[level-membership-for-internal-medicine-category]
Chapter 16 Critical care medicine
Introduction
Critical care medicine (or ‘intensive care medicine’) is concerned predominantly with the management of patients with acute life-threatening conditions (’the critically ill’) in specialized units. As well as emergency cases, such units admit high-risk patients electively after major surgery (Table 16.1). Intensive care medicine also encompasses the resuscitation and transport of those who become acutely ill, or are injured in the community. Management of seriously ill patients throughout the hospital (e.g. in coronary care units, acute admissions wards, postoperative recovery areas or emergency units), including critically ill patients who have been discharged to the ward (‘outreach care’), is also undertaken. Teamwork and a multidisciplinary approach are central to the provision of intensive care and are most effective when directed and coordinated by committed specialists.
Table 16.1 Some common indications for admission to intensive care
General aspects of managing the critically ill
Recognition and diagnosis of critical illness
Early recognition and immediate resuscitation are fundamental to the successful management of the critically ill. In order to facilitate identification of ‘at risk’ patients on the ward and early referral to the critical care emergency or outreach team a number of early warning systems have been devised (e.g. the Modified Early Warning Score, MEWS; see Box 16.1). These are based primarily on bedside recognition of deteriorating physiological variables and can be used to supplement clinical intuition. A MEWS score of ≥5 is associated with an increased risk of death and warrants immediate admission to ICU. Another example of a system used to trigger referral to a Medical Emergency Team (MET) is also shown in Box 16.1 (see also ‘Management of shock and sepsis’ and ‘Clinical assessment of respiratory failure’, below).
 Box 16.1
Box 16.1
Early warning systems for referral of ‘at risk’ patients to the critical care team
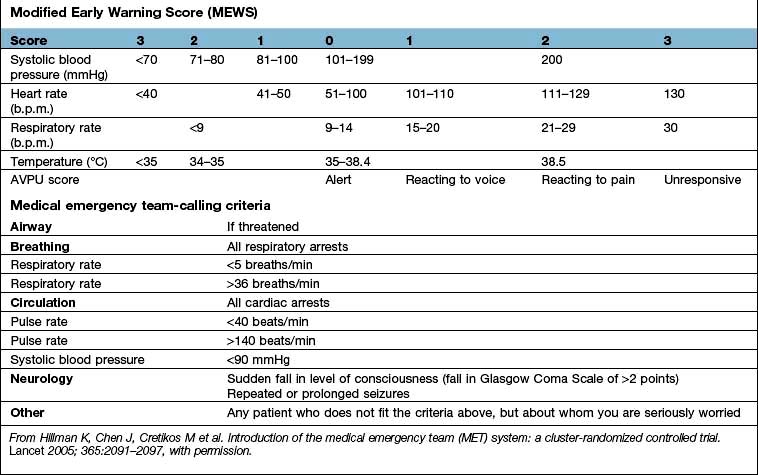
Medical emergency team-calling criteria
|
Airway |
If threatened |
|
Breathing |
All respiratory arrests |
|
Respiratory rate |
<5 breaths/min |
|
Respiratory rate |
>36 breaths/min |
|
Circulation |
All cardiac arrests |
|
Pulse rate |
<40 beats/min |
|
Pulse rate |
>140 beats/min |
|
Systolic blood pressure |
<90 mmHg |
|
Neurology |
Sudden fall in level of consciousness (fall in Glasgow Coma Scale of >2 points) |
|
|
Repeated or prolonged seizures |
|
Other |
Any patient who does not fit the criteria above, but about whom you are seriously worried |
From Hillman K, Chen J, Cretikos M et al. Introduction of the medical emergency team (MET) system: a cluster-randomized controlled trial. Lancet 2005; 365:2091–2097, with permission.
In some of the most seriously ill patients, the precise underlying diagnosis is initially unclear but in all cases, the immediate objective is to preserve life and prevent, reverse or minimize damage to vital organs such as the lungs, brain, kidneys and liver. This involves a rapid assessment of the physiological derangement followed by prompt institution of measures to support cardiovascular and respiratory function in order to restore perfusion of vital organs, improve delivery of oxygen to the tissues and encourage the removal of carbon dioxide and other waste products of metabolism (following the ABC approach: Airway, Breathing, Circulation, see Fig. 16.25, below). The patient’s condition and response to treatment should be closely monitored throughout. The underlying diagnosis may only become clear as the results of investigations become available, a more detailed history is obtained and a more thorough physical examination is performed. In practice resuscitation, assessment and diagnosis usually proceed in parallel.
Critically ill patients require multidisciplinary care with:
 Intensive skilled nursing care (usually 1 : 1 or 1 : 2 nurse/patient ratio in the UK).
Intensive skilled nursing care (usually 1 : 1 or 1 : 2 nurse/patient ratio in the UK).
 Specialized physiotherapy including mobilization and rehabilitation.
Specialized physiotherapy including mobilization and rehabilitation.
 Management of pain and distress with judicious administration of analgesics and sedatives (see p. 893).
Management of pain and distress with judicious administration of analgesics and sedatives (see p. 893).



 Care of the mouth, prevention of constipation and of pressure sores.
Care of the mouth, prevention of constipation and of pressure sores.
 Nutritional support (see p. 222). Protein energy malnutrition is common in critically ill patients and is associated with muscle wasting, weakness, delayed mobilization, difficulty weaning from ventilation, immune compromise and impaired wound healing. There is also an association between malnutrition and increased mortality. It is therefore recommended that nutritional support should be instituted as soon as is practicable in those unable to meet their nutritional needs orally, ideally within 1–2 days of the acute episode. Enteral nutrition, which is less expensive, preserves gut mucosal integrity, is more physiological and is associated with fewer complications, is preferred. Recently, the value of early feeding has been questioned, apart from giving small amounts to ensure gut viability. Parenteral nutrition is sometimes indicated at a later stage for those unable to tolerate or absorb enteral nutrition and should be initiated without delay, at least within 3 days. Persistent attempts at enteral nutrition in those with gastrointestinal intolerance leads to underfeeding and malnutrition.
Nutritional support (see p. 222). Protein energy malnutrition is common in critically ill patients and is associated with muscle wasting, weakness, delayed mobilization, difficulty weaning from ventilation, immune compromise and impaired wound healing. There is also an association between malnutrition and increased mortality. It is therefore recommended that nutritional support should be instituted as soon as is practicable in those unable to meet their nutritional needs orally, ideally within 1–2 days of the acute episode. Enteral nutrition, which is less expensive, preserves gut mucosal integrity, is more physiological and is associated with fewer complications, is preferred. Recently, the value of early feeding has been questioned, apart from giving small amounts to ensure gut viability. Parenteral nutrition is sometimes indicated at a later stage for those unable to tolerate or absorb enteral nutrition and should be initiated without delay, at least within 3 days. Persistent attempts at enteral nutrition in those with gastrointestinal intolerance leads to underfeeding and malnutrition.
 Critically ill patients commonly require intravenous insulin infusions, often in high doses, to combat hyperglycaemia and insulin resistance (see p. 1006). Although the use of intensive insulin therapy to achieve ‘tight glycaemic control’ (blood glucose level between 4.4 and 6.1 mmol/L) was shown to improve outcome (at least when combined with aggressive nutritional support), more recent studies have failed to confirm this finding and have indicated that this approach is associated with an unacceptably high incidence of hypoglycaemia, and possibly an increase in mortality. Current recommendations suggest that blood glucose levels should be maintained at <8–10 mmol/L.
Critically ill patients commonly require intravenous insulin infusions, often in high doses, to combat hyperglycaemia and insulin resistance (see p. 1006). Although the use of intensive insulin therapy to achieve ‘tight glycaemic control’ (blood glucose level between 4.4 and 6.1 mmol/L) was shown to improve outcome (at least when combined with aggressive nutritional support), more recent studies have failed to confirm this finding and have indicated that this approach is associated with an unacceptably high incidence of hypoglycaemia, and possibly an increase in mortality. Current recommendations suggest that blood glucose levels should be maintained at <8–10 mmol/L.
FURTHER READING
Casaer MP, Mesotten O, Hermans G et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med 2011; 365:506–517.
The NICE-SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1293–1297.
Ziegler TR. Parenteral nutrition in the critically ill patient. N Engl J Med 2009; 361:1088–1097.
Applied cardiorespiratory physiology
Oxygen delivery and consumption
Oxygen delivery (DO2) (Fig. 16.1) is defined as the total amount of oxygen delivered to the tissues per unit time. It is dependent on the volume of blood flowing through the microcirculation per minute (i.e. the total cardiac output, 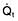 ) and the amount of oxygen contained in that blood (i.e. the arterial oxygen content, CaO2). Oxygen is transported both in combination with haemoglobin and dissolved in plasma. The amount combined with haemoglobin is determined by the oxygen capacity of haemoglobin (usually taken as 1.34 mL of oxygen per gram of haemoglobin) and its percentage saturation with oxygen (SO2), while the volume dissolved in plasma depends on the partial pressure of oxygen (PO2). Except when hyperbaric oxygen is administered, the amount of dissolved oxygen in plasma is insignificant.
) and the amount of oxygen contained in that blood (i.e. the arterial oxygen content, CaO2). Oxygen is transported both in combination with haemoglobin and dissolved in plasma. The amount combined with haemoglobin is determined by the oxygen capacity of haemoglobin (usually taken as 1.34 mL of oxygen per gram of haemoglobin) and its percentage saturation with oxygen (SO2), while the volume dissolved in plasma depends on the partial pressure of oxygen (PO2). Except when hyperbaric oxygen is administered, the amount of dissolved oxygen in plasma is insignificant.
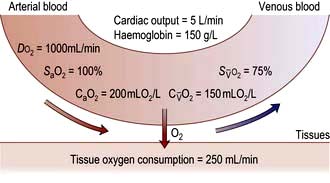
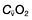 , mixed venous oxygen content;
, mixed venous oxygen content; 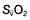 , mixed venous oxygen saturation; CaO2, arterial oxygen content.
, mixed venous oxygen saturation; CaO2, arterial oxygen content.Oxygenation of the blood
Oxyhaemoglobin dissociation curve
The saturation of haemoglobin with oxygen is determined by the partial pressure of oxygen (PO2) in the blood, the relationship between the two being described by the oxyhaemoglobin dissociation curve (Fig. 16.2). The sigmoid shape of this curve is significant for a number of reasons:
 Modest falls in the partial pressure of oxygen in the arterial blood (PaO2) may be tolerated (since oxygen content is relatively unaffected) provided that the percentage saturation remains above 92%.
Modest falls in the partial pressure of oxygen in the arterial blood (PaO2) may be tolerated (since oxygen content is relatively unaffected) provided that the percentage saturation remains above 92%.


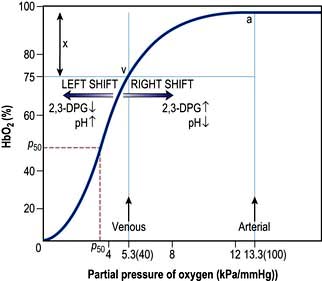
Figure 16.2 The oxyhaemoglobin dissociation curve. a, arterial point; v, venous point; x, arteriovenous oxygen content difference. HbO2 (%) is the percentage saturation of haemoglobin with oxygen. The curve will move to the right in the presence of acidosis (metabolic or respiratory), pyrexia or an increased red cell 2,3-DPG concentration. For a given arteriovenous oxygen content difference, the mixed venous PO2 will then be higher. Furthermore, if the mixed venous PO2 is unchanged, the arteriovenous oxygen content difference increases and more oxygen is off-loaded to the tissues (see p. 374). P50 (the PO2 at which haemoglobin is half saturated with O2) is a useful index of these shifts – the higher the P50 (i.e. shift to the right), the lower the affinity of haemoglobin for O2.
 .
.Alveolar oxygen tension (PAO2)
The partial pressures of inspired gases are shown in Figure 16.3. By the time the inspired gases reach the alveoli they are fully saturated with water vapour at body temperature (37°C), which has a partial pressure of 6.3 kPa (47 mmHg) and contain CO2 at a partial pressure of approximately 5.3 kPa (40 mmHg); the PAO2 is thereby reduced to approximately 13.4 kPa (100 mmHg).
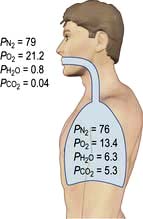
The clinician can influence PAO2 by administering oxygen or by increasing the barometric pressure.
Pulmonary gas exchange
In normal subjects there is a small alveolar-arterial oxygen difference (PA–aO2). This is due to:



