[level-membership-for-pulmolory-and-respiratory-category]
Chapter 44 Cystic Fibrosis
Genetics
Cystic fibrosis is caused by mutations in a gene on chromosome 7 encoding the protein subsequently termed the CFTR gene. More than 1800 mutations have been reported to the Cystic Fibrosis Genetic Analysis Consortium. Most of these mutations are rare, and only four mutations occur in a frequency of more than 1%. CFTR mutations can be grouped into five classes: CFTR is not synthesized (I), is inadequately processed (II), is not regulated (III), shows abnormal conductance (IV), or has partially defective production or processing (V). Class I, II, and III mutations are more common and associated with pancreatic insufficiency, whereas patients with the less common class IV and V mutations often are “pancreatic sufficient” (Figure 44-1).
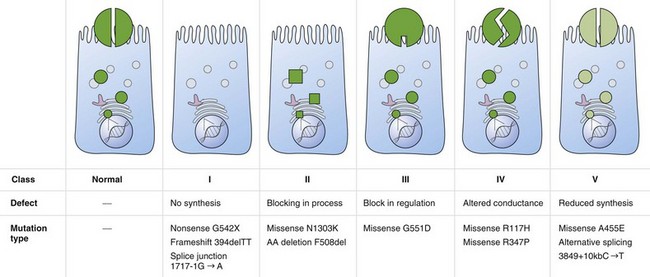
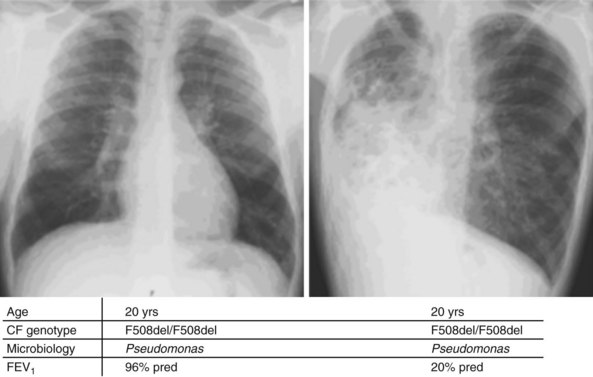
Whereas classes IV and V CFTR mutations are linked with pancreatic sufficiency, attempts to link specific mutations to the severity of lung disease have shown large phenotypic variability. This is best documented for patients homozygous for the F508del mutation who exhibit a wide spectrum in lung disease severity. This wide phenotypic variation suggests that environmental factors and genes other than CFTR influence the development, progression, and disease severity of CF (Figure 44-2).
Pathophysiology
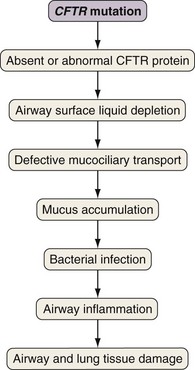
Airway epithelial cells secrete chloride and absorb sodium chloride (NaCl), the balance of which is regulated through apical channels, including CFTR (Figure 44-3). Ion secretion and absorption affect water transport, and a balance between secretion and absorption is thought to be important to maintain an adequate layer of airway surface liquid (ASL). The ASL supports the thin mucous layer on top of epithelial cells, which is constantly transported out of the lungs through ciliary movement. Lack or dysfunction of CFTR leads to reduced chloride secretion and NaCl hyperabsorption with depletion of ASL. In the absence of adequate ASL, respiratory cilia collapse, leading to breakdown of mucociliary transport. Mucus accumulates in the lower airways, and inhaled bacteria are trapped in this viscous mucous layer on top of respiratory epithelial cells.
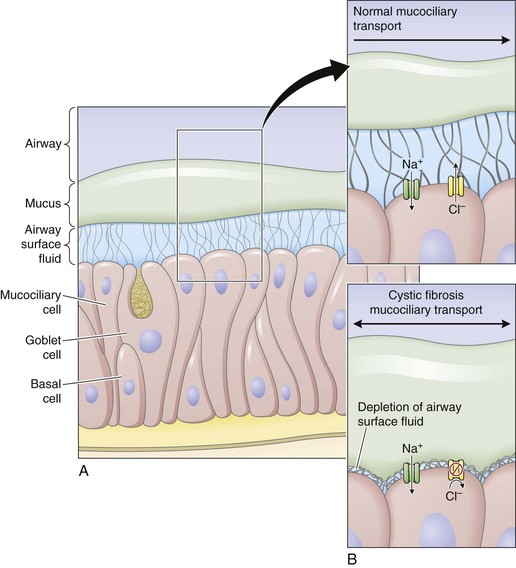
Figure 44-3 Restoring airway surface liquid in cystic fibrosis.
