[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 111 Crohn’s Disease
Idiopathic inflammatory bowel disease (IBD) comprises conditions characterized by chronic or relapsing immune activation and inflammation within the gastrointestinal tract. Crohn’s disease and ulcerative colitis (UC) are the two major forms of idiopathic IBD; less common, but increasingly recognized, are the microscopic colitides, primarily collagenous colitis and lymphocytic colitis (see Chapter 124). Other chronic inflammatory conditions of the intestine share some features of presentation and pathogenesis with idiopathic IBD, but they have identifiable etiologies. These disorders include diversion colitis, bypass enteropathy, radiation colitis, and drug-induced colitides. The two major forms of IBD share many clinical and epidemiologic characteristics, suggesting that underlying causes may be similar. Indeed, more than occasionally, Crohn’s disease cannot be distinguished from UC on clinical grounds, yet the two diseases are distinct syndromes with divergent treatment and prognosis.
HISTORY OF CROHN’S DISEASE
Although the eponym Crohn’s disease has gained general acceptance in recent decades, clear clinicopathologic reports of the same process date back at least two centuries. Morgagni provided a description of intestinal inflammation characteristic of Crohn’s disease in 1761.1 Only after the identification of the tubercle bacillus by Koch in 1882 was it possible to describe persons with ileocecal disease similar to intestinal tuberculosis but lacking the organism. Such reports were provided by Fenwick (1889),1 Dalziel (1913),2 Weiner (1914), Moschcowitz and Wilensky (1923 and 1927), and Goldfarb and Suissman (1931).3 In 1932, the landmark publication of Crohn, Ginzburg, and Oppenheimer called attention to “terminal ileitis” as a distinct entity and chronic disease.4 This term was soon deemed unsuitable, however, when it became apparent that the disease process might involve the colon. Patients, too, misunderstood and were frightened by the “terminal” nature of their illness. The term “regional enteritis” embraced the focal nature of the process, but failed to incorporate knowledge of the possibility of disparate sites of involvement within the gastrointestinal tract, including the small and large bowel in combination5 and large bowel in isolation.6 The term “granulomatous enterocolitis” lost acceptance when it became clear that granulomas were not a sine qua non of the diagnosis. In the end, the name “Crohn’s disease” has been adopted to encompass the many clinical presentations of this pathologic entity. But for the alphabetic priority these authors chose, Crohn’s disease might well have been Ginzburg’s or Oppenheimer’s disease.
EPIDEMIOLOGY
Misclassification of disease is problematic. Historically, unidentified infections, later recognized by improved culture and diagnostic techniques, might have accounted for some portion of cases, particularly among persons with a single episode of disease. At times, differentiating Crohn’s disease from UC may be difficult, especially at the time of diagnosis, and before the passage of time has allowed distinctive disease characteristics to become manifest. Reassignment of a diagnosis of Crohn’s disease or UC may be as high as 9% in the first two years after diagnosis.7
Despite these methodologic limitations, distinct and reproducible geographic and temporal trends in incidence have been observed. In both North America and Europe, higher incidence rates have been noted in more northern latitudes. For example, age-adjusted annual incidence rates of 9 per 100,000 persons were reported in Olmsted County, Minnesota,8 and Copenhagen County, Denmark,9 and as high as 20 per 100,000 in Nova Scotia.10 Comparatively, estimates of incidence rates reached only 0.9 per 100,000 in Spain11 and 3.4 per 100,000 in Italy.12 A north-south gradient similar to that observed in Europe13 has been noted in the United States14 and even within the state of California itself, with estimated incidence rates of 7.0 and 3.6 per 100,000 in northern and southern California, respectively.15,16 In Asian countries, the incidence rate has remained low, with a mean estimated incidence of 0.5 per 100,000 persons in Korea17 and a similar incidence in Japan,18,19 whereas in Australia and New Zealand, incidence rates have ranged from 1.75 to 2.1 per 100,000.20,21 Crohn’s disease is thought to be extremely rare in much of South America and Africa22 with the exception of South Africa, where the most recent estimate of the incidence rate for the white population is 2.6 per 100,000; it is considerably lower among the nonwhite population.23
In some regions of the world where Crohn’s disease was rare, although still low compared with western countries, incidence is rising dramatically. For example, in Seoul, South Korea, the incidence increased from 0.05 per 100,000 in the early 1980s to 1.34 per 100,000 between 2001 and 2005.17 This trend has been seen throughout other regions as well.24–27 Estimates from less-affluent nations may be influenced by increasing access to health care, and therefore, genetic and environmental factors in these regions are difficult to disentangle.
Incidence rates have continued to rise in some regions, such as in Denmark,9,28 whereas in others they appear to be stabilizing. In Olmsted County, Minnesota, rates had been steadily increasing from approximately 3 per 100,000 (1954-1963) to nearly 8 per 100,000 (1964-1973), but since the late 1970s these rates have not changed significantly.8,29 Mortality trends for Crohn’s disease have followed a similar pattern, with rising mortality until the mid-1970s and stable rate since.30 Although improved diagnostic capabilities might have played some role in the rising incidence leading up to the mid-1970s, the fact that Crohn’s-related mortality was rising in parallel argues against the theory that rising Crohn’s diagnoses merely represented detection bias involving mild cases. Most recently, the prevalence of Crohn’s disease in the United States is estimated to be 201 per 100,000 adults and 43 per 100,000 in children, adolescents, and adults younger than 20 years.14
Studies throughout the world have shown a small excess risk of Crohn’s disease among women. Most reports show a female-to-male ratio in adult patients between unity and 1.3 : 1.10,14,28 In the pediatric population this is reversed, with more boys having Crohn’s disease than girls.14 This slight difference in risk in adult-onset disease may be explained by hormonal or life-style factors and stands in contrast to the nearly equal or even slight male predominance seen in UC.
Crohn’s disease is diagnosed most often among persons 15 to 30 years of age, although the age of diagnosis can range from early childhood throughout an entire lifespan. Population-based studies have shown the median age of diagnosis to be approximately 30 years.8,10 Conflicting information may be found regarding trends in the age of diagnosis. In Olmsted County, Minnesota, younger age groups have had a fairly stable incidence over the past 20 years, with rising rates in patients aged 60 and older.8,10 This trend of older-age diagnoses also was seen in population-based studies in Copenhagen, Denmark,31 and in Stockholm, Sweden,32 the median age of diagnosis increased from 25 years in 1960 to 1964 to 32 years in 1985 to 1989. These findings reflect diagnosis in a larger proportion of patients older than 60 years. Indeed, many, though not all,10 studies have shown a smaller second peak in incidence later in life, generally in the seventh decade.33 This second peak may be the result of ascertainment bias because of more frequent contact with medical care and more frequent evaluation of older patients. Differences in clinical presentation among younger and older patients suggest that distinct risk factors are operative at different ages at onset.34 The pathologic findings in young and old patients are not different, although some studies have identified a greater proportion of colonic and distal disease among older patients,33 compared with a predominance of ileocolonic disease in younger patients.35
ETIOLOGY AND PATHOGENESIS
INITIATING EVENTS
In light of the nature of the pathologic findings in Crohn’s disease (see later) and UC, it long has been clear that IBD represents a state of sustained immune response. The question arises as to whether this is an appropriate response to an unrecognized pathogen or an inappropriate response to an innocuous stimulus. Many infectious agents have been proposed as the cause of Crohn’s disease including chlamydia, Listeria monocytogenes, cell-wall–deficient Pseudomonas species, reovirus, and many others. Paramyxovirus (measles virus) has been implicated etiologically in Crohn’s disease as a cause of granulomatous vasculitis and microinfarcts of the intestine,36 but a proposed association between early measles vaccination and Crohn’s disease largely has been disproved.37 Another suggestion has been that the commensal flora, although normal in speciation, possess more subtle virulence factors, such as enteroadherence, that cause or contribute to IBD.38
Among the most enduring hypotheses is that Mycobacterium paratuberculosis is the causative agent of Crohn’s disease. This notion dates to Dalziel’s observation in 1913 that idiopathic granulomatous enterocolitis in humans is similar to Johne’s disease, a granulomatous bowel disease of ruminants caused by M. paratuberculosis.39 M. paratuberculosis is extremely fastidious in its culture requirements, and some proponents of this hypothesis have speculated that the presence of M. paratuberculosis as a spheroplast may have confounded efforts to confirm this hypothesis by culture of the organism; demonstrating it by immunohistochemistry, in situ hybridization, and polymerase chain reaction methodology; and empirical treatment with antimycobacterial antibiotics (see “Medical Therapy,” later). Most investigation in this area has been inconclusive, providing insufficient evidence to prove or reject the hypothesis.
In light of the diversity of substances and bacteria within the intestinal lumen, it is remarkable that the intestine is not perpetually inflamed. The presence of low-level physiologic inflammation within the healthy intestinal mucosa represents a state of preparedness to deal with potentially harmful agents. A more vigorous response would not be appropriate if directed toward the innocuous commensal flora of the intestine. Experiments in animal models of IBD suggest that in a genetically susceptible host, a classic pathogen is not necessary to cause IBD, but rather nonpathogenic commensal enteric flora are sufficient to induce an inappropriate chronic inflammatory response. In diverse models, animals raised under germ-free conditions show diminished or delayed expression of the IBD phenotype.40 On introduction of defined bacterial flora, however, the expected phenotype of bowel inflammation becomes manifest.40 Such models suggest that a diversity of genetic alterations, including those that affect intestinal barrier function and regulation of mucosal immunity, can result in intestinal inflammation. As in the animal models of IBD, evidence in patients with Crohn’s disease also points to an over-responsiveness of mucosal T cells to the enteric flora, manifest in part by the presence of antibodies against an array of bacterial antigens, including Escherichia coli outer membrane porin C (OmpC) and flagellin. Advances in the genetic bases of Crohn’s disease confirm that diverse bacterial agents are capable of fueling the inflammation of Crohn’s disease (see later).
GENETICS
The argument for a genetic predisposition to IBD begins with the observation that family members of affected persons are at greatly increased risk for developing IBD. The relative risk among first-degree relatives is 14 to 15 times higher than that of the general population.41 Roughly one of five patients with Crohn’s disease report having at least one affected relative. Many families have more than one affected member, and although there is a tendency within families for either UC or Crohn’s disease to be present exclusively, mixed kindreds also occur, suggesting the presence of some shared genetic traits as a basis for both diseases, as has recently been confirmed.
Ethnicity also plays a role. Eastern European (Ashkenazi) Jews are at a two- to four-fold higher risk of developing IBD than non-Jews of the same geographic location, and they are at greater risk of having multiple affected family members. Studies of monozygotic and dizygotic twins suggest that genetic composition is a more powerful determinant of disease for Crohn’s disease than for UC: The concordance rate among monozygotic twins is as high as 67% for Crohn’s disease but only 13% to 20% for UC. Most studies have suggested that concordance of disease location and disease behavior are higher than one would expect by chance.42 Finally, some subclinical markers of Crohn’s disease, including anti-OmpC antibodies, are more common among apparently healthy family members of Crohn’s disease probands than among the general population.43
Early studies of IBD genetics were limited by the slow speed of DNA sequencing and an incomplete understanding of the human genome’s structure. Studies of IBD genetics depended upon selecting a candidate gene based upon the then-current understanding of pathogenesis. With automated, rapid DNA sequencing, and mapping of the common genetic variants that occur in humans (the HapMap, or haplotype map), genome-wide association (GWA) studies became feasible. GWA studies can simultaneously explore many hundred thousands of genetic markers, providing a broad and unbiased approach to assessing the association of genomic loci to a specific disease without prior hypothesis about a candidate gene. GWA studies have been more successful in Crohn’s disease than in any other complex disease, and they have accelerated the pace of gene discovery, providing unexpected insights into pathogenesis.44 More than 30 genetic loci have been convincingly associated with Crohn’s disease in a large and powerful GWA study, confirming some known loci and discovering many loci that had not been described previously (Table 111-1).45 Thus far, genetic studies have highlighted three pathways of fundamental importance in the pathogenesis of Crohn’s disease.
