First-Generation Nonsteroidal Antiinflammatory Drugs (NSAIDs)
Aspirin
Ibuprofen
Second-Generation NSAID (Selective Cyclooxygenase-2 Inhibitor)
Celecoxib
Drug That Lacks Antiinflammatory Actions
Acetaminophen
Aspirin
Aspirin is a highly valuable and effective medication. The drug provides excellent relief of mild to moderate pain, reduces fever, protects against thrombotic disorders, and remains a drug of choice for rheumatoid arthritis and other inflammatory conditions. Despite the introduction of many new NSAIDs, aspirin remains one of the most widely used members of the group and is the standard against which the others must be compared.
Chemistry
Aspirin belongs to a chemical family known as salicylates. All members of this group are derivatives of salicylic acid. Aspirin is produced by substituting an acetyl group onto salicylic acid. Because of this acetyl group, aspirin is commonly known as acetylsalicylic acid, or simply ASA.
Mechanism of Action
Aspirin is a nonselective inhibitor of cyclooxygenase. Most beneficial effects—reductions of inflammation, pain, and fever—result from inhibiting COX-2. One beneficial effect—protection against MI and ischemic stroke—results from inhibiting COX-1. Major adverse effects—gastric ulceration, bleeding, and renal impairment—result from inhibiting COX-1.
It is important to note that aspirin is an irreversible inhibitor of cyclooxygenase. In contrast, all other NSAIDs are reversible (competitive) inhibitors. Because inhibition of cyclooxygenase by aspirin is irreversible, duration of action depends on how quickly specific tissues can synthesize new molecules of COX-1 and COX-2. With other NSAIDs, effects decline as soon as drug levels fall.
Pharmacokinetics
Absorption
Aspirin is absorbed rapidly and completely after oral dosing. The principal site of absorption is the small intestine. When administered by rectal suppository, aspirin is absorbed slowly, and blood levels are lower than with oral dosing.
Metabolism
Aspirin has a very short half-life (15–20 minutes) owing to rapid conversion to salicylic acid, an active metabolite. The rate of inactivation of salicylic acid depends on the amount present: at low therapeutic levels, salicylic acid has a half-life of approximately 2 hours, but at high therapeutic levels, the half-life may exceed 20 hours.
Distribution
Salicylic acid is extensively bound to plasma albumin. At therapeutic levels, binding is between 80% and 90%. Aspirin undergoes distribution to all body tissues and fluids, including breast milk, fetal tissues, and the central nervous system (CNS).
Excretion
Salicylic acid and its metabolites are excreted by the kidneys. Excretion of salicylic acid is highly dependent on urinary pH. Accordingly, by raising the pH of urine from 6 to 8, we can increase the rate of excretion fourfold.
Plasma Drug Levels
Low therapeutic doses of aspirin produce plasma salicylate levels less than100 mcg/mL. Antiinflammatory doses produce salicylate levels of about 150 to 300 mcg/mL. Signs of salicylism (toxicity) begin when plasma salicylate levels exceed 200 mcg/mL. Severe toxicity occurs at levels above 400 mcg/mL.
Therapeutic Uses
Suppression of Inflammation
Aspirin is an initial drug of choice for rheumatoid arthritis, osteoarthritis, and juvenile arthritis. Aspirin is also indicated for other inflammatory disorders, including rheumatic fever, tendinitis, and bursitis. The dosages employed to suppress inflammation are considerably larger than dosages used for analgesia or reduction of fever. The use of aspirin and other NSAIDs to treat arthritis is discussed further in Chapter 57.
The precise mechanisms by which aspirin decreases inflammation have not been established. We do know that prostanoids contribute to several components of the inflammatory process. Hence inhibition of COX-2 provides a partial explanation of antiinflammatory effects. Other possible mechanisms include modulation of T-cell function, suppression of inflammatory cell infiltration, and stabilization of lysosomes.
Analgesia
Aspirin is used widely to relieve mild to moderate pain. The degree of analgesia produced depends on the type of pain. Aspirin is most active against joint pain, muscle pain, and headache. For some forms of postoperative pain, aspirin can be more effective than opioids. However, aspirin is relatively ineffective against severe pain of visceral origin. In contrast to opioid analgesics, aspirin produces neither tolerance nor physical dependence. In addition, aspirin is safer than opioids.
Aspirin relieves pain primarily through actions in the periphery. At sites of injury, prostanoids sensitize pain receptors to mechanical and chemical stimulation. Aspirin reduces pain by inhibiting COX-2, thereby suppressing prostanoid production. In addition to this peripheral mechanism, aspirin works in the CNS to help relieve pain.
Reduction of Fever
Aspirin is a drug of choice for reducing temperature in febrile adults. However, because of the risk for Reye syndrome (see later), aspirin should not be used to treat fever in children. Although aspirin readily reduces fever, it will not lower normal body temperature, nor will it lower temperature that has become elevated in response to physical activity or to a rise in environmental temperature.
Body temperature is regulated by the hypothalamus, which maintains a balance between heat production and heat loss. Fever occurs when the set point of the hypothalamus becomes elevated, causing the hypothalamus to increase heat production and decrease heat loss. Set-point elevation is triggered by local synthesis of prostaglandins in response to endogenous pyrogens (fever-promoting substances). Aspirin lowers the set point by inhibiting COX-2 and thereby inhibits pyrogen-induced synthesis of prostaglandins.
Dysmenorrhea
Aspirin can provide relief from primary dysmenorrhea. Benefits derive from inhibiting prostaglandin synthesis in uterine smooth muscle. (Prostaglandins promote uterine contraction, so suppression of prostaglandin synthesis relieves cramping.) Some of the newer aspirin-like drugs (e.g., ibuprofen, naproxen) are superior to aspirin for dysmenorrhea. The efficacy of the newer drugs is attributed to a greater ability to inhibit COX in the uterus.
Suppression of Platelet Aggregation
Synthesis of TXA2 in platelets promotes aggregation. Aspirin suppresses platelet aggregation by causing irreversible inhibition of COX-1, the enzyme that makes TXA2. Because platelets lack the machinery to synthesize new COX-1, the effects of a single dose persist for the life of the platelet (about 8 days).
There is a large body of evidence demonstrating that aspirin, through its antiplatelet actions, can benefit a variety of patients. Accordingly, the U.S. Food and Drug Administration (FDA) recommended wider use of aspirin for antiplatelet effects. Professional labeling now recommends daily aspirin for men and women with the following:
• Ischemic stroke (to reduce the risk for death and nonfatal stroke)
• Transient ischemic attacks (to reduce the risk for death and nonfatal stroke)
• Acute MI (to reduce the risk for vascular mortality)
• Previous MI (to reduce the combined risk for death and nonfatal MI)
• Chronic stable angina (to reduce the risk of MI and sudden death)
• Unstable angina (to reduce the combined risk for death and nonfatal MI)
• Angioplasty and other revascularization procedures (in patients who have a preexisting condition for which aspirin is already indicated)
According to a review published in the Journal of the American Medical Association—“Aspirin Dose for the Prevention of Cardiovascular Disease”—a dose of 75 to 81 mg/day for these indications is adequate. Higher doses, which are commonly prescribed in these circumstances, offer no greater protection but will increase the risk for gastrointestinal (GI) bleeding.
In addition to these applications, aspirin can be taken by healthy people for primary prevention of MI and stroke. However, more recent studies show that aspirin provides less protection against cardiovascular disease than once thought. The potential small benefit must be weighed against the major risk of aspirin use, namely, GI hemorrhage. Hence, to determine the net benefit of primary prevention for any man or woman, we must determine his or her individual risk for a GI bleed and compare that risk with his or her individual risk for a cardiovascular event (i.e., the risk for an MI in men, or the risk for ischemic stroke in women).a Many organizations, including the American Heart Association (AHA), the American Thoracic Society, and the European Society of Cardiology, recommend against the use of aspirin for primary prevention of cardiovascular disease unless the patient has a 10-year risk greater than 10%.
How do we calculate 10-year risk for a cardiovascular event? Risk for an MI or stroke can be assessed using the calculator at http://cvdrisk.nhlbi.nih.gov/.
Cancer Prevention
Colorectal Cancer.
There is good evidence that regular use of aspirin decreases the risk for colorectal cancer, even when the dosage is low. Results from the Nurses’ Health Study showed that regular use of high-dose aspirin (650 mg/day or more) reduces the risk for colorectal cancer. This dosage is much greater than that used to prevent cardiovascular disease and hence poses a significant risk for bleeding. In fact, for every one or two cancers prevented, high-dose aspirin would cause eight additional serious bleeds. Fortunately, more recent studies indicate that low-dose aspirin is effective, too. For example, results of a study reported in The Lancet in 2010 indicate that taking low-dose aspirin (75–300 mg/day) for more than 5 years reduces the incidence of colorectal cancer (by 24%) as well as mortality from colon cancer (by 35%). At these low doses, the benefits of cancer protection may well outweigh the risk for possible bleeding and other adverse events.
Aspirin protects against colorectal cancer probably by inhibiting COX-2. In animal models, COX-2 promotes tumor growth and metastases, and inhibition of COX-2 slows tumor growth. In humans, most colorectal cancers express COX-2. Furthermore, protection by aspirin is limited to colon cancers that have high COX-2 levels. Aspirin does not protect against colon cancers with little or no COX-2.
Other Cancers.
Available data suggest that protection may not be limited to colorectal cancer. Results of a meta-analysis reported in The Lancet, “Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials,” show that daily low-dose aspirin reduces the risk for death from all solid tumors (by 34%), but does not reduce the risk for death from hematologic cancers. In addition, The Lancet published an additional article in 2012 that analyzed 5 randomized controlled trials. This analysis determined that use of aspirin may also prevent distant metastasis of tumors that already exist. Earlier studies have shown protection against specific cancers. In a study involving men older than 60 years, daily use of aspirin and other NSAIDs was associated with a 50% decrease in the incidence of prostate cancer. In a study involving 2884 women, aspirin appeared to reduce the risk for breast cancer, especially among women with hormone receptor–positive tumors and among those who took 7 or more aspirin tablets a week. In another study, taking aspirin at least 3 times a week for at least 6 months was associated with a 40% reduction in the incidence of ovarian cancer. In contrast to these positive results, results from the Women’s Health Study found no protection with low-dose aspirin against cancer of the breast, colon, or any other tissue. The reasons for this discrepancy are not clear. Four additional studies are currently underway to examine various effects of aspirin on cancer prevention.