Pathologically, there are three possible causes of an increased PA–aO2 difference:
 Diffusion defect. This is not a major cause of hypoxaemia even in conditions such as lung fibrosis, in which the alveolar-capillary membrane is considerably thickened. Carbon dioxide is also not affected, as it is more soluble than oxygen.
Diffusion defect. This is not a major cause of hypoxaemia even in conditions such as lung fibrosis, in which the alveolar-capillary membrane is considerably thickened. Carbon dioxide is also not affected, as it is more soluble than oxygen.
 Right-to-left shunts. In certain congenital cardiac lesions or when a segment of lung is completely collapsed, a proportion of venous blood passes to the left side of the heart without taking part in gas exchange, causing arterial hypoxaemia. This hypoxaemia cannot be corrected by administering oxygen to increase the PAO2, because blood leaving normal alveoli is already fully saturated; further increases in PO2 will not, therefore, significantly affect its oxygen content. On the other hand, because of the shape of the carbon dioxide dissociation curve (Fig. 16.4), the high PCO2 of the shunted blood can be compensated for by over-ventilating patent alveoli, thus lowering the CO2 content of the effluent blood. Indeed, many patients with acute right-to-left shunts hyperventilate in response to the hypoxia and/or to stimulation of mechanoreceptors in the lung, so that their PaCO2 is normal or low.
Right-to-left shunts. In certain congenital cardiac lesions or when a segment of lung is completely collapsed, a proportion of venous blood passes to the left side of the heart without taking part in gas exchange, causing arterial hypoxaemia. This hypoxaemia cannot be corrected by administering oxygen to increase the PAO2, because blood leaving normal alveoli is already fully saturated; further increases in PO2 will not, therefore, significantly affect its oxygen content. On the other hand, because of the shape of the carbon dioxide dissociation curve (Fig. 16.4), the high PCO2 of the shunted blood can be compensated for by over-ventilating patent alveoli, thus lowering the CO2 content of the effluent blood. Indeed, many patients with acute right-to-left shunts hyperventilate in response to the hypoxia and/or to stimulation of mechanoreceptors in the lung, so that their PaCO2 is normal or low.
 Ventilation/perfusion mismatch (see p. 796). Diseases of the lung parenchyma (e.g. pulmonary oedema, acute lung injury) result in
Ventilation/perfusion mismatch (see p. 796). Diseases of the lung parenchyma (e.g. pulmonary oedema, acute lung injury) result in 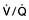 mismatch, producing an increase in alveolar deadspace and hypoxaemia. The increased deadspace can be compensated for by increasing overall ventilation. In contrast to the hypoxia resulting from a true right-to-left shunt, that due to areas of low
mismatch, producing an increase in alveolar deadspace and hypoxaemia. The increased deadspace can be compensated for by increasing overall ventilation. In contrast to the hypoxia resulting from a true right-to-left shunt, that due to areas of low 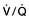 can be partially corrected by administering oxygen and thereby increasing the PAO2 even in poorly ventilated areas of lung.
can be partially corrected by administering oxygen and thereby increasing the PAO2 even in poorly ventilated areas of lung.
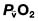 and saturation (
and saturation (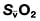 )
)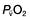 is the partial pressure of oxygen in pulmonary arterial blood that has been thoroughly mixed during its passage through the right heart. Assuming PaO2 remains constant,
is the partial pressure of oxygen in pulmonary arterial blood that has been thoroughly mixed during its passage through the right heart. Assuming PaO2 remains constant, 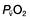 and
and 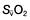 will fall if more oxygen has to be extracted from each unit volume of blood arriving at the tissues. A low
will fall if more oxygen has to be extracted from each unit volume of blood arriving at the tissues. A low 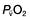 therefore indicates either that oxygen delivery has fallen or that tissue oxygen requirements have increased without a compensatory rise in cardiac output. If
therefore indicates either that oxygen delivery has fallen or that tissue oxygen requirements have increased without a compensatory rise in cardiac output. If 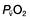 falls, the effect of a given degree of pulmonary shunting on arterial oxygenation will be exacerbated. Thus, worsening arterial hypoxaemia does not necessarily indicate a deterioration in pulmonary function but might instead reflect a fall in cardiac output and/or a rise in oxygen consumption.
falls, the effect of a given degree of pulmonary shunting on arterial oxygenation will be exacerbated. Thus, worsening arterial hypoxaemia does not necessarily indicate a deterioration in pulmonary function but might instead reflect a fall in cardiac output and/or a rise in oxygen consumption.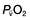 and
and 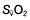 may reflect impaired tissue oxygen extraction (due to microcirculatory dysfunction) and/or reduced oxygen utilization (e.g. due to mitochondrial dysfunction) as seen in severe sepsis (see below).
may reflect impaired tissue oxygen extraction (due to microcirculatory dysfunction) and/or reduced oxygen utilization (e.g. due to mitochondrial dysfunction) as seen in severe sepsis (see below).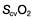 ), rather than pulmonary artery blood is less invasive and has been shown to be a valuable guide to the resuscitation of critically ill patients (see
), rather than pulmonary artery blood is less invasive and has been shown to be a valuable guide to the resuscitation of critically ill patients (see Cardiac output
Cardiac output is the product of heart rate and stroke volume, and is affected by changes in either of these (see Fig. 16.5).
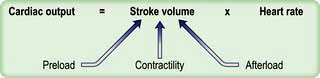
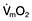 ) and this may precipitate ischaemia in areas of the myocardium with restricted coronary perfusion. When the heart rate falls, a point is reached at which the increase in stroke volume is insufficient to compensate for bradycardia and again cardiac output falls.
) and this may precipitate ischaemia in areas of the myocardium with restricted coronary perfusion. When the heart rate falls, a point is reached at which the increase in stroke volume is insufficient to compensate for bradycardia and again cardiac output falls.Stroke volume
Three interdependent factors determine the stroke volume (see p. 671).
Preload
This is defined as the tension of the myocardial fibres at the end of diastole, just before the onset of ventricular contraction, and is therefore related to the degree of stretch of the fibres. As the end-diastolic volume of the ventricle increases, tension in the myocardial fibres is increased and stroke volume rises (Fig. 16.6). Myocardial oxygen consumption (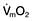 ) increases only slightly with an increase in preload (produced, for example, by a ‘fluid challenge’, see below) and this is therefore the most efficient way of improving cardiac output.
) increases only slightly with an increase in preload (produced, for example, by a ‘fluid challenge’, see below) and this is therefore the most efficient way of improving cardiac output.
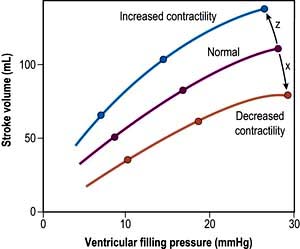
Myocardial contractility
This refers to the ability of the heart to perform work, independent of changes in preload and afterload. The state of myocardial contractility determines the response of the ventricles to changes in preload and afterload. Contractility is often reduced in critically ill patients, as a result of either pre-existing myocardial damage (e.g. ischaemic heart disease), or the acute disease process itself (e.g. sepsis). Changes in myocardial contractility alter the slope and position of the Starling curve; worsening ventricular performance is manifested as a depressed, flattened curve (Fig. 16.6 and Fig. 14.5). Inotropic drugs can be used to increase myocardial contractility (see below).
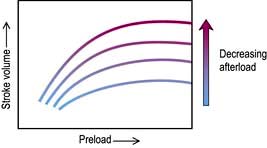
Afterload
Decreasing the afterload (exercise, sepsis, vasodilator agents) can increase the stroke volume achieved at a given preload (Fig. 16.7), while reducing 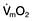 . The reduction in wall tension also leads to an increase in coronary blood flow, thereby improving the myocardial oxygen supply/demand ratio. Excessive reductions in afterload will cause hypotension.
. The reduction in wall tension also leads to an increase in coronary blood flow, thereby improving the myocardial oxygen supply/demand ratio. Excessive reductions in afterload will cause hypotension.
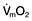 . Right ventricular afterload is normally negligible because the resistance of the pulmonary circulation is very low but is increased in pulmonary hypertension.
. Right ventricular afterload is normally negligible because the resistance of the pulmonary circulation is very low but is increased in pulmonary hypertension.Monitoring critically ill patients
Assessment of tissue perfusion
 Pale, cold skin, delayed capillary refill and the absence of visible veins in the hands and feet indicate poor perfusion. Although peripheral skin temperature measurements can help clinical evaluation, the earliest compensatory response to hypovolaemia or a low cardiac output, and the last to resolve after resuscitation is vasoconstriction in the splanchnic region.
Pale, cold skin, delayed capillary refill and the absence of visible veins in the hands and feet indicate poor perfusion. Although peripheral skin temperature measurements can help clinical evaluation, the earliest compensatory response to hypovolaemia or a low cardiac output, and the last to resolve after resuscitation is vasoconstriction in the splanchnic region.

 Urinary flow is a sensitive indicator of renal perfusion and haemodynamic performance.
Urinary flow is a sensitive indicator of renal perfusion and haemodynamic performance.
Blood pressure
Hypotension jeopardizes perfusion of vital organs. The adequacy of blood pressure in an individual patient must always be assessed in relation to the premorbid value. Blood pressure is traditionally measured using a sphygmomanometer but if rapid alterations are anticipated, continuous monitoring using an intra-arterial cannula is indicated (Practical Box 16.1; Fig. 16.8).
 Practical Box 16.1
Practical Box 16.1
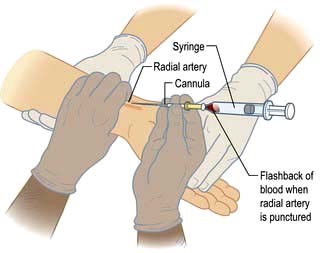
Radial artery cannulation
Technique
1. The procedure is explained to the patient and, if possible, consent obtained.
2. The arm is supported, with the wrist extended, by an assistant. (Gloves should be worn.)
3. The skin should be cleaned with chlorhexidine.
4. The radial artery is palpated where it arches over the head of the radius.
5. In conscious patients, local anaesthetic is injected to raise a weal over the artery, taking care not to puncture the vessel or obscure its pulsation.
6. A small skin incision is made over the proposed puncture site.
7. A small parallel-sided cannula (20 gauge for adults, 22 gauge for children) is used in order to allow blood flow to continue past the cannula.
8. The cannula is inserted over the point of maximal pulsation and advanced in line with the direction of the vessel at an angle of approximately 30°.
9. ‘Flashback’ of blood into the cannula indicates that the radial artery has been punctured.
10. To ensure that the shoulder of the cannula enters the vessel, the needle and cannula are lowered and advanced a few millimetres into the vessel.
11. The cannula is threaded off the needle into the vessel and the needle withdrawn.
12. The cannula is connected to a non-compliant manometer line filled with saline. This is then connected via a transducer and continuous flush device to a monitor, which records the arterial pressure.






Central venous pressure (CVP)
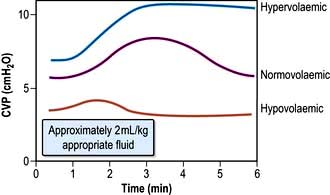
This provides a fairly simple, but approximate method of gauging the adequacy of a patient’s circulating volume and the contractile state of the myocardium. The absolute value of the CVP is not as useful as its response to a fluid challenge (the infusion of 100–200 mL of fluid over a few minutes) (Fig. 16.9). The hypovolaemic patient will initially respond to transfusion with little or no change in CVP, together with some improvement in cardiovascular function (falling heart rate, rising blood pressure, increased peripheral temperature and urine output). As the normovolaemic state is approached, the CVP usually rises slightly and reaches a plateau, while other cardiovascular values begin to stabilize. At this stage, volume replacement should be slowed, or even stopped, in order to avoid overtransfusion (indicated by an abrupt and sustained rise in CVP, often accompanied by some deterioration in the patient’s condition). In cardiac failure, the venous pressure is usually high; the patient will not improve in response to volume replacement, which will cause a further, sometimes dramatic, rise in CVP.
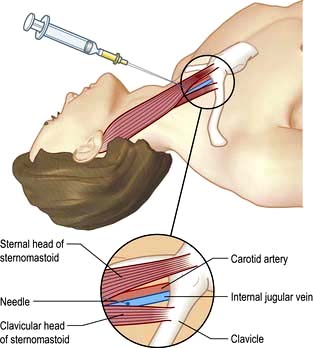
Central venous catheters are usually inserted via a percutaneous puncture of the subclavian or internal jugular vein using a guidewire technique (Practical Box 16.2; Figs 16.10, 16.11). The guidewire techniques can also be used in conjunction with a vein dilator for inserting multilumen catheters, double lumen cannulae for haemofiltration or pulmonary artery catheter introducers. The routine use of ultrasound to guide central venous cannulation reduces complication rates.
 Practical Box 16.2
Practical Box 16.2
Internal jugular vein cannulation
Technique
1. The procedure is explained to the patient and, if possible, consent obtained.
2. The patient is placed head-down to distend the central veins (this facilitates cannulation and minimizes the risk of air embolism but may exacerbate respiratory distress and is dangerous in those with raised intracranial pressure).
3. The skin is cleaned with an antiseptic solution such as chlorhexidine. Sterile precautions are taken throughout the procedure.
4. Local anaesthetic (1% plain lidocaine) is injected intradermally to raise a weal at the apex of a triangle formed by the two heads of sternomastoid with the clavicle at its base.
5. A small incision is made through the weal.
6. The cannula or needle is inserted through the incision and directed laterally downwards and backwards in the direction of the nipple until the vein is punctured just beneath the skin and deep to the lateral head of sternomastoid.
Ultrasound-guided puncture is recommended to reduce the incidence of complications.
7. Check that venous blood is easily aspirated.
8. The cannula is threaded off the needle into the vein or the guidewire is passed through the needle (see Fig. 16.11).
9. The CVP manometer line is connected to a manometer/transducer.
10. A chest X-ray should be taken to verify that the tip of the catheter is in the superior vena cava and to exclude pneumothorax.







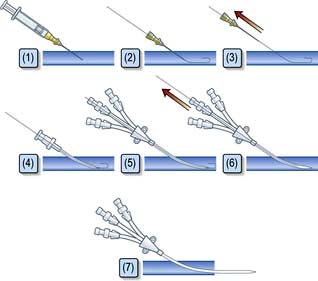
The following are common pitfalls in interpreting central venous pressure readings:
 Blocked catheter. This results in a sustained high reading, with a damped or absent waveform, which often does not correlate with clinical assessment.
Blocked catheter. This results in a sustained high reading, with a damped or absent waveform, which often does not correlate with clinical assessment.
 Transducer wrongly positioned. Failure to level the system is a common cause of erroneous readings.
Transducer wrongly positioned. Failure to level the system is a common cause of erroneous readings.

