[level-membership-for-neurosurgery-category]
Chapter 2 Current Understanding of Spinal Pain and the Nomenclature of Lumbar Disc Pathology
Any structure to be deemed a cause of back pain should:
Tissues capable of transmitting pain in the back are as follows:
General pain mechanism
The two categories of pain are as follows:
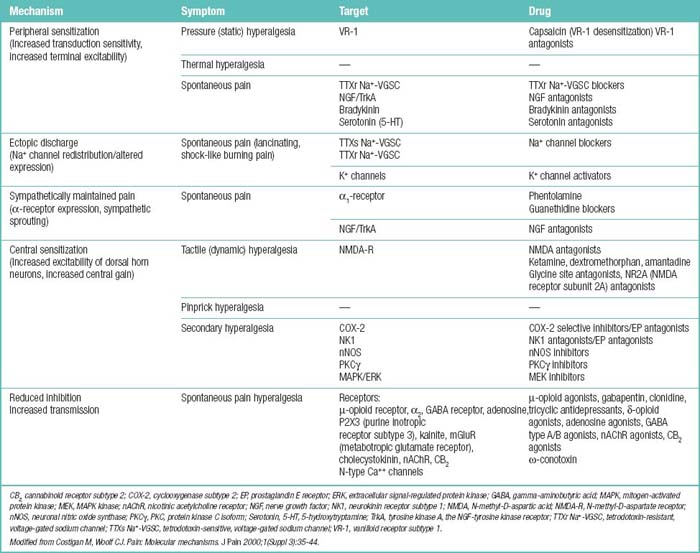
Potential associations between particular mechanisms and particular symptoms are as follows:
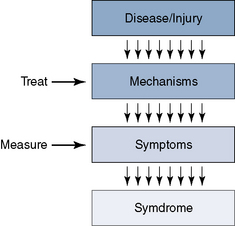
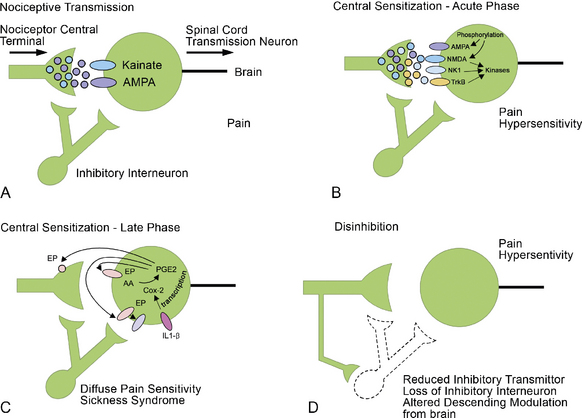
A number of different input channels can lead to the pain sensation. These should be the first anatomic targets for treatment (Fig. 2-1; Table 2.1), as follows:
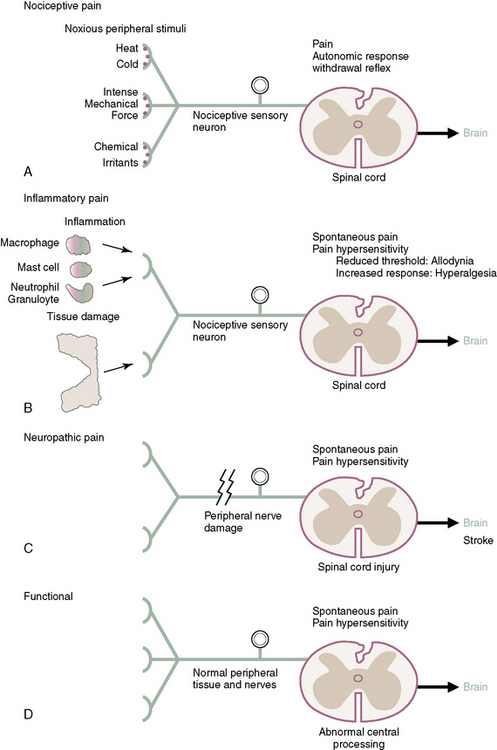
The four primary types of pain (Fig. 2-2) are as follows:
BOX 2.1 Positive and Negative Symptoms of Peripheral Neuropathic Pain
Woolf CJ: Dissecting out mechanisms responsible for peripheral neuropathic pain: implications for diagnosis and therapy. Life Sci 2004;74:2605-2610.