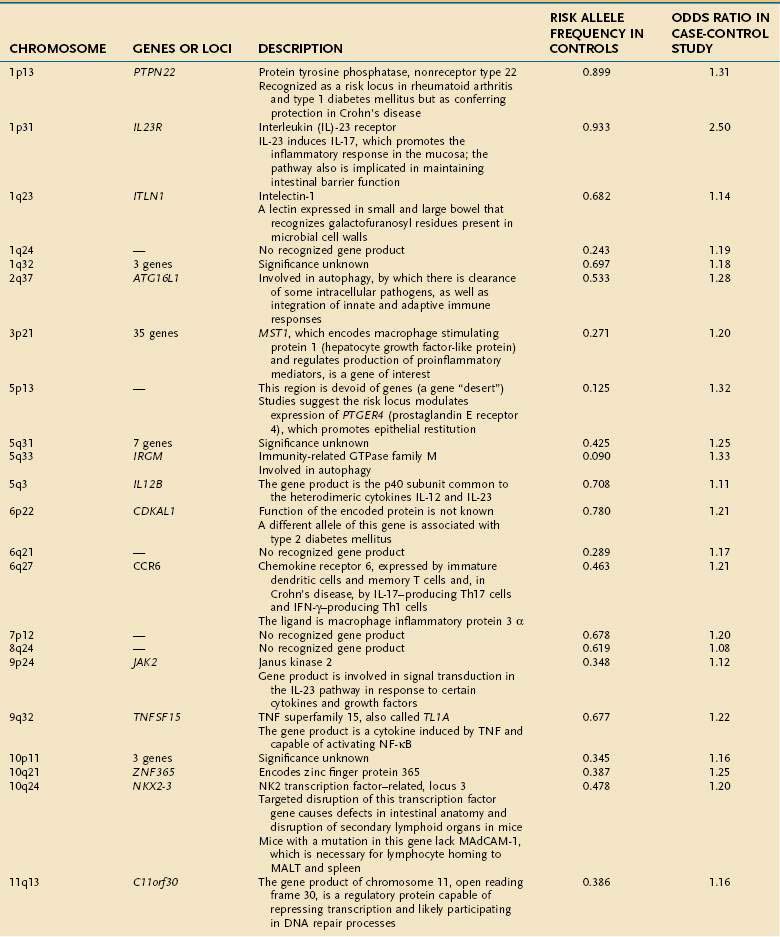
The first susceptibility locus for Crohn’s disease was identified in 2001 as the NOD2 (nucleotide-binding oligomerization domain 2) gene, also known as CARD15 (caspase-recruitment domain 15).46,47 The allelic variants most commonly associated with Crohn’s disease in European and American populations include one frameshift insertion leading to early truncation of the protein (Leu1007fsinsC) and two missense mutations (Arg702Trp, Gly908Arg). Carriage of disease-associated allelic variants on both chromosomes confers an odds ratio for Crohn’s disease of 17.1 (95% confidence interval [CI], 10.7-27.2), and heterozygous persons have an odds ratio of 2.5 (95% CI, 2.0-2.9) for the disease.48 Studies have associated genetic polymorphisms of NOD2/CARD15 with younger onset and ileal location of disease and increased likelihood of stricture formation.49–52 It has been estimated that as many as 20% to 30% of patients with Crohn’s disease bear abnormal NOD2/CARD15. Nevertheless, the penetrance of NOD2/CARD15 is estimated to be less than 1%53; that is, disease-related allelic variants of the gene may be found in a large number of persons who do not have Crohn’s disease; this strongly suggests that environmental factors, as yet incompletely elucidated, play a significant role in the expression of the Crohn’s disease phenotype (see later).
The discovery of the association of NOD2/CARD15 with Crohn’s disease has opened a remarkable window into the pathogenesis of Crohn’s disease. The gene product of NOD2/CARD15 is a cytosolic protein that functions as an intracellular sensor of bacteria. Specifically, the protein binds to muramyl dipeptide (MDP; MurNAc-L-Ala-D-isoGln), a component of bacterial peptidoglycan, found in Gram-positive and Gram-negative bacteria.54,55 The NOD2/CARD15 protein is expressed in monocytes and enterocytes, specifically in Paneth cells,56 which lie within the crypts and produce the endogenous antimicrobial peptides called defensins. The NOD2/CARD15 gene consists of two CARD domains, a nucleotide binding domain (NBD), and 10 leucine-rich repeats (LRR). NOD2/CARD15 variants associated with Crohn’s disease lie within the LRR and interfere with binding to MDP. In mononuclear cells, mutations in NOD2 result in decreased activation of nuclear factor (NF)-κB, whereas an excess of NF-κB expression is observed in tissue inflamed by Crohn’s disease. This apparent paradox has yet to be unraveled completely, but it is clear that defects in NOD2 impair antibacterial responses, particularly to oral exposure to pathogens. Notably, the production of β-defensins, which are antibacterial proteins produced by Paneth cells, is defective in Crohn’s patients with variant NOD2.57 These findings strongly implicate defects in innate immunity—the immediate and nonspecific immune responses to microbial infection—in a subset of patients with Crohn’s disease, with subsequent chronic activation of adaptive immunity, the antigen-specific responses mediated by antigen presenting cells (APCs) and T cells.
Subsequent discoveries have implicated defects in multiple genes in the autophagy pathway in the pathogenesis of Crohn’s disease.58,59 Autophagy is an ancient cellular process, highly conserved in evolution, by which segments of cytoplasm are isolated within a membrane and delivered to lysosomes by mechanisms that do not involve transport through endocytic or vacuolar sorting pathways. This unique process plays a role in cellular homeostasis by clearing proteins that are long-lived, misfolded, or aggregated, and by clearing apoptotic bodies, which might otherwise trigger inflammation and autoimmunity. Autophagy has been shown to contribute directly to innate immunity through direct killing of pathogens, activation of Toll-like receptors and Nodd-like receptors, and elaboration of immunomodulatory cytokines such as interferon (IFN)-γ. Autophagy also stands at the interface of innate and adaptive immune responses, delivering antigen to human leukocyte antigen (HLA) class II molecules in APCs for antigen-specific binding.59
GWA studies have identified variants that predispose to Crohn’s disease in at least two autophagy-related genes. The first, the autophagy-related 16-like 1 (ATG16L1) gene, was noted as having a disease-associated single nucleotide polymorphism (SNP) encoding an amino acid substitution in exon 8, resulting in a change from alanine to threonine58–60; this minor allele is protective against Crohn’s disease. ATG16L1 is expressed by intestinal epithelial cells, APCs, and various subsets of human T cells. The second autophagy gene associated with Crohn’s disease is the IRGM (immunity-related GTPase [guanosine triphosphatase] family member M) gene on chromosome 5q33.1.61 Careful study suggests that the disease-associated variants of this gene do not affect the amino acid sequence of its product, but they more likely alter its expression.61 IRGM appears to be important in resistance to intracellular pathogens such as mycobacteria, Listeria monocytogenes, and Toxoplasma gondii.59
A third pathway associated with Crohn’s disease is interleukin (IL)-23 and other gene products associated with this protein.62 IL-23 is a heterodimeric cytokine comprising two linked subunits (p19 and p40). IL-23 is produced by many cell types, including dendritic cells and macrophages, in response to diverse microbial signals. Naïve CD4+ T cells up-regulate IL-23 receptor when exposed to IL-6 and transforming growth factor (TGF)-β, completing an autocrine loop in the generation of Th17 T cells, effector T cells that produce IL-17.63,64 A rare variant of the IL23R gene leading to a glutamine at position 381 rather than an arginine is strongly protective for Crohn’s disease, with an odds ratio of 0.26 to 0.45; other, more common SNPs are associated with increased risk for Crohn’s disease and UC.65 In the same pathway, variants of the IL12B gene, encoding the p40 subunit common to IL-12 and IL-23, and of the JAK2 and STAT3 genes, with roles in IL23R signaling, as well as in Th17 differentiation in the case of STAT3, also have been associated with Crohn’s disease susceptibility.45 Together, these findings support the pivotal role of this pathway in maintaining mucosal homeostasis in the normal intestine. As the functional alterations associated with the many other identified genetic risk loci are elucidated, it is certain that new insights into the causes of Crohn’s disease will arise.
ENVIRONMENT
Although the greatest relative risk of Crohn’s disease is found among first-degree relatives of affected persons, particularly siblings of the proband, environmental factors also are important. As noted earlier, the rising incidence of Crohn’s disease over many decades highly suggests an environmental contribution to the expression of disease. Epidemiologic studies have examined numerous risk factors for Crohn’s disease. Most studies have found breast-feeding to be protective for IBD, presumably by playing a role in early programming of immune responses in the developing gastrointestinal tract. Occupations associated with outdoor physical labor are relatively under-represented among Crohn’s patients. Crohn’s disease has been associated with higher socioeconomic status,66 presumably because of relative underexposure to diverse environmental antigens in the course of childhood—the hygiene hypothesis as it relates to intestinal mucosal immunity in IBD. Many, but not all, studies have discerned an increased risk of Crohn’s disease among women who use oral contraceptives. Nonsteroidal anti-inflammatory drugs (NSAIDs) have been implicated not only in exacerbations of IBD but also as a potential precipitant of new cases, perhaps by increasing intestinal permeability. Increased intake of refined sugars and a paucity of fresh fruits and vegetables in the diet have been associated with the development of Crohn’s disease. It is conceivable that this observation may be confounded by exacerbation of symptoms in patients with mild disease because of increased dietary fiber intake and subsequent avoidance of these food items before diagnosis.
Smoking is one of the more notable environmental factors for IBD. UC is largely a disease of ex-smokers and nonsmokers, whereas Crohn’s disease is more prevalent among smokers. In addition, smokers have more surgery for their disease and a greater risk of relapse after resection. The reasons for the divergent effect of smoking on Crohn’s disease and UC are poorly understood, but they might include effects on intestinal permeability, cytokine production, and clotting in the microvasculature. More recently, studies have focused on the role of carbon monoxide in stimulating immunosuppressive effects mediated by heme oxygenase-1.67
Many patients report a correlation between disease exacerbations and stress. Although depression and anxiety are a common reaction to illness, Crohn’s disease has not been shown to be caused by stress or by an anxious personality. The mind-body connection between emotional states or stress and intestinal inflammation in IBD is slowly being revealed, however, and studies indicate that stress may be associated with risk of relapse in Crohn’s disease.68
ADAPTIVE IMMUNE RESPONSE AND INFLAMMATION
The interaction between effector T cells and APCs is critical to the pathogenesis of Crohn’s disease (Fig. 111-1). The antigens that perpetuate the inflammatory response are taken up by APC. Degradation of antigen within proteasomes results in presentation of an epitope in the context of major histocompatibility complex (MHC) class II. Interaction between MHC class II and the T-cell receptor (CD3) results in antigen-specific interaction between the macrophage and the CD4+ T cell. This event is necessary, but not sufficient, to activate the T cell. A second costimulatory signal is needed as well, because binding of CD3 to MHC class II without a costimulatory signal can result in anergy or apoptosis. Important costimulatory signals include binding of tumor necrosis factor (TNF) to TNF receptor, CD40 to CD40 ligand, and B7 to CD28. Activation of T cells leads to production of IL-2, an important growth factor for T cells.
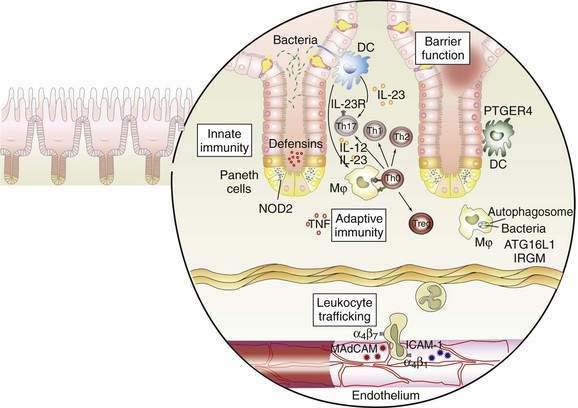
As noted earlier, the p40 subunit is common to IL-12 and IL-23, each of which, in turn, is critical in shaping the Th1 and Th17 responses that characterize Crohn’s disease. In addition to IL-23, the presence of TGF-β and IL-6 facilitate differentiation of naïve T cells into pathogenic Th17 cells.63 Activated APCs further shape and amplify the immune response by producing the T cell growth factor IL-2 and the proinflammatory cytokines IL-1 and TNF. Within mononuclear cells, the key nuclear transcription factor is NF-κB, which regulates the transcription of IL-1, IL-6, IL-8, TNF, and other peptides central to the inflammatory response.69
NF-κB is regulated tightly within the cell. In the inactive state, NF-κB is held in the cytoplasm, bound to inhibitory κBα. During cell activation after receptor binding, various kinases phosphorylate inhibitory κBα, thereby leading to its degradation. NF-κB is then released, permitting translocation to the nucleus, where it binds to the promoter regions of numerous genes that support the inflammatory response. Such genes include those that encode proinflammatory cytokines such as TNF, adhesion molecules, and chemokines.69
Expression of adhesion molecules is critical to amplify the immune response, because the resident populations of granulocytes and mononuclear cells alone do not account for the vigorous inflammatory reaction characterizing IBD. Adhesion molecules on the leukocyte surface and their ligands on the endothelium of venules in the lamina propria interact in a coordinated multistep process that permits trafficking of inflammatory cells into the mucosa. First, a weak interaction between selectins on the leukocyte surface and the endothelium leads to rolling of the leukocytes along the endothelium. Second, in the presence of chemokines such as IL-8, activation occurs, and integrins are expressed on the leukocyte surface. Third, interactions between leukocyte integrins and immunoglobulin-like cellular adhesion molecules on the endothelial surface lead to spreading of the cell and diapedesis.70 Specificity is conferred by the presence of tissue-specific cellular adhesion molecules. The integrins α4β7 and αEβ7 are of special importance in IBD, because the corresponding ligands—mucosal addressin cellular adhesion molecule and E-cadherin—are intestine specific. Mucosal addressin cellular adhesion molecule is expressed constitutively on the endothelium of venules in the lamina propria,70 whereas binding of αEβ7 on intestinal lymphocytes to E-cadherin on intestinal epithelium permits localization of intraepithelial lymphocytes. Antibodies to the α4 subunit of integrin have proved to be therapeutic in Crohn’s disease.71
Once recruited to the lamina propria, mononuclear cells and granulocytes elaborate a variety of injurious and proinflammatory substances that ultimately cause tissue destruction. These substances include prostaglandins, reactive oxygen metabolites, nitric oxide, leukotrienes, and proteases. Collagenase and matrix metalloproteinases play a pivotal role in the tissue destruction seen in IBD.72 Counterbalancing these destructive substances are other substances that promote epithelial restitution and repair, including IL-11, trefoil peptides, and growth factors such as epidermal growth factor and keratinocyte growth factor.