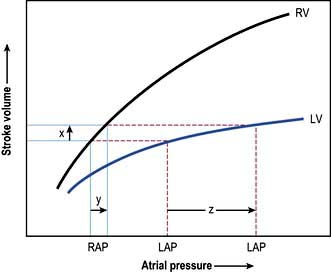
Left atrial pressure
In uncomplicated cases, careful interpretation of the CVP provides a reasonable guide to the filling pressures of both sides of the heart. In many critically ill patients, however, there is a disparity in function between the two ventricles. Most commonly, left ventricular performance is worse, so that the left ventricular function curve is displaced downward and to the right (Fig. 16.12). High right ventricular filling pressures, with normal or low left atrial pressures, are less common but occur with right ventricular dysfunction and in cases where the pulmonary vascular resistance (i.e. right ventricular afterload) is raised, such as in acute respiratory failure and pulmonary embolism.
Pulmonary artery pressure
A ‘balloon flotation catheter’ enables reliable catheterization of the pulmonary artery. These ‘Swan–Ganz’ catheters can be inserted centrally (Fig. 16.10) or through the femoral vein, or via a vein in the antecubital fossa. Passage of the catheter from the major veins, through the chambers of the heart, into the pulmonary artery and into the wedged position is monitored and guided by the pressure waveforms recorded from the distal lumen (Practical Box 16.3; Fig. 16.13). A chest X-ray should always be obtained to check the final position of the catheter. In difficult cases, screening with an image intensifier may be required.
 Practical Box 16.3
Practical Box 16.3
Passage of a pulmonary artery balloon flotation catheter through the chambers of the heart into the ‘wedged’ position
Consent should be obtained if possible from the patient after explanation of the procedure.
Note: (a), (b), (c), (d) refer to Figure 16.13.
1. A balloon flotation catheter is inserted through a large vein (see text).
2. Once in the thorax, respiratory oscillations are seen. The catheter should be advanced further towards the lower superior vena cava/right atrium (a), where pressure oscillations become more pronounced. The balloon should then be inflated and the catheter advanced.
3. When the catheter is in the right ventricle (b), there is no dicrotic notch and the diastolic pressure is close to zero. The patient should be returned to the horizontal, or slightly head-up, position before advancing the catheter further.
4. When the catheter reaches the pulmonary artery (c) a dicrotic notch appears and there is elevation of the diastolic pressure. The catheter should be advanced further with the balloon inflated.
5. Reappearance of a venous waveform indicates that the catheter is ‘wedged’. The balloon is deflated to obtain the pulmonary artery pressure. The balloon is inflated intermittently to obtain the pulmonary artery occlusion (also known as pulmonary artery, or capillary, ‘wedge’) pressure (d).
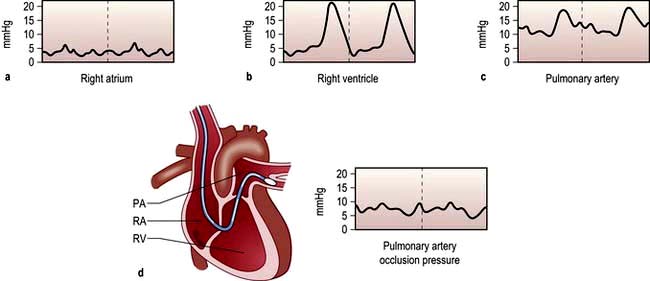
Figure 16.13 Passage of pulmonary artery balloon flotation catheter through the chambers of the heart into the ‘wedged’ position to measure the pulmonary artery occlusion pressure. (See Practical Box 16.3.)
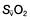 .
.Less invasive techniques for assessing cardiac function and guiding volume replacement
Arterial pressure variation as a guide to hypovolaemia
Systolic arterial pressure decreases during the inspiratory phase of intermittent positive pressure ventilation (p. 894). The magnitude of this cyclical variability has been shown to correlate more closely with hypovolaemia than other monitored variables, including CVP. Systolic pressure (or pulse pressure) variation during mechanical ventilation can therefore be used as a simple and reliable guide to the adequacy of the circulatory volume. The response to fluid loading can also easily be predicted by observing the changes in pulse pressure during passive leg raising.
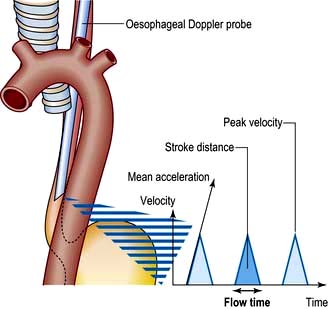
Oesophageal Doppler
Stroke volume, cardiac output and myocardial function can be assessed non-invasively using Doppler ultrasonography. A probe is passed into the oesophagus to continuously monitor velocity waveforms from the descending aorta (Fig. 16.14). Although reasonable estimates of stroke volume, and hence cardiac output can be obtained, the technique is best used for trend analysis rather than for making absolute measurements. Oesophageal Doppler probes can be inserted quickly and easily and are particularly valuable for perioperative optimization of the circulating volume and cardiac performance in the unconscious patient. They are contraindicated in patients with oropharyngeal/oesophageal pathology.
Echocardiography
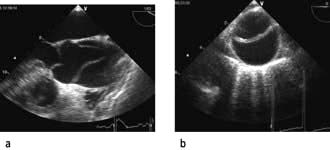
Echocardiography is being used increasingly often to provide immediate diagnostic information about cardiac structure and function (myocardial contractility, ventricular filling) in the critically ill patient. Transoesophageal echocardiography (TOE) is preferred because of its superior image clarity (Fig. 16.15).
Disturbances of acid–base balance
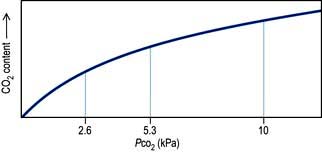
The physiology of acid–base control is discussed on page 660. Acid–base disturbances can be described in relation to the diagram illustrated in Figure 13.13, p. 663 (which shows PaCO2 plotted against arterial [H+]).
Blood gas and acid–base values (normal ranges) are shown in Table 16.2. (For blood gas analysis, see p. 891.)
Table 16.2 Arterial blood gas and acid–base values (normal ranges)
|
H+ |
35–45 nmol/L |
pH 7.35–7.45 |
|
PO2 (breathing room air) |
10.6–13.3 kPa |
(80–100 mmHg) |
|
PCO2 |
4.8–6.1 kPa |
(36–46 mmHg) |
|
Base deficit |
±2.5 |
|
|
Plasma HCO3– |
22–26 mmol/L |
|
|
O2 saturation |
95–100% |
|
Respiratory acidosis. This is caused by retention of carbon dioxide. The PaCO2 and [H+] rise. A chronically raised PaCO2 is compensated by renal retention of bicarbonate, and the [H+] returns towards normal. A constant arterial bicarbonate concentration is then usually established within 2–5 days. This represents a primary respiratory acidosis with a compensatory metabolic alkalosis (see p. 666). Common causes of respiratory acidosis include ventilatory failure and COPD (type II respiratory failure where there is a high PaCO2 and a low PaO2, see p. 814).
Respiratory alkalosis. In this case, the reverse occurs and there is a fall in PaCO2 and [H+], often with a small reduction in bicarbonate concentration. If hypocarbia persists, some degree of renal compensation may occur, producing a metabolic acidosis, although in practice this is unusual. A respiratory alkalosis may be produced, intentionally or unintentionally, when patients are mechanically ventilated; it may also be seen with hypoxaemic (type I) respiratory failure (see Ch. 15, p. 817), spontaneous hyperventilation and in those living at high altitudes.
Metabolic acidosis (p. 664). This may be due to excessive acid production, most commonly lactate and H+ (lactic acidosis) as a consequence of anaerobic metabolism during an episode of shock or following cardiac arrest. A metabolic acidosis may also develop in chronic renal failure or in diabetic ketoacidosis. It can also follow the loss of bicarbonate from the gut or from the kidney in renal tubular acidosis. Respiratory compensation for a metabolic acidosis is usually slightly delayed because the blood–brain barrier initially prevents the respiratory centre from sensing the increased blood [H+]. Following this short delay, however, the patient hyperventilates and ‘blows off’ carbon dioxide to produce a compensatory respiratory alkalosis. There is a limit to this respiratory compensation, since in practice values for PaCO2 less than about 1.4 kPa (11 mmHg) are rarely achieved. Spontaneous respiratory compensation cannot occur if the patient’s ventilation is controlled or if the respiratory centre is depressed, for example by drugs or head injury.
Shock, sepsis and acute disturbances of haemodynamic function
Causes of shock
Abnormalities of tissue perfusion can result from:




The causes of shock are shown in Table 16.3. Often shock can result from a combination of these factors (e.g. in sepsis, distributive shock is frequently complicated by hypovolaemia and myocardial depression).
| Hypovolaemic | Obstructive |
|---|---|
Pathophysiology
The sympatho-adrenal response to shock (Fig. 16.16)
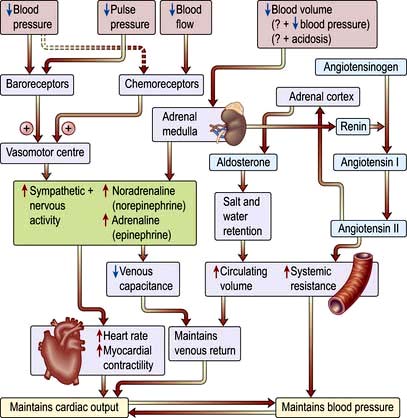
Reduction in perfusion of the renal cortex stimulates the juxtaglomerular apparatus to release renin. This converts angiotensinogen to angiotensin I, which in turn is converted in the lungs and by the vascular endothelium to the potent vasoconstrictor angiotensin II. Angiotensin II also stimulates secretion of aldosterone by the adrenal cortex, causing sodium and water retention (p. 566). This helps to restore the circulating volume (see p. 639).
Neuroendocrine response
 There is release of pituitary hormones such as adrenocorticotrophic hormone (ACTH), vasopressin (antidiuretic hormone, ADH) and endogenous opioid peptides. (In septic shock there may be a relative deficiency of vasopressin.)
There is release of pituitary hormones such as adrenocorticotrophic hormone (ACTH), vasopressin (antidiuretic hormone, ADH) and endogenous opioid peptides. (In septic shock there may be a relative deficiency of vasopressin.)
 There is release of cortisol, which causes fluid retention and antagonizes insulin.
There is release of cortisol, which causes fluid retention and antagonizes insulin.
 There is release of glucagon, which raises the blood sugar level.
There is release of glucagon, which raises the blood sugar level.
Release of pro- and anti-inflammatory mediators
Severe infection (often with bacteraemia or endotoxaemia), the presence of large areas of damaged tissue (e.g. following trauma or extensive surgery), hypoxia or prolonged/repeated episodes of hypoperfusion can trigger an exaggerated inflammatory response with systemic activation of leucocytes and release of a variety of potentially damaging ‘mediators’ (see also Ch. 3). Although beneficial when targeted against local areas of infection or necrotic tissue, dissemination of this ‘innate immune’ response can produce shock and widespread tissue damage. Characteristically the initial episode of overwhelming inflammation is followed by a period of immune suppression, which in some cases may be profound and during which the patient is at increased risk of developing secondary infections. It also seems that pro- and anti-inflammatory elements of the host response may co-exist.
Microorganisms and their toxic products (Fig. 16.17)
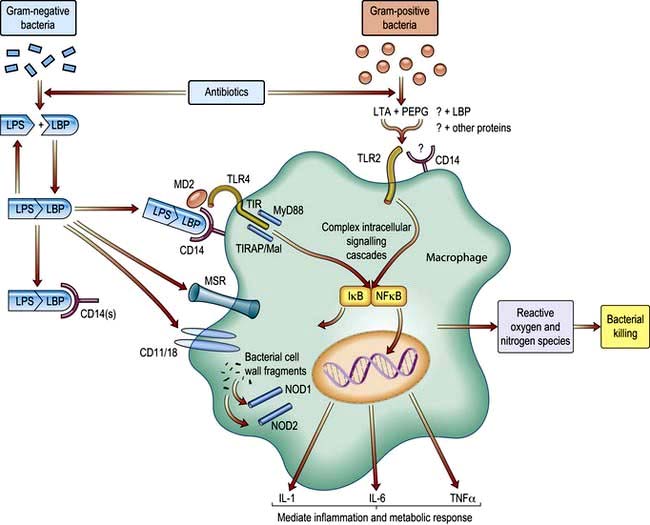
Endotoxin is a lipopolysaccharide (LPS) derived from the cell wall of Gram-negative bacteria and is a potent trigger of the inflammatory response. The lipid A portion of LPS can be bound by a protein normally present in human serum known as lipopolysaccharide binding protein (LBP). The LBP/LPS complex attaches to the cell surface marker CD14 and, combined with a secreted protein (MD2), this complex then binds to a member of the toll-like receptor family (TLR4), which transduces the activation signal into the cell. These receptors act through a critical adaptor molecule, myeloid differentiation factor 88 (MyD 88), to regulate the activity of NFκB pathways. Intracellular pattern recognition receptors such as nucleotide-binding oligomerization domain (NOD) 1 may also be involved. Another mechanism in this complex area involves TREM-I (triggering receptor expressed in myeloid cells, see p. 54), which triggers secretion of pro-inflammatory cytokines.
Specific kinases then phosphorylate inhibitory kappa B (IκB), releasing the nuclear transcription factor NFκB, which passes into the nucleus where it binds to DNA and promotes the synthesis of a wide variety of inflammatory mediators. Gram-positive bacteria have cell wall components which are similar in structure to LPS (e.g. lipoteichoic acid), and can also trigger a systemic inflammatory response, probably through similar (TLR2) but not identical pathways (Fig. 16.17). Following traumatic or surgical tissue injury, inflammatory pathways may be triggered by damage-associated molecular patterns (DAMPS) such as DNA fragments.
Activation of complement cascade
Fragments of C3 act as opsonins and co-stimulatory molecules that assist lymphocytes with the adaptive immune response, while small peptides derived from C3, C4 and C5 cause leucocyte chemotaxis, release of cytokines and increased vascular permeability (see p. 51).
Cytokines
Pro-inflammatory cytokines (see also p. 49) such as the interleukins (ILs) and tumour necrosis factor (TNF) are also mediators of the systemic inflammatory response. TNF release initiates many of the responses to endotoxin, for example, and acts synergistically with IL-1, in part through induction of cyclo-oxygenase, platelet-activating factor (PAF) and nitric oxide synthase (see below). Subsequently, other cytokines including IL-6 and IL-8 appear in the circulation. IL-6 is the major stimulant for hepatic synthesis of acute phase proteins and is involved in the induction of fever, anaemia and cachexia, while IL-8 is a chemoattractant. The cytokine network is extremely complex, with many endogenous self-regulating mechanisms. For example, naturally occurring soluble TNF receptors are shed from cell surfaces during the inflammatory response, binding to TNF and thereby reducing its biological activity. An endogenous inhibitory protein that binds competitively to the IL-1 receptor has also been identified.
Products of arachidonic acid metabolism
Arachidonic acid, derived from the breakdown of membrane phospholipid, is metabolized to form prostaglandins and leukotrienes, which are key inflammatory mediators (see Fig. 15.30) and p. 826).
Adhesion molecules
Adhesion of activated leucocytes to the vessel wall and their subsequent extravascular migration is a key component of the sequence of events leading to endothelial injury, tissue damage and organ dysfunction (see also p. 23). This process is mediated by inducible intercellular adhesion molecules (ICAMs) found on the surface of leucocytes and endothelial cells. Expression of these molecules can be induced by endotoxin and pro-inflammatory cytokines. Several families of molecules are involved in promoting leucocyte-endothelial interaction. The selectins are ‘capture’ molecules and initiate the process of leucocyte rolling on vascular endothelium, while members of the immunoglobulin superfamily (ICAM-1 and vascular cell adhesion molecule-1) are involved in the formation of a more secure bond which leads to leucocyte migration into the tissues (see Fig. 3.13).
Endothelium-derived vasoactive mediators
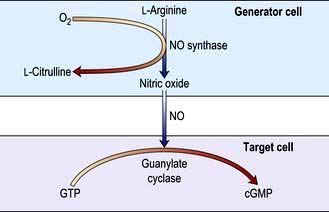
Endothelial cells synthesize a number of mediators which contribute to the regulation of blood vessel tone and the fluidity of the blood; these include nitric oxide, prostacyclin and endothelin (a potent vasoconstrictor). Nitric oxide (NO) is synthesized from the terminal guanidino-nitrogen atoms of the amino acid L-arginine under the influence of nitric oxide synthase (NOS). NO inhibits platelet aggregation and adhesion and produces vasodilatation by activating guanylate cyclase in the underlying vascular smooth muscle to form cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP) (Fig. 16.18). There are several distinct NOS enzymes.