(From Ratjen F: Restoring airway surface liquid in cystic fibrosis, N Engl J Med 354:291–293, 2006.)
The spectrum of bacteria that are relevant for CF lung disease is relatively limited. Overall, Pseudomonas aeruginosa is the most common isolate, followed by Staphylococcus aureus and Haemophilus influenzae. Later in the course of disease, multiresistant organisms such as Stenotrophomonas maltophilia, Achromobacter (Alcaligenes) xylosoxidans, and Burkholderia cepacia complex may be isolated. As in other chronic pulmonary diseases, nontuberculous mycobacteria (usually Mycobacterium avium-intracellulare or M. abscessus) may be isolated. It is challenging to prove whether these organisms are causing ongoing disease requiring treatment, or if they are colonizing only the damaged lung. For a more detailed discussion of nontuberculous mycobacteria (NTM) infections, see Chapter 31.
Evidence indicates that inflammation is dysregulated in CF airways. Neutrophilic airway inflammation has been detected in infants with CF in the first months of life, as well as in CF fetal lung tissue. Whether or not inflammation is directly related to the CFTR defect is still disputed. However, an exaggerated, sustained, and prolonged inflammatory response to bacterial and viral pathogens is an accepted feature of CF lung disease. The persistent endobronchial inflammation is deleterious for the course of lung disease (Figure 44-4).
Clinical Features
Typical signs and symptoms for CF are listed in Box 44-1. Symptoms of CF may vary, with monosymptomatic cases often diagnosed late. It is therefore important to be aware of the spectrum of symptoms that may arise and to initiate adequate diagnostic steps.
Diagnostic Approach
The diagnosis of cystic fibrosis is established by clinical manifestations (see Box 44-1), a history of CF in a sibling, or a positive newborn screening result, in conjunction with laboratory evidence of CFTR dysfunction. CFTR dysfunction is documented by elevated sweat chloride or characteristic abnormalities in nasal potential difference or by CF-causing mutations in the CFTR gene.
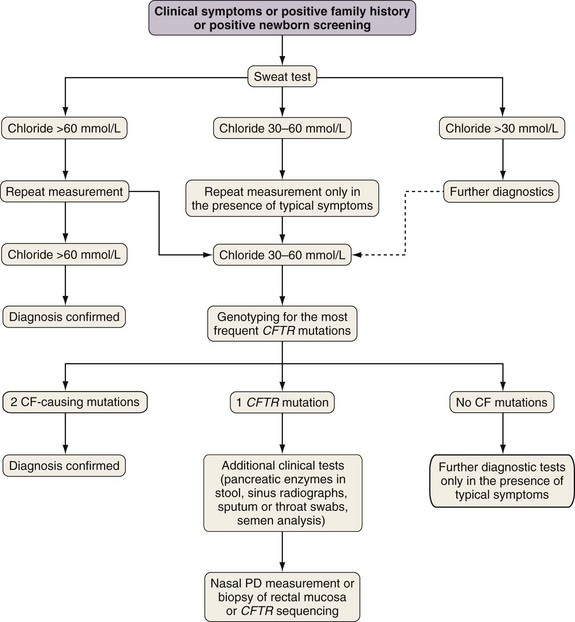
A diagnostic algorithm is presented in Figure 44-5. Abnormal ion transport is reflected in high sweat NaCl levels, and measurement of chloride concentration in sweat after iontophoresis of pilocarpine is used for diagnosis. Sweat testing must be done using standardized methods, by qualified staff in an experienced laboratory. A sweat chloride concentration greater than 60 mmol/L on repeated analysis is diagnostic for CF; 30 to 60 mmol/L is considered a “borderline” result but may be seen in patients with CF.