Adaptive pain and maladaptive pain are defined in Box 2.2.
BOX 2.2 Two Broad Classes of Pain
Modified from Woolf CJ; American College of Physicians; American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med 2004;140:441-451.
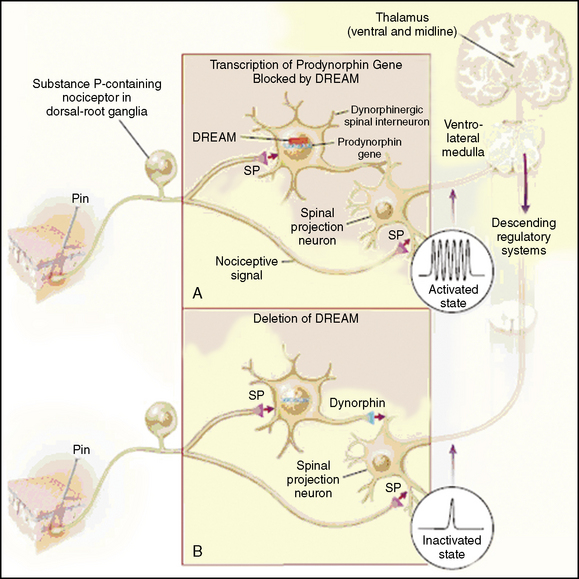
Multiple mechanisms that can produce pain, as follows:
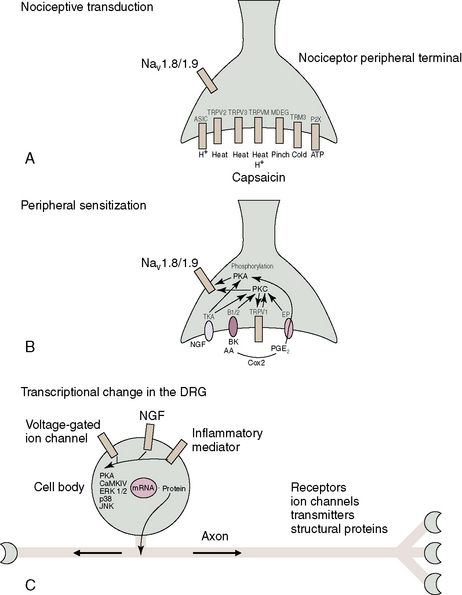
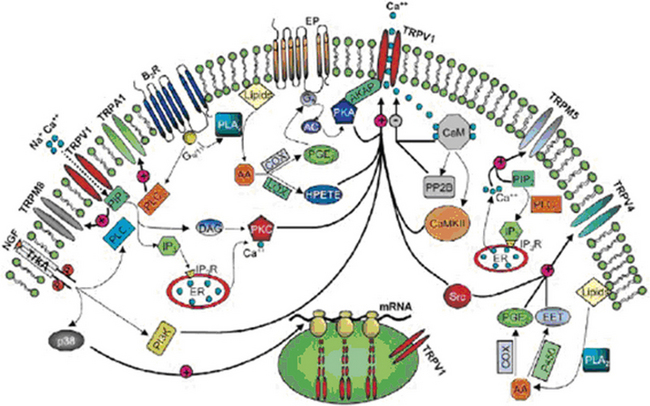
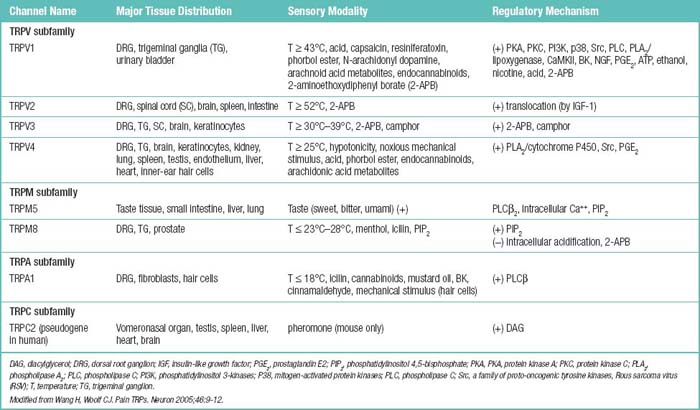
Pain transient receptor potential (TRP) ion channels are listed in Table 2.2 and illustrated in Figure 2-6. TRP ion channels are molecular gateways in sensory systems, an interface between the environment and the nervous system. Several TRPs transduce thermal, chemical, and mechanical stimuli into inward currents, an essential first step in eliciting thermal and pain sensations.