PATHOLOGY
Focal intestinal inflammation is the hallmark pathologic finding in Crohn’s disease. This tendency for focal inflammation is evident in focal crypt inflammation, focal areas of marked chronic inflammation, the presence of aphthae and ulcers on a background of little or no chronic inflammation, and the interspersing of segments of involved bowel with segments of uninvolved bowel. Even within a single biopsy specimen one can see a pronounced variability in the degree of inflammation. The presence of focally enhanced gastritis, characterized by a focal perifoveolar or periglandular lymphomonocytic infiltrate, is a common finding that occurs in 43% of unselected patients with Crohn’s disease.73 This finding underscores the focal nature of the inflammation, despite the strong potential for inflammation to occur anywhere along the entire longitudinal axis of the gastrointestinal tract. To a certain extent, the nature of the findings and the depth of inflammatory changes depend on the chronicity of the inflammation.
EARLY FINDINGS
Because of the variable and often long delay between the onset of the disease process and its diagnosis, it rarely is possible to observe the evolution of pathology from the earliest events. Studies of recurrent Crohn’s disease after ileal resection have offered a window into the sequence of pathologic changes in the disease.74
Aphthous Ulcers
The earliest characteristic lesion of Crohn’s disease is the aphthous ulcer. These superficial ulcers are minute, ranging in size from barely visible to three millimeters, and are surrounded by a halo of erythema. In the small intestine, aphthous ulcers arise most often over lymphoid aggregates, with destruction of the overlying M cells. In the colon, aphthae can occur without an endoscopically visible central erosion and may be associated with lymphoepithelial complexes. Crohn’s aphthae typically occur in normal mucosa, although villus blunting may be seen in the surrounding small intestinal mucosa.75
Aphthous ulcers represent focal areas of immune activation. The M cells and underlying lymphoid aggregates are primary locations for antigen sampling and antigen presentation, so it is not surprising that human leukocyte antigen (HLA)-DR is strongly expressed on the follicle-associated epithelium of the aphthous ulcer.76 Contact with luminal contents is a key factor in the development of aphthous ulcers in Crohn’s disease.
Aphthous ulcers heal in bowel excluded from the fecal stream by ileostomy, whereas re-establishing intestinal continuity leads to their recurrence77; these observations provide strong evidence for the role of luminal factors in the early pathogenesis of Crohn’s disease.
Granulomas
The presence of granulomas (Fig. 111-2), while highly characteristic of Crohn’s disease, is neither unique to Crohn’s disease nor universally found.78 Noncaseating granulomas, like aphthous lesions, are believed to be an early finding. Estimates of the prevalence of granulomas in Crohn’s disease have varied greatly, ranging from 15% in endoscopic series to as high as 70% in surgical series.79 Whether granulomas are found appears to be, in part, a matter of how hard one looks and how much tissue is available to examine; the more tissue sampled, the larger the specimen, and the more levels taken for histopathology, the more likely granulomas will be found.
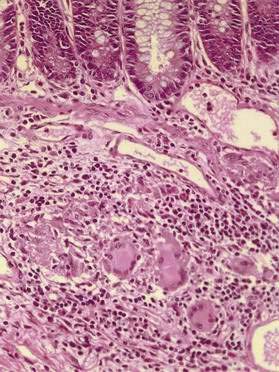
Granulomas may be discovered in involved and uninvolved bowel, in any layer of the intestine, and in mesenteric lymph nodes. Granulomas also may be found outside the intestinal tract—for example, in skin, eye, and liver—but extraintestinal granulomas are rare; occasionally, they may be recognized as millet seed-like nodules on the serosal surface of the bowel at laparotomy. The granulomas of Crohn’s disease are sarcoid-like, consisting of collections of epithelioid histiocytes and a mixture of other inflammatory cells, including lymphocytes and eosinophils; giant cells occasionally are seen. The granulomas usually are sparse, scattered, and not well formed. In contrast to the granulomas of tuberculosis, there is little or no central necrosis, and acid-fast stains and mycobacterial cultures are negative. It also is important to distinguish the granulomas of Crohn’s disease from those that can occur in association with an injured crypt. The latter represent a response to mucin released from injured goblet cells and may be found in UC and other conditions.79
LATER FINDINGS
A prevailing generalization is that intestinal inflammation in Crohn’s disease is a transmural process, in contrast to the more superficial inflammation of UC. The transmural nature of the inflammation, however, cannot be appreciated on superficial endoscopic biopsy, and in resected specimens it tends to be focal. Transmural involvement is observed less commonly than is disease of the mucosa and submucosa, but to the extent that transmural disease is noted, it is highly consistent with a diagnosis of Crohn’s disease. Dense lymphoid aggregates can enlarge the submucosa. At times, lymphoid aggregates also may be seen just outside the muscularis propria. The presence of lymphoid aggregates in the submucosa and external to the muscularis propria is a reliable sign of Crohn’s disease even when granulomas are not seen.79 Lymphoid aggregates occasionally may be seen within the muscularis propria, most often adjacent to the myenteric plexus.
Fibrosis is another transmural aspect of the disease. Fibrosis may be evident grossly as irregular thickening of the bowel wall and, along with hypertrophy of the muscularis mucosa, can contribute to the development of strictures. TGF-β is released locally in the presence of inflammation and is a cytokine critical for restitution and healing. In Crohn’s disease, however, TGF-β may be a double-edged sword. Fibroblasts isolated from the lamina propria produce primarily type III collagen in response to TGF-β1, and in the inflamed tissues of Crohn’s disease, significantly greater amounts of type III collagen are produced in response to this cytokine.80 Thus, a cytokine essential to the healing process also is implicated in fibrogenesis in Crohn’s disease.
OTHER FINDINGS
At the anatomic level, one of the most characteristic findings of Crohn’s disease is the presence of fat wrapping, a term that refers to the creeping of mesenteric fat onto the serosal surface of the bowel. Surgeons have long taken fat wrapping as a reliable indicator of the presence of diseased tissue. Mesenteric adipose tissue hypertrophy and creeping fat are recognized early in the course of disease at laparotomy or laparoscopy. Locally, fat wrapping correlates with the presence of underlying acute and chronic inflammation, as well as transmural inflammation in the form of lymphoid aggregates.81 Expression of peroxisome proliferator-activated receptor γ (PPARγ), a pivotal mediator in the regulation of adipose tissue homeostasis, is increased greatly in Crohn’s tissues.82 In turn, adipocytes may participate in the inflammatory process of Crohn’s disease by producing TNF and other inflammatory mediators.
At the microscopic level, the finding of pyloric metaplasia, normally a response to peptic ulcer disease when found in the duodenum, strongly suggests a diagnosis of Crohn’s disease when found in the terminal ileum. Careful descriptive immunopathology of areas of pyloric metaplasia reveals the presence of an ulcer-associated cell lineage. Bud-like glandular structures arise adjacent to areas of ulceration and are distinguished by production of epidermal growth factor in acinar cells of the nascent gland and by trefoil proteins (see Chapter 1) in the more superficial cells lining the tract. Epidermal growth factor and trefoil proteins, in turn, can promote restitution of the epithelium in adjacent mucosal ulceration.
CLINICAL FEATURES
DISEASE LOCATION
Crohn’s disease has a predilection for the distal small intestine and proximal colon. One third to one half of all patients have disease affecting both ileum and colon. Another one third have disease confined to the small intestine, primarily the terminal ileum, and there may be an increasing group with isolated colonic disease.83–85 Isolated jejunal involvement is rare. Gross involvement of the esophagus, stomach, or duodenum also is rare and almost always is seen in association with disease of the distal small intestine or colon. Focally enhanced acute and chronic inflammation may be seen in gastric biopsies in patients with Crohn’s disease either with or without gross involvement of the stomach.86 The discontinuous nature of the disease makes possible many variations in disease location, leading to considerable differences in clinical presentation. The disease usually stays confined to the segment in which it begins, but anatomic localization can vary over time, generally by involvement of additional segments of the alimentary tract, reflecting gross involvement with a disease that has the potential to affect any segment of the gastrointestinal tract.
CLINICAL PRESENTATION
The presentation of Crohn’s disease may be subtle and varies considerably. Factors contributing to this variability include the location of disease, the intensity of inflammation, and presence of specific intestinal and extraintestinal complications. Compared with UC, abdominal pain is a more frequent and persistent complaint. Pain is attributable to inflammation, abscess, or obstruction and may be intermittent and colicky or sustained and severe. Some patients experience symptoms that are mild but long-standing or that are atypical. Such patients are more likely to experience a delay in diagnosis in excess of a year. In the past, a mean delay in diagnosis of 3.3 years from the onset of symptoms was reported,87 but with improved diagnostic methods, and perhaps heightened awareness of the disease, more recent series have described delays of less than one year.84 A prodromal period is common in Crohn’s disease (not typically seen in UC) and might contribute to a delayed diagnosis,88 as does a prior diagnosis of irritable bowel syndrome and older age at onset of symptoms.89 Occasionally, radiologic and endoscopic findings are subtle, precluding definitive diagnosis even among patients with typical symptoms. Fecal occult blood may be found in approximately one half of patients, but in contrast to UC, gross rectal bleeding is uncommon, and acute hemorrhage is rare.90 Constitutional symptoms, particularly weight loss and fever, or growth retardation in children, may be prominent and occasionally are the sole presenting features of Crohn’s disease.
Typical Presentations
Although most patients with Crohn’s colitis have relative or complete sparing of the rectum, proctitis may be the initial presentation in some cases. Among a series of 96 patients with idiopathic proctitis, 13.6% were diagnosed with Crohn’s disease, usually within three years of initial presentation.91
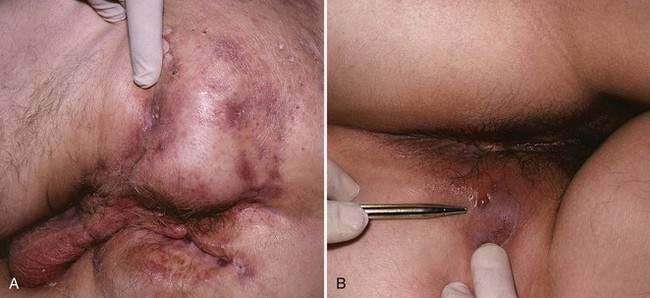
Perianal disease is another common presentation of Crohn’s disease. In as many as 24% of patients with Crohn’s disease, perianal disease precedes intestinal manifestations, with a mean lead time of four years.92 More often, however, perianal disease occurs concomitantly with or after the onset of symptoms of luminal disease. Perianal findings may be categorized as skin lesions, anal canal lesions, and perianal fistulas.93 Skin lesions include maceration, superficial ulcers, abscesses, and skin tags. Skin tags are generally of two types: type 1 (elephant ears) are typically soft and painless and can be quite large; type 2, which often arise from healed fissures, ulcers or hemorrhoids, are typically edematous, hard, and tender.94 Anal canal lesions include fissures, ulcers, and stenosis. The anal fissures of Crohn’s disease tend to be placed more eccentrically than the usual idiopathic fissures, which generally occur in the midline. In most cases, anal stricture is asymptomatic, but occasionally obstruction occurs, particularly if stool consistency improves in the course of treatment. Deeper abscesses can arise secondary to fistulas, especially when the internal os is located high in the rectum.
