FIGURE 44-3 Acute epiglottitis. In this lateral soft tissue radiograph of the neck, the arrow indicates the enlarged edematous epiglottis (the “thumbprint sign”).
INFECTIONS OF DEEP NECK STRUCTURES
Deep neck infections are usually extensions of infection from other primary sites, most often within the pharynx or oral cavity. Many of these infections are life threatening but are difficult to detect at early stages, when they may be more easily managed. Three of the most clinically relevant spaces in the neck are the submandibular (and sublingual) space, the lateral pharyngeal (or parapharyngeal) space, and the retropharyngeal space. These spaces communicate with one another and with other important structures in the head, neck, and thorax, providing pathogens with easy access to areas that include the mediastinum, carotid sheath, skull base, and meninges. Once infection reaches these sensitive areas, mortality rates can be as high as 20–50%.
Infection of the submandibular and/or sublingual space typically originates from an infected or recently extracted lower tooth. The result is the severe, life-threatening infection referred to as Ludwig’s angina (see “Oral Infections,” above). Infection of the lateral pharyngeal (or parapharyngeal) space is most often a complication of common infections of the oral cavity and upper respiratory tract, including tonsillitis, peritonsillar abscess, pharyngitis, mastoiditis, and periodontal infection. This space, situated deep in the lateral wall of the pharynx, contains a number of sensitive structures, including the carotid artery, internal jugular vein, cervical sympathetic chain, and portions of cranial nerves IX through XII; at its distal end, it opens into the posterior mediastinum. Involvement of this space with infection can therefore be rapidly fatal. Examination may reveal some tonsillar displacement, trismus, and neck rigidity, but swelling of the lateral pharyngeal wall can easily be missed. The diagnosis can be confirmed by CT. Treatment consists of airway management, operative drainage of fluid collections, and at least 10 days of IV therapy with an antibiotic active against streptococci and oral anaerobes (e.g., ampicillin/sulbactam). A particularly severe form of this infection involving the components of the carotid sheath (postanginal septicemia, Lemierre’s disease) is described above (see “Oral Infections”). Infection of the retropharyngeal space also can be extremely dangerous, as this space runs posterior to the pharynx from the skull base to the superior mediastinum. Infections in this space are more common among children <5 years old because of the presence of several small retropharyngeal lymph nodes that typically atrophy by age 4 years. Infection is usually a consequence of extension from another site of infection—most commonly, acute pharyngitis. Other sources include otitis media, tonsillitis, dental infections, Ludwig’s angina, and anterior extension of vertebral osteomyelitis. Retropharyngeal space infection also can follow penetrating trauma to the posterior pharynx (e.g., from an endoscopic procedure). Infections are commonly polymicrobial, involving a mixture of aerobes and anaerobes; group A β-hemolytic streptococci and S. aureus are the most common pathogens. M. tuberculosis was a common cause in the past but now is rarely involved in the United States.
Patients with retropharyngeal abscess typically present with sore throat, fever, dysphagia, and neck pain and are often drooling because of difficulty and pain with swallowing. Examination may reveal tender cervical adenopathy, neck swelling, and diffuse erythema and edema of the posterior pharynx as well as a bulge in the posterior pharyngeal wall that may not be obvious on routine inspection. A soft tissue mass is usually demonstrable by lateral neck radiography or CT. Because of the risk of airway obstruction, treatment begins with securing of the airway, followed by a combination of surgical drainage and IV antibiotic administration. Initial empirical therapy should cover streptococci, oral anaerobes, and S. aureus; ampicillin/sulbactam, clindamycin plus ceftriaxone, or meropenem is usually effective. Complications result primarily from extension to other areas (e.g., rupture into the posterior pharynx may lead to aspiration pneumonia and empyema). Extension may also occur to the lateral pharyngeal space and mediastinum, resulting in mediastinitis and pericarditis, or into nearby major blood vessels. All these events are associated with a high mortality rate.
45 |
Oral Manifestations of Disease |
As primary care physicians and consultants, internists are often asked to evaluate patients with disease of the oral soft tissues, teeth, and pharynx. Knowledge of the oral milieu and its unique structures is necessary to guide preventive services and recognize oral manifestations of local or systemic disease (Chap. 46e). Furthermore, internists frequently collaborate with dentists in the care of patients who have a variety of medical conditions that affect oral health or who undergo dental procedures that increase their risk of medical complications.
DISEASES OF THE TEETH AND PERIODONTAL STRUCTURES
Tooth formation begins during the sixth week of embryonic life and continues through 17 years of age. Teeth start to develop in utero and continue to develop until after the tooth erupts. Normally, all 20 deciduous teeth have erupted by age 3 and have been shed by age 13. Permanent teeth, eventually totaling 32, begin to erupt by age 6 and have completely erupted by age 14, though third molars (“wisdom teeth”) may erupt later.
The erupted tooth consists of the visible crown covered with enamel and the root submerged below the gum line and covered with bonelike cementum. Dentin, a material that is denser than bone and exquisitely sensitive to pain, forms the majority of the tooth substance, surrounding a core of myxomatous pulp containing the vascular and nerve supply. The tooth is held firmly in the alveolar socket by the periodontium, supporting structures that consist of the gingivae, alveolar bone, cementum, and periodontal ligament. The periodontal ligament tenaciously binds the tooth’s cementum to the alveolar bone. Above this ligament is a collar of attached gingiva just below the crown. A few millimeters of unattached or free gingiva (1–3 mm) overlap the base of the crown, forming a shallow sulcus along the gum-tooth margin.
Dental Caries, Pulpal and Periapical Disease, and Complications Dental caries usually begin asymptomatically as a destructive infectious process of the enamel. Bacteria—principally Streptococcus mutans—colonize the organic buffering biofilm (plaque) on the tooth surface. If not removed by brushing or by the natural cleansing and antibacterial action of saliva, bacterial acids can demineralize the enamel. Fissures and pits on the occlusal surfaces are the most frequent sites of early decay. Surfaces between the teeth, adjacent to tooth restorations and exposed roots, are also vulnerable, particularly as individuals age. Over time, dental caries extend to the underlying dentin, leading to cavitation of the enamel. Without management, the caries will penetrate to the tooth pulp, producing acute pulpitis. At this stage, when the pulp infection is limited, the tooth may become sensitive to percussion and to hot or cold, and pain resolves immediately when the irritating stimulus is removed. Should the infection spread throughout the pulp, irreversible pulpitis occurs, leading to pulp necrosis. At this later stage, pain can be severe and has a sharp or throbbing visceral quality that may be worse when the patient lies down. Once pulp necrosis is complete, pain may be constant or intermittent, but cold sensitivity is lost.
Treatment of caries involves removal of the softened and infected hard tissue and restoration of the tooth structure with silver amalgam, glass ionomer, composite resin, or gold. Once irreversible pulpitis occurs, root canal therapy becomes necessary; removal of the contents of the pulp chamber and root canals is followed by thorough cleaning and filling with an inert material. Alternatively, the tooth may be extracted.
Pulpal infection leads to periapical abscess formation, which can produce pain on chewing. If the infection is mild and chronic, a periapical granuloma or eventually a periapical cyst forms, either of which produces radiolucency at the root apex. When unchecked, a periapical abscess can erode into the alveolar bone, producing osteomyelitis; penetrate and drain through the gingivae, producing a parulis (gumboil); or track along deep fascial planes, producing virulent cellulitis (Ludwig’s angina) involving the submandibular space and floor of the mouth (Chap. 201). Elderly patients, patients with diabetes mellitus, and patients taking glucocorticoids may experience little or no pain or fever as these complications develop.
Periodontal Disease Periodontal disease and dental caries are the primary causes of tooth loss. Like dental caries, chronic infection of the gingiva and anchoring structures of the tooth begins with formation of bacterial plaque. The process begins at the gum line. Plaque and calculus (calcified plaque) are preventable by appropriate daily oral hygiene, including periodic professional cleaning. Left undisturbed, chronic inflammation can ensue and produce hyperemia of the free and attached gingivae (gingivitis), which then typically bleed with brushing. If this issue is ignored, severe periodontitis can develop, leading to deepening of the physiologic sulcus and destruction of the periodontal ligament. Gingival pockets develop around the teeth. As the periodontium (including the supporting bone) is destroyed, the teeth loosen. A role for chronic inflammation due to chronic periodontal disease in promoting coronary heart disease and stroke has been proposed. Epidemiologic studies have demonstrated a moderate but significant association between chronic periodontal inflammation and atherogenesis, though a causal role remains unproven.
Acute and aggressive forms of periodontal disease are less common than the chronic forms described above. However, if the host is stressed or exposed to a new pathogen, rapidly progressive and destructive disease of the periodontal tissue can occur. A virulent example is acute necrotizing ulcerative gingivitis. Stress and poor oral hygiene are risk factors. The presentation includes sudden gingival inflammation, ulceration, bleeding, interdental gingival necrosis, and fetid halitosis. Localized juvenile periodontitis, which is seen in adolescents, is particularly destructive and appears to be associated with impaired neutrophil chemotaxis. AIDS-related periodontitis resembles acute necrotizing ulcerative gingivitis in some patients and a more destructive form of adult chronic periodontitis in others. It may also produce a gangrene-like destructive process of the oral soft tissues and bone that resembles noma, an infectious condition seen in severely malnourished children in developing nations.
Prevention of Tooth Decay and Periodontal Infection Despite the reduced prevalences of dental caries and periodontal disease in the United States (due in large part to water fluoridation and improved dental care, respectively), both diseases constitute a major public health problem worldwide, particularly in certain groups. The internist should promote preventive dental care and hygiene as part of health maintenance. Populations at high risk for dental caries and periodontal disease include those with hyposalivation and/or xerostomia, diabetics, alcoholics, tobacco users, persons with Down syndrome, and those with gingival hyperplasia. Furthermore, patients lacking access to dental care (e.g., as a result of low socioeconomic status) and patients with a reduced ability to provide self-care (e.g., individuals with disabilities, nursing home residents, and persons with dementia or upper-extremity disability) suffer at a disproportionate rate. It is important to provide counseling regarding regular dental hygiene and professional cleaning, use of fluoride-containing toothpaste, professional fluoride treatments, and (for patients with limited dexterity) use of electric toothbrushes and also to instruct persons caring for those who are not capable of self-care. Cost, fear of dental care, and differences in language and culture create barriers that prevent some people from seeking preventive dental services.
Developmental and Systemic Disease Affecting the Teeth and Periodontium In addition to posing cosmetic issues, malocclusion, the most common developmental oral problem, can interfere with mastication unless corrected through orthodontic and surgical techniques. Impacted third molars are common and can become infected or erupt into an insufficient space. Acquired prognathism due to acromegaly may also lead to malocclusion, as may deformity of the maxilla and mandible due to Paget’s disease of the bone. Delayed tooth eruption, a receding chin, and a protruding tongue are occasional features of cretinism and hypopituitarism. Congenital syphilis produces tapering, notched (Hutchinson’s) incisors and finely nodular (mulberry) molar crowns. Enamel hypoplasia results in crown defects ranging from pits to deep fissures of primary or permanent teeth. Intrauterine infection (syphilis, rubella), vitamin deficiency (A, C, or D), disorders of calcium metabolism (malabsorption, vitamin D–resistant rickets, hypoparathyroidism), prematurity, high fever, and rare inherited defects (amelogenesis imperfecta) are all causes. Tetracycline, given in sufficiently high doses during the first 8 years of life, may produce enamel hypoplasia and discoloration. Exposure to endogenous pigments can discolor developing teeth; etiologies include erythroblastosis fetalis (green or bluish-black), congenital liver disease (green or yellow-brown), and porphyria (red or brown that fluoresces with ultraviolet light). Mottled enamel occurs if excessive fluoride is ingested during development. Worn enamel is seen with age, bruxism, or excessive acid exposure (e.g., chronic gastric reflux or bulimia). Celiac disease is associated with nonspecific enamel defects in children but not in adults.
Total or partial tooth loss resulting from periodontitis is seen with cyclic neutropenia, Papillon-Lefévre syndrome, Chédiak-Higashi syndrome, and leukemia. Rapid focal tooth loosening is most often due to infection, but rarer causes include Langerhans cell histiocytosis, Ewing’s sarcoma, osteosarcoma, and Burkitt’s lymphoma. Early loss of primary teeth is a feature of hypophosphatasia, a rare congenital error of metabolism.
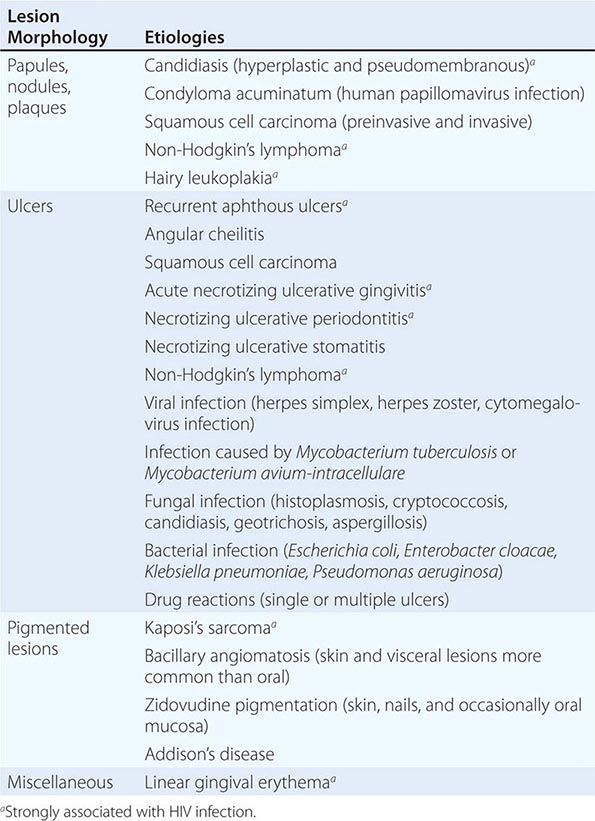
Pregnancy may produce gingivitis and localized pyogenic granulomas. Severe periodontal disease occurs in uncontrolled diabetes mellitus. Gingival hyperplasia may be caused by phenytoin, calcium channel blockers (e.g., nifedipine), and cyclosporine, though excellent daily oral care can prevent or reduce its occurrence. Idiopathic familial gingival fibromatosis and several syndrome-related disorders cause similar conditions. Discontinuation of the medication may reverse the drug-induced form, though surgery may be needed to control both of the latter entities. Linear gingival erythema is variably seen in patients with advanced HIV infection and probably represents immune deficiency and decreased neutrophil activity. Diffuse or focal gingival swelling may be a feature of early or late acute myelomonocytic leukemia as well as of other lymphoproliferative disorders. A rare but pathognomonic sign of granulomatosis with polyangiitis is a red-purplish, granular gingivitis (strawberry gums).
DISEASES OF THE ORAL MUCOSA
Infections Most oral mucosal diseases involve microorganisms (Table 45-1).
|
VESICULAR, BULLOUS, OR ULCERATIVE LESIONS OF THE ORAL MUCOSA |
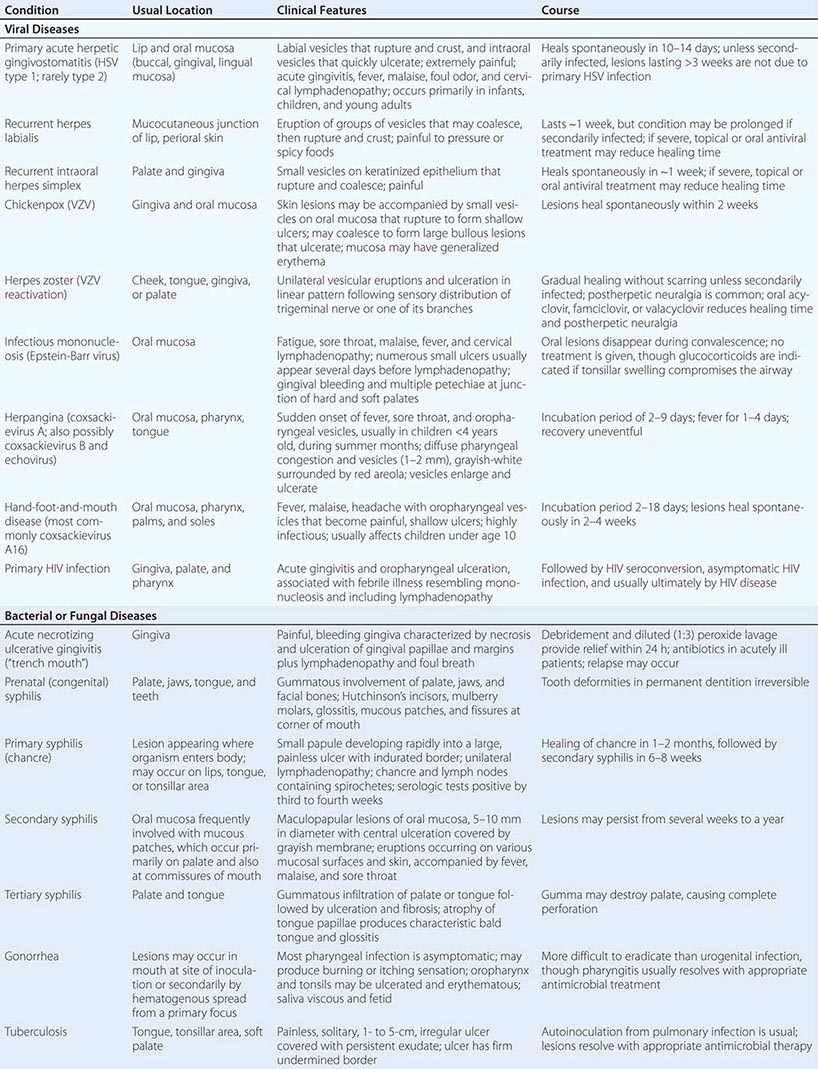
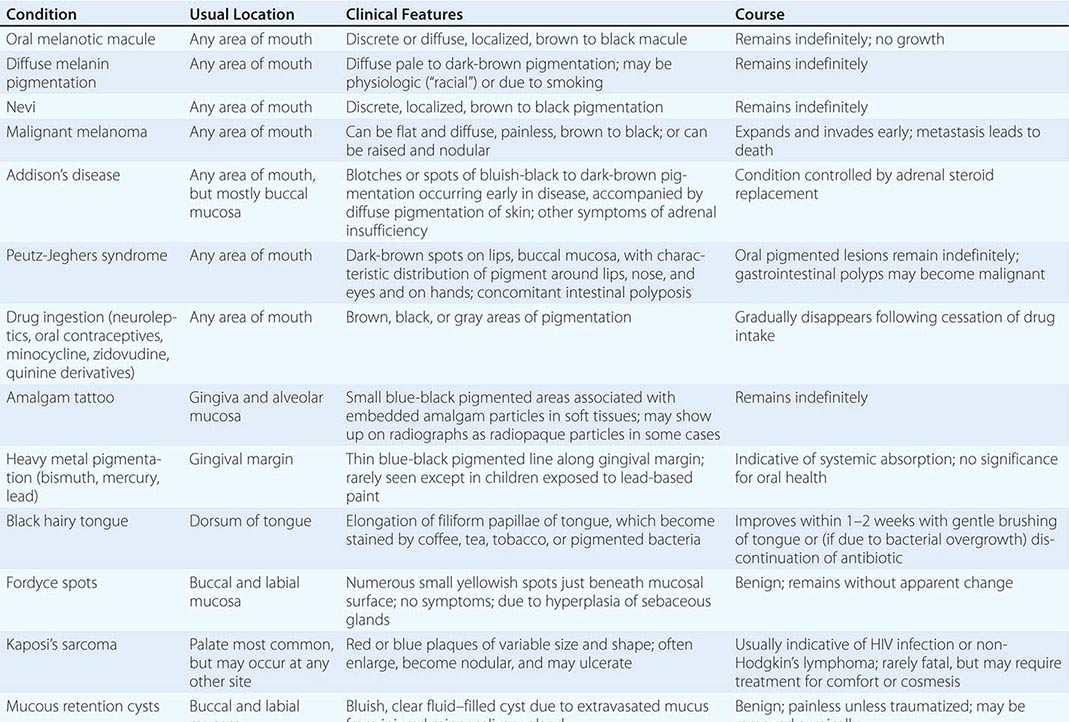
Pigmented Lesions See Table 45-2.
|
PIGMENTED LESIONS OF THE ORAL MUCOSA |
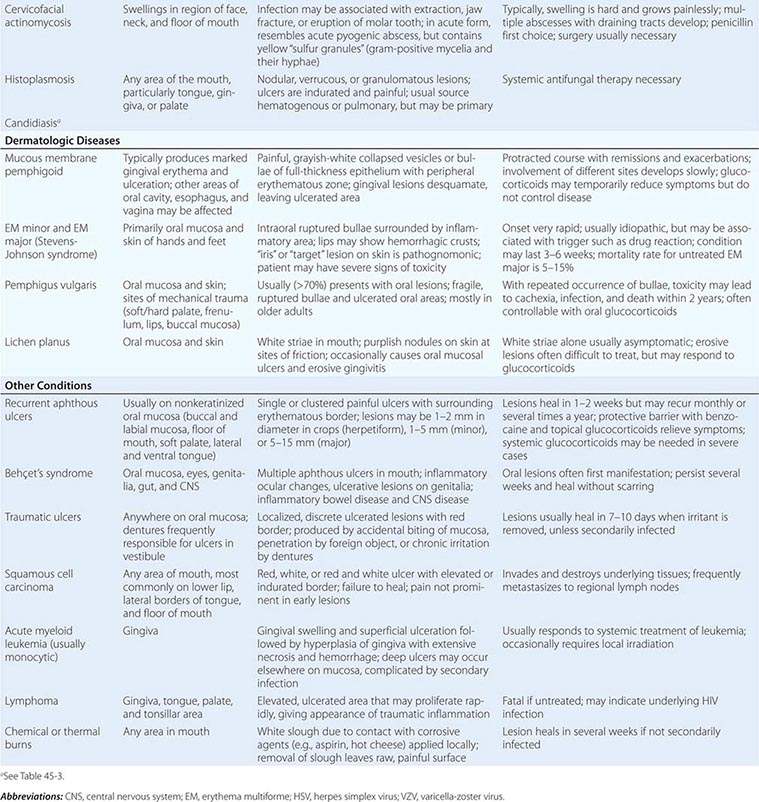
Dermatologic Diseases See Tables 45-1, 45-2, and 45-3 and Chaps. 70–74.