Redox imbalance
 lipid and protein peroxidation
lipid and protein peroxidation

 increased capillary permeability
increased capillary permeability
 impaired mitochondrial respiration
impaired mitochondrial respiration

 apoptosis (see p. 32) (which may contribute to the organ damage and immune hyporesponsiveness associated with sepsis).
apoptosis (see p. 32) (which may contribute to the organ damage and immune hyporesponsiveness associated with sepsis).
Haemodynamic and microcirculatory changes
The dominant haemodynamic feature of severe sepsis/septic shock is peripheral vascular failure with:



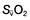 and a lactic acidosis (so-called ‘tissue dysoxia’). Vasodilatation and increased vascular permeability also occur in anaphylactic shock.
and a lactic acidosis (so-called ‘tissue dysoxia’). Vasodilatation and increased vascular permeability also occur in anaphylactic shock.Activation of the coagulation system
Initially the production of PGI2 by the capillary endothelium is impaired. Cell damage (e.g. to the vascular endothelium) leads to exposure to tissue factor (p. 416), which triggers coagulation. In severe cases these changes are compounded by elevated levels of plasminogen activation inhibitor type 1, which impairs fibrinolysis, as well as by deficiencies in physiological inhibitors of coagulation (including antithrombin, proteins C and S and tissue factor-pathway inhibitor). Antithrombin and protein C have a number of anti-inflammatory properties, whereas thrombin is pro-inflammatory.
Plasminogen is converted to plasmin, which breaks down thrombus, liberating fibrin/fibrinogen degradation products (FDPs). In some cases there is increased fibrinolysis. Circulating levels of FDPs are therefore increased, the thrombin time, PTT and PT are prolonged and platelet and fibrinogen levels fall. Activation of the coagulation cascade can be confirmed by demonstrating increased plasma levels of D-dimers. The development of disseminated intravascular coagulation (DIC) often heralds the onset of multiple organ failure. Because clotting factors and platelets are consumed in DIC, they are unavailable for haemostasis elsewhere and a coagulation defect results – hence the alternative name for DIC is ‘consumption coagulopathy’. DIC presents with microvascular bleeding or generalized ‘oozing’ of blood, e.g. from surgical or traumatic wounds and skin puncture sites. In some cases, a microangiopathic haemolytic anaemia develops. DIC is relatively uncommon but is particularly associated with septic shock, especially when due to meningococcal infection (see p. 127). Management of the underlying cause is most urgent. Supportive treatment may include infusions of fresh frozen plasma, platelets, cryoprecipitate when fibrinogen levels are low and occasionally factor VIII concentrates.
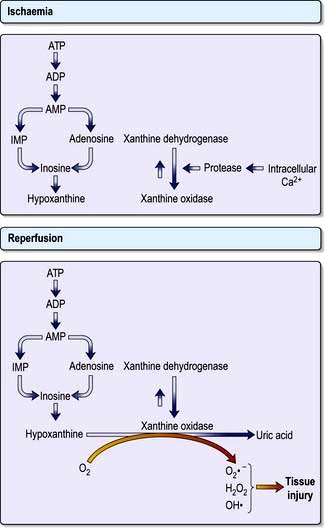
Reperfusion injury
Restoration of flow to previously hypoxic tissues can exacerbate cell damage through the generation of large quantities of reactive oxygen species and activation of polymorphonuclear leucocytes (see above) (Fig. 16.20). The gut mucosa seems to be especially vulnerable to this ‘ischaemia-reperfusion injury’.
Metabolic response to trauma, major surgery and severe infection
This is initiated and controlled by the neuroendocrine system and various cytokines (e.g. IL-6) acting in concert, and is characterized initially by an increase in energy expenditure (‘hypermetabolism’) (see also p. 201). Gluconeogenesis is stimulated by increased glucagon and catecholamine levels, while hepatic mobilization of glucose from glycogen is increased. Catecholamines inhibit insulin release and reduce peripheral glucose uptake. Combined with elevated circulating levels of other insulin antagonists such as cortisol, and downregulation of insulin receptors, these changes mean that the majority of patients are hyperglycaemic (‘insulin resistance’). Later hypoglycaemia may be precipitated by depletion of hepatic glycogen stores and inhibition of gluconeogenesis. Free fatty acid synthesis is also increased, leading to hypertriglyceridaemia.
Clinical features of shock and sepsis
Although many clinical features are common to all types of shock, there are certain aspects in which they differ (Box 16.2).
 Box 16.2
Box 16.2
Haemodynamic changes in shock
Hypovolaemic shock



Extreme hypovolaemia may be associated with bradycardia.
Additional clinical features may occur in the following types of shock.



Anaphylactic shock (see p. 69)







Sepsis, severe sepsis and septic shock
 Pyrexia and rigors, or hypothermia (unusual)
Pyrexia and rigors, or hypothermia (unusual)

 Vasodilatation, warm peripheries
Vasodilatation, warm peripheries





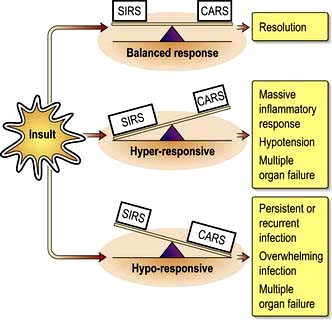
The clinical signs of sepsis (triggered by PAMPS) are not always associated with bacteraemia and can occur with non-infectious processes such as pancreatitis, cardiopulmonary bypass or severe trauma (triggered by DAMPS). The term ‘systemic inflammatory response syndrome’ (SIRS) describes the disseminated inflammation that can complicate this diverse range of disorders (Box 16.3). Patterns of systemic inflammatory response are shown in Figure 16.21, which illustrates the pro-inflammatory response (SIRS) and the counter-regulatory anti-inflammatory response syndrome (CARS).
 Box 16.3
Box 16.3



Sepsis and multiple organ failure (MOF) (also known as multiple organ dysfunction syndrome, MODS)
Sepsis is being diagnosed with increasing frequency and is now the commonest cause of death in non-coronary adult intensive care units. The estimated incidence of severe sepsis has varied from 77 to 300 cases per 100 000 of the population. Mortality rates are high (between 20% and 60%) and are closely related to the severity of illness and the number of organs that fail. Those who die are overwhelmed by persistent or recurrent sepsis, with fever, intractable hypotension and failure of several organs (Fig. 16.22).
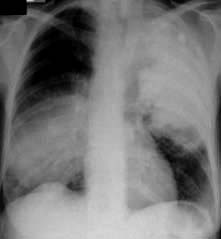
Sequential failure of vital organs occurs progressively over weeks, although the pattern of organ dysfunction is variable. In most cases the lungs are the first to be affected (acute lung injury, ALI; acute respiratory distress syndrome, ARDS; see below) in association with cardiovascular instability and deteriorating renal function. Damage to the mucosal lining of the gastrointestinal tract, as a result of reduced splanchnic flow followed by reperfusion, allows bacteria within the gut lumen, or their cell wall components, to gain access to the circulation. The liver defences, which are often compromised by poor perfusion, are overwhelmed and the lungs and other organs are exposed to bacterial toxins and inflammatory mediators released by liver macrophages. Some have therefore called the gut the ‘motor of multiple organ failure’. Secondary pulmonary infection, complicating ALI/ARDS, also frequently acts as a further stimulus to the inflammatory response. Later, kidney injury and liver dysfunction develop (see p. 884). Gastrointestinal failure, with an inability to tolerate enteral feeding and paralytic ileus, is common. Ischaemic colitis, acalculous cholecystitis, pancreatitis and gastrointestinal haemorrhage may also occur. Features of central nervous system dysfunction include impaired consciousness and disorientation, progressing to coma. Characteristically, these patients initially have a hyperdynamic circulation with vasodilatation and a high cardiac output, associated with an increased metabolic rate. Eventually, however, cardiovascular collapse supervenes. It is now often possible to support such patients for weeks or months; many now die following a decision to withdraw or not to escalate treatment (see p. 897).
Acute lung injury/acute respiratory distress syndrome
Definition and causes (Table 16.4)

 Stiff lungs (reduced pulmonary compliance resulting in high inflation pressures)
Stiff lungs (reduced pulmonary compliance resulting in high inflation pressures)
 Chest radiograph: new bilateral, diffuse, patchy or homogeneous pulmonary infiltrates
Chest radiograph: new bilateral, diffuse, patchy or homogeneous pulmonary infiltrates


