Craniosynostosis, Selected Craniofacial Syndromes, and Other Abnormalities of the Skull
The basic clinical and radiologic features of craniosynostosis result either from lack of sutural formation or from premature fusion of contiguous portions of calvarial bones across the membranous sutures between them. The prevalence of premature sutural closures is displayed in e-Table 20-1.1 Normal sutures permit growth of the skull in a direction perpendicular to their long axes. With normal endocranial stimulus to growth, cessation of growth in one suture is compensated by increased growth in others, with resulting craniofacial deformity (e-Table 20-2; Figs. 20-1 through 20-4).
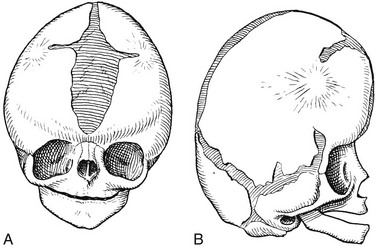
Figure 20-2 Premature synostosis of both coronal sutures.
Schematic representation as seen in Apert syndrome. The suture is obliterated except for short, open terminal segments and open segments near the sagittal suture and anterior fontanel. The metopic, sagittal, lambdoid, and temporoparietal sutures all are widely open. The calvaria is shortened in the anteroposterior direction and elongated in the craniocaudal plane. A, Frontal aspect. B, Lateral aspect.
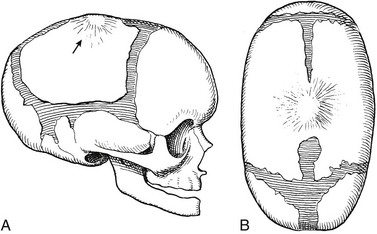
Figure 20-3 Schematic drawings of premature synostosis of the sagittal suture.
A, Lateral aspect. B, Superior aspect.
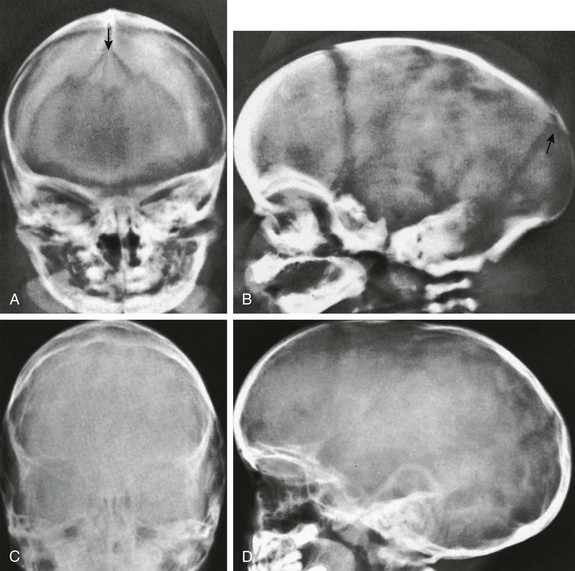
Figure 20-4 Premature synostosis of the sagittal suture with elongation of the calvaria in the occipital-frontal direction, a decrease in the transverse axis of the calvaria, and relative (to length) decrease in vertical height.
The sagittal suture is ridged externally. A and B, Frontal (A) and lateral (B) projections in a 2-day-old neonate. A sutural bone is present in the region of the posterior fontanel (arrows). The coronal, lambdoid, and squamosal sutures are normally wide, as is the squamocondyloid synchondrosis at the base of the occipital squamosa. C and D, On frontal (C) and lateral (D) projections, similar changes are evident in the more mature skull of an 11-month-old girl.
e-Table 20-1
Prevalence of Individual Premature Suture Closure per 1 Million Live Births
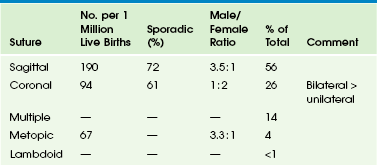
Modified from Cohen Jr MM. Epidemiology of craniosynostosis. In: Cohen Jr MM, MacLean RE, eds. Craniosynostosis. New York: Oxford University Press; 2000:113.
e-Table 20-2
Calvarial Configurations in Primary Craniosynostosis
| Suture | Calvarial Configuration | Descriptive Terms |
| Sagittal | Long, narrow head | Scaphocephaly or dolichocephaly |
| Bilateral coronal | Short, wide head; hypertelorism; proptosis; small anterior fossa | Brachycephaly or bradycephaly |
| Metopic | Frontal wedging or keel-shaped head | Trigonocephaly |
| Bilateral lambdoid | Shallow posterior fossa, prominent bregma | Turricephaly |
| Unilateral coronal | Unilateral frontal flattening, uptilting of orbit and tilting of nasal septum | Plagiocephaly |
| Unilateral lambdoid | Unilateral posterior flattening | Plagiocephaly |
| All sutures | Small, round head | Microcephaly |
From Vannier MW. Radiologic evaluation of craniosynostosis. In: Cohen Jr MM, MacLean RE, eds. Craniosynostosis. New York: Oxford University Press; 2000:148.
The deformity of the shape of the head is present before the bony sutural changes are seen. Only a portion of the bony suture needs to be closed to have craniosynostosis (e-Fig. 20-5). The suture-associated dura mater is responsible for determining the development of the cranial suture. The dura supplies osteoinductive growth factors (e.g., transforming growth factor-β or fibroblast growth factor-2) and cellular elements. Abnormal head shape secondary to abnormal suture development can be diagnosed in utero at 13 weeks’ gestational age.2,3 Craniosynostosis is associated with genetic abnormalities (e-Box 20-1) and is a secondary finding in many systemic disorders (e-Box 20-2).4
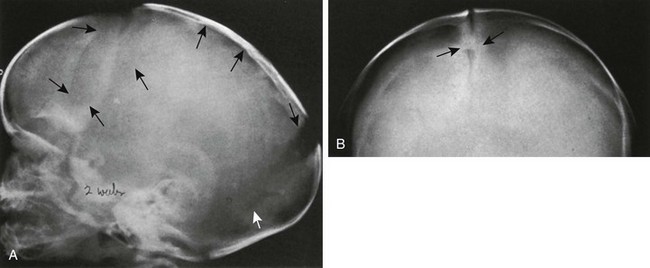
e-Figure 20-5 Severe craniosynostosis of the scaphocephalic type caused by premature synostosis of only a short segment of the sagittal suture of a 2-week-old infant.
A, In lateral projection, deformity resulting from the craniostenosis is clearly seen, but the sagittal suture itself is invisible (superior black arrows). Coronal (black arrows) and lambdoid sutures (white arrow) are widened. B, In frontal projection, the sagittal suture is visible in its entirety and is open except for a short segment (arrows) only a few millimeters long.
Specific head shapes are associated with sutural synostoses (e-Fig. 20-6). The normal head has an egg shape, being widest in the parietal area posterior to the ears with a narrower, gently rounded forehead (Fig. 20-7).
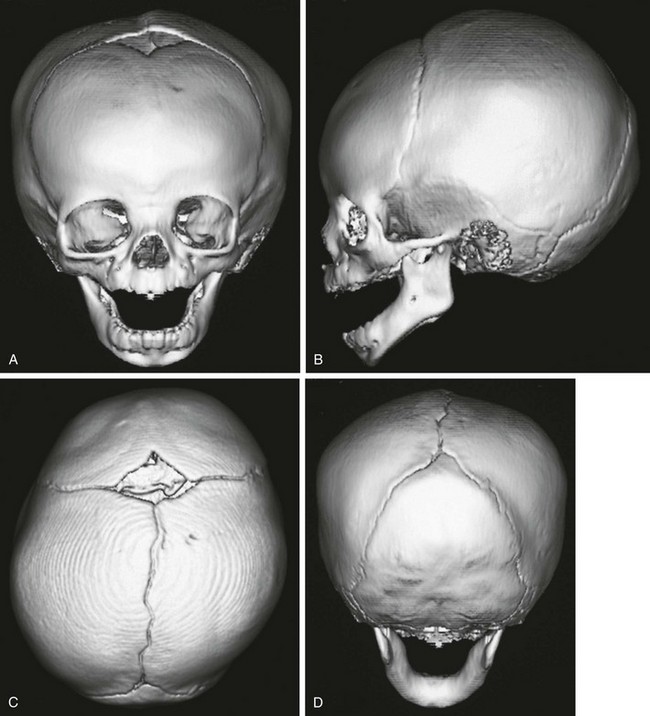
Figure 20-7 A normal three-dimensional computed tomography scan of the head in a 7-month-old infant.
A, Frontal view clearly shows the anterior fontanel and coronal sutures. The metopic suture has closed. B, Lateral view shows the lambdoid and squamosal sutures and the normal coronal sutures. C, Bird’s eye view reveals the anterior fontanel and the coronal, sagittal, and lambdoid sutures. D, Posterior view shows the posterior sagittal suture and the lambdoid sutures. The normal head is egg shaped, widest at the biparietal area (C).
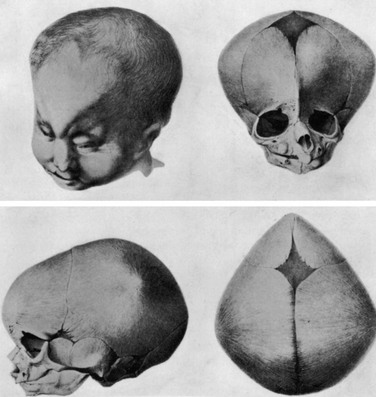
e-Figure 20-6 Trigonocephaly in a newborn.
A photograph of the head and face and three views of the skull—frontal, lateral, and superior. Note the ridge on the forehead. It appears similar to a keel of a boat. (From Welcker H. Untersuchungen über Wachstum und Bau des menschlichen Schädels. Leipzig: W. Engelmann; 1862.)