|
WHITE LESIONS OF ORAL MUCOSA |
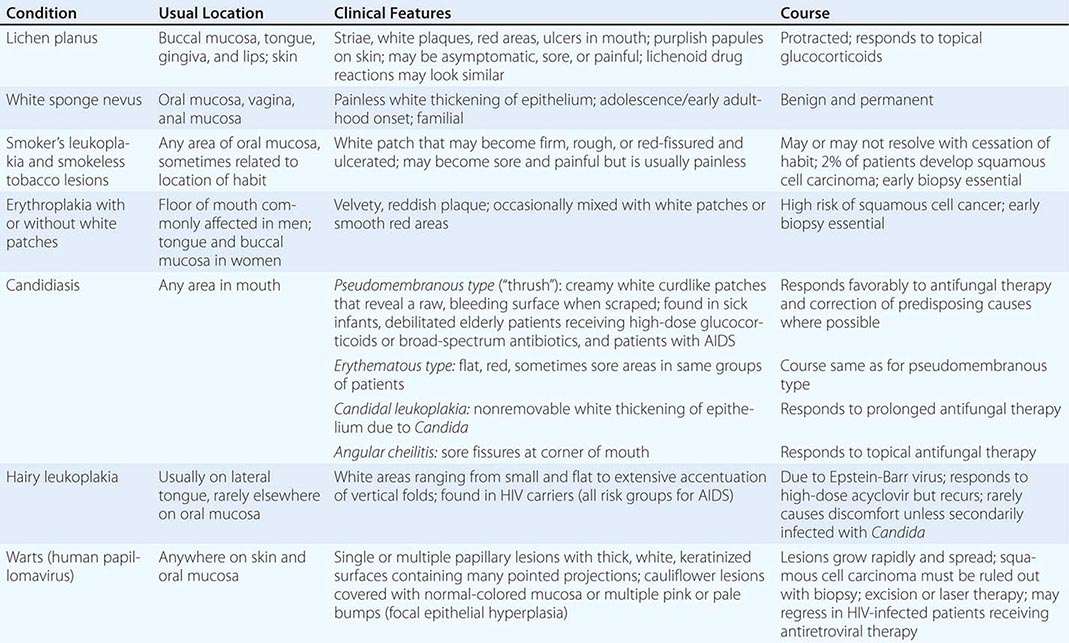
Diseases of the Tongue See Table 45-4.
|
ALTERATIONS OF THE TONGUE |

HIV Disease and AIDS See Tables 45-1, 45-2, 45-3, and 45-5; Chap. 226; and Fig. 218-3.
|
ORAL LESIONS ASSOCIATED WITH HIV INFECTION |
Ulcers Ulceration is the most common oral mucosal lesion. Although there are many causes, the host and the pattern of lesions, including the presence of organ system features, narrow the differential diagnosis (Table 45-1). Most acute ulcers are painful and self-limited. Recurrent aphthous ulcers and herpes simplex account for the majority. Persistent and deep aphthous ulcers can be idiopathic or can accompany HIV/AIDS. Aphthous lesions are often the presenting symptom in Behçet’s syndrome (Chap. 387). Similar-appearing, though less painful, lesions may occur in reactive arthritis, and aphthous ulcers are occasionally present during phases of discoid or systemic lupus erythematosus (Chap. 382). Aphthous-like ulcers are seen in Crohn’s disease (Chap. 351), but, unlike the common aphthous variety, they may exhibit granulomatous inflammation on histologic examination. Recurrent aphthae are more prevalent in patients with celiac disease and have been reported to remit with elimination of gluten.
Of major concern are chronic, relatively painless ulcers and mixed red/white patches (erythroplakia and leukoplakia) of >2 weeks’ duration. Squamous cell carcinoma and premalignant dysplasia should be considered early and a diagnostic biopsy performed. This awareness and this procedure are critically important because early-stage malignancy is vastly more treatable than late-stage disease. High-risk sites include the lower lip, floor of the mouth, ventral and lateral tongue, and soft palate–tonsillar pillar complex. Significant risk factors for oral cancer in Western countries include sun exposure (lower lip), tobacco and alcohol use, and human papillomavirus infection. In India and some other Asian countries, smokeless tobacco mixed with betel nut, slaked lime, and spices is a common cause of oral cancer. Rarer causes of chronic oral ulcer, such as tuberculosis, fungal infection, granulomatosis with polyangiitis, and midline granuloma may look identical to carcinoma. Making the correct diagnosis depends on recognizing other clinical features and performing a biopsy of the lesion. The syphilitic chancre is typically painless and therefore easily missed. Regional lymphadenopathy is invariably present. The syphilitic etiology is confirmed with appropriate bacterial and serologic tests.
Disorders of mucosal fragility often produce painful oral ulcers that fail to heal within 2 weeks. Mucous membrane pemphigoid and pemphigus vulgaris are the major acquired disorders. While their clinical features are often distinctive, a biopsy or immunohistochemical examination should be performed to diagnose these entities and to distinguish them from lichen planus and drug reactions.
Hematologic and Nutritional Disease Internists are more likely to encounter patients with acquired, rather than congenital, bleeding disorders. Bleeding should stop 15 min after minor trauma and within an hour after tooth extraction if local pressure is applied. More prolonged bleeding, if not due to continued injury or rupture of a large vessel, should lead to investigation for a clotting abnormality. In addition to bleeding, petechiae and ecchymoses are prone to occur at the vibrating line between the soft and hard palates in patients with platelet dysfunction or thrombocytopenia.
All forms of leukemia, but particularly acute myelomonocytic leukemia, can produce gingival bleeding, ulcers, and gingival enlargement. Oral ulcers are a feature of agranulocytosis, and ulcers and mucositis are often severe complications of chemotherapy and radiation therapy for hematologic and other malignancies. Plummer-Vinson syndrome (iron deficiency, angular stomatitis, glossitis, and dysphagia) raises the risk of oral squamous cell cancer and esophageal cancer at the postcricoidal tissue web. Atrophic papillae and a red, burning tongue may occur with pernicious anemia. Deficiencies in B-group vitamins produce many of these same symptoms as well as oral ulceration and cheilosis. Consequences of scurvy include swollen, bleeding gums; ulcers; and loosening of the teeth.
NONDENTAL CAUSES OF ORAL PAIN
Most, but not all, oral pain emanates from inflamed or injured tooth pulp or periodontal tissues. Nonodontogenic causes are often overlooked. In most instances, toothache is predictable and proportional to the stimulus applied, and an identifiable condition (e.g., caries, abscess) is found. Local anesthesia eliminates pain originating from dental or periodontal structures, but not referred pains. The most common nondental source of pain is myofascial pain referred from muscles of mastication, which become tender and ache with increased use. Many sufferers exhibit bruxism (grinding of the teeth) secondary to stress and anxiety. Temporomandibular joint disorder is closely related. It affects both sexes, with a higher prevalence among women. Features include pain, limited mandibular movement, and temporomandibular joint sounds. The etiologies are complex; malocclusion does not play the primary role once attributed to it. Osteoarthritis is a common cause of masticatory pain. Anti-inflammatory medication, jaw rest, soft foods, and heat provide relief. The temporomandibular joint is involved in 50% of patients with rheumatoid arthritis, and its involvement is usually a late feature of severe disease. Bilateral preauricular pain, particularly in the morning, limits range of motion.
Migrainous neuralgia may be localized to the mouth. Episodes of pain and remission without an identifiable cause and a lack of relief with local anesthesia are important clues. Trigeminal neuralgia (tic douloureux) can involve the entire branch or part of the mandibular or maxillary branch of the fifth cranial nerve and can produce pain in one or a few teeth. Pain may occur spontaneously or may be triggered by touching the lip or gingiva, brushing the teeth, or chewing. Glossopharyngeal neuralgia produces similar acute neuropathic symptoms in the distribution of the ninth cranial nerve. Swallowing, sneezing, coughing, or pressure on the tragus of the ear triggers pain that is felt in the base of the tongue, pharynx, and soft palate and may be referred to the temporomandibular joint. Neuritis involving the maxillary and mandibular divisions of the trigeminal nerve (e.g., maxillary sinusitis, neuroma, and leukemic infiltrate) is distinguished from ordinary toothache by the neuropathic quality of the pain. Occasionally, phantom pain follows tooth extraction. Pain and hyperalgesia behind the ear and on the side of the face in the day or so before facial weakness develops often constitute the earliest symptom of Bell’s palsy. Likewise, similar symptoms may precede visible lesions of herpes zoster infecting the seventh nerve (Ramsey-Hunt syndrome) or trigeminal nerve. Postherpetic neuralgia may follow either condition. Coronary ischemia may produce pain exclusively in the face and jaw; as in typical angina pectoris, this pain is usually reproducible with increased myocardial demand. Aching in several upper molar or premolar teeth that is unrelieved by anesthetizing the teeth may point to maxillary sinusitis.
Giant cell arteritis is notorious for producing headache, but it may also produce facial pain or sore throat without headache. Jaw and tongue claudication with chewing or talking is relatively common. Tongue infarction is rare. Patients with subacute thyroiditis often experience pain referred to the face or jaw before the tenderness of the thyroid gland and transient hyperthyroidism are appreciated.
“Burning mouth syndrome” (glossodynia) occurs in the absence of an identifiable cause (e.g., vitamin B12 deficiency, iron deficiency, diabetes mellitus, low-grade Candida infection, food sensitivity, or subtle xerostomia) and predominantly affects postmenopausal women. The etiology may be neuropathic. Clonazepam, α-lipoic acid, and cognitive behavioral therapy have benefited some patients. Some cases associated with an angiotensin-converting enzyme inhibitor have remitted when treatment with the drug was discontinued.
DISEASES OF THE SALIVARY GLANDS
Saliva is essential to oral health. Its absence leads to dental caries, periodontal disease, and difficulties in wearing dental prostheses, masticating, and speaking. Its major components, water and mucin, serve as a cleansing solvent and lubricating fluid. In addition, saliva contains antimicrobial factors (e.g., lysozyme, lactoperoxidase, secretory IgA), epidermal growth factor, minerals, and buffering systems. The major salivary glands secrete intermittently in response to autonomic stimulation, which is high during a meal but low otherwise. Hundreds of minor glands in the lips and cheeks secrete mucus continuously throughout the day and night. Consequently, oral function becomes impaired when salivary function is reduced. The sensation of a dry mouth (xerostomia) is perceived when salivary flow is reduced by 50%. The most common etiology is medication, especially drugs with anticholinergic properties but also alpha and beta blockers, calcium channel blockers, and diuretics. Other causes include Sjögren’s syndrome, chronic parotitis, salivary duct obstruction, diabetes mellitus, HIV/AIDS, and radiation therapy that includes the salivary glands in the field (e.g., for Hodgkin’s disease and for head and neck cancer). Management involves the elimination or limitation of drying medications, preventive dental care, and supplementation with oral liquid or salivary substitutes. Sugarless mints or chewing gum may stimulate salivary secretion if dysfunction is mild. When sufficient exocrine tissue remains, pilocarpine or cevimeline has been shown to increase secretions. Commercial saliva substitutes or gels relieve dryness. Fluoride supplementation is critical to prevent caries.
Sialolithiasis presents most often as painful swelling but in some instances as only swelling or only pain. Conservative therapy consists of local heat, massage, and hydration. Promotion of salivary secretion with mints or lemon drops may flush out small stones. Antibiotic treatment is necessary when bacterial infection in suspected. In adults, acute bacterial parotitis is typically unilateral and most commonly affects postoperative, dehydrated, and debilitated patients. Staphylococcus aureus (including methicillin-resistant strains) and anaerobic bacteria are the most common pathogens. Chronic bacterial sialadenitis results from lowered salivary secretion and recurrent bacterial infection. When suspected bacterial infection is not responsive to therapy, the differential diagnosis should be expanded to include benign and malignant neoplasms, lymphoproliferative disorders, Sjögren’s syndrome, sarcoidosis, tuberculosis, lymphadenitis, actinomycosis, and granulomatosis with polyangiitis. Bilateral nontender parotid enlargement occurs with diabetes mellitus, cirrhosis, bulimia, HIV/AIDS, and drugs (e.g., iodide, propylthiouracil).
Pleomorphic adenoma comprises two-thirds of all salivary neoplasms. The parotid is the principal salivary gland affected, and the tumor presents as a firm, slow-growing mass. Although this tumor is benign, its recurrence is common if resection is incomplete. Malignant tumors such as mucoepidermoid carcinoma, adenoid cystic carcinoma, and adenocarcinoma tend to grow relatively fast, depending upon grade. They may ulcerate and invade nerves, producing numbness and facial paralysis. Surgical resection is the primary treatment. Radiation therapy (particularly neutron-beam therapy) is used when surgery is not feasible and as post-resection for certain histologic types with a high risk of recurrence. Malignant salivary gland tumors have a 5-year survival rate of ~68%.
DENTAL CARE FOR MEDICALLY COMPLEX PATIENTS
Routine dental care (e.g., uncomplicated extraction, scaling and cleaning, tooth restoration, and root canal) is remarkably safe. The most common concerns regarding care of dental patients with medical disease are excessive bleeding for patients taking anticoagulants, infection of the heart valves and prosthetic devices from hematogenous seeding by the oral flora, and cardiovascular complications resulting from vasopressors used with local anesthetics during dental treatment. Experience confirms that the risk of any of these complications is very low.
Patients undergoing tooth extraction or alveolar and gingival surgery rarely experience uncontrolled bleeding when warfarin anticoagulation is maintained within the therapeutic range currently recommended for prevention of venous thrombosis, atrial fibrillation, or mechanical heart valve. Embolic complications and death, however, have been reported during subtherapeutic anticoagulation. Therapeutic anticoagulation should be confirmed before and continued through the procedure. Likewise, low-dose aspirin (e.g., 81–325 mg) can safely be continued. For patients taking aspirin and another antiplatelet medication (e.g., clopidogrel), the decision to continue the second antiplatelet medication should be based on individual consideration of the risks of thrombosis and bleeding.
Patients at risk for bacterial endocarditis (Chap. 155) should maintain optimal oral hygiene, including flossing, and have regular professional cleanings. Currently, guidelines recommend that prophylactic antibiotics be restricted to those patients at high risk for bacterial endocarditis who undergo dental and oral procedures involving significant manipulation of gingival or periapical tissue or penetration of the oral mucosa. If unexpected bleeding occurs, antibiotics given within 2 h after the procedure provide effective prophylaxis.
Hematogenous bacterial seeding from oral infection can undoubtedly produce late prosthetic-joint infection and therefore requires removal of the infected tissue (e.g., drainage, extraction, root canal) and appropriate antibiotic therapy. However, evidence that late prosthetic-joint infection follows routine dental procedures is lacking. For this reason, antibiotic prophylaxis is not recommended before dental surgery for patients with orthopedic pins, screws, and plates. Antibiotic prophylaxis is recommended for patients within the first 2 years after joint replacement who have inflammatory arthropathies, immunosuppression, type 1 diabetes mellitus, previous prosthetic-joint infection, hemophilia, or malnourishment.
Concern often arises regarding the use of vasoconstrictors to treat patients with hypertension and heart disease. Vasoconstrictors enhance the depth and duration of local anesthesia, thus reducing the anesthetic dose and potential toxicity. If intravascular injection is avoided, 2% lidocaine with 1:100,000 epinephrine (limited to a total of 0.036 mg of epinephrine) can be used safely in patients with controlled hypertension and stable coronary heart disease, arrhythmia, or congestive heart failure. Precautions should be taken with patients taking tricyclic antidepressants and nonselective beta blockers because these drugs may potentiate the effect of epinephrine.
Elective dental treatments should be postponed for at least 1 month and preferably for 6 months after myocardial infarction, after which the risk of reinfarction is low provided the patient is medically stable (e.g., stable rhythm, stable angina, and no heart failure). Patients who have suffered a stroke should have elective dental care deferred for 6 months. In both situations, effective stress reduction requires good pain control, including the use of the minimal amount of vasoconstrictor necessary to provide good hemostasis and local anesthesia.
Bisphosphonate therapy is associated with osteonecrosis of the jaw. However, the risk with oral bisphosphonate therapy is very low. Most patients affected have received high-dose aminobisphosphonate therapy for multiple myeloma or metastatic breast cancer and have undergone tooth extraction or dental surgery. Intraoral lesions, of which two-thirds are painful, appear as exposed yellow-white hard bone involving the mandible or maxilla. Screening tests for determining risk of osteonecrosis are unreliable. Patients slated for aminobisphosphonate therapy should receive preventive dental care that reduces the risk of infection and the need for future dentoalveolar surgery.
HALITOSIS
Halitosis typically emanates from the oral cavity or nasal passages. Volatile sulfur compounds resulting from bacterial decay of food and cellular debris account for the malodor. Periodontal disease, caries, acute forms of gingivitis, poorly fitting dentures, oral abscess, and tongue coating are common causes. Treatment includes correcting poor hygiene, treating infection, and tongue brushing. Hyposalivation can produce and exacerbate halitosis. Pockets of decay in the tonsillar crypts, esophageal diverticulum, esophageal stasis (e.g., achalasia, stricture), sinusitis, and lung abscess account for some instances. A few systemic diseases produce distinctive odors: renal failure (ammoniacal), hepatic (fishy), and ketoacidosis (fruity). Helicobacter pylori gastritis can also produce ammoniacal breath. If a patient presents because of concern about halitosis but no odor is detectable, then pseudohalitosis or halitophobia must be considered.
AGING AND ORAL HEALTH
While tooth loss and dental disease are not normal consequences of aging, a complex array of structural and functional changes that occur with age can affect oral health. Subtle changes in tooth structure (e.g., diminished pulp space and volume, sclerosis of dentinal tubules, and altered proportions of nerve and vascular pulp content) result in the elimination or diminution of pain sensitivity and a reduction in the reparative capacity of the teeth. In addition, age-associated fatty replacement of salivary acini may reduce physiologic reserve, thus increasing the risk of hyposalivation. In healthy older adults, there is minimal, if any, reduction in salivary flow.
Poor oral hygiene often results when general health fails or when patients lose manual dexterity and upper-extremity flexibility. This situation is particularly common among frail older adults and nursing home residents and must be emphasized because regular oral cleaning and dental care reduce the incidence of pneumonia and oral disease as well as the mortality risk in this population. Other risks for dental decay include limited lifetime fluoride exposure. Without assiduous care, decay can become quite advanced yet remain asymptomatic. Consequently, much of a tooth—or the entire tooth—can be destroyed before the patient is aware of the process.
Periodontal disease, a leading cause of tooth loss, is indicated by loss of alveolar bone height. More than 90% of the U.S. population has some degree of periodontal disease by age 50. Healthy adults who have not had significant alveolar bone loss by the sixth decade of life do not typically experience significant worsening with advancing age.
Complete edentulousness with advanced age, though less common than in previous decades, still affects <50% of the U.S. population ≥85 years of age. Speech, mastication, and facial contours are dramatically affected. Edentulousness may also exacerbate obstructive sleep apnea, particularly in asymptomatic individuals who wear dentures. Dentures can improve verbal articulation and restore diminished facial contours. Mastication can also be restored; however, patients expecting dentures to facilitate oral intake are often disappointed. Accommodation to dentures requires a period of adjustment. Pain can result from friction or traumatic lesions produced by loose dentures. Poor fit and poor oral hygiene may permit the development of candidiasis. This fungal infection may be either asymptomatic or painful and is suggested by erythematous smooth or granular tissue conforming to an area covered by the appliance. Individuals with dentures and no natural teeth need regular (annual) professional oral examinations.
46e |
Atlas of Oral Manifestations of Disease |
The health status of the oral cavity is linked to cardiovascular disease, diabetes, and other systemic illnesses. Thus, examining the oral cavity for signs of disease is a key part of the physical exam. This chapter presents numerous outstanding clinical photographs illustrating many of the conditions discussed in Chap. 45, Oral Manifestations of Disease. Conditions affecting the teeth, periodontal tissues, and oral mucosa are all represented.
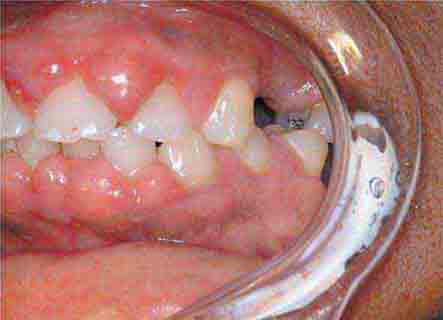
FIGURE 46e-1 Gingival overgrowth secondary to calcium channel blocker use.