Adverse Effects
When administered short term in analgesic or antipyretic (fever-reducing) doses, aspirin rarely causes serious adverse effects. However, toxicity is common when treating inflammatory disorders, which require long-term high-dose treatment.
Gastrointestinal Effects
The most common side effects are gastric distress, heartburn, and nausea. These can be reduced by taking aspirin with food or a full glass of water.
Occult GI bleeding occurs often. In most cases, the amount of blood lost each day is insignificant. However, with chronic aspirin use, cumulative blood loss can produce anemia.
Long-term aspirin—even in low doses—can cause life-threatening gastric ulceration, perforation, and bleeding. Ulcers result from four causes:
• Increased secretion of acid and pepsin
• Decreased production of cytoprotective mucus and bicarbonate
• Decreased submucosal blood flow
• The direct irritant action of aspirin on the gastric mucosa
The first three occur secondary to inhibition of COX-1. Direct injury to the stomach is most likely with aspirin preparations that dissolve slowly: owing to slow dissolution, particulate aspirin becomes entrapped in folds of the stomach wall, causing prolonged exposure to high concentrations of the drug. Because aspirin-induced ulcers are often asymptomatic, perforation and upper GI hemorrhage can occur without premonitory signs. (Hemorrhage is due in part to erosion of the stomach wall and in part to suppression of platelet aggregation.) Factors that increase the risk for ulceration include the following:
• A history of peptic ulcer disease
• Previous intolerance to aspirin or other NSAIDs
• History of alcohol abuse (Alcohol intensifies the irritant effects of aspirin and should not be consumed.)
What can we do to prevent ulcers? According to an expert panel—convened in 2008 by the American College of Gastroenterology, the AHA, and the American College of Cardiology—prophylaxis with a proton pump inhibitor (PPI) is recommended for patients at risk, including those with a history of peptic ulcers, those taking glucocorticoids, and older adults. Proton pump inhibitors (e.g., omeprazole, lansoprazole) reduce ulcer generation by suppressing production of gastric acid. In addition to PPIs, other drugs that may be considered include histamine-2 receptor antagonists (H2RAs) and misoprostol. COX-2 inhibitors may also be tried instead of traditional NSAIDs because they are thought to produce fewer GI side effects. Because many ulcers are caused by infection with Helicobacter pylori (see Chapter 62), the panel recommends that patients with ulcer histories undergo testing and treatment for H. pylori before starting long-term aspirin use. Treatment of NSAID-induced ulcers is discussed in Chapter 62.
Bleeding
Aspirin promotes bleeding by inhibiting platelet aggregation. Taking just two 325-mg aspirin tablets can double bleeding time for about 1 week. (Recall that platelets are unable to replace aspirin-inactivated cyclooxygenase, and hence bleeding time is prolonged for the life of the platelet.) Because of its effects on platelets, aspirin is contraindicated for patients with bleeding disorders (e.g., hemophilia, vitamin K deficiency, hypoprothrombinemia). To minimize blood loss during childbirth and elective surgery, high-dose aspirin should be discontinued at least 1 week before these procedures. There is no need to stop aspirin before procedures with a low risk for bleeding (e.g., dental, dermatologic, or cataract surgery). In most cases, use of low-dose aspirin to protect against thrombosis should not be interrupted for elective surgery and dental procedures. Caution is needed when aspirin is used in conjunction with anticoagulants.
In patients taking daily aspirin, high blood pressure increases the risk for hemorrhagic stroke, even though aspirin protects against ischemic stroke. To reduce risk for hemorrhagic stroke, blood pressure should be 150/90 mm Hg (and preferably lower) before starting daily aspirin.
Renal Impairment
Aspirin can cause acute, reversible impairment of renal function, resulting in salt and water retention and edema. Clinically significant effects are most likely in patients with additional risk factors: advanced age, existing renal impairment, hypovolemia, hepatic cirrhosis, or heart failure. Aspirin impairs renal function by inhibiting COX-1, thereby depriving the kidney of prostaglandins needed for normal function.
Development of renal impairment is signaled by reduced urine output, weight gain despite use of diuretics, and a rapid rise in serum creatinine and blood urea nitrogen. If any of these occurs, aspirin should be withdrawn immediately. In most cases, kidney function then returns to baseline level.
The risk for acute renal impairment can be reduced by identifying high-risk patients and treating them with the smallest dosages possible.
In addition to its acute effects on renal function, aspirin may pose a risk for renal papillary necrosis and other types of renal injury when used long term.
Salicylism
Salicylism is a syndrome that begins to develop when aspirin levels climb just slightly above therapeutic. Overt signs include tinnitus, sweating, headache, and dizziness. Acid-base disturbance may also occur (see later). If salicylism develops, aspirin should be withheld until symptoms subside. Aspirin should then resume, but with a small reduction in dosage. In some cases, development of tinnitus can be used to adjust aspirin dosage: when tinnitus occurs, the maximal acceptable dose has been achieved. However, this guideline may be inappropriate for older patients because they may fail to develop tinnitus even when aspirin levels become toxic.
Acid-base disturbance results from the effects of aspirin on respiration. When administered in high therapeutic doses, aspirin acts on the CNS to stimulate breathing. The resultant increase in CO2 loss produces respiratory alkalosis. In response, the kidneys excrete more bicarbonate. There is also a subsequent buildup of acids, producing a resultant metabolic acidosis. Thus many patients that present with salicylate toxicity will have a mixed acid-base imbalance.
Reye Syndrome
Use of aspirin in children younger than 18 years is associated with Reye syndrome.
This syndrome is a rare but serious illness of childhood that has a mortality rate of 20% to 30%. Characteristic symptoms are encephalopathy and fatty liver degeneration. Epidemiologic data suggested a relationship between Reye syndrome and use of aspirin by children who have influenza or chickenpox. Although a direct causal link between aspirin and Reye syndrome was never established, the Centers for Disease Control and Prevention recommended that aspirin (and other NSAIDs) be avoided by children and teenagers suspected of having influenza or chickenpox. In response to this recommendation, aspirin was removed from most products intended for children, and aspirin use by children declined sharply. As a result, Reye syndrome essentially vanished: the incidence declined from a high of 555 cases in 1980 to no more than 2 cases per year since 1994. If a child with chickenpox or influenza needs an analgesic-antipyretic, acetaminophen can be used safely.
Adverse Effects Associated With Use During Pregnancy
Aspirin poses risks to the pregnant patient and her fetus. Accordingly, the drug is classified in FDA Pregnancy Risk Category D: there is evidence of human fetal risk, but the potential benefits from use of the drug during pregnancy may outweigh the potential for harm. The principal risks to pregnant women are (1) anemia (from GI blood loss) and (2) postpartum hemorrhage. In addition, by inhibiting prostaglandin synthesis, aspirin may suppress spontaneous uterine contractions and may thereby prolong labor.
Aspirin crosses the placenta and may adversely affect the fetus. Because prostaglandins help keep the ductus arteriosus patent, inhibition of prostaglandin synthesis by aspirin may induce premature closure of the ductus arteriosus. Aspirin use has also been associated with low birth weight, stillbirth, renal toxicity, intracranial hemorrhage in preterm infants, and neonatal death.
Hypersensitivity Reactions
Hypersensitivity develops in about 0.3% of aspirin users. Reactions are most likely in adults with a history of asthma, rhinitis, and nasal polyps. Hypersensitivity reactions are uncommon in children. The aspirin hypersensitivity reaction begins with profuse, watery rhinorrhea and may progress to generalized urticaria, bronchospasm, laryngeal edema, and shock. Despite its resemblance to severe anaphylaxis, this reaction is not allergic and is not mediated by the immune system. What does cause these reactions? Because individuals who react to aspirin are also sensitive to most other NSAIDs, we believe that the reactions are due to inhibition of COX-1, which triggers production of leukotrienes, which in turn causes bronchospasm, hives, and other signs of hypersensitivity. However, if this is the mechanism, it remains unclear why hypersensitivity is limited mainly to adults with the predisposing conditions noted earlier. As with severe anaphylactic reactions, epinephrine is the treatment of choice.
Hypersensitivity to aspirin is considered a contraindication to using other drugs with aspirin-like properties. Nonetheless, if an aspirin-like drug must be taken, four such drugs are probably safe. One of these—celecoxib—is selective for COX-2. Another—meloxicam—is somewhat selective for COX-2, but only at low doses. The other two—acetaminophen and salsalate—are only weak inhibitors of COX-1.
Cardiovascular Events
In contrast to all other NSAIDs, aspirin does NOT increase the risk for thrombotic events, including MI and ischemic stroke. In fact, when taken in low doses, aspirin protects against these events.
Erectile Dysfunction
Daily use of aspirin and other NSAIDs is associated with a 22% increase in the risk for erectile dysfunction, as shown in a study of 80,966 men in California. However, a causal relationship has not been established.
Summary of Precautions and Contraindications
Aspirin is contraindicated in patients with peptic ulcer disease, bleeding disorders (e.g., hemophilia, vitamin K deficiency, hypoprothrombinemia), and hypersensitivity to aspirin itself or other NSAIDs. In addition, the drug should be used with extreme caution by pregnant women and by children who have chickenpox or influenza. Caution should also be exercised when treating older-adult patients, patients who smoke cigarettes, and patients with H. pylori infection, heart failure, hepatic cirrhosis, hypovolemia, renal dysfunction, asthma, hay fever, chronic urticaria, nasal polyps, or a history of alcoholism. Aspirin should be withdrawn 1 week before elective surgery or the anticipated date of childbirth.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
Nonsteroidal Antiinflammatory Drugs
| Life Stage | Patient Care Concerns |
| Infants | Because of the risk for Reye syndrome, aspirin should be avoided in infants. Acetaminophen and ibuprofen can be used safely in small doses for fever. |
| Children/adolescents | Because of the risk for Reye syndrome, aspirin should be avoided in children and adolescents. Acetaminophen and ibuprofen can be used safely in small doses for fever. |
| Pregnant women | NSAIDs may result in premature closure of the ductus arteriosus. Therefore their use is contraindicated in the third trimester of pregnancy. |
| Breastfeeding women | NSAIDs and acetaminophen appear safe for use in breastfeeding mothers. |
| Older adults | NSAIDs are the most common drug used to treat chronic pain in older adults. These drugs have been shown to increase hospital admissions in this population. Caution should be used with NSAIDs in older adults. |
Drug Interactions
Because of its widespread use, aspirin has been reported to interact with many other medications. However, most of these interactions have little clinical significance. Significant interactions are discussed next.