Clinical Course of Lung Disease and Principles of Therapy
Pulmonary exacerbations in patients with CF are often triggered by viral infections but require prompt treatment with antibiotics directed against pathogens present in respiratory cultures. It is important to recognize the signs of a pulmonary exacerbation early to avoid permanent damage to the lung (Box 44-2). In general, any increase in symptoms lasting more than a few days, associated with a decline in FEV1, requires antibiotic therapy. Because most CF patients are chronically infected with bacteria, suppression rather than eradication of organisms is the primary goal of therapy. Duration of therapy is not guided by changes in sputum bacteriology but rather by improvement in symptoms and pulmonary function.
Box 44-2
Symptoms and Signs of Pulmonary Exacerbation in Cystic Fibrosis Patients
Laboratory Findings
Data from Gibson RL, Burns JL, Ramsey BW: Am J Respir Crit Care Med 168:918–951, 2003.
Treatment
Symptomatic Therapy
Treatment of Airway Infection
Aggressive treatment of airway infection is a main reason for the increased life expectancy of patients with CF achieved over recent decades. As mentioned, bacterial pathogens in patients with CF are usually limited to a relatively small spectrum, with S. aureus or H. influenzae the most prominent in younger patients and P. aeruginosa in older patients. Most patients go through phases of clinical stability with intermittent pulmonary exacerbations. A set of criteria is used to diagnose pulmonary exacerbations (see Box 44-2), but the threshold for initiating targeted antibiotic therapy should be rather low. Genotyping has shown that the organisms present at exacerbation are the same as when the patient is clinically stable, but with higher bacterial density. Thus, choice of antimicrobial therapy on the basis of the most recent sputum cultures is indicated.
Overall, Pseudomonas aeruginosa is the major pathogen in CF lung disease. Its prevalence increases with age, and most adult patients are chronically infected with this organism. After an initial transient colonization period with nonmucoid strains, untreated patients generally become chronically infected with mucoid strains of P. aeruginosa. Antibiotic therapy usually fails to eradicate mucoid P. aeruginosa from the airways. High bacterial counts, low metabolic rate of pathogens in biofilms, poor penetration of antibiotics into airway secretions, and anaerobic conditions in sputum are considered responsible for this finding. Chronic infection with mucoid strains has a negative impact on the subsequent course of lung disease. Although eradication is virtually impossible in chronic infection, treatment can be effective in the early phase of P. aeruginosa infection; a major improvement in patients with CF is early antibiotic therapy. Both inhaled antibiotic therapy with tobramycin alone and combined inhaled antibiotics and oral ciprofloxacin have been used, but more recent evidence suggests that adding ciprofloxacin does not increase therapeutic success. Although the optimal treatment regimen for early P. aeruginosa infection has yet to be determined, inhaled tobramycin successfully reduces the incidence of chronic airway infection with P. aeruginosa in patients with CF (Table 44-1).
| Antibiotic: Choose One | Pediatric Dose | Adult Dose |
|---|---|---|
| Staphylococcus aureus | ||
| Dicloxacillin | 6.25-12.5 mg/kg four times daily | 250-500 mg four times daily |
| Cephalexin | 12.5-25 mg/kg four times daily | 500 mg four times daily |
| Amoxicillin/clavulanate | 12.5-22.5 mg/kg amoxicillin* | 400-875 mg amoxicillin* |
| Haemophilus influenzae | ||
| Amoxicillin | 25-50 mg/kg twice daily | 500-875 mg twice daily |
| Amoxicillin/clavulanate | 12.5-22.5 mg/kg amoxicillin* | 400-875 mg of amoxicillin* |
| Cefuroxime axetil | 15-20 mg/kg twice daily | 250-500 mg twice daily |
| Pseudomonas aeruginosa | ||
| Ciprofloxacin | 10-15 mg/kg twice daily | 750 mg twice daily |
| Tobramycin† | 300 mg by nebulizer, twice daily | 300 mg by nebulizer, twice daily |
| Colistin† | 150 mg by nebulizer, twice daily | 150 mg by nebulizer, twice daily |
Modified from Gibson RL, Burns JL, Ramsey BW: Am J Respir Crit Care Med 168:918–951, 2003.