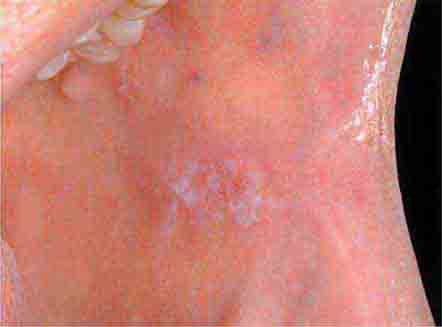
FIGURE 46e-2 Oral lichen planus.
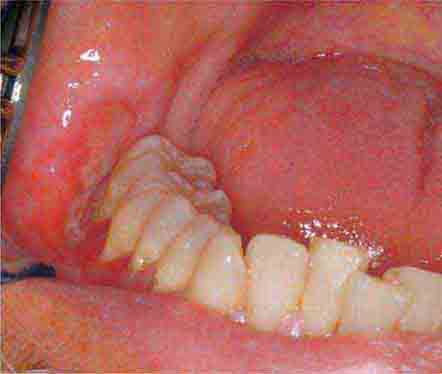
FIGURE 46e-3 Erosive lichen planus.
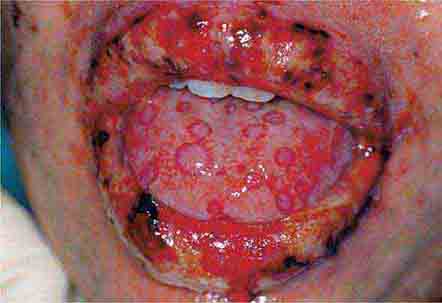
FIGURE 46e-4 Stevens-Johnson syndrome—reaction to nevirapine.
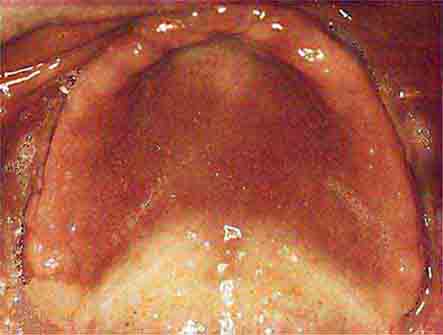
FIGURE 46e-5 Erythematosus candidiasis under a denture (i.e., the patient should be treated for this fungal infection).
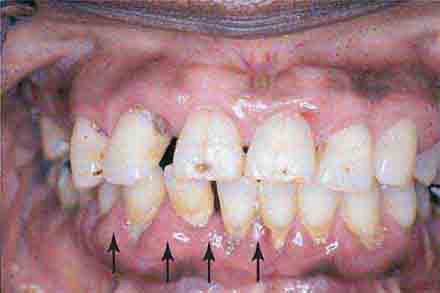
FIGURE 46e-6 Severe periodontitis.
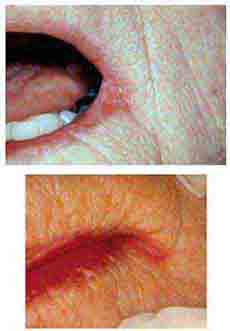
FIGURE 46e-7 Angular cheilitis.
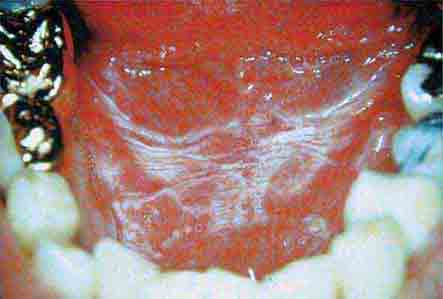
FIGURE 46e-8 Sublingual leukoplakia.
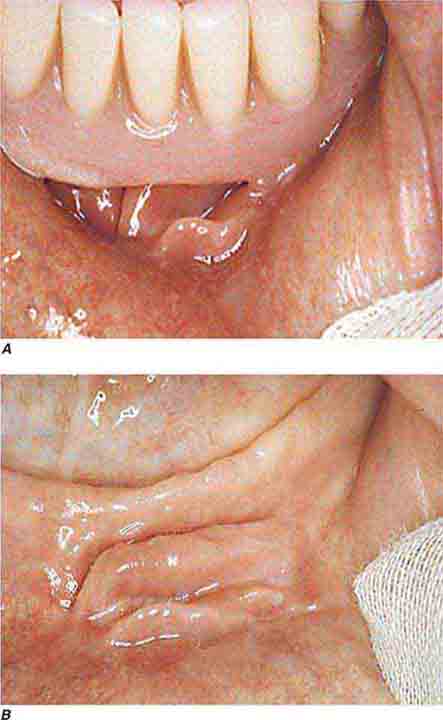
FIGURE 46e-9 A. Epulis (gingival hypertrophy) under denture. B. Epulis fissuratum.
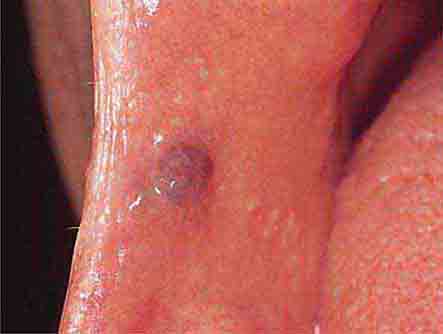
FIGURE 46e-10 Traumatic lesion inside of cheek.
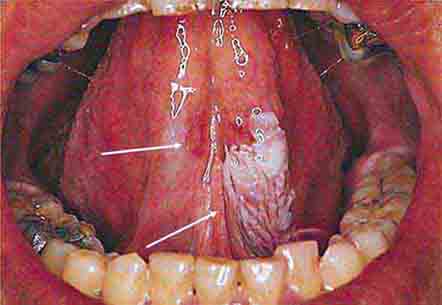
FIGURE 46e-11 Oral leukoplakia, subtype homogenous leukoplakia.
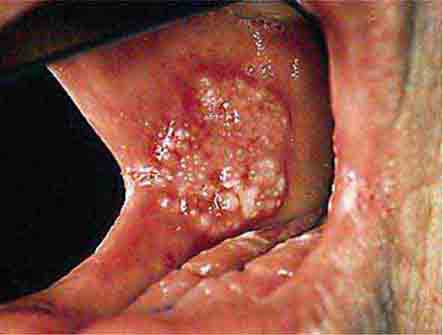
FIGURE 46e-12 Oral carcinoma.
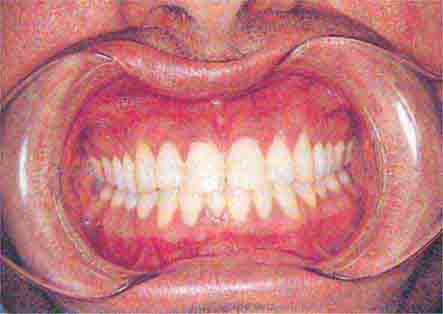
FIGURE 46e-13 Healthy mouth.
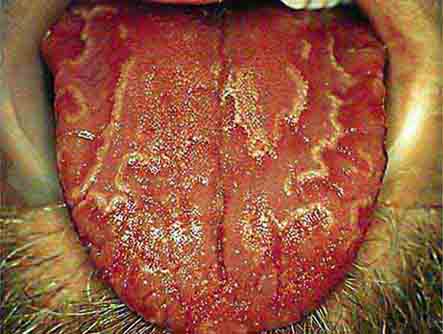
FIGURE 46e-14 Geographic tongue.
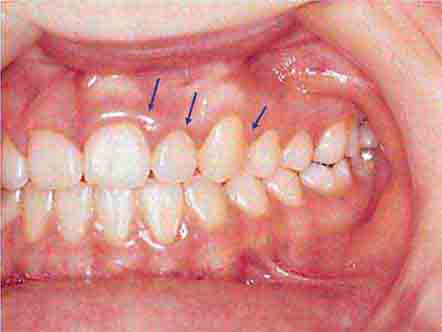
FIGURE 46e-15 Moderate gingivitis.
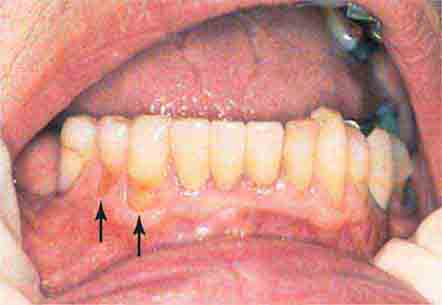
FIGURE 46e-16 Gingival recession.
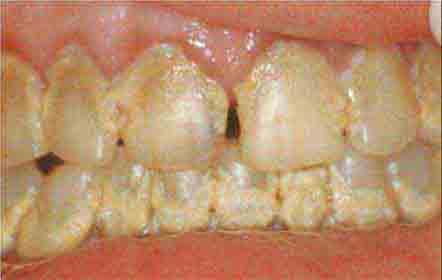
FIGURE 46e-17 Heavy calculus and gingival inflammation.
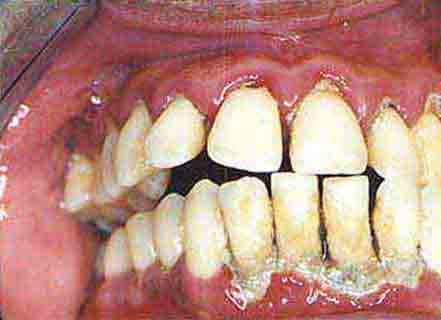
FIGURE 46e-18 Severe gingival inflammation and heavy calculus.
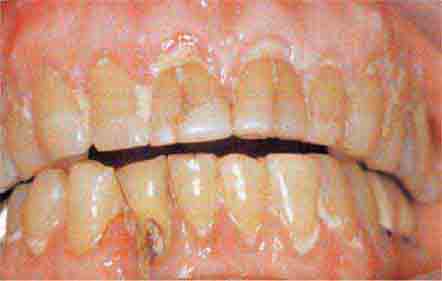
FIGURE 46e-19 Root cavity in presence of severe periodontal disease.
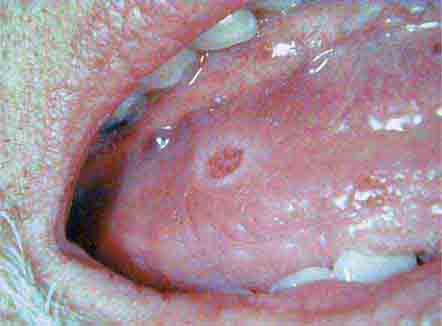
FIGURE 46e-20 Ulcer on lateral border of tongue—potential carcinoma.
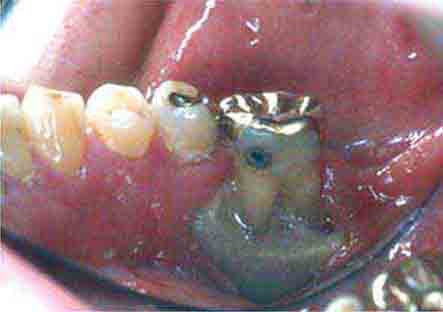
FIGURE 46e-21 Osteonecrosis.
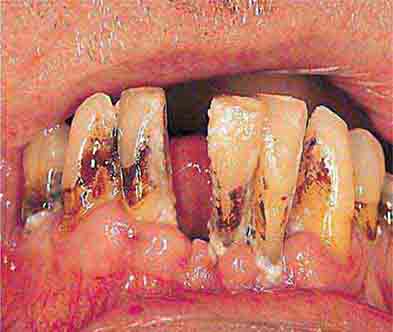
FIGURE 46e-22 Severe periodontal disease, missing tooth, very mobile teeth.
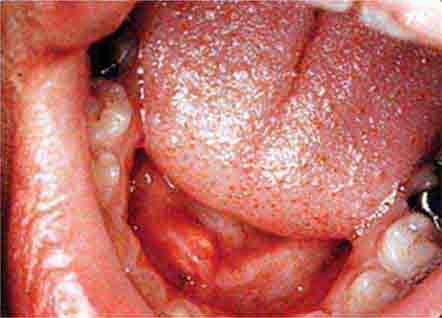
FIGURE 46e-23 Salivary stone.
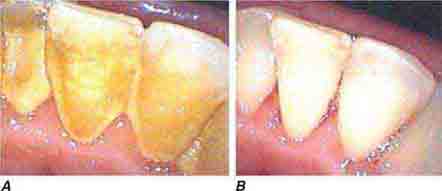
FIGURE 46e-24 A. Calculus. B. Teeth cleaned.
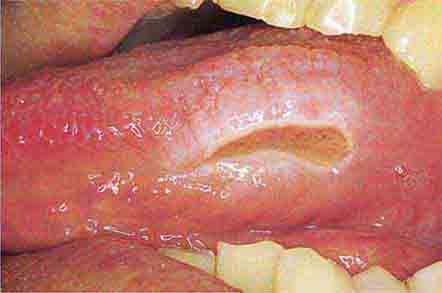
FIGURE 46e-25 Traumatic ulcer.
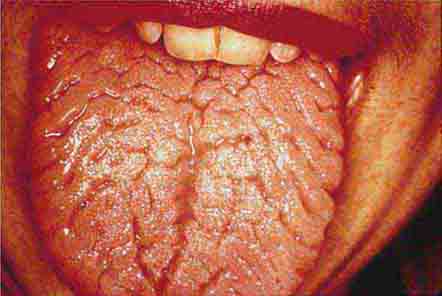
FIGURE 46e-26 Fissured tongue.
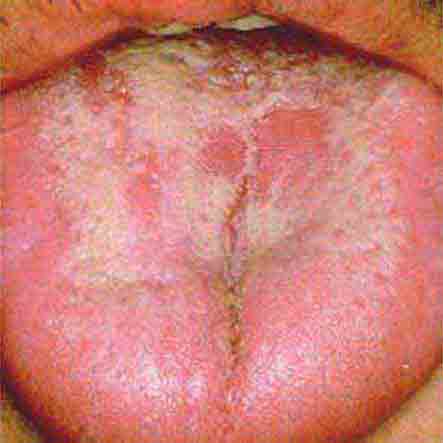
FIGURE 46e-27 White coated tongue—likely candidiasis.
ACKNOWLEDGMENT
Dr. Jane Atkinson was a co-author of this chapter in the 17th edition. Some of the materials have been carried over into this edition.
SECTION 5 |
ALTERATIONS IN CIRCULATORY AND RESPIRATORY FUNCTIONS |
47e |
Dyspnea |
DYSPNEA
The American Thoracic Society defines dyspnea as a “subjective experience of breathing discomfort that consists of qualitatively distinct sensations that vary in intensity. The experience derives from interactions among multiple physiological, psychological, social, and environmental factors and may induce secondary physiological and behavioral responses.” Dyspnea, a symptom, can be perceived only by the person experiencing it and must be distinguished from the signs of increased work of breathing.
MECHANISMS OF DYSPNEA
Respiratory sensations are the consequence of interactions between the efferent, or outgoing, motor output from the brain to the ventilatory muscles (feed-forward) and the afferent, or incoming, sensory input from receptors throughout the body (feedback) as well as the integrative processing of this information that we infer must be occurring in the brain (Fig. 47e-1). In contrast to painful sensations, which can often be attributed to the stimulation of a single nerve ending, dyspnea sensations are more commonly viewed as holistic, more akin to hunger or thirst. A given disease state may lead to dyspnea by one or more mechanisms, some of which may be operative under some circumstances (e.g., exercise) but not others (e.g., a change in position).
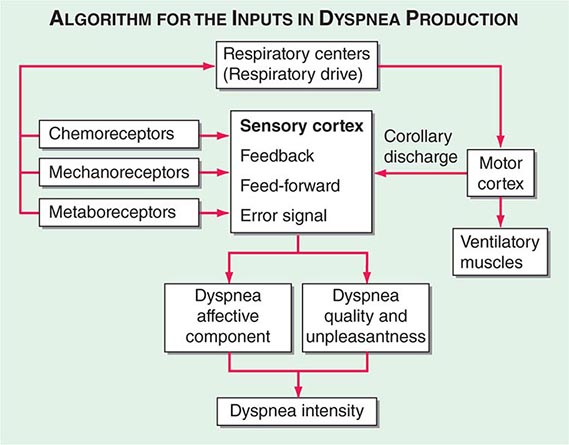
FIGURE 47e-1 Hypothetical model for integration of sensory inputs in the production of dyspnea. Afferent information from the receptors throughout the respiratory system projects directly to the sensory cortex to contribute to primary qualitative sensory experiences and to provide feedback on the action of the ventilatory pump. Afferents also project to the areas of the brain responsible for control of ventilation. The motor cortex, responding to input from the control centers, sends neural messages to the ventilatory muscles and a corollary discharge to the sensory cortex (feed-forward with respect to the instructions sent to the muscles). If the feed-forward and feedback messages do not match, an error signal is generated and the intensity of dyspnea increases. An increasing body of data supports the contribution of affective inputs to the ultimate perception of unpleasant respiratory sensations. (Adapted from MA Gillette, RM Schwartzstein, in SH Ahmedzai, MF Muer [eds]. Supportive Care in Respiratory Disease. Oxford, UK, Oxford University Press, 2005.)
Motor Efferents Disorders of the ventilatory pump—most commonly, increased airway resistance or stiffness (decreased compliance) of the respiratory system—are associated with increased work of breathing or the sense of an increased effort to breathe. When the muscles are weak or fatigued, greater effort is required, even though the mechanics of the system are normal. The increased neural output from the motor cortex is sensed via a corollary discharge, a neural signal that is sent to the sensory cortex at the same time that motor output is directed to the ventilatory muscles.
Sensory Afferents Chemoreceptors in the carotid bodies and medulla are activated by hypoxemia, acute hypercapnia, and acidemia. Stimulation of these receptors and of others that lead to an increase in ventilation produce a sensation of “air hunger.” Mechanoreceptors in the lungs, when stimulated by bronchospasm, lead to a sensation of chest tightness. J-receptors, which are sensitive to interstitial edema, and pulmonary vascular receptors, which are activated by acute changes in pulmonary artery pressure, appear to contribute to air hunger. Hyperinflation is associated with the sensation of increased work of breathing, an inability to get a deep breath, or an unsatisfying breath. Metaboreceptors, which are located in skeletal muscle, are believed to be activated by changes in the local biochemical milieu of the tissue active during exercise and, when stimulated, contribute to breathing discomfort.
Integration: Efferent-Reafferent Mismatch A discrepancy or mismatch between the feed-forward message to the ventilatory muscles and the feedback from receptors that monitor the response of the ventilatory pump increases the intensity of dyspnea. This mismatch is particularly important when there is a mechanical derangement of the ventilatory pump, as in asthma or chronic obstructive pulmonary disease (COPD).
Contribution of Emotional or Affective Factors to Dyspnea Acute anxiety or fear may increase the severity of dyspnea either by altering the interpretation of sensory data or by leading to patterns of breathing that heighten physiologic abnormalities in the respiratory system. In patients with expiratory flow limitation, for example, the increased respiratory rate that accompanies acute anxiety leads to hyperinflation, increased work and effort of breathing, and the sense of an unsatisfying breath.
ASSESSING DYSPNEA
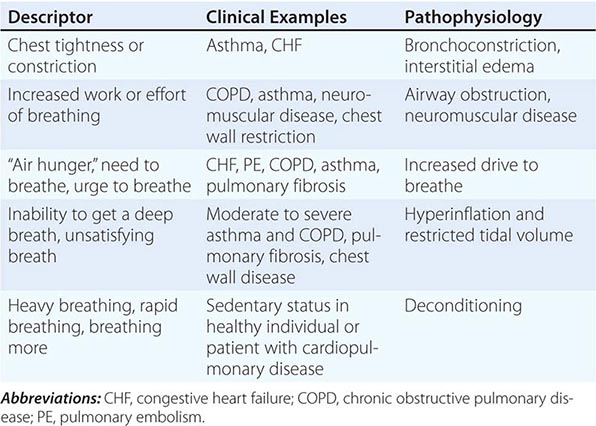
Quality of Sensation Like pain assessment, dyspnea assessment begins with a determination of the quality of the patient’s discomfort (Table 47e-1). Dyspnea questionnaires or lists of phrases commonly used by patients assist those who have difficulty describing their breathing sensations.
|
ASSOCIATION OF QUALITATIVE DESCRIPTORS, CLINICAL CHARACTERISTICS, AND PATHOPHYSIOLOGIC MECHANISMS OF SHORTNESS OF BREATH |
Sensory Intensity A modified Borg scale or visual analogue scale can be utilized to measure dyspnea at rest, immediately following exercise, or on recall of a reproducible physical task, such as climbing the stairs at home. An alternative approach is to gain a sense of the patient’s disability by inquiring about what activities are possible. These methods indirectly assess dyspnea and may be affected by nonrespiratory factors, such as leg arthritis or weakness. The Baseline Dyspnea Index and the Chronic Respiratory Disease Questionnaire are commonly used tools for this purpose.
Affective Dimension For a sensation to be reported as a symptom, it must be perceived as unpleasant and interpreted as abnormal. Laboratory studies have demonstrated that air hunger evokes a stronger affective response than does increased effort or work of breathing. Some therapies for dyspnea, such as pulmonary rehabilitation, may reduce breathing discomfort, in part, by altering this dimension.