Table 16.4 Disorders associated with acute respiratory distress syndrome
| Direct lung injury | Indirect lung injury |
|---|---|
|
Common causes |
|
Less common causes
ALI/ARDS can occur as a nonspecific reaction of the lungs to a wide variety of direct pulmonary and indirect non-pulmonary insults. By far the commonest predisposing factor is sepsis, and 20–40% of patients with severe sepsis will develop ALI/ARDS (Table 16.4).
Pathogenesis and pathophysiology of ALI/ARDS
Acute lung injury can be viewed as an early manifestation of a generalized inflammatory response with endothelial dysfunction and is therefore frequently associated with the development of multiple organ dysfunction syndrome (MODS) (see p. 882).
Non-cardiogenic pulmonary oedema
This is the cardinal feature of ALI and is the first and clinically most evident sign of a generalized increase in vascular permeability caused by the microcirculatory changes and release of inflammatory mediators described previously (see p. 877), with activated neutrophils playing a particularly key role. The pulmonary epithelium is also damaged in the early stages, reducing surfactant production and lowering the threshold for alveolar flooding.
Pulmonary hypertension
Pulmonary hypertension sometimes complicated by right ventricular failure (p. 762) is a common feature of ALI/ARDS. Initially, mechanical obstruction of the pulmonary circulation may occur as a result of vascular compression by interstitial oedema, while local activation of the coagulation cascade leads to thrombosis and obstruction in the pulmonary microvasculature. Later, pulmonary vasoconstriction may develop in response to increased autonomic nervous activity and circulating substances such as catecholamines, serotonin, thromboxane and complement. Those vessels supplying alveoli with low oxygen tensions constrict (the ‘hypoxic vasoconstrictor response’), diverting pulmonary blood flow to better oxygenated areas of lung, thus limiting the degree of shunt.
Physiological changes
Shunt and deadspace increase, compliance falls, and there is evidence of airflow limitation. Although the lungs in ALI and ARDS are diffusely injured, the pulmonary lesions, when identified as densities on a CT scan, are predominantly located in dependent regions (Fig. 16.23). This is partly explained by the effects of gravity on the distribution of extravascular lung water and areas of lung collapse. Pleural effusions are common.
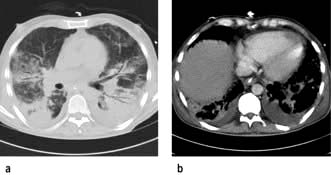
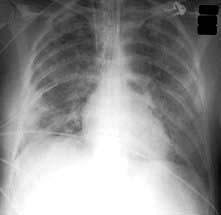
Clinical presentation of ali/ards
The first sign of the development of ALI/ARDS is often an unexplained tachypnoea, followed by increasing hypoxaemia, with central cyanosis, and breathlessness. Fine crackles are heard throughout both lung fields. Later, the chest X-ray shows bilateral diffuse shadowing, interstitial at first, but subsequently with an alveolar pattern and air bronchograms (Fig. 16.24). The differential diagnosis includes cardiac failure and lung fibrosis.
Management of ali/ards
 matching by increasing perfusion of ventilated lung units, as well as reducing pulmonary hypertension. It has been shown to improve oxygenation in so-called ‘responders’ with ALI/ARDS but has not been shown to increase survival. Its administration requires specialized monitoring equipment, as products of its combination with oxygen include toxic nitrogen dioxide.
matching by increasing perfusion of ventilated lung units, as well as reducing pulmonary hypertension. It has been shown to improve oxygenation in so-called ‘responders’ with ALI/ARDS but has not been shown to increase survival. Its administration requires specialized monitoring equipment, as products of its combination with oxygen include toxic nitrogen dioxide.Prognosis
FURTHER READING
Hall JP, Kress JP. The burden of functional recovery from ARDS. N Engl J Med 2011; 364:1360–1366.
Herridge MS et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
The RENAL Replacement Therapy Study Investigators. Intensity of continuous renal-replacement therapy in critically ill patients. N Engl J Med 2009; 361:1627–1638.
Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 2007; 369:1556–1565.
Acute kidney injury
Acute kidney injury (AKI) is a common and serious complication of critical illness which adversely affects the prognosis. A modification of the RIFLE classification has been proposed and covers the spectrum of severity and consequences of acute kidney injury (see Ch. 12; Box 12.5).
The importance of preventing renal injury by rapid and effective resuscitation, as well as the avoidance of nephrotoxic drugs (especially NSAIDs), and control of infection cannot be overemphasized. Shock and sepsis are the most common causes of AKI in the critically ill, but diagnosis of the cause of renal dysfunction is necessary to exclude reversible pathology, especially obstruction (see Ch. 12).
Oliguria is usually the first indication of renal impairment and immediate attempts should be made to optimize cardiovascular function, particularly by expanding the circulating volume and restoring blood pressure. Restoration of the urine output is a good indicator of successful resuscitation. Evidence now suggests that dopamine is not an effective means of preventing or reversing renal impairment and this agent should not be used for renal protection in sepsis (p. 888). If these measures fail to reverse oliguria, administration of diuretics such as furosemide by bolus or infusion, or less often mannitol (for example in rhabdomyolysis) may be indicated (see Ch. 12). If oliguria persists, it is necessary to reduce fluid intake and review drug doses.
Intermittent haemodialysis has a number of theoretical disadvantages in the critically ill. In particular, it is frequently complicated by hypotension and it may be difficult to remove sufficient volumes of fluid. Nevertheless, provided that strict guidelines are used to improve tolerance and metabolic control, almost all patients with acute kidney injury can be managed successfully with daily haemodialysis. The use of continuous veno-venous haemofiltration, often with dialysis (CVVHD), is however generally preferred in the critically ill (see Ch. 12) and is indicated for fluid overload, electrolyte disturbances (especially hyperkalaemia), severe acidosis and, less often uraemia. The intensity of renal replacement therapy does not seem to influence outcome. Peritoneal dialysis is unsatisfactory in critically ill patients and is contraindicated in those who have undergone intra-abdominal surgery.
Management of shock and sepsis (Fig. 16.25)
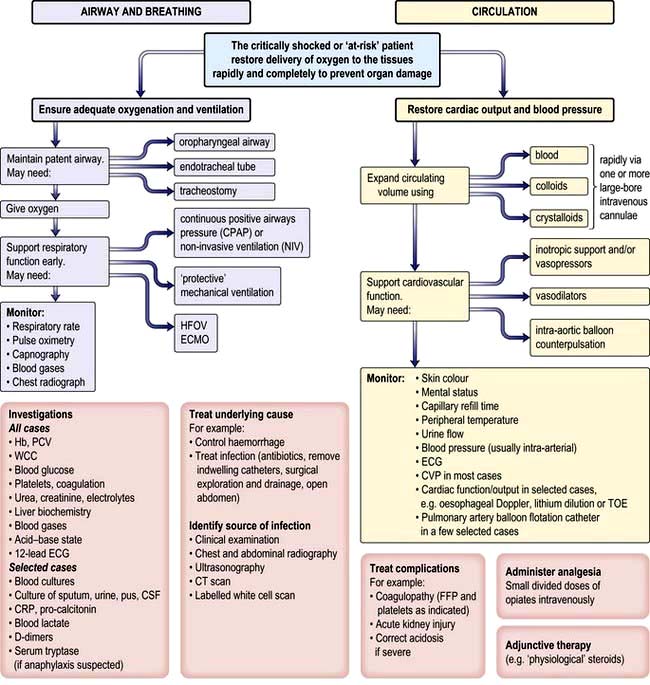
The underlying cause of shock should be corrected, e.g. haemorrhage should be controlled or infection eradicated. In patients with septic shock, every effort must be made to identify the source of infection and isolate the causative organism. As well as a thorough history and clinical examination, X-rays, ultrasonography or CT scanning may be required to locate the origin of the infection. Appropriate samples (urine, sputum, cerebrospinal fluid, pus drained from abscesses) should be sent to the laboratory for microscopy, culture and sensitivities. Several blood cultures should be performed and empirical, broad-spectrum antibiotic therapy (p. 85) should be commenced within the first hour of recognition of sepsis. If an organism is isolated later, therapy can be adjusted appropriately. The choice of antibiotic depends on the likely source of infection, previous antibiotic therapy and known local resistance patterns, as well as on whether infection was acquired in hospital or in the community. Abscesses must be drained and infected indwelling catheters removed.
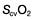 ≥70% (or
≥70% (or 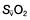 ≥65%) as the initial goals of resuscitation. Administration of fluids can also be targeted at abolishing arterial pressure variation and/or optimizing stroke volume.
≥65%) as the initial goals of resuscitation. Administration of fluids can also be targeted at abolishing arterial pressure variation and/or optimizing stroke volume.Preload and volume replacement
Choice of fluid for volume replacement
Blood
This is conventionally given for haemorrhagic shock as soon as it is available. In extreme emergencies, group-specific crossmatch can be performed in minutes (see p. 408). When available and not contraindicated, blood salvage should be employed for those with severe on-going bleeding.
Although red cell transfusion will augment oxygen-carrying capacity, and hence global oxygen delivery, tissue oxygenation is also dependent on microcirculatory flow. This is influenced by the viscosity of the blood and hence the packed cell volume (PCV). Conventionally, a PCV of 30–35% has been considered to provide the optimal balance between oxygen-carrying capacity and tissue flow, although it is well recognized that previously fit people with haemorrhagic shock can tolerate extremely low Hb concentrations, provided their circulating volume and cardiac output are maintained. Transfusion of old stored red cells, which become spherical rather than biconcave and poorly deformable, with increased adhesiveness, can compromise microvascular flow and worsen tissue hypoxia. Whole blood has now been largely replaced by red cell concentrates (see p. 412)
Complications of blood transfusion are discussed on page 408.
Special problems arise as a result of massive transfusion:
 Temperature changes. Bank blood is stored at 4°C; transfusion may result in hypothermia, peripheral venoconstriction (which slows the rate of the infusion) and arrhythmias. If possible, blood should be warmed during massive transfusion and in those at risk of hypothermia (e.g. during prolonged major surgery with open body cavity).
Temperature changes. Bank blood is stored at 4°C; transfusion may result in hypothermia, peripheral venoconstriction (which slows the rate of the infusion) and arrhythmias. If possible, blood should be warmed during massive transfusion and in those at risk of hypothermia (e.g. during prolonged major surgery with open body cavity).





Myocardial contractility and inotropic agents
Severe lactic acidosis conventionally is said to depress myocardial contractility and limit the response to vasopressor agents. Attempted correction of acidosis with intravenous sodium bicarbonate, however, generates additional carbon dioxide which diffuses across cell membranes, producing or exacerbating intracellular acidosis. Other disadvantages of bicarbonate therapy include sodium overload and a left shift of the oxyhaemoglobin dissociation curve. Ionized calcium levels may be reduced and, combined with the fall in intracellular pH, this may impair myocardial performance. Treatment of lactic acidosis should therefore concentrate on correcting the cause. Bicarbonate should only be administered to correct extreme persistent metabolic acidosis (see Chapter 13).
Many of the most seriously ill patients become increasingly resistant to the effects of pressor agents, an observation attributed to ‘downregulation’ of adrenergic receptors and NO-induced ‘vasoplegia’ (p. 879).
All inotropic agents should be administered via a large central vein, and their effects continually monitored (Table 16.5).
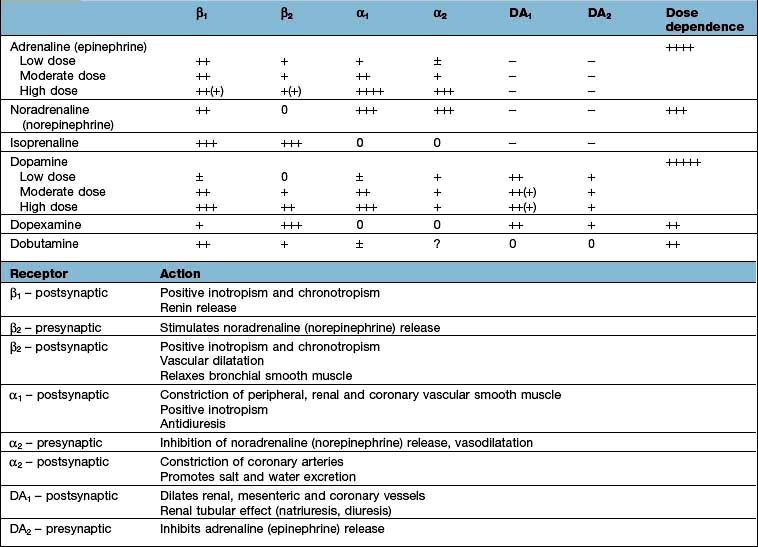
Noradrenaline (norepinephrine)
This is predominantly an α-adrenergic agonist. It is particularly useful in those with hypotension associated with a low systemic vascular resistance, e.g. in septic shock. There is a risk of producing excessive vasoconstriction with impaired organ perfusion and increased afterload. Noradrenaline administration should normally therefore be guided by comprehensive haemodynamic monitoring, including invasive or non-invasive determination of cardiac output (see p. 875) and calculation of systemic vascular resistance.
FURTHER READING
Annane D, Vignon P, Renault A et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomized trial. Lancet 2007; 370:676–684.
De Backer D, Biston P, Devriendt J et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
Russell JA, Walley KR, Singer J et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
High-risk surgical patients (Box 16.4)
 Box 16.4
Box 16.4
Patients at risk of developing perioperative multiorgan failure

 Patients with co-morbidity, especially limited cardiorespiratory reserve
Patients with co-morbidity, especially limited cardiorespiratory reserve
 Patients with trauma to two body cavities requiring multiple blood transfusions
Patients with trauma to two body cavities requiring multiple blood transfusions


Modified from Shoemaker WC, Appel PL, Kram HB et al. Prospective trial of supranormal values of survivors as therapeutic goals in high-risk surgical patients. Chest 1988; 94:1176–1186.
Vasodilator therapy
In selected cases, afterload reduction is used to increase stroke volume and decrease myocardial oxygen requirements by reducing the systolic ventricular wall tension. Vasodilatation (see p. 720) also decreases heart size and the diastolic ventricular wall tension so that coronary blood flow is improved. The relative magnitude of the falls in preload and afterload depends on the pre-existing haemodynamic disturbance, concurrent volume replacement and the agent selected (see below). Vasodilators also improve microcirculatory flow.
Vasodilator therapy can be particularly helpful in patients with cardiac failure in whom the ventricular function curve is flat (Fig. 16.6) so that falls in preload have only a limited effect on stroke volume. This form of treatment, combined in selected cases with inotropic support, is therefore useful in cardiogenic shock and in the management of patients with cardiogenic pulmonary oedema or mitral regurgitation.
Mechanical support of the myocardium
Intra-aortic balloon counterpulsation (IABCP) is the technique used most widely for mechanical support of the failing myocardium. It is discussed on page 696.
Adjunctive treatment
Initial attempts to combat the high mortality associated with sepsis concentrated on cardiovascular and respiratory support in the hope that survival could be prolonged until surgery, antibiotics and the patient’s own defences had eradicated the infection and injured tissues were repaired. Despite some success, mortality rates remained unacceptably high. So far, attempts to improve outcome by modulating the inflammatory response (including high-dose steroids) or neutralizing endotoxin (Table 16.6) have proved disappointing and in some cases may even have been harmful.
Table 16.6 Some of the therapeutic strategies tested in randomized, controlled phase II or III trials in human sepsis
The aim of current sepsis guidelines is to combine these, and other evidence-based interventions, with early effective resuscitation (aimed especially at achieving an adequate circulating volume, combined with the rational use of inotropes and/or vasoactive agents to maintain blood pressure, cardiac output and oxygen transport) in order to create ‘bundles of care’ delivered within specific time limits (see http://www.survivingsepsis.org).
FURTHER READING
Dellinger RP, Levy MM, Carlet JM et al. for the International Surviving Sepsis Campaign Guidelines Committee. Surviving Sepsis Campaign: International Guidelines for Management of Severe Sepsis and Septic Shock. Crit Care Med 2008; 36:296–327.
Sprung CL, Annane D, Keh D et al. for the CORTICUS Study Group. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
Toussaint S, Gerlach H. Activated protein C for sepsis. N Engl J Med 2009; 361:2640–2652.
Respiratory failure (see Ch. 15)


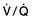 mismatch. Common causes include cardiogenic pulmonary oedema, pneumonia, acute lung injury and lung fibrosis.
mismatch. Common causes include cardiogenic pulmonary oedema, pneumonia, acute lung injury and lung fibrosis.Clinical assessment of respiratory failure











Monitoring of respiratory failure
Blood gas analysis
Normal values of blood gas analysis are shown in Table 16.2.
 The sample should be analysed immediately. Alternatively, the syringe should be immersed in iced water (the end having first been sealed with a cap) to prevent the continuing metabolism of white cells causing a reduction in PO2 and a rise in PCO2.
The sample should be analysed immediately. Alternatively, the syringe should be immersed in iced water (the end having first been sealed with a cap) to prevent the continuing metabolism of white cells causing a reduction in PO2 and a rise in PCO2.

