, the IPG is programmed in the outpatient clinic. At the initial programming session, the neurologist or psychiatrist seeks to determine the settings that provide the most therapeutic benefit, minimize undesirable side effects, and limit the stimulator’s power consumption to extend battery life.12 When treating PD, for example, most experienced programmers first stimulate in a monopolar configuration using an electrode contact as the cathode (negative voltage) and the IPG’s outer casing as the anode (positive voltage). The applied stimulation pulse train consists of a pulse width of 60 to 90 µsec, a pulse frequency of 130 Hz, and an amplitude of 0.5 to 4 V or more. Adverse effects may occur at higher voltages, but if they occur at voltages within or before the therapeutic range, one can use a bipolar stimulation configuration (i.e., one of the contacts acts as the cathode, another as the anode) to restrict the spread of current into adjacent regions of the brain responsible for inducing the side effects.
Anatomic Targets
Identifying anatomic targets for DBS initially relied on the experience and knowledge gained from surgical ablative procedures. Although the therapeutic correlation between the lesion target and the DBS target is true in most cases, it is not able to be generalized. For example, DBS of the external globus pallidus (GPe) can improve PD motor symptoms,13 but lesioning the GPe can worsen PD motor symptoms.14 Positron emission tomography (PET) has proved useful for establishing which brain regions show abnormal activity preoperatively15 and whether DBS reverses these abnormalities postoperatively.16 Intraoperative microelectrode recordings also support the concept that brain regions with abnormal neuronal activity coherent with the patient’s symptoms are potential target areas for DBS. Other investigators have used computational models to examine the inhomogeneity of voltage fields produced by DBS and how these distributions can lead to suprathreshold currents in multiple neuronal and non-neuronal elements, some of which may be adjacent to the presumed target nucleus or fiber pathway.17,18 Correlation analysis between model predictions and clinical outcomes of DBS can then be used to determine which stimulated neural elements improve symptoms or generate undesirable side effects. These considerations are addressed in more detail in the following sections on the clinical indications for DBS. Remarkably, DBS can produce functional benefit by targeting any of a number of different interconnected brain regions, which emphasizes the role of a common dysfunctional network in many neurological disorders (Fig. 83-3).
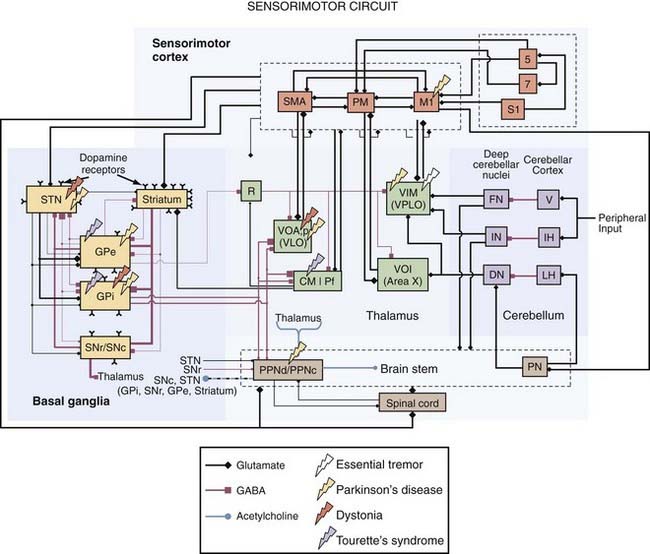
Tremor
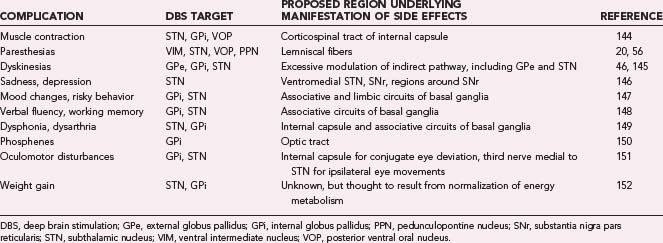
The most common surgical target for treating medication-refractory ET with DBS is the ventral intermediate (VIM) nucleus of the thalamus.19 The VIM nucleus rests in the posterior third of the thalamus, 0 to 15 mm dorsal to the anterior commissure–posterior commissure (AC-PC) line and approximately 10 to 20 mm from the midline. Intraoperative microelectrode mapping of the VIM nucleus is often necessary to define the implantation site because the minimal current intensity necessary to relieve tremor usually depends on the electrode’s precise location within the nucleus. Correct placement of the lead can be ascertained not only by suppression of tremor in the upper limb but also by transient paresthesias in the fingers or face.20 The lead is generally placed at the border of the VIM nucleus and the posterior ventral oral (VOP) nucleus of the thalamus to prevent current from spreading too posteriorly into the sensory nucleus (ventral caudal nucleus [VC]). If paresthesias persist with stimulation, current is likely spreading into the VC nucleus and the lead is too posterior (Table 83-1). Leads placed too medially elicit intraoral paresthesias with stimulation, and paresthesias in the leg usually indicate that the lead is too lateral. Placing the lead in the anterior portion of the VIM nucleus allows one to avoid continued activation of the VC nucleus; however, it is likely that the VOP nucleus is also affected by stimulation under these conditions. Given the reported relationships among the VOP nucleus, neuronal activity, and tremor,21 it is possible that some of the benefit obtained with stimulation in this area occurs because of the effects of stimulation on both the VIM nucleus and the VOP nucleus.
Because efferents from the VIM nucleus project directly to the primary motor cortex and, to a lesser extent, the premotor and supplementary motor cortices,22,23 the VIM nucleus is in a critical position to affect motor output. In patients with ET, pathologic spike activity in the VIM nucleus is characterized by increased “burstiness” (with 5 to 10 spikes/burst), which for many cells is coherent with the frequency of tremor.24 Whether this activity originates from within the VIM nucleus, arises from recurrent thalamocortical loops, or derives from tremor-related activity occurring in the cerebellum,25 or indirectly from the inferior olivary nucleus26 is unclear. However, it is clear that high-frequency stimulation in regions of the VIM nucleus where burst cells tuned to the tremor frequency are found can have dramatic suppressive effects on tremor. VIM-DBS is typically indicated for suppressing hand tremor, but improvement in vocalization and head tremor has also been observed.27 Leg tremor can be more difficult to treat with thalamic DBS because the relevant region is found laterally in the thalamus near the internal capsule. Patients with PD exhibiting considerable resting tremor have also benefited from VIM-DBS, but the therapy appears to have little beneficial effect on other motor signs.28 Stimulation in regions anterior to the VIM nucleus, including the VOP nucleus and the anterior ventral oral (VOA) nucleus, can improve rigidity and drug-induced dyskinesia similar to that reported for lesions in this region; however, it is unlikely to improve bradykinesia. VIM-DBS can also produce some benefit for patients whose tremor is associated with cerebellar dysfunction or multiple sclerosis, but the magnitude of the effect is often less than that obtained by patients with ET or PD.29 This may be due to the proximal nature of such tremor and the requirement that a greater area of the VIM nucleus be stimulated to cover the somatotopic region contributing to the tremor.30 Using two leads in such cases may provide greater therapeutic benefit by stimulating a greater area of the thalamus.31,32
Parkinson’s Disease
DBS is an effective therapy for PD patients who have responded well to levodopa therapy in the past but now exhibit motor fluctuations, drug-induced dyskinesias, and an unpredictable response to antiparkinsonian medication. In these cases, the therapeutic effects of DBS can be produced by targeting one of a number of different nuclei or fiber pathways (see Fig. 83-3). The most common DBS target for PD patients is the STN, which is positioned 0 to 6 mm below the AC-PC line, in the middle third of the intercommissural distance, and 9 to 15 mm lateral to the midline. To avoid implanting through motor cortical areas and passing through the lateral ventricle, the implantation trajectory is usually oblique in both the coronal (about 15 degrees off the vertical axis) and sagittal (about 20 degrees anterior to the vertical axis) planes. Compared with the STN of normal nonhuman primates, human STN neurons in the parkinsonian state have several atypical characteristics, including increased burstiness (30% to 40% versus 4% to 5%), higher firing rates (25 to 40 versus 15 to 25 spikes/sec), and augmented oscillatory activity in the beta frequencies (15 to 30 Hz).33 During microelectrode mapping, STN neuronal activity is easily identified owing to the density of neurons in the STN compared with the surrounding structures. Neurons in the dorsolateral STN are responsive to passive sensorimotor manipulation and are thought to be one of the primary cellular targets for DBS. However, recent studies suggest that the therapeutic target for STN-DBS also includes regions dorsomedial to the STN,34,35 an area that encompasses the zona incerta,36 pallidofugal fibers passing through the fields of Forel en route to the thalamus and brainstem,18 cerebellothalamic fibers,37 and dopaminergic nigrostriatal fibers projecting to the striatum, pallidum, and STN.38 When the spread of suprathreshold currents envelops nonmotor regions of the STN and other adjacent structures, mood alterations and difficulty performing working memory tasks have been reported (see Table 83-1).39,40
The sensorimotor (posteroventrolateral) internal globus pallidus (GPi) is another therapeutic target for DBS.40,41 The GPi resides in the anterior two thirds of the intercommissural distance, 4 mm below to 8 mm above the intercommissural plane and 7 to 23 mm lateral to the midline. The GPi is a much larger target than the STN (478 versus 158 mm3),42 which may necessitate the use of higher stimulation voltages to produce the same therapeutic benefit; however, it may also provide more opportunities to limit current spread to adjacent regions implicated in the generation of adverse side effects (see Table 83-1). In PD patients, microelectrode mapping studies have demonstrated that spike activity in the GPi is characterized by a higher than normal firing rate (80 to 100 Hz), increased burstiness, and elevated power in the beta oscillation frequency.33,43
The external globus pallidus (GPe) is also an effective target for managing parkinsonian motor symptoms,13 and it is an even larger structure than the GPi (808 mm3).42 Neurons in the GPe exhibit increased burstiness with long pauses in between,43 whereas regions between the pallidal segments are identifiable by the presence of large, regularly firing neurons called border cells. Studies suggest that contact electrodes near the border of the GPe-GPi or entirely within the GPe may be more effective at relieving bradykinesia than stimulation with contacts entirely within the GPi, a site considered optimal for mitigating levodopa-induced dyskinesias.44–46 These studies found that in contrast to bradykinesia, rigidity was reduced no matter which pallidal segment was stimulated, suggesting that the underlying therapeutic mechanism for rigidity may be different from that for bradykinesia. It is possible, however, that the lack of benefit on akinesia observed with GPi-DBS resulted from stimulation current spreading to the internal capsule. Indeed, studies in the nonhuman primate model of PD have demonstrated worsening bradykinesia in the face of improving rigidity during stimulation at voltages near the threshold for activation of the corticospinal tract.47 Another possibility, although less likely, is that these effects stem from the modulation of different motor subcircuits within the pallidum. Hoover and Strick22,48 showed through transsynaptic anatomic tracers that the motor cortex and supplementary motor areas target different subregions of the sensorimotor pallidum. Along these lines, electrical stimulation of the motor cortex modulated neural activity in the posteroventral globus pallidus, which is related to the execution of movement, whereas stimulation of the supplementary motor areas affected neurons in the anterodorsal pallidum, which is related to the planning of movement.49,50