DIFFERENTIAL DIAGNOSIS
Dyspnea most often results from deviations from normal function in the cardiovascular and respiratory systems. These deviations produce breathlessness as a consequence of increased drive to breathe; increased effort or work of breathing; and/or stimulation of receptors in the heart, lungs, or vascular system. Most diseases of the respiratory system are associated with alterations in the mechanical properties of the lungs and/or chest wall, and some stimulate pulmonary receptors. In contrast, disorders of the cardiovascular system more commonly lead to dyspnea by causing gas-exchange abnormalities or stimulating pulmonary and/or vascular receptors (Table 47e-2).
|
MECHANISMS OF DYSPNEA IN COMMON DISEASES |
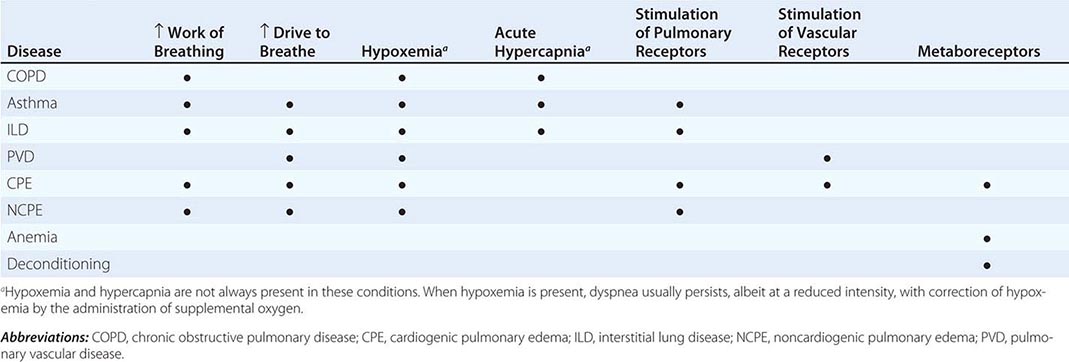
Respiratory System Dyspnea • DISEASES OF THE AIRWAYS Asthma and COPD, the most common obstructive lung diseases, are characterized by expiratory airflow obstruction, which typically leads to dynamic hyperinflation of the lungs and chest wall. Patients with moderate to severe disease have both increased resistive and elastic loads (a term that relates to the stiffness of the system) on the ventilatory muscles and experience increased work of breathing. Patients with acute bronchoconstriction also report a sense of tightness, which can exist even when lung function is still within the normal range. These patients are commonly tachypneic; this condition leads to hyperinflation and reduced respiratory system compliance and also limits tidal volume. Both the chest tightness and the tachypnea are probably due to stimulation of pulmonary receptors. Both asthma and COPD may lead to hypoxemia and hypercapnia from ventilation-perfusion (![]() /Q) mismatch (and diffusion limitation during exercise with emphysema); hypoxemia is much more common than hypercapnia as a consequence of the different ways in which oxygen and carbon dioxide bind to hemoglobin.
/Q) mismatch (and diffusion limitation during exercise with emphysema); hypoxemia is much more common than hypercapnia as a consequence of the different ways in which oxygen and carbon dioxide bind to hemoglobin.
DISEASES OF THE CHEST WALL Conditions that stiffen the chest wall, such as kyphoscoliosis, or that weaken ventilatory muscles, such as myasthenia gravis or the Guillain-Barré syndrome, are also associated with an increased effort to breathe. Large pleural effusions may contribute to dyspnea, both by increasing the work of breathing and by stimulating pulmonary receptors if there is associated atelectasis.
DISEASES OF THE LUNG PARENCHYMA Interstitial lung diseases, which may arise from infections, occupational exposures, or autoimmune disorders, are associated with increased stiffness (decreased compliance) of the lungs and increased work of breathing. In addition, ![]() /Q mismatch and the destruction and/or thickening of the alveolar-capillary interface may lead to hypoxemia and an increased drive to breathe. Stimulation of pulmonary receptors may further enhance the hyperventilation characteristic of mild to moderate interstitial disease.
/Q mismatch and the destruction and/or thickening of the alveolar-capillary interface may lead to hypoxemia and an increased drive to breathe. Stimulation of pulmonary receptors may further enhance the hyperventilation characteristic of mild to moderate interstitial disease.
Cardiovascular System Dyspnea • DISEASES OF THE LEFT HEART Diseases of the myocardium resulting from coronary artery disease and nonischemic cardiomyopathies cause a greater left-ventricular end-diastolic volume and an elevation of the left-ventricular end-diastolic as well as pulmonary capillary pressures. These elevated pressures lead to interstitial edema and stimulation of pulmonary receptors, thereby causing dyspnea; hypoxemia due to ![]() /Q mismatch may also contribute to breathlessness. Diastolic dysfunction, characterized by a very stiff left ventricle, may lead to severe dyspnea with relatively mild degrees of physical activity, particularly if it is associated with mitral regurgitation.
/Q mismatch may also contribute to breathlessness. Diastolic dysfunction, characterized by a very stiff left ventricle, may lead to severe dyspnea with relatively mild degrees of physical activity, particularly if it is associated with mitral regurgitation.
DISEASES OF THE PULMONARY VASCULATURE Pulmonary thromboembolic disease and primary diseases of the pulmonary circulation (primary pulmonary hypertension, pulmonary vasculitis) cause dyspnea via increased pulmonary-artery pressure and stimulation of pulmonary receptors. Hyperventilation is common, and hypoxemia may be present. However, in most cases, use of supplemental oxygen has only a minimal impact on the severity of dyspnea and hyperventilation.
DISEASES OF THE PERICARDIUM Constrictive pericarditis and cardiac tamponade are both associated with increased intracardiac and pulmonary vascular pressures, which are the likely cause of dyspnea in these conditions. To the extent that cardiac output is limited (at rest or with exercise) metaboreceptors may be stimulated if cardiac output is compromised to the degree that lactic acidosis develops; chemoreceptors will also be activated.
Dyspnea with Normal Respiratory and Cardiovascular Systems Mild to moderate anemia is associated with breathing discomfort during exercise. This symptom is thought to be related to stimulation of metaboreceptors; oxygen saturation is normal in patients with anemia. The breathlessness associated with obesity is probably due to multiple mechanisms, including high cardiac output and impaired ventilatory pump function (decreased compliance of the chest wall). Cardiovascular deconditioning (poor fitness) is characterized by the early development of anaerobic metabolism and the stimulation of chemoreceptors and metaboreceptors. Dyspnea that is medically unexplained has been associated with increased sensitivity to the unpleasantness of acute hypercapnia.
Dyspnea (see Fig. 47e-2)
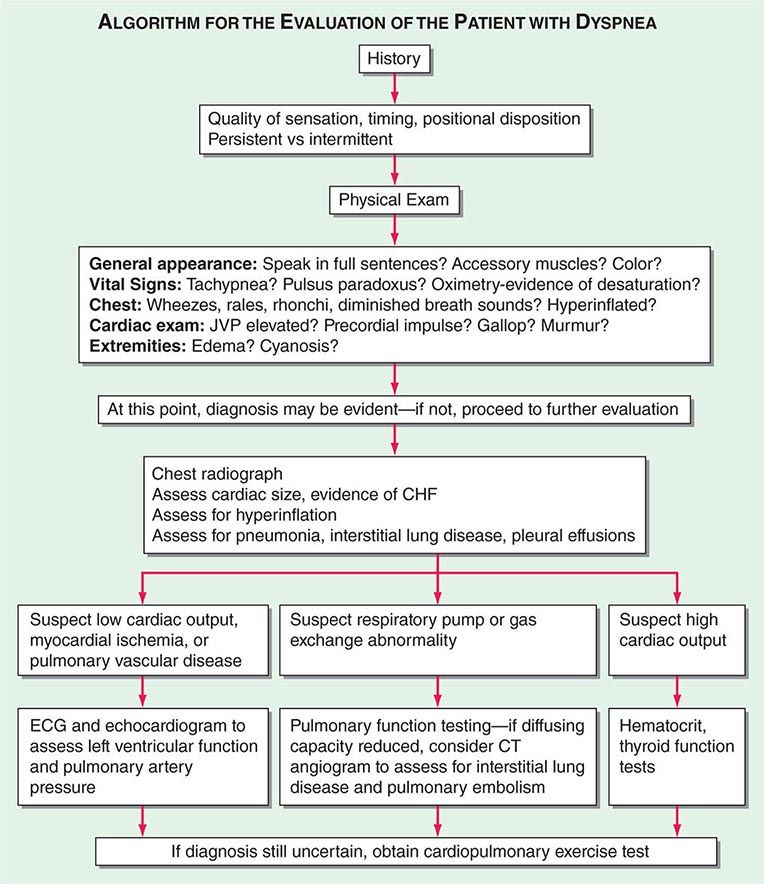
FIGURE 47e-2 Algorithm for the evaluation of the patient with dyspnea. JVP, jugular venous pulse; CHF, congestive heart failure; ECG, electrocardiogram; CT, computed tomography. (Adapted from RM Schwartzstein, D Feller-Kopman, in E Braunwald, L Goldman [eds]. Primary Cardiology, 2nd ed. Philadelphia, WB Saunders, 2003.)
HISTORY
The patient should be asked to describe in his/her own words what the discomfort feels like as well as the effect of position, infections, and environmental stimuli on the dyspnea. Orthopnea is a common indicator of congestive heart failure (CHF), mechanical impairment of the diaphragm associated with obesity, or asthma triggered by esophageal reflux. Nocturnal dyspnea suggests CHF or asthma. Acute, intermittent episodes of dyspnea are more likely to reflect episodes of myocardial ischemia, bronchospasm, or pulmonary embolism, while chronic persistent dyspnea is typical of COPD, interstitial lung disease, and chronic thromboembolic disease. Information on risk factors for occupational lung disease and for coronary artery disease should be elicited. Left atrial myxoma or hepatopulmonary syndrome should be considered when the patient complains of platypnea—i.e., dyspnea in the upright position with relief in the supine position.
PHYSICAL EXAMINATION
The physical examination should begin during the interview of the patient. Inability of the patient to speak in full sentences before stopping to get a deep breath suggests a condition that leads to stimulation of the controller or impairment of the ventilatory pump with reduced vital capacity. Evidence of increased work of breathing (supraclavicular retractions; use of accessory muscles of ventilation; and the tripod position, characterized by sitting with the hands braced on the knees) is indicative of increased airway resistance or stiffness of the lungs and the chest wall. When measuring the vital signs, the physician should accurately assess the respiratory rate and measure the pulsus paradoxus (Chap. 288); if the systolic pressure decreases by >10 mmHg, the presence of COPD, acute asthma, or pericardial disease should be considered. During the general examination, signs of anemia (pale conjunctivae), cyanosis, and cirrhosis (spider angiomata, gynecomastia) should be sought. Examination of the chest should focus on symmetry of movement; percussion (dullness is indicative of pleural effusion; hyperresonance is a sign of emphysema); and auscultation (wheezes, rhonchi, prolonged expiratory phase, and diminished breath sounds are clues to disorders of the airways; rales suggest interstitial edema or fibrosis). The cardiac examination should focus on signs of elevated right heart pressures (jugular venous distention, edema, accentuated pulmonic component to the second heart sound); left ventricular dysfunction (S3 and S4 gallops); and valvular disease (murmurs). When examining the abdomen with the patient in the supine position, the physician should note whether there is paradoxical movement of the abdomen: inward motion during inspiration is a sign of diaphragmatic weakness, and rounding of the abdomen during exhalation is suggestive of pulmonary edema. Clubbing of the digits may be an indication of interstitial pulmonary fibrosis, and joint swelling or deformation as well as changes consistent with Raynaud’s disease may be indicative of a collagen-vascular process that can be associated with pulmonary disease.
Patients with exertional dyspnea should be asked to walk under observation in order to reproduce the symptoms. The patient should be examined during and at the end of exercise for new findings that were not present at rest and for changes in oxygen saturation.
CHEST IMAGING
After the history elicitation and the physical examination, a chest radiograph should be obtained. The lung volumes should be assessed: hyperinflation indicates obstructive lung disease, whereas low lung volumes suggest interstitial edema or fibrosis, diaphragmatic dysfunction, or impaired chest wall motion. The pulmonary parenchyma should be examined for evidence of interstitial disease and emphysema. Prominent pulmonary vasculature in the upper zones indicates pulmonary venous hypertension, while enlarged central pulmonary arteries suggest pulmonary arterial hypertension. An enlarged cardiac silhouette suggests dilated cardiomyopathy or valvular disease. Bilateral pleural effusions are typical of CHF and some forms of collagen-vascular disease. Unilateral effusions raise the specter of carcinoma and pulmonary embolism but may also occur in heart failure. CT of the chest is generally reserved for further evaluation of the lung parenchyma (interstitial lung disease) and possible pulmonary embolism.
LABORATORY STUDIES
Laboratory studies should include electrocardiography to seek evidence of ventricular hypertrophy and prior myocardial infarction. Echocardiography is indicated when systolic dysfunction, pulmonary hypertension, or valvular heart disease is suspected. Bronchoprovocation testing is useful in patients with intermittent symptoms suggestive of asthma but normal physical examination and lung function; up to one-third of patients with the clinical diagnosis of asthma do not have reactive airways disease when formally tested. Measurement of brain natriuretic peptide levels in serum is increasingly used to assess for CHF in patients presenting with acute dyspnea but may be elevated in the presence of right ventricular strain as well.
DISTINGUISHING CARDIOVASCULAR FROM RESPIRATORY SYSTEM DYSPNEA
If a patient has evidence of both pulmonary and cardiac disease, a cardiopulmonary exercise test should be carried out to determine which system is responsible for the exercise limitation. If, at peak exercise, the patient achieves predicted maximal ventilation, demonstrates an increase in dead space or hypoxemia, or develops bronchospasm, the respiratory system is probably the cause of the problem. Alternatively, if the heart rate is >85% of the predicted maximum, if the anaerobic threshold occurs early, if the blood pressure becomes excessively high or decreases during exercise, if the O2 pulse (O2 consumption/heart rate, an indicator of stroke volume) falls, or if there are ischemic changes on the electrocardiogram, an abnormality of the cardiovascular system is likely the explanation for the breathing discomfort.
PULMONARY EDEMA
MECHANISMS OF FLUID ACCUMULATION
The extent to which fluid accumulates in the interstitium of the lung depends on the balance of hydrostatic and oncotic forces within the pulmonary capillaries and in the surrounding tissue. Hydrostatic pressure favors movement of fluid from the capillary into the interstitium. The oncotic pressure, which is determined by the protein concentration in the blood, favors movement of fluid into the vessel. Levels of albumin, the primary protein in the plasma, may be low in conditions such as cirrhosis and nephrotic syndrome. While hypoalbuminemia favors movement of fluid into the tissue for any given hydrostatic pressure in the capillary, it is usually not sufficient by itself to cause interstitial edema. In a healthy individual, the tight junctions of the capillary endothelium are impermeable to proteins, and the lymphatics in the tissue carry away the small amounts of protein that may leak out; together, these factors result in an oncotic force that maintains fluid in the capillary. Disruption of the endothelial barrier, however, allows protein to escape the capillary bed and enhances the movement of fluid into the tissue of the lung.
CARDIOGENIC PULMONARY EDEMA
(See also Chap. 326) Cardiac abnormalities that lead to an increase in pulmonary venous pressure shift the balance of forces between the capillary and the interstitium. Hydrostatic pressure is increased and fluid exits the capillary at an increased rate, resulting in interstitial and, in more severe cases, alveolar edema. The development of pleural effusions may further compromise respiratory system function and contribute to breathing discomfort.
Early signs of pulmonary edema include exertional dyspnea and orthopnea. Chest radiographs show peribronchial thickening, prominent vascular markings in the upper lung zones, and Kerley B lines. As the pulmonary edema worsens, alveoli fill with fluid; the chest radiograph shows patchy alveolar filling, typically in a perihilar distribution, which then progresses to diffuse alveolar infiltrates. Increasing airway edema is associated with rhonchi and wheezes.
NONCARDIOGENIC PULMONARY EDEMA
In noncardiogenic pulmonary edema, lung water increases due to damage of the pulmonary capillary lining with consequent leakage of proteins and other macromolecules into the tissue; fluid follows the protein as oncotic forces are shifted from the vessel to the surrounding lung tissue. This process is associated with dysfunction of the surfactant lining the alveoli, increased surface forces, and a propensity for the alveoli to collapse at low lung volumes. Physiologically, noncardiogenic pulmonary edema is characterized by intrapulmonary shunt with hypoxemia and decreased pulmonary compliance leading to lower functional residual capacity. On pathologic examination, hyaline membranes are evident in the alveoli, and inflammation leading to pulmonary fibrosis may be seen. Clinically, the picture ranges from mild dyspnea to respiratory failure. Auscultation of the lungs may be relatively normal despite chest radiographs that show diffuse alveolar infiltrates. CT scans demonstrate that the distribution of alveolar edema is more heterogeneous than was once thought. Although normal intracardiac pressures are considered by many to be part of the definition of noncardiogenic pulmonary edema, the pathology of the process, as described above, is distinctly different, and a combination of cardiogenic and noncardiogenic pulmonary edema is observed in some patients.
It is useful to categorize the causes of noncardiogenic pulmonary edema in terms of whether the injury to the lung is likely to result from direct, indirect, or pulmonary vascular causes (Table 47e-3). Direct injuries are mediated via the airways (e.g., aspiration) or as the consequence of blunt chest trauma. Indirect injury is the consequence of mediators that reach the lung via the bloodstream. The third category includes conditions that may result from acute changes in pulmonary vascular pressures, possibly due to sudden autonomic discharge (in the case of neurogenic and high-altitude pulmonary edema) or sudden swings of pleural pressure as well as transient damage to the pulmonary capillaries (in the case of reexpansion pulmonary edema).
|
COMMON CAUSES OF NONCARDIOGENIC PULMONARY EDEMA |
DISTINGUISHING CARDIOGENIC FROM NONCARDIOGENIC PULMONARY EDEMA
The history is essential for assessing the likelihood of underlying cardiac disease as well as for identification of one of the conditions associated with noncardiogenic pulmonary edema. The physical examination in cardiogenic pulmonary edema is notable for evidence of increased intracardiac pressures (S3 gallop, elevated jugular venous pulse, peripheral edema) and rales and/or wheezes on auscultation of the chest. In contrast, the physical examination in noncardiogenic pulmonary edema is dominated by the findings of the precipitating condition; pulmonary findings may be relatively normal in the early stages. The chest radiograph in cardiogenic pulmonary edema typically shows an enlarged cardiac silhouette, vascular redistribution, interstitial thickening, and perihilar alveolar infiltrates; pleural effusions are common. In noncardiogenic pulmonary edema, heart size is normal, alveolar infiltrates are distributed more uniformly throughout the lungs, and pleural effusions are uncommon. Finally, the hypoxemia of cardiogenic pulmonary edema is due largely to ![]() /Q to mismatch and responds to the administration of supplemental oxygen. In contrast, hypoxemia in noncardiogenic pulmonary edema is due primarily to intrapulmonary shunting and typically persists despite high concentrations of inhaled oxygen.
/Q to mismatch and responds to the administration of supplemental oxygen. In contrast, hypoxemia in noncardiogenic pulmonary edema is due primarily to intrapulmonary shunting and typically persists despite high concentrations of inhaled oxygen.
48 |
Cough and Hemoptysis |
COUGH
Cough performs an essential protective function for human airways and lungs. Without an effective cough reflex, we are at risk for retained airway secretions and aspirated material predisposing to infection, atelectasis, and respiratory compromise. At the other extreme, excessive coughing can be exhausting; can be complicated by emesis, syncope, muscular pain, or rib fractures; and can aggravate abdominal or inguinal hernias and urinary incontinence. Cough is often a clue to the presence of respiratory disease. In many instances, cough is an expected and accepted manifestation of disease, as in acute respiratory tract infection. However, persistent cough in the absence of other respiratory symptoms commonly causes patients to seek medical attention.
COUGH MECHANISM
Spontaneous cough is triggered by stimulation of sensory nerve endings that are thought to be primarily rapidly adapting receptors and C fibers. Both chemical (e.g., capsaicin) and mechanical (e.g., particulates in air pollution) stimuli may initiate the cough reflex. A cationic ion channel—the type 1 vanilloid receptor—found on rapidly adapting receptors and C fibers is the receptor for capsaicin, and its expression is increased in patients with chronic cough. Afferent nerve endings richly innervate the pharynx, larynx, and airways to the level of the terminal bronchioles and extend into the lung parenchyma. They may also be located in the external auditory meatus (the auricular branch of the vagus nerve, or the Arnold nerve) and in the esophagus. Sensory signals travel via the vagus and superior laryngeal nerves to a region of the brainstem in the nucleus tractus solitarius vaguely identified as the “cough center.” The cough reflex involves a highly orchestrated series of involuntary muscular actions, with the potential for input from cortical pathways as well. The vocal cords adduct, leading to transient upper-airway occlusion. Expiratory muscles contract, generating positive intrathoracic pressures as high as 300 mmHg. With sudden release of the laryngeal contraction, rapid expiratory flows are generated, exceeding the normal “envelope” of maximal expiratory flow seen on the flow-volume curve (Fig. 48-1). Bronchial smooth-muscle contraction together with dynamic compression of airways narrows airway lumens and maximizes the velocity of exhalation. The kinetic energy available to dislodge mucus from the inside of airway walls is directly proportional to the square of the velocity of expiratory airflow. A deep breath preceding a cough optimizes the function of the expiratory muscles; a series of repetitive coughs at successively lower lung volumes sweeps the point of maximal expiratory velocity progressively further into the lung periphery.