Sagittal synosotosis is characterized by a long and narrow head (see Figs. 20-3 and 20-4). The back is usually narrower than the front, and anterior or posterior bossing may exist. Metopic synostosis (trigonocephaly) results in a triangular shape of the whole forehead (not just a rounded forehead with a ridge superimposed) (Fig. 20-8; see e-Fig. 20-6).
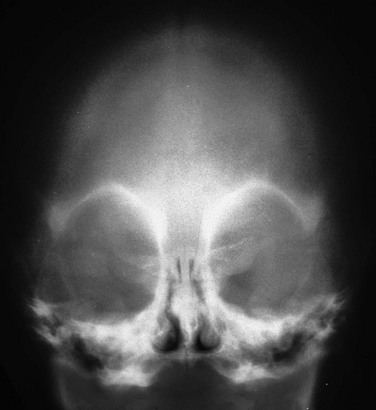
Figure 20-8 Trigonocephaly.
Frontal projection shows the characteristic orbital hypotelorism and narrowing of the forehead. The metopic suture is invariably closed.
Unicoronal synostosis results in flattening of the ipsilateral forehead, flattening of the ipsilateral occipital area, the “harlequin eye” (the sphenoid is drawn up toward the closed suture and is thickened), ipsilateral temporal bulging and cheek protrusion, contralateral forehead bossing, and deviation of the nose away from the synostosed side (Fig. 20-9). Features that distinguish unicoronal synostosis from anterior deformational plagiocephaly are listed in e-Table 20-3.
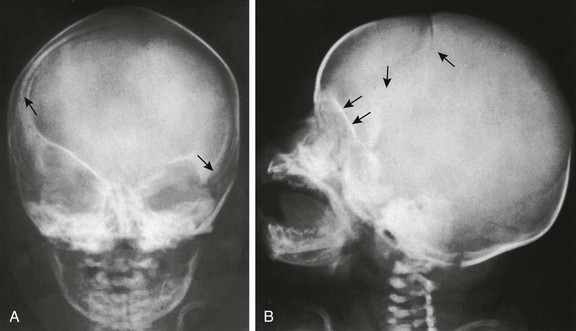
Figure 20-9 Premature synostosis of the caudal segment of the right limb of the coronal suture in a 3-week-old infant.
A, In frontal projection, arrows are directed at the caudal ends of the right and left limbs of the coronal suture. The roof of the right orbit is lifted into a more oblique position, as is the right wing of the sphenoid. B, In lateral projection, the right limb of the coronal suture stops abruptly a few centimeters below the anterior fontanel. The lifting of the right orbital roof also is well seen (two lower arrows).
e-Table 20-3
Features that Differentiate Unilateral Coronal Synostosis from Anterior Deformational Plagiocephaly
| Features | Synostotic | Deformational |
| Ipsilateral superior orbital rim | Up | Down |
| Ipsilateral ear | Anterior, high | Posterior, low |
| Nasal root | Ipsilateral | Midline |
| Ipsilateral malar eminence | Forward | Backward |
| Chin deviation | Contralateral | Ipsilateral |
| Ipsilateral palpebral fissure | Wide, low | Narrow, high |
| Anterior fontanel deviation | Contralateral | None |
Modified from Cohen Jr MM, MacLean RE. Anatomic, genetic, nasologic, diagnostic, and psychosocial considerations. In: Cohen Jr MM, MacLean RE, eds. Craniosynostosis. New York: Oxford University Press; 2000:126.
Bicoronal synostosis causes a brachycephalic head (i.e., the head is wide and short). The supraorbital rims and forehead are recessed with bitemporal and upper forehead bulging (Fig. 20-10).
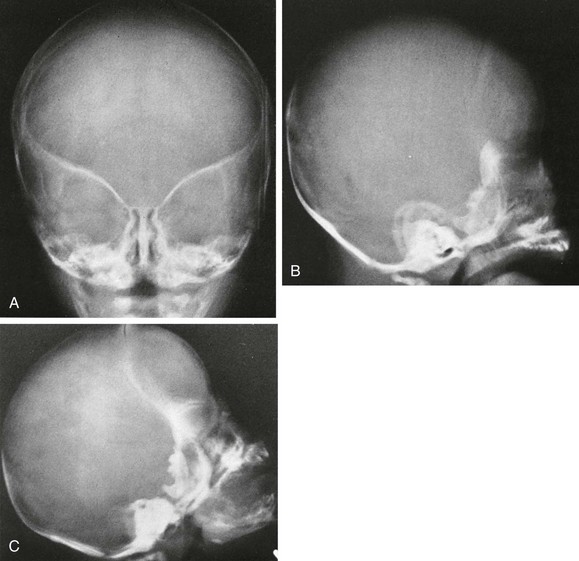
Figure 20-10 Bilateral coronal synostosis.
A, Frontal view shows the elevated orbital roofs. B, The height of the skull is increased from caudad to cephalad and decreased in the anteroposterior dimension. The coronal suture is not seen. C, Other lateral projection showing the absence of a complete coronal suture. The head is tall (towering) and short in the anteroposterior direction.
Lambdoid synostosis results in ipsilateral occipital flattening with compensatory bulging at the superior and inferior axes where the suture should have been (e-Fig. 20-11). The features differentiating unilateral lambdoid synostosis from posterior deformational plagiocephaly are listed in e-Table 20-4.
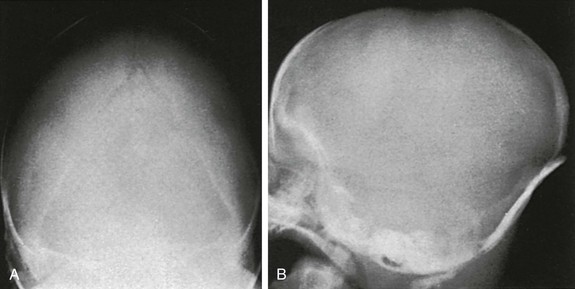
e-Figure 20-11 Premature closure and ridging of the caudal two thirds of both lambdoid sutures and flattening of the occipital squamosa in a 1-month-old infant.
The uppermost segment of the lambdoid suture is still open, and a small sutural bone is visible at the lambda itself. The sides of the occipital squamosa are straight, rather than convex, and the squamosa is narrower than is normal. A and B, Anteroposterior (A) and left lateral (B) projections.
e-Table 20-4
| Features | Synostotic | Deformational |
| Contralateral posterior bossing | Parieto-occipital | Occipital |
| Forehead | Contralateral frontal bossing | Ipsilateral frontal bossing |
| Ipsilateral occipitomastoid bossing | Present | Absent |
| Ipsilateral ear | Anterior, anteroinferior, posterior, or symmetric | Anterior |
| Skull base and face | Ipsilateral inferior tilt | Normal* |
| Head shape, vertex view | Trapezoid-shaped or parallelogram-shaped depending on severity | Parallelogram |
| Head shape, posterior view | Parallelogram | Normal |
| Lambdoid suture | Unilateral synostosis† | Patent suture |
†Bulges at inferior and superior axes of location where suture should be.
Modified from Cohen Jr MM, MacLean RE. Anatomic, genetic, nasologic, diagnostic, and psychosocial considerations. In: Cohen Jr MM, MacLean RE, eds. Craniosynostosis. New York: Oxford University Press; 2000:127.
Synostosis of multiple sutures occurs in 14%, and the resultant head shape depends on which sutures are closed.1 The kleeblattschädel (“cloverleaf skull”) anomaly may result when all sutures except the squamosal are closed, resulting in severe temporal and vertex bulging with exophthalmos (Fig. 20-12). Unusual minor synostosis may exist, causing abnormal skull appearance.5–8
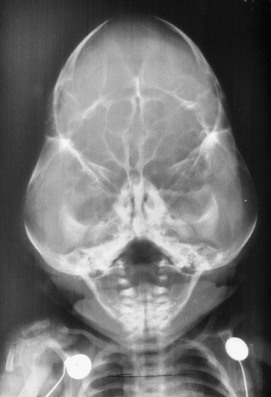
Figure 20-12 Kleeblattschädel (cloverleaf skull) anomaly.
Multisuture closure has occurred, and a bizarre configuration of the face and head with bulging of the temporal region is seen.
Microcrania, or a small neurocranium, may result when all sutures are closed. This usually occurs with failure of brain growth. Rarely, it may occur without failure of brain growth, and the child may develop increased intracranial pressure (Fig. 20-13).
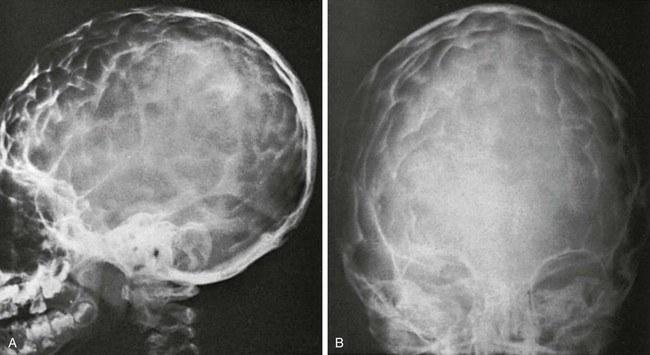
Figure 20-13 Radiographic findings in the microcephalic type of craniostenosis in a 7-year-old child.
A and B, Lateral (A) and frontal (B) projections. All sutures of the cranium are obliterated, and the skull is shortened in all its diameters. The long-standing increased intracranial pressure is indicated by the heavy convolutional markings. Detailed anatomy of the sellae is not seen.