Facet joint pain
Definition
Facet joints have been implicated as responsible for spinal pain in 39% of patients with neck pain, 34% of patients with thoracic pain, and 27% of patients with low back pain [1].
Innervations
The nerve roots are invested by pia mater and covered by arachnoid and dura as far as the spinal nerve. The dura of the dural sac continues around the roots as their dural sleeve, which blends with the epineurium of the spinal nerve (Fig. 2-7).
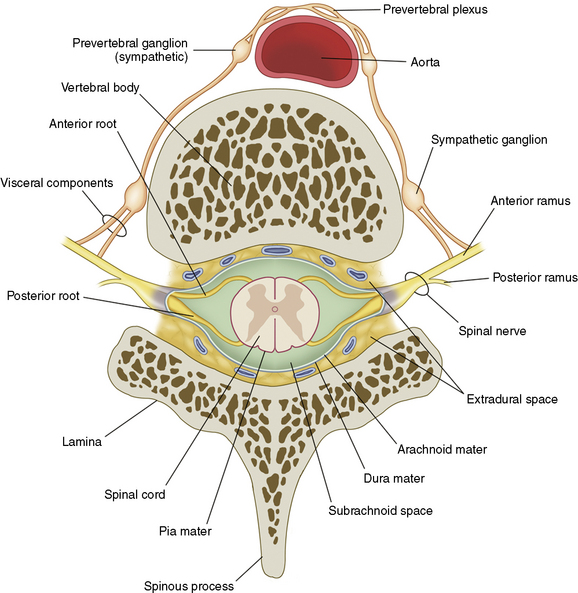
Figure 2–7 Spinal nerves.
(From Schuenke M, Schulte E, Schumacher U, et al [eds]: Thieme Atlas of Anatomy: General Anatomy and Musculoskeletal System. Stuttgart, Thieme, 2006, p 62.)
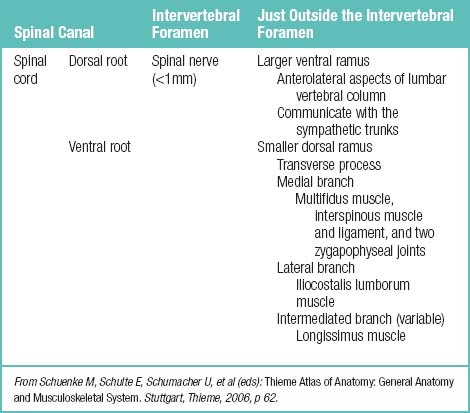
Facet joints are well innervated by the medial branches of the dorsal rami, which contain free and encapsulated nerve endings as well as nociceptors and mechanoreceptors. Each segmental medial branch of the dorsal ramus supplies at least two (in humans, monkeys, and cats) or three (in rats) facet joints (Table 2.3).
Intervertebral disc
The three components of the intervertebral disc (IVD) are as follows:
Distinguishing “Normal Aging” from Disease of the Intervertebral Disc
Figures 2-8 and 2-9 and Table 2.4 contain information pertinent to this issue.