Gait initiation and balance disturbances are often observed in patients with PD, but neither STN-DBS nor GPi-DBS provides uniform benefits for all patients, particularly for medically refractory gait and balance disorders. Recent reports suggest that the pedunculopontine nucleus (PPN) may be a potential DBS target for improving drug-resistant gait and balance problems.51 The PPN has a central role in the mesencephalic locomotor region, and during continuous electrical stimulation in animal models (20 to 60 Hz), locomotion and muscle tone are increased.52,53 A few patients have now been implanted with PPN-DBS systems, but improvement in gait has not been consistent.54 This variability may stem from the fact that in humans we do not know what pathologic activity exists in the PPN, which regions of the PPN when stimulated provide a therapeutic benefit, and what stereotactic coordinates and stimulation parameters to use to minimize paresthesias and other side effects.55,56 Moreover, the capacity for DBS to modulate PPN output in some patients may be confounded by progressive PPN degeneration over time. Histologic analysis, for example, indicates that approximately 50% of the large cholinergic neurons in the PPN pars compacta degenerate in PD.51
Dystonia
In patients with medication-resistant primary generalized dystonia, segmental dystonia, and complex cervical dystonia, DBS of the posteroventrolateral (sensorimotor) GPi is the preferred surgical treatment, largely displacing pallidotomy.57 At present, it is unclear what neural elements underlie the therapeutic benefit for either phasic or tonic dystonia, especially because DBS alleviates phasic dystonia more readily and more quickly than tonic dystonia. One likely hypothesis is that DBS affects the output of GPi efferents projecting to the thalamus and brainstem. Indeed, these cells have a significant degree of abnormal firing patterns in patients with dystonia, which is also observed in the GPi of PD patients. In both cases, there is increased burstiness, but in dystonia, GPi neurons exhibit, on average, lower than normal firing rates and increased oscillatory activity in the 4- to 10-Hz band.58,59
Other anatomic targets for treating dystonia with DBS include the VIM nucleus, the VOP nucleus, and the VOA nucleus. Given the success of GPi-DBS for treating primary dystonia, however, there has been little movement toward thalamic targets, despite the report of some success with thalamotomy for dystonia.60,61 Moreover, when thalamic DBS has been attempted, the results have been mixed. This may be due to the fact that most of these patients had secondary dystonia,62 which, other than tardive dystonia,63 does not respond well to DBS.57,64 For focal dystonia, one group in Italy reported that extradural motor cortex stimulation can provide some therapeutic benefit.65 Sun and colleagues66 reported that STN-DBS can be effective for managing dystonia, arguing that in their patients, STN-DBS provided faster improvement of dystonic symptoms, with lower voltages necessary to elicit the improvement, compared with GPi-DBS. Although control of symptoms was no better with STN-DBS than with GPi-DBS, their GPi-DBS patients did not do as well as others reported in the literature. Double-blind randomized clinical trials comparing the long-term effects of STN- and GPi-DBS in dystonia patients may be necessary to determine the validity of their assertion.
Neuropsychiatric Indications
The pathophysiology of Tourette’s syndrome is thought to include dysfunction of both associative-limbic and sensorimotor circuits. Not surprisingly, anatomic targets for DBS have followed this multimodal composition. Vandewalle and colleagues67 first reported that bilateral stimulation of the medial thalamus reduced tic severity in a patient with Tourette’s syndrome. Their coordinates (5 mm lateral of the AC-PC line and 4 mm posterior of the midcommissural point, at the level of the AC-PC plane) were based on those described by Hassler and Dieckmann68 for partially suppressing tics with bilateral lesions of the median and rostral intralaminar thalamic nuclei and the internal VOA nucleus.68 Other regions in the associative-limbic network have also proved effective, including the centromedian-parafascicular complex of the thalamus and the associative-limbic ventromedial GPi.69,70 The motor territory of the GPi and GPe is reportedly an effective stimulation target as well,71,72 which parallels the improvement seen with sensorimotor GPi-DBS in patients with hyperkinetic and hypokinetic movement disorders.
Two other emerging indications for DBS are medication-refractory obsessive-compulsive disorder and depression—both of which have targets in the anterior limb of the internal capsule. Nuttin and colleagues73 were the first to report the use of DBS at the anterior limb of the internal capsule for treating obsessive-compulsive disorder in 1999. This fiber tract contains numerous projections to and from frontal cortical areas. The limbic basal ganglia (caudate nucleus, nucleus accumbens, GPi, STN) are also thought to be involved in obsessive-compulsive disorder, and stimulation in these regions has produced therapeutic effects in some patients. DBS of the anterior limb of the internal capsule as well as the anterior cingulate cortex (Cg25) reportedly improves the symptoms of depression.16 The trajectory used for Cg25 places the distal and proximal contacts in cortical gray matter, with the remaining two middle contacts in white matter. Interestingly, the contacts used in these studies tended to be the two middle contacts, which, when activated, would be expected to affect both afferent and efferent projections of Cg25.
Neural Responses
The similarity in therapeutic outcomes derived from surgical ablation and DBS at the same targets led to the hypothesis that high-frequency stimulation inhibits output from the stimulated nucleus.8,10 Recent studies have challenged this view, however, suggesting that instead of suppressing output, DBS overdrives the output of neurons and fibers of passage near the active electrode.74 This process occurs because the threshold for eliciting action potentials is significantly lower in the axon than in the cell body,75 which means that even if somatic and initial segment processes are suppressed by electrical stimulation, action potentials can still be evoked in axonal efferents downstream.76 The dissociation between somatic and axonal responses to DBS has been confirmed experimentally for a number of different DBS target structures.
Changes in Somatic Activity in the Stimulated Nucleus
Inhibition of somatic activity during DBS has been observed via microelectrode recordings in the STN77,78 and GPi79,80 of human patients. Several hypotheses have been proposed to explain this inhibition, including (1) depolarization block resulting from the inactivation of sodium channels81,82 or an increase in potassium current,83 (2) presynaptic depression of excitatory afferents,84 and (3) stimulation-induced activation of inhibitory afferents.85 Evidence to support the depolarization block hypothesis is derived primarily from brain slice recordings. Following the onset of high-frequency stimulation in the STN of rat brain slices, for example, neurons in the STN initially show an elevated firing rate, after which these cells fail to be driven by the stimulation.86 Brain slices, however, inevitably sever connections from afferent input structures. Several in vivo studies have noted that although STN or GPi neurons are directly inhibited by high-frequency stimulation, their firing probabilities are not completely suppressed.78,79,87 Other studies have indicated that somatic inhibition can occur even after a single stimulation pulse,77 with both inhibition and recovery from inhibition occurring at latencies consistent with the kinetics of GABAergic synapses.87 If one considers the hypothesis of driving afferent input with DBS, the observation of decreased activity in the stimulated nucleus (STN or GPi) is not surprising. The majority of afferents to these structures are GABAergic (ratio of 9 : 1 for the GPi),88 forming dense synaptic baskets around the soma and proximal dendrites. In the VIM nucleus, which contains mostly excitatory input, it was reported that approximately half of all recorded neurons had higher than normal firing rates during stimulation.89
Yet, simply driving synaptic input does not explain the variety of somatic responses elicited by DBS. For instance, a small fraction of cells in the STN or GPi shows an excitatory response to local high-frequency stimulation.90,91 Another set of STN and GPi cells is completely suppressed, even at microelectrode recording distances at which synaptic elements would theoretically not be activated.80 Computational models indicate that the former could result from driving membrane-bound sodium channels in conjunction with synaptic depression, whereas the latter could be explained by activating afferent axonal collaterals distal to their somatic target.92 Even more striking is the pattern of neuronal firing during stimulation. This pattern consists of the generation of action potentials that are time-locked to the stimulation train. In other words, when a neuron near the active electrode fires an action potential, it does so at a fixed latency from the preceding stimulus pulse. Such entrainment has been shown in the STN87 and GPi,90 and it is believed to derive from the repetitive activation of somatic and dendritic membrane-bound ion channels.92,93 In the pallidum, for instance, high-frequency stimulation generated a firing pattern in 70% of the recorded cells; this pattern consisted of two excitatory phases at 3 msec and 7 msec, which were separated by inhibitory phases.90 A similar firing pattern was observed with neurons in the STN adjacent to the active electrode.87 Neuronal activity was initially inhibited after each DBS pulse, but discharge probabilities became higher at 3 msec and returned to baseline after 7 msec.