In addition to inhaled antibiotics, azithromycin can improve pulmonary function and reduce pulmonary exacerbations in patients with P. aeruginosa –positive disease and more recently has been shown to reduce exacerbations in P. aeruginosa–negative patients as well. Although macrolides have no efficacy against P. aeruginosa when tested in routine cultures, some evidence suggests macrolides may affect P. aeruginosa growing in biofilms. Whether this explains their efficacy or whether this is caused by antiinflammatory properties of macrolides is still unclear (Table 44-1).
Complications
Pulmonary Complications
Hemoptysis
Chronic pulmonary infection leads to enlargement of the bronchial artery circulation supplying the lung. Hemoptysis can occur even in patients with mild lung disease and usually indicates infection. Vitamin K deficiency caused by pancreatic insufficiency can contribute to the problem. Massive hemoptysis (>250 mL of blood in 24 hours) is less common but occasionally can be life-threatening (see Chapter 20). Hemoptysis is usually treated with antibiotic therapy. Vitamin K and tranexamic acid may also be used. Ongoing massive hemoptysis requires bronchial artery embolization. Angiography is used to locate the abnormal bronchial artery vessels. These vessels are aberrant, and arteries arising from one side may supply contralateral segments of the lung. Because location of the bleeding does not always correlate with origin of abnormal bronchial arteries, bronchoscopy to locate source of bleeding is not helpful. However, bronchoscopy may be necessary to manage the airway in life-threatening hemoptysis. Embolization should be performed by experienced interventional radiologists because of potential complications of bronchial artery embolization, including infarction of the esophagus, lung parenchyma, or chest wall (causing dysphagia or severe chest pain), and transverse myelitis caused by accidental embolization of the spinal arteries.
Allergic Bronchopulmonary Aspergillosis
Although Aspergillus is frequently detected in sputum cultures, most of these patients do not have symptoms that can be attributed to the presence of the fungus. About 5% of patients with CF have symptoms caused by hypersensitivity. This entity is called allergic bronchopulmonary aspergillosis (ABPA) and should be suspected in patients who have respiratory deterioration but do not respond well to antibiotic therapy. In addition to symptoms of chronic productive cough, ABPA is usually accompanied by asthmatic features (wheezing), and patients may expectorate gritty brown sputum (“sandy” sputum). Some features of ABPA are difficult to assess in CF, because bronchiectasis, wheezing, productive cough, and presence of Aspergillus in the sputum are common features of CF lung disease. Diagnosis is supported by elevated IgE (usually >1000 IU/mL), a positive skin test, and serum precipitins against Aspergillus (see aspergilloma, Chapter 53). Treatment of symptoms and decline in lung function requires corticosteroids; high doses (1-2 mg/kg) may be required initially before the dose can be tapered. Adjunctive antifungal therapy with itraconazole may be efficacious in patients without CF who have ABPA, but its efficacy in patients with CF remains unproven. Duration of ABPA therapy is tailored to clinical response, improvement of FEV1, and normalization of serum IgE levels.
Borowitz D, Baker RD, Stallings V. Consensus Report on Nutrition for Pediatric Patients with Cystic Fibrosis. J Pediatr Gastroenterol Nutr. 2002;35:246–259.
Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Del Rev. 2002;54:1359–1371.
Davis PB. CF since 1938. Am J Respir Crit Care Med. 2006;173:475–482.
Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:918–951.
Knowles MR, Durie PR. What is cystic fibrosis? N Engl J Med. 2002;347:439–442.
Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689.
Farrell PM, Rosenstein BJ, White TB. Guidelines for diagnosis of cystic fibrosis in newborns through older adults. Cystic Fibrosis Foundation consensus report. J Pediatr. 2008;153:S4–S14.