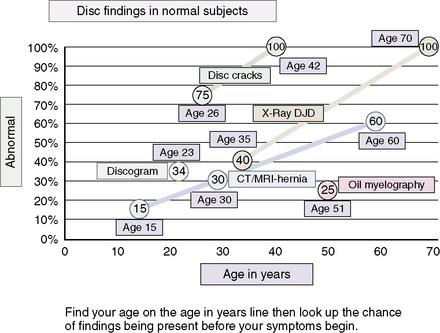
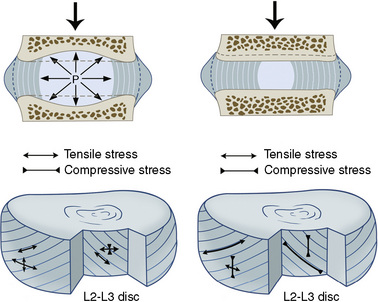
Table 2.4 Pathophysiology of Intervertebral Disc Aging and Degeneration
| Process | Effects |
|---|---|
| Diminished cellular response | Senescence (alteration in gene expression and transcription factors) |
| Apoptosis (programmed cell death) | |
| Biochemical processes | Imbalance between catabolic and anabolic activity: |
From Biyani A, Andersson GBJ: Low back pain: Pathophysiology and management.J Am Acad Orthop Surg 2004;12:106-115.
Nerve and Blood Vessel Ingrowth
Although the normal adult IVD is avascular and aneural, nerves and blood vessels grow into diseased IVD. One avenue of investigation has been the local production of angiogenic and neurogenic molecules within degenerate IVD (Fig. 2-10), which has yielded the following findings:
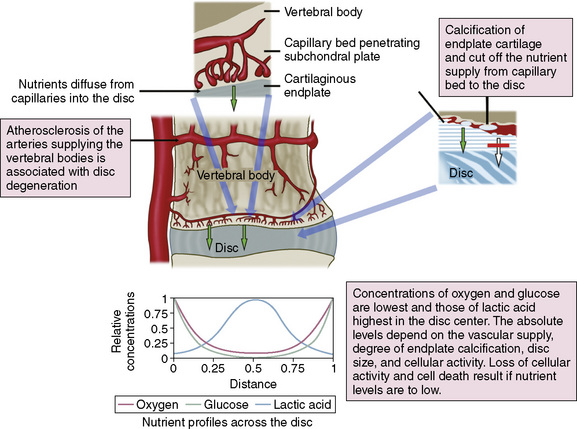
Figure 2–10 Schematic view of the routes for nutrient transport into the avascular disc and resulting nutrient profiles.
(Reprinted from Urban JPG, Roberts S: The intervertebral disc: Normal, aging, and pathologic. In Herkowitz HN, Garfin SR, Eismont FJ, et al [eds]: Rothman-Simeone The Spine, 5th ed. Philadelphia, Saunders, 2006, p 77.)
Cytokines as regulators of disease processes
There has also been growing interest in the possible role of cytokines in regulating the connective tissue degradation, nerve and vessel ingrowth, and macrophage accumulation that characterize IVD degeneration (Table 2.5) [4,5]. A number of cytokines have been implicated, including TNF (tumor necrosis factor) (Fig. 2-11), IL-1 (interleukin-1), IL-6, and IL-10, PDGF (platelet-derived growth factor), VEGF (vascular endothelial growth factor), IGF-1 (insulin-like growth factor-1), TGF-β (transforming growth factor-β), EGF (epidermal growth factor), and FGF (fibroblast growth factor) [6].
| Chemical Substance | Function |
|---|---|
| Phospholipase A2 | Mediates mechanical hyperalgesia |
| Nitric oxide | Inhibits mechanical hyperalgesia and produces thermal hyperalgesia |
| MMP-2 (gelatinase A) and MMP-9 (gelatinase) |
IL, interleukin; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinase.
From Biyani A, Andersson GBJ: Low back pain: Pathophysiology and management.J Am Acad Orthop Surg 2004;12:106-115.