Dissociated Output from the Stimulated Nucleus
Recent experiments and computational models indicate that changes in the firing rate and pattern of somatic activity do not necessarily translate to similar changes in efferent output.17,94 Although it is extremely difficult to record extracellular activity from axons, it is possible to record from downstream nuclei innervated by the stimulated axons. Hashimoto and colleagues94 showed that neuronal firing rates in the GPe and GPi increased during therapeutic STN-DBS in parkinsonian monkeys (Fig. 83-4). They also noted that GPe and GPi somatic activity became entrained to each stimulus pulse, such that the firing probability was increased at 3 and 6.5 msec after each pulse; periods in between showed pronounced inhibition, especially in the GPi. In contrast, during therapeutically ineffective stimulation, the firing rate and pattern in the GPi did not change significantly. A complementary follow-up study in these monkeys using computational models of STN neurons showed that for those neurons affected by the stimulation, action potentials developed first in the axon (see Fig. 83-4).18 For therapeutic DBS settings in these monkeys, approximately 50% of STN axons were entrained to the stimulus train at least 80% of the time. Furthermore, this percentage was significantly higher for clinically effective compared with clinically ineffective stimulation.
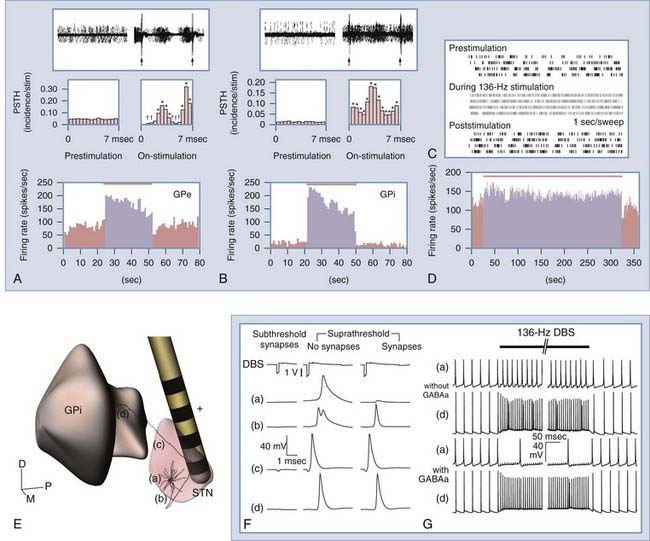
The output activation hypothesis also applies to other nuclei. A study investigating bradykinesia and rigidity in parkinsonian monkeys during GPe high-frequency stimulation demonstrated that 67% of recorded STN cells exhibited a decrease in their firing rate and burstiness during therapeutic stimulation, but the percentage dropped to 31% for clinically ineffective stimulation.95 Similarly, based on recordings in the pallidal receiving areas of the thalamus during GPi high-frequency stimulation, Anderson and colleagues96 noted that 77% of thalamic neurons were inhibited in normal monkeys. Studies in PD patients97 and in patients with dystonia98 have confirmed that GPi high-frequency stimulation decreases activity in the VOP nucleus; however, they also described time-locked responses of thalamic neurons with elevated firing probabilities at 3.5 to 5 msec and pronounced inhibitory phases at the other times during the interpulse interval.
In addition to activating axonal output from the stimulated nucleus, DBS can affect other fibers passing through the target. For instance, a majority of fibers from the putamen and GPe pass through the GPi en route to the STN and substantia nigra pars reticularis (SNr),99,100 and a large proportion of STN fibers targeting the GPe projects through the GPi.101 Computational models predict that these fibers are activated during GPi-DBS, indicating that the therapeutic mechanisms of DBS may involve multiple pathways.92 Dopaminergic fibers from the substantia nigra pars compacta (SNc), serotoninergic fibers from the dorsal raphe nucleus, and cholinergic fibers from the PPN also have trajectories that pass through the GPi en route to the GPe and putamen.102 Suprathreshold currents from the DBS electrode can extend beyond the borders of the stimulated nucleus and affect these fibers of passage. This is especially relevant to DBS in the STN and PPN, which are small nuclei surrounded by large fiber tracts. The PPN, for instance, is bordered laterally by the medial lemniscus; medially by the superior cerebellar peduncle; rostrally by the posterolateral substantia nigra; rostrodorsally by the retrorubral field; caudally by the pontine cuneiform, subcuneiform nuclei, and locus caeruleus; and ventrally by the pontine reticular formation.51 It is therefore not surprising that stimulation currents can and often do exceed the boundaries of the PPN, as revealed by the presence of paresthesias during PPN-DBS in PD patients.56 In the case of STN-DBS, should suprathreshold currents spread lateral to the STN, activation of the corticospinal tract within the internal capsule can become problematic because it evokes strong contralateral muscle contractions and diminishes any improvement in bradykinesia gained by the stimulation. It has also been suggested that modulation of the cerebellothalamic fibers adjacent to the STN may underlie the improvement in parkinsonian tremor with STN-DBS.37
Therapeutic Mechanisms
Regularization of Pathologic Activity
One proposed mechanism of DBS, which is consistent with the activation of axonal output, is that stimulation overrides pathologic network activity by generating more regular firing patterns in downstream nuclei.74,103 According to this theory, the stimulation frequency needs to be faster than the baseline somatic firing rate in order to reset somatic activity and drive axonal activity before the neuron can resume its pathologic firing pattern. In support of this argument, PD patients with GPi-DBS implants typically require stimulation frequencies greater than 100 Hz, which is higher than the average baseline firing rate in the parkinsonian GPi (70 to 100 Hz). When pathologic GPi activity exhibits lower rates, such as in dystonia (40 to 60 Hz), there is evidence that lower stimulation frequencies can be therapeutic (>60 Hz).104 PPN neurons exhibit much lower discharge rates (about 15 Hz), and for this target the therapeutic stimulation frequency is between 20 and 60 Hz in PD patients.54 Interestingly, in the case of PPN-DBS, frequencies less than 20 Hz and greater than 60 Hz can worsen freezing of gait; this suggests that for low-frequency stimulation, additional spikes may be overlaid on an already pathologic rhythm, whereas for high-frequency stimulation, cholinergic signaling may not be able to keep up with the stimulus frequency. Unlike the examples in which therapeutic DBS frequencies are only slightly higher than the nucleus’s baseline firing rate, the STN exhibits an average firing rate of 35 to 40 Hz in PD patients, but therapeutic STN-DBS usually requires frequencies greater than 100 Hz. One possible explanation is that the therapeutic outcome derived from STN-DBS requires overdriving the fastest nodal point in the affected circuit—in this case, pallidofugal fibers of passage (70 to 100 Hz) coursing through the fields of Forel. Another possibility is that regulating downstream glutamatergic synaptic signaling requires different activation patterns than for GABAergic synapses.
In addition to regularizing neuronal discharge with DBS, stimulation can affect the nuclei’s ability to synchronize on what are seemingly pathologic oscillatory rhythms. In PD patients, synchronized oscillations throughout the basal ganglia occur at frequencies between 15 and 30 Hz (the so-called beta range)105,106 and are markedly reduced following dopaminergic therapy (e.g., levodopa treatment).107 Similarly, clinically effective STN-DBS has been shown in humans to decrease beta-band oscillations in the STN108 and globus pallidus,109 and this decrease correlates with clinical improvement. Decreased beta as well as increased gamma (>30 Hz) activity has also been reported during STN-DBS in the thalamus of parkinsonian monkeys.110 In comparison to PD patients, dystonia patients have predominant pallidal oscillations in the 4- to 10-Hz range and less in the beta range.111,112 These oscillations, which are minimal at rest, are enhanced during dystonic movements and correlate with electromyographic activity in the dystonic limb. It has been posited that networks in the brain naturally use these oscillations to synchronize activity among interconnected nuclei to enhance the transmission of information and promote synaptic plasticity.113 Disturbances in the generation and dynamic modulation of these oscillations would thus hinder the ability of the affected networks to function normally, as in the case of PD, where synchronization of prokinetic oscillations in the basal ganglia may be impaired.114 This may explain why low-frequency stimulation in the GPi or STN can worsen the expression of parkinsonian motor symptoms.