Interpretation of the results of blood gas analysis can be considered in two separate parts:


Correct interpretation requires a knowledge of the clinical history, the age of the patient, the inspired oxygen concentration and any other relevant treatment (e.g. the ventilator settings for those on mechanical ventilation or the administration of sodium bicarbonate). The oxygen content of the arterial blood is determined by the percentage saturation of haemoglobin with oxygen. The relationship between the latter and the PaO2 is determined by the oxyhaemoglobin dissociation curve (Fig. 16.2).




Management of respiratory failure
Standard management of patients with respiratory failure includes:
 administration of supplemental oxygen through a patent airway
administration of supplemental oxygen through a patent airway
 treatment for distal airways obstruction
treatment for distal airways obstruction



Oxygen therapy
Methods of oxygen administration
Oxygen is initially given via a facemask. In the majority of patients (except those with COPD and chronically elevated PaCO2) the concentration of oxygen given is not vital and oxygen can therefore be given by a ‘variable performance’ device such as a simple facemask or nasal cannulae (Fig. 16.26).
With these devices, the inspired oxygen concentration varies from about 35% to 55%, with oxygen flow rates of between 6 and 10 L/min. Nasal cannulae are often preferred because they are less claustrophobic and do not interfere with feeding or speaking, but they can cause ulceration of the nasal or pharyngeal mucosa. Higher concentrations of oxygen can be administered by using a mask with a reservoir bag attached (Fig. 16.26c). Figure 16.26 should be compared with the fixed-performance mask shown in Figure 15.25, with which the oxygen concentration can be controlled. This latter type of mask is used in patients with COPD and chronic type II failure; the hazards of reducing hypoxic drive can be overemphasized and are less dangerous when the patient is in a critical care unit – remember, severe hypoxaemia is more dangerous than hypercapnia.
Respiratory support
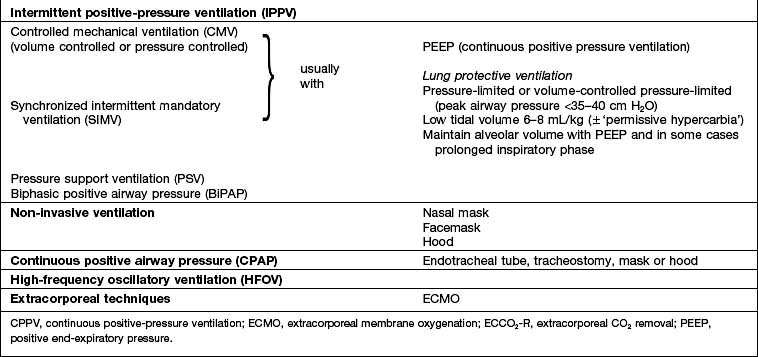
If, despite the above measures, the patient continues to deteriorate or fails to improve, the institution of some form of respiratory support is necessary (Table 16.7). Non-invasive ventilation via a mask or hood (see p. 895) can be used, particularly in respiratory failure due to COPD, but in critically ill patients invasive ventilation through an endotracheal tube or tracheostomy is more usual.
 |
Intermittent positive pressure ventilation (IPPV) is achieved by intermittently inflating the lungs with a positive pressure delivered by a mechanical ventilator. Over the last few decades there have been a number of refinements and modifications to the manner in which positive pressure is applied to the airway and in the interplay between the patient’s respiratory efforts and mechanical assistance (p. 894).
Beneficial effects of mechanical ventilation


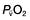 and thus PaO2 may improve. Because ventilated patients are connected to a leak-free circuit, it is possible to administer high concentrations of oxygen (up to 100%) accurately and to apply a positive end-expiratory pressure (PEEP). In selected cases, the latter may reduce shunting and increase PaO2 (see below).
and thus PaO2 may improve. Because ventilated patients are connected to a leak-free circuit, it is possible to administer high concentrations of oxygen (up to 100%) accurately and to apply a positive end-expiratory pressure (PEEP). In selected cases, the latter may reduce shunting and increase PaO2 (see below).
Indications for mechanical ventilation
 Acute respiratory failure, with signs of severe respiratory distress (e.g. respiratory rate >40/min, inability to speak, patient exhausted) persisting despite maximal therapy. Confusion, restlessness, agitation, a decreased conscious level, a rising PaCO2 (>8 kPa) and extreme hypoxaemia (<8 kPa), despite oxygen therapy, are further indications.
Acute respiratory failure, with signs of severe respiratory distress (e.g. respiratory rate >40/min, inability to speak, patient exhausted) persisting despite maximal therapy. Confusion, restlessness, agitation, a decreased conscious level, a rising PaCO2 (>8 kPa) and extreme hypoxaemia (<8 kPa), despite oxygen therapy, are further indications.






Institution of invasive respiratory support
This requires tracheal intubation. If the patient is conscious, the procedure must be fully explained and consent obtained before anaesthesia is induced. The complications of tracheal intubation are given in Table 16.8.
| Complication | Comments |
|---|---|
|
Immediate |
|
|
Trauma to the upper airway |
Lips, teeth, gums, trachea |
|
Tube in one or other (usually the right) main bronchus |
Avoid by checking both lungs are being inflated, i.e. both sides of the chest move and air entry is heard bilaterally on auscultation |
|
Early |
|
|
Migration of the tube out of the trachea |
Dangerous complications |
|
Manual inflation with 100% oxygen |
|
|
Tracheal suction |
|
|
Check position of tube |
|
|
Deflate cuff |
|
|
Check tube for ‘kinks’ or blockage with secretions or blood (common) |
|
|
If no improvement, remove tube, ventilate with facemask and then insert new endotracheal tube |
|
|
Late |
|
|
Sinusitis |
|
Sedation, analgesia and muscle relaxation
Most critically ill patients will require analgesia and many will receive sedatives. The combination of an opiate with a benzodiazepine or propofol is often used to facilitate mechanical ventilation and to obtund the physiological response to stress. Heavy sedation is indicated in those with severe respiratory failure, especially since ‘lung protective’ ventilatory strategies (see p. 895) are inherently uncomfortable. A few may require neuromuscular blockade, indeed evidence suggests that early administration of atracurium improves outcomes for mechanically ventilated patients with severe ARDS. It is now recognized, however, that minimizing sedation levels using ‘sedation scores’ and ‘daily wakening’, or even the avoidance of sedatives altogether, often in combination with spontaneous breathing modes of respiratory support (see p. 895) is associated with reductions in the duration of mechanical ventilation and more rapid discharge from the ICU and hospital. It is also now recognized that benzodiazepines predispose to the development of delirium (which is an independent predictor of increased mortality and length of hospital stay) in critically ill patients. The use of dexmedetomidine (an α2 agonist) rather than a benzodiazepine has been shown to be associated with less delirium and reduced time on the ventilator.
Tracheostomy
Tracheostomy has a small but significant mortality rate. Complications of tracheostomy are shown in Table 16.9.
Table 16.9 Complications of tracheostomy
(As for tracheal intubation, see Table 16.8), plus:
|
|
 mismatch, fluid retention and collapse of peripheral alveoli. Traditionally, the latter was prevented by using high tidal volumes (10–12 mL/kg) but high inflation pressures, with overdistension of compliant alveoli, perhaps exacerbated by the repeated opening and closure of distal airways, can disrupt the alveolar–capillary membrane. There is an increase in microvascular permeability and release of inflammatory mediators leading to ‘ventilator-associated lung injury’. Extreme overdistension of the lungs during mechanical ventilation with high tidal volumes and PEEP can rupture alveoli and cause air to dissect centrally along the perivascular sheaths. This ‘barotrauma’ may be complicated by pneumomediastinum, subcutaneous emphysema, pneumoperitoneum, pneumothorax, and intra-abdominal air. The risk of pneumothorax is increased in those with destructive lung disease (e.g. necrotizing pneumonia, emphysema), asthma or fractured ribs.
mismatch, fluid retention and collapse of peripheral alveoli. Traditionally, the latter was prevented by using high tidal volumes (10–12 mL/kg) but high inflation pressures, with overdistension of compliant alveoli, perhaps exacerbated by the repeated opening and closure of distal airways, can disrupt the alveolar–capillary membrane. There is an increase in microvascular permeability and release of inflammatory mediators leading to ‘ventilator-associated lung injury’. Extreme overdistension of the lungs during mechanical ventilation with high tidal volumes and PEEP can rupture alveoli and cause air to dissect centrally along the perivascular sheaths. This ‘barotrauma’ may be complicated by pneumomediastinum, subcutaneous emphysema, pneumoperitoneum, pneumothorax, and intra-abdominal air. The risk of pneumothorax is increased in those with destructive lung disease (e.g. necrotizing pneumonia, emphysema), asthma or fractured ribs.Techniques for respiratory support (Table 16.7)
Controlled mechanical ventilation (CMV)
This technique is used in patients in whom respiratory efforts are absent or have been abolished.


Continuous positive airway pressure (CPAP)
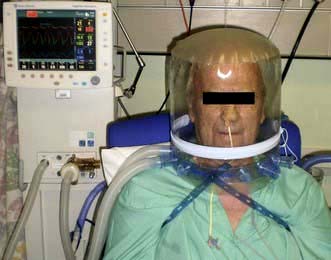
The application of CPAP achieves for the spontaneously breathing patient what PEEP does for the ventilated patient. Oxygen and air are delivered under pressure via an endotracheal tube, a tracheostomy, a tightly fitting facemask or a hood (Fig 16.27). Not only can CPAP improve oxygenation, but the lungs become more compliant, and the work of breathing is reduced.
‘Lung-protective’ ventilation
This is designed to avoid exacerbating or perpetuating lung injury by avoiding overdistension of alveoli, minimizing airway pressures and preventing the repeated opening and closure of distal airways. Alveolar volume is maintained with PEEP, and sometimes by prolonging the inspiratory phase, while tidal volumes are limited to 6–8 mL/kg ideal bodyweight. Peak airway pressures should not exceed 35–40 cmH2O. An alternative is to deliver a constant preset inspiratory pressure for a prescribed time in order to generate a low tidal volume at reduced airway pressures (‘pressure-limited’ mechanical ventilation). Respiratory rate can be increased to improve CO2 removal and avoid severe acidosis (pH <7.2), but hypercarbia is frequent and should be accepted (‘permissive hypercarbia’). Both techniques can be used with SIMV. Ventilation with low tidal volumes has been shown to improve outcome in patients with acute lung injury (ALI) or the acute respiratory distress syndrome (ARDS) (see p. 884). Lung protective ventilation should now be used in almost all patients undergoing mechanical ventilation.
FURTHER READING
Nava S, Hill N. Non-invasive ventilation in acute respiratory failure. Lancet 2009; 374:250–259.
Peek GJ, Mugford M, Tinwoipati R et al. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): a multicentre randomised controlled trial. Lancet 2009; 374:1351–1363.
Non-invasive ventilation (NIV)
NIV is suitable for patients who are conscious, cooperative and able to protect their airway; they must also be able to expectorate effectively. Positive pressure is applied to the airways using a tight-fitting full-face/nasal mask or a hood. The most popular ventilators for this purpose are those that deliver bilevel positive airway pressure (BiPAP), which are simple to use, cheap and flexible. With the latter technique, inspiratory and expiratory pressure levels and times are set independently and unrestricted spontaneous respiration is possible throughout the respiratory cycle. BiPAP can also be patient triggered. There is a reduced risk of ventilator-associated pneumonia and improved patient comfort, with preservation of airway defence mechanisms, speech and swallowing (which allows better nutrition). Spontaneous coughing and expectoration are not hampered, allowing effective physiotherapy, and sedation is usually unnecessary. Institution of non-invasive respiratory support can rest the respiratory muscles, reduce respiratory acidosis and breathlessness, improve clearance of secretions and re-expand collapsed lung segments. The intubation rate, length of ICU and hospital stay and, in some categories of patient, mortality, may all be reduced. NIV is particularly useful in acute hypercapnic respiratory failure associated with COPD, provided the patient is not profoundly hypoxic. NIV may also be useful as a means of avoiding tracheal intubation in immunocompromised patients with acute respiratory failure. Evidence suggests that early NIV after extubation of hypercapnic patients with respiratory disorders can reduce the risk of subsequent respiratory failure and mortality. Box 16.5 shows some indications for the use of NIV when standard medical treatment has failed. Remember, NIV should not be used as a substitute for invasive ventilation when the latter is clearly more appropriate.
 Box 16.5
Box 16.5
Some indications for the use of non-invasive ventilation (NIV)
 Acute exacerbation of COPD (pH <7.35)
Acute exacerbation of COPD (pH <7.35)

 Chest wall deformity/neuromuscular disease (hypercapnic respiratory failure)
Chest wall deformity/neuromuscular disease (hypercapnic respiratory failure)




Modified from BTS guidelines after 1997, http://www.british thoracic society.co.uk.
Weaning
Techniques for weaning
 The traditional method is to allow the patient to breathe entirely spontaneously for a short time, following which respiratory support is reinstituted. The periods of spontaneous breathing are gradually increased and the periods of respiratory support are progressively reduced. Initially it is usually advisable to ventilate the patient throughout the night. This method can be stressful and tiring for both patients and staff, although it is sometimes successful when other methods have failed.
The traditional method is to allow the patient to breathe entirely spontaneously for a short time, following which respiratory support is reinstituted. The periods of spontaneous breathing are gradually increased and the periods of respiratory support are progressively reduced. Initially it is usually advisable to ventilate the patient throughout the night. This method can be stressful and tiring for both patients and staff, although it is sometimes successful when other methods have failed.