Deformities Mimicking Craniosynostosis
Cranial deformities mimicking synostosis may result from static forces such as intrauterine crowding or prolonged recumbency. The term plagiocephaly refers to any flattening of the calvarium without denoting an etiology and is preceded by terms that describe the location and side (e.g., right posterior plagiocephaly). Postnatal deformational plagiocephaly may affect primarily the forehead or the occipital area. Since the 1993 recommendation by the American Academy of Pediatrics to put infants to sleep on their backs, a marked increase is seen in occipital deformational plagiocephaly (e-Fig. 20-14). The ipsilateral occipital area is flattened, with flattening of the contralateral forehead, malposition of the ipsilateral external ear, protrusion of the ipsilateral cheek, and compensatory bulging of the ipsilateral forehead and contralateral occipital area. Viewed from above, the head has a parallelogram shape (Fig. 20-15).
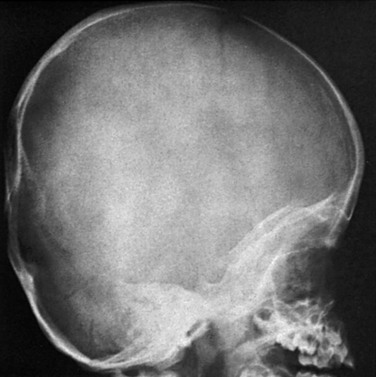
Perisutural sclerosis of the lambdoid may be seen on a plain radiograph; however, the suture is open. The concept of a “sticky lambdoid suture” is no longer considered valid.9,10 Plagiocephaly also may occur with bony, muscular, or ocular torticollis.
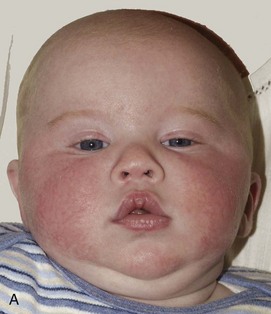
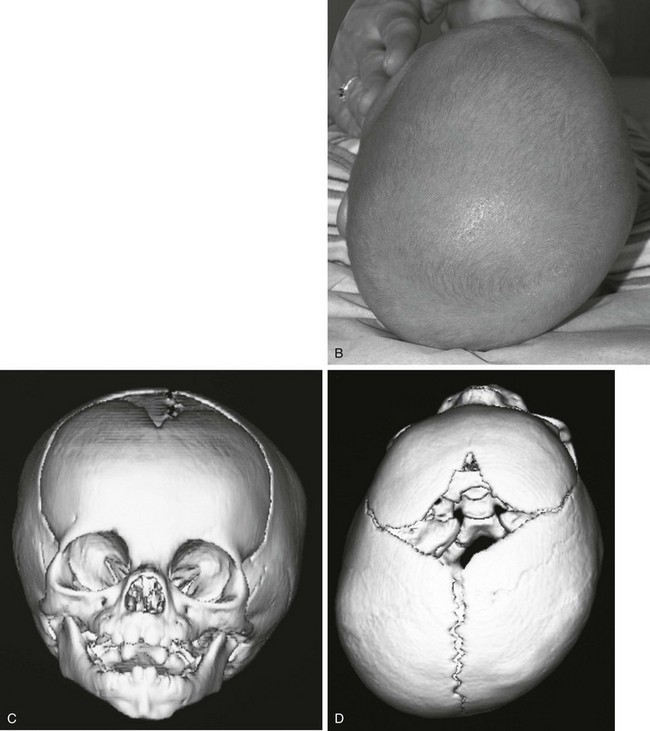
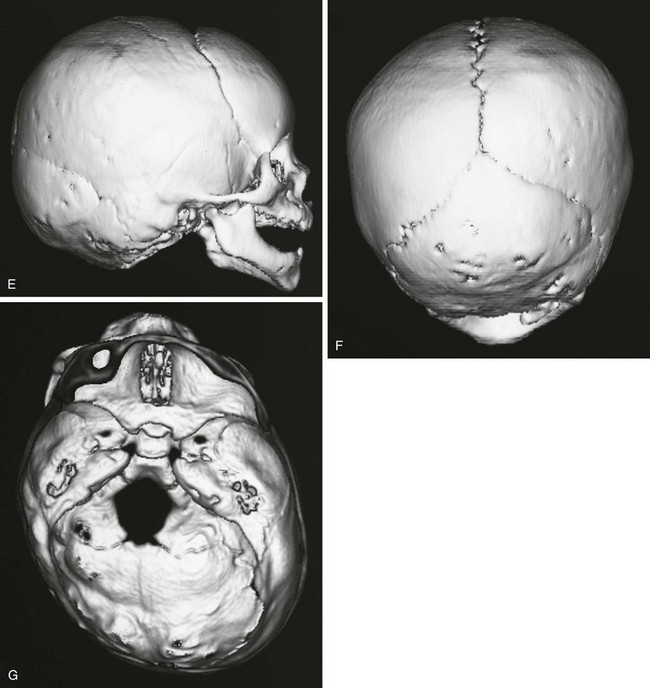
Figure 20-15 Deformational plagiocephaly.
A and B, Frontal view and bird’s eye view of an infant reveal the parallelogram shape of the skull and face. C and D, Frontal view and bird’s eye view three-dimensional computed tomography reveals the abnormal-shaped head mimicking the clinical picture. E to G, Lateral, posterior, and basal views show all of the sutures are open despite the abnormal head shape. Right occipital flattening and left frontal flattening with compensatory right frontal and left occipital bulging are seen. This gives the parallelogram shape. No skull base angulation is evident.
In deformational plagiocephaly, the skull base (i.e., aligned from the crista galli through the foramen magnum) remains straight, with less than 7 degrees angulation, whereas in unilateral coronal or lambdoid synostosis, the skull base curves (Figs. 20-15 through 20-17). Pseudoscaphocephaly occurs when premature infants lie on the sides of their heads in the neonatal intensive care unit. Although the head is long and narrow, the sagittal suture is open, and the widest part of the skull is in the biparietal area.
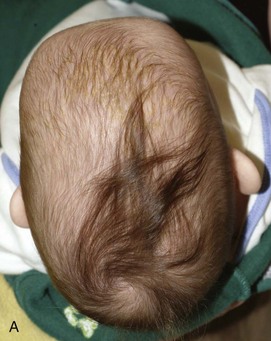
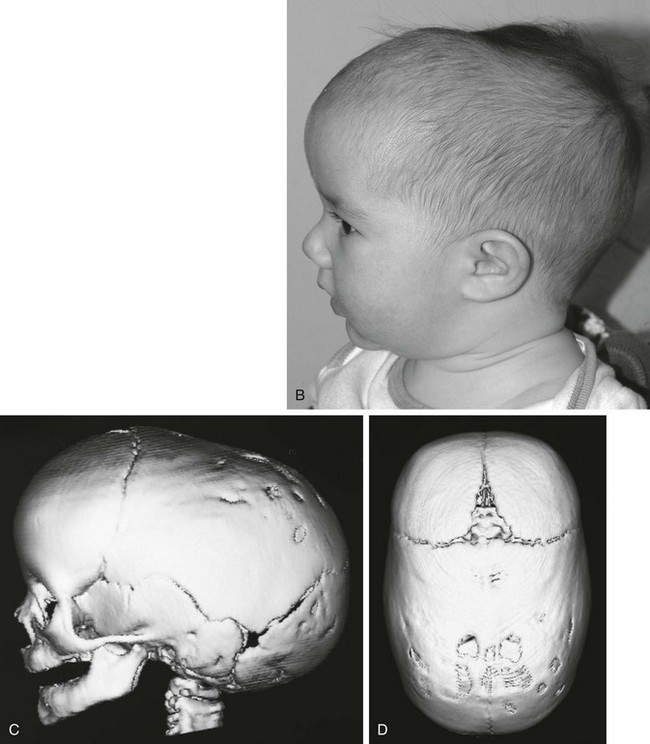
Figure 20-16 Sagittal synostosis.
A and B, Bird’s eye view and lateral view of an infant’s head reveal a narrow, elongated calvaria from front to back. C and D, Lateral view and bird’s eye view three-dimensional computed tomography shows the elongated (anteroposterior), narrow calvaria with no sagittal suture present and frontal and occipital bulging.
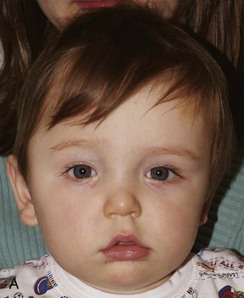
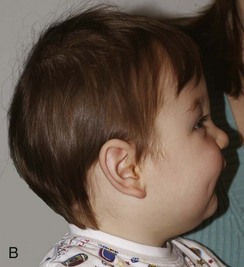
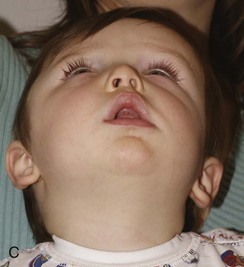
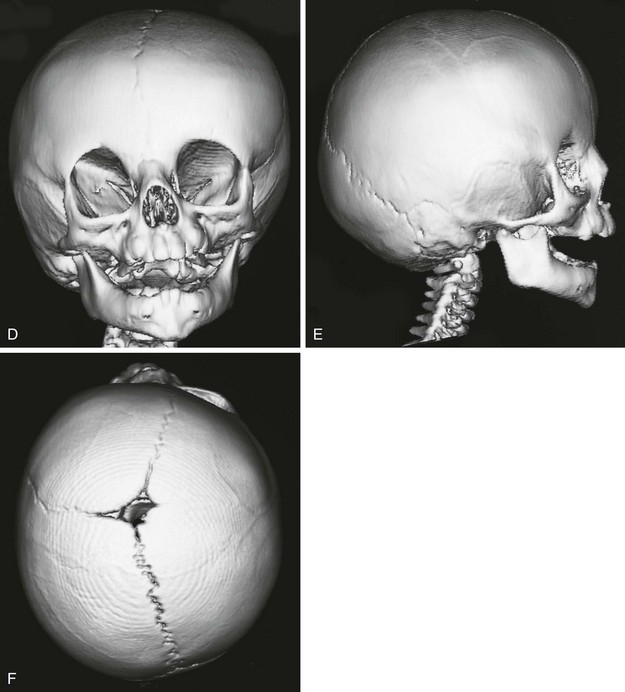
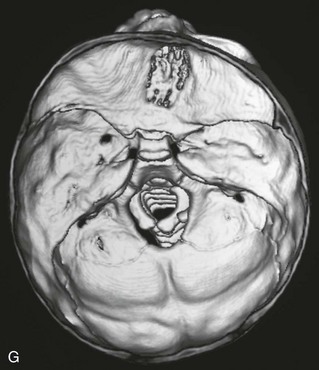
Figure 20-17 Unilateral right coronal synostosis.