There are a couple of mechanisms by which DBS at a frequency higher than a neuron’s own spontaneous discharge rate could override a neuron’s intrinsic output. When stimulation directly evokes action potentials in the axon, these spikes propagate antidromically back along the axon and collide with orthodromic spikes initiated in the soma or dendrites. Moreover, because of the refractory period following an action potential, successive antidromic spikes may prevent the neuron from resuming its pathologic firing patterns. Suppressing the capacity for somatodendritic spikes to propagate down the axon may also facilitate the replacement of pathologically generated somatic activity with axonally initiated firing patterns entrained to the stimulation, thereby increasing the rate and regularity of synaptic signaling in downstream targets. Although this tonic, high-frequency firing pattern is not considered natural, it is devoid of dynamic information that is of value to downstream nuclei. The resulting “informational lesion” may thus prevent pathologic activity from being transmitted and amplified within networks implicated in the manifestation of symptoms.115
Compensatory Responses
It is counterintuitive that stimulating (or lesioning) a region of the brain would improve symptoms of a neurological disorder without causing additional disabilities. To understand this apparent contradiction, it is useful to consider what pathologic activity actually means. Imaging studies have indicated that abnormal neural activity is present in both patients expressing symptoms of a disorder and nonmanifesting carriers of genes implicated in the disorder (e.g., DYT1 dystonia).116 In the STN, nonhuman primate models of PD suggest that changes in neuronal activity patterns begin to appear before the expression of parkinsonian motor symptoms.117 When symptoms do develop, one possibility is that the abnormal activity exceeds some threshold for generating symptoms; alternatively, the pathologic firing patterns may reflect an excessive compensatory process that can no longer suppress symptoms of the disorder. In PD patients performing complex movements, PET studies have demonstrated that in conjunction with hypoactivity along the striatofrontal pathways, the lateral premotor and inferolateral parietal regions become overactive,118 and as the sequence, duration, or complexity of the task expands, the overactivity progressively increases.119 The parietal cortex is thought to integrate sensory, motivational, and attentional inputs and could act as a compensatory pathway for generating sensory-guided movements in PD.120 In the case of DBS, swapping pathologic with regularized activity, even if it means disabling the majority of functional information the pathway encodes, could liberate other pathways to compensate for the remaining pathology in the network and the loss of function in the stimulated pathway. DBS may also facilitate more normal firing patterns in nuclei at varying distances downstream of the stimulated target or within the stimulated target at subthreshold distances. With STN-DBS, for instance, activity in the parietal cortex positively correlates with improvement in freezing of gait, whereas improvement in other motor signs correlates with metabolic increases in frontal cortical areas.121
For many anatomic targets, unilateral DBS can elicit bilateral therapeutic effects, and bilateral stimulation produces additional therapeutic benefit on the side affected by unilateral stimulation.122–124 These clinical observations indicate that at some level, unilateral DBS modulates neuronal activity on the contralateral side.125,126 Fukuda and colleagues127 reported that improvement in motor performance in PD patients following GPi-DBS correlated with bilateral changes in regional cerebral blood flow in the supplementary motor cortex and cerebellum. This group also found bilateral changes in regional cerebral blood flow in the cerebellum with VIM-DBS in patients with severe tremor.128 PET studies have shown that STN-DBS produces a marked metabolic decrease in the contralateral GPi, which may facilitate activity in ventrolateral thalamic areas implicated in sensorimotor performance.124 In addition to the contralateral GPi, neuronal activity in the contralateral STN is inhibited immediately following cessation of unilateral STN-DBS.129 Pallidothalamic axons send collaterals to the contralateral VOA, VOP, centromedian-parafascicular complex, and PPN. STN neurons have been found to project to the contralateral centromedian nucleus and globus pallidus, the latter of which could explain the decrease in contralateral STN activity observed with ipsilateral STN-DBS.130 The ipsilateral cerebellum also has connections with the striatum through the parafascicular nucleus of the thalamus.131 The bilateral effects of unilateral DBS may also result from bilateral PPN projections or higher level corticocortical and corticothalamic cross-connectivity.
Therapeutic Latencies
DBS can induce both short- and long-term alterations in network activity (Fig. 83-5). These changes are embodied in the temporal latency between the initiation of stimulation and full therapeutic benefit and the persistent therapeutic effect once stimulation is ended.132,133 PD patients with DBS implants, for instance, often find that improvement in axial symptoms takes hours to manifest completely, whereas upper limb tremor fades away within seconds.134,135 A similar symptom-dependent disparity has been observed for dystonia patients. Krauss and colleagues57 reported that DBS improves phasic dystonic movements within minutes of stimulation onset, whereas relief from tonic posturing requires several months to take full effect. Similarly, Houeto and colleagues69 reported that improvement in tic severity in patients with Tourette’s syndrome took several weeks after the initiation of DBS, with the benefit lasting at least a month after discontinuation of stimulation.
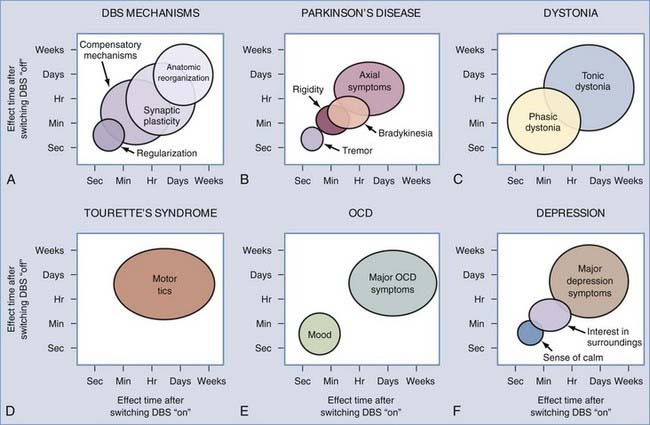
These observations suggest that multiple network changes are occurring within the brain over different time frames. Correcting abnormal firing rates or patterns may occur within seconds of DBS onset, but structural changes such as synapse modification or axonal sprouting or pruning likely require much longer. In brain slices, high-frequency stimulation in the STN was found to induce three types of plastic changes at glutamatergic synapses: (1) short-term potentiation of the evoked postsynaptic current, which increased glutamate release from the presynaptic terminals over a 5-minute period; (2) long-term potentiation of the evoked postsynaptic current, indicative of changes in postsynaptic protein expression that required more than 30 minutes; and (3) long-term depression of the evoked postsynaptic current, which modified presynaptic regulation over a period exceeding 30 minutes. Determining whether these changes are also important in vivo will require further experimental investigation. Future studies are also needed to investigate the physiologic mechanisms underlying the neural responses to DBS that evolve over milliseconds to seconds.90,93
There is mounting evidence that the mechanisms underlying the therapeutic latency may depend on the DBS target and parameters of stimulation. Vitek and colleagues13 reported that improvement in bradykinesia in PD patients occurred within seconds for GPe-DBS, but the equivalent therapeutic effects took half a minute or more for GPi-DBS (and they were often preceded by a period of aggravated symptoms). In that study, GPe-DBS sometimes induced dyskinesias that started in the hand and spread from the upper extremity to the leg over minutes. Wu and associates80 reported that the latency of onset for dyskinesia suppression during GPi-DBS decreased from 5 seconds to 1 second as the frequency of stimulation increased from 80 to 100 Hz. Higher frequency stimulation (185 versus 135 Hz) also appeared to decrease the therapeutic latency for rest tremor in PD patients, which may reflect a temporal tightening of the downstream responses to the stimulation with higher frequency.94 In the case of motor signs that take days to weeks to improve following the onset of stimulation and worsen after the cessation of stimulation, the mechanisms underlying these delays and the effects of varying stimulation parameter settings remain to be determined.
Long-Term Effects
In some patients, symptoms take many months to return after a period of chronic stimulation, whereas in others, the same symptoms reappear almost immediately after turning off the stimulator. In a series of tremor patients with VIM-DBS implants reported by Lozano,136 40% experienced a significant reduction in tremor by the mere implantation of the lead, with stimulation adding to the benefit, a finding that suggests a “lesion effect” in the VIM nucleus during the surgical process. Another 30% of patients responded well to DBS initially but eventually developed tolerance to the treatment. A number of possibilities can explain the results observed in these patients, including plasticity in motor cortical regions targeted by VIM nucleus projections; generation of tremor rhythms through alternative pathways, such as the basal ganglia–thalamocortical network, that VIM-DBS cannot control; and mechanical failure of the implant. The remainder of patients (30%) showed substantial benefit with DBS that persisted even after the stimulator was turned off. A similar phenomenon has been observed in dystonia patients with GPi-DBS, some of whom continued to be symptom free only to discover that the stimulator battery had been drained for months (Vitek, personal communication). Although there is a possibility that the stimulation therapy inadvertently created a lesion in the tissue surrounding the electrode contacts in these cases, postmortem histology around DBS leads generally does not support an ablative effect.137,138
Following the cessation of DBS, restoration of abnormal synchronization within the brain is likely to be a fast process. One explanation for the long-term persistence of benefit is that DBS enables or facilitates an anatomic reorganization of the networks it affects. Yianni and colleagues133 posited that the seemingly pathologic low-frequency oscillations observed in patients with tremor, familial myoclonic dystonia, parkinsonian dyskinesias, and multiple sclerosis promoted axonal sprouting. Long-term DBS that disrupts these rhythms could reverse these connections and thus allow the affected networks to resynchronize on normal rhythms. In support of this hypothesis, a histologic study showed that thalamocortical neurons exhibit reorganized afferent connections for at least 1 year after lesioning the GPi and SNr,139 with different effects on each GABAergic subtype.140 Because long-term therapeutic effects can differ among patients, it is possible that axonal sprouting or pruning, as well as synaptic plasticity, may be more tractable in certain patients or certain DBS targets.