Anticoagulants: Warfarin, Heparin, and Others
Aspirin’s most important interactions are with anticoagulants. Because aspirin suppresses platelet function and can decrease prothrombin production, aspirin can intensify the effects of warfarin, heparin, and other anticoagulants. Furthermore, because aspirin can initiate gastric bleeding, augmenting anticoagulant effects can increase the risk for gastric hemorrhage. Accordingly, the combination of aspirin with anticoagulants must be used with care—even when aspirin is taken in low doses to reduce the risk for thrombotic events.
Glucocorticoids
Like aspirin, glucocorticoids promote gastric ulceration. As a result, the risk for ulcers is greatly increased when these drugs are combined—as may happen when treating arthritis. To reduce the risk for gastric ulceration, patients can be given a PPI or H2RA for prophylaxis.
Alcohol
Combining alcohol with aspirin and other NSAIDs increases the risk for gastric bleeding. To alert the public to this risk, the FDA now requires that labels for aspirin include the following statement: Alcohol Warning: If you consume three or more alcoholic drinks every day, ask your doctor whether you should take aspirin or other pain relievers/fever reducers. Aspirin [and related drugs] may cause stomach bleeding. A similar label is required for all other NSAIDs and acetaminophen.
Nonaspirin NSAIDs
Ibuprofen, naproxen, and other nonaspirin NSAIDs can reduce the antiplatelet effects of aspirin by blocking access of aspirin to COX-1 in platelets. This interaction is important: in patients taking low-dose aspirin to prevent MI or ischemic stroke, other NSAIDs could negate aspirin’s benefits. Because immediate-release aspirin produces complete platelet inhibition about 1 hour after dosing, we can prevent interference by giving aspirin about 2 hours before giving other NSAIDs. Of course, we could eliminate interference entirely by using high-dose aspirin, rather than another NSAID, when conditions call for NSAID therapy.
ACE Inhibitors and ARBs
Like aspirin, angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) can impair renal function. In susceptible patients, combining aspirin with drugs in either class can increase the risk for acute renal failure. High-dose aspirin should be avoided in patients taking these drugs. However, low-dose aspirin taken for antiplatelet effects should be continued.
Vaccines
Aspirin and other NSAIDs may blunt the immune response to vaccines. Accordingly, these drugs should not be used routinely to prevent vaccination-associated fever and pain.
Acute Poisoning
Aspirin overdose is a common cause of poisoning. Although rarely fatal in adults, aspirin poisoning may be lethal in children. The lethal dose for adults is 20 to 25 g. In contrast, as little as 4000 mg (4 g) can kill a child.
Signs and Symptoms
Initially, aspirin overdose produces a state of compensated respiratory alkalosis—the same state seen in mild salicylism. As poisoning progresses, respiratory excitation is replaced with respiratory depression. Acidosis, hyperthermia, sweating, and dehydration are prominent, and electrolyte imbalance is likely. Stupor and coma result from effects in the CNS. Death usually results from respiratory failure.
Treatment
Aspirin poisoning is an acute medical emergency that requires hospitalization. The immediate threats to life are respiratory depression, hyperthermia, dehydration, and acidosis. Treatment is largely supportive. If respiration is inadequate, mechanical ventilation should be instituted. External cooling (e.g., sponging with tepid water) can help reduce hyperthermia. Intravenous fluids are given to correct dehydration; the composition of these fluids is determined by electrolyte and acid-base status. Slow infusion of bicarbonate is given to reverse acidosis. Several measures (e.g., gastric lavage, giving activated charcoal) can reduce further GI absorption of aspirin. Alkalinization of the urine with bicarbonate accelerates excretion of aspirin and salicylate. If necessary, hemodialysis or peritoneal dialysis can be used to remove salicylates.
Formulations
Aspirin is available in multiple formulations, including plain and buffered tablets, enteric-coated preparations, and tablets used to produce a buffered solution. These different formulations reflect efforts to increase rates of absorption and decrease gastric irritation. For the most part, the clinical utility of the more complex formulations is no greater than that of plain aspirin tablets.
Aspirin Tablets, Plain
All brands are essentially the same with respect to analgesic efficacy, onset, and duration. Some less expensive tablets have greater particle size, which results in slower dissolution and prolonged contact with the gastric mucosa, which increases gastric irritation. When aspirin tablets decompose, they smell like vinegar (acetic acid) and should be discarded.
Aspirin Tablets, Buffered
The amount of buffer in buffered aspirin tablets is too small to produce significant elevation of gastric pH. An equivalent effect on pH can be achieved by taking plain aspirin tablets with food or a glass of water. Buffered aspirin tablets are no different from plain tablets with respect to analgesic effects and gastric distress. Buffered tablets may dissolve faster than plain tablets, resulting in somewhat faster onset.
Buffered Aspirin Solution
A buffered aspirin solution is produced by dissolving effervescent aspirin tablets [Alka-Seltzer] in a glass of water. This solution has considerable buffering capacity owing to its high content of sodium bicarbonate. Effects on gastric pH are sufficient to decrease the incidence of gastric irritation and bleeding. In addition, aspirin absorption is accelerated and peak blood levels are raised. Unfortunately, these benefits come with a price. The sodium content of buffered aspirin solution can be detrimental to individuals on a sodium-restricted diet. Also, absorption of bicarbonate can elevate urinary pH, which will accelerate aspirin excretion. Lastly, this highly buffered preparation is expensive. Because of this combination of benefits and drawbacks, the buffered aspirin solution is well suited for occasional use but is generally inappropriate for long-term therapy.
Enteric-Coated Preparations
Enteric-coated preparations dissolve in the intestine rather than the stomach, thereby reducing gastric irritation. Unfortunately, absorption from these formulations can be delayed and erratic. Patients should be advised not to crush or chew them.
Timed-Release Tablets
Timed-release tablets offer no advantage over plain aspirin tablets. Because the half-life of salicylic acid is long to begin with, and because aspirin produces irreversible inhibition of cyclooxygenase, timed-release tablets cannot prolong effects.
Rectal Suppositories
Rectal suppositories have been employed for patients who cannot take aspirin orally. Absorption can be variable, resulting in plasma drug levels that are insufficient in some patients and excessive in others. Also, rectal irritation can occur. Because of these undesirable properties, aspirin suppositories are not generally recommended.
Dosage and Administration
Aspirin is almost always administered by mouth. Gastric irritation can be minimized by dosing with water or food. Dosage depends on the age of the patient and the condition being treated. Adult and pediatric dosages for major indications are shown in Table 55.3.
TABLE 55.3
Aspirin Dosage
| Indication | Adult Dosage | Pediatric Dosage* |
| Aches and pains, fever | 325–650 mg every 4 h as needed |
2–3 yr: 160 mg 4–5 yr: 240 mg 6–8 yr: 325 mg 9–10 yr: 405 mg 11 yr: 485 mg Over 11 yr: 650 mg All of these doses are administered every 4 h as needed |
| Acute rheumatic fever | 5–8 g/day in divided doses | 100 mg/kg/day (initially), then 75 mg/kg/day for 4–6 wk |
| Rheumatoid arthritis | 3.6–5.4 g/day in divided doses | 90–130 mg/kg/day in divided doses every 4–6 h |
| SUPPRESSION OF PLATELET AGGREGATION | ||
| Initial therapy | 325 mg once a day | |
| Chronic therapy | 80 mg once a day | |
Nonaspirin First-Generation Nonsteroidal Antiinflammatory Drugs
In attempts to produce an aspirin-like drug with fewer GI, renal, and hemorrhagic effects than aspirin, the pharmaceutical industry has produced a large number of drugs with actions much like those of aspirin. In the United States more than 20 nonaspirin NSAIDs are available (Table 55.4). Like aspirin, all other first-generation NSAIDs inhibit both COX-1 and COX-2. However, in contrast to aspirin, which causes irreversible inhibition of cyclooxygenase, the other traditional NSAIDs cause reversible inhibition. All of these drugs have antiinflammatory, analgesic, and antipyretic properties. In addition, they all can cause gastric ulceration, bleeding, and renal impairment—although the intensity of these effects may be less with some agents. Patients who are hypersensitive to aspirin are likely to experience cross-hypersensitivity with other NSAIDs. For most NSAIDs, safety during pregnancy has not been established, and hence use by pregnant women is discouraged.
TABLE 55.4
Clinical Pharmacology of the Oral Nonsteroidal Antiinflammatory Drugs
| Drug | Maximal Daily Dosage (mg) | Plasma Half-Life (h) | Major Indicationsa | ||||
| Arthritis | Moderate Pain | Fever | Dysmenorrhea | Bursitis/Tendinitis | |||
| FIRST-GENERATION NSAIDs | |||||||
| Salicylates | |||||||
| Aspirin (many trade names) | 8000 | 0.2–0.3 | A | A | A | ||
| Magnesium salicylate [Doan’s Tablets, others] | 4640 | 2–30b | A | A | A | ||
| Sodium salicylate* (generic) | 3900 | 2–30b | A | A | A | ||
| Salsalate | 3000 | 2–30b | A | A | A | ||
| Propionic Acid Derivatives | |||||||
| Fenoprofen [Nalfon] | 3200 | 3 | A | A | |||
| Flurbiprofen (generic) | 300 | 5.7 | A | I | I | I | I |
| Ibuprofen [Advil, Motrin, others]c | 3200 | 1.8–2 | A | A | A | A | |
| Ketoprofen (generic) | 300 | 2 | A | A | A | ||
| Naproxen [Aleve, others]d | 1375 | 12–17 | A | A | A | A | A |
| Oxaprozin [Daypro] | 1800 | 42–50 | A | ||||
| Others | |||||||
| Diclofenac [Cambia, Cataflam, Voltaren XR, Zipsor, Zorvolex]e | 200 | 2 | A | A | A | ||
| Diflunisal (generic) | 1500 | 11–15 | A | A | |||
| Etodolac (generic) | 1200 | 7.3 | A | A | I | ||
| Indomethacin [Indocin] | 200 | 4.5 | A | A | |||
| Ketorolac (generic)f |
40 PO 120 IV |
5–6 | Ag | ||||
| Meclofenamate (generic) | 400 | 1.3 | A | A | A | ||
Mefenamic acid [Ponstel, Ponstan  ] ] |
1000 | 2 | A | A | |||
Meloxicam [Mobic, Mobicox  ] ] |
15 | 15–20 | A | ||||
| Nabumetone (generic) | 2000 | 22 | A | ||||
| Piroxicam [Feldene] | 20 | 50 | A | ||||
| Sulindac [Clinoril] | 400 | 7.8 | A | A | |||
| Tolmetin (generic) | 1800 | 2–7 | A | ||||
| SECOND-GENERATION NSAIDs (COX-2 INHIBITORS) | |||||||
| Celecoxib [Celebrex] | 800 | 11 | A | A | A | ||
The principal indications for the nonaspirin NSAIDs are rheumatoid arthritis and osteoarthritis. In addition, certain NSAIDs are used to treat fever, bursitis, tendinitis, mild to moderate pain, and dysmenorrhea (see Table 55.4).