A to C, Three views of an infant show flattening of the right frontal and occipital regions and some recession of the right orbit. Note the elongated shape of the head from caudad to cephalad and shortening in the anteroposterior dimension. Unilateral right coronal synostosis. D to G, Three-dimensional computed tomography shows the absence of the right coronal suture, elevation of the sphenoid seen through the right orbit, right frontal and occipital flattening with a compensatory ipsilateral and temporal and contralateral frontal bulging on the left side, angulation of skull base toward side of coronal synostoses with decreased intracranial volume on affected side.
Radiographic Findings
Radiographic findings reflect the deformities of the cranium observed clinically. The initial examination is a skull series consisting of anteroposterior or Caldwell, Towne, and both lateral projections. The shape of the head is ascertained. The anterior fontanel should be visible at least until 7 months of age. The sagittal, coronal, and lambdoid sutures are all identified. The metopic suture closes any time from before birth to after 3 months of age.11 As many as 10% may remain open into adulthood. Closure of only a short segment of a suture is as effective in preventing separation of the opposing bones as total obliteration (see e-Fig. 20-5). The key findings to the diagnosis of craniosynostosis on skull series are (1) abnormal head shape and (2) obliteration of a portion of a suture.
According to Jane and Persing, cranial restructuring techniques have focused on (1) release of sutural synostosis, (2) remodeling of cranial bone, (3) active reduction of an abnormally long dimension of the skull, and (4) active expansion of abnormally narrow areas.12
In most instances, three-dimensional computed tomography (CT) is required in planning for cranial restructuring. It is important to keep the radiation dose as low as reasonably achievable. This is quite easy in examination for bone changes and is accomplished by lowering both the kilovolt (kV) and milliampere (mA). Exams performed at 40 mA and 100 to 120 kV at 1 second with a slice thickness of 1.25 millimeters (mm) give a CT dose index “dose” of approximately 5 mGy (500 millirads). Images are reconfigured to 0.625 mm for reformatted images and three-dimensional reconstruction (Figs. 20-16 through 20-20 and e-Fig. 20-21). In syndromic children, magnetic resonance imaging (MRI) of the brain may be performed for developmental anomalies.

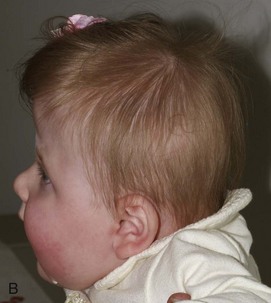
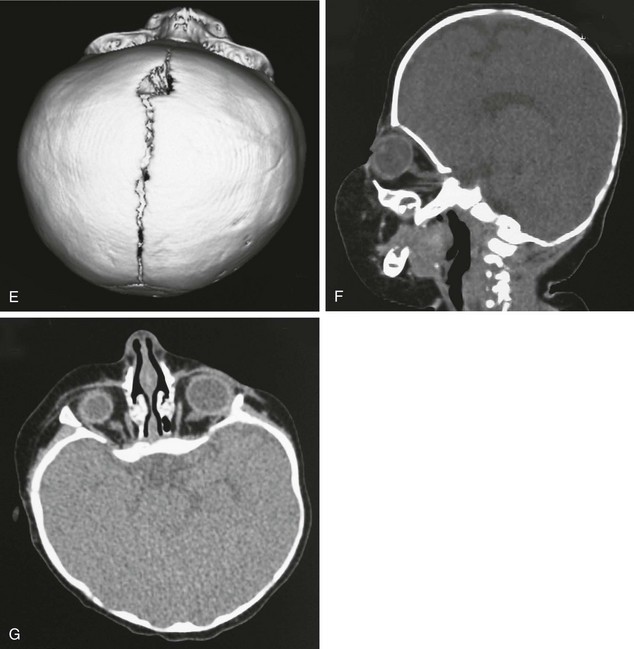
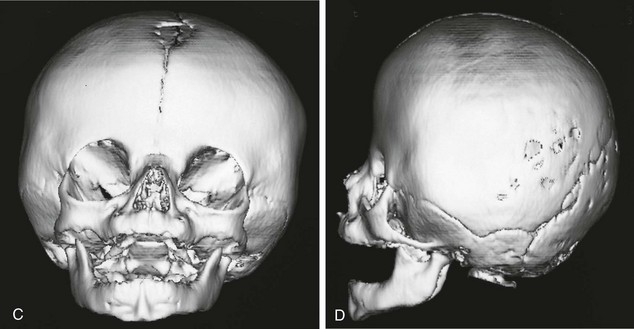
Figure 20-18 Bilateral coronal synostosis.
A and B, Frontal and lateral views of a child show the towering calvaria, with some temporal bulging and orbital recession. C to G, Computed tomography and three-dimensional reconstructed images of the child reveal the absence of the coronal sutures and the increased dimension from inferior to superior of the skull (towering skull). Decreased anteroposterior dimension of the skull, temporal bulging, and orbital recession are seen.
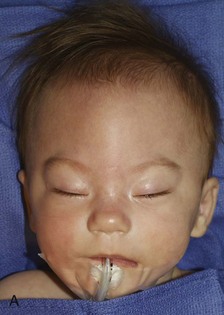
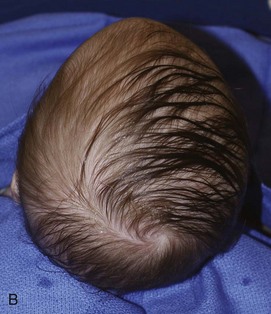
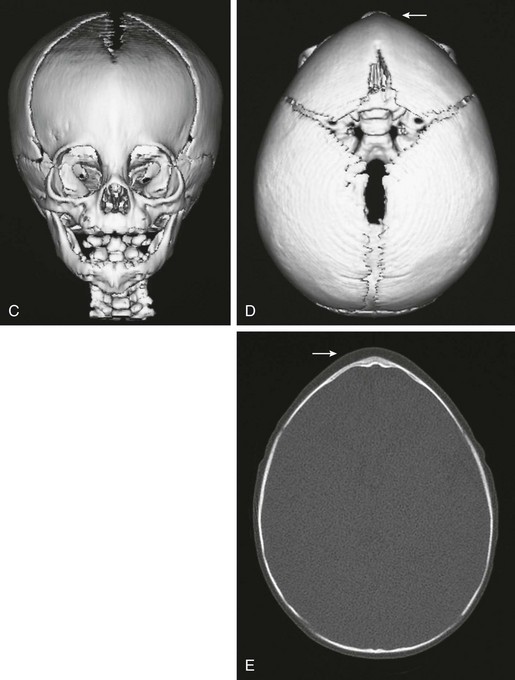
Figure 20-19 Metopic synostosis.
A and B, View of a patient showing a keel-shaped forehead. It is not round, but rather triangular, and some hypotelorism is present. C to E, Triangular or keel-shaped forehead and the closure of the metopic suture. This diagnosis cannot be made on just closure of the suture, but the patient must have the appropriate configuration of the forehead (arrow) (i.e., triangular shape, not rounded).
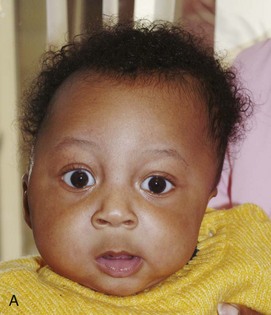
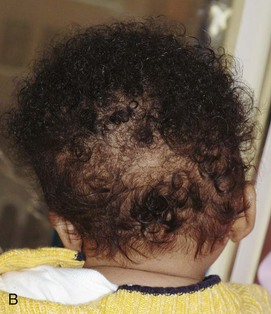
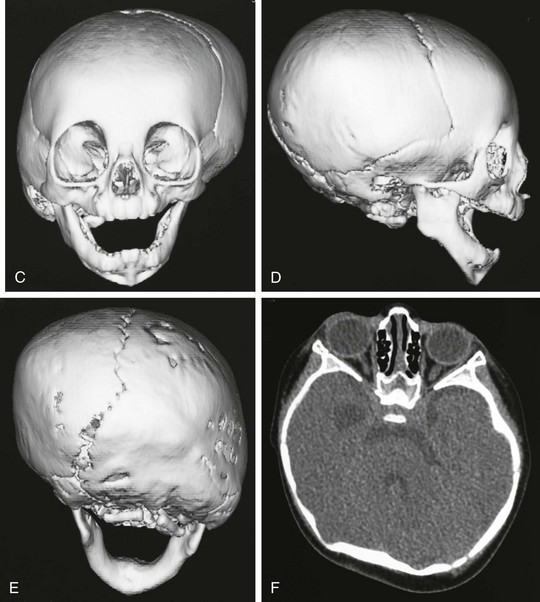
Figure 20-20 Unilateral right lambdoid synostosis.
A and B, Note the asymmetry of the infant’s head, with bulging of the inferior occipital region on the right. Compensatory left parietal bulging is seen superiorly. C to F, Computed tomography and reconstructed three-dimensional images show the absence of the right lambdoid suture with compensatory bulging of the superior and inferior ends of the synostosal suture (i.e., left parietal and right occipital region). Skull-base angulation is evident.