Other groups have hypothesized that DBS, particularly in the STN, may be neuroprotective.141,142 In monkeys, several weeks of continuous high-frequency stimulation in the STN, preceding or following systemic injection of the dopaminergic neurotoxin 1-methyl, 4-phenyl, 1,2,3,6-tetrahydropyridine (MPTP), resulted in 20% more tyrosine hydroxylase–positive reactivity (a signal for dopamine content) in the ipsilateral SNc.142 Likewise, STN lesions with kainic acid were shown to shield dopaminergic SNc neurons during subsequent injections of MPTP. Surgical ablation of the STN following injection of MPTP could also recover damaged dopaminergic neurons in the SNc. However, when the DBS implant or the kainic acid injection volume extended into fiber pathways around the STN, there was a notable reduction in dopaminergic reactivity, which may indicate that nigrostriatal axotomy occurred.142 Although it is possible that, as the authors suggested, these results derived from a reduction of glutamate excitotoxicity in the SNc, other mechanisms may be more congruent with the increased STN output with STN-DBS,94,143 including the release of neurotrophic factors or stimulation of GABAergic fibers of passage that innervate the SNc.
Conclusion
Unlike the immutable effects of surgical ablation, DBS is reversible. It can be performed bilaterally with relative safety, and the treatment parameters can be tailored on an ongoing basis to optimize the clinical effect. Contrary to initial hypotheses, the physiologic mechanisms underlying the responses to ablation and DBS appear to be significantly different. A growing number of electrophysiologic, biochemical, imaging, and computer modeling studies now suggest that DBS elicits dissociated effects between somatic and axonal elements. Current hypotheses on the mechanisms of DBS point toward overdriving output to downstream nuclei, thereby regularizing their activity and reorganizing network activity in a fashion that enables other pathways to compensate for the loss of function in the stimulated pathway. Remarkably, the same anatomic targets have proved efficacious for multiple neurological disorders, and the stimulation of different anatomic targets can generate similar therapeutic benefits for the same disorder. These observations emphasize the concept of network dysfunction in neurological disorders and point toward additional targets that may treat symptoms more effectively. Developing a better understanding of the neural processes underlying the therapeutic responses to DBS is critical for improving efficacy, expediting programming sessions, and accelerating the development of this therapy. DBS is poised for several important advances that will likely increase its efficacy and reliability on a patient-by-patient basis (Table 83-2), but many of these innovations require a better understanding of the mechanisms by which DBS generates its therapeutic benefit.
TABLE 83-2 Future Advances in Deep Brain Stimulation Therapies
| TOPIC | ADVANCE | OUTCOME |
|---|---|---|
| Refined targeting | Higher-resolution imaging, computer models for predicting volumes of activation with a particular set of stimulation parameters | More effective therapy with fewer side effects, faster programming sessions |
| New anatomic targets | PPN for improving freezing of gait in PD patients, STN for better dystonia management, GPe for faster therapeutic onset in PD and dystonia patients | Treatment of symptoms that are currently resistant to DBS in existing targets |
| New indications | Treatment of Alzheimer’s disease, schizophrenia, addiction, minimally conscious state, pain, stroke and traumatic brain injury rehabilitation, migraines, tinnitus, eating disorders, obesity | Therapy option for patients with untreated, medication-refractory neurological disorders |
| Current- versus voltage-controlled stimulation | Current-controlled IPGs | Less affected by impedance changes at electrode-tissue interface, fewer programming sessions |
| Current steering | Independent-channel IPGs, DBS leads with segmented electrode contacts | More effective therapy with fewer side effects requiring fewer reimplantation procedures |
| Battery life | IPGs with rechargeable batteries | No need to replace IPG every 3-5 yr—especially important for new indications involving very high current intensities, such as OCD and dystonia |
| Adaptive closed-loop systems | Identification of changes in neural activity that correlate with improvement in disease symptoms | Ability to titrate therapy according to patient’s behavior, mood, and brain state |
| Alternatives to electrical stimulation | Optical stimulation, transcranial magnetic stimulation | Ability to induce reversible excitation or inhibition of a targeted pathway with less invasive approaches |
DBS, deep brain stimulation; GPe, external globus pallidus; IPG, implantable pulse generator; OCD, obsessive-compulsive disorder; PD, Parkinson’s disease; PPN, pedunculopontine nucleus; STN, subthalamic nucleus.
Benabid A, Piallat B, Wallace B, et al. Might deep brain stimulation of the subthalamic nucleus be neuroprotective in patients with Parkinson’s disease? Thal Relat Syst. 2003;2:95-102.
Benabid AL, Pollak P, Louveau A, et al. Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl Neurophysiol. 1987;50:344-346.
Dostrovsky JO, Levy R, Wu JP, et al. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J Neurophysiol. 2000;84:570-574.
Gildenberg PL. Evolution of neuromodulation. Stereotact Funct Neurosurg. 2005;83:71.
Grill WM, Snyder AN, Miocinovic S. Deep brain stimulation creates an informational lesion of the stimulated nucleus. Neuroreport. 2004;15:1137-1140.
Hashimoto T, Elder CM, Okun MS, et al. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci. 2003;23:1916-1923.
Hassler R, Riechert T, Mundinger F, et al. Physiological observations in stereotaxic operations in extrapyramidal motor disturbances. Brain. 1960;83:337-350.
Kringelbach ML, Jenkinson N, Owen SL, et al. Translational principles of deep brain stimulation. Nat Rev Neurosci. 2007;8:623.
Maks CB, Butson CR, Walter BL, et al. Deep brain stimulation activation volumes and their association with neurophysiological mapping and therapeutic outcomes. J Neurol Neurosurg Psychiatry. 2009;80:659-666.
McIntyre CC, Grill WM, Sherman DL, et al. Cellular effects of deep brain stimulation: model-based analysis of activation and inhibition. J Neurophysiol. 2004;91:1457-1469.
Meissner W, Leblois A, Hansel D, et al. Subthalamic high frequency stimulation resets subthalamic firing and reduces abnormal oscillations. Brain. 2005;128:2372-2382.
Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50:381.
Miocinovic S, Parent M, Butson CR, et al. Computational analysis of subthalamic nucleus and lenticular fasciculus activation during therapeutic deep brain stimulation. J Neurophysiol. 2006;96:1569-1580.
Montgomery E. Dynamically coupled, high-frequency reentrant, non-linear oscillators embedded in scale-free basal ganglia-thalamic-cortical networks mediating function and deep brain stimulation effects. Nonlinear Studies. 2004;11:385.
Nowak LG, Bullier J. Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. II. Evidence from selective inactivation of cell bodies and axon initial segments. Exp Brain Res. 1998;118:489-500.
Parent M, Parent A. Axonal collateralization in primate basal ganglia and related thalamic nuclei. Thal Relat Syst. 2002;2:71-86.
Perlmutter JS, Mink JW. Deep brain stimulation. Annu Rev Neurosci. 2006;29:229.
Siegfried J, Lippitz B. Chronic electrical stimulation of the VL-VPL complex and of the pallidum in the treatment of movement disorders: personal experience since 1982. Stereotact Funct Neurosurg. 1994;62:71-75.
Spiegel EA, Wycis HT, Marks M, et al. Stereotaxic apparatus for operations on the human brain. Science. 1947;106:349-350.
Toth S, Tomka I. Responses of the human thalamus and pallidum to high frequency stimulations. Confin Neurol. 1968;30:17-40.
1 Spiegel EA, Wycis HT, Marks M, Lee AJ. Stereotaxic apparatus for operations on the human brain. Science. 1947;106(2754):349-350.
2 Horsley V, Clarke RH. The structure and functions of the cerebellum examined by a new method. Brain. 1908;31(1):45-124.
3 Albe-Fessard D, Arfel G, Guiot G, et al. Identification et délimitation precise de cértaines structures souscorticales de l’homme par l’electro-physiologie. CR Acad Sci (Paris). 1961;243:2412-2414.
4 Hassler R, Riechert T, Mundinger F, et al. Physiological observations in stereotaxic operations in extrapyramidal motor disturbances. Brain. 1960;83:337-350.
5 Alberts WW, Feinstein B, Levin G, Wright EWJr. Electrical stimulation of therapeutic targets in waking dyskinetic patients. Electroencephalogr Clin Neurophysiol. 1966;20(6):559-566.
6 Bergstrom MR, Johansson GG, Laitinen LV, Sipponen P. Electrical stimulation of the thalamic and subthalamic area in cerebral palsy. Acta Physiol Scand. 1966;67(2):208-213.
7 Spiegel EA, Wycis HT, Szekely EG, et al. Stimulation of Forel’s field during stereotaxic operations in the human brain. Electroencephalogr Clin Neurophysiol. 1964;16:537-548.
8 Toth S, Tomka I. Responses of the human thalamus and pallidum to high frequency stimulations. Confin Neurol. 1968;30(1):17-40.
9 Bechtereva NP, Bondartchuk AN, Smirnov VM. Therapeutic electrostimulations of the deep brain structures. Vopr Neirokhir. 1972;1:7-12.
10 Benabid AL, Pollak P, Louveau A, et al. Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl Neurophysiol. 1987;50(1-6):344-346.
11 Siegfried J, Lippitz B. Chronic electrical stimulation of the VL-VPL complex and of the pallidum in the treatment of movement disorders: personal experience since 1982. Stereotact Funct Neurosurg. 1994;62(1-4):71-75.
12 Volkmann J, Moro E, Pahwa R. Basic algorithms for the programming of deep brain stimulation in Parkinson’s disease. Mov Disord. 2006;21(Suppl 14):S284-S289.
13 Vitek JL, Hashimoto T, Peoples J, et al. Acute stimulation in the external segment of the globus pallidus improves parkinsonian motor signs. Mov Disord. 2004;19(8):907-915.