Unusual Presentations
Upper gastrointestinal tract Crohn’s disease is uncommon in the absence of disease beyond the ligament of Treitz. Approximately one third of patients with proximal Crohn’s disease do not have evidence of distal Crohn’s disease at the time of diagnosis, but virtually all develop distal disease in time.95 Patients with proximal Crohn’s disease tend to be younger at the time of diagnosis and more often present with abdominal pain and malaise95; they do not undergo surgery more often than do patients with lower tract disease alone, but the length of bowel that is resected tends to be greater.95
Esophageal Crohn’s disease is rare, occurring in less than 2% of patients. Presenting symptoms can include dysphagia, odynophagia, substernal chest pain, and heartburn. These symptoms may be progressive and lead to profound weight loss.96 Aphthous ulcers sometimes are found in the mouth and posterior pharynx. Esophageal stricture and even esophagobronchial fistula can complicate the course. An intriguing observation is that HLA-DR expression often is seen in the esophageal epithelium of patients with Crohn’s disease, even when the disease is located more distally in the gastrointestinal tract, perhaps indicating widespread immunologic activation of the gastrointestinal mucosa.97
Crohn’s disease confined solely to the jejunum and ileum is unusual and may be impossible to differentiate from ulcerative jejunoileitis, a distinct condition that occasionally responds to a gluten-free diet (Chapter 115). Frank malabsorption and steatorrhea often occur. When the disease is confined to a short segment of intestine or has features consistent with Crohn’s disease, initial management should be based on the presumed diagnosis of Crohn’s disease.
Controversy continues to surround the diagnosis of Crohn’s disease of the appendix. When idiopathic granulomatous inflammation is confined to the appendix, the presentation most often resembles that of acute appendicitis and occasionally periappendiceal abscess. The condition is rare, but the lack of disease in other locations of bowel portends a favorable prognosis, with a postoperative recurrence rate as low as 6%.98 Some authors suggest that granulomatous appendicitis should be considered an entity separate from Crohn’s disease.98
DISEASE BEHAVIOR
Clinical observation suggests that disease behavior in Crohn’s disease may be divided roughly into two categories: aggressive fistulizing disease and indolent cicatrizing disease99; a third subset of patients appear to develop neither of these behaviors over long periods of observation. Moreover, these distinctions are not always neat. Both fistula and stricture can occur simultaneously in the same patient, such as in the patient with a fistula arising behind a terminal ileal stricture, or at different times.
Genetic factors are important in determining disease behavior, with NOD2 variants being associated with fibrostenotic disease.49 In addition, serologic antibody responses to microbrial antigens and carbohydrates are associated with certain disease phenotypes.100–105 Specifically, the presence of antiglycan antibodies to mannan (a constituent of the cell wall of baker’s yeast anti-Saccharomyces cerevisiae antibody [ASCA]) correlates with small intestinal disease; identification of anti-CBir1 (antiflagellin) is associated with internal penetrating and stricturing disease; and anti-Escherichia coli outer membrane porin C (anti-OmpC) predicts internal perforations. When perinuclear antineutrophil cytoplasmic antibodies (pANCA) are present in a patient with Crohn’s disease, the phenotype is often that of an inflammatory “UC-like” Crohn’s disease.
Fistula and Abscess
Fistulas are frequent manifestations of the transmural nature of Crohn’s disease. Immune activation triggers the release of a variety of proteases and matrix metalloproteinases106 that can contribute directly to tissue destruction, sinus tract formation, and, finally, penetration to adjacent tissues. Perianal fistulas are common and are estimated to occur in 15% to 35% of patients (Fig. 111-3). When the fistula arises from an anal gland, a low-lying perianal fistula is the most common result. Such fistulas often are minimally symptomatic and can resolve with local care alone. Surprisingly, not all perianal fistulas occur in the setting of active rectal inflammation. In some cases, perianal fistulization may be extensive, forming a network of passages and extending to multiple openings that can include not only the perianal region but also the labia or scrotum, buttocks, or thighs.
It has been estimated that as many as one fourth of all patients with Crohn’s disease present with an intra-abdominal abscess at some time in their lives107; this figure is much less than one would imagine in light of the high incidence of fistulas. For the most part, inflamed serosal surfaces adhere to innocent serosa, thereby containing what would be an otherwise free perforation. Another common scenario is a perforation and abscess around the site of a surgical anastomosis. The classic presentation of an intra-abdominal abscess is that of a patient with spiking fevers and focal abdominal tenderness or localized peritoneal signs. Unfortunately, many of the patients at highest risk for perforation or abscess also are taking glucocorticoids, which are notorious for suppressing peritoneal signs and fever and masking the presentations of infection; therefore, a high level of suspicion must be maintained. When free perforation and peritonitis occur, the situation is life-threatening.
Stricture
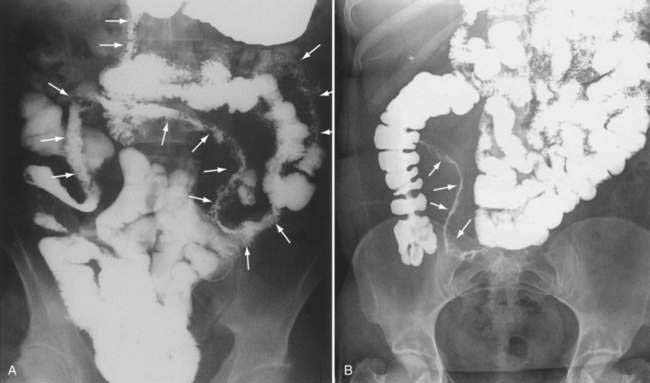
However, not all obstructive presentations are caused by fibrotic strictures. The classic radiologic string sign of a markedly narrowed bowel segment amid widely spaced bowel loops (Fig. 111-4) is a result of spasm and edema associated with active inflammation rather than fibrostenosis; the typical string sign transiently resolves with administration of glucagon, which relieves smooth muscle spasm.
CLASSIFICATION OF DISEASE
The Vienna Classification of Crohn’s Disease is one proposed scheme that incorporates the patient’s age at onset, disease location, and disease behavior into a schema with 24 potential subgroups.108 It is not surprising that in this scheme, significant associations are noted between age at diagnosis and location, and between disease behavior and location, along with a trend toward an association between age at diagnosis and disease behavior.109
Increasingly, subclinical characteristics such as serologic markers and genetic profiles will be used for their prognostic value in projecting outcomes in this heterogeneous disease. The Montreal classification is a first attempt at integrating serotype, genotype, and clinical phenotype as a structure to subclassify disease, and systems such as this will be used more as further supportive data emerge.110
PATHOPHYSIOLOGY OF COMMON SYMPTOMS AND SIGNS
Diarrhea
Diarrhea is the most common complaint among patients with Crohn’s disease. Increased stool frequency and decreased stool consistency arise through alterations in mucosal function and intestinal motility. In any given patient, multiple factors are likely to contribute to diarrhea. Altered fluid and electrolyte absorption and secretion can decrease stool consistency. Increased mucosal permeability from mucosal inflammation can result in exudation of protein and fluids. Increased production of prostaglandins, biogenic amines, cytokines, neuropeptides, and reactive oxygen metabolites all contribute to these alterations. An imbalance in the luminal concentration of bile salts relative to dietary fat can result in either bile salt-induced diarrhea or steatorrhea in the setting of ileal dysfunction or resection (see Chapter 101). Bacterial overgrowth can occur behind strictured bowel and contribute to malabsorption (Chapter 102). Disordered colonic motility is seen in the setting of chronic inflammation and also contributes to diarrhea. Occasionally, medications used to treat Crohn’s disease can exacerbate diarrhea. Secretory diarrhea can occur with olsalazine, and any of the 5-aminosalicylates rarely can induce a paradoxical increase in diarrhea, usually from salicylate sensitivity.
Abdominal Pain
The pathophysiology of abdominal pain in Crohn’s disease is not well understood. Numerous lines of investigation have provided tantalizing clues about the connection between the nervous system and Crohn’s disease, although the relationship among the enteric nervous system, inflammation, and immune activation in Crohn’s disease is quite complex. Stretch receptors in the intestinal wall may be stimulated as a food bolus passes through stenotic bowel, leading to abdominal pain and possibly vomiting. Visceral pain can result from serosal inflammation. The ganglia of the myenteric plexuses in the intestine in Crohn’s disease are increased in size and number, possibly indicating neural dysfunction.111
Substance P receptors have been found in increased numbers around the lymphoid follicles, in the microvasculature, and on enteric neurons in Crohn’s disease, even in locations distant from active inflammation,112 and there is increased binding of substance P to its receptors in the setting of an inflamed mucosa.113,114 Substance P binding can participate in the expression of pain. Enteroglia, support cells of the enteric nervous system, express MHC class II antigens in Crohn’s disease, raising the possibility that they also participate in the inflammatory process as APCs.115
Weight Loss and Malnutrition
Weight loss and malnutrition often are seen in patients with Crohn’s disease and contribute to the complaints of weakness, irritability, malaise, and easy fatigability that are so common. In children, malnutrition can manifest as growth retardation. A host of specific nutritional deficiencies may be found even among patients in long-standing remission,116,117 including iron, folic acid, vitamin B12, calcium, magnesium, zinc, and, particularly in the setting of malabsorption from small intestinal disease, fat-soluble vitamins. Potential contributing factors for these deficiencies are numerous and include inadequate intestinal absorption among patients with extensive small intestinal disease or resection and increased protein losses through exudation from inflamed intestine. Specific medications can cause absorption problems, including decreased calcium absorption with glucocorticoids; malabsorption of fat, fat-soluble vitamins, and calcium with cholestyramine; and folate malabsorption with sulfasalazine.
Anorexia, nausea, and vomiting also can contribute to weight loss and poor nutrition. As with other symptoms of Crohn’s disease, diverse mechanisms may be contributory. TNF originally was discovered as a cytokine capable of inducing cachexia in patients with malignancy and sepsis. Indeed, serum levels of TNF in severely ill patients with Crohn’s disease may be high enough to contribute to anorexia. Delayed gastric emptying may be a causative factor for their symptoms in as many as one third of children with Crohn’s disease,118 and it reflects an unexpectedly high rate of gastroduodenal involvement. Anorexia, nausea, or vomiting also may be caused by drugs used to treat the disease, including metronidazole, sulfasalazine, 6-mercaptopurine, azathioprine, and methotrexate.
Anemia
Anemia is found in one third of patients with Crohn’s disease, primarily as a consequence of iron deficiency from blood loss. Macrocytic anemia can result from vitamin B12 deficiency because of ileal disease or resection, from bacterial overgrowth or, less commonly, from folate deficiency because of proximal small intestinal disease or sulfasalazine therapy. Overproduction of IFN-γ, TNF, or IL-1 can inhibit erythropoietin production, contributing to anemia resistant to iron supplementation.119
EXTRAINTESTINAL MANIFESTATIONS
In addition to penetrating and cicatrizing complications that can arise in patients with Crohn’s disease, numerous complications can occur distant from the bowel. Depending on the definition, it is estimated that between 6% and 25% of all patients with Crohn’s disease have an extraintestinal manifestation of IBD.120–122 Many of these complications are common to Crohn’s disease and UC and indeed to other nonidiopathic inflammatory conditions of the intestine. For example, patients with ileal Crohn’s disease are at increased risk for cholelithiasis, but patients with extensive UC are at nearly the same risk.123 In Crohn’s disease, however, the major risk factor for this complication appears to be the number of prior ileal resections.116 In large series, extraintestinal manifestations are found to occur more often in Crohn’s disease than in UC and are more common among patients with colonic involvement than in patients with no colonic inflammation. One fourth of those affected have more than one manifestation.83,124 Some complications occur as a direct result of the bowel disease, such as nephrolithiasis resulting from oxalate malabsorption. In the case of inflammatory mucocutaneous, joint, and ocular manifestations, the pathogenesis is an influx of mononuclear cells activated in the intestine but homing aberrantly to the involved extraintestinal organs.125
Musculoskeletal
Among the most common extraintestinal manifestations are disorders of the bones and joints. Clubbing of the fingernails is a common and innocuous finding. More consequential are arthritic manifestations, which are observed more commonly in patients with Crohn’s disease than in those with UC. In a study of 976 patients with UC and 483 patients with Crohn’s disease, pauciarticular arthropathy (type I, affecting four or fewer joints) occurred in 3.6% of patients with UC and in 6.0% of those with Crohn’s disease.126 In most patients, joint symptoms occurred in the setting of a relapse of intestinal symptoms. Polyarticular arthropathy (type II, with five or more joints affected) occurred in 2.5% of patients with UC and 4.0% of those with Crohn’s disease.126 Among patients with Crohn’s disease, nearly one half had joint symptoms in association with a relapse in intestinal disease. Distinct HLA genotypes are associated with these two types of peripheral arthropathy. Type I is associated with HLA-DRB1*0103, B*35, and B*27; type II is associated with HLA-B*44.127
Other reports indicate that peripheral arthralgias occur in 16% to 20% of patients with Crohn’s disease,128 most strongly in association with colonic disease.83 Patients tend to have waxing and waning joint pain and stiffness in association with flares of intestinal disease. Joints may be involved in an asymmetrical or migratory fashion. With rare exception, the disease is nondeforming and often is accompanied by skin (erythema nodosum) and eye (uveitis) complications. Rheumatoid factor typically is negative. Knee and ankle joints often are affected first, but elbows, wrists, proximal interphalangeal, metacarpophalangeal, and metatarsophalangeal joints may be involved subsequently.128 Patients who have undergone ileocecal resection for their disease tend to have fewer arthritic complications after their surgery.129
Axial arthropathies are less common than peripheral arthropathies and occur in 3% to 10% of patients with IBD.130 Spondylitis associated with IBD, like idiopathic ankylosing spondylitis, manifests as insidious low back pain and morning stiffness that is improved by exercise. As many as 75% of patients with Crohn’s disease and spondylitis are positive for HLA-B27. Iritis can occur in association with this manifestation. Bilateral symmetrical sacroiliitis without progression to spondylitis is more common than spondylitis and is reported to occur in 4% to 18% of patients.128 In one study, radiologic findings of sacroiliitis were detected in 29% of patients with Crohn’s disease, although only 3% had symptoms of sacroileitis.131
Rarer rheumatologic complications include granulomatous vasculitis,132 periostitis, and amyloidosis. In addition, a septic joint, although a rare complication of Crohn’s disease, should be kept in mind. A septic hip joint is a striking, devastating, and fortunately rare complication of a psoas abscess that extends directly to the acetabular capsule.