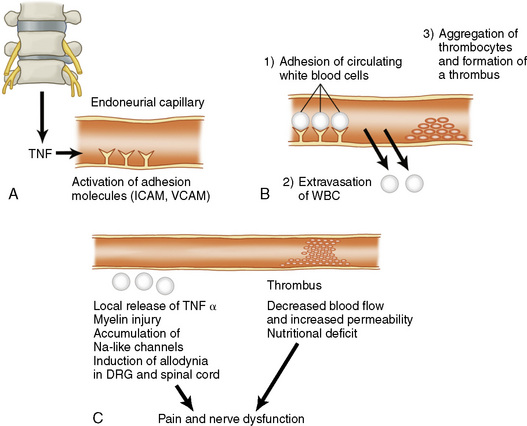
Studies of IL-1 have yielded the following findings:
As IL-1 is the regulator of cartilage catabolism, the TGF-β superfamily is the regulator of cartilage anabolism. A twofold increase was seen in proteoglycan synthesis by rabbit nucleus pulposus cells after injection of rabbit intervertebral discs with an adenoviral vector carrying a human TGF-β transgene [6].
Nomenclature and classification of lumbar disc pathology
General classifications of disc lesions are listed in Box 2.3 and described here:
BOX 2.3 General Classification of Disc Lesions*
Modified from Milette PC: Classification, diagnostic imaging, and imaging characterization of a lumbar herniated disc. Radiol Clin North AM 2000;38:1267-1292.
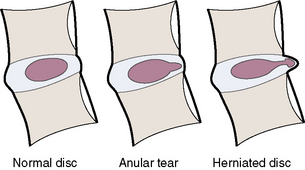
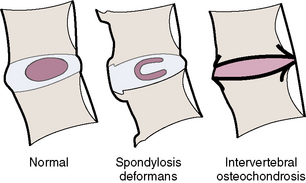

Classifications of Disc Herniation
Figure 2-15 illustrates, and Box 2.4 summarizes, the proposed categories for the description and classification of disc herniation, which are as follows [7]:
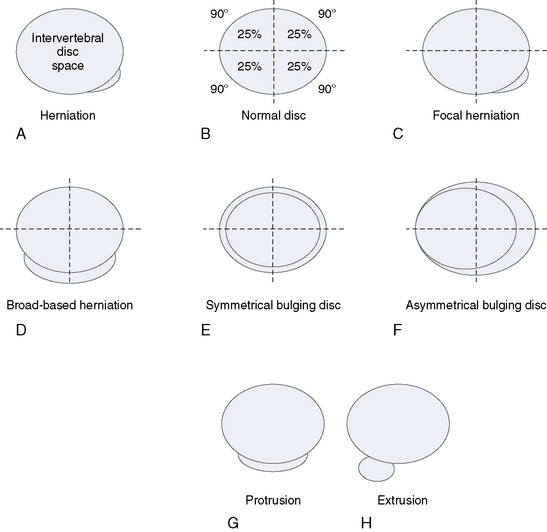
BOX 2.4 Proposed Categories for Description and Classification of a Disc Herniation
Modified from Fardon DF, Milette PC; Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology: Nomenclature and classification of lumbar disc pathology: Recommendations of the Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology. Spine 2003;26:E93-E113.
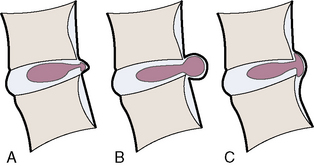
The distinction between protrusion and extrusion of a disc herniation is clarified by Figures 2-16 and 2-17.
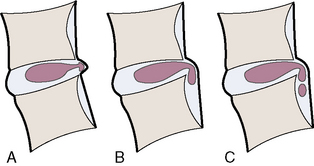
Complications of herniated discs are illustrated in Figure 2-18.
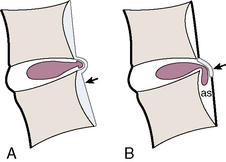
Figures 2-19 through 2-22 summarize the nomenclature for the anatomic zones and levels of the spine.
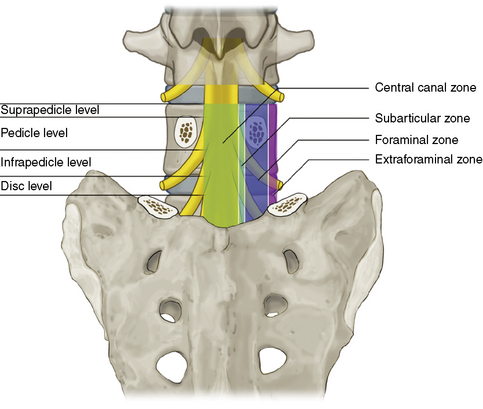
Figure 2–19 Coronal drawing illustrating the main anatomic zones and levels of a spinal segment.