14 Zhang J, Russo GS, Mewes K, et al. Lesions in monkey globus pallidus externus exacerbate parkinsonian symptoms. Exp Neurol. 2006;199(2):446-453.
15 Mayberg HS, Brannan SK, Mahurin RK, et al. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. 1997;8(4):1057-1061.
16 Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-660.
17 McIntyre CC, Grill WM, Sherman DL, Thakor NV. Cellular effects of deep brain stimulation: model-based analysis of activation and inhibition. J Neurophysiol. 2004;91(4):1457-1469.
18 Miocinovic S, Parent M, Butson CR, et al. Computational analysis of subthalamic nucleus and lenticular fasciculus activation during therapeutic deep brain stimulation. J Neurophysiol. 2006;96(3):1569-1580.
19 Benabid AL, Pollak P, Gervason C, et al. Long-term suppression of tremor by chronic stimulation of the ventral intermediate thalamic nucleus. Lancet. 1991;337(8738):403-406.
20 Benabid AL, Lebas JF, Grand S, et al. Deep brain stimulation for movement disorders. In: Winn HR, editor. Youmans Neurological Surgery. 5th ed. Philadelphia: Saunders; 2004:2803.
21 Garonzik IM, Hua SE, Ohara S, Lenz FA. Intraoperative microelectrode and semi-microelectrode recording during the physiological localization of the thalamic nucleus ventral intermediate. Mov Disord. 2002;17(Suppl 3):S135-S144.
22 Hoover JE, Strick PL. The organization of cerebellar and basal ganglia outputs to primary motor cortex as revealed by retrograde transneuronal transport of herpes simplex virus type 1. J Neurosci. 1999;19(4):1446-1463.
23 Strick PL. How do the basal ganglia and cerebellum gain access to the cortical motor areas? Behav Brain Res. 1985;18(2):107-123.
24 Hua SE, Lenz FA, Zirh TA, et al. Thalamic neuronal activity correlated with essential tremor. J Neurol Neurosurg Psychiatry. 1998;64(2):273-276.
25 Jenkins IH, Bain PG, Colebatch JG, et al. A positron emission tomography study of essential tremor: evidence for overactivity of cerebellar connections. Ann Neurol. 1993;34(1):82-90.
26 Hallett M, Dubinsky RM. Glucose metabolism in the brain of patients with essential tremor. J Neurol Sci. 1993;114(1):45-48.
27 Lyons KE, Pahwa R. Deep brain stimulation and essential tremor. J Clin Neurophysiol. 2004;21(1):2-5.
28 Benabid AL, Pollak P, Seigneuret E, et al. Chronic VIM thalamic stimulation in Parkinson’s disease, essential tremor and extra-pyramidal dyskinesias. Acta Neurochir Suppl (Wien). 1993;58:39-44.
29 Lozano AM. Vim thalamic stimulation for tremor. Arch Med Res. 2000;31(3):266-269.
30 Hirai T, Miyazaki M, Nakajima H, et al. The correlation between tremor characteristics and the predicted volume of effective lesions in stereotaxic nucleus ventralis intermedius thalamotomy. Brain. 1983;106(Pt 4):1001-1018.
31 Vitek JL, Ashe J, DeLong MR, Alexander GE. Physiologic properties and somatotopic organization of the primate motor thalamus. J Neurophysiol. 1994;71(4):1498-1513.
32 Foote KD, Okun MS. Ventralis intermedius plus ventralis oralis anterior and posterior deep brain stimulation for posttraumatic Holmes tremor: two leads may be better than one: technical note. Neurosurgery. 2005;56(2 Suppl):E445.
33 Boraud T, Bezard E, Bioulac B, Gross CE. From single extracellular unit recording in experimental and human parkinsonism to the development of a functional concept of the role played by the basal ganglia in motor control. Prog Neurobiol. 2002;66(4):265-283.
34 Butson CR, Cooper SE, Henderson JM, McIntyre CC. Patient-specific analysis of the volume of tissue activated during deep brain stimulation. Neuroimage. 2007;34(2):661-670.
35 Maks CB, Butson CR, Walter BL, et al. Deep brain stimulation activation volumes and their association with neurophysiological mapping and therapeutic outcomes. J Neurol Neurosurg Psychiatry. 2009;80:659-666.
36 Plaha P, Ben-Shlomo Y, Patel NK, Gill SS. Stimulation of the caudal zona incerta is superior to stimulation of the subthalamic nucleus in improving contralateral parkinsonism. Brain. 2006;129(Pt 7):1732-1747.
37 Stover NP, Okun MS, Evatt ML, et al. Stimulation of the subthalamic nucleus in a patient with Parkinson disease and essential tremor. Arch Neurol. 2005;62(1):141-143.
38 Cossette M, Levesque M, Parent A. Extrastriatal dopaminergic innervation of human basal ganglia. Neurosci Res. 1999;34(1):51-54.
39 Funkiewiez A, Ardouin C, Krack P, et al. Acute psychotropic effects of bilateral subthalamic nucleus stimulation and levodopa in Parkinson’s disease. Mov Disord. 2003;18(5):524-530.
40 Vitek JL. Deep brain stimulation for Parkinson’s disease. A critical re-evaluation of STN versus GPi DBS. Stereotact Funct Neurosurg. 2002;78(3-4):119-131.
41 Kumar R, Lang AE, Rodriguez-Oroz MC, et al. Deep brain stimulation of the globus pallidus pars interna in advanced Parkinson’s disease. Neurology. 2000;55(12 Suppl 6):S34-S39.
42 Yelnik J. Functional anatomy of the basal ganglia. Mov Disord. 2002;17(Suppl 3):S15-S21.
43 Hutchison WD, Lozano AM, Davis KD, et al. Differential neuronal activity in segments of globus pallidus in Parkinson’s disease patients. Neuroreport. 1994;5(12):1533-1537.
44 Bejjani B, Damier P, Arnulf I, et al. Pallidal stimulation for Parkinson’s disease. Two targets? Neurology. 1997;49(6):1564-1569.
45 Krack P, Pollak P, Limousin P, et al. Opposite motor effects of pallidal stimulation in Parkinson’s disease. Ann Neurol. 1998;43(2):180-192.
46 Yelnik J, Damier P, Bejjani BP, et al. Functional mapping of the human globus pallidus: contrasting effect of stimulation in the internal and external pallidum in Parkinson’s disease. Neuroscience. 2000;101(1):77-87.
47 Xu W, Miocinovic S, Zhang J, et al. Dissociation of motor symptoms during deep brain stimulation of the subthalamic nucleus in the region of the internal capsule. Paper presented at: Society for Neuroscience Conference; November 3-7, 2007; San Diego, CA. Poster 693.17.
48 Hoover JE, Strick PL. Multiple output channels in the basal ganglia. Science. 1993;259(5096):819-821.
49 Nambu A, Yoshida S, Jinnai K. Discharge patterns of pallidal neurons with input from various cortical areas during movement in the monkey. Brain Res. 1990;519(1-2):183-191.
50 Yoshida S, Nambu A, Jinnai K. The distribution of the globus pallidus neurons with input from various cortical areas in the monkeys. Brain Res. 1993;611(1):170-174.
51 Pahapill PA, Lozano AM. The pedunculopontine nucleus and Parkinson’s disease. Brain. 2000;123(Pt 9):1767-1783.
52 Lai YY, Siegel JM. Muscle tone suppression and stepping produced by stimulation of midbrain and rostral pontine reticular formation. J Neurosci. 1990;10(8):2727-2734.
53 Garcia-Rill E, Houser CR, Skinner RD, et al. Locomotion-inducing sites in the vicinity of the pedunculopontine nucleus. Brain Res Bull. 1987;18(6):731-738.
54 Stefani A, Lozano AM, Peppe A, et al. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain. 2007;130(Pt 6):1596-1607.
55 Zrinzo L, Zrinzo LV, Tisch S, et al. Stereotactic localization of the human pedunculopontine nucleus: atlas-based coordinates and validation of a magnetic resonance imaging protocol for direct localization. Brain. 2008;131(Pt 6):1588-1598.
56 Weinberger M, Hamani C, Hutchison WD, et al. Pedunculopontine nucleus microelectrode recordings in movement disorder patients. Exp Brain Res. 2008;188(2):165-174.
57 Krauss JK, Yianni J, Loher TJ, Aziz TZ. Deep brain stimulation for dystonia. J Clin Neurophysiol. 2004;21(1):18-30.
58 Starr PA, Rau GM, Davis V, et al. Spontaneous pallidal neuronal activity in human dystonia: comparison with Parkinson’s disease and normal macaque. J Neurophysiol. 2005;93(6):3165-3176.
59 Vitek JL, Chockkan V, Zhang JY, et al. Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann Neurol. 1999;46(1):22-35.
60 Tasker RR, Doorly T, Yamashiro K. Thalamotomy in generalized dystonia. Adv Neurol. 1988;50:615-631.
61 Cooper IS. Effect of thalamic lesions upon torticollis. N Engl J Med. 1964;270:567-572.
62 Vercueil L, Pollak P, Fraix V, et al. Deep brain stimulation in the treatment of severe dystonia. J Neurol. 2001;248(8):695-700.
63 Trottenberg T, Volkmann J, Deuschl G, et al. Treatment of severe tardive dystonia with pallidal deep brain stimulation. Neurology. 2005;64(2):344-346.