In contrast to aspirin, the nonaspirin NSAIDs do not protect against MI and stroke. In fact, they increase the risk for thrombotic events. For the NSAIDs as a group, the increase in cardiovascular risk is relatively low—about 12%. Risk is highest with indomethacin (71%), sulindac (41%), and meloxicam (37%). However, although the increase in risk with these drugs appears high, it pales in comparison with smoking, which increases cardiovascular risk by 200% to 300%. To minimize cardiovascular risk, nonaspirin NSAIDs should be used in the lowest effective dosage for the shortest time needed. Also, these drugs should not be used before coronary artery bypass graft (CABG) surgery or for 14 days after. Other measures to reduce risk are discussed later under “American Heart Association Statement on Cyclooxygenase Inhibitors in Chronic Pain.”
Although individual NSAIDs differ chemically, pharmacokinetically, and to some extent pharmacodynamically, all are similar clinically: they all produce essentially equivalent antiinflammatory effects, and they all present a similar risk for serious adverse effects (gastric ulceration, bleeding, renal impairment, MI, and stroke). However, for reasons that are not understood, individual patients may respond better to one agent than another. Furthermore, individual patients may tolerate one NSAID better than another. Therefore to optimize therapy for each patient, trials with more than one NSAID may be needed.
Ibuprofen
Basic Pharmacology
Ibuprofen [Advil, Motrin, Caldolor, others] is the prototype of the propionic acid derivatives. Other members of the family are shown in Table 55.4 and discussed individually below. Like aspirin, ibuprofen inhibits cyclooxygenase and has antiinflammatory, analgesic, and antipyretic actions. The drug is used to treat fever, mild to moderate pain, and arthritis. In addition, ibuprofen appears superior to most other NSAIDs for relief of primary dysmenorrhea, presumably because it produces good inhibition of cyclooxygenase in uterine smooth muscle. In clinical trials, ibuprofen was highly effective at promoting closure of the ductus arteriosus in preterm infants, a condition for which indomethacin is the current treatment of choice.
Ibuprofen is generally well tolerated, and the incidence of adverse effects is low. The drug produces less gastric bleeding than aspirin and less inhibition of platelet aggregation as well. Consequently, ibuprofen is among the safer NSAIDs for use with anticoagulants. Very rarely, ibuprofen has been associated with Stevens-Johnson syndrome, a severe hypersensitivity reaction that causes blistering of the skin and mucous membranes and can result in scarring, blindness, and even death. Like other nonaspirin NSAIDs, ibuprofen may pose a risk for MI and stroke.
Oral Preparations and Dosages
Ibuprofen, by itself, is available in five oral formulations: (1) standard tablets (100, 200, 400, 600, and 800 mg); (2) chewable tablets (50 and 100 mg); (3) capsules (200 mg); (4) a 20-mg/mL oral suspension [Children’s Advil, Children’s Motrin, PediaCare Fever]; and (5) a 40-mg/mL oral suspension [Advil Pediatric Drops, Motrin Infant’s, PediaCare Fever]. Administration with meals or milk can reduce gastric distress.
Dosages for adults are as follows:
• Arthritis—1.2 to 3.2 g/day administered in three or four divided doses
Dosages for children are as follows:
• Juvenile arthritis—30 to 40 mg/kg/day in three or four divided doses
• Fever reduction—5 mg/kg every 6 to 8 hours (for temperatures up to 102.5° F) or 10 mg/kg every 6 to 8 hours (for temperatures above 102.5° F) as needed. The total daily dose should not exceed 40 mg/kg.
Oral ibuprofen is also available in three fixed-dose combinations: (1) ibuprofen/oxycodone [Combunox] for short-term oral therapy of moderate to severe pain, (2) ibuprofen/hydrocodone [Vicoprofen] for short-term oral therapy of moderate to severe pain, and (3) ibuprofen/famotidine [Duexis] for treatment of rheumatoid arthritis and osteoarthritis, while reducing the risk for GI ulcers.
Nonacetylated Salicylates: Magnesium Salicylate, Sodium Salicylate, and Salsalate
Similarities to Aspirin
The nonacetylated salicylates are similar to aspirin (an acetylated salicylate) in most respects. Like aspirin, these drugs inhibit COX-1 and COX-2 and are employed to treat arthritis, moderate pain, and fever. The most common adverse effects are GI disturbances. As with aspirin, these drugs should not be given to children with chickenpox or influenza owing to the possibility of precipitating Reye syndrome.
Contrasts With Aspirin
In contrast to aspirin, the nonacetylated salicylates cause little or no suppression of platelet aggregation. As a result, these drugs cannot protect against MI and stroke and may actually increase risk.
Because of its sodium content, sodium salicylate should be avoided in patients on a sodium-restricted diet (e.g., patients with hypertension or heart failure).
Magnesium salicylate may accumulate to toxic levels in patients with chronic renal insufficiency and hence should not be used by these people.
Salsalate is a prodrug that breaks down to release two molecules of salicylate in the alkaline environment of the small intestine. Because the stomach is not exposed to salicylate, salsalate produces less gastric irritation than aspirin.
Preparations, Dosage, and Administration
Magnesium salicylate [Doan’s Tablets, others] is supplied in 580-mg caplets and tablets for oral use. The usual dosage is 1090 mg every 6 hours. The maximal dosage is 4640 mg/day, administered in three or four doses.
Sodium salicylate (generic) is supplied in a combination pill with methenamine marketed as Cystex (162.5 mg/162 mg). Dosing is 2 tablets with a full glass of water 4 times a day for treatment of urinary pain.
Salsalate is supplied in capsules (500 mg) and tablets (500 and 750 mg) for oral use. The usual dosage is 3000 mg/day in divided doses.
Fenoprofen
Fenoprofen [Nalfon] belongs to the propionic acid family of NSAIDs. Like other NSAIDs, the drug inhibits synthesis of prostanoids, thereby causing antiinflammatory, analgesic, and antipyretic effects. Fenoprofen is indicated for arthritis and mild to moderate pain. The most common adverse effects are GI disturbances. Fenoprofen is supplied in tablets (600 mg) and capsules (200 and 400 mg). The usual dosage for mild to moderate pain is 200 mg every 4 to 6 hours as needed. The dosage range for rheumatoid arthritis and osteoarthritis is 300 to 600 mg every 6 to 8 hours, but should not exceed 3.2 g/day.
Flurbiprofen
Flurbiprofen is chemically related to ibuprofen and the other derivatives of propionic acid. The drug is approved for arthritis and has been used investigationally for bursitis, tendinitis, moderate pain, fever, and primary dysmenorrhea. The most common adverse effects are GI disturbances (dyspepsia, nausea, diarrhea, abdominal pain). The risk for serious GI effects (ulceration, perforation, hemorrhage) may be greater than with ibuprofen. Like other NSAIDs, flurbiprofen can exacerbate renal impairment and may pose a risk for MI and stroke. The drug is supplied in tablets (50 and 100 mg) for oral use. The usual dosage for rheumatoid arthritis is 200 to 300 mg/day administered in two to four divided doses.
Ketoprofen
Ketoprofen belongs to the propionic acid family of NSAIDs. The drug inhibits synthesis of prostanoids and has antiinflammatory, analgesic, and antipyretic effects. Indications are rheumatoid arthritis, osteoarthritis, mild to moderate pain, and primary dysmenorrhea. The most common adverse effects are dyspepsia, nausea, vomiting, and abdominal pain. Ketoprofen is supplied in immediate-release capsules (50 and 75 mg) and extended-release capsules (100, 150, and 200 mg). The usual dosage for rheumatoid arthritis is 200 to 225 mg/day administered in three or four divided doses. The dosage for moderate pain or primary dysmenorrhea is 25 to 50 mg every 6 to 8 hours as needed.
Naproxen
Actions and Uses
Naproxen [Aleve, Anaprox, Naprelan, Naprosyn], a member of the propionic acid family of NSAIDs, is highly selective for COX-1. The drug has a prolonged half-life and so can be administered less frequently than other propionic acid derivatives (e.g., ibuprofen). Naproxen is approved for arthritis, bursitis, tendinitis, primary dysmenorrhea, fever, and mild to moderate pain. Like other NSAIDs, the drug acts primarily by inhibiting cyclooxygenase.
Adverse Effects
Naproxen is among the better tolerated NSAIDs. The most common adverse effects are GI disturbances. Like other NSAIDs, the drug can compromise renal function and may increase the risk for MI and stroke. However, because naproxen is COX-1 selective, the risk for MI and stroke appears lower than with other traditional NSAIDs. Bleeding time can be prolonged secondary to reversible inhibition of platelet aggregation.
Preparations, Dosage, and Administration
Naproxen is supplied in immediate-release tablets (220, 250, 275, 375, and 500 mg) sold as Aleve, Anaprox, and Naprosyn; a 550-mg double-strength tablet sold as Anaprox DS; delayed-release enteric-coated tablets (375 and 500 mg) sold as EC-Naprosyn; controlled-release tablets (375, 500, and 750 mg) sold as Naprelan; and an oral suspension (25 mg/mL) sold as Naprosyn and Naproxen. For all products, the usual dosage for rheumatoid arthritis is 250 to 500 mg of naproxen twice daily. The dosage for mild to moderate pain is 500 mg initially followed by 250 mg every 6 to 8 hours as needed.