 Tracheostomy is often used to facilitate weaning from mechanical ventilation.
Tracheostomy is often used to facilitate weaning from mechanical ventilation.

Outcomes: withholding and withdrawing treatment
 avoid admitting patients who cannot benefit from intensive care and
avoid admitting patients who cannot benefit from intensive care and
 limit further aggressive therapy when the prognosis is clearly hopeless.
limit further aggressive therapy when the prognosis is clearly hopeless.
Decisions to limit therapy, not to resuscitate or to withdraw treatment should be made jointly by the medical staff of the unit, the primary physician or surgeon, the nurses and if possible the patient, normally in consultation with the patient’s family. Limitation of active treatment is not the cessation of medical or nursing care: rather, a caring approach must be adopted to ensure a dignified death, free of pain and distress, with support for family and friends (see Ch. 10).
Scoring systems
FURTHER READING
Herridge MS, Tansey CM, Matte A et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med 2011; 364:1293–1304.
Lautrette A, Darmen M, Megarlane B et al. A communication strategy and brochure for relatives of patients dying in the ICU. N Engl J Med 2007; 356:469–478.
Brain death






Diagnostic tests for the confirmation of brainstem death
All brainstem reflexes are absent in brainstem death.
Tests
The following tests should not be performed in the presence of seizures or abnormal postures.
 Oculocephalic reflexes should be absent. In a comatose patient whose brainstem is intact, the eyes will rotate relative to the orbit (i.e. doll’s eye movements will be present). In a brainstem dead patient, when the head is rotated from side to side, the eyes move with the head and therefore remain stationary relative to the orbit.
Oculocephalic reflexes should be absent. In a comatose patient whose brainstem is intact, the eyes will rotate relative to the orbit (i.e. doll’s eye movements will be present). In a brainstem dead patient, when the head is rotated from side to side, the eyes move with the head and therefore remain stationary relative to the orbit.


 There are no vestibulo-ocular reflexes on caloric testing (see p. 1078).
There are no vestibulo-ocular reflexes on caloric testing (see p. 1078).

 There is no gag or cough reflex in response to pharyngeal, laryngeal or tracheal stimulation.
There is no gag or cough reflex in response to pharyngeal, laryngeal or tracheal stimulation.