(Adapted from Fardon DF, Milette PC; Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology: Nomenclature and classification of lumbar disc pathology: Recommendations of the Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology. Spine 2001;26:E93-E113.)
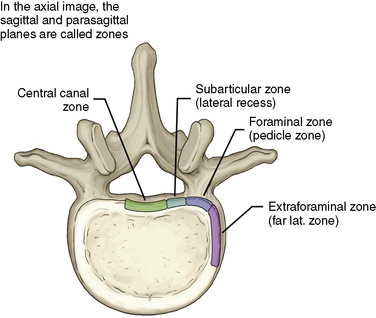
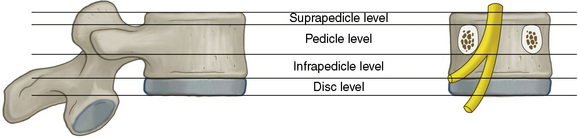
Figure 2–21 Schematic representation of the anatomic levels of a spinal segment identified on craniocaudal images.
(Adapted from Fardon DF, Milette PC; Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology: Nomenclature and classification of lumbar disc pathology: Recommendations of the Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology. Spine 2001;26:E93-E113.)
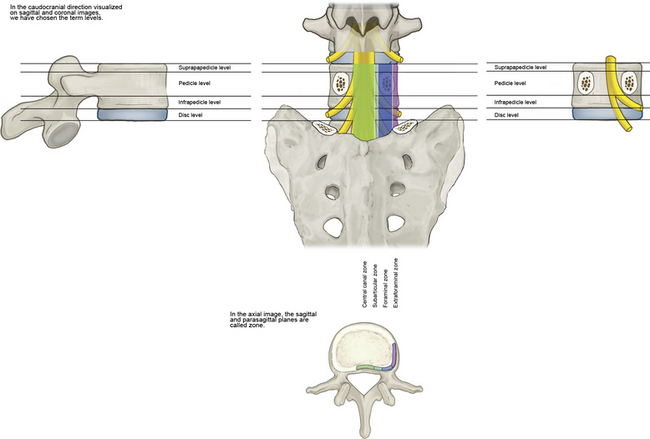
Figure 2–22 Schematic summative representation of anatomic levels and zones of a spinal segment.
(Adapted from Fardon DF, Milette PC; Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology: Nomenclature and classification of lumbar disc pathology: Recommendations of the Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology. Spine 2001;26:E93-E113.)
Dorsal root ganglion
Postlaminectomy syndrome
Facet joint involvement in chronic pain following lumbar surgery has been shown to be present in approximately 8% to 16% of patients. Through the use of a single nerve block, one study found the prevalence of sacroiliac joint pain following lumbar fusion to be 35% [8].
Epidural fibrosis may occur after an annular tear, disc herniation, hematoma, infection, surgical trauma, vascular abnormalities, or intrathecal injection of contrast media. Epidural fibrosis may account for as much as 20% to 36% of all cases of failed back surgery syndrome. Alternatively, there may be a final common pathway for all these etiologies that results in peripheral and central facilitation potentiated by inflammatory and nerve injury mechanisms. Paraspinal muscles may also become denervated and involved in the pathogenesis of failed back surgery syndrome [8].