Naproxen/Esomeprazole [Vimovo]
Vimovo is a fixed-dose combination of naproxen plus esomeprazole, a PPI that blocks production of gastric acid (see Chapter 62), and thereby protects against naproxen-induced ulcers. Vimovo delayed-release tablets are available in two naproxen/esomeprazole strengths: 375 mg/20 mg and 500 mg/20 mg. For patients with osteoarthritis, rheumatoid arthritis, or ankylosing spondylitis, the usual dosage is 1 tablet twice daily.
Oxaprozin
Oxaprozin [Daypro] belongs to the propionic acid family of NSAIDs. Approved uses are rheumatoid arthritis and osteoarthritis. As with other NSAIDs, benefits derive from inhibiting synthesis of prostanoids. Like other propionic acid derivatives, oxaprozin is generally well tolerated. The drug has an unusually long half-life (42–50 hours) and hence can be administered just once a day. Oxaprozin is available in 600-mg tablets. The dosage for arthritis is 1200 mg once a day. The maximal dosage is 1800 mg/day.
Diclofenac
Oral
Oral diclofenac [Voltaren XR, Cataflam, Cambia, Zipsor, Zorvolex] is approved for rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, mild pain, primary dysmenorrhea, and migraine. As with other NSAIDs, antiinflammatory, analgesic, and antipyretic effects result from inhibiting cyclooxygenase. Diclofenac is well absorbed after oral administration but undergoes extensive (40%–50%) metabolism on its first pass through the liver. In the blood, about 99.5% of the drug is protein bound, primarily to albumin. Diclofenac is metabolized by the liver and excreted in the urine.
The most common adverse effects are abdominal pain, dyspepsia, and nausea. By impairing renal function, diclofenac can cause fluid retention, which can exacerbate hypertension and heart failure. Diclofenac can cause severe liver injury, even with topical therapy. Accordingly, patients should receive periodic tests of liver function and should be instructed to report manifestations of liver injury (e.g., jaundice, fatigue, nausea). If liver injury is diagnosed, diclofenac should be discontinued.
Diclofenac is supplied in immediate-release tablets (50 mg) as Cataflam, generic enteric-coated delayed-release tablets (25, 50, and 75 mg), extended-release tablets (100 mg) as Voltaren XR, liquid-filled capsules (25 mg) as Zipsor, immediate-release tablets (18 and 35 mg) as Zorvolex, and a powder for oral solution (50 mg) as Cambia. The dosage for rheumatoid arthritis is 150 to 200 mg/day administered in two or three divided doses. The dosage for osteoarthritis is 100 to 150 mg/day administered in two or three divided doses. The Zorvolex dose for mild to moderate pain is 54 to 105 mg/day administered in three divided doses.
Topical
Diclofenac is available in three topical formulations—Voltaren Gel, Flector Patch, and Pennsaid (solution)—for treatment of pain and inflammation. Voltaren Gel is for osteoarthritis, Flector Patch is for minor pain, and Pennsaid is for osteoarthritis of the knee. A fourth topical formulation—Solaraze—is used for actinic keratoses (see Chapter 85). Topical diclofenac is more expensive than oral diclofenac, but also safer: with topical therapy, blood levels are only about 5% of those achieved with oral therapy, and hence the risk for systemic toxicity is low. Efficacy of topical diclofenac appears about equal to that of oral therapy. Whether topical diclofenac shares the drug interactions of oral diclofenac has not been determined.
Voltaren Gel (1% diclofenac sodium) was the first prescription topical NSAID for treating pain and inflammation. Application is done to the knees, elbows, and other amenable joints. For joints of the lower extremity (knees, ankles, feet), the dosage is 4 g of gel applied 4 times a day. For joints of the upper extremity (elbows, wrists, hands), the dosage is 2 g of gel applied 4 times a day. Total body exposure should not exceed 32 g/day. Treated areas should be protected from sunlight (natural or artificial). Local dermatitis is the principal adverse effect.
Flector Patch (1.3% diclofenac epolamine) is the first prescription NSAID patch indicated for pain of strains, sprains, and contusions. One patch is applied over the injury twice a day—but only to skin that is intact. The patch should not be worn while bathing or showering. Local reactions—pruritus, dermatitis, burning—are the principal adverse effects.
Pennsaid is a 1.5% or 2% solution of diclofenac sodium, formulated with 45% dimethyl sulfoxide (DMSO) to facilitate skin penetration. The product has only one indication: osteoarthritis of the knee. Efficacy equals that of oral diclofenac. Application is done 4 times a day. For each application, 40 drops are spread around the entire knee (front, back, and sides). Application-site reactions are common. Among these are dry skin, erythema and induration, pruritus, and contact dermatitis with vesicles. Systemic effects are minimal. Patients may experience a garlicky odor or taste owing to the DMSO. As with Voltaren Gel, the treated area should not be exposed to sunlight, natural or artificial.
Diclofenac/Misoprostol [Arthrotec]
Oral diclofenac, in combination with misoprostol, is available under the trade name Arthrotec. Misoprostol is a prostaglandin analog that can protect against NSAID-induced ulcers. The combination product is approved for patients with rheumatoid arthritis or osteoarthritis who are at high risk for NSAID-induced gastric or duodenal ulcers. In patients with arthritis, the combination is as effective as diclofenac alone and produces significantly less GI ulceration. The most bothersome side effect is diarrhea (caused by misoprostol). Misoprostol can induce uterine contraction, and hence the product is contraindicated for use during pregnancy. Diclofenac/misoprostol is supplied in two strengths: 50 mg/200 mcg and 75 mg/200 mcg. For rheumatoid arthritis or osteoarthritis, the usual dosage is 50 mg/200 mcg 3 or 4 times a day.
Diflunisal
Diflunisal is a derivative of salicylic acid. However, unlike the salicylates, diflunisal is not converted to salicylic acid in the body. The drug is indicated for mild to moderate pain, rheumatoid arthritis, and osteoarthritis. Like other NSAIDs, the drug inhibits prostaglandin synthesis and can cause GI disturbances, suppression of platelet aggregation, and renal impairment and may increase the risk for MI and stroke. Diflunisal has a prolonged half-life (11–15 hours) and hence can be administered only 2 or 3 times a day. Diflunisal is supplied in tablets (250 and 500 mg) for oral use. For treatment of arthritis and mild to moderate pain, the initial dose is 500 to 1000 mg. Maintenance doses of 250 to 500 mg are administered every 8 to 12 hours.
Etodolac
Etodolac is indicated for rheumatoid arthritis, osteoarthritis, and moderate pain. Investigational uses include bursitis and tendinitis. Like other NSAIDs, etodolac produces many of its effects by suppressing the synthesis of prostanoids. The most common adverse effects are dyspepsia, nausea, vomiting, diarrhea, and abdominal pain. Etodolac may cause less gastric ulceration and bleeding than other NSAIDs. The drug is supplied in immediate-release tablets (400 and 500 mg), extended-release tablets (400, 500, and 600 mg), and capsules (200 and 300 mg). The recommended dosage for arthritis is 800 to 1200 mg/day of the extended-release medication or 400 to 1000 mg/day of the immediate-release medication in divided doses. The dosage for moderate pain is 200 to 400 mg every 6 to 8 hours.
Indomethacin
Actions and Uses
Indomethacin [Indocin, Tivorbex] is an effective antiinflammatory agent approved for arthritis, bursitis, tendinitis, and, as discussed in Chapter 58, acute gouty arthritis. Although indomethacin is able to reduce pain and fever, it is not routinely used for these effects, owing to potential toxicity.
Pharmacokinetics
Indomethacin is well absorbed after oral administration and distributes to all body fluids and tissues. The drug is metabolized in the liver. Metabolites and parent drug are excreted in the urine and feces.
Adverse Effects
Untoward effects are seen in 35% to 50% of patients, causing about 20% to discontinue treatment. The most common adverse effect is severe frontal headache, which occurs in 25% to 50% of patients. Other CNS effects (dizziness, vertigo, confusion) are also common. Seizures and psychiatric changes (e.g., depression, psychosis) have occurred. Mild GI reactions (nausea, vomiting, indigestion) develop in 3% to 9% of users. More severe GI effects (ulceration with perforation, hemorrhage) may also occur. Hematologic reactions (neutropenia, thrombocytopenia, aplastic anemia) have occurred but are rare. Indomethacin suppresses platelet aggregation.
Precautions and Contraindications
Because of its adverse effects, indomethacin is generally contraindicated for infants and children younger than 14 years, patients with peptic ulcer disease, and women who are pregnant or breastfeeding. Caution is required in patients with seizures and psychiatric disorders, in patients involved in hazardous activities, and in patients receiving anticoagulant therapy.
Preparations, Dosage, and Administration
Indomethacin [Indocin] is available in immediate-release capsules (25 and 50 mg), extended-release capsules (75 mg), an oral suspension (5 mg/mL), and rectal suppositories (50 mg). For treatment of rheumatoid arthritis, the initial dosage is 25 mg 2 or 3 times a day. The maximal daily dosage is 200 mg. Gastrointestinal reactions can be reduced by dosing with meals.
A new form of indomethacin was recently approved and is available in 20- and 40-mg capsules sold as Tivorbex. Tivorbex is unique because the capsules contain particles that are 20 times smaller than traditional indomethacin particles. This allows for increased dissolution, thus producing an equianalgesic effect at smaller doses and therefore less toxic side effects.
Ketorolac
Actions and Uses
Ketorolac is a powerful analgesic with minimal antiinflammatory actions. Pain relief is equivalent to that produced by morphine and other opioids. Although ketorolac lacks the serious adverse effects associated with opioids (respiratory depression, tolerance, dependence, abuse potential), it nonetheless has serious adverse effects of its own. Accordingly, use should be short term and restricted to managing acute pain of moderate to severe intensity. Ketorolac is not indicated for chronic pain or for minor aches and discomfort. The usual indication is postoperative pain, for which ketorolac can be as effective as morphine. Like other NSAIDs, ketorolac suppresses prostaglandin synthesis. This action is thought to underlie analgesic effects.