The examination should be performed and repeated by two senior doctors.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
Chapter 16 Critical care medicine
Introduction
Critical care medicine (or ‘intensive care medicine’) is concerned predominantly with the management of patients with acute life-threatening conditions (’the critically ill’) in specialized units. As well as emergency cases, such units admit high-risk patients electively after major surgery (Table 16.1). Intensive care medicine also encompasses the resuscitation and transport of those who become acutely ill, or are injured in the community. Management of seriously ill patients throughout the hospital (e.g. in coronary care units, acute admissions wards, postoperative recovery areas or emergency units), including critically ill patients who have been discharged to the ward (‘outreach care’), is also undertaken. Teamwork and a multidisciplinary approach are central to the provision of intensive care and are most effective when directed and coordinated by committed specialists.
Table 16.1 Some common indications for admission to intensive care
General aspects of managing the critically ill
Recognition and diagnosis of critical illness
Early recognition and immediate resuscitation are fundamental to the successful management of the critically ill. In order to facilitate identification of ‘at risk’ patients on the ward and early referral to the critical care emergency or outreach team a number of early warning systems have been devised (e.g. the Modified Early Warning Score, MEWS; see Box 16.1). These are based primarily on bedside recognition of deteriorating physiological variables and can be used to supplement clinical intuition. A MEWS score of ≥5 is associated with an increased risk of death and warrants immediate admission to ICU. Another example of a system used to trigger referral to a Medical Emergency Team (MET) is also shown in Box 16.1 (see also ‘Management of shock and sepsis’ and ‘Clinical assessment of respiratory failure’, below).
Box 16.1
Early warning systems for referral of ‘at risk’ patients to the critical care team
Medical emergency team-calling criteria
|
Airway |
If threatened |
|
Breathing |
All respiratory arrests |
|
Respiratory rate |
<5 breaths/min |
|
Respiratory rate |
>36 breaths/min |
|
Circulation |
All cardiac arrests |
|
Pulse rate |
<40 beats/min |
|
Pulse rate |
>140 beats/min |
|
Systolic blood pressure |
<90 mmHg |
|
Neurology |
Sudden fall in level of consciousness (fall in Glasgow Coma Scale of >2 points) |
|
|
Repeated or prolonged seizures |
|
Other |
Any patient who does not fit the criteria above, but about whom you are seriously worried |
From Hillman K, Chen J, Cretikos M et al. Introduction of the medical emergency team (MET) system: a cluster-randomized controlled trial. Lancet 2005; 365:2091–2097, with permission.
In some of the most seriously ill patients, the precise underlying diagnosis is initially unclear but in all cases, the immediate objective is to preserve life and prevent, reverse or minimize damage to vital organs such as the lungs, brain, kidneys and liver. This involves a rapid assessment of the physiological derangement followed by prompt institution of measures to support cardiovascular and respiratory function in order to restore perfusion of vital organs, improve delivery of oxygen to the tissues and encourage the removal of carbon dioxide and other waste products of metabolism (following the ABC approach: Airway, Breathing, Circulation, see Fig. 16.25, below). The patient’s condition and response to treatment should be closely monitored throughout. The underlying diagnosis may only become clear as the results of investigations become available, a more detailed history is obtained and a more thorough physical examination is performed. In practice resuscitation, assessment and diagnosis usually proceed in parallel.
Critically ill patients require multidisciplinary care with:
Intensive skilled nursing care (usually 1 : 1 or 1 : 2 nurse/patient ratio in the UK).
Specialized physiotherapy including mobilization and rehabilitation.
Management of pain and distress with judicious administration of analgesics and sedatives (see p. 893).
Care of the mouth, prevention of constipation and of pressure sores.
Nutritional support (see p. 222). Protein energy malnutrition is common in critically ill patients and is associated with muscle wasting, weakness, delayed mobilization, difficulty weaning from ventilation, immune compromise and impaired wound healing. There is also an association between malnutrition and increased mortality. It is therefore recommended that nutritional support should be instituted as soon as is practicable in those unable to meet their nutritional needs orally, ideally within 1–2 days of the acute episode. Enteral nutrition, which is less expensive, preserves gut mucosal integrity, is more physiological and is associated with fewer complications, is preferred. Recently, the value of early feeding has been questioned, apart from giving small amounts to ensure gut viability. Parenteral nutrition is sometimes indicated at a later stage for those unable to tolerate or absorb enteral nutrition and should be initiated without delay, at least within 3 days. Persistent attempts at enteral nutrition in those with gastrointestinal intolerance leads to underfeeding and malnutrition.
Critically ill patients commonly require intravenous insulin infusions, often in high doses, to combat hyperglycaemia and insulin resistance (see p. 1006). Although the use of intensive insulin therapy to achieve ‘tight glycaemic control’ (blood glucose level between 4.4 and 6.1 mmol/L) was shown to improve outcome (at least when combined with aggressive nutritional support), more recent studies have failed to confirm this finding and have indicated that this approach is associated with an unacceptably high incidence of hypoglycaemia, and possibly an increase in mortality. Current recommendations suggest that blood glucose levels should be maintained at <8–10 mmol/L.
FURTHER READING
Casaer MP, Mesotten O, Hermans G et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med 2011; 365:506–517.
The NICE-SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1293–1297.
Ziegler TR. Parenteral nutrition in the critically ill patient. N Engl J Med 2009; 361:1088–1097.
Applied cardiorespiratory physiology
Oxygen delivery and consumption
Oxygen delivery (DO2) (Fig. 16.1) is defined as the total amount of oxygen delivered to the tissues per unit time. It is dependent on the volume of blood flowing through the microcirculation per minute (i.e. the total cardiac output, ) and the amount of oxygen contained in that blood (i.e. the arterial oxygen content, CaO2). Oxygen is transported both in combination with haemoglobin and dissolved in plasma. The amount combined with haemoglobin is determined by the oxygen capacity of haemoglobin (usually taken as 1.34 mL of oxygen per gram of haemoglobin) and its percentage saturation with oxygen (SO2), while the volume dissolved in plasma depends on the partial pressure of oxygen (PO2). Except when hyperbaric oxygen is administered, the amount of dissolved oxygen in plasma is insignificant.
Oxygenation of the blood
Oxyhaemoglobin dissociation curve
The saturation of haemoglobin with oxygen is determined by the partial pressure of oxygen (PO2) in the blood, the relationship between the two being described by the oxyhaemoglobin dissociation curve (Fig. 16.2). The sigmoid shape of this curve is significant for a number of reasons:
Modest falls in the partial pressure of oxygen in the arterial blood (PaO2) may be tolerated (since oxygen content is relatively unaffected) provided that the percentage saturation remains above 92%.
Figure 16.2 The oxyhaemoglobin dissociation curve. a, arterial point; v, venous point; x, arteriovenous oxygen content difference. HbO2 (%) is the percentage saturation of haemoglobin with oxygen. The curve will move to the right in the presence of acidosis (metabolic or respiratory), pyrexia or an increased red cell 2,3-DPG concentration. For a given arteriovenous oxygen content difference, the mixed venous PO2 will then be higher. Furthermore, if the mixed venous PO2 is unchanged, the arteriovenous oxygen content difference increases and more oxygen is off-loaded to the tissues (see p. 374). P50 (the PO2 at which haemoglobin is half saturated with O2) is a useful index of these shifts – the higher the P50 (i.e. shift to the right), the lower the affinity of haemoglobin for O2.
Alveolar oxygen tension (PAO2)
The partial pressures of inspired gases are shown in Figure 16.3. By the time the inspired gases reach the alveoli they are fully saturated with water vapour at body temperature (37°C), which has a partial pressure of 6.3 kPa (47 mmHg) and contain CO2 at a partial pressure of approximately 5.3 kPa (40 mmHg); the PAO2 is thereby reduced to approximately 13.4 kPa (100 mmHg).
The clinician can influence PAO2 by administering oxygen or by increasing the barometric pressure.
Pulmonary gas exchange
In normal subjects there is a small alveolar-arterial oxygen difference (PA–aO2). This is due to:
Pathologically, there are three possible causes of an increased PA–aO2 difference:
Diffusion defect. This is not a major cause of hypoxaemia even in conditions such as lung fibrosis, in which the alveolar-capillary membrane is considerably thickened. Carbon dioxide is also not affected, as it is more soluble than oxygen.
Right-to-left shunts. In certain congenital cardiac lesions or when a segment of lung is completely collapsed, a proportion of venous blood passes to the left side of the heart without taking part in gas exchange, causing arterial hypoxaemia. This hypoxaemia cannot be corrected by administering oxygen to increase the PAO2, because blood leaving normal alveoli is already fully saturated; further increases in PO2 will not, therefore, significantly affect its oxygen content. On the other hand, because of the shape of the carbon dioxide dissociation curve (Fig. 16.4), the high PCO2 of the shunted blood can be compensated for by over-ventilating patent alveoli, thus lowering the CO2 content of the effluent blood. Indeed, many patients with acute right-to-left shunts hyperventilate in response to the hypoxia and/or to stimulation of mechanoreceptors in the lung, so that their PaCO2 is normal or low.
Ventilation/perfusion mismatch (see p. 796). Diseases of the lung parenchyma (e.g. pulmonary oedema, acute lung injury) result in mismatch, producing an increase in alveolar deadspace and hypoxaemia. The increased deadspace can be compensated for by increasing overall ventilation. In contrast to the hypoxia resulting from a true right-to-left shunt, that due to areas of low can be partially corrected by administering oxygen and thereby increasing the PAO2 even in poorly ventilated areas of lung.
Cardiac output
Cardiac output is the product of heart rate and stroke volume, and is affected by changes in either of these (see Fig. 16.5).
Stroke volume
Three interdependent factors determine the stroke volume (see p. 671).
Preload
This is defined as the tension of the myocardial fibres at the end of diastole, just before the onset of ventricular contraction, and is therefore related to the degree of stretch of the fibres. As the end-diastolic volume of the ventricle increases, tension in the myocardial fibres is increased and stroke volume rises (Fig. 16.6). Myocardial oxygen consumption () increases only slightly with an increase in preload (produced, for example, by a ‘fluid challenge’, see below) and this is therefore the most efficient way of improving cardiac output.
Myocardial contractility
This refers to the ability of the heart to perform work, independent of changes in preload and afterload. The state of myocardial contractility determines the response of the ventricles to changes in preload and afterload. Contractility is often reduced in critically ill patients, as a result of either pre-existing myocardial damage (e.g. ischaemic heart disease), or the acute disease process itself (e.g. sepsis). Changes in myocardial contractility alter the slope and position of the Starling curve; worsening ventricular performance is manifested as a depressed, flattened curve (Fig. 16.6 and Fig. 14.5). Inotropic drugs can be used to increase myocardial contractility (see below).
Afterload
Decreasing the afterload (exercise, sepsis, vasodilator agents) can increase the stroke volume achieved at a given preload (Fig. 16.7), while reducing . The reduction in wall tension also leads to an increase in coronary blood flow, thereby improving the myocardial oxygen supply/demand ratio. Excessive reductions in afterload will cause hypotension.
Monitoring critically ill patients
Assessment of tissue perfusion
Pale, cold skin, delayed capillary refill and the absence of visible veins in the hands and feet indicate poor perfusion. Although peripheral skin temperature measurements can help clinical evaluation, the earliest compensatory response to hypovolaemia or a low cardiac output, and the last to resolve after resuscitation is vasoconstriction in the splanchnic region.
Urinary flow is a sensitive indicator of renal perfusion and haemodynamic performance.
Blood pressure
Hypotension jeopardizes perfusion of vital organs. The adequacy of blood pressure in an individual patient must always be assessed in relation to the premorbid value. Blood pressure is traditionally measured using a sphygmomanometer but if rapid alterations are anticipated, continuous monitoring using an intra-arterial cannula is indicated (Practical Box 16.1; Fig. 16.8).
Practical Box 16.1
Radial artery cannulation
Technique
1. The procedure is explained to the patient and, if possible, consent obtained.
2. The arm is supported, with the wrist extended, by an assistant. (Gloves should be worn.)
3. The skin should be cleaned with chlorhexidine.
4. The radial artery is palpated where it arches over the head of the radius.
5. In conscious patients, local anaesthetic is injected to raise a weal over the artery, taking care not to puncture the vessel or obscure its pulsation.
6. A small skin incision is made over the proposed puncture site.
7. A small parallel-sided cannula (20 gauge for adults, 22 gauge for children) is used in order to allow blood flow to continue past the cannula.
8. The cannula is inserted over the point of maximal pulsation and advanced in line with the direction of the vessel at an angle of approximately 30°.
9. ‘Flashback’ of blood into the cannula indicates that the radial artery has been punctured.
10. To ensure that the shoulder of the cannula enters the vessel, the needle and cannula are lowered and advanced a few millimetres into the vessel.
11. The cannula is threaded off the needle into the vessel and the needle withdrawn.
12. The cannula is connected to a non-compliant manometer line filled with saline. This is then connected via a transducer and continuous flush device to a monitor, which records the arterial pressure.
Central venous pressure (CVP)
This provides a fairly simple, but approximate method of gauging the adequacy of a patient’s circulating volume and the contractile state of the myocardium. The absolute value of the CVP is not as useful as its response to a fluid challenge (the infusion of 100–200 mL of fluid over a few minutes) (Fig. 16.9). The hypovolaemic patient will initially respond to transfusion with little or no change in CVP, together with some improvement in cardiovascular function (falling heart rate, rising blood pressure, increased peripheral temperature and urine output). As the normovolaemic state is approached, the CVP usually rises slightly and reaches a plateau, while other cardiovascular values begin to stabilize. At this stage, volume replacement should be slowed, or even stopped, in order to avoid overtransfusion (indicated by an abrupt and sustained rise in CVP, often accompanied by some deterioration in the patient’s condition). In cardiac failure, the venous pressure is usually high; the patient will not improve in response to volume replacement, which will cause a further, sometimes dramatic, rise in CVP.
Central venous catheters are usually inserted via a percutaneous puncture of the subclavian or internal jugular vein using a guidewire technique (Practical Box 16.2; Figs 16.10, 16.11). The guidewire techniques can also be used in conjunction with a vein dilator for inserting multilumen catheters, double lumen cannulae for haemofiltration or pulmonary artery catheter introducers. The routine use of ultrasound to guide central venous cannulation reduces complication rates.
Practical Box 16.2
Internal jugular vein cannulation
Technique
1. The procedure is explained to the patient and, if possible, consent obtained.
2. The patient is placed head-down to distend the central veins (this facilitates cannulation and minimizes the risk of air embolism but may exacerbate respiratory distress and is dangerous in those with raised intracranial pressure).
3. The skin is cleaned with an antiseptic solution such as chlorhexidine. Sterile precautions are taken throughout the procedure.
4. Local anaesthetic (1% plain lidocaine) is injected intradermally to raise a weal at the apex of a triangle formed by the two heads of sternomastoid with the clavicle at its base.
5. A small incision is made through the weal.
6. The cannula or needle is inserted through the incision and directed laterally downwards and backwards in the direction of the nipple until the vein is punctured just beneath the skin and deep to the lateral head of sternomastoid.
Ultrasound-guided puncture is recommended to reduce the incidence of complications.
7. Check that venous blood is easily aspirated.
8. The cannula is threaded off the needle into the vein or the guidewire is passed through the needle (see Fig. 16.11).
9. The CVP manometer line is connected to a manometer/transducer.
10. A chest X-ray should be taken to verify that the tip of the catheter is in the superior vena cava and to exclude pneumothorax.
The following are common pitfalls in interpreting central venous pressure readings:
Blocked catheter. This results in a sustained high reading, with a damped or absent waveform, which often does not correlate with clinical assessment.
Transducer wrongly positioned. Failure to level the system is a common cause of erroneous readings.
Left atrial pressure
In uncomplicated cases, careful interpretation of the CVP provides a reasonable guide to the filling pressures of both sides of the heart. In many critically ill patients, however, there is a disparity in function between the two ventricles. Most commonly, left ventricular performance is worse, so that the left ventricular function curve is displaced downward and to the right (Fig. 16.12). High right ventricular filling pressures, with normal or low left atrial pressures, are less common but occur with right ventricular dysfunction and in cases where the pulmonary vascular resistance (i.e. right ventricular afterload) is raised, such as in acute respiratory failure and pulmonary embolism.
Pulmonary artery pressure
A ‘balloon flotation catheter’ enables reliable catheterization of the pulmonary artery. These ‘Swan–Ganz’ catheters can be inserted centrally (Fig. 16.10) or through the femoral vein, or via a vein in the antecubital fossa. Passage of the catheter from the major veins, through the chambers of the heart, into the pulmonary artery and into the wedged position is monitored and guided by the pressure waveforms recorded from the distal lumen (Practical Box 16.3; Fig. 16.13). A chest X-ray should always be obtained to check the final position of the catheter. In difficult cases, screening with an image intensifier may be required.
Practical Box 16.3
Passage of a pulmonary artery balloon flotation catheter through the chambers of the heart into the ‘wedged’ position
Consent should be obtained if possible from the patient after explanation of the procedure.
Note: (a), (b), (c), (d) refer to Figure 16.13.
1. A balloon flotation catheter is inserted through a large vein (see text).
2. Once in the thorax, respiratory oscillations are seen. The catheter should be advanced further towards the lower superior vena cava/right atrium (a), where pressure oscillations become more pronounced. The balloon should then be inflated and the catheter advanced.
3. When the catheter is in the right ventricle (b), there is no dicrotic notch and the diastolic pressure is close to zero. The patient should be returned to the horizontal, or slightly head-up, position before advancing the catheter further.
4. When the catheter reaches the pulmonary artery (c) a dicrotic notch appears and there is elevation of the diastolic pressure. The catheter should be advanced further with the balloon inflated.
5. Reappearance of a venous waveform indicates that the catheter is ‘wedged’. The balloon is deflated to obtain the pulmonary artery pressure. The balloon is inflated intermittently to obtain the pulmonary artery occlusion (also known as pulmonary artery, or capillary, ‘wedge’) pressure (d).
Figure 16.13 Passage of pulmonary artery balloon flotation catheter through the chambers of the heart into the ‘wedged’ position to measure the pulmonary artery occlusion pressure. (See Practical Box 16.3.)
Less invasive techniques for assessing cardiac function and guiding volume replacement
Arterial pressure variation as a guide to hypovolaemia
Systolic arterial pressure decreases during the inspiratory phase of intermittent positive pressure ventilation (p. 894). The magnitude of this cyclical variability has been shown to correlate more closely with hypovolaemia than other monitored variables, including CVP. Systolic pressure (or pulse pressure) variation during mechanical ventilation can therefore be used as a simple and reliable guide to the adequacy of the circulatory volume. The response to fluid loading can also easily be predicted by observing the changes in pulse pressure during passive leg raising.
Oesophageal Doppler
Stroke volume, cardiac output and myocardial function can be assessed non-invasively using Doppler ultrasonography. A probe is passed into the oesophagus to continuously monitor velocity waveforms from the descending aorta (Fig. 16.14). Although reasonable estimates of stroke volume, and hence cardiac output can be obtained, the technique is best used for trend analysis rather than for making absolute measurements. Oesophageal Doppler probes can be inserted quickly and easily and are particularly valuable for perioperative optimization of the circulating volume and cardiac performance in the unconscious patient. They are contraindicated in patients with oropharyngeal/oesophageal pathology.
Echocardiography
Echocardiography is being used increasingly often to provide immediate diagnostic information about cardiac structure and function (myocardial contractility, ventricular filling) in the critically ill patient. Transoesophageal echocardiography (TOE) is preferred because of its superior image clarity (Fig. 16.15).
Disturbances of acid–base balance
The physiology of acid–base control is discussed on page 660. Acid–base disturbances can be described in relation to the diagram illustrated in Figure 13.13, p. 663 (which shows PaCO2 plotted against arterial [H+]).
Blood gas and acid–base values (normal ranges) are shown in Table 16.2. (For blood gas analysis, see p. 891.)
Table 16.2 Arterial blood gas and acid–base values (normal ranges)
|
H+ |
35–45 nmol/L |
pH 7.35–7.45 |
|
PO2 (breathing room air) |
10.6–13.3 kPa |
(80–100 mmHg) |
|
PCO2 |
4.8–6.1 kPa |
(36–46 mmHg) |
|
Base deficit |
±2.5 |
|
|
Plasma HCO3– |
22–26 mmol/L |
|
|
O2 saturation |
95–100% |
|
Respiratory acidosis. This is caused by retention of carbon dioxide. The PaCO2 and [H+] rise. A chronically raised PaCO2 is compensated by renal retention of bicarbonate, and the [H+] returns towards normal. A constant arterial bicarbonate concentration is then usually established within 2–5 days. This represents a primary respiratory acidosis with a compensatory metabolic alkalosis (see p. 666). Common causes of respiratory acidosis include ventilatory failure and COPD (type II respiratory failure where there is a high PaCO2 and a low PaO2, see p. 814).
Respiratory alkalosis. In this case, the reverse occurs and there is a fall in PaCO2 and [H+], often with a small reduction in bicarbonate concentration. If hypocarbia persists, some degree of renal compensation may occur, producing a metabolic acidosis, although in practice this is unusual. A respiratory alkalosis may be produced, intentionally or unintentionally, when patients are mechanically ventilated; it may also be seen with hypoxaemic (type I) respiratory failure (see Ch. 15, p. 817), spontaneous hyperventilation and in those living at high altitudes.
Metabolic acidosis (p. 664). This may be due to excessive acid production, most commonly lactate and H+ (lactic acidosis) as a consequence of anaerobic metabolism during an episode of shock or following cardiac arrest. A metabolic acidosis may also develop in chronic renal failure or in diabetic ketoacidosis. It can also follow the loss of bicarbonate from the gut or from the kidney in renal tubular acidosis. Respiratory compensation for a metabolic acidosis is usually slightly delayed because the blood–brain barrier initially prevents the respiratory centre from sensing the increased blood [H+]. Following this short delay, however, the patient hyperventilates and ‘blows off’ carbon dioxide to produce a compensatory respiratory alkalosis. There is a limit to this respiratory compensation, since in practice values for PaCO2 less than about 1.4 kPa (11 mmHg) are rarely achieved. Spontaneous respiratory compensation cannot occur if the patient’s ventilation is controlled or if the respiratory centre is depressed, for example by drugs or head injury.
Shock, sepsis and acute disturbances of haemodynamic function
Causes of shock
Abnormalities of tissue perfusion can result from:
The causes of shock are shown in Table 16.3. Often shock can result from a combination of these factors (e.g. in sepsis, distributive shock is frequently complicated by hypovolaemia and myocardial depression).
| Hypovolaemic | Obstructive |
|---|---|
Pathophysiology
The sympatho-adrenal response to shock (Fig. 16.16)
Reduction in perfusion of the renal cortex stimulates the juxtaglomerular apparatus to release renin. This converts angiotensinogen to angiotensin I, which in turn is converted in the lungs and by the vascular endothelium to the potent vasoconstrictor angiotensin II. Angiotensin II also stimulates secretion of aldosterone by the adrenal cortex, causing sodium and water retention (p. 566). This helps to restore the circulating volume (see p. 639).
Neuroendocrine response
There is release of pituitary hormones such as adrenocorticotrophic hormone (ACTH), vasopressin (antidiuretic hormone, ADH) and endogenous opioid peptides. (In septic shock there may be a relative deficiency of vasopressin.)
There is release of cortisol, which causes fluid retention and antagonizes insulin.
There is release of glucagon, which raises the blood sugar level.
Release of pro- and anti-inflammatory mediators
Severe infection (often with bacteraemia or endotoxaemia), the presence of large areas of damaged tissue (e.g. following trauma or extensive surgery), hypoxia or prolonged/repeated episodes of hypoperfusion can trigger an exaggerated inflammatory response with systemic activation of leucocytes and release of a variety of potentially damaging ‘mediators’ (see also Ch. 3
[/not-level-membership-for-internal-medicine-category]