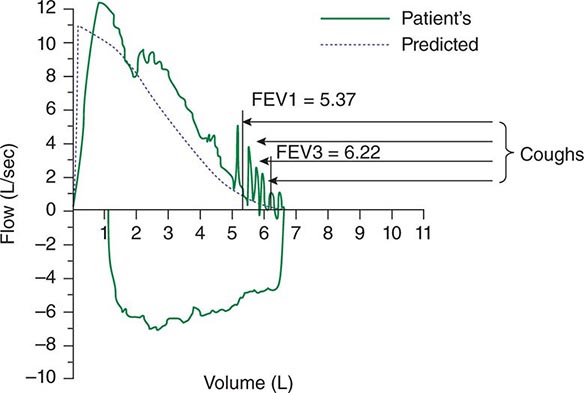
FIGURE 48-1 Flow-volume curve shows spikes of high expiratory flow achieved with cough. FEV1, forced expiratory volume in 1 s.
IMPAIRED COUGH
Weak or ineffective cough compromises the ability to clear lower respiratory tract infections, predisposing to more serious infections and their sequelae. Weakness, paralysis, or pain of the expiratory (abdominal and intercostal) muscles is foremost on the list of causes of impaired cough (Table 48-1). Cough strength is generally assessed qualitatively; peak expiratory flow or maximal expiratory pressure at the mouth can be used as a surrogate marker for cough strength. A variety of assistive devices and techniques have been developed to improve cough strength, running the gamut from simple (splinting of the abdominal muscles with a tightly-held pillow to reduce postoperative pain while coughing) to complex (a mechanical cough-assist device supplied via face mask or tracheal tube that applies a cycle of positive pressure followed rapidly by negative pressure). Cough may fail to clear secretions despite a preserved ability to generate normal expiratory velocities; such failure may be due to either abnormal airway secretions (e.g., bronchiectasis due to cystic fibrosis) or structural abnormalities of the airways (e.g., tracheomalacia with expiratory collapse during cough).
|
CAUSES OF IMPAIRED COUGH |
SYMPTOMATIC COUGH
The cough of chronic bronchitis in long-term cigarette smokers rarely leads the patient to seek medical advice. It lasts for only seconds to a few minutes, is productive of benign-appearing mucoid sputum, and generally does not cause discomfort. Cough may occur in the context of other respiratory symptoms that together point to a diagnosis; for example, cough accompanied by wheezing, shortness of breath, and chest tightness after exposure to a cat or other sources of allergens suggests asthma. At times, however, cough is the dominant or sole symptom of disease, and it may be of sufficient duration and severity that relief is sought. The duration of cough is a clue to its etiology. Acute cough (<3 weeks) is most commonly due to a respiratory tract infection, aspiration, or inhalation of noxious chemicals or smoke. Subacute cough (3–8 weeks in duration) is a common residuum of tracheobronchitis, as in pertussis or “postviral tussive syndrome.” Chronic cough (>8 weeks) may be caused by a wide variety of cardiopulmonary diseases, including those of inflammatory, infectious, neoplastic, and cardiovascular etiologies. When initial assessment with chest examination and radiography is normal, cough-variant asthma, gastroesophageal reflux, nasopharyngeal drainage, and medications (angiotensin-converting enzyme [ACE] inhibitors) are the most common causes of chronic cough.
ASSESSMENT OF CHRONIC COUGH
Details as to the sound, the time of occurrence during the day, and the pattern of coughing infrequently provide useful etiologic clues. Regardless of cause, cough often worsens upon first lying down at night, with talking, or with the hyperpnea of exercise; it frequently improves with sleep. An exception may involve the cough that occurs only with certain allergic exposures or exercise in cold air, as in asthma. Useful historical questions include what circumstances surround the onset of cough, what makes the cough better or worse, and whether or not the cough produces sputum.
The physical examination seeks clues suggesting the presence of cardiopulmonary disease, including findings such as wheezing or crackles on chest examination. Examination of the auditory canals and tympanic membranes (for irritation of the latter resulting in stimulation of Arnold’s nerve), the nasal passageways (for rhinitis or polyps), and the nails (for clubbing) may also provide etiologic clues. Because cough can be a manifestation of a systemic disease such as sarcoidosis or vasculitis, a thorough general examination is equally important.
In virtually all instances, evaluation of chronic cough merits a chest radiograph. The list of diseases that can cause persistent cough without other symptoms and without detectable abnormalities on physical examination is long. It includes serious illnesses such as sarcoidosis or Hodgkin’s disease in young adults, lung cancer in older patients, and (worldwide) pulmonary tuberculosis. An abnormal chest film prompts an evaluation aimed at explaining the cough. In a patient with chronic productive cough, examination of expectorated sputum is warranted. Purulent-appearing sputum should be sent for routine bacterial culture and, in certain circumstances, mycobacterial culture as well. Cytologic examination of mucoid sputum may be useful to assess for malignancy and to distinguish neutrophilic from eosinophilic bronchitis. Expectoration of blood—whether streaks of blood, blood mixed with airway secretions, or pure blood—deserves a special approach to assessment and management (see “Hemoptysis,” below).
CHRONIC COUGH WITH A NORMAL CHEST RADIOGRAPH
It is commonly held that (alone or in combination) the use of an ACE inhibitor; postnasal drainage; gastroesophageal reflux; and asthma account for more than 90% of cases of chronic cough with a normal or noncontributory chest radiograph. However, clinical experience does not support this contention, and strict adherence to this concept discourages the search for alternative explanations by both clinicians and researchers. ACE inhibitor–induced cough occurs in 5–30% of patients taking these agents and is not dose dependent. ACE metabolizes bradykinin and other tachykinins, such as substance P. The mechanism of ACE inhibitor–associated cough may involve sensitization of sensory nerve endings due to accumulation of bradykinin. In support of this hypothesis, polymorphisms in the neurokinin-2 receptor gene are associated with ACE inhibitor–induced cough. Any patient with chronic unexplained cough who is taking an ACE inhibitor should have a trial period off the medication, regardless of the timing of the onset of cough relative to the initiation of ACE inhibitor therapy. In most instances, a safe alternative is available; angiotensin-receptor blockers do not cause cough. Failure to observe a decrease in cough after 1 month off medication argues strongly against this etiology. Postnasal drainage of any etiology can cause cough as a response to stimulation of sensory receptors of the cough-reflex pathway in the hypopharynx or aspiration of draining secretions into the trachea. Clues suggesting this etiology include postnasal drip, frequent throat clearing, and sneezing and rhinorrhea. On speculum examination of the nose, excess mucoid or purulent secretions, inflamed and edematous nasal mucosa, and/or polyps may be seen; in addition, secretions or a cobblestoned appearance of the mucosa along the posterior pharyngeal wall may be noted. Unfortunately, there is no means by which to quantitate postnasal drainage. In many instances, this diagnosis must rely on subjective information provided by the patient. This assessment must also be counterbalanced by the fact that many people who have chronic postnasal drainage do not experience cough.
Linking gastroesophageal reflux to chronic cough poses similar challenges. It is thought that reflux of gastric contents into the lower esophagus may trigger cough via reflex pathways initiated in the esophageal mucosa. Reflux to the level of the pharynx (laryngopharyngeal reflux), with consequent aspiration of gastric contents, causes a chemical bronchitis and possibly pneumonitis that can elicit cough for days afterward. Retrosternal burning after meals or on recumbency, frequent eructation, hoarseness, and throat pain may be indicative of gastroesophageal reflux. Nevertheless, reflux may also elicit minimal or no symptoms. Glottic inflammation detected on laryngoscopy may be a manifestation of recurrent reflux to the level of the throat, but it is a nonspecific finding. Quantification of the frequency and level of reflux requires a somewhat invasive procedure to measure esophageal pH directly (either nasopharyngeal placement of a catheter with a pH probe into the esophagus for 24 h or endoscopic placement of a radiotransmitter capsule into the esophagus). The precise interpretation of test results that permits an etiologic linking of reflux events and cough remains debated. Again, assigning the cause of cough to gastroesophageal reflux must be weighed against the observation that many people with symptomatic reflux do not experience chronic cough.
Cough alone as a manifestation of asthma is common among children but not among adults. Cough due to asthma in the absence of wheezing, shortness of breath, and chest tightness is referred to as “cough-variant asthma.” A history suggestive of cough-variant asthma ties the onset of cough to exposure to typical triggers for asthma and the resolution of cough to discontinuation of exposure. Objective testing can establish the diagnosis of asthma (airflow obstruction on spirometry that varies over time or reverses in response to a bronchodilator) or exclude it with certainty (a negative response to a bronchoprovocation challenge—e.g., with methacholine). In a patient capable of taking reliable measurements, home expiratory peak flow monitoring can be a cost-effective method to support or discount a diagnosis of asthma.
Chronic eosinophilic bronchitis causes chronic cough with a normal chest radiograph. This condition is characterized by sputum eosinophilia in excess of 3% without airflow obstruction or bronchial hyperresponsiveness and is successfully treated with inhaled glucocorticoids.
Treatment of chronic cough in a patient with a normal chest radiograph is often empirical and is targeted at the most likely cause(s) of cough as determined by history, physical examination, and possibly pulmonary-function testing. Therapy for postnasal drainage depends on the presumed etiology (infection, allergy, or vasomotor rhinitis) and may include systemic antihistamines; antibiotics; nasal saline irrigation; and nasal pump sprays with glucocorticoids, antihistamines, or anticholinergics. Antacids, histamine type 2 (H2) receptor antagonists, and proton-pump inhibitors are used to neutralize or decrease the production of gastric acid in gastroesophageal reflux disease; dietary changes, elevation of the head and torso during sleep, and medications to improve gastric emptying are additional therapeutic measures. Cough-variant asthma typically responds well to inhaled glucocorticoids and intermittent use of inhaled β-agonist bronchodilators.
Patients who fail to respond to treatment targeting the common causes of chronic cough or who have had these causes excluded by appropriate diagnostic testing should undergo chest CT. Diseases causing cough that may be missed on chest x-ray include tumors, early interstitial lung disease, bronchiectasis, and atypical mycobacterial pulmonary infection. On the other hand, patients with chronic cough who have normal findings on chest examination, lung function testing, oxygenation assessment, and chest CT can be reassured as to the absence of serious pulmonary pathology.
SYMPTOM-BASED TREATMENT OF COUGH
Chronic idiopathic cough, also called cough hypersensitivity syndrome, is distressingly common. It is often experienced as a tickle or sensitivity in the throat, occurs more often in women, and is typically “dry” or at most productive of scant amounts of mucoid sputum. It can be exhausting, interfere with work, and cause social embarrassment. Once serious underlying cardiopulmonary pathology has been excluded, an attempt at cough suppression is appropriate. Most effective are narcotic cough suppressants, such as codeine or hydrocodone, which are thought to act in the “cough center” in the brainstem. The tendency of narcotic cough suppressants to cause drowsiness and constipation and their potential for addictive dependence limit their appeal for long-term use. Dextromethorphan is an over-the-counter, centrally acting cough suppressant with fewer side effects and less efficacy than the narcotic cough suppressants. Dextromethorphan is thought to have a different site of action than narcotic cough suppressants and can be used in combination with them if necessary. Benzonatate is thought to inhibit neural activity of sensory nerves in the cough-reflex pathway. It is generally free of side effects; however, its effectiveness in suppressing cough is variable and unpredictable. Case series have reported benefit from off-label use of gabapentin or amitriptyline for chronic idiopathic cough. Novel cough suppressants without the limitations of currently available agents are greatly needed. Approaches that are being explored include the development of neurokinin receptor antagonists, type 1 vanilloid receptor antagonists, and novel opioid and opioid-like receptor agonists.
HEMOPTYSIS
Hemoptysis, the expectoration of blood from the respiratory tract, can arise at any location from the alveoli to the glottis. It is important to distinguish hemoptysis from epistaxis (bleeding from the nasopharynx) and hematemesis (bleeding from the upper gastrointestinal tract). Hemoptysis can range from the expectoration of blood-tinged sputum to that of life-threatening large volumes of bright red blood. For most patients, any degree of hemoptysis can cause anxiety and often prompts medical evaluation.
While precise epidemiologic data are lacking, the most common etiology of hemoptysis is infection of the medium-sized airways. In the United States, the cause is usually viral or bacterial bronchitis. Hemoptysis can arise in the setting of acute bronchitis or during an exacerbation of chronic bronchitis. Worldwide, the most common cause of hemoptysis is infection with Mycobacterium tuberculosis, presumably because of the high prevalence of tuberculosis and its predilection for cavity formation. While these are the most common causes, the differential diagnosis for hemoptysis is extensive, and a step-wise approach to evaluation is appropriate.
ETIOLOGY
One way to approach the source of hemoptysis is to search systematically for potential sites of bleeding from the alveolus to the mouth. Diffuse bleeding in the alveolar space, often referred to as diffuse alveolar hemorrhage (DAH), may present as hemoptysis. Causes of DAH can be inflammatory or noninflammatory. Inflammatory DAH is due to small-vessel vasculitis/capillaritis from a variety of diseases, including granulomatosis with polyangiitis and microscopic polyangiitis. Similarly, systemic autoimmune diseases such as systemic lupus erythematosus can manifest as pulmonary capillaritis. Antibodies to the alveolar basement membrane, as are seen in Goodpasture’s disease, can also result in alveolar hemorrhage. In the early period after bone marrow transplantation, patients can develop a form of inflammatory DAH that can be catastrophic and life-threatening. The exact pathophysiology of this process is not well understood, but DAH should be suspected in patients with sudden-onset dyspnea and hypoxemia in the first 100 days after bone marrow transplantation.
Alveoli can also bleed due to direct inhalational injury, including thermal injury from fires, inhalation of illicit substances (e.g., cocaine), and inhalation of toxic chemicals. If alveoli are irritated from any process, patients with thrombocytopenia, coagulopathy, or antiplatelet or anticoagulant use will be at increased risk of hemoptysis.
Bleeding in hemoptysis most commonly arises from the small- to medium-sized airways. Irritation and injury of the bronchial mucosa can lead to small-volume bleeding. More significant hemoptysis can result from the proximity of the bronchial artery and vein to the airway, with these vessels and the bronchus running together in what is often referred to as the bronchovascular bundle. In the smaller airways, these blood vessels are close to the airspace, and lesser degrees of inflammation or injury can therefore result in their rupture into the airways. While alveolar hemorrhage arises from capillaries that are part of the low-pressure pulmonary circulation, bronchial bleeding generally originates from bronchial arteries, which are under systemic pressure and thus are predisposed to larger-volume bleeding.
Any infection of the airways can result in hemoptysis, although acute bronchitis is most commonly caused by viral infection. In patients with a history of chronic bronchitis, bacterial superinfection with organisms such as Streptococcus pneumoniae, Haemophilus influenzae, or Moraxella catarrhalis can also result in hemoptysis. Patients with bronchiectasis (a permanent dilation of the airways with loss of mucosal integrity) are particularly prone to hemoptysis due to chronic inflammation and anatomic abnormalities that bring the bronchial arteries closer to the mucosal surface. One common presentation of patients with advanced cystic fibrosis—the prototypical bronchiectatic lung disease—is hemoptysis, which can be life-threatening.
Pneumonias of any sort can cause hemoptysis. Tuberculous infection, which can lead to bronchiectasis or cavitary pneumonia, is a very common cause of hemoptysis worldwide. Patients may present with a chronic cough productive of blood-streaked sputum or with larger-volume bleeding. Rasmussen’s aneurysm (the dilation of a pulmonary artery in a cavity formed by previous tuberculous infection) remains a source of massive, life-threatening hemoptysis in the developing world. Community-acquired pneumonia and lung abscess can also result in bleeding. Once again, if the infection results in cavitation, there is a greater likelihood of bleeding due to erosion into blood vessels. Infections with Staphylococcus aureus and gram-negative rods (e.g., Klebsiella pneumoniae) are especially likely to cause necrotizing lung infections and thus to be associated with hemoptysis.
![]() While not common in North America, pulmonary paragonimiasis (i.e., infection with the lung fluke Paragonimus westermani) often presents as fever, cough, and hemoptysis. This infection is a public health issue in Southeast Asia and China and is frequently confused with active tuberculosis, in which the clinical picture can be similar. Paragonimiasis should be considered in recent immigrants from endemic areas who have new or recurrent hemoptysis. In addition, pulmonary paragonimiasis has been reported secondary to ingestion of crayfish or small crabs in the United States.
While not common in North America, pulmonary paragonimiasis (i.e., infection with the lung fluke Paragonimus westermani) often presents as fever, cough, and hemoptysis. This infection is a public health issue in Southeast Asia and China and is frequently confused with active tuberculosis, in which the clinical picture can be similar. Paragonimiasis should be considered in recent immigrants from endemic areas who have new or recurrent hemoptysis. In addition, pulmonary paragonimiasis has been reported secondary to ingestion of crayfish or small crabs in the United States.
Other causes of airway irritation resulting in hemoptysis include inhalation of toxic chemicals, thermal injury, and direct trauma from suctioning of the airways (particularly in intubated patients). All of these etiologies should be considered in light of the individual patient’s history and exposures.
Perhaps the most feared cause of hemoptysis is bronchogenic lung cancer, although hemoptysis is a presenting symptom in only ~10% of patients. Cancers arising in the proximal airways are much more likely to cause hemoptysis, but any malignancy in the chest can do so. Because both squamous cell carcinomas and small-cell carcinomas are more commonly in or adjacent to the proximal airways, and large at presentation, they are more often a cause of hemoptysis. These cancers can present with large-volume and life-threatening hemoptysis because of erosion into the hilar vessels. Carcinoid tumors, which are found almost exclusively as endobronchial lesions with friable mucosa, can also present with hemoptysis.
In addition to cancers arising in the lung, metastatic disease in the pulmonary parenchyma can bleed. Malignancies that commonly metastasize to the lungs include renal cell, breast, colon, testicular, and thyroid cancers as well as melanoma. While hemoptysis is not a common manifestation of pulmonary metastases, the combination of multiple pulmonary nodules and hemoptysis should raise suspicion of this etiology. Finally, disease of the pulmonary vasculature can cause hemoptysis. Perhaps most frequently, congestive heart failure with transmission of elevated left atrial pressures can lead to rupture of small alveolar capillaries. These patients rarely present with bright red blood but more commonly have pink, frothy sputum or blood-tinged secretions. Patients with a focal jet of mitral regurgitation can present with an upper-lobe opacity on chest radiography together with hemoptysis. This finding is thought to be due to focal increases in pulmonary capillary pressure due to the regurgitant jet. Pulmonary arteriovenous malformations are prone to bleeding. Pulmonary embolism can also lead to the development of hemoptysis, which is generally associated with pulmonary infarction. Pulmonary arterial hypertension from other causes rarely results in hemoptysis.
EVALUATION
As with most signs of possible illness, the initial step in the evaluation of hemoptysis is a thorough history and physical examination (Fig. 48-2). As already mentioned, initial questioning should focus on ascertaining whether the bleeding is truly from the respiratory tract and not the nasopharynx or gastrointestinal tract; bleeding from the latter sources requires different approaches to evaluation and treatment.
FIGURE 48-2 Decision tree for evaluation of hemoptysis. CBC, complete blood count; CT, computed tomography; CXR, chest x-ray; UA, urinalysis.
History and Physical Examination The specific characteristics of hemoptysis may be helpful in determining an etiology, such as whether the expectorated material consists of blood-tinged, purulent secretions; pink, frothy sputum; or pure blood. Information on specific triggers of the bleeding (e.g., recent inhalation exposures) as well as any previous episodes of hemoptysis should be elicited during history-taking. Monthly hemoptysis in a woman suggests catamenial hemoptysis from pulmonary endometriosis. Moreover, the volume of blood expectorated is important not only in determining the cause but also in gauging the urgency for further diagnostic and therapeutic maneuvers. Patients rarely exsanguinate from hemoptysis but can effectively “drown” in aspirated blood. Large-volume hemoptysis, referred to as massive hemoptysis, is variably defined as hemoptysis of >200–600 mL in 24 h. Massive hemoptysis should be considered a medical emergency. All patients should be asked about current or former cigarette smoking; this behavior predisposes to chronic bronchitis and increases the likelihood of bronchogenic cancer. Practitioners should inquire about symptoms and signs suggestive of respiratory tract infection (including fever, chills, and dyspnea), recent inhalation exposures, recent use of illicit substances, and risk factors for venous thromboembolism.
A medical history of malignancy or treatment thereof, rheumatologic disease, vascular disease, or underlying lung disease (e.g., bronchiectasis) may be relevant to the cause of hemoptysis. Because many causes of DAH can be part of a pulmonary-renal syndrome, specific inquiry into a history of renal insufficiency is important.
The physical examination begins with an assessment of vital signs and oxygen saturation to gauge whether there is evidence of life-threatening bleeding. Tachycardia, hypotension, and decreased oxygen saturation mandate a more expedited evaluation of hemoptysis. A specific focus on respiratory and cardiac examinations is important; these examinations should include inspection of the nares, auscultation of the lungs and heart, assessment of the lower extremities for symmetric or asymmetric edema, and evaluation for jugular venous distention. Clubbing of the digits may suggest underlying lung diseases such as bronchogenic carcinoma or bronchiectasis, which predispose to hemoptysis. Similarly, mucocutaneous telangiectasias should raise the specter of pulmonary arterial-venous malformations.
Diagnostic Evaluation For most patients, the next step in evaluation of hemoptysis should be a standard chest radiograph. If a source of bleeding is not identified on plain film, CT of the chest should be performed. CT allows better delineation of bronchiectasis, alveolar filling, cavitary infiltrates, and masses than does chest radiograph. The practitioner should consider a CT protocol to assess for pulmonary embolism if the history or examination suggests venous thromboembolism as a cause of bleeding.