64 Kupsch A, Kuehn A, Klaffke S, et al. Deep brain stimulation in dystonia. J Neurol. 2003;250(Suppl 1):I47-I52.
65 Pagni CA, Albanese A, Bentivoglio A, et al. Results by motor cortex stimulation in treatment of focal dystonia, Parkinson’s disease and post-ictal spasticity. The experience of the Italian Study Group of the Italian Neurosurgical Society. Acta Neurochir Suppl. 2008;101:13-21.
66 Sun B, Chen S, Zhan S, et al. Subthalamic nucleus stimulation for primary dystonia and tardive dystonia. Acta Neurochir Suppl. 2007;97(Pt 2):207-214.
67 Vandewalle V, van der Linden C, Groenewegen HJ, Caemaert J. Stereotactic treatment of Gilles de la Tourette syndrome by high frequency stimulation of thalamus. Lancet. 1999;353(9154):724.
68 Hassler R, Dieckmann G. [Stereotaxic treatment of tics and inarticulate cries or coprolalia considered as motor obsessional phenomena in Gilles de la Tourette’s disease]. Rev Neurol (Paris). 1970;123(2):89-100.
69 Houeto JL, Karachi C, Mallet L, et al. Tourette’s syndrome and deep brain stimulation. J Neurol Neurosurg Psychiatry. 2005;76(7):992-995.
70 Welter ML, Mallet L, Houeto JL, et al. Internal pallidal and thalamic stimulation in patients with Tourette syndrome. Arch Neurol. 2008;65(7):952-957.
71 Ackermans L, Temel Y, Cath D, et al. Deep brain stimulation in Tourette’s syndrome: two targets? Mov Disord. 2006;21(5):709-713.
72 Diederich NJ, Kalteis K, Stamenkovic M, et al. Efficient internal pallidal stimulation in Gilles de la Tourette syndrome: a case report. Mov Disord. 2005;20(11):1496-1499.
73 Nuttin B, Cosyns P, Demeulemeester H, et al. Electrical stimulation in anterior limbs of internal capsules in patients with obsessive-compulsive disorder. Lancet. 1999;354(9189):1526.
74 Vitek JL. Mechanisms of deep brain stimulation: excitation or inhibition. Mov Disord. 2002;17(Suppl 3):S69-S72.
75 Nowak LG, Bullier J. Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. I. Evidence from chronaxie measurements. Exp Brain Res. 1998;118(4):477-488.
76 Nowak LG, Bullier J. Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. II. Evidence from selective inactivation of cell bodies and axon initial segments. Exp Brain Res. 1998;118(4):489-500.
77 Filali M, Hutchison WD, Palter VN, et al. Stimulation-induced inhibition of neuronal firing in human subthalamic nucleus. Exp Brain Res. 2004;156(3):274-281.
78 Welter ML, Houeto JL, Bonnet AM, et al. Effects of high-frequency stimulation on subthalamic neuronal activity in parkinsonian patients. Arch Neurol. 2004;61(1):89-96.
79 Dostrovsky JO, Levy R, Wu JP, et al. Microstimulation-induced inhibition of neuronal firing in human globus pallidus. J Neurophysiol. 2000;84(1):570-574.
80 Wu YR, Levy R, Ashby P, et al. Does stimulation of the GPi control dyskinesia by activating inhibitory axons? Mov Disord. 2001;16(2):208-216.
81 Benazzouz A, Piallat B, Pollak P, Benabid AL. Responses of substantia nigra pars reticulata and globus pallidus complex to high frequency stimulation of the subthalamic nucleus in rats: electrophysiological data. Neurosci Lett. 1995;189(2):77-80.
82 Beurrier C, Bioulac B, Audin J, Hammond C. High-frequency stimulation produces a transient blockade of voltage-gated currents in subthalamic neurons. J Neurophysiol. 2001;85(4):1351-1356.
83 Shin DS, Samoilova M, Cotic M, et al. High frequency stimulation or elevated K+ depresses neuronal activity in the rat entopeduncular nucleus. Neuroscience. 2007;149(1):68-86.
84 Anderson TR, Hu B, Iremonger K, Kiss ZH. Selective attenuation of afferent synaptic transmission as a mechanism of thalamic deep brain stimulation-induced tremor arrest. J Neurosci. 2006;26(3):841-850.
85 Dostrovsky JO, Lozano AM. Mechanisms of deep brain stimulation. Mov Disord. 2002;17(Suppl 3):S63-S68.
86 Magarinos-Ascone C, Pazo JH, Macadar O, Buno W. High-frequency stimulation of the subthalamic nucleus silences subthalamic neurons: a possible cellular mechanism in Parkinson’s disease. Neuroscience. 2002;115(4):1109-1117.
87 Meissner W, Leblois A, Hansel D, et al. Subthalamic high frequency stimulation resets subthalamic firing and reduces abnormal oscillations. Brain. 2005;128(Pt 10):2372-2382.
88 Shink E, Smith Y. Differential synaptic innervation of neurons in the internal and external segments of the globus pallidus by the GABA- and glutamate-containing terminals in the squirrel monkey. J Comp Neurol. 1995;358(1):119-141.
89 Agnesi F, Lin J, Goerss SJ, et al. Resolving local extracellular unit activity during deep brain stimulation at a site of stimulation in human patients. Soc Neurosci Abstr. 414(18), 2007.
90 Bar-Gad I, Elias S, Vaadia E, Bergman H. Complex locking rather than complete cessation of neuronal activity in the globus pallidus of a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primate in response to pallidal microstimulation. J Neurosci. 2004;24(33):7410-7419.
91 Tai CH, Boraud T, Bezard E, et al. Electrophysiological and metabolic evidence that high-frequency stimulation of the subthalamic nucleus bridles neuronal activity in the subthalamic nucleus and the substantia nigra reticulata. FASEB J. 2003;17(13):1820-1830.
92 Johnson MD, McIntyre CC. Quantifying the neural elements activated and inhibited by globus pallidus deep brain stimulation. J Neurophysiol. 2008;100:2549-2563.
93 Garcia L, D’Alessandro G, Fernagut PO, et al. Impact of high-frequency stimulation parameters on the pattern of discharge of subthalamic neurons. J Neurophysiol. 2005;94(6):3662-3669.
94 Hashimoto T, Elder CM, Okun MS, et al. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J Neurosci. 2003;23(5):1916-1923.
95 Zhang J, Russo GS, Chen XX, et al. Deep brain stimulation of monkey globus pallidus externus in experimental parkinsonism. Paper presented at: Society for Neuroscience Conference; November 8-12; 2003; New Orleans, LA. Poster 273.12.
96 Anderson ME, Postupna N, Ruffo M. Effects of high-frequency stimulation in the internal globus pallidus on the activity of thalamic neurons in the awake monkey. J Neurophysiol. 2003;89(2):1150-1160.
97 Montgomery EBJr. Effects of GPi stimulation on human thalamic neuronal activity. Clin Neurophysiol. 2006;117(12):2691-2702.
98 Pralong E, Debatisse D, Maeder M, et al. Effect of deep brain stimulation of GPI on neuronal activity of the thalamic nucleus ventralis oralis in a dystonic patient. Neurophysiol Clin. 2003;33(4):169-173.
99 Parent M, Parent A. Axonal collateralization in primate basal ganglia and related thalamic nuclei. Thal Relat Syst. 2002;2:71-86.
100 Sato F, Lavallee P, Levesque M, Parent A. Single-axon tracing study of neurons of the external segment of the globus pallidus in primate. J Comp Neurol. 2000;417(1):17-31.
101 Sato F, Parent M, Levesque M, Parent A. Axonal branching pattern of neurons of the subthalamic nucleus in primates. J Comp Neurol. 2000;424(1):142-152.
102 Charara A, Parent A. Brainstem dopaminergic, cholinergic and serotoninergic afferents to the pallidum in the squirrel monkey. Brain Res. 1994;640(1-2):155-170.
103 Montgomery EBJr, Baker KB. Mechanisms of deep brain stimulation and future technical developments. Neurol Res. 2000;22(3):259-266.
104 Alterman RL, Shils JL, Miravite J, Tagliati M. Lower stimulation frequency can enhance tolerability and efficacy of pallidal deep brain stimulation for dystonia. Mov Disord. 2007;22(3):366-368.
105 Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72(2):507-520.
106 Magnin M, Morel A, Jeanmonod D. Single-unit analysis of the pallidum, thalamus and subthalamic nucleus in parkinsonian patients. Neuroscience. 2000;96(3):549-564.
107 Brown P, Oliviero A, Mazzone P, et al. Dopamine dependency of oscillations between subthalamic nucleus and pallidum in Parkinson’s disease. J Neurosci. 2001;21(3):1033-1038.
108 Kuhn AA, Kempf F, Brucke C, et al. High-frequency stimulation of the subthalamic nucleus suppresses oscillatory beta activity in patients with Parkinson’s disease in parallel with improvement in motor performance. J Neurosci. 2008;28(24):6165-6173.
109 Brown P, Mazzone P, Oliviero A, et al. Effects of stimulation of the subthalamic area on oscillatory pallidal activity in Parkinson’s disease. Exp Neurol. 2004;188(2):480-490.
110 Xu W, Russo G, Hashimoto T, et al. Subthalamic nucleus stimulation modulates thalamic neuronal activity. J Neurosci. 2008;28:11916-11924.
111 Liu X, Yianni J, Wang S, et al. Different mechanisms may generate sustained hypertonic and rhythmic bursting muscle activity in idiopathic dystonia. Exp Neurol. 2006;198(1):204-213.
