20. Cystic Fibrosis
Definition
Cystic fibrosis (CF) is an ultimately lethal inherited disorder. It predominately affects the function of endocrine glands, with wide-ranging effects on multiple organ systems, including the lungs, pancreas, intestines, and liver. CF is characterized by chronic pulmonary infections and inadequate pancreatic enzymes, along with other associated complications.
Incidence
In the United States, the incidence of cystic fibrosis in Caucasians of northern European heritage is 1:3200; in the African American population, 1:15,000; in the Hispanic population, 1:9200; and in the Asian American population, 1:30,000. Internationally, the occurrence ranges from 1:620 in a specific group of Dutch heritage to 1:90,000 among Asian populations.
Etiology
Cystic fibrosis is an autosomal recessive disease, the expression of which is a defect in the gene for the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR encodes a protein that is a chloride channel regulated by cyclic adenosine monophosphate (cAMP). This mutation produces cAMP-regulated chloride transport abnormalities in epithelial cells’ mucosal surfaces. The subsequent chloride transport failure, along with the concomitant water transport abnormalities, culminates in very viscous secretions within the respiratory system, pancreas, gastrointestinal tract, sweat glands, and other exocrine tissues. The viscous secretions are tenacious and difficult to clear.
Lung disease is the most common reason for death among CF patients. The lungs develop quite normally in utero, are normal at birth, and for a period of time after birth. The thick, tenacious mucous secretions establish a scenario for infection that is followed by a neutrophilic inflammatory response. This cycle of infection followed by neutrophilic inflammation continues throughout the patient’s life. It eventually produces structural damage, impaired gas exchange, end-stage lung disease, and finally, early death.
Signs and Symptoms
• Abdominal distention
• Cheilosis
• Cough (dry and productive)
• Cyanosis
• Digit clubbing
• Dry skin
• Hepatosplenomegaly
• Hyperresonant chest
• Increased anteroposterior chest diameter
• Kyphosis
• Nasal polyps
• Parotid gland swelling
• Rectal prolapse
• Respiratory distress
• Rhinitis
• Scoliosis
• Submandibular gland swelling
• Tachypnea
• Wheezing
Medical Management
Cystic fibrosis affects multiple organs. Management is thus a multidisciplinary effort that may involve surgery (general, cardiothoracic, or transplant), endocrinology, otolaryngology, pulmonology, cardiology, and gastroenterology. Most often the patient with cystic fibrosis is treated primarily by a pulmonologist because of the prevalence of pulmonary symptoms. The viscous secretions commonly seen in the patient with cystic fibrosis create a persistent, rich growth medium for infectious organisms, such as Haemophilus influenza, Klebsiella pneumoniae, Escherichia coli, and Staphylococcus aureus. The patient typically experiences recurrent bouts of respiratory/pulmonary infections that are treated with antibiotics. They are usually very well educated about the disease, even at a young age, and receive respiratory treatments every day. One goal of the treatments is to dramatically reduce the viscosity of the secretions and facilitate expectoration of the mucus. Daily chest physiotherapy and postural drainage are often required to help facilitate expectoration of the mucus.
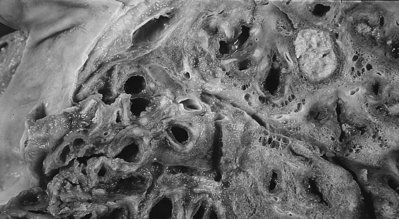 |
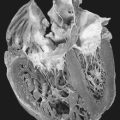
| Cystic Fibrosis. This lung from a 20-year-old man shows diffuse bronchiectasis. |
The patient with cystic fibrosis may be small for his or her age. The effects of this disease are far reaching. Because of the dysfunction of the pancreas caused by the disease, growth and development are impaired. Fat-soluble vitamins are not properly absorbed, which also interferes with growth and development. A high-energy, high-fat diet with fat-soluble vitamin and mineral supplementation may be required to compensate for these inadequacies. The patient with cystic fibrosis is encouraged to exercise regularly to improve physical fitness. Upper-body exercises help increase the endurance of accessory respiratory muscles.
The ultimate goals in medically managing the patient with cystic fibrosis are to: (1) maintain pulmonary function as close to normal as possible; (2) provide adequate nutrition via a high-energy, high-fat diet with supplementation of vitamins, minerals, phytonutrients, and enzymes to promote growth and development; and (3) manage any complications that develop because of the disease. As a result of advances in knowledge and understanding of cystic fibrosis, patients are living longer, more normal lives, but they still have a limited life expectancy. The most recent estimated median survival is about 30 years of age; male patients seem to survive slightly longer than female patients.
Complications
• Atelectasis
• Bronchiectasis
• Bronchiolitis
• Bronchitis
• Cholecystitis
• Chronic sinusitis
• Coagulation irregularities
• Cor pulmonale
• Cystic fibrosis–related diabetes mellitus
• Delayed puberty
• Distal intestinal obstruction syndrome
• Fatty liver
• Focal biliary cirrhosis
• Gallstones
• Gastroesophageal reflux
• Heatstroke
• Hemoptysis
• Hypertrophic pulmonary osteoarthropathy
• Liver failure
• Meconium ileus
• Mucocele
• Mucopyocele
• Nasal polyps
• Osteoporosis
• Pancreatic tissue damage
• Pneumothorax
• Portal hypertension
• Rectal prolapse
• Reduced fertility
• Rickets
• Salt depletion
• Vitamin deficiencies
Anesthesia Implications
The foremost anesthesia consideration revolves around the patient’s respiratory status. The respiratory function of a patient with cystic fibrosis can deteriorate very significantly in the course of a single day. A chest x-ray should be obtained to determine whether the patient has a pneumothorax or if there are any pneumonic processes or bullous disease present.
Coagulation time should be obtained preoperatively. Determining this value gives two pieces of information. First, it reflects the patient’s nutritional status—specifically, prolonged PT with normal PTT times may be the result of a vitamin K deficiency, which is a fat-soluble vitamin frequently missing from the patient’s dietary intake. Second, the prolonged coagulation times alert the anesthetist to the possibility of greater-than-expected blood loss from the proposed surgical procedure. It may be necessary to administer vitamin K or fresh frozen plasma, or to consult with a hematologist to reduce the patient’s possiblity of bleeding.
The effects of cystic fibrosis on the gastrointestinal tract may provoke signs and symptoms of gastroesophageal reflux disease (GERD). In such cases the patient should receive preoperative medications consistent with GERD, such as non-particulate antacids and/or H 2-receptor antagonists. Rapid sequence induction may be utilized despite the patient’s higher probability for rapid desaturation when apneic.
The nature and scope of the surgical procedure proposed has considerable bearing on the anesthesia technique selected. Both general and regional techniques offer advantages. Regional anesthesia avoids manipulation of the airway, which reduces the risk of aspiration and bronchospasm. A regional technique also eliminates the requirement for positive pressure ventilation, which decreases potential for pneumothorax during the perioperative period. General anesthesia with slower respiratory rates, slightly smaller tidal volumes, humidification of inspired gases, and longer expiratory phases are beneficial because patients with cystic fibrosis are easily fatigued by the work of breathing and are capable only of marginal tidal volume exchanges on a consistent basis.
Regional anesthesia lessens the need for analgesics, particularly opioids, in the immediate postoperative period. Thus another potential source of postoperative airway compromise can be avoided.
Because of the propensity for spontaneous pneumothorax in the patient with cystic fibrosis, nitrous oxide should be avoided. Nitrous oxide can rapidly expand a pneumothorax, should one actually occur.