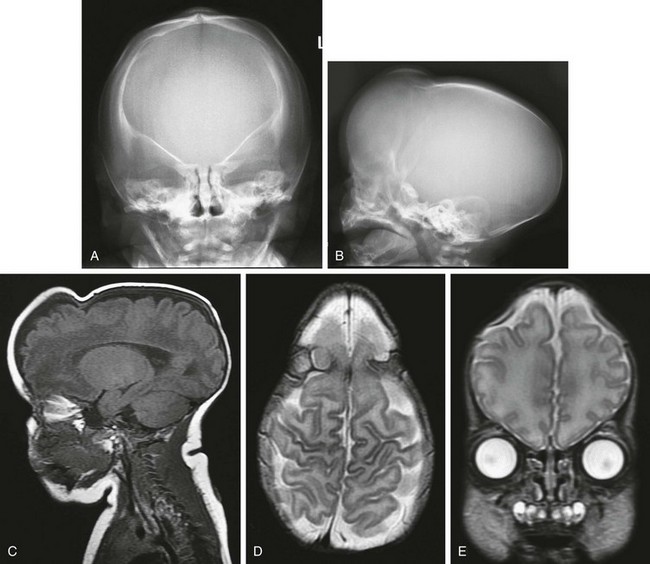
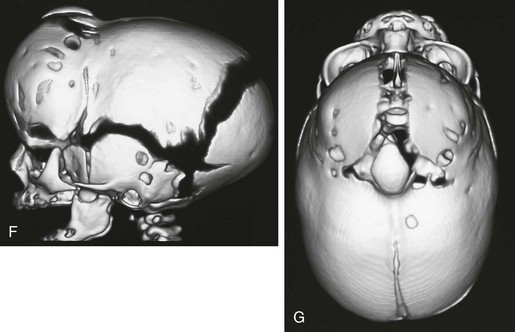
e-Figure 20-21 Multiple suture closure.
A and B, Frontal and lateral radiographs show elevated orbital ridge, upper coronal suture closure, and ridging and closure of the sagittal suture. C to E, Magnetic resonance imaging sequences—sagittal T1-weighted (C), axial T2-weighted (D), and coronal T2-weighted (E)—reveal normal brain, but severe deformity. F and G, Three-dimensional computed tomography shows the multiple (coronal and sagittal) suture closure.
Associated Abnormalities
Limb defects are the most common feature of syndromes associated with craniosynostosis, occurring in 84%.1 Syndactyly and polysyndactyly constitute 30% of the limb defects, and deficiencies account for 22%.
The best known acrocephalosyndactyly is Apert syndrome (acrocephalosyndactyly type I), in which usually bicoronal synostosis is associated with symmetric complex syndactyly of at least the second, third, and fourth digits, resulting in the “mitten-hand” and “sock-foot” appearance (e-Fig. 20-22). Mental retardation is present in varying degrees. Evaluation of craniofacial deformities may be assisted by CT (e-Fig. 20-23, Fig. 20-24, and e-Fig. 20-25).
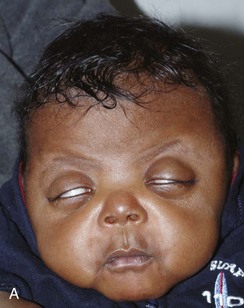
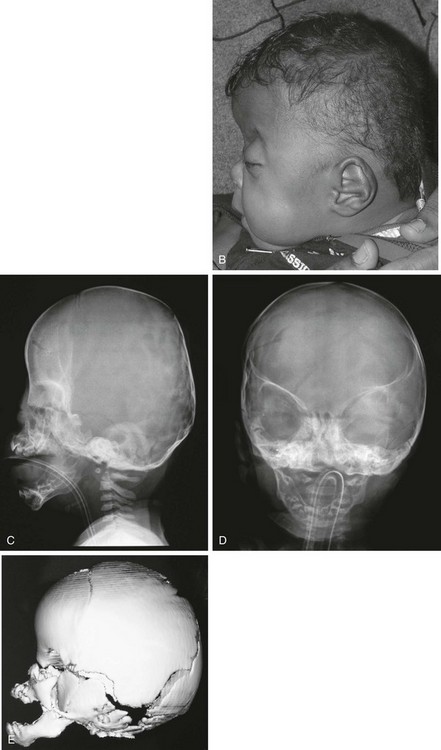
Figure 20-24 Apert syndrome.
A and B, View of the child in frontal and lateral projection. The child has a shortened anteroposterior diameter and increased inferior caudad-to-cephalad dimension consistent with bilateral coronal synostosis. Exophthalmos, hypertelorism, and maxillary retrusion are present. C and D, A radiograph of the same patient shows the bilateral coronal synostosis and the expected contour of the calvaria. E, A three-dimensional computed tomography lateral view confirms closure of inferior coronal suture and maxillary retrusion.
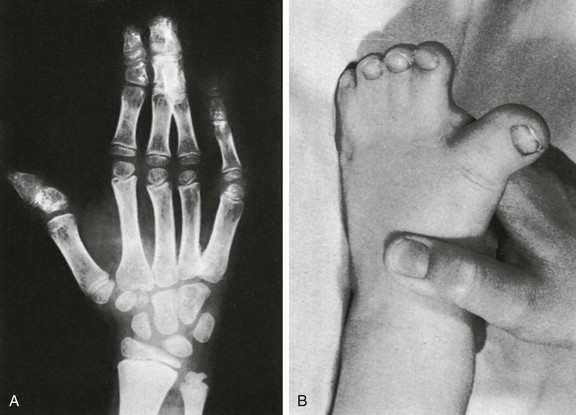
e-Figure 20-22 Acrocephalosyndactyly, type I (Apert syndrome).
A, The middle three digits of the hand are affected by bony and soft tissue syndactyly. B, Typical appearance of a “sock-foot” deformity. (From Kozlowski K, Maroteaux P, Silverman F, et al. Classification of osseous dysplasia. Ann Radiol. 1969;12:965-1007.)
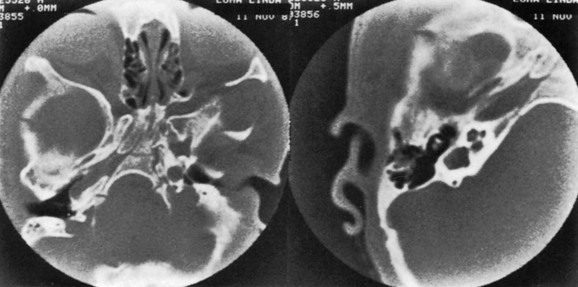
e-Figure 20-23 A computed tomography scan of bizarre deformity of the skull base caused by asymmetric craniosynostosis in a patient with Apert syndrome.
Left, Image through the lower portion of the body of the sphenoid, the spheno-occipital synchondrosis, and the basiocciput shows basilar asymmetry and shows a partial cleft in the sphenoid body. Right, A magnified view of the right inner ear shows dysplasia with a large cavity containing the vestibule and semicircular canals. The ossicles are unremarkable. (Courtesy Anton N. Hasso, MD.)
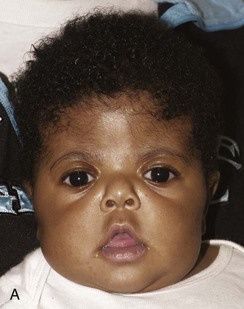
e-Figure 20-25 Apert syndrome.
A and B, This child also has a shortened anteroposterior dimension of the calvaria and increased caudad-to-cephalad dimension consistent with bilateral coronal synostosis. C and D, Three-dimensional computed tomography shows closure of the coronal synostosis.
Acrocephalosyndactyly types II, III, and IV are known as Apert-Crouzon disease, Saethre-Chotzen syndrome, and Waardenburg syndrome; they involve varying degrees of facial abnormality and syndactylies in patterns generally repetitive for each type. Acrocephalosyndactyly type V, Pfeiffer syndrome, has only soft tissue syndactyly, which is not marked, but the thumbs and great toes are deformed and broad. All forms are transmitted by dominant inheritance (Fig. 20-26).
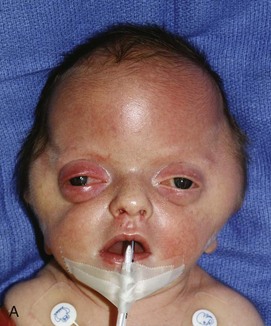
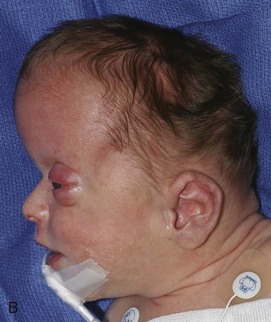
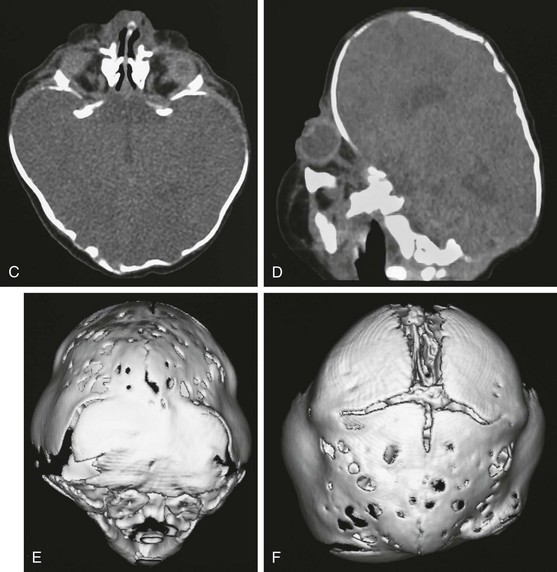
Figure 20-26 Pfeiffer syndrome.