Pharmacokinetics
Ketorolac is administered orally and parenterally (intramuscularly or intravenously). The drug is eliminated by hepatic metabolism and urinary excretion. In young adults, ketorolac has a half-life of 4 to 6 hours. The half-life may be prolonged in older adults and in those with renal impairment.
Adverse Effects and Contraindications
Ketorolac can cause all of the adverse effects associated with other NSAIDs, including peptic ulcers, GI bleeding or perforation, prolonged bleeding time, renal impairment, hypersensitivity reactions, suppression of uterine contractions, and premature closure of the ductus arteriosus. Concurrent use with other NSAIDs increases the risk for these effects and hence is contraindicated. Other contraindications include active peptic ulcer disease, history of peptic ulcer disease or recent GI bleeding, advanced renal impairment, confirmed or suspected intracranial bleeding, use before major surgery, history of NSAID hypersensitivity reactions, and use during labor and delivery.
Preparations, Dosage, and Administration
Oral Therapy.
Ketorolac is available in 10-mg tablets for oral dosing. Dosing is parenteral initially, followed by oral dosing if needed. Owing to risks associated with prolonged use, treatment (parenteral plus oral) should not exceed 5 days.
Oral ketorolac is indicated only as a follow-up to parenteral therapy. Initial oral doses are based on preceding parenteral doses. The usual oral maintenance dosage is 10 mg every 4 to 6 hours. Combined oral and parenteral treatment should not exceed 5 days.
Intranasal Therapy.
Ketorolac [Sprix] is available in a metered-dose spray device (15.75 mg/actuation) for short-term, intranasal treatment of moderate to moderately severe pain. As with oral and parenteral therapy, treatment should not exceed 5 days. Dosage depends on weight, age, and renal function. For patients with normal renal function who weigh more than 50 kg, the usual dosage is 2 sprays (one 15.75-mg spray in each nostril) every 6 to 8 hours as needed. The dosage is lower—one 15.75-mg spray in one nostril every 6 to 8 hours as needed—for older adults and those who have impaired kidney function or weigh less than 50 kg.
Meclofenamate
Meclofenamate is indicated for rheumatoid arthritis, osteoarthritis, mild to moderate pain, and dysmenorrhea. As with other NSAIDs, benefits derive from inhibiting cyclooxygenase. Therapeutic effects are no better than with other NSAIDs, but adverse GI effects are greater: 3% to 9% of patients experience nausea, vomiting, abdominal pain, and cramps; worse yet, 10% to 33% develop diarrhea. Because of this poor benefit-to-risk profile, meclofenamate is not a drug of first choice. Meclofenamate is available in 50- and 100-mg capsules. Dosages are as follows: arthritis, 200 to 400 mg/day in three or four divided doses; moderate pain, 50 mg every 4 to 6 hours; and dysmenorrhea, 100 mg 3 times a day for up to 6 days.
Mefenamic Acid
Mefenamic acid [Ponstel, Ponstan  ] is indicated for relief of primary dysmenorrhea and moderate pain. The principal adverse effect is diarrhea, which can be severe. Mefenamic acid is supplied in 250-mg capsules. The dosage for primary dysmenorrhea is 500 mg initially followed by 250 mg every 6 hours as needed. The drug should be administered with food or milk to reduce gastric distress. Usual treatment duration is 2 to 3 days.
] is indicated for relief of primary dysmenorrhea and moderate pain. The principal adverse effect is diarrhea, which can be severe. Mefenamic acid is supplied in 250-mg capsules. The dosage for primary dysmenorrhea is 500 mg initially followed by 250 mg every 6 hours as needed. The drug should be administered with food or milk to reduce gastric distress. Usual treatment duration is 2 to 3 days.
Nabumetone
Nabumetone is a prodrug that undergoes hepatic conversion to its active form: 6-MNA. In contrast to most traditional NSAIDs, 6-MNA inhibits COX-2 more than COX-1. Although nabumetone has antipyretic, analgesic, and antiinflammatory properties, the drug is approved only for osteoarthritis and rheumatoid arthritis. Principal adverse effects are diarrhea, abdominal cramps, dyspepsia, and nausea. Nabumetone causes much less GI ulceration than other first-generation NSAIDs, possibly because it preferentially inhibits COX-2. Nabumetone is supplied in 500- and 750-mg tablets. Dosing with food increases the rate of absorption. Treatment of arthritis begins with a single 1000-mg dose. After this, the daily dosage is 1500 to 2000 mg administered in one or two doses. Dosage should be reduced in patients with renal impairment.
Piroxicam
Piroxicam [Feldene] has antiinflammatory, analgesic, and antipyretic properties but is approved only for rheumatoid arthritis and osteoarthritis. The drug’s most outstanding feature is its long half-life (about 50 hours). Because piroxicam is eliminated so slowly, therapeutic effects can be maintained with once-a-day dosing. In general, piroxicam is better tolerated than aspirin. Undesired effects are seen in 11% to 46% of patients, causing between 4% and 12% to discontinue therapy. Gastrointestinal reactions are most common, occurring in about 20% of patients. The incidence of gastric ulceration is about 1%. Like aspirin, piroxicam inhibits platelet aggregation and prolongs bleeding time. The drug is supplied in 10- and 20-mg capsules for oral use. The usual dosage is 20 mg once a day or 10 mg twice daily.
Sulindac
Sulindac [Clinoril] is a prodrug that undergoes conversion to its active form in the body. The drug is approved for rheumatoid arthritis, osteoarthritis, tendinitis, bursitis, and acute gouty arthritis. Principal adverse effects are abdominal distress, dyspepsia, nausea, vomiting, and diarrhea. Gastric ulceration is less common than with some other NSAIDs. Like other NSAIDs, sulindac causes reversible inhibition of platelet aggregation, prolongs bleeding time, and impairs renal function. The drug is supplied in 150- and 200-mg tablets. The usual dosage is 150 mg administered twice daily with meals. The maximal daily dosage is 400 mg.
Tolmetin
Tolmetin is approved for rheumatoid arthritis and osteoarthritis. The drug has analgesic and antipyretic properties but is not employed to relieve fever or pain unrelated to inflammation. Adverse effects occur in 25% to 40% of patients, causing between 5% and 10% to discontinue treatment. Gastrointestinal effects (nausea, vomiting, indigestion) are most common. Gastric ulceration has occurred, but less frequently than with aspirin. Nonetheless, caution should be exercised in patients with a history of peptic ulcer disease. Hypersensitivity reactions are more common than with aspirin. Effects on the CNS (headache, dizziness, anxiety, drowsiness) are less severe and less frequent than with indomethacin. Unlike most other NSAIDs, tolmetin does not augment the effects of warfarin, an oral anticoagulant. The drug is supplied in tablets (200 and 600 mg) and capsules (400 mg). For rheumatoid arthritis, the initial dosage is 400 mg 3 times a day. The maximal daily dosage is 1.8 g. Gastrointestinal distress can be minimized by dosing with food.
Meloxicam
Meloxicam [Mobic, Vivlodex, Mobicox  ] can inhibit COX-1 and COX-2 but shows selectivity for COX-2 at low doses. Like other NSAIDs, the drug has analgesic, antiinflammatory, and antipyretic actions. Approved indications are osteoarthritis, rheumatoid arthritis, and pauciarticular/polyarticular-course juvenile rheumatoid arthritis (JRA). For osteoarthritis, meloxicam is as effective as first-generation NSAIDs. Direct comparison with true COX-2 inhibitors (e.g., celecoxib) has not been made. Despite its COX-2 selectivity, meloxicam has a side-effect profile like that of the first-generation NSAIDs. Gastrointestinal effects (abdominal pain, constipation, diarrhea, dyspepsia, flatulence, nausea, and vomiting) occur in 20% to 25% of patients. More serious effects—GI ulceration, bleeding, perforation, and death—have also occurred. Meloxicam does not suppress platelet aggregation. The drug has a long half-life (15–20 hours) and undergoes elimination in the urine (50%) and feces (50%). Meloxicam is available in tablets (7.5 and 15 mg) and an oral solution (7.5 mg/5 mL). Both formulations can be taken with or without food. Because of its long half-life, meloxicam can be administered just once a day. The recommended dose for initial and maintenance therapy of osteoarthritis and rheumatoid arthritis is 7.5 mg/day. For patients with JRA, the recommended daily dosage is 0.125 mg/kg (but no more than 7.5 mg).
] can inhibit COX-1 and COX-2 but shows selectivity for COX-2 at low doses. Like other NSAIDs, the drug has analgesic, antiinflammatory, and antipyretic actions. Approved indications are osteoarthritis, rheumatoid arthritis, and pauciarticular/polyarticular-course juvenile rheumatoid arthritis (JRA). For osteoarthritis, meloxicam is as effective as first-generation NSAIDs. Direct comparison with true COX-2 inhibitors (e.g., celecoxib) has not been made. Despite its COX-2 selectivity, meloxicam has a side-effect profile like that of the first-generation NSAIDs. Gastrointestinal effects (abdominal pain, constipation, diarrhea, dyspepsia, flatulence, nausea, and vomiting) occur in 20% to 25% of patients. More serious effects—GI ulceration, bleeding, perforation, and death—have also occurred. Meloxicam does not suppress platelet aggregation. The drug has a long half-life (15–20 hours) and undergoes elimination in the urine (50%) and feces (50%). Meloxicam is available in tablets (7.5 and 15 mg) and an oral solution (7.5 mg/5 mL). Both formulations can be taken with or without food. Because of its long half-life, meloxicam can be administered just once a day. The recommended dose for initial and maintenance therapy of osteoarthritis and rheumatoid arthritis is 7.5 mg/day. For patients with JRA, the recommended daily dosage is 0.125 mg/kg (but no more than 7.5 mg).
Meloxicam is now available in a “Solumatrix” form using “fine particle technology” sold as Vivlodex. Capsules (5 and 10 mg) of Vivlodex contain particles of meloxicam that are 10 times smaller than traditional tablets. This allows for adequate pain control in osteoarthritis at 33% lower doses. Using less drug can decrease the toxic side effects of meloxicam.
 Black Box Warning: First-Generation Nonsteroidal Antiinflammatory Drugs
Black Box Warning: First-Generation Nonsteroidal Antiinflammatory Drugs
All first-generation NSAIDs are associated with increased risk for gastrointestinal bleeding and cardiovascular events that can lead to hospitalization or death.