Spinal stenosis
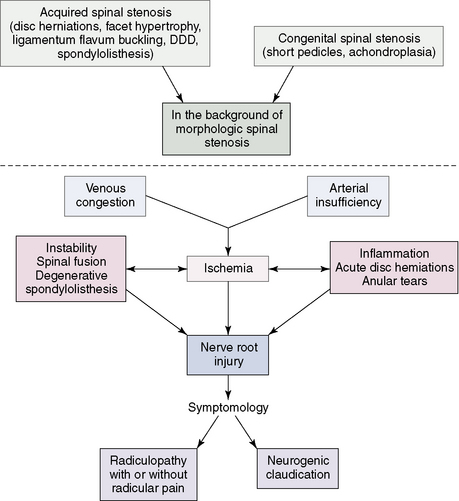
Spinal stenosis can be defined as a narrowing of the spinal canal that results in symptoms and signs due to entrapment and compression of the intraspinal vascular and nervous structures. Disc bulging, protrusion, and herniation in the cervical as well as lumbar areas, combined with osteophytes and arthritic changes of the facet joints, can cause narrowing of the spinal canal, encroachment on the contents of the dural sac, or localized nerve root canal stenosis (Fig. 2-23).
1 Manchukonda R., Manchikanti K.N., Cash K.A., et al. Facet joint pain in chronic spinal pain: An evaluation of prevalence and false-positive rate of diagnostic blocks. J Spinal Disord Tech. 2007;20:539-545.
2 Kallakuri S., Singh A., Chen C., Cavanaugh J.M. Demonstration of substance P, calcitonin gene-related peptide, and protein gene product 9.5 containing nerve fibers in human cervical facet joint capsules. Spine. 2004;29:1182-1186.
3 Freemont A.J., Watkins A., Le Maitre C., et al. Nerve growth factor expression and innervation of the painful intervertebral disc. J Pathol. 2002;197:282-286.
4 Biyani A., Andersson G.B.J. Low back pain: Pathophysiology and management. J Am Acad Orthop Surg. 2004;12:106-115.
5 Freemont A.J., Watkins A., Le Maitre C., et al. Current understanding of cellular and molecular events in intervertebral disc degeneration: Implications for therapy. J Pathol. 2002;196:374-379.
6 Nishida K., Kang J.D., Gilbertson L.G., et al. Modulation of the biologic activity of the rabbit intervertebral disc by gene therapy: An in vivo study of adenovirus-mediated transfer of the human transforming growth factor beta 1 encoding gene. Spine. 1999;24:2419-2425.
7 Fardon D.F., Milette P.C., Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology. Nomenclature and classification of lumbar disc pathology: Recommendations of the Combined Task Forces of the North American Spine Society, American Society of Spine Radiology, and American Society of Neuroradiology. Spine. 2003;26:E93-E113.
8 Boswell M.V., Shah R.V., Everett C.R., et al. Interventional techniques in the management of chronic spinal pain: Evidence-based practice guidelines. Pain Physician. 2005;8:1-47.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 2 Current Understanding of Spinal Pain and the Nomenclature of Lumbar Disc Pathology
Any structure to be deemed a cause of back pain should:
Tissues capable of transmitting pain in the back are as follows:
General pain mechanism
The two categories of pain are as follows:
Potential associations between particular mechanisms and particular symptoms are as follows:
A number of different input channels can lead to the pain sensation. These should be the first anatomic targets for treatment (Fig. 2-1; Table 2.1), as follows:
The four primary types of pain (Fig. 2-2) are as follows:
BOX 2.1 Positive and Negative Symptoms of Peripheral Neuropathic Pain
Woolf CJ: Dissecting out mechanisms responsible for peripheral neuropathic pain: implications for diagnosis and therapy. Life Sci 2004;74:2605-2610.
Adaptive pain and maladaptive pain are defined in Box 2.2.
BOX 2.2 Two Broad Classes of Pain
Modified from Woolf CJ; American College of Physicians; American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med 2004;140:441-451.
Multiple mechanisms that can produce pain, as follows:
Pain transient receptor potential (TRP) ion channels are listed in Table 2.2 and illustrated in Figure 2-6. TRP ion channels are molecular gateways in sensory systems, an interface between the environment and the nervous system. Several TRPs transduce thermal, chemical, and mechanical stimuli into inward currents, an essential first step in eliciting thermal and pain sensations.
Facet joint pain
Definition
Facet joints have been implicated as responsible for spinal pain in 39% of patients with neck pain, 34% of patients with thoracic pain, and 27% of patients with low back pain [1].