Glucocorticoids used to treat Crohn’s disease may be a cause of joint pain. Withdrawal of glucocorticoids can lead to pseudoarthritis, with diffuse joint aches that gradually resolve; adrenal insufficiency should be considered in such patients. Aseptic necrosis of the hip and other joints can occur with or without the use of glucocorticoids and may be disabling.133 Osteomyelitis can occur as a result of direct extension by a fistula, usually to the pelvis, or it may be a recurrent problem distant to the site of inflammation, presumably through hematogenous spread of bacteria.134
Metabolic bone disease is common in Crohn’s disease; osteopenia (T score on dual energy x-ray absorptiometry between −2.49 and −1.0) or osteoporosis (T score no more than −2.5) occurs in 30% to 60% of patients. Morbidity, as a consequence of increased susceptibility to bone fractures, includes debilitating and painful vertebral crush fractures, which can occur even in children with Crohn’s disease. Although glucocorticoid use is the main risk factor for this metabolic bone disease in UC, low bone mineral density is a feature of Crohn’s disease even at diagnosis in both adults and children.135,136 Contributing factors include malabsorption of calcium and vitamin D, smoking,137 and perhaps the effects of proinflammatory cytokines such as TNF, IL-1, and IL-6 on osteoclasts, some of which may be genetically determined.138 Low body mass index may be the most important risk factor for developing osteoporosis.139 Sarcopenia (decreased muscle mass) is closely associated with decreased bone density and is seen in up to 60% of patients with Crohn’s disease.140
Mucocutaneous
The most common skin lesions associated with IBD are pyoderma gangrenosum and erythema nodosum. Neither condition is found solely in IBD, and the finding of one or the other lesion is not specific for either major form of IBD.141
Pyoderma gangrenosum appears first as a papule, pustule, or nodule. It can occur virtually anywhere on the body but most often it occurs on the leg or occasionally around a stoma, and progresses to an ulcer with undermined borders. The ulcer typically has a violaceous rim and crater-like holes pitting the base. The phenomenon of pathergy, or the development of large ulcers in response to minor trauma, is characteristic of pyoderma gangrenosum and the skin lesions of Behçet’s syndrome.142 Healing classically is associated with a cribriform, or pocked, scar. In Crohn’s disease, pyoderma gangrenosum often occurs without an associated flare of intestinal symptoms.
Aphthous ulcers of the mouth are common among patients with Crohn’s disease and UC but also are often seen among otherwise healthy persons.143 As the most cephalad part of the gastrointestinal tract, the mouth rarely may be involved directly by the granulomatous inflammation of Crohn’s disease. Angular cheilitis is seen in nearly 8% of patients with Crohn’s disease.143
A rare manifestation is metastatic Crohn’s disease, granulomatous inflammation of the skin remote from the gastrointestinal tract but histologically identical to the primary intestinal lesion.144 Described cases have included lesions behind the ears, in the perineum, or on the feet, legs, penis, and vulva. Other rare skin manifestations of Crohn’s disease include leukocytoclastic vasculitis,145 Sweet’s syndrome (neutrophilic dermatosis),146 cutaneous polyarteritis nodosa, and epidermolysis bullosa acquisita. Some reports have suggested an increased occurrence of psoriasis among patients with Crohn’s disease.147
Ocular
Ocular manifestations are estimated to occur in 6% of patients with Crohn’s disease.148 Episcleritis is more common in Crohn’s disease than in UC, consists of injection of the sclera and conjunctiva, and does not affect visual acuity. Episodes tend to occur in association with active intestinal disease. Scleritis involves deeper layers of the eye and also occurs most often in parallel with active intestinal disease, but it can cause lasting damage if untreated. Uveitis usually manifests with headache, deep eye pain, lacrimation, blurred vision, and photophobia, as a consequence of iridospasm. Physical examination findings include meiosis and ciliary flush. Visual acuity is preserved unless the posterior segment becomes involved. In contrast to the uveitis associated with ankylosing spondylitis, the presentation of uveitis in patients with IBD often is insidious, with bilateral involvement and extension to the posterior segment.149 Slit-lamp examination demonstrates an inflammatory flare in the anterior chamber. At least one report suggests that children with Crohn’s colitis often have asymptomatic anterior chamber inflammation.150 Other ocular complications of Crohn’s disease include a particular corneal injury referred to as keratopathy and night blindness resulting from malabsorption of vitamin A.
Hepatobiliary
Gallstones are found in over 25% of men and women with Crohn’s disease, representing a relative risk of 1.8 compared with the general population.151 Asymptomatic and mild elevations of liver biochemical tests often are seen in Crohn’s disease, but few of these patients develop clinical evidence of cirrhosis. Primary sclerosing cholangitis more often is associated with UC, but it occurs in 4% of patients with Crohn’s disease, usually those with colonic involvement.152 No genetic risk factors have yet been identified.153 In patients with Crohn’s disease, the inflammatory changes most often are confined to the small biliary radicals, and therefore the presentation is usually one of abnormal liver biochemical tests, pericholangitis on liver biopsy, and a normal cholangiogram.152 Other hepatobiliary complications of Crohn’s disease include fatty liver and autoimmune hepatitis.
Renal and Genitourinary
In addition to the direct complications of perforating Crohn’s disease with encroachment on the bladder and other genitourinary structures, and inflammatory entrapment of the ureter, uric acid and oxalate stones are common in patients with Crohn’s disease.154 In the setting of fat malabsorption resulting from intestinal resection or extensive small intestinal disease, the malabsorbed free fatty acids bind luminal calcium, thereby decreasing the calcium that is available to bind and clear oxalate. Increased oxalate is absorbed as the sodium salt, resulting in hyperoxaluria and calcium oxalate stone formation. Uric acid stones are believed to result from volume depletion and a hypermetabolic state. Rare intrinsic renal complications include membranous nephropathy, glomerulonephritis, and renal amyloidosis. Interstitial nephritis has been associated with mesalamine use, but it is not clear if it is a direct result of the medication or of the disease itself. Penile and vulvar edema also have been reported, but the mechanism for these occurrences is unknown.
Vascular
A prothrombotic tendency has been noted in both major forms of IBD. Patients might present with venous thromboembolism or, much less commonly, arterial thrombosis.155 The hypercoagulable state can arise from many possible causes. Contributing factors can include thrombocytosis, increased levels of fibrinogen, fibrinopeptide A, factor V, and factor VIII, antithrombin III deficiency, and free protein S deficiency, all related to active bowel inflammation. Circulating immune complexes, increased levels of plasminogen activator inhibitors, decreased levels of tissue plasminogen activator, and spontaneous platelet aggregation may be present independent of bowel inflammation. Defective methylenetetrahydrofolate reductase (MTHFR), along with folate and vitamin B12 deficiency, is linked to hyperhomocysteinemia, which in turn predisposes to thrombosis. Increased prevalence of the factor V Leiden mutation156 and MTHFR157 have been observed by some but not other investigators.158 In more than half of patients who experience thrombosis, no predisposing factor can be identified.159
Other
Clinically significant disease of the lungs,160 heart, pancreas, and nervous system161 in association with Crohn’s disease is unusual but reported. Subclinical lung involvement may be much more common than is apparent, perhaps reflecting the commonality of bronchus-associated lymphoid tissue and gut-associated lymphoid tissue.160 Patients with IBD are more at risk to develop asthma, and there also may be an association with chronic obstructive pulmonary disease.147,162 Cardiomyopathy can result from a variety of nutrient deficiencies in patients with marked malabsorption. Pleuropericarditis, myocarditis, and endocarditis occur rarely.163 Acute pancreatitis,164 granulomatous pancreatitis,165 and pancreatic insufficiency166 also have been reported.
DIFFERENTIAL DIAGNOSIS
There are a number of clinical situations in which Crohn’s disease should enter the differential diagnosis, including diarrhea or abdominal pain, especially when localized to the right lower quadrant; evidence of intestinal inflammation on radiologic or endoscopic studies; discovery of an intestinal stricture or fistula arising from the intestine; and evidence of inflammation or granulomas on intestinal histology. Categories of causation that overlap with Crohn’s disease in clinical presentation include functional bowel disorders, primarily irritable bowel syndrome; immune-mediated diseases, particularly other colitides and most importantly UC; medications, especially NSAIDs; vascular disorders, notably ischemic bowel disease and collagen vascular diseases; neoplasia, including carcinoma and lymphoma; infectious diarrheas, intestinal inflammation, or granulomas; and miscellaneous other diseases and syndromes, including diverticular disease. Once the presence of bowel inflammation has been confirmed, the differential diagnosis may focus on presentation according to the anatomic location of the findings (Table 111-2).
| Differential Diagnosis of Ileitis |
NSAID, nonsteroidal anti-inflammatory drug.
From Sands BE. From symptom to diagnosis: Clinical distinctions among various forms of intestinal inflammation. Gastroenterology 2004; 126:1518, with permission.
ESTABLISHING THE DIAGNOSIS AND EVALUATING DISEASE ACTIVITY
Ultimately, the diagnosis of Crohn’s disease is confirmed by findings on imaging studies, endoscopy, and usually histopathology. Barium studies had been the mainstay of Crohn’s imaging for many years, and they accurately define the anatomic location of disease and can reveal evidence of active inflammation. Although a small bowel follow-through study is being replaced at many centers with computed tomography (CT) or magnetic resonance imaging (MRI) (see later), it is still often the primary modality when small intestinal disease is suspected (see Fig. 111-4). Barium studies are especially useful to delineate the late transmural complications of Crohn’s disease, but typical findings may be seen early in the disease as well. Early findings include aphthous ulcers, a coarse villus mucosal pattern, and thickened folds.167 Submucosal edema may be evident as thickening or flattening of the valvulae conniventes, whereas transmural edema manifests as widening of the separation between bowel loops. Ulcers most often occur on the mesenteric border, with consequent pseudosacculation of the antimesenteric border because of shortening of the mesenteric portion.167 Later findings include a cobblestone appearance resulting from edema and inflammation of relatively spared islands of mucosa separated by intersecting longitudinal and transverse knife-like clefts of ulceration.167 Still later, one can discern fistulas, sinus tracts, and fixed strictures.