A and B, Frontal and lateral views of the child reflect the multisutural synostosis (bilateral coronal, bilateral lambdoid, and sagittal synostosis) with the resultant cloverleaf configuration. C and D, Computed tomography with bone windows shows the temporal bulging, bony fenestrations, maxillary retrusion, and exophthalmos. The characteristic deformity of the calvaria is noted. E and F, Posterior view (E) and vertex view (F), three-dimensional computed tomography shows multiple suture closures (coronal, both lambdoid and sagittal sutures).
In Crouzon syndrome, the cardinal elements originally included (1) brachycephaly, (2) facial dysostosis with a hooked parrot nose and small maxilla with class III malocclusion, (3) bilateral exophthalmos, and (4) genetic transmission and familial incidence. It usually does not result in mental retardation (e-Fig. 20-27 and Fig. 20-28).
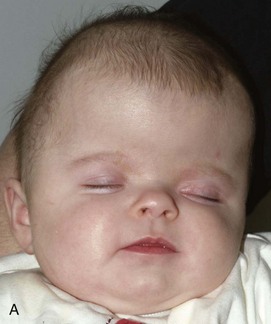
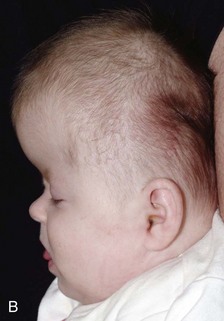
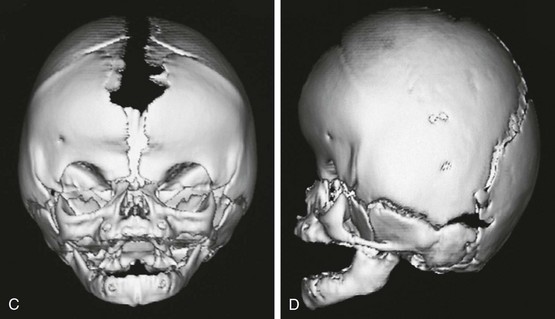
Figure 20-28 Crouzon syndrome.
A and B, Frontal and lateral views of an infant reveal a head shape consistent with bilateral coronal synostosis. The dimension is increased from caudad to cranial and shortened in the anteroposterior dimension. C and D, Three-dimensional computed tomography reveals the bilateral coronal synostosis with the characteristic findings.
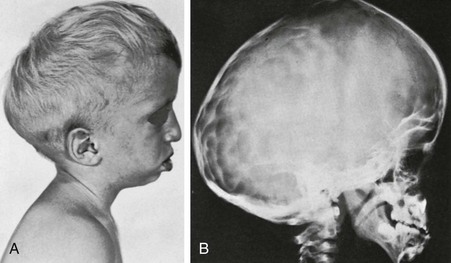
e-Figure 20-27 Crouzon disease (craniofacial dysostosis).
A, The lateral photograph shows the large calvaria and cranial cavity elongated ventrodorsally. The face is small, and the beaked nose is not seen completely in this view. Marked bilateral exophthalmos is also present. B, In the lateral radiograph, the frontal squamosa is large, and the edges of the frontal and parietal bones bulge externally at the anterior fontanel. Convolutional markings are heavy in the occipital squamosa. The facial segment is relatively and absolutely small, with a small maxilla and relatively larger mandible; the latter has a markedly increased obtuse angle. The anterior cranial fossa is small and shallow compared with the middle and posterior fossae.
Kleeblattschädel (cloverleaf skull) results from closure of all sutures except the squamosal sutures, leading to severe temporal and vertex bulging with exophthalmos (see Fig. 20-12). Hydrocephalus develops in utero, deforming the very plastic skull into a superior portion related to the position of the dilated frontal lobes and bilateral inferolateral portions corresponding to the dilated temporal lobes. Most patients do not survive infancy (see Fig. 20-12). Kleeblattschädel may be found in thanatophoric dysplasia type II.
Craniofacial Syndromes
Goldenhar Syndrome and Hemifacial Microsomia
Goldenhar syndrome is part of the oculoauricular vertebral spectrum, which includes hemifacial microsomia.13,14 Most reported cases are sporadic. An increased incidence of Goldenhar syndrome is present in infants of mothers with diabetes. The phenotype has been reported in association with other conditions, including trisomy 18, and with maternal thalidomide, primidone, and retinoic acid use.
The hallmarks of Goldenhar syndrome are epibulbar dermoids, preauricular appendages, mandibular hypoplasia, microtia, and vertebral anomalies. Extreme variability of expression is characteristic of this anomaly. Most frequently, the orbital lesions, mandibular hypoplasia (Fig. 20-29), and microtia are unilateral and on the same side.15 Colobomas of the upper eyelid occur in 60% of patients and may be large, requiring immediate repair to prevent corneal ulceration. Deafness is common because of associated anomalies of the middle and inner ear.16 Vertebral anomalies occur in 60% of patients and are most often cervical; these anomalies include basilar invagination, occipitalization of the atlas, C1-2 instability, cervical synostosis, hemivertebra, butterfly vertebrae, scoliosis, kyphosis, and Sprengel deformities (e-Fig. 20-30).17 Verterbral anomalies below the cervical spine are found in only 10% of patients. The anomalies may be severe, however, and associated with costal malformations similar to Jarcho-Levin syndrome.
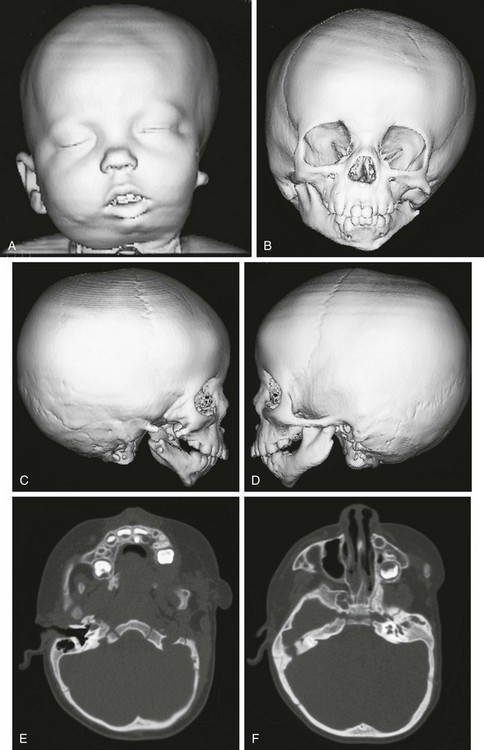
Figure 20-29 Goldenhar syndrome.
A, Three-dimensional surface rendering image shows left hemifacial microsomia and left ear microtia. B, Three-dimensional computed tomography reconstruction shows hypoplastic left mandible and zygomatic arch. C and D, Lateral three-dimensional image confirms asymmetry and left microsomia. E and F, Bone window computed tomography view at skull base reveals normal right external canal and temporal bone. The left face is hypoplastic. The left external auditory canal is absent.
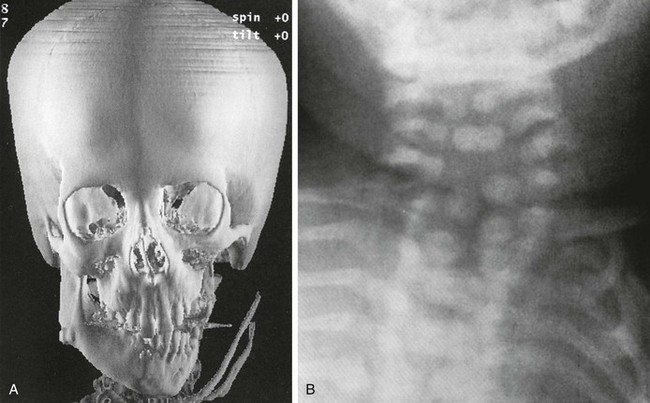
e-Figure 20-30 Goldenhar syndrome (hemifacial microsomia).
A, Marked left hemimandibular hypoplasia is present on this three-dimensional computed tomography image. B, The cervical spine radiograph shows formation and segmentation errors.
Hemifacial microsomia is a variable complex malformation of asymmetric hypoplasia of the face and ear with microsomia, unilateral microtia, and ipsilateral hypoplasis of the mandibular ramus and condyle. Hemifacial microsomia implies unilateral involvement, but the structures affected are bilateral and are only affected to different degrees. Primate and rodent studies have suggested that hemifacial microsomia may be caused by a hemorrhagic event involving the stapedial artery during early stages of craniofacial development.18 Patients with hemifacial microsomia resemble those with Goldenhar syndrome, but the presence of epibulbar dermoids, lipodermoids, auricular appendices, pretragal blind-ending fissures, and vertebral anomalies favors a diagnosis of Goldenhar syndrome.
Treacher Collins Syndrome
The condition has been diagnosed in utero by ultrasonography and is autosomal dominant, with variable penetrance and expression. The facial features of Treacher Collins syndrome are characteristic.19,20 Abnormalities are typically bilateral and symmetric: micrognathia, narrow face, depressed cheekbones, antimongoloid slant of eyes, malformed small ears, large downturned mouth, high-arched or cleft palate, and conductive hearing loss. The features are characterized by abnormalities in the derivatives of the first and second branchial arches.20 Mutations in the TCOF1 gene are associated with Treacher Collins syndrome, and numerous other associated mutations may be present.21 The features demonstrated on imaging are primarily facial, consisting of mandibular, zygomatic, maxillary, and supraorbital hypoplasia. The orbits are described as being egg shaped. The mandibular shape of a short body and ramus is characteristic and varies with the patient’s age. The mandibular condyle and coronoid process may be severely hypoplastic, flat, or even absent. Mandibular growth is severely affected (Fig. 20-31). Ear abnormalities include hypoplasia or absence of the middle ear. The ossicles and cochlea and vestibular apparatus may be severely malformed. Radiographic detection of zygomatic hypoplasia or aplasia is an important supporting finding for the diagnosis. Craniofacial three-dimensional CT for morphologic mapping has become invaluable for planning surgical treatment.22
Pierre Robin Sequence
Pierre Robin sequence, or Robin sequence, represents a nonrandom association of micrognathia, cleft palate, and glossoptosis.23 Pierre Robin sequence is causally heterogeneous and pathogenetically and phenotypically variable.24 Patients with Pierre Robin sequence are classified as isolated (most common), syndromic, or with associated anomalies. Numerous syndromes, including Stickler and velocardiofacial syndromes, are associated with Pierre Robin sequence.25 Respiratory compromise from mechanical and central nervous system causes is common. The mandible may be small or may be normal sized and retrognathic in position as a result of a large cranial base (Fig. 20-32). Typically, a reduction in cranial base and maxillary and mandibular lengths is seen.26 Mandibular deficiency is most pronounced in the body.