Laboratory studies should include a complete blood count to assess both the hematocrit and the platelet count as well as coagulation studies. Renal function should be evaluated and urinalysis conducted because of the possibility of pulmonary-renal syndromes presenting with hemoptysis. The documentation of acute renal insufficiency or the detection of red blood cells or their casts on urinalysis should elevate suspicion of small-vessel vasculitis, and studies such as antineutrophil cytoplasmic antibody, antiglomerular basement membrane antibody, and antinuclear antibody should be considered. If a patient is producing sputum, Gram’s and acid-fast staining as well as culture should be undertaken.
If all of these studies are unrevealing, bronchoscopy should be considered. In any patient with a history of cigarette smoking, airway inspection should be part of the evaluation of new-onset hemoptysis as endobronchial lesions are not reliably visualized on CT.
49 |
Hypoxia and Cyanosis |
HYPOXIA
The fundamental purpose of the cardiorespiratory system is to deliver O2 and nutrients to cells and to remove CO2 and other metabolic products from them. Proper maintenance of this function depends not only on intact cardiovascular and respiratory systems, but also on an adequate number of red blood cells and hemoglobin and a supply of inspired gas containing adequate O2.
RESPONSES TO HYPOXIA
Decreased O2 availability to cells results in an inhibition of oxidative phosphorylation and increased anaerobic glycolysis. This switch from aerobic to anaerobic metabolism, the Pasteur effect, maintains some, albeit reduced, adenosine 5′-triphosphate (ATP) production. In severe hypoxia, when ATP production is inadequate to meet the energy requirements of ionic and osmotic equilibrium, cell membrane depolarization leads to uncontrolled Ca2+ influx and activation of Ca2+-dependent phospholipases and proteases. These events, in turn, cause cell swelling, activation of apoptotic pathways, and, ultimately, cell death.
The adaptations to hypoxia are mediated, in part, by the upregulation of genes encoding a variety of proteins, including glycolytic enzymes, such as phosphoglycerate kinase and phosphofructokinase, as well as the glucose transporters Glut-1 and Glut-2; and by growth factors, such as vascular endothelial growth factor (VEGF) and erythropoietin, which enhance erythrocyte production. The hypoxia-induced increase in expression of these key proteins is governed by the hypoxia-sensitive transcription factor, hypoxia-inducible factor-1 (HIF-1).
During hypoxia, systemic arterioles dilate, at least in part, by opening of KATP channels in vascular smooth-muscle cells due to the hypoxia-induced reduction in ATP concentration. By contrast, in pulmonary vascular smooth-muscle cells, inhibition of K+ channels causes depolarization which, in turn, activates voltage-gated Ca2+ channels raising the cytosolic [Ca2+] and causing smooth-muscle cell contraction. Hypoxia-induced pulmonary arterial constriction shunts blood away from poorly ventilated portions toward better ventilated portions of the lung; however, it also increases pulmonary vascular resistance and right ventricular afterload.
Effects on the Central Nervous System Changes in the central nervous system (CNS), particularly the higher centers, are especially important consequences of hypoxia. Acute hypoxia causes impaired judgment, motor incoordination, and a clinical picture resembling acute alcohol intoxication. High-altitude illness is characterized by headache secondary to cerebral vasodilation, gastrointestinal symptoms, dizziness, insomnia, fatigue, or somnolence. Pulmonary arterial and sometimes venous constriction causes capillary leakage and high-altitude pulmonary edema (HAPE) (Chap. 47e), which intensifies hypoxia, further promoting vasoconstriction. Rarely, high-altitude cerebral edema (HACE) develops, which is manifest by severe headache and papilledema and can cause coma. As hypoxia becomes more severe, the regulatory centers of the brainstem are affected, and death usually results from respiratory failure.
Effects on the Cardiovascular System Acute hypoxia stimulates the chemoreceptor reflex arc to induce venoconstriction and systemic arterial vasodilation. These acute changes are accompanied by transiently increased myocardial contractility, which is followed by depressed myocardial contractility with prolonged hypoxia.
CAUSES OF HYPOXIA
Respiratory Hypoxia When hypoxia occurs from respiratory failure, PaO2 declines, and when respiratory failure is persistent, the hemoglobin-oxygen (Hb-O2) dissociation curve (see Fig. 127-2) is displaced to the right, with greater quantities of O2 released at any level of tissue PO2. Arterial hypoxemia, i.e., a reduction of O2 saturation of arterial blood (SaO2), and consequent cyanosis are likely to be more marked when such depression of PaO2 results from pulmonary disease than when the depression occurs as the result of a decline in the fraction of oxygen in inspired air (FIO2). In this latter situation, PaCO2 falls secondary to anoxia-induced hyperventilation and the Hb-O2 dissociation curve is displaced to the left, limiting the decline in SaO2 at any level of PaO2.
The most common cause of respiratory hypoxia is ventilation-perfusion mismatch resulting from perfusion of poorly ventilated alveoli. Respiratory hypoxemia may also be caused by hypoventilation, in which case it is associated with an elevation of PaCO2 (Chap. 306e). These two forms of respiratory hypoxia are usually correctable by inspiring 100% O2 for several minutes. A third cause of respiratory hypoxia is shunting of blood across the lung from the pulmonary arterial to the venous bed (intrapulmonary right-to-left shunting) by perfusion of nonventilated portions of the lung, as in pulmonary atelectasis or through pulmonary arteriovenous connections. The low PaO2 in this situation is only partially corrected by an FIO2 of 100%.
Hypoxia Secondary to High Altitude As one ascends rapidly to 3000 m (~10,000 ft), the reduction of the O2 content of inspired air (FIO2) leads to a decrease in alveolar PO2 to approximately 60 mmHg, and a condition termed high-altitude illness develops (see above). At higher altitudes, arterial saturation declines rapidly and symptoms become more serious; and at 5000 m, unacclimated individuals usually cease to be able to function normally owing to the changes in CNS function described above.
Hypoxia Secondary to Right-to-Left Extrapulmonary Shunting From a physiologic viewpoint, this cause of hypoxia resembles intrapulmonary right-to-left shunting but is caused by congenital cardiac malformations, such as tetralogy of Fallot, transposition of the great arteries, and Eisenmenger’s syndrome (Chap. 282). As in pulmonary right-to-left shunting, the PaO2 cannot be restored to normal with inspiration of 100% O2.
Anemic Hypoxia A reduction in hemoglobin concentration of the blood is accompanied by a corresponding decline in the O2-carrying capacity of the blood. Although the PaO2 is normal in anemic hypoxia, the absolute quantity of O2 transported per unit volume of blood is diminished. As the anemic blood passes through the capillaries and the usual quantity of O2 is removed from it, the PO2 and saturation in the venous blood decline to a greater extent than normal.
Carbon Monoxide (CO) Intoxication (See also Chap. 472e) Hemoglobin that binds with CO (carboxy-hemoglobin, COHb) is unavailable for O2 transport. In addition, the presence of COHb shifts the Hb-O2 dissociation curve to the left (see Fig. 127-2) so that O2 is unloaded only at lower tensions, further contributing to tissue hypoxia.
Circulatory Hypoxia As in anemic hypoxia, the PaO2 is usually normal, but venous and tissue PO2 values are reduced as a consequence of reduced tissue perfusion and greater tissue O2 extraction. This pathophysiology leads to an increased arterial-mixed venous O2 difference (a-v-O2 difference), or gradient. Generalized circulatory hypoxia occurs in heart failure (Chap. 279) and in most forms of shock (Chap. 324).
Specific Organ Hypoxia Localized circulatory hypoxia may occur as a result of decreased perfusion secondary to arterial obstruction, as in localized atherosclerosis in any vascular bed, or as a consequence of vasoconstriction, as observed in Raynaud’s phenomenon (Chap. 302). Localized hypoxia may also result from venous obstruction and the resultant expansion of interstitial fluid causing arteriolar compression and, thereby, reduction of arterial inflow. Edema, which increases the distance through which O2 must diffuse before it reaches cells, can also cause localized hypoxia. In an attempt to maintain adequate perfusion to more vital organs in patients with reduced cardiac output secondary to heart failure or hypovolemic shock, vasoconstriction may reduce perfusion in the limbs and skin, causing hypoxia of these regions.
Increased O2 Requirements If the O2 consumption of tissues is elevated without a corresponding increase in perfusion, tissue hypoxia ensues and the PO2 in venous blood declines. Ordinarily, the clinical picture of patients with hypoxia due to an elevated metabolic rate, as in fever or thyrotoxicosis, is quite different from that in other types of hypoxia: the skin is warm and flushed owing to increased cutaneous blood flow that dissipates the excessive heat produced, and cyanosis is usually absent.
Exercise is a classic example of increased tissue O2 requirements. These increased demands are normally met by several mechanisms operating simultaneously: (1) increase in the cardiac output and ventilation and, thus, O2 delivery to the tissues; (2) a preferential shift in blood flow to the exercising muscles by changing vascular resistances in the circulatory beds of exercising tissues, directly and/or reflexly; (3) an increase in O2 extraction from the delivered blood and a widening of the arteriovenous O2 difference; and (4) a reduction in the pH of the tissues and capillary blood, shifting the Hb-O2 curve to the right (see Fig. 127-2), and unloading more O2 from hemoglobin. If the capacity of these mechanisms is exceeded, then hypoxia, especially of the exercising muscles, will result.
Improper Oxygen Utilization Cyanide (Chap. 473e) and several other similarly acting poisons cause cellular hypoxia. The tissues are unable to use O2, and, as a consequence, the venous blood tends to have a high O2 tension. This condition has been termed histotoxic hypoxia.
ADAPTATION TO HYPOXIA
An important component of the respiratory response to hypoxia originates in special chemosensitive cells in the carotid and aortic bodies and in the respiratory center in the brainstem. The stimulation of these cells by hypoxia increases ventilation, with a loss of CO2, and can lead to respiratory alkalosis. When combined with the metabolic acidosis resulting from the production of lactic acid, the serum bicarbonate level declines (Chap. 66).
With the reduction of PaO2, cerebrovascular resistance decreases and cerebral blood flow increases in an attempt to maintain O2 delivery to the brain. However, when the reduction of PaO2 is accompanied by hyperventilation and a reduction of PaCO2, cerebrovascular resistance rises, cerebral blood flow falls, and tissue hypoxia intensifies.
The diffuse, systemic vasodilation that occurs in generalized hypoxia increases the cardiac output. In patients with underlying heart disease, the requirements of peripheral tissues for an increase of cardiac output with hypoxia may precipitate congestive heart failure. In patients with ischemic heart disease, a reduced PaO2 may intensify myocardial ischemia and further impair left ventricular function.
One of the important compensatory mechanisms for chronic hypoxia is an increase in the hemoglobin concentration and in the number of red blood cells in the circulating blood, i.e., the development of polycythemia secondary to erythropoietin production (Chap. 131). In persons with chronic hypoxemia secondary to prolonged residence at a high altitude (>13,000 ft, 4200 m), a condition termed chronic mountain sickness develops. This disorder is characterized by a blunted respiratory drive, reduced ventilation, erythrocytosis, cyanosis, weakness, right ventricular enlargement secondary to pulmonary hypertension, and even stupor.
CYANOSIS
Cyanosis refers to a bluish color of the skin and mucous membranes resulting from an increased quantity of reduced hemoglobin (i.e., deoxygenated hemoglobin) or of hemoglobin derivatives (e.g., methemoglobin or sulfhemoglobin) in the small blood vessels of those tissues. It is usually most marked in the lips, nail beds, ears, and malar eminences. Cyanosis, especially if developed recently, is more commonly detected by a family member than the patient. The florid skin characteristic of polycythemia vera (Chap. 131) must be distinguished from the true cyanosis discussed here. A cherry-colored flush, rather than cyanosis, is caused by COHb (Chap. 473e).
The degree of cyanosis is modified by the color of the cutaneous pigment and the thickness of the skin, as well as by the state of the cutaneous capillaries. The accurate clinical detection of the presence and degree of cyanosis is difficult, as proved by oximetric studies. In some instances, central cyanosis can be detected reliably when the SaO2 has fallen to 85%; in others, particularly in dark-skinned persons, it may not be detected until it has declined to 75%. In the latter case, examination of the mucous membranes in the oral cavity and the conjunctivae rather than examination of the skin is more helpful in the detection of cyanosis.
The increase in the quantity of reduced hemoglobin in the mucocutaneous vessels that produces cyanosis may be brought about either by an increase in the quantity of venous blood as a result of dilation of the venules (including precapillary venules) or by a reduction in the Sao2 in the capillary blood. In general, cyanosis becomes apparent when the concentration of reduced hemoglobin in capillary blood exceeds 40 g/L (4 g/dL).
It is the absolute, rather than the relative, quantity of reduced hemoglobin that is important in producing cyanosis. Thus, in a patient with severe anemia, the relative quantity of reduced hemoglobin in the venous blood may be very large when considered in relation to the total quantity of hemoglobin in the blood. However, since the concentration of the latter is markedly reduced, the absolute quantity of reduced hemoglobin may still be low, and, therefore, patients with severe anemia and even marked arterial desaturation may not display cyanosis. Conversely, the higher the total hemoglobin content, the greater the tendency toward cyanosis; thus, patients with marked polycythemia tend to be cyanotic at higher levels of SaO2 than patients with normal hematocrit values. Likewise, local passive congestion, which causes an increase in the total quantity of reduced hemoglobin in the vessels in a given area, may cause cyanosis. Cyanosis is also observed when nonfunctional hemoglobin, such as methemoglobin (consequential or acquired) or sulfhemoglobin (Chap. 127), is present in blood.
Cyanosis may be subdivided into central and peripheral types. In central cyanosis, the SaO2 is reduced or an abnormal hemoglobin derivative is present, and the mucous membranes and skin are both affected. Peripheral cyanosis is due to a slowing of blood flow and abnormally great extraction of O2 from normally saturated arterial blood; it results from vasoconstriction and diminished peripheral blood flow, such as occurs in cold exposure, shock, congestive failure, and peripheral vascular disease. Often in these conditions, the mucous membranes of the oral cavity or those beneath the tongue may be spared. Clinical differentiation between central and peripheral cyanosis may not always be simple, and in conditions such as cardiogenic shock with pulmonary edema, there may be a mixture of both types.
DIFFERENTIAL DIAGNOSIS
Central Cyanosis (Table 49-1) Decreased SaO2 results from a marked reduction in the PaO2. This reduction may be brought about by a decline in the FIO2 without sufficient compensatory alveolar hyperventilation to maintain alveolar PO2. Cyanosis usually becomes manifest in an ascent to an altitude of 4000 m (13,000 ft).
|
CAUSES OF CYANOSIS |
Seriously impaired pulmonary function, through perfusion of unventilated or poorly ventilated areas of the lung or alveolar hypoventilation, is a common cause of central cyanosis (Chap. 306e). This condition may occur acutely, as in extensive pneumonia or pulmonary edema, or chronically, with chronic pulmonary diseases (e.g., emphysema). In the latter situation, secondary polycythemia is generally present and clubbing of the fingers (see below) may occur. Another cause of reduced SaO2 is shunting of systemic venous blood into the arterial circuit. Certain forms of congenital heart disease are associated with cyanosis on this basis (see above and Chap. 282).
Pulmonary arteriovenous fistulae may be congenital or acquired, solitary or multiple, microscopic or massive. The severity of cyanosis produced by these fistulae depends on their size and number. They occur with some frequency in hereditary hemorrhagic telangiectasia. Sao2 reduction and cyanosis may also occur in some patients with cirrhosis, presumably as a consequence of pulmonary arteriovenous fistulae or portal vein–pulmonary vein anastomoses.
In patients with cardiac or pulmonary right-to-left shunts, the presence and severity of cyanosis depend on the size of the shunt relative to the systemic flow as well as on the Hb-O2 saturation of the venous blood. With increased extraction of O2 from the blood by the exercising muscles, the venous blood returning to the right side of the heart is more unsaturated than at rest, and shunting of this blood intensifies the cyanosis. Secondary polycythemia occurs frequently in patients in this setting and contributes to the cyanosis.
Cyanosis can be caused by small quantities of circulating methemoglobin (Hb Fe3+) and by even smaller quantities of sulfhemoglobin (Chap. 127); both of these hemoglobin derivatives impair oxygen delivery to the tissues. Although they are uncommon causes of cyanosis, these abnormal hemoglobin species should be sought by spectroscopy when cyanosis is not readily explained by malfunction of the circulatory or respiratory systems. Generally, digital clubbing does not occur with them.
Peripheral Cyanosis Probably the most common cause of peripheral cyanosis is the normal vasoconstriction resulting from exposure to cold air or water. When cardiac output is reduced, cutaneous vasoconstriction occurs as a compensatory mechanism so that blood is diverted from the skin to more vital areas such as the CNS and heart, and cyanosis of the extremities may result even though the arterial blood is normally saturated.
Arterial obstruction to an extremity, as with an embolus, or arteriolar constriction, as in cold-induced vasospasm (Raynaud’s phenomenon) (Chap. 302), generally results in pallor and coldness, and there may be associated cyanosis. Venous obstruction, as in thrombophlebitis or deep venous thrombosis, dilates the subpapillary venous plexuses and thereby intensifies cyanosis.
CLUBBING
The selective bulbous enlargement of the distal segments of the fingers and toes due to proliferation of connective tissue, particularly on the dorsal surface, is termed clubbing; there is also increased sponginess of the soft tissue at the base of the clubbed nail. Clubbing may be hereditary, idiopathic, or acquired and associated with a variety of disorders, including cyanotic congenital heart disease (see above), infective endocarditis, and a variety of pulmonary conditions (among them primary and metastatic lung cancer, bronchiectasis, asbestosis, sarcoidosis, lung abscess, cystic fibrosis, tuberculosis, and mesothelioma), as well as with some gastrointestinal diseases (including inflammatory bowel disease and hepatic cirrhosis). In some instances, it is occupational, e.g., in jackhammer operators.
Clubbing in patients with primary and metastatic lung cancer, mesothelioma, bronchiectasis, or hepatic cirrhosis may be associated with hypertrophic osteoarthropathy. In this condition, the subperiosteal formation of new bone in the distal diaphyses of the long bones of the extremities causes pain and symmetric arthritis-like changes in the shoulders, knees, ankles, wrists, and elbows. The diagnosis of hypertrophic osteoarthropathy may be confirmed by bone radiograph or magnetic resonance imaging (MRI). Although the mechanism of clubbing is unclear, it appears to be secondary to humoral substances that cause dilation of the vessels of the distal digits as well as growth factors released from platelet precursors in the digital circulation. In certain circumstances, clubbing is reversible, such as following lung transplantation for cystic fibrosis.
50 |
Edema |
STARLING FORCES AND FLUID EXCHANGE
About one-third of total-body water is confined to the extracellular space. Approximately 75% of the latter is interstitial fluid, and the remainder is the plasma. The forces that regulate the disposition of fluid between these two components of the extracellular compartment frequently are referred to as the Starling forces. The hydrostatic pressure within the capillaries and the colloid oncotic pressure in the interstitial fluid tend to promote movement of fluid from the vascular to the extravascular space. By contrast, the colloid oncotic pressure contributed by plasma proteins and the hydrostatic pressure within the interstitial fluid promote the movement of fluid into the vascular compartment. As a consequence, there is movement of water and diffusible solutes from the vascular space at the arteriolar end of the capillaries. Fluid is returned from the interstitial space into the vascular system at the venous end of the capillaries and by way of the lymphatics. These movements are usually balanced so that there is a steady state in the sizes of the intravascular and interstitial compartments, yet a large exchange between them occurs. However, if either the capillary hydrostatic pressure is increased and/or the oncotic pressure is reduced, a further net movement of fluid from intravascular to the interstitial spaces will take place.
Edema is defined as a clinically apparent increase in the interstitial fluid volume, which develops when Starling forces are altered so that there is increased flow of fluid from the vascular system into the interstitium. Edema due to an increase in capillary pressure may result from an elevation of venous pressure caused by obstruction to venous and/or lymphatic drainage. An increase in capillary pressure may be generalized, as occurs in heart failure, or it may be localized to one extremity when venous pressure is elevated due to unilateral thrombophlebitis (see below). The Starling forces also may be imbalanced when the colloid oncotic pressure of the plasma is reduced owing to any factor that may induce hypoalbuminemia, as when large quantities of protein are lost in the urine such as in the nephrotic syndrome (see below), or when synthesis is reduced in a severe catabolic state.
CAPILLARY DAMAGE
Edema may also result from damage to the capillary endothelium, which increases its permeability and permits the transfer of proteins into the interstitial compartment. Injury to the capillary wall can result from drugs (see below), viral or bacterial agents, and thermal or mechanical trauma. Increased capillary permeability also may be a consequence of a hypersensitivity reaction and of immune injury. Damage to the capillary endothelium is presumably responsible for inflammatory edema, which is usually nonpitting, localized, and accompanied by other signs of inflammation—i.e., erythema, heat, and tenderness.
REDUCTION OF EFFECTIVE ARTERIAL VOLUME
In many forms of edema, despite the increase in extracellular fluid volume, the effective arterial blood volume, a parameter that represents the filling of the arterial tree and that effectively perfuses the tissues, is reduced. Underfilling of the arterial tree may be caused by a reduction of cardiac output and/or systemic vascular resistance, by pooling of blood in the splanchnic veins (as in cirrhosis), and by hypoalbuminemia (Fig. 50-1A). As a consequence of underfilling, a series of physiologic responses designed to restore the effective arterial volume to normal are set into motion. A key element of these responses is the renal retention of sodium and, therefore, water, thereby restoring effective arterial volume, but sometimes also leading to or intensifying edema.