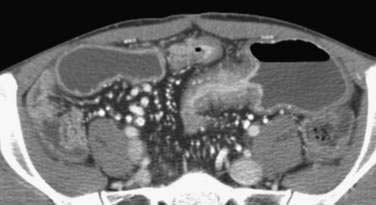
Standard CT studies do not demonstrate mucosal detail and often appear normal early in the course of the disease. The advent of CT enterography, however, has allowed fine mucosal changes to be evaluated along with extraluminal features (and complications of Crohn’s disease). CT enterography varies from routine CT by the use of a high-resolution multidetector scanner, intravenous contrast, and large volumes of oral contrast (either dilute barium or negative water-based contrast) to improve visualization of the small intestinal wall and reveal luminal details (Fig. 111-5). Radiologic findings that are significantly correlated with endoscopic evidence of Crohn’s activity include mural enhancement (segmental enhancement of all or part of the small intestinal wall); increased density of perienteric fat (focal increased inhomogeneous attenuation in the perienteric fat, compared with the appearance of subcutaneous or perienteric fat in adjacent noninflamed intestinal loops), and the comb sign (segmental dilatation of the vasa recta involving an intestinal loop).168 Mural enhancement may be the most useful finding and can be quantified in a semiautomated fashion using dedicated software.168,169 When compared to a consensus diagnosis of Crohn’s disease based on clinical presentation and four different imaging modalities, the sensitivity of CT enterography was 82%, specificity was 89%, and accuracy was 85%.170 The safety of radiation exposure associated with the routine use of CT is a matter of much debate, but safety needs to be taken into careful consideration if this technology is to replace other diagnostic modalities, especially in children.171,172
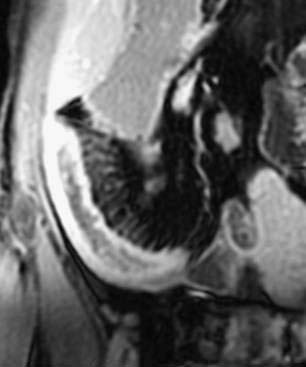
As an alternative to CT, MRI has made substantial strides in evaluating the intestine. MR enterography has the advantages of providing high soft tissue contrast, obtaining static and dynamic images, and avoiding ionizing radiation.173 Similar to CT enterography, patients drink an oral contrast agent before the procedure. Some European centers incorporate enteroclysis with nasoduodenal intubation to administer the contrast, which might increase the yield for subtle mucosal lesions but is likely to be less acceptable to most patients.173 Findings of intestinal wall thickening, submucosal edema, vasa recta engorgement, and lymphadenopathy are signs of active disease (Fig. 111-6). Using dynamic FIESTA (fast imaging employing steady state acquisition), images can add information regarding the functional status of fibrotic segments. A scoring system was developed for assessing small intestinal Crohn’s disease and gives higher scores for details such as increased wall thickness and contrast enhancement, stenosis and mucosal abnormalities, absence of peristalsis and distensibility, and extraintestinal findings.174 Using these criteria, compared with the gold standard of ileocolonoscopy with biopsies, the MRI images yielded a diagnostic accuracy of 91%.
Other potentially useful diagnostic modalities include ultrasound and scintigraphy. Transabdominal ultrasound is used mainly to exclude other causes of abdominal pain, including biliary and gynecologic causes, but it can also be effective in evaluating disease activity of luminal Crohn’s disease.175 There has been some interest in using endoscopic ultrasound to differentiate Crohn’s disease (transmural) from UC, but its major value is still to help evaluate and guide therapy of perianal disease.176,177 Doppler vascular flow studies to evaluate Crohn’s disease activity have been investigated with mixed results.178,179 Ultrasound- and CT-guided percutaneous drainage of intra-abdominal abscesses is a safe and effective alternative to surgical drainage in well-selected patients.180 A growing body of evidence suggests that leukocyte scintigraphy may be a useful diagnostic study in Crohn’s disease. Among children with suspected IBD, 99mTc leukocyte scintigraphy was highly sensitive in identifying abnormalities in patients with just mild inflammation on biopsy and a normal small bowel follow-through.181 Compared with the gold standard of intraoperative findings, it had an accuracy of 84%.182 Early data suggest a possible role for positron emission tomography (PET)/CT scan to help in evaluating the activity level and distribution of Crohn’s disease.183,184
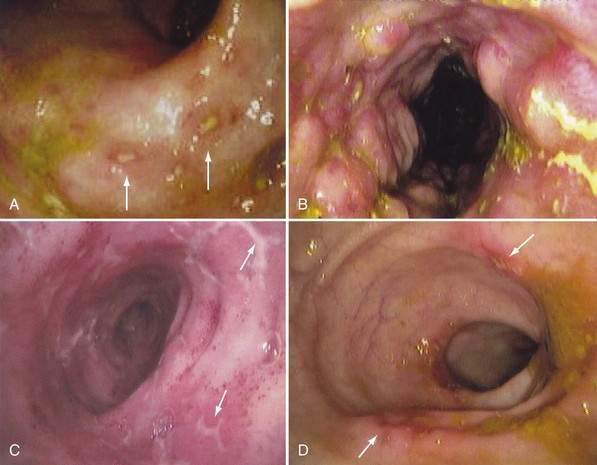
Because of its ability to visualize the mucosa directly and permit biopsy for histopathology, endoscopy complements radiologic techniques. Typical mucosal features recognized on endoscopy include aphthous ulcers, mucosal edema, cobblestoning, and luminal narrowing (Fig. 111-7). The visual impression of demarcated lesions on a background of normal mucosa is most easily recognized in early or mild disease. Rectal sparing is more specific before treatment has been initiated. The discontinuous segmental nature of the disease is an important clue to the diagnosis and has a high positive predictive value.185 Intubation and biopsy of the terminal ileum should be attempted in all patients having colonoscopy and greatly increase the sensitivity and specificity of the examination.186 In general, the diagnostic accuracy of colonoscopy and histologic interpretation is increased substantially by obtaining multiple biopsies from both involved and uninvolved sites. The use of jumbo forceps should be considered to improve sampling of the submucosa. Balloon dilation of strictures is another application of endoscopy in Crohn’s disease that might delay surgery or eliminate the need for it, but balloon dilation is associated with a measurable complication rate.
Wireless capsule endoscopy has become routine for detecting the small intestinal lesions of Crohn’s disease. Although wireless capsule endoscopy may be very sensitive in identifying lesions (even if standard endoscopy has been unrevealing),187,188 its low specificity limits its use as a first-line study to diagnose small intestinal Crohn’s disease.170 The presence of significant bowel stricture should be excluded radiologically before attempting capsule endoscopy, because the rate of capsule retention may be as high as 25% and obstruction is possible.189 Nonetheless, capsule retention can be clinically useful to localize occult strictures pre- and intra-operatively.190
DIFFERENTIATING CROHN’S DISEASE FROM ULCERATIVE COLITIS
When IBD is confined to the colon, the main diagnostic distinction is between Crohn’s colitis and UC. As noted earlier, UC and Crohn’s disease share many similarities in epidemiology and clinical manifestations, and the distinction between them is becoming increasingly important with regard to choices of surgical and medical therapies. Patients with features of both diseases are said to have indeterminate colitis (also referred to as IBD, unclassified), a vague term applied in various ways among different centers. As many as 10% of patients presenting with IBD are considered to have indeterminate colitis. A diagnosis of indeterminate colitis has particular implications for surgical therapy. Patients undergoing ileoanal pouch construction for indeterminate colitis have a relatively high likelihood of developing Crohn’s-like complications of the pouch, although the rate of pouch failure is not significantly different from those with UC.191,192 Histology, when applied without attention to clinical features, is highly likely to be unable to differentiate Crohn’s disease from UC.193,194 Therefore, the entire clinical picture must be considered for accurate diagnosis (Table 111-3). Discriminating features for Crohn’s disease include the presence of small intestinal disease, predominantly right-sided colonic disease, rectal sparing, fistulization (with the exception of rare rectovaginal or perianal fistulas in UC), major perineal complications, and granulomas. In cases initially labeled as indeterminate, the true diagnosis usually becomes clear with the passage of time.
Table 111-3 Differentiating Crohn’s Colitis from Ulcerative Colitis
| FEATURE | CROHN’S COLITIS | ULCERATIVE COLITIS |
|---|---|---|
| Mucosal lesions | Aphthous ulcers are common in early disease; late disease is notable for stellate, rake, bear-claw, linear, or serpiginous ulcers and cobblestoning |
ASCA, anti-Saccharomyces cerevisiae antibody; pANCA, perinuclear antineutrophil cytoplasmic antibody.
From Sands BE. From symptom to diagnosis: clinical distinctions among various forms of intestinal inflammation. Gastroenterology 2004; 126:1518, with permission.
With an incomplete understanding of the environmental and genetic determinants that produce a clinical phenotype of Crohn’s disease or UC, the immunologic markers noted previously are being explored as a means of differentiating the two diseases. pANCA and ASCA were the first such markers shown to correlate with the diagnosis of UC and Crohn’s disease, respectively.103,195,196 To improve the test characteristics, the prediction model has added anti-OmpC99 and anti-CBir1 to pANCA and ASCA.104 When combined, and applying a diagnostic algorithm in a high-prevalence (59%) group, the sensitivity for diagnosing Crohn’s disease is 88% and for diagnosing UC is 93%, with a specificity greater than 95% for both diseases. The positive predictive value (PPV) and negative predictive value (NPV) for Crohn’s disease is 96% and 93%, respectively. The PPV and NPV for UC is 89% and 98%, respectively. The pretest probability is high in this group, and when applying this algorithm to a lower-prevalence group (15%), which is more appropriate for the pretest probability associated with indeterminate colitis, the PPV and NPV for Crohn’s disease drop to 74% and 73%, respectively.197 Thus, serologic testing for pANCA, ASCA, OmpC, and anti-CBir1 currently is an adjunct to diagnosis in selected cases—one additional piece of evidence to be considered but not definitive in establishing the diagnosis. Genetic research has led to great strides over the past few years, and CARD15/NOD2 testing is available commercially; due to its low diagnostic accuracy, however, this test is not currently recommended as part of the diagnostic algorithm for Crohn’s disease.
MEASURING DISEASE ACTIVITY
Composite scoring systems, most commonly the Crohn’s Disease Activity Index (CDAI, Table 111-4), are used in an attempt to integrate the many possible features of the disease. Other disease activity indices include the van Hees index,198 the Cape Town index,199 the Harvey-Bradshaw index,200 the International Organization of IBD (or Oxford) index,201 the St. Marks Crohn’s index,202 De Dombal’s index,203 the Talstad index,204 and a Crohn’s disease activity index for survey research.205 Specialized indices have been developed for use in children with Crohn’s disease.206,207 All these indices vary in the features included in the scoring system, but most include a combination of subjective symptoms and objective findings on examination and laboratory testing. A great deal of interobserver and methodologic variation has been noted, even among experienced researchers,208 and in fibrostenotic disease, indices that rely more heavily on subjective measurements can poorly reflect bowel inflammation as a cause of symptoms.209 Other approaches have included use of disease-activity indices that focus on a specific outcome, such as perianal disease,210,211 endoscopic findings,212,213 or achieving an individual goal of therapy.214 Each of these approaches has advantages and disadvantages, but all have their application in research rather than in clinical practice.
| VARIABLE | SCALE | WEIGHT |
|---|---|---|
| Liquid or very soft stools | Stool count summed daily for 7 days | 2 |
| Abdominal pain | Sum of 7 days of daily ratings as: 0 = none, 1 = mild, 2 = moderate, 3 = severe |
5 |
| General well-being | Sum of 7 days of daily ratings as: 0 = generally well, 1 = slightly below par, 2 = poor, 3 = very poor, 4 = terrible |
7 |
| Features of extraintestinal disease | Any of the following present during the 7 days: |
* To calculate the CDAI, the scale is multiplied by the weighting factor for each variable, and then all eight weighted variables are added.
From Best WR, Becktel JM, Singleton JW, et al. Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. Gastroenterology 1976; 70:439, with permission.