Second-Generation Nonsteroidal Antiinflammatory Drugs (Cyclooxygenase-2 Inhibitors, Coxibs)
The COX-2 inhibitors, also known as coxibs, were developed on the theory that selective inhibition of COX-2 should be able to suppress pain and inflammation while posing little or no risk for gastric ulceration. To some degree, theory and reality agree: coxibs are just as effective as traditional NSAIDs at suppressing inflammation and pain, and they pose a somewhat lower risk for GI side effects. However, even with coxibs, patients can develop clinically significant gastroduodenal ulceration and bleeding. Furthermore, like traditional NSAIDs, coxibs can impair renal function and can thereby cause hypertension and edema. Coxibs also increase the risk for MI and stroke.
Celecoxib
Therapeutic Use
Celecoxib [Celebrex] was the first selective COX-2 inhibitor to reach the market. The drug is indicated for osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, juvenile idiopathic arthritis, acute pain, and dysmenorrhea. In addition, celecoxib is used off-label for a rare genetic disorder known as familial adenomatous polyposis, which predisposes to development of colorectal cancer. For patients with arthritis, celecoxib is equal to naproxen (an NSAID) at relieving joint pain, stiffness, and swelling. Owing to concerns about cardiovascular safety, celecoxib is considered a last-choice drug for long-term management of pain (see later under “American Heart Association Statement on Cyclooxygenase Inhibitors in Chronic Pain”).
It is important to note that celecoxib does not provide the cardiovascular benefits of aspirin because celecoxib does not inhibit COX-1 in platelets and hence does not suppress platelet aggregation.
Mechanism of Action
Celecoxib causes selective inhibition of COX-2, the COX isoform whose products mediate inflammation and pain. At therapeutic doses, celecoxib does not inhibit COX-1, the COX isoform whose products protect the stomach, help maintain renal function, and promote platelet aggregation.
Pharmacokinetics
Celecoxib is well absorbed after oral administration. Plasma levels peak in 3 hours. Binding to plasma proteins is extensive (97%). The drug undergoes hepatic metabolism followed by renal excretion. The half-life is 11 hours.
Adverse Effects
In premarketing trials, celecoxib was well tolerated. The discontinuation rate owing to adverse effects was 7.1% for celecoxib versus 6.1% for placebo. The most common complaints were dyspepsia and abdominal pain. Celecoxib does not decrease platelet aggregation and hence does not promote bleeding. Possible cardiovascular events are the biggest concern.
Gastroduodenal Ulceration
Because celecoxib does not inhibit COX-1, the isoform of COX that protects the stomach, a low incidence of gastroduodenal ulceration would be expected. Some data support this expectation; others do not. When celecoxib was first approved, conclusions about its safety were based on 6-month data from the Celecoxib Arthritis Safety Study (CLASS), which indicated that celecoxib caused less GI toxicity than conventional NSAIDs (diclofenac, naproxen, ibuprofen). However, longer term (12-month) data from the same study showed no difference in GI toxicity between celecoxib and conventional NSAIDs. Other studies have shown that, compared with patients taking conventional NSAIDs, those taking celecoxib had a lower incidence of endoscopically detectable ulcers and a lower incidence of hospitalization for GI bleeding. What’s the bottom line? Celecoxib may be safer than conventional NSAIDs, especially when used short term. However, convincing data of superior safety are lacking. Like traditional NSAIDs, celecoxib can be combined with a PPI to reduce GI complications.
Cardiovascular Events
There is strong evidence that coxibs, like other nonaspirin NSAIDs, increase the risk for MI, stroke, and other serious cardiovascular events. In the Adenoma Prevention with Celecoxib (APC) trial, patients who took 400 mg or 800 mg of celecoxib a day experienced more major fatal or nonfatal cardiovascular events than did patients who took placebo. To minimize risk, celecoxib should be used in the lowest effective dosage for the shortest time needed. Also, the drug should be avoided in patients with existing heart disease and those who have just undergone CABG surgery, and should be used with caution in patients with cardiovascular risk factors, such as hypertension, diabetes, and dyslipidemia. Other measures to reduce risk are discussed later under “American Heart Association Statement on Cyclooxygenase Inhibitors in Chronic Pain.”
Why is the risk for MI and stroke increased? First, because celecoxib does not inhibit COX-1, platelet aggregation is not suppressed. Second, because celecoxib inhibits COX-2 in blood vessels, vasoconstriction is increased. These two factors—unimpeded platelet aggregation and increased vasoconstriction—increase the likelihood of vessel blockage after the process of thrombosis has begun.
Renal Impairment
Like conventional NSAIDs, celecoxib can impair renal function, thereby posing a risk to patients with hypertension, edema, heart failure, or kidney disease. Renal impairment apparently results from inhibiting COX-2.
Sulfonamide Allergy
Celecoxib contains a sulfur molecule and hence can precipitate an allergic reaction in patients allergic to sulfonamides. Accordingly, the drug should be avoided by patients with sulfa allergy.
Use in Pregnancy
Celecoxib and other NSAIDs can cause premature closure of the ductus arteriosus. Accordingly, these drugs are contraindicated during the third trimester of pregnancy.
Drug Interactions
Warfarin
Celecoxib may increase the anticoagulant effects of warfarin and may thereby increase the risk for bleeding. Celecoxib itself does not inhibit platelet aggregation and does not promote bleeding. However, the drug may enhance the anticoagulant effects of warfarin (perhaps by increasing warfarin levels). Celecoxib may be combined with warfarin, but effects of warfarin should be monitored closely, especially during the first few days of treatment.
Other Interactions
Information on the interactions of celecoxib with other drugs is limited. Celecoxib may decrease the diuretic effects of furosemide as well as the antihypertensive effects of ACE inhibitors. Conversely, celecoxib may increase levels of lithium (a drug for bipolar disorder). Levels of celecoxib may be increased by fluconazole (an antifungal drug).
Preparations, Dosage, and Administration
Celecoxib [Celebrex] is available in capsules (50, 100, 200, and 400 mg). To minimize cardiovascular risk, the drug should be used in the lowest effective dosage for the shortest time needed. Approved dosages are as follows:

Acetaminophen
Acetaminophen [Tylenol, Ofirmev, many others] is like aspirin in some respects but different in others. Acetaminophen has analgesic and antipyretic properties equivalent to those of aspirin. However, in contrast to aspirin and the other NSAIDs, acetaminophen is devoid of clinically useful antiinflammatory and antirheumatic actions. In addition, acetaminophen does not suppress platelet aggregation, does not cause gastric ulceration, and does not decrease renal blood flow or cause renal impairment. However, acetaminophen overdose can cause severe liver injury. In the United States acetaminophen is used more than any other analgesic.
Mechanism of Action
Differences between the effects of acetaminophen and aspirin are thought to result from selective inhibition of cyclooxygenase, the enzyme needed to make prostaglandins and related compounds. Whereas aspirin can inhibit cyclooxygenase in both the CNS and the periphery, inhibition by acetaminophen is limited to the CNS; acetaminophen has only minimal effects on cyclooxygenase at peripheral sites. By decreasing prostaglandin synthesis in the CNS, acetaminophen is able to reduce fever and pain. The inability to inhibit prostaglandin synthesis outside the CNS may explain the absence of antiinflammatory effects, gastric ulceration, and adverse effects on the kidneys and platelets.
Pharmacokinetics
Acetaminophen is readily absorbed after oral dosing and undergoes wide distribution. Most of each dose is metabolized by the liver, and the metabolites are excreted in the urine. The plasma half-life is approximately 2 hours.
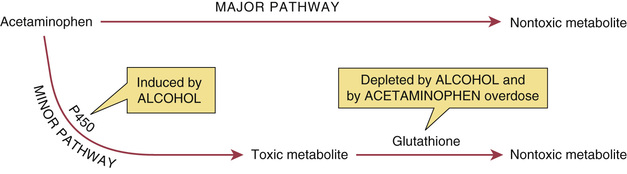
Acetaminophen can be metabolized by two pathways; one is major, and the other is minor (Fig. 55.1). In the major pathway, acetaminophen undergoes conjugation with glucuronic acid and other compounds to form nontoxic metabolites. In the minor pathway, acetaminophen is oxidized by a cytochrome P450–containing enzyme into a highly reactive toxic metabolite: N-acetyl-p-benzoquinone imine. At therapeutic doses, practically all of the drug is converted to nontoxic metabolites through the major pathway. Only a small fraction is converted into the toxic metabolite through the minor pathway. Furthermore, under normal conditions, the toxic metabolite undergoes rapid conversion to a nontoxic form; glutathione is required for the conversion. In the event of acetaminophen overdose, a larger than normal amount is processed through the minor pathway, and hence a large quantity of the toxic metabolite is produced. As the liver attempts to clear the metabolite, glutathione is rapidly depleted, and further detoxification stops. As a result, the toxic metabolite accumulates, causing damage to the liver (see later).
Adverse Effects
Adverse effects are extremely rare at therapeutic doses. Acetaminophen does not cause gastric ulceration or renal impairment and does not inhibit platelet aggregation. In addition, there is no evidence linking acetaminophen with Reye syndrome. Individuals who are hypersensitive to aspirin only rarely experience cross-hypersensitivity with acetaminophen. Overdose can cause severe liver injury (see later).
Data from the Nurses’ Health Study show an association between daily use of acetaminophen (500 mg or more/day) and development of hypertension. Additional studies that examined a possible relationship between acetaminophen and hypertension in both men and women found conflicting results. Until more data become available, it would be prudent to monitor blood pressure in patients who take acetaminophen daily. The mechanism by which acetaminophen might raise blood pressure is unknown.
Studies have shown an association between acetaminophen and development of asthma. However, as with hypertension, a causal relationship has not been established. In fact, regarding asthma, the association may well be the other way around. That is, people may be taking acetaminophen because they have respiratory symptoms, rather than having respiratory symptoms because they took acetaminophen. To prove that acetaminophen actually does cause asthma, stronger data are needed.
Rarely, patients experience anaphylaxis, a severe hypersensitivity reaction characterized by breathing difficulty associated with swelling of the face, mouth, and throat. If these symptoms develop, patients should seek immediate medical help.