112 Sharott A, Grosse P, Kuhn AA, et al. Is the synchronization between pallidal and muscle activity in primary dystonia due to peripheral afferance or a motor drive? Brain. 2008;131(Pt 2):473-484.
113 Gray CM. Synchronous oscillations in neuronal systems: mechanisms and functions. J Comput Neurosci. 1994;1(1-2):11-38.
114 Brown P. Bad oscillations in Parkinson’s disease. J Neural Transm Suppl. 2006;70:27-30.
115 Grill WM, Snyder AN, Miocinovic S. Deep brain stimulation creates an informational lesion of the stimulated nucleus. Neuroreport. 2004;15(7):1137-1140.
116 Eidelberg D, Moeller JR, Antonini A, et al. Functional brain networks in DYT1 dystonia. Ann Neurol. 1998;44(3):303-312.
117 Bezard E, Boraud T, Bioulac B, Gross CE. Involvement of the subthalamic nucleus in glutamatergic compensatory mechanisms. Eur J Neurosci. 1999;11(6):2167-2170.
118 Samuel M, Ceballos-Baumann AO, Blin J, et al. Evidence for lateral premotor and parietal overactivity in Parkinson’s disease during sequential and bimanual movements. A PET study. Brain. 1997;120(Pt 6):963-976.
119 Catalan MJ, Ishii K, Honda M, et al. A PET study of sequential finger movements of varying length in patients with Parkinson’s disease. Brain. 1999;122(Pt 3):483-495.
120 Ceballos-Baumann AO. Functional imaging in Parkinson’s disease: activation studies with PET, fMRI and SPECT. J Neurol. 2003;250(Suppl 1):I15-I23.
121 Lyoo CH, Aalto S, Rinne JO, et al. Different cerebral cortical areas influence the effect of subthalamic nucleus stimulation on parkinsonian motor deficits and freezing of gait. Mov Disord. 2007;22(15):2176-2182.
122 Kumar R, Lozano AM, Sime E, et al. Comparative effects of unilateral and bilateral subthalamic nucleus deep brain stimulation. Neurology. 1999;53(3):561-566.
123 Chung SJ, Jeon SR, Kim SR, et al. Bilateral effects of unilateral subthalamic nucleus deep brain stimulation in advanced Parkinson’s disease. Eur Neurol. 2006;56(2):127-132.
124 Arai N, Yokochi F, Ohnishi T, et al. Mechanisms of unilateral STN-DBS in patients with Parkinson’s disease: a PET study. J Neurol. 2008;255:1236-1243.
125 Alberts JL, Okun MS, Vitek JL. The persistent effects of unilateral pallidal and subthalamic deep brain stimulation on force control in advanced Parkinson’s patients. Parkinsonism Relat Disord. 2008;14:481-488.
126 Tabbal SD, Ushe M, Mink JW, et al. Unilateral subthalamic nucleus stimulation has a measurable ipsilateral effect on rigidity and bradykinesia in Parkinson disease. Exp Neurol. 2008;211(1):234-242.
127 Fukuda M, Mentis M, Ghilardi MF, et al. Functional correlates of pallidal stimulation for Parkinson’s disease. Ann Neurol. 2001;49(2):155-164.
128 Fukuda M, Barnes A, Simon ES, et al. Thalamic stimulation for parkinsonian tremor: correlation between regional cerebral blood flow and physiological tremor characteristics. Neuroimage. 2004;21(2):608-615.
129 Walker HC, Guthrie BL, Guthrie SL, et al. Subthalamic neuronal activity is altered by contralateral deep brain stimulation in Parkinson’s disease. Paper presented at: Movement Disorders Society Conference; June 22-26, 2008; Chicago, IL. Poster 318.
130 Castle M, Aymerich MS, Sanchez-Escobar C, et al. Thalamic innervation of the direct and indirect basal ganglia pathways in the rat: Ipsi- and contralateral projections. J Comp Neurol. 2005;483(2):143-153.
131 Hoshi E, Tremblay L, Feger J, et al. The cerebellum communicates with the basal ganglia. Nat Neurosci. 2005;8(11):1491-1493.
132 Temperli P, Ghika J, Villemure JG, et al. How do parkinsonian signs return after discontinuation of subthalamic DBS? Neurology. 2003;60(1):78-81.
133 Yianni J, Bain PG, Gregory RP, et al. Post-operative progress of dystonia patients following globus pallidus internus deep brain stimulation. Eur J Neurol. 2003;10(3):239-247.
134 Gross C, Rougier A, Guehl D, et al. High-frequency stimulation of the globus pallidus internalis in Parkinson’s disease: a study of seven cases. J Neurosurg. 1997;87(4):491-498.
135 Hristova A, Lyons K, Troster AI, et al. Effect and time course of deep brain stimulation of the globus pallidus and subthalamus on motor features of Parkinson’s disease. Clin Neuropharmacol. 2000;23(4):208-211.
136 Lozano A. Deep brain stimulation: challenges to integrating stimulation technology with human neurobiology, neuroplasticity, and neural repair. J Rehabil Res Dev. 2001;38(6):x-xix.
137 Haberler C, Alesch F, Mazal PR, et al. No tissue damage by chronic deep brain stimulation in Parkinson’s disease. Ann Neurol. 2000;48(3):372-376.
138 Nielsen MS, Bjarkam CR, Sorensen JC, et al. Chronic subthalamic high-frequency deep brain stimulation in Parkinson’s disease—a histopathological study. Eur J Neurol. 2007;14(2):132-138.
139 Kultas-Ilinsky K, De Boom T, Ilinsky IA. Synaptic reorganization in the feline ventral anterior thalamic nucleus induced by lesions in the basal ganglia. Exp Neurol. 1992;116(3):312-329.
140 Ambardekar AV, Surin A, Parts K, et al. Distribution and binding parameters of GABAA receptors in the thalamic nuclei of Macaca mulatta and changes caused by lesioning in the globus pallidus and reticular thalamic nucleus. Neuroscience. 2003;118(4):1033-1043.
141 Benabid A, Piallat B, Wallace B, et al. Might deep brain stimulation of the subthalamic nucleus be neuroprotective in patients with Parkinson’s disease? Thal Relat Syst. 2003;2(2):95-102.
142 Wallace BA, Ashkan K, Heise CE, et al. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP-treated monkeys. Brain. 2007;130(Pt 8):2129-2145.
143 Windels F, Bruet N, Poupard A, et al. Influence of the frequency parameter on extracellular glutamate and gamma-aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in rats. J Neurosci Res. 2003;72(2):259-267.
144 Namba S, Wani T, Shimizu Y, et al. Sensory and motor responses to deep brain stimulation. Correlation with anatomical structures. J Neurosurg. 1985;63(2):224-234.
145 Bejjani BP, Damier P, Arnulf I, et al. Deep brain stimulation in Parkinson’s disease: opposite effects of stimulation in the pallidum. Mov Disord. 1998;13(6):969-970.
146 Bejjani BP, Damier P, Arnulf I, et al. Transient acute depression induced by high-frequency deep-brain stimulation. N Engl J Med. 1999;340(19):1476-1480.
147 Smeding HM, Goudriaan AE, Foncke EM, et al. Pathological gambling after bilateral subthalamic nucleus stimulation in Parkinson disease. J Neurol Neurosurg Psychiatry. 2007;78(5):517-519.
148 Saint-Cyr JA, Trepanier LL, Kumar R, et al. Neuropsychological consequences of chronic bilateral stimulation of the subthalamic nucleus in Parkinson’s disease. Brain. 2000;123(Pt 10):2091-2108.
149 Klostermann F, Ehlen F, Vesper J, et al. Effects of subthalamic deep brain stimulation on dysarthrophonia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2008;79(5):522-529.
150 Vitek JL, Bakay RA, Hashimoto T, et al. Microelectrode-guided pallidotomy: technical approach and its application in medically intractable Parkinson’s disease. J Neurosurg. 1998;88(6):1027-1043.
151 Anagnostou E, Sporer B, Steude U, et al. Contraversive eye deviation during deep brain stimulation of the globus pallidus internus. Neurology. 2001;56(10):1396-1399.
152 Montaurier C, Morio B, Bannier S, et al. Mechanisms of body weight gain in patients with Parkinson’s disease after subthalamic stimulation. Brain. 2007;130(Pt 7):1808-1818.
153 Bartholow R. Experimental investigations into the functions of the human brain. Am J Med Sci. 1874;67:305-313.
154 Spiegel EA, Wycis HT. Stereoencephalotomy. New York: Grune & Stratton; 1952.
155 Heath RG. Psychiatry. Annu Rev Med. 1954;5:223-236.
156 Pool JL. Psychosurgery in older people. J Am Geriatr Soc. 1954;2(7):456-466.
157 Hosobuchi Y, Adams JE, Rutkin B. Chronic thalamic stimulation for the control of facial anesthesia dolorosa. Arch Neurol. 1973;29(3):158-161.
158 Richardson DE, Akil H. Long term results of periventricular gray self-stimulation. Neurosurgery. 1977;1(2):199-202.
159 Benabid AL, Pollak P, Gross C, et al. Acute and long-term effects of subthalamic nucleus stimulation in Parkinson’s disease. Stereotact Funct Neurosurg. 1994;62(1-4):76-84.
160 Schiff ND, Giacino JT, Kalmar K, et al. Behavioural improvements with thalamic stimulation after severe traumatic brain injury. Nature. 2007;448(7153):600-603.