Another approach with some merit is the measurement of biologic markers of disease inflammation. The erythrocyte sedimentation rate and serum acute phase response proteins (e.g., C-reactive protein and orosomucoid) may be useful in tracking disease activity, but they lack sensitivity and specificity. Another approach is to measure the serum levels of cytokines, cytokine receptors, and adhesion molecules that are proximate to the expression of acute phase reactants; examples include IL-6, IL-1, soluble IL-2 receptor, and soluble intercellular adhesion molecule-1.215 As with the acute phase reactants, these tests also lack sensitivity and specificity. Direct measurements of intestinal immune activation in a mucosal sample could enhance sensitivity and specificity but are inconvenient, invasive, and, if dependent on biopsy, subject to variability and poor standardization. Quantification of radiolabeled leukocytes in stool appears to be a sensitive and fairly specific indicator of mucosal inflammation, but it is cumbersome to perform and exposes the patient to radiation. Fecal excretion of calprotectin (a calcium- and zinc-binding protein found in neutrophils) and lactoferrin (an iron-binding glycoprotein secreted by most mucosal membranes), has been shown to be sensitive markers of intestinal inflammation216 that also might correlate with relapse of quiescent disease217 and response to biologic therapy.218
Ultimately, it is desirable to measure the patient’s overall state of well being, or subjective health status. Health-related quality of life may be measured with generic instruments, which focus on various domains of health common to many disease states, or with disease-specific instruments, which focus on specific domains relevant to the disease of interest. The Inflammatory Bowel Disease Questionnaire is the most widely accepted disease-specific instrument and measures separate domains for bowel, social, systemic, and emotional function.219
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 111 Crohn’s Disease
Idiopathic inflammatory bowel disease (IBD) comprises conditions characterized by chronic or relapsing immune activation and inflammation within the gastrointestinal tract. Crohn’s disease and ulcerative colitis (UC) are the two major forms of idiopathic IBD; less common, but increasingly recognized, are the microscopic colitides, primarily collagenous colitis and lymphocytic colitis (see Chapter 124). Other chronic inflammatory conditions of the intestine share some features of presentation and pathogenesis with idiopathic IBD, but they have identifiable etiologies. These disorders include diversion colitis, bypass enteropathy, radiation colitis, and drug-induced colitides. The two major forms of IBD share many clinical and epidemiologic characteristics, suggesting that underlying causes may be similar. Indeed, more than occasionally, Crohn’s disease cannot be distinguished from UC on clinical grounds, yet the two diseases are distinct syndromes with divergent treatment and prognosis.
HISTORY OF CROHN’S DISEASE
Although the eponym Crohn’s disease has gained general acceptance in recent decades, clear clinicopathologic reports of the same process date back at least two centuries. Morgagni provided a description of intestinal inflammation characteristic of Crohn’s disease in 1761.1 Only after the identification of the tubercle bacillus by Koch in 1882 was it possible to describe persons with ileocecal disease similar to intestinal tuberculosis but lacking the organism. Such reports were provided by Fenwick (1889),1 Dalziel (1913),2 Weiner (1914), Moschcowitz and Wilensky (1923 and 1927), and Goldfarb and Suissman (1931).3 In 1932, the landmark publication of Crohn, Ginzburg, and Oppenheimer called attention to “terminal ileitis” as a distinct entity and chronic disease.4 This term was soon deemed unsuitable, however, when it became apparent that the disease process might involve the colon. Patients, too, misunderstood and were frightened by the “terminal” nature of their illness. The term “regional enteritis” embraced the focal nature of the process, but failed to incorporate knowledge of the possibility of disparate sites of involvement within the gastrointestinal tract, including the small and large bowel in combination5 and large bowel in isolation.6 The term “granulomatous enterocolitis” lost acceptance when it became clear that granulomas were not a sine qua non of the diagnosis. In the end, the name “Crohn’s disease” has been adopted to encompass the many clinical presentations of this pathologic entity. But for the alphabetic priority these authors chose, Crohn’s disease might well have been Ginzburg’s or Oppenheimer’s disease.
EPIDEMIOLOGY
Misclassification of disease is problematic. Historically, unidentified infections, later recognized by improved culture and diagnostic techniques, might have accounted for some portion of cases, particularly among persons with a single episode of disease. At times, differentiating Crohn’s disease from UC may be difficult, especially at the time of diagnosis, and before the passage of time has allowed distinctive disease characteristics to become manifest. Reassignment of a diagnosis of Crohn’s disease or UC may be as high as 9% in the first two years after diagnosis.7
Despite these methodologic limitations, distinct and reproducible geographic and temporal trends in incidence have been observed. In both North America and Europe, higher incidence rates have been noted in more northern latitudes. For example, age-adjusted annual incidence rates of 9 per 100,000 persons were reported in Olmsted County, Minnesota,8 and Copenhagen County, Denmark,9 and as high as 20 per 100,000 in Nova Scotia.10 Comparatively, estimates of incidence rates reached only 0.9 per 100,000 in Spain11 and 3.4 per 100,000 in Italy.12 A north-south gradient similar to that observed in Europe13 has been noted in the United States14 and even within the state of California itself, with estimated incidence rates of 7.0 and 3.6 per 100,000 in northern and southern California, respectively.15,16 In Asian countries, the incidence rate has remained low, with a mean estimated incidence of 0.5 per 100,000 persons in Korea17 and a similar incidence in Japan,18,19 whereas in Australia and New Zealand, incidence rates have ranged from 1.75 to 2.1 per 100,000.20,21 Crohn’s disease is thought to be extremely rare in much of South America and Africa22 with the exception of South Africa, where the most recent estimate of the incidence rate for the white population is 2.6 per 100,000; it is considerably lower among the nonwhite population.23
In some regions of the world where Crohn’s disease was rare, although still low compared with western countries, incidence is rising dramatically. For example, in Seoul, South Korea, the incidence increased from 0.05 per 100,000 in the early 1980s to 1.34 per 100,000 between 2001 and 2005.17 This trend has been seen throughout other regions as well.24–27 Estimates from less-affluent nations may be influenced by increasing access to health care, and therefore, genetic and environmental factors in these regions are difficult to disentangle.
Incidence rates have continued to rise in some regions, such as in Denmark,9,28 whereas in others they appear to be stabilizing. In Olmsted County, Minnesota, rates had been steadily increasing from approximately 3 per 100,000 (1954-1963) to nearly 8 per 100,000 (1964-1973), but since the late 1970s these rates have not changed significantly.8,29 Mortality trends for Crohn’s disease have followed a similar pattern, with rising mortality until the mid-1970s and stable rate since.30 Although improved diagnostic capabilities might have played some role in the rising incidence leading up to the mid-1970s, the fact that Crohn’s-related mortality was rising in parallel argues against the theory that rising Crohn’s diagnoses merely represented detection bias involving mild cases. Most recently, the prevalence of Crohn’s disease in the United States is estimated to be 201 per 100,000 adults and 43 per 100,000 in children, adolescents, and adults younger than 20 years.14
Studies throughout the world have shown a small excess risk of Crohn’s disease among women. Most reports show a female-to-male ratio in adult patients between unity and 1.3 : 1.10,14,28 In the pediatric population this is reversed, with more boys having Crohn’s disease than girls.14 This slight difference in risk in adult-onset disease may be explained by hormonal or life-style factors and stands in contrast to the nearly equal or even slight male predominance seen in UC.
Crohn’s disease is diagnosed most often among persons 15 to 30 years of age, although the age of diagnosis can range from early childhood throughout an entire lifespan. Population-based studies have shown the median age of diagnosis to be approximately 30 years.8,10 Conflicting information may be found regarding trends in the age of diagnosis. In Olmsted County, Minnesota, younger age groups have had a fairly stable incidence over the past 20 years, with rising rates in patients aged 60 and older.8,10 This trend of older-age diagnoses also was seen in population-based studies in Copenhagen, Denmark,31 and in Stockholm, Sweden,32 the median age of diagnosis increased from 25 years in 1960 to 1964 to 32 years in 1985 to 1989. These findings reflect diagnosis in a larger proportion of patients older than 60 years. Indeed, many, though not all,10 studies have shown a smaller second peak in incidence later in life, generally in the seventh decade.33 This second peak may be the result of ascertainment bias because of more frequent contact with medical care and more frequent evaluation of older patients. Differences in clinical presentation among younger and older patients suggest that distinct risk factors are operative at different ages at onset.34 The pathologic findings in young and old patients are not different, although some studies have identified a greater proportion of colonic and distal disease among older patients,33 compared with a predominance of ileocolonic disease in younger patients.35
ETIOLOGY AND PATHOGENESIS
INITIATING EVENTS
In light of the nature of the pathologic findings in Crohn’s disease (see later) and UC, it long has been clear that IBD represents a state of sustained immune response. The question arises as to whether this is an appropriate response to an unrecognized pathogen or an inappropriate response to an innocuous stimulus. Many infectious agents have been proposed as the cause of Crohn’s disease including chlamydia, Listeria monocytogenes, cell-wall–deficient Pseudomonas species, reovirus, and many others. Paramyxovirus (measles virus) has been implicated etiologically in Crohn’s disease as a cause of granulomatous vasculitis and microinfarcts of the intestine,36 but a proposed association between early measles vaccination and Crohn’s disease largely has been disproved.37 Another suggestion has been that the commensal flora, although normal in speciation, possess more subtle virulence factors, such as enteroadherence, that cause or contribute to IBD.38
Among the most enduring hypotheses is that Mycobacterium paratuberculosis is the causative agent of Crohn’s disease. This notion dates to Dalziel’s observation in 1913 that idiopathic granulomatous enterocolitis in humans is similar to Johne’s disease, a granulomatous bowel disease of ruminants caused by M. paratuberculosis.39 M. paratuberculosis is extremely fastidious in its culture requirements, and some proponents of this hypothesis have speculated that the presence of M. paratuberculosis as a spheroplast may have confounded efforts to confirm this hypothesis by culture of the organism; demonstrating it by immunohistochemistry, in situ hybridization, and polymerase chain reaction methodology; and empirical treatment with antimycobacterial antibiotics (see “Medical Therapy,” later). Most investigation in this area has been inconclusive, providing insufficient evidence to prove or reject the hypothesis.
In light of the diversity of substances and bacteria within the intestinal lumen, it is remarkable that the intestine is not perpetually inflamed. The presence of low-level physiologic inflammation within the healthy intestinal mucosa represents a state of preparedness to deal with potentially harmful agents. A more vigorous response would not be appropriate if directed toward the innocuous commensal flora of the intestine. Experiments in animal models of IBD suggest that in a genetically susceptible host, a classic pathogen is not necessary to cause IBD, but rather nonpathogenic commensal enteric flora are sufficient to induce an inappropriate chronic inflammatory response. In diverse models, animals raised under germ-free conditions show diminished or delayed expression of the IBD phenotype.40 On introduction of defined bacterial flora, however, the expected phenotype of bowel inflammation becomes manifest.40 Such models suggest that a diversity of genetic alterations, including those that affect intestinal barrier function and regulation of mucosal immunity, can result in intestinal inflammation. As in the animal models of IBD, evidence in patients with Crohn’s disease also points to an over-responsiveness of mucosal T cells to the enteric flora, manifest in part by the presence of antibodies against an array of bacterial antigens, including Escherichia coli outer membrane porin C (OmpC) and flagellin. Advances in the genetic bases of Crohn’s disease confirm that diverse bacterial agents are capable of fueling the inflammation of Crohn’s disease (see later).
GENETICS
The argument for a genetic predisposition to IBD begins with the observation that family members of affected persons are at greatly increased risk for developing IBD. The relative risk among first-degree relatives is 14 to 15 times higher than that of the general population.41 Roughly one of five patients with Crohn’s disease report having at least one affected relative. Many families have more than one affected member, and although there is a tendency within families for either UC or Crohn’s disease to be present exclusively, mixed kindreds also occur, suggesting the presence of some shared genetic traits as a basis for both diseases, as has recently been confirmed.
Ethnicity also plays a role. Eastern European (Ashkenazi) Jews are at a two- to four-fold higher risk of developing IBD than non-Jews of the same geographic location, and they are at greater risk of having multiple affected family members. Studies of monozygotic and dizygotic twins suggest that genetic composition is a more powerful determinant of disease for Crohn’s disease than for UC: The concordance rate among monozygotic twins is as high as 67% for Crohn’s disease but only 13% to 20% for UC. Most studies have suggested that concordance of disease location and disease behavior are higher than one would expect by chance.42 Finally, some subclinical markers of Crohn’s disease, including anti-OmpC antibodies, are more common among apparently healthy family members of Crohn’s disease probands than among the general population.43
Early studies of IBD genetics were limited by the slow speed of DNA sequencing and an incomplete understanding of the human genome’s structure. Studies of IBD genetics depended upon selecting a candidate gene based upon the then-current understanding of pathogenesis. With automated, rapid DNA sequencing, and mapping of the common genetic variants that occur in humans (the HapMap, or haplotype map), genome-wide association (GWA) studies became feasible. GWA studies can simultaneously explore many hundred thousands of genetic markers, providing a broad and unbiased approach to assessing the association of genomic loci to a specific disease without prior hypothesis about a candidate gene. GWA studies have been more successful in Crohn’s disease than in any other complex disease, and they have accelerated the pace of gene discovery, providing unexpected insights into pathogenesis.44 More than 30 genetic loci have been convincingly associated with Crohn’s disease in a large and powerful GWA study, confirming some known loci and discovering many loci that had not been described previously (Table 111-1).45 Thus far, genetic studies have highlighted three pathways of fundamental importance in the pathogenesis of Crohn’s disease.