FIGURE 50-1 Clinical conditions in which a decrease in cardiac output (A) and systemic vascular resistance (B) cause arterial underfilling with resulting neurohumoral activation and renal sodium and water retention. In addition to activating the neurohumoral axis, adrenergic stimulation causes renal vasoconstriction and enhances sodium and fluid transport by the proximal tubule epithelium. RAAS, renin-angiotensin aldosterone system; SNS, sympathetic nervous system. (Modified from RW Schrier: Ann Intern Med 113:155, 1990.)
RENAL FACTORS AND THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM
The diminished renal blood flow characteristic of states in which the effective arterial blood volume is reduced is translated by the renal juxtaglomerular cells (specialized myoepithelial cells surrounding the afferent arteriole) into a signal for increased renin release. Renin is an enzyme with a molecular mass of about 40,000 Da that acts on its substrate, angiotensinogen, an α2-globulin synthesized by the liver, to release angiotensin I, a decapeptide, which in turn is converted to angiotensin II (AII), an octapeptide. AII has generalized vasoconstrictor properties, particularly on the renal efferent arterioles. This action reduces the hydrostatic pressure in the peritubular capillaries, whereas the increased filtration fraction raises the colloid osmotic pressure in these vessels, thereby enhancing salt and water reabsorption in the proximal tubule as well as in the ascending limb of the loop of Henle.
The renin-angiotensin-aldosterone system (RAAS) operates as both a hormonal and paracrine system. Its activation causes sodium and water retention and thereby contributes to edema formation. Blockade of the conversion of angiotensin I to AII and blockade of the AII receptor enhance sodium and water excretion and reduce many forms of edema. AII that enters the systemic circulation stimulates the production of aldosterone by the zona glomerulosa of the adrenal cortex. Aldosterone in turn enhances sodium reabsorption (and potassium excretion) by the collecting tubule, further favoring edema formation. In patients with heart failure, not only is aldosterone secretion elevated but the biologic half-life of the hormone is prolonged secondary to the depression of hepatic blood flow, which reduces its hepatic catabolism and increases further the plasma level of the hormone. Blockade of the action of aldosterone by spironolactone or eplerenone (aldosterone antagonists) or by amiloride (a blocker of epithelial sodium channels) often induces a moderate diuresis in edematous states.
ARGININE VASOPRESSIN
(See also Chap. 404) The secretion of arginine vasopressin (AVP) occurs in response to increased intracellular osmolar concentration, and, by stimulating V2 receptors, AVP increases the reabsorption of free water in the distal tubules and collecting ducts of the kidneys, thereby increasing total-body water. Circulating AVP is elevated in many patients with heart failure secondary to a nonosmotic stimulus associated with decreased effective arterial volume and reduced compliance of the left atrium. Such patients fail to show the normal reduction of AVP with a reduction of osmolality, contributing to edema formation and hyponatremia.
ENDOTHELIN-1
This potent peptide vasoconstrictor is released by endothelial cells. Its concentration in the plasma is elevated in patients with severe heart failure and contributes to renal vasoconstriction, sodium retention, and edema.
NATRIURETIC PEPTIDES
Atrial distention causes release into the circulation of atrial natriuretic peptide (ANP), a polypeptide; a high-molecular-weight precursor of ANP is stored in secretory granules within atrial myocytes. The closely related brain natriuretic peptide (pre-prohormone BNP) is stored primarily in ventricular myocytes and is released when ventricular diastolic pressure rises. Released ANP and BNP (which is derived from its precursor) bind to the natriuretic receptor-A, which causes: (1) excretion of sodium and water by augmenting glomerular filtration rate, inhibiting sodium reabsorption in the proximal tubule, and inhibiting release of renin and aldosterone; and (2) dilation of arterioles and venules by antagonizing the vasoconstrictor actions of AII, AVP, and sympathetic stimulation. Thus, elevated levels of natriuretic peptides have the capacity to oppose sodium retention in hypervolemic and edematous states.
Although circulating levels of ANP and BNP are elevated in heart failure and in cirrhosis with ascites, the natriuretic peptides are not sufficiently potent to prevent edema formation. Indeed, in edematous states, resistance to the actions of natriuretic peptides may be increased, further reducing their effectiveness.
Further discussion of the control of sodium and water balance is found in Chap. 64e.
CLINICAL CAUSES OF EDEMA
A weight gain of several kilograms usually precedes overt manifestations of generalized edema, and a similar weight loss from diuresis can be induced in a slightly edematous patient before “dry weight” is achieved. Anasarca refers to gross, generalized edema. Ascites (Chap. 59) and hydrothorax refer to accumulation of excess fluid in the peritoneal and pleural cavities, respectively, and are considered special forms of edema.
Edema is recognized by the persistence of an indentation of the skin after pressure; this is known as “pitting” edema. In its more subtle form, edema may be detected by noting that after the stethoscope is removed from the chest wall, the rim of the bell leaves an indentation on the skin of the chest for a few minutes. Edema may be present when the ring on a finger fits more snugly than in the past or when a patient complains of difficulty putting on shoes, particularly in the evening. Edema may also be recognized by puffiness of the face, which is most readily apparent in the periorbital areas.
GENERALIZED EDEMA
The differences among the major causes of generalized edema are shown in Table 50-1. Cardiac, renal, hepatic, or nutritional disorders are responsible for a majority of patients with generalized edema. Consequently, the differential diagnosis of generalized edema should be directed toward identifying or excluding these several conditions.
|
PRINCIPAL CAUSES OF GENERALIZED EDEMA: HISTORY, PHYSICAL EXAMINATION, AND LABORATORY FINDINGS |
Heart Failure (See also Chap. 279) In heart failure, the impaired systolic emptying of the ventricle(s) and/or the impairment of ventricular relaxation promotes an accumulation of blood in the venous circulation at the expense of the effective arterial volume. In addition, the heightened tone of the sympathetic nervous system causes renal vasoconstriction and reduction of glomerular filtration. In mild heart failure, a small increment of total blood volume may repair the deficit of effective arterial volume through the operation of Starling’s law of the heart, in which an increase in ventricular diastolic volume promotes a more forceful contraction and may thereby maintain the cardiac output. However, if the cardiac disorder is more severe, sodium and water retention continue, and the increment in blood volume accumulates in the venous circulation, raising venous pressure and causing edema (Fig. 50-1).
The presence of heart disease, as manifested by cardiac enlargement and/or ventricular hypertrophy, together with evidence of cardiac failure, such as dyspnea, basilar rales, venous distention, and hepatomegaly, usually indicates that edema results from heart failure. Noninvasive tests such as echocardiography may be helpful in establishing the diagnosis of heart disease. The edema of heart failure typically occurs in the dependent portions of the body.
Edema of Renal Disease (See also Chap. 338) The edema that occurs during the acute phase of glomerulonephritis is characteristically associated with hematuria, proteinuria, and hypertension. Although some evidence supports the view that the fluid retention is due to increased capillary permeability, in most instances, the edema results from primary retention of sodium and water by the kidneys owing to renal insufficiency. This state differs from most forms of heart failure in that it is characterized by a normal (or sometimes even increased) cardiac output. Patients with edema due to acute renal failure commonly have arterial hypertension as well as pulmonary congestion on chest roentgenogram, often without considerable cardiac enlargement, but they may not develop orthopnea. Patients with chronic renal failure may also develop edema due to primary renal retention of sodium and water.
Nephrotic Syndrome and other Hypoalbuminemic States The primary alteration in the nephrotic syndrome is a diminished colloid oncotic pressure due to losses of large quantities (≥3.5 g/d) of protein into the urine. With severe hypoalbuminemia (<35 g/L) and the consequent reduced colloid osmotic pressure, the sodium and water that are retained cannot be restrained within the vascular compartment, and total and effective arterial blood volumes decline. This process initiates the edema-forming sequence of events described above, including activation of the RAAS. The nephrotic syndrome may occur during the course of a variety of kidney diseases, which include glomerulonephritis, diabetic glomerulosclerosis, and hypersensitivity reactions. The edema is diffuse, symmetric, and most prominent in the dependent areas; as a consequence, periorbital edema is most prominent in the morning.
Hepatic Cirrhosis (See also Chap. 365) This condition is characterized in part by hepatic venous outflow blockade, which in turn expands the splanchnic blood volume and increases hepatic lymph formation. Intrahepatic hypertension acts as a stimulus for renal sodium retention and causes a reduction of effective arterial blood volume. These alterations are frequently complicated by hypoalbuminemia secondary to reduced hepatic synthesis of albumin, as well as peripheral arterial vasodilation. These effects reduce the effective arterial blood volume further, leading to activation of the RAAS and renal sympathetic nerves and to release of AVP, endothelin, and other sodium-and water-retaining mechanisms (Fig. 50-1B). The concentration of circulating aldosterone often is elevated by the failure of the liver to metabolize this hormone. Initially, the excess interstitial fluid is localized preferentially proximal (upstream) to the congested portal venous system and obstructed hepatic lymphatics, i.e., in the peritoneal cavity (causing ascites, Chap. 59). In later stages, particularly when there is severe hypoalbuminemia, peripheral edema may develop. A sizable accumulation of ascitic fluid may increase intraabdominal pressure and impede venous return from the lower extremities and contribute to the accumulation of edema of the lower extremities.
The excess production of prostaglandins (PGE2 and PGI2) in cirrhosis attenuates renal sodium retention. When the synthesis of these substances is inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs), renal function may deteriorate, and this may increase sodium retention further.
Drug-Induced Edema A large number of widely used drugs can cause edema (Table 50-2). Mechanisms include renal vasoconstriction (NSAIDs and cyclosporine), arteriolar dilation (vasodilators), augmented renal sodium reabsorption (steroid hormones), and capillary damage.
|
DRUGS ASSOCIATED WITH EDEMA FORMATION |
Source: Modified from Chertow GM: Approach to the patient with edema, in Primary Cardiology, 2nd ed, E Braunwald, L Goldman (eds). Philadelphia, Saunders, 2003, pp 117–128.
Edema of Nutritional Origin A diet grossly deficient in protein over a prolonged period may produce hypoproteinemia and edema. The latter may be intensified by the development of beriberi heart disease, which also is of nutritional origin, in which multiple peripheral arteriovenous fistulae result in reduced effective systemic perfusion and effective arterial blood volume, thereby enhancing edema formation (Chap. 96e) (Fig. 50-1B). Edema may actually become intensified when famished subjects are first provided with an adequate diet. The ingestion of more food may increase the quantity of sodium ingested, which is then retained along with water. So-called refeeding edema also may be linked to increased release of insulin, which directly increases tubular sodium reabsorption. In addition to hypoalbuminemia, hypokalemia and caloric deficits may be involved in the edema of starvation.
LOCALIZED EDEMA
In this condition, the hydrostatic pressure in the capillary bed upstream (proximal) of the obstruction increases so that an abnormal quantity of fluid is transferred from the vascular to the interstitial space. Since the alternative route (i.e., the lymphatic channels) also may be obstructed or maximally filled, an increased volume of interstitial fluid in the limb develops (i.e., there is trapping of fluid in the interstitium of the extremity). The displacement of large quantities of fluid into a limb may occur at the expense of the blood volume in the remainder of the body, thereby reducing effective arterial blood volume and leading to the retention of NaCl and H2O until the deficit in plasma volume has been corrected.
Localized edema due to venous or lymphatic obstruction may be caused by thrombophlebitis, chronic lymphangitis, resection of regional lymph nodes, and filariasis, among other causes. Lymphedema is particularly intractable because restriction of lymphatic flow results in increased protein concentration in the interstitial fluid, a circumstance that aggravates fluid retention.
Other Causes of Edema These causes include hypothyroidism (myxedema) and hyperthyroidism (pretibial myxedema secondary to Graves’ disease), the edema in which is typically nonpitting and due to deposition of hyaluronic acid and, in Graves’ disease, lymphocytic infiltration and inflammation; exogenous hyperadrenocortism; pregnancy; and administration of estrogens and vasodilators, particularly dihydropyridines such as nifedipine.
DISTRIBUTION OF EDEMA
The distribution of edema is an important guide to its cause. Edema associated with heart failure tends to be more extensive in the legs and to be accentuated in the evening, a feature also determined largely by posture. When patients with heart failure are confined to bed, edema may be most prominent in the presacral region. Severe heart failure may cause ascites that may be distinguished from the ascites caused by hepatic cirrhosis by the jugular venous pressure, which is usually elevated in heart failure and normal in cirrhosis.
Edema resulting from hypoproteinemia, as occurs in the nephrotic syndrome, characteristically is generalized, but it is especially evident in the very soft tissues of the eyelids and face and tends to be most pronounced in the morning owing to the recumbent posture assumed during the night. Less common causes of facial edema include trichinosis, allergic reactions, and myxedema. Edema limited to one leg or to one or both arms is usually the result of venous and/or lymphatic obstruction. Unilateral paralysis reduces lymphatic and venous drainage on the affected side and may also be responsible for unilateral edema. In patients with obstruction of the superior vena cava, edema is confined to the face, neck, and upper extremities in which the venous pressure is elevated compared with that in the lower extremities.
51e |
Approach to the Patient with a Heart Murmur |
The differential diagnosis of a heart murmur begins with a careful assessment of its major attributes and response to bedside maneuvers. The history, clinical context, and associated physical examination findings provide additional clues by which the significance of a heart murmur can be established. Accurate bedside identification of a heart murmur can inform decisions regarding the indications for noninvasive testing and the need for referral to a cardiovascular specialist. Preliminary discussions can be held with the patient regarding antibiotic or rheumatic fever prophylaxis, the need to restrict various forms of physical activity, and the potential role for family screening.
Heart murmurs are caused by audible vibrations that are due to increased turbulence from accelerated blood flow through normal or abnormal orifices, flow through a narrowed or irregular orifice into a dilated vessel or chamber, or backward flow through an incompetent valve, ventricular septal defect, or patent ductus arteriosus. They traditionally are defined in terms of their timing within the cardiac cycle (Fig. 51e-1). Systolic murmurs begin with or after the first heart sound (S1) and terminate at or before the component (A2 or P2) of the second heart sound (S2) that corresponds to their site of origin (left or right, respectively). Diastolic murmurs begin with or after the associated component of S2 and end at or before the subsequent S1. Continuous murmurs are not confined to either phase of the cardiac cycle but instead begin in early systole and proceed through S2 into all or part of diastole. The accurate timing of heart murmurs is the first step in their identification. The distinction between S1 and S2 and, therefore, systole and diastole is usually a straightforward process but can be difficult in the setting of a tachyarrhythmia, in which case the heart sounds can be distinguished by simultaneous palpation of the carotid upstroke, which should closely follow S1.
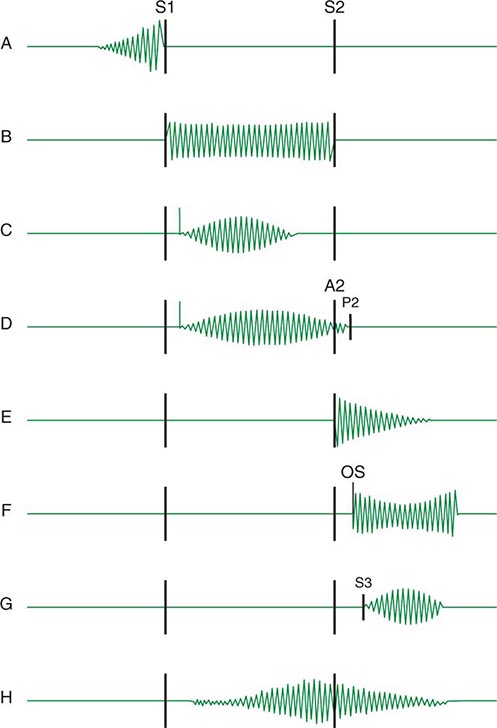
FIGURE 51e-1 Diagram depicting principal heart murmurs. A. Presystolic murmur of mitral or tricuspid stenosis. B. Holosystolic (pansystolic) murmur of mitral or tricuspid regurgitation or of ventricular septal defect. C. Aortic ejection murmur beginning with an ejection click and fading before the second heart sound. D. Systolic murmur in pulmonic stenosis spilling through the aortic second sound, pulmonic valve closure being delayed. E. Aortic or pulmonary diastolic murmur. F. Long diastolic murmur of mitral stenosis after the opening snap (OS). G. Short mid-diastolic inflow murmur after a third heart sound. H. Continuous murmur of patent ductus arteriosus. (Adapted from P Wood: Diseases of the Heart and Circulation, London, Eyre & Spottiswood, 1968. Permission granted courtesy of Antony and Julie Wood.)
Duration and Character The duration of a heart murmur depends on the length of time over which a pressure difference exists between two cardiac chambers, the left ventricle and the aorta, the right ventricle and the pulmonary artery, or the great vessels. The magnitude and variability of this pressure difference, coupled with the geometry and compliance of the involved chambers or vessels, dictate the velocity of flow; the degree of turbulence; and the resulting frequency, configuration, and intensity of the murmur. The diastolic murmur of chronic aortic regurgitation (AR) is a blowing, high-frequency event, whereas the murmur of mitral stenosis (MS), indicative of the left atrial–left ventricular diastolic pressure gradient, is a low-frequency event, heard as a rumbling sound with the bell of the stethoscope. The frequency components of a heart murmur may vary at different sites of auscultation. The coarse systolic murmur of aortic stenosis (AS) may sound higher pitched and more acoustically pure at the apex, a phenomenon eponymously referred to as the Gallavardin effect. Some murmurs may have a distinct or unusual quality, such as the “honking” sound appreciated in some patients with mitral regurgitation (MR) due to mitral valve prolapse (MVP).
The configuration of a heart murmur may be described as crescendo, decrescendo, crescendo-decrescendo, or plateau. The decrescendo configuration of the murmur of chronic AR (Fig. 51e-1E) can be understood in terms of the progressive decline in the diastolic pressure gradient between the aorta and the left ventricle. The crescendo-decrescendo configuration of the murmur of AS reflects the changes in the systolic pressure gradient between the left ventricle and the aorta as ejection occurs, whereas the plateau configuration of the murmur of chronic MR (Fig. 51e-1B) is consistent with the large and nearly constant pressure difference between the left ventricle and the left atrium.
Intensity The intensity of a heart murmur is graded on a scale of 1–6 (or I–VI). A grade 1 murmur is very soft and is heard only with great effort. A grade 2 murmur is easily heard but not particularly loud. A grade 3 murmur is loud but is not accompanied by a palpable thrill over the site of maximal intensity. A grade 4 murmur is very loud and is accompanied by a thrill. A grade 5 murmur is loud enough to be heard with only the edge of the stethoscope touching the chest, whereas a grade 6 murmur is loud enough to be heard with the stethoscope slightly off the chest. Murmurs of grade 3 or greater intensity usually signify important structural heart disease and indicate high blood flow velocity at the site of murmur production. Small ventricular septal defects (VSDs), for example, are accompanied by loud, usually grade 4 or greater, systolic murmurs as blood is ejected at high velocity from the left ventricle to the right ventricle. Low-velocity events, such as left-to-right shunting across an atrial septal defect (ASD), are usually silent. The intensity of a heart murmur may be diminished by any process that increases the distance between the intracardiac source and the stethoscope on the chest wall, such as obesity, obstructive lung disease, and a large pericardial effusion. The intensity of a murmur also may be misleadingly soft when cardiac output is reduced significantly or when the pressure gradient between the involved cardiac structures is low.
Location and Radiation Recognition of the location and radiation of the murmur helps facilitate its accurate identification (Fig. 51e-2). Adventitious sounds, such as a systolic click or diastolic snap, or abnormalities of S1 or S2 may provide additional clues. Careful attention to the characteristics of the murmur and other heart sounds during the respiratory cycle and the performance of simple bedside maneuvers complete the auscultatory examination. These features, along with recommendations for further testing, are discussed below in the context of specific systolic, diastolic, and continuous heart murmurs (Table 51e-1).