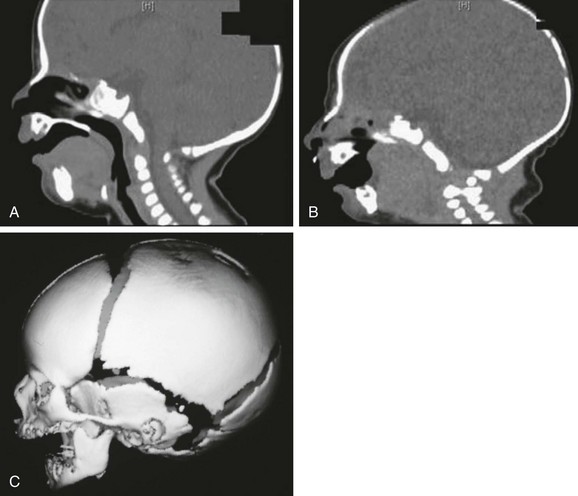
Figure 20-32 Pierre Robin sequence.
A and B, Sagittal reconstructed computed tomography scans show the large tongue obstructing the oropharynx as well as the retropositioned small mandible. C, Three-dimensional reconstruction shows the retropositioned small mandible.
Cardiovascular anomalies include septal defects and patent ductus arteriosus. Numerous skeletal anomalies include anomalies of the ribs, sternum, and spine, and limb reductions. Airway management is similar in both nonsyndromic and syndromic Pierre Robin sequence. The infant may be managed with positioning, nasal pharyngeal airway, tie-tongue adhesions, or mandibular distraction.27–30 Cine MRI or CT gives dynamic and three-dimensional information that may be useful in the evaluation of the airways of these patients.
Other Abnormalities of the Skull
Cranioschisis (Cranium Bifidum)
Cranioschisis, or cranium bifidum, usually occurs in the median sagittal plane, anteriorly or posteriorly (Figs. 20-32 and 20-33). Both sites are characterized by bony defects and may accompany meningocele or meningoencephalocele.31 Meningoceles are characterized by a hernia sac, which is covered with skin and contains only meninges and cerebrospinal fluid (e-Fig. 20-34). Meningoencephaloceles also contain brain (e-Fig. 20-35). Rarely, cranioschisis may be associated with only a scalp nodule, with or without intracranial communication. Occasionally, small cranial defects—for example, cranium bifidum occultum—through which no herniation occurs are encountered (e-Fig. 20-36). MRI is most effective for evaluation of the sac contents.
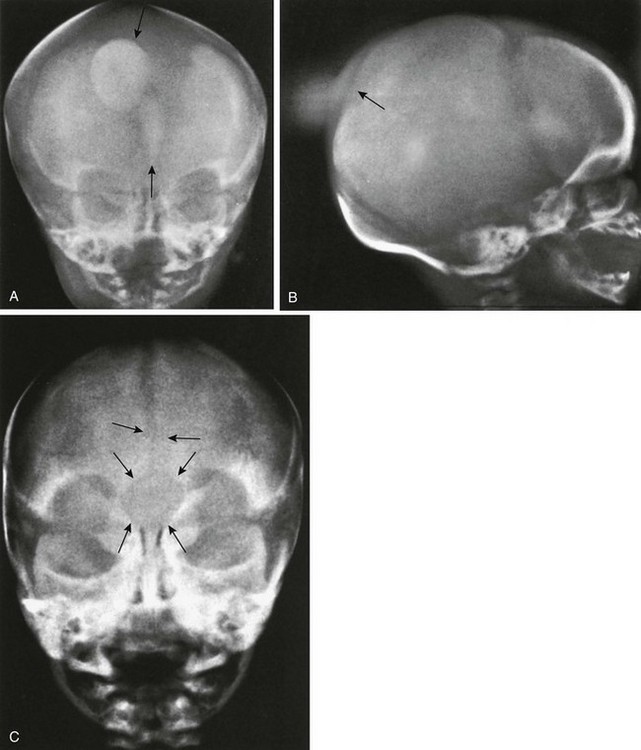
Figure 20-33 Cranium bifidum.
A and B, Frontal (A) and lateral (B) projections show intrafrontal and interparietal cranium bifidum in the metopic suture and in the sagittal suture, with protrusion of a soft tissue sac (meningoencephalocele) at anterior and posterior sites, in a 2-day-old girl. The metopic suture also is widely open, and a smaller mass of soft tissue bulges externally between the two sides of the frontal squamosa. In A, the larger superior patch of increased density (upper arrow) represents the interparietal protrusion, and the lower small patch (lower arrow) represents the smaller and shallower intrafrontal protrusion. C, Large circular bone defect (cranium bifidum) at the glabella in a widely open metopic suture. A lump of soft tissue bulged externally at this site.
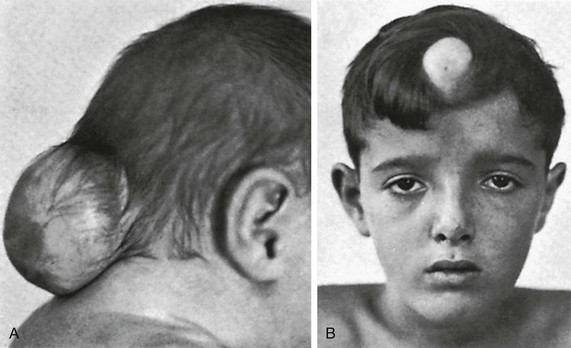
e-Figure 20-34 Examples of meningoceles.
A, Occipital meningocele in a 1-week-old neonate. B, Frontal meningocele in a 9-year-old boy.
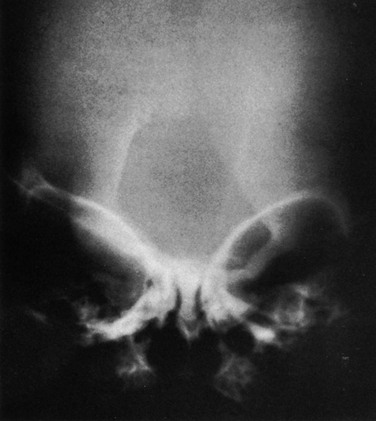
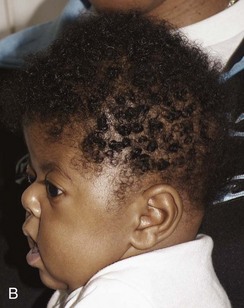
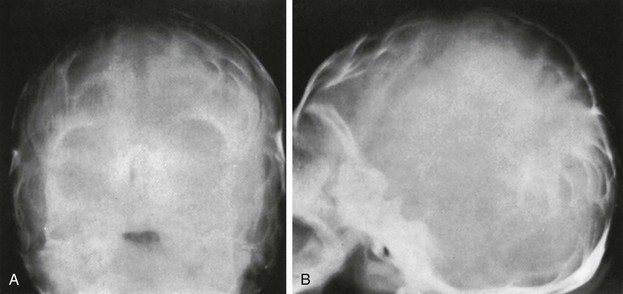
Lacunar Skull of the Newborn (Lückenschädel, Craniolacunia)
Lacunar skull develops during fetal life and is present at birth.32 It is nearly always associated with meningomyelocele, myelocele, or encephalocele and Chiari II malformation. The cause is unknown, but it is probably a dysplasia of the calvarium and the internal periosteum (i.e., the dura). It is not caused by fetal increased intracranial pressure because it is found in infants whose skulls are normal or are small in size without evidence of hydrocephalus. The characteristic “soap bubble” rarefactions in the upper part of the calvarium are easily recognized (Fig. 20-37). These begin to fade after birth and usually disappear by 4 to 5 months of age, even in the presence of progressive hydrocephalus in some instances. Normal convolutional rarefactions differ from lacunar rarefactions in that they are not obvious until the end of the first year and tend to appear first in the posterior and lower lateral portions of the calvarium.
Sinus Pericranii
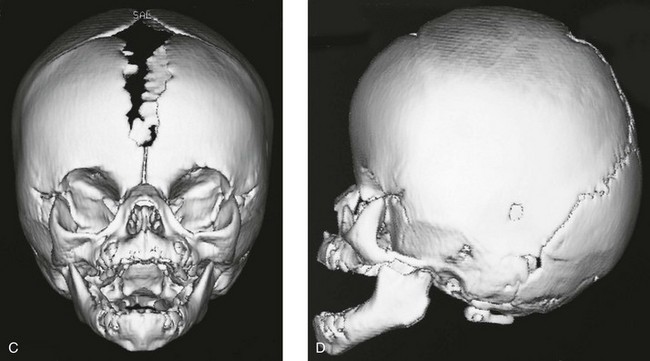
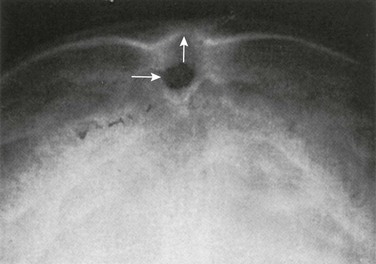
The term sinus pericranii is used for a soft, fluctuant mass, often of a red-to-blue color, observed in the scalp over the region of the sagittal or transverse sinuses. It responds in size to maneuvers that tend to increase intracranial pressure and may be associated with an underlying bony defect of the calvarium. It results from an abnormal communication between the intracranial and extracranial venous systems, and its significance is cosmetic.33–35 It must be differentiated from more serious lesions such as arteriovenous malformations, angiomas and hemangiomas, sebaceous or dermoid cysts, meningocele, encephalocele, and abscess. MRI is the best imaging modality for demonstrating the presence of an abnormal vascular communication (Fig. 20-38).