Acetaminophen use has also been associated with Stevens-Johnson Syndrome (SJS), acute generalized exanthematous pustulosis (AGEP), and toxic epidermal necrolysis (TEN). SJS and TEN are characterized by painful rash, blistering of the skin and mucous membranes, and detachment of the epidermis. These are considered medical emergencies because they can result in death. Recovery can take weeks to months. AGEP is characterized by pustular lesions that predominately affect the upper trunk and body folds. AGEP usually resolves within 2 weeks of onset. These reactions can occur at any time, even if the patient has taken acetaminophen previously. If rash appears while taking acetaminophen, the drug should be stopped and the patient should seek medical attention.
Drug and Vaccine Interactions
Alcohol
Regular alcohol consumption increases the risk for liver injury from acetaminophen—but only if acetaminophen dosage is excessive. Three mechanisms are involved. First, alcohol induces synthesis of the P450-containing enzyme in the minor metabolic pathway, thereby increasing production of acetaminophen’s toxic metabolite (see Fig. 55.1). Second, stores of glutathione are depleted in chronic alcoholics. As a result, the liver is unable to convert the toxic metabolite to a nontoxic form. Third, chronic alcohol abusers often have preexisting liver damage, which renders them less able to tolerate injury from acetaminophen.
Alcohol in combination with acetaminophen can increase the risk for liver and kidney damage. Although information regarding liver disease and acetaminophen has existed for some time, newer data reveal that even low-dose combinations of alcohol and acetaminophen can lead to renal dysfunction. Authorities recommend that, if you drink alcohol on a regular basis, you should consume no more than 2000 mg of acetaminophen a day (one half the normal maximum).
Although therapeutic doses of acetaminophen may be safe for alcohol drinkers, high doses certainly are not. Accordingly, to alert the public to the potential risk of combining alcohol with acetaminophen, the FDA requires that acetaminophen labels bear the following statement: Alcohol Warning: If you consume three or more alcoholic drinks every day, ask your doctor whether you should take acetaminophen or other pain relievers/fever reducers.
Warfarin
There is evidence that acetaminophen may increase the risk for bleeding in patients taking warfarin. This is surprising because, unlike NSAIDs, acetaminophen does not suppress platelet aggregation and so should not promote bleeding. Yes, acetaminophen may inhibit warfarin metabolism, which would cause warfarin levels to rise. Although this interaction has not been proven, caution is advised. Accordingly, for patients taking more than 1 g of acetaminophen daily for several days, responses to warfarin should be monitored closely. Occasional use of acetaminophen is not a concern.
Vaccines
Acetaminophen and other analgesic-antipyretics can blunt the immune response to childhood vaccines. Accordingly, routine use of these drugs to prevent vaccination-associated pain or fever should be discouraged.
Therapeutic Uses
Acetaminophen is indicated for relief of pain and fever. Because acetaminophen is not associated with Reye syndrome, the drug is preferred to NSAIDs for use by children suspected of having chickenpox or influenza. Because it does not cause GI injury, acetaminophen is preferred to NSAIDs for patients with peptic ulcer disease. In addition, acetaminophen may be a safe alternative to aspirin for patients who have experienced aspirin hypersensitivity reactions. Because of its weak antiinflammatory actions, acetaminophen is not useful for treating arthritis or rheumatic fever.
Acute Toxicity: Liver Damage
Overdose with acetaminophen can cause severe liver injury and death. The cause is accumulation of the toxic metabolite discussed earlier. In the United States acetaminophen overdose—intentional or unintentional—is the leading cause of acute liver failure, accounting for about 50% of all cases. Risk for liver injury is increased by fasting, chronic alcohol use, and taking more than 4000 mg of acetaminophen a day.
Signs and Symptoms
The principal feature of acetaminophen overdose is hepatic necrosis. Severe poisoning can progress to hepatic failure, coma, and death. Early symptoms of poisoning (nausea, vomiting, diarrhea, sweating, abdominal discomfort) belie the severity of intoxication. It is not until 48 to 72 hours after drug ingestion that overt indications of hepatic injury appear.
Treatment
Liver damage can be minimized by giving acetylcysteine [Mucomyst  , Acetadote], a specific antidote to acetaminophen. Acetylcysteine reduces injury by substituting for depleted glutathione in the reaction that converts the toxic metabolite of acetaminophen to its nontoxic form. When given within 8 to 10 hours of acetaminophen overdose, acetylcysteine is 100% effective at preventing severe liver injury. And even when administered as much as 24 hours after poisoning, it can still provide significant protection. Acetylcysteine may be administered orally or intravenously.
, Acetadote], a specific antidote to acetaminophen. Acetylcysteine reduces injury by substituting for depleted glutathione in the reaction that converts the toxic metabolite of acetaminophen to its nontoxic form. When given within 8 to 10 hours of acetaminophen overdose, acetylcysteine is 100% effective at preventing severe liver injury. And even when administered as much as 24 hours after poisoning, it can still provide significant protection. Acetylcysteine may be administered orally or intravenously.
For oral therapy, acetylcysteine is supplied in solution (100 and 200 mg/mL) and should be diluted to 50 mg/mL with water, fruit juice, or a cola beverage. Conventional treatment consists of a loading dose (140 mg/kg) followed by 17 more doses (70 mg/kg) given every 4 hours for 72 hours. However, for most patients, treatment can be stopped after just 20 hours. Oral acetylcysteine has an extremely unpleasant odor and may induce vomiting. If the patient is unable to tolerate oral dosing, acetylcysteine can be administered intravenously or through a nasogastric tube.
Minimizing Risk
Risk for liver failure is very low with normal therapeutic doses (up to 4000 mg/day), except in people who drink alcohol, are undernourished, or have liver disease. Patient education can help reduce injury. Accordingly, you should do the following:
• Inform patients about the risk for liver toxicity.
• Advise patients to consume no more than 4000 mg of acetaminophen a day, including the amount in combination prescription products (e.g., Vicodin, Percocet) as well as over-the-counter products.
• Advise patients who are undernourished (e.g., owing to fasting or illness) to consume no more than 3000 mg of acetaminophen a day. Undernourished people are at risk because they have low stores of glutathione, the cofactor needed to convert the toxic metabolite of acetaminophen to a nontoxic form.
• Advise patients not to drink alcohol while taking acetaminophen.
• Advise patients who won’t stop drinking alcohol (more than 3 drinks a day) to take no more than 2000 mg of acetaminophen a day.
• Advise patients with liver disease to ask their prescriber if acetaminophen is safe.
To help reduce overdosage, McNeil Consumer Healthcare, maker of the Tylenol brand of acetaminophen, changed the dosing recommendations on Tylenol labels. On the new labels, issued in 2011, the maximal daily dose of Extra-Strength Tylenol (500 mg/tablet) is stated as 3000 mg (6 tablets), and the maximal daily dose of Regular Strength Tylenol (325 mg/tablet) is stated as 3250 mg (10 tablets). Other manufacturers are expected to make similar changes. Please note, however, that the maximal daily dose recommended by the FDA is still 4000 mg, even though these product labels recommend a lower dose.
Preparations, Dosage, and Administration
Preparations
Numerous acetaminophen-containing products are on the market, including a wide assortment of fixed-dose combinations. The drug is available in rectal suppositories, solution for intravenous dosing, and multiple oral formulations (standard tablets, chewable tablets, effervescent granules, capsules, liquids, elixirs, and solutions). Many products are available over the counter, and many others require a prescription. Furthermore, product strengths vary widely. All these products are mentioned here because they create a significant risk for overdose—either from taking two or more products that both contain acetaminophen or from taking too much of a single-ingredient product (owing to failure to carefully read the label). You should alert patients to these dangers.
Dosage and Administration
Oral.
The recommended oral dosage for adults and children older than 12 years is 325 to 650 mg every 4 to 6 hours, up to a maximum of 4000 mg/day. Dosages for younger children are based either on body weight—10 to 15 mg/kg/dose—or on age as follows:
• Up to 3 months—40 mg every 4 hours
• 4 to 11 months—80 mg every 4 hours
• 12 to 23 months—120 mg every 4 hours
• 2 to 3 years—160 mg every 4 hours
• 4 to 5 years—240 mg every 4 hours
• 6 to 8 years—320 mg every 4 to 6 hours
• 9 to 10 years—400 mg every 4 to 6 hours
Rectal.
Acetaminophen suppositories [FeverAll, Acephen] are available in four strengths: 80, 120, 325, and 650 mg. The recommended dosage for adults and children over 12 years is 650 mg every 4 to 6 hours, up to a maximum of 3900 mg/day. Dosages for younger children vary with age as follows:
American Heart Association (AHA) Statement on Cyclooxygenase Inhibitors in Chronic Pain
Because most COX inhibitors—and especially COX-2 inhibitors—increase the risk for MI and stroke, the AHA recommends a stepped-care approach to their use, as discussed in an article titled “Use of Nonsteroidal Anti-inflammatory Drugs: An Update for Clinicians: A Scientific Statement from the American Heart Association.” Recommendations in the article apply specifically to managing musculoskeletal pain in patients with or at high risk for cardiovascular disease. However, the recommendations may also apply to patients who lack documented cardiovascular risk. The approach has four basic steps:
Step 1. Begin with nondrug measures. Options include physical therapy, exercise, weight loss, orthotics, and application of heat or cold.
Step 2. If nondrug measures don’t work, initiate drug therapy using acetaminophen or aspirin, which do not increase cardiovascular risk. If these drugs can’t control pain, an opioid or tramadol can be tried, but only short term.
Step 3. If step 2 drugs are ineffective or intolerable, try other nonselective NSAIDs, such as naproxen, ibuprofen, or a nonacetylated salicylate (e.g., magnesium salicylate).
Step 4. As a last resort, try the selective COX-2 inhibitor celecoxib. Of all the NSAIDs, COX-2 inhibitors pose the greatest risk for cardiovascular harm, and hence celecoxib is a last-choice drug for chronic pain.
Whenever these drugs are employed, patients should use the lowest effective dosage for the shortest time required. During steps 2, 3, and 4, if the patient is considered at high risk for a thrombotic event, low-dose aspirin (81 mg/day) plus a PPI or H2RA should be added to the regimen (except, of course, if high-dose aspirin is already in use).