Rowe SM, Miller S, Sorscher EJ. Mechanisms of disease: cystic fibrosis. N Engl J Med. 2005;352:1992–2001.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 44 Cystic Fibrosis
Genetics
Cystic fibrosis is caused by mutations in a gene on chromosome 7 encoding the protein subsequently termed the CFTR gene. More than 1800 mutations have been reported to the Cystic Fibrosis Genetic Analysis Consortium. Most of these mutations are rare, and only four mutations occur in a frequency of more than 1%. CFTR mutations can be grouped into five classes: CFTR is not synthesized (I), is inadequately processed (II), is not regulated (III), shows abnormal conductance (IV), or has partially defective production or processing (V). Class I, II, and III mutations are more common and associated with pancreatic insufficiency, whereas patients with the less common class IV and V mutations often are “pancreatic sufficient” (Figure 44-1).
Whereas classes IV and V CFTR mutations are linked with pancreatic sufficiency, attempts to link specific mutations to the severity of lung disease have shown large phenotypic variability. This is best documented for patients homozygous for the F508del mutation who exhibit a wide spectrum in lung disease severity. This wide phenotypic variation suggests that environmental factors and genes other than CFTR influence the development, progression, and disease severity of CF (Figure 44-2).
Pathophysiology
Airway epithelial cells secrete chloride and absorb sodium chloride (NaCl), the balance of which is regulated through apical channels, including CFTR (Figure 44-3). Ion secretion and absorption affect water transport, and a balance between secretion and absorption is thought to be important to maintain an adequate layer of airway surface liquid (ASL). The ASL supports the thin mucous layer on top of epithelial cells, which is constantly transported out of the lungs through ciliary movement. Lack or dysfunction of CFTR leads to reduced chloride secretion and NaCl hyperabsorption with depletion of ASL. In the absence of adequate ASL, respiratory cilia collapse, leading to breakdown of mucociliary transport. Mucus accumulates in the lower airways, and inhaled bacteria are trapped in this viscous mucous layer on top of respiratory epithelial cells.
Figure 44-3 Restoring airway surface liquid in cystic fibrosis.
(From Ratjen F: Restoring airway surface liquid in cystic fibrosis, N Engl J Med 354:291–293, 2006.)
The spectrum of bacteria that are relevant for CF lung disease is relatively limited. Overall, Pseudomonas aeruginosa is the most common isolate, followed by Staphylococcus aureus and Haemophilus influenzae. Later in the course of disease, multiresistant organisms such as Stenotrophomonas maltophilia, Achromobacter (Alcaligenes) xylosoxidans, and Burkholderia cepacia complex may be isolated. As in other chronic pulmonary diseases, nontuberculous mycobacteria (usually Mycobacterium avium-intracellulare or M. abscessus) may be isolated. It is challenging to prove whether these organisms are causing ongoing disease requiring treatment, or if they are colonizing only the damaged lung. For a more detailed discussion of nontuberculous mycobacteria (NTM) infections, see Chapter 31.
Evidence indicates that inflammation is dysregulated in CF airways. Neutrophilic airway inflammation has been detected in infants with CF in the first months of life, as well as in CF fetal lung tissue. Whether or not inflammation is directly related to the CFTR defect is still disputed. However, an exaggerated, sustained, and prolonged inflammatory response to bacterial and viral pathogens is an accepted feature of CF lung disease. The persistent endobronchial inflammation is deleterious for the course of lung disease (Figure 44-4).
Clinical Features
Typical signs and symptoms for CF are listed in Box 44-1. Symptoms of CF may vary, with monosymptomatic cases often diagnosed late. It is therefore important to be aware of the spectrum of symptoms that may arise and to initiate adequate diagnostic steps.
Diagnostic Approach
The diagnosis of cystic fibrosis is established by clinical manifestations (see Box 44-1), a history of CF in a sibling, or a positive newborn screening result, in conjunction with laboratory evidence of CFTR dysfunction. CFTR dysfunction is documented by elevated sweat chloride or characteristic abnormalities in nasal potential difference or by CF-causing mutations in the CFTR gene.
A diagnostic algorithm is presented in Figure 44-5. Abnormal ion transport is reflected in high sweat NaCl levels, and measurement of chloride concentration in sweat after iontophoresis of pilocarpine is used for diagnosis. Sweat testing must be done using standardized methods, by qualified staff in an experienced laboratory. A sweat chloride concentration greater than 60 mmol/L on repeated analysis is diagnostic for CF; 30 to 60 mmol/L is considered a “borderline” result but may be seen in patients with CF.