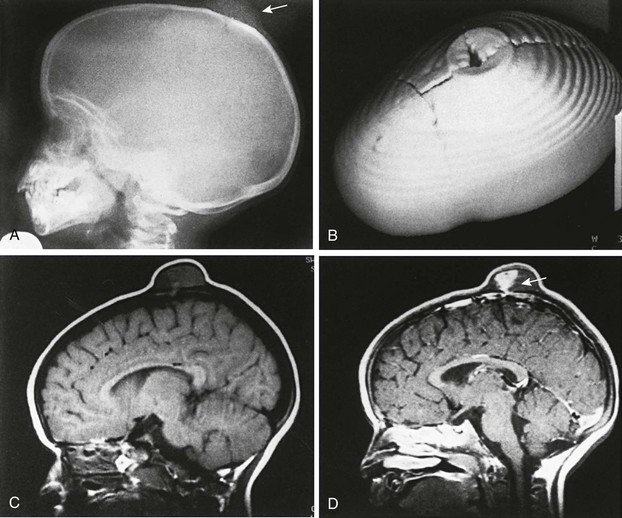
Figure 20-38 Sinus pericranii.
A, Lateral view of the skull showing a soft tissue mass and a well-defined bone defect in the midline of the parietal region. B, Three-dimensional computed tomography showing erosion of both tables similar to the crater of a volcano. C and D, Magnetic resonance images (TR/TE 400/20) before (C) and after (D) gadolinium injection. C, The tumor shows mixed signal intensity and areas of signal void indicating flowing blood. D, After administration of intravenous gadolinium, heterogeneous enhancement is evident, and an emissary vein is seen extending through the parietal bone to the superior sagittal sinus (arrow). (From Bigot J-L, Iacona C, Lepreux A, et al. Sinus pericranii: advantages of MR imaging. Pediatr Radiol. 2000;30:710-712.)
Cohen, MM, Jr. Perspectives on the face. New York: Oxford University Press; 2006.
Eteson, DJ, Steward, RE. Craniofacial defects in the human skeletal dysplasias. Birth Defects. 1984;20:14–45.
Glass, RB, Fernbach, SK, Norton, KI, et al. The infant skull: a vault of information. Radiographics. 2004;24:507–522.
Kirmi, O, Lo, SJ, Johnson, D, et al. Craniosynostosis: a radiological and surgical perspective. Semin Ultrasound CT MRI. 2009;30:492–512.
Lachman RS, ed. Taybi and Lachman’s radiology of syndromes, metabolic disorders and skeletal dysplasias, 5th ed, Philadelphia, PA: Elsevier, 2006.
Sood, S, Rozzelle, A, Shaqiri, B, et al. Effect of molding helmet on head shape in nonsurgically treated sagittal craniosynostosis. J Neurosurg Pediatr. 2011;7:1–6.
References
1. Cohen MM, Jr., MacLean RE, eds. Craniosynostosis. New York: Oxford University Press, 2000.
2. Wong, GB, Mulliken, JB, Benacerraf, BR. Prenatal sonographic diagnosis of major craniofacial anomalies. Plast Reconstr Surg. 2001;108:1316–1333.
3. Fjortoft, MI, Sevely, A, Boetto, S, et al. Prenatal diagnosis of craniosynostosis: value of MR imaging. Neuroradiology. 2007;49:515–521.
4. Currarino, G. Sagittal synostosis in X-linked hypophosphatemic rickets and related diseases. Pediatr Radiol. 2007;37:805–812.
5. Greene, AK, Mulliken, JB, Proctor, MR, et al. Phenotypically unusual combined craniosynostoses: presentation and management. Plast Reconstr Surg. 2008;122:853–862.
6. Rogers, GF, Greene, AK, Oh, AK, et al. Zygomaticotemporal synostosis: a rare cause of progressive facial asymmetry. Cleft Palate Craniofac J. 2007;44:106–111.
7. Rogers, GF, Proctor, MR, Mulliken, JB. Unilateral fusion of the frontosphenoidal suture: a rare cause of synostotic frontal plagiocephaly. Plast Reconstr Surg. 2002;110:1011–1021.
8. Currarino, G. Premature closure of the frontozygomatic suture: unusual fronto-orbital dysplasia mimicking unilateral coronal synostosis. AJNR Am J Neuroradiol. 1985;7:643–646.
9. Rollins, N, Sklar, F. Factitious lambdoid perisutural sclerosis: does the “sticky suture” exist? Pediatr Radiol. 1996;26:356–358.
10. Regelsberger, J, Schmidt, T, Busse, B, et al. Synchrotron—microcomputed tomography studies of normal and pathological cranial sutures: further insight. J Neurosurg Pediatr. 2010;5:238–242.
11. Vu, HL, Panchal, J, Parker, EE, et al. The timing of physiologic closure of the metopic suture: a review of 159 patients using reconstructed 3D CT scans of the craniofacial region. J Craniofac Surg. 2001;12:527–532.
12. Jane, JA, Persing, JA. Neurosurgical treatment of craniosynostosis. In: Cohen MM, Jr., MacLean RE, eds. Craniosynostosis. New York: Oxford University Press; 2000:209–227.
13. Martelli-Junior, H, de Miranda, RT, Fernandes, CM, et al. Goldenhar syndrome: clinical features with orofacial emphasis. J Appl Oral Sci. 2010;18:646–649.
14. Tasse, C, Bohringer, S, Fischer, S, et al. Oculo-auriculo-vetebral spectrum (OAVS): clinical evaluation and severity scoring of 53 patients and proposal for a new classification. Eur J Med Genet. 2005;48:397–411.
15. Santos, DT, Miyazaki, O, Cavalcanti, MGP. Clinical-embryological and radiological correlations of oculo-auriculo-vertebral spectrum using 3D-CT. Dentomaxillofac Radiol. 2003;32:8–14.
16. Johnson, JM, Moonis, G, Green, GE, et al. Syndromes of the first and second branchial arches, part 1: Embryology and characteristic defects. AJNR Am J Neurol Radiol. 2011;32:14–19.
17. Anderson, PJ, David, DJ. Spinal anomalies in Goldenhar syndrome. Cleft Palate Craniofac J. 2005;42:477–480.
18. Werler, MM, Starr, JR, Cloonan, YK, et al. Hemifacial microsomia: from gestation to childhood. J Craniofac Surg. 2009;20(supp 1):664–669.
19. Thompson, JT, Anderson, PJ, David, DJ. Treacher Collins syndrome: protocol management from birth to maturity. J Craniofac Surg. 2009;20:2028–2035.
20. Trainor, PA, Dixon, J, Dixon, MJ. Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur J Hum Genet. 2009;17:275–283.
21. Fisher, E. Exploring the genetic origins of Treacher Collins syndrome. Clin Genet. 2001;79:330–332.
22. Senggen, E, Laswed, T, Meuwiy, JY, et al. First and second branchial arch syndromes: multimodality approach. Pediatr Radiol. 2011;41:549–561.
23. Bütow, KW, Koogendijk, CF, Zwahlen, RA. Pierre Robin sequence: appearances and 25 years of experience with an innovative treatment protocol. J Pediatr Surg. 2009;44:2112–2118.
24. Evans, AK, Rahbar, R, Rogers, GE, et al. Robin sequence: a retrospective review of 115 patients. Int J Pediatr Othrhinolaryngol. 2006;70:973–980.
25. Cohen, MM. Robin sequences and complexes. Editorial comment: causal heterogeneity and pathogenetic/phenotypic variabililty. Am J Med Genet. 1999;83:311–315.
26. Suri, S, Ross, RB, Tompson, BD. Craniofacial morphology and adolescent facial growth in Pierre Robin sequence. Am J Orthod Dentofacial Orthop. 2010;137:763–774.
27. Al-Samkari, HT, Kane, AA, Molter, DW, et al. Neonatal outcomes of Pierre Robin sequence: an institutional experience. Clin Pediatr (Phila). 2010;49:1117–1122.
28. Mackay, DR. Controversies in the diagnosis and management of the Robin sequence. J Craniofac Surg. 2011;22:415–420.
29. Evans, KN, Sie, KC, Hopper, RA, et al. Robin sequence: from diagnosis to development of an effective management plan. Pediatrics. 2011;127:936–948.
30. Pruzansky, S. Not all dwarfed mandibles are alike. Birth Defects. 1969;1:120–129.
31. Diebler, D, Dulac, O. Cephaloceles: clinical and neuroradiological appearance: associated cerebral malformation. Neuroradiology. 1983;25:188–216.
32. McRae, DL. Observations on craniolacunia. Acta Radiol. 1966;5:55–64.
33. Bigot, JL, Iacona, C, Lepreux, A, et al. Sinus pericranii: advantages of MR imaging. Pediatr Radiol. 2000;30:710–712.
34. Carnes, JT. Sinus pericranii: demonstration using three-dimensional surface shading. J Comput Assist Tomogr. 2002;26:285–286.
35. Yanik, B, Keyik, B, Conk Bayir, I, et al. Sinus pericranii: color Doppler ultrasonographic findings. J Ultrasound Med. 2006;25:679–682.