The first susceptibility locus for Crohn’s disease was identified in 2001 as the NOD2 (nucleotide-binding oligomerization domain 2) gene, also known as CARD15 (caspase-recruitment domain 15).46,47 The allelic variants most commonly associated with Crohn’s disease in European and American populations include one frameshift insertion leading to early truncation of the protein (Leu1007fsinsC) and two missense mutations (Arg702Trp, Gly908Arg). Carriage of disease-associated allelic variants on both chromosomes confers an odds ratio for Crohn’s disease of 17.1 (95% confidence interval [CI], 10.7-27.2), and heterozygous persons have an odds ratio of 2.5 (95% CI, 2.0-2.9) for the disease.48 Studies have associated genetic polymorphisms of NOD2/CARD15 with younger onset and ileal location of disease and increased likelihood of stricture formation.49–52 It has been estimated that as many as 20% to 30% of patients with Crohn’s disease bear abnormal NOD2/CARD15. Nevertheless, the penetrance of NOD2/CARD15 is estimated to be less than 1%53; that is, disease-related allelic variants of the gene may be found in a large number of persons who do not have Crohn’s disease; this strongly suggests that environmental factors, as yet incompletely elucidated, play a significant role in the expression of the Crohn’s disease phenotype (see later).
The discovery of the association of NOD2/CARD15 with Crohn’s disease has opened a remarkable window into the pathogenesis of Crohn’s disease. The gene product of NOD2/CARD15 is a cytosolic protein that functions as an intracellular sensor of bacteria. Specifically, the protein binds to muramyl dipeptide (MDP; MurNAc-L-Ala-D-isoGln), a component of bacterial peptidoglycan, found in Gram-positive and Gram-negative bacteria.54,55 The NOD2/CARD15 protein is expressed in monocytes and enterocytes, specifically in Paneth cells,56 which lie within the crypts and produce the endogenous antimicrobial peptides called defensins. The NOD2/CARD15 gene consists of two CARD domains, a nucleotide binding domain (NBD), and 10 leucine-rich repeats (LRR). NOD2/CARD15 variants associated with Crohn’s disease lie within the LRR and interfere with binding to MDP. In mononuclear cells, mutations in NOD2 result in decreased activation of nuclear factor (NF)-κB, whereas an excess of NF-κB expression is observed in tissue inflamed by Crohn’s disease. This apparent paradox has yet to be unraveled completely, but it is clear that defects in NOD2 impair antibacterial responses, particularly to oral exposure to pathogens. Notably, the production of β-defensins, which are antibacterial proteins produced by Paneth cells, is defective in Crohn’s patients with variant NOD2.57 These findings strongly implicate defects in innate immunity—the immediate and nonspecific immune responses to microbial infection—in a subset of patients with Crohn’s disease, with subsequent chronic activation of adaptive immunity, the antigen-specific responses mediated by antigen presenting cells (APCs) and T cells.
Subsequent discoveries have implicated defects in multiple genes in the autophagy pathway in the pathogenesis of Crohn’s disease.58,59 Autophagy is an ancient cellular process, highly conserved in evolution, by which segments of cytoplasm are isolated within a membrane and delivered to lysosomes by mechanisms that do not involve transport through endocytic or vacuolar sorting pathways. This unique process plays a role in cellular homeostasis by clearing proteins that are long-lived, misfolded, or aggregated, and by clearing apoptotic bodies, which might otherwise trigger inflammation and autoimmunity. Autophagy has been shown to contribute directly to innate immunity through direct killing of pathogens, activation of Toll-like receptors and Nodd-like receptors, and elaboration of immunomodulatory cytokines such as interferon (IFN)-γ. Autophagy also stands at the interface of innate and adaptive immune responses, delivering antigen to human leukocyte antigen (HLA) class II molecules in APCs for antigen-specific binding.59
GWA studies have identified variants that predispose to Crohn’s disease in at least two autophagy-related genes. The first, the autophagy-related 16-like 1 (ATG16L1) gene, was noted as having a disease-associated single nucleotide polymorphism (SNP) encoding an amino acid substitution in exon 8, resulting in a change from alanine to threonine58–60; this minor allele is protective against Crohn’s disease. ATG16L1 is expressed by intestinal epithelial cells, APCs, and various subsets of human T cells. The second autophagy gene associated with Crohn’s disease is the IRGM (immunity-related GTPase [guanosine triphosphatase] family member M) gene on chromosome 5q33.1.61 Careful study suggests that the disease-associated variants of this gene do not affect the amino acid sequence of its product, but they more likely alter its expression.61 IRGM appears to be important in resistance to intracellular pathogens such as mycobacteria, Listeria monocytogenes, and Toxoplasma gondii.59
A third pathway associated with Crohn’s disease is interleukin (IL)-23 and other gene products associated with this protein.62 IL-23 is a heterodimeric cytokine comprising two linked subunits (p19 and p40). IL-23 is produced by many cell types, including dendritic cells and macrophages, in response to diverse microbial signals. Naïve CD4+ T cells up-regulate IL-23 receptor when exposed to IL-6 and transforming growth factor (TGF)-β, completing an autocrine loop in the generation of Th17 T cells, effector T cells that produce IL-17.63,64 A rare variant of the IL23R gene leading to a glutamine at position 381 rather than an arginine is strongly protective for Crohn’s disease, with an odds ratio of 0.26 to 0.45; other, more common SNPs are associated with increased risk for Crohn’s disease and UC.65 In the same pathway, variants of the IL12B gene, encoding the p40 subunit common to IL-12 and IL-23, and of the JAK2 and STAT3 genes, with roles in IL23R signaling, as well as in Th17 differentiation in the case of STAT3, also have been associated with Crohn’s disease susceptibility.45 Together, these findings support the pivotal role of this pathway in maintaining mucosal homeostasis in the normal intestine. As the functional alterations associated with the many other identified genetic risk loci are elucidated, it is certain that new insights into the causes of Crohn’s disease will arise.
ENVIRONMENT
Although the greatest relative risk of Crohn’s disease is found among first-degree relatives of affected persons, particularly siblings of the proband, environmental factors also are important. As noted earlier, the rising incidence of Crohn’s disease over many decades highly suggests an environmental contribution to the expression of disease. Epidemiologic studies have examined numerous risk factors for Crohn’s disease. Most studies have found breast-feeding to be protective for IBD, presumably by playing a role in early programming of immune responses in the developing gastrointestinal tract. Occupations associated with outdoor physical labor are relatively under-represented among Crohn’s patients. Crohn’s disease has been associated with higher socioeconomic status,66 presumably because of relative underexposure to diverse environmental antigens in the course of childhood—the hygiene hypothesis as it relates to intestinal mucosal immunity in IBD. Many, but not all, studies have discerned an increased risk of Crohn’s disease among women who use oral contraceptives. Nonsteroidal anti-inflammatory drugs (NSAIDs) have been implicated not only in exacerbations of IBD but also as a potential precipitant of new cases, perhaps by increasing intestinal permeability. Increased intake of refined sugars and a paucity of fresh fruits and vegetables in the diet have been associated with the development of Crohn’s disease. It is conceivable that this observation may be confounded by exacerbation of symptoms in patients with mild disease because of increased dietary fiber intake and subsequent avoidance of these food items before diagnosis.
Smoking is one of the more notable environmental factors for IBD. UC is largely a disease of ex-smokers and nonsmokers, whereas Crohn’s disease is more prevalent among smokers. In addition, smokers have more surgery for their disease and a greater risk of relapse after resection. The reasons for the divergent effect of smoking on Crohn’s disease and UC are poorly understood, but they might include effects on intestinal permeability, cytokine production, and clotting in the microvasculature. More recently, studies have focused on the role of carbon monoxide in stimulating immunosuppressive effects mediated by heme oxygenase-1.67
Many patients report a correlation between disease exacerbations and stress. Although depression and anxiety are a common reaction to illness, Crohn’s disease has not been shown to be caused by stress or by an anxious personality. The mind-body connection between emotional states or stress and intestinal inflammation in IBD is slowly being revealed, however, and studies indicate that stress may be associated with risk of relapse in Crohn’s disease.68
ADAPTIVE IMMUNE RESPONSE AND INFLAMMATION
The interaction between effector T cells and APCs is critical to the pathogenesis of Crohn’s disease (Fig. 111-1). The antigens that perpetuate the inflammatory response are taken up by APC. Degradation of antigen within proteasomes results in presentation of an epitope in the context of major histocompatibility complex (MHC) class II. Interaction between MHC class II and the T-cell receptor (CD3) results in antigen-specific interaction between the macrophage and the CD4+ T cell. This event is necessary, but not sufficient, to activate the T cell. A second costimulatory signal is needed as well, because binding of CD3 to MHC class II without a costimulatory signal can result in anergy or apoptosis. Important costimulatory signals include binding of tumor necrosis factor (TNF) to TNF receptor, CD40 to CD40 ligand, and B7 to CD28. Activation of T cells leads to production of IL-2, an important growth factor for T cells.
As noted earlier, the p40 subunit is common to IL-12 and IL-23, each of which, in turn, is critical in shaping the Th1 and Th17 responses that characterize Crohn’s disease. In addition to IL-23, the presence of TGF-β and IL-6 facilitate differentiation of naïve T cells into pathogenic Th17 cells.63 Activated APCs further shape and amplify the immune response by producing the T cell growth factor IL-2 and the proinflammatory cytokines IL-1 and TNF. Within mononuclear cells, the key nuclear transcription factor is NF-κB, which regulates the transcription of IL-1, IL-6, IL-8, TNF, and other peptides central to the inflammatory response.69
NF-κB is regulated tightly within the cell. In the inactive state, NF-κB is held in the cytoplasm, bound to inhibitory κBα. During cell activation after receptor binding, various kinases phosphorylate inhibitory κBα, thereby leading to its degradation. NF-κB is then released, permitting translocation to the nucleus, where it binds to the promoter regions of numerous genes that support the inflammatory response. Such genes include those that encode proinflammatory cytokines such as TNF, adhesion molecules, and chemokines.69
Expression of adhesion molecules is critical to amplify the immune response, because the resident populations of granulocytes and mononuclear cells alone do not account for the vigorous inflammatory reaction characterizing IBD. Adhesion molecules on the leukocyte surface and their ligands on the endothelium of venules in the lamina propria interact in a coordinated multistep process that permits trafficking of inflammatory cells into the mucosa. First, a weak interaction between selectins on the leukocyte surface and the endothelium leads to rolling of the leukocytes along the endothelium. Second, in the presence of chemokines such as IL-8, activation occurs, and integrins are expressed on the leukocyte surface. Third, interactions between leukocyte integrins and immunoglobulin-like cellular adhesion molecules on the endothelial surface lead to spreading of the cell and diapedesis.70 Specificity is conferred by the presence of tissue-specific cellular adhesion molecules. The integrins α4β7 and αEβ7 are of special importance in IBD, because the corresponding ligands—mucosal addressin cellular adhesion molecule and E-cadherin—are intestine specific. Mucosal addressin cellular adhesion molecule is expressed constitutively on the endothelium of venules in the lamina propria,70 whereas binding of αEβ7 on intestinal lymphocytes to E-cadherin on intestinal epithelium permits localization of intraepithelial lymphocytes. Antibodies to the α4 subunit of integrin have proved to be therapeutic in Crohn’s disease.71
Once recruited to the lamina propria, mononuclear cells and granulocytes elaborate a variety of injurious and proinflammatory substances that ultimately cause tissue destruction. These substances include prostaglandins, reactive oxygen metabolites, nitric oxide, leukotrienes, and proteases. Collagenase and matrix metalloproteinases play a pivotal role in the tissue destruction seen in IBD.72 Counterbalancing these destructive substances are other substances that promote epithelial restitution and repair, including IL-11, trefoil peptides, and growth factors such as epidermal growth factor and keratinocyte growth factor.
PATHOLOGY
Focal intestinal inflammation is the hallmark pathologic finding in Crohn’s disease. This tendency for focal inflammation is evident in focal crypt inflammation, focal areas of marked chronic inflammation, the presence of aphthae and ulcers on a background of little or no chronic inflammation, and the interspersing of segments of involved bowel with segments of uninvolved bowel. Even within a single biopsy specimen one can see a pronounced variability in the degree of inflammation. The presence of focally enhanced gastritis, characterized by a focal perifoveolar or periglandular lymphomonocytic infiltrate, is a common finding that occurs in 43% of unselected patients with Crohn’s disease.73 This finding underscores the focal nature of the inflammation, despite the strong potential for inflammation to occur anywhere along the entire longitudinal axis of the gastrointestinal tract. To a certain extent, the nature of the findings and the depth of inflammatory changes depend on the chronicity of the inflammation.
EARLY FINDINGS
Because of the variable and often long delay between the onset of the disease process and its diagnosis, it rarely is possible to observe the evolution of pathology from the earliest events. Studies of recurrent Crohn’s disease after ileal resection have offered a window into the sequence of pathologic changes in the disease.74