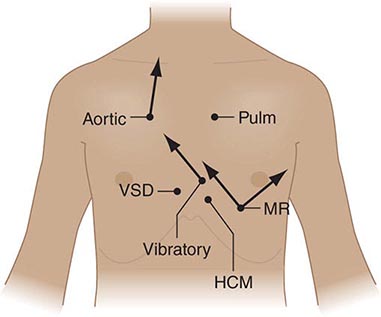
FIGURE 51e-2 Maximal intensity and radiation of six isolated systolic murmurs. HOCM, hypertrophic obstructive cardiomyopathy; MR, mitral regurgitation; Pulm, pulmonary stenosis; Aortic, aortic stenosis; VSD, ventricular septal defect. (From JB Barlow: Perspectives on the Mitral Valve. Philadelphia, FA Davis, 1987, p 140.)
|
PRINCIPAL CAUSES OF HEART MURMURS |

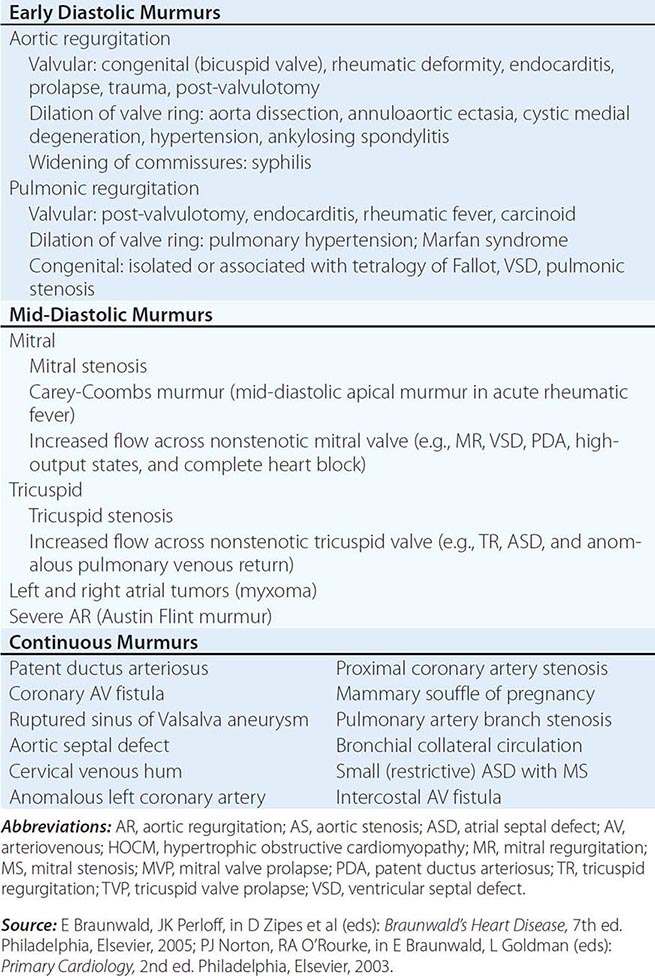
SYSTOLIC HEART MURMURS
Early Systolic Murmurs Early systolic murmurs begin with S1 and extend for a variable period, ending well before S2. Their causes are relatively few in number. Acute, severe MR into a normal-sized, relatively noncompliant left atrium results in an early, decrescendo systolic murmur best heard at or just medial to the apical impulse. These characteristics reflect the progressive attenuation of the pressure gradient between the left ventricle and the left atrium during systole owing to the rapid rise in left atrial pressure caused by the sudden volume load into an unprepared, noncompliant chamber and contrast sharply with the auscultatory features of chronic MR. Clinical settings in which acute, severe MR occur include (1) papillary muscle rupture complicating acute myocardial infarction (MI) (Chap. 295), (2) rupture of chordae tendineae in the setting of myxomatous mitral valve disease (MVP, Chap. 283), (3) infective endocarditis (Chap. 155), and (4) blunt chest wall trauma.
Acute, severe MR from papillary muscle rupture usually accompanies an inferior, posterior, or lateral MI and occurs 2–7 days after presentation. It often is signaled by chest pain, hypotension, and pulmonary edema, but a murmur may be absent in up to 50% of cases. The posteromedial papillary muscle is involved 6 to 10 times more frequently than the anterolateral papillary muscle. The murmur is to be distinguished from that associated with post-MI ventricular septal rupture, which is accompanied by a systolic thrill at the left sternal border in nearly all patients and is holosystolic in duration. A new heart murmur after an MI is an indication for transthoracic echocardiography (TTE) (Chap. 270e), which allows bedside delineation of its etiology and pathophysiologic significance. The distinction between acute MR and ventricular septal rupture also can be achieved with right heart catheterization, sequential determination of oxygen saturations, and analysis of the pressure waveforms (tall v wave in the pulmonary artery wedge pressure in MR). Post-MI mechanical complications of this nature mandate aggressive medical stabilization and prompt referral for surgical repair.
Spontaneous chordal rupture can complicate the course of myxomatous mitral valve disease (MVP) and result in new-onset or “acute on chronic” severe MR. MVP may occur as an isolated phenomenon, or the lesion may be part of a more generalized connective tissue disorder as seen, for example, in patients with Marfan syndrome. Acute, severe MR as a consequence of infective endocarditis results from destruction of leaflet tissue, chordal rupture, or both. Blunt chest wall trauma is usually self-evident but may be disarmingly trivial; it can result in papillary muscle contusion and rupture, chordal detachment, or leaflet avulsion. TTE is indicated in all cases of suspected acute, severe MR to define its mechanism and severity, delineate left ventricular size and systolic function, and provide an assessment of suitability for primary valve repair.
A congenital, small muscular VSD (Chap. 282) may be associated with an early systolic murmur. The defect closes progressively during septal contraction, and thus, the murmur is confined to early systole. It is localized to the left sternal border (Fig. 51e-2) and is usually of grade 4 or 5 intensity. Signs of pulmonary hypertension or left ventricular volume overload are absent. Anatomically large and uncorrected VSDs, which usually involve the membranous portion of the septum, may lead to pulmonary hypertension. The murmur associated with the left-to-right shunt, which earlier may have been holosystolic, becomes limited to the first portion of systole as the elevated pulmonary vascular resistance leads to an abrupt rise in right ventricular pressure and an attenuation of the interventricular pressure gradient during the remainder of the cardiac cycle. In such instances, signs of pulmonary hypertension (right ventricular lift, loud and single or closely split S2) may predominate. The murmur is best heard along the left sternal border but is softer. Suspicion of a VSD is an indication for TTE.
Tricuspid regurgitation (TR) with normal pulmonary artery pressures, as may occur with infective endocarditis, may produce an early systolic murmur. The murmur is soft (grade 1 or 2), is best heard at the lower left sternal border, and may increase in intensity with inspiration (Carvallo’s sign). Regurgitant “c–v” waves may be visible in the jugular venous pulse. TR in this setting is not associated with signs of right heart failure.
Mid-Systolic Murmurs Mid-systolic murmurs begin at a short interval after S1, end before S2 (Fig. 51e-1C), and are usually crescendo-decrescendo in configuration. Aortic stenosis is the most common cause of a mid-systolic murmur in an adult. The murmur of AS is usually loudest to the right of the sternum in the second intercostal space (aortic area, Fig. 51e-2) and radiates into the carotids. Transmission of the mid-systolic murmur to the apex, where it becomes higher-pitched, is common (Gallavardin effect; see above).
Differentiation of this apical systolic murmur from MR can be difficult. The murmur of AS will increase in intensity, or become louder, in the beat after a premature beat, whereas the murmur of MR will have constant intensity from beat to beat. The intensity of the AS murmur also varies directly with the cardiac output. With a normal cardiac output, a systolic thrill and a grade 4 or higher murmur suggest severe AS. The murmur is softer in the setting of heart failure and low cardiac output. Other auscultatory findings of severe AS include a soft or absent A2, paradoxical splitting of S2, an apical S4, and a late-peaking systolic murmur. In children, adolescents, and young adults with congenital valvular AS, an early ejection sound (click) is usually audible, more often along the left sternal border than at the base. Its presence signifies a flexible, noncalcified bicuspid valve (or one of its variants) and localizes the left ventricular outflow obstruction to the valvular (rather than sub- or supravalvular) level.
Assessment of the volume and rate of rise of the carotid pulse can provide additional information. A small and delayed upstroke (parvus et tardus) is consistent with severe AS. The carotid pulse examination is less discriminatory, however, in older patients with stiffened arteries. The electrocardiogram (ECG) shows signs of left ventricular hypertrophy (LVH) as the severity of the stenosis increases. TTE is indicated to assess the anatomic features of the aortic valve, the severity of the stenosis, left ventricular size, wall thickness and function, and the size and contour of the aortic root and proximal ascending aorta.
The obstructive form of hypertrophic cardiomyopathy (HOCM) is associated with a mid-systolic murmur that is usually loudest along the left sternal border or between the left lower sternal border and the apex (Chap. 287, Fig. 51e-2). The murmur is produced by both dynamic left ventricular outflow tract obstruction and MR, and thus, its configuration is a hybrid between ejection and regurgitant phenomena. The intensity of the murmur may vary from beat to beat and after provocative maneuvers but usually does not exceed grade 3. The murmur classically will increase in intensity with maneuvers that result in increasing degrees of outflow tract obstruction, such as a reduction in preload or afterload (Valsalva, standing, vasodilators), or with an augmentation of contractility (inotropic stimulation). Maneuvers that increase preload (squatting, passive leg raising, volume administration) or afterload (squatting, vasopressors) or that reduce contractility (β-adrenoreceptor blockers) decrease the intensity of the murmur. In rare patients, there may be reversed splitting of S2. A sustained left ventricular apical impulse and an S4 may be appreciated. In contrast to AS, the carotid upstroke is rapid and of normal volume. Rarely, it is bisferiens or bifid in contour (see Fig. 267-2D) due to mid-systolic closure of the aortic valve. LVH is present on the ECG, and the diagnosis is confirmed by TTE. Although the systolic murmur associated with MVP behaves similarly to that due to HOCM in response to the Valsalva maneuver and to standing/squatting (Fig. 51e-3), these two lesions can be distinguished on the basis of their associated findings, such as the presence of LVH in HOCM or a nonejection click in MVP.
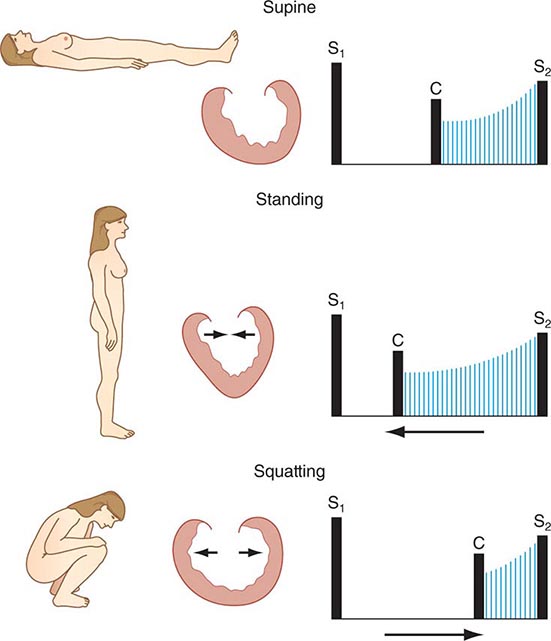
FIGURE 51e-3 A mid-systolic nonejection sound (C) occurs in mitral valve prolapse and is followed by a late systolic murmur that crescendos to the second heart sound (S2). Standing decreases venous return; the heart becomes smaller; C moves closer to the first heart sound (S1), and the mitral regurgitant murmur has an earlier onset. With prompt squatting, venous return and afterload increase; the heart becomes larger; C moves toward S2; and the duration of the murmur shortens. (From JA Shaver, JJ Leonard, DF Leon: Examination of the Heart, Part IV, Auscultation of the Heart. Dallas, American Heart Association, 1990, p 13. Copyright, American Heart Association.)
The mid-systolic, crescendo-decrescendo murmur of congenital pulmonic stenosis (PS, Chap. 282) is best appreciated in the second and third left intercostal spaces (pulmonic area) (Figs. 51e-2 and 51e-4). The duration of the murmur lengthens and the intensity of P2 diminishes with increasing degrees of valvular stenosis (Fig. 51e-1D). An early ejection sound, the intensity of which decreases with inspiration, is heard in younger patients. A parasternal lift and ECG evidence of right ventricular hypertrophy indicate severe pressure overload. If obtained, the chest x-ray may show poststenotic dilation of the main pulmonary artery. TTE is recommended for complete characterization.
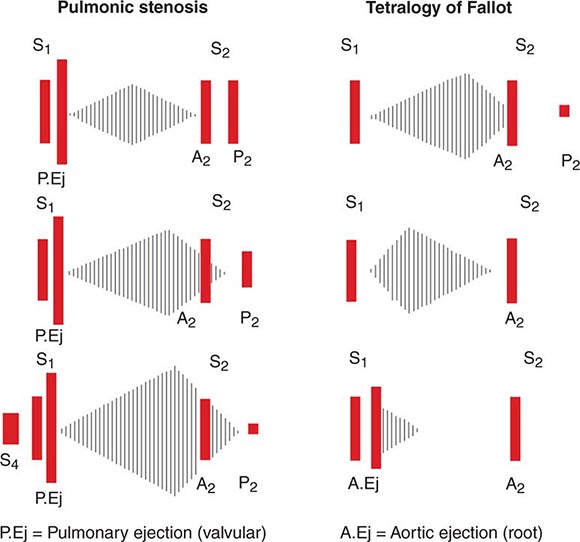
FIGURE 51e-4 Left. In valvular pulmonic stenosis with intact ventricular septum, right ventricular systolic ejection becomes progressively longer, with increasing obstruction to flow. As a result, the murmur becomes longer and louder, enveloping the aortic component of the second heart sound (A2). The pulmonic component (P2) occurs later, and splitting becomes wider but more difficult to hear because A2 is lost in the murmur and P2 becomes progressively fainter and lower pitched. As the pulmonic gradient increases, the isometric contraction phase shortens until the pulmonic valve ejection sound fuses with the first heart sound (S1). In severe pulmonic stenosis with concentric hypertrophy and decreasing right ventricular compliance, a fourth heart sound appears. Right. In tetralogy of Fallot with increasing obstruction at the pulmonic infundibular area, an increasing amount of right ventricular blood is shunted across the silent ventricular septal defect and flow across the obstructed outflow tract decreases. Therefore, with increasing obstruction the murmur becomes shorter, earlier, and fainter. P2 is absent in severe tetralogy of Fallot. A large aortic root receives almost all cardiac output from both ventricular chambers, and the aorta dilates and is accompanied by a root ejection sound that does not vary with respiration. (From JA Shaver, JJ Leonard, DF Leon: Examination of the Heart, Part IV, Auscultation of the Heart. Dallas, American Heart Association, 1990, p 45. Copyright, American Heart Association.)
Significant left-to-right intracardiac shunting due to an ASD (Chap. 282) leads to an increase in pulmonary blood flow and a grade 2–3 mid-systolic murmur at the middle to upper left sternal border attributed to increased flow rates across the pulmonic valve with fixed splitting of S2. Ostium secundum ASDs are the most common cause of these shunts in adults. Features suggestive of a primum ASD include the coexistence of MR due to a cleft anterior mitral valve leaflet and left axis deviation of the QRS complex on the ECG. With sinus venosus ASDs, the left-to-right shunt is usually not large enough to result in a systolic murmur, although the ECG may show abnormalities of sinus node function. A grade 2 or 3 mid-systolic murmur may also be heard best at the upper left sternal border in patients with idiopathic dilation of the pulmonary artery; a pulmonary ejection sound is also present in these patients. TTE is indicated to evaluate a grade 2 or 3 mid-systolic murmur when there are other signs of cardiac disease.
An isolated grade 1 or 2 mid-systolic murmur, heard in the absence of symptoms or signs of heart disease, is most often a benign finding for which no further evaluation, including TTE, is necessary. The most common example of a murmur of this type in an older adult patient is the crescendo-decrescendo murmur of aortic valve sclerosis, heard at the second right interspace (Fig. 51e-2). Aortic sclerosis is defined as focal thickening and calcification of the aortic valve to a degree that does not interfere with leaflet opening. The carotid upstrokes are normal, and electrocardiographic LVH is not present. A grade 1 or 2 mid-systolic murmur often can be heard at the left sternal border with pregnancy, hyperthyroidism, or anemia, physiologic states that are associated with accelerated blood flow. Still’s murmur refers to a benign grade 2, vibratory or musical mid-systolic murmur at the mid or lower left sternal border in normal children and adolescents, best heard in the supine position (Fig. 51e-2).
Late Systolic Murmurs A late systolic murmur that is best heard at the left ventricular apex is usually due to MVP (Chap. 283). Often, this murmur is introduced by one or more nonejection clicks. The radiation of the murmur can help identify the specific mitral leaflet involved in the process of prolapse or flail. The term flail refers to the movement made by an unsupported portion of the leaflet after loss of its chordal attachment(s). With posterior leaflet prolapse or flail, the resultant jet of MR is directed anteriorly and medially, as a result of which the murmur radiates to the base of the heart and masquerades as AS. Anterior leaflet prolapse or flail results in a posteriorly directed MR jet that radiates to the axilla or left infrascapular region. Leaflet flail is associated with a murmur of grade 3 or 4 intensity that can be heard throughout the precordium in thin-chested patients. The presence of an S3 or a short, rumbling mid-diastolic murmur due to enhanced flow signifies severe MR.
Bedside maneuvers that decrease left ventricular preload, such as standing, will cause the click and murmur of MVP to move closer to the first heart sound, as leaflet prolapse occurs earlier in systole. Standing also causes the murmur to become louder and longer. With squatting, left ventricular preload and afterload are increased abruptly, leading to an increase in left ventricular volume, and the click and murmur move away from the first heart sound as leaflet prolapse is delayed; the murmur becomes softer and shorter in duration (Fig. 51e–3). As noted above, these responses to standing and squatting are directionally similar to those observed in patients with HOCM.
A late, apical systolic murmur indicative of MR may be heard transiently in the setting of acute myocardial ischemia; it is due to apical tethering and malcoaptation of the leaflets in response to structural and functional changes of the ventricle and mitral annulus. The intensity of the murmur varies as a function of left ventricular afterload and will increase in the setting of hypertension. TTE is recommended for assessment of late systolic murmurs.
Holosystolic Murmurs (Figs. 51e-1B and 51e-5) Holosystolic murmurs begin with S1 and continue through systole to S2. They are usually indicative of chronic mitral or tricuspid valve regurgitation or a VSD and warrant TTE for further characterization. The holosystolic murmur of chronic MR is best heard at the left ventricular apex and radiates to the axilla (Fig. 51e-2); it is usually high-pitched and plateau in configuration because of the wide difference between left ventricular and left atrial pressure throughout systole. In contrast to acute MR, left atrial compliance is normal or even increased in chronic MR. As a result, there is only a small increase in left atrial pressure for any increase in regurgitant volume.
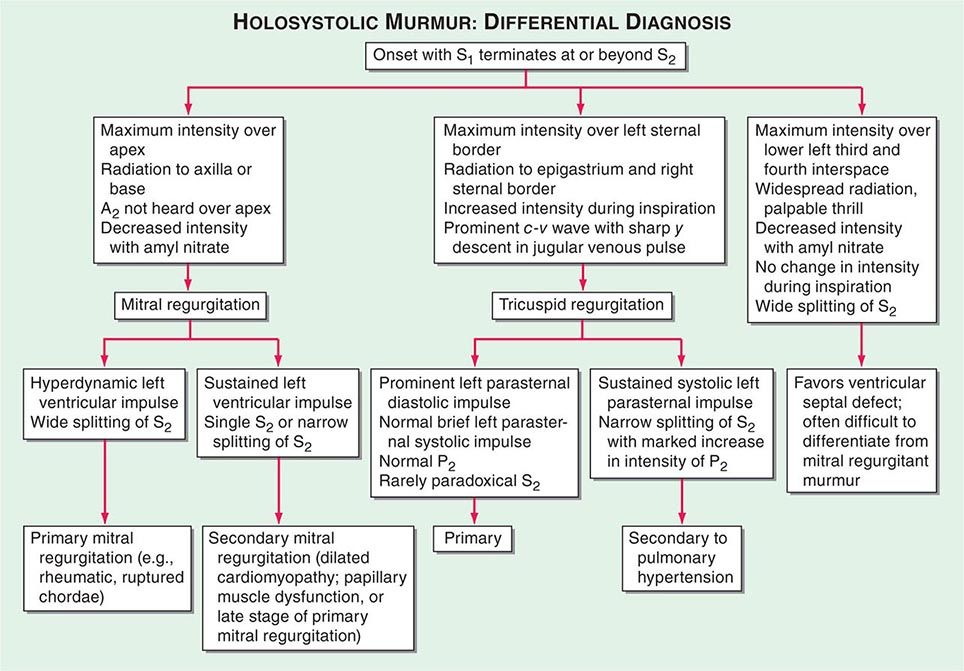
FIGURE 51e-5 Differential diagnosis of a holosystolic murmur.
Several conditions are associated with chronic MR and an apical holosystolic murmur, including rheumatic scarring of the leaflets, mitral annular calcification, postinfarction left ventricular remodeling, and severe left ventricular chamber enlargement. The circumference of the mitral annulus increases as the left ventricle enlarges and leads to failure of leaflet coaptation with central MR in patients with dilated cardiomyopathy (Chap. 287). The severity of the MR is worsened by any contribution from apical displacement of the papillary muscles and leaflet tethering (remodeling). Because the mitral annulus is contiguous with the left atrial endocardium, gradual enlargement of the left atrium from chronic MR will result in further stretching of the annulus and more MR; thus, “MR begets MR.” Chronic severe MR results in enlargement and leftward displacement of the left ventricular apex beat and, in some patients, a diastolic filling complex, as described previously.
The holosystolic murmur of chronic TR is generally softer than that of MR, is loudest at the left lower sternal border, and usually increases in intensity with inspiration (Carvallo’s sign). Associated signs include c–v waves in the jugular venous pulse, an enlarged and pulsatile liver, ascites, and peripheral edema. The abnormal jugular venous waveforms are the predominant finding and are seen very often in the absence of an audible murmur despite Doppler echocardiographic verification of TR. Causes of primary TR include myxomatous disease (prolapse), endocarditis, rheumatic disease, radiation, carcinoid, Ebstein’s anomaly, and chordal detachment as a complication of right ventricular endomyocardial biopsy. TR is more commonly a passive process that results secondarily from annular enlargement due to right ventricular dilatation in the face of volume or pressure overload.
The holosystolic murmur of a VSD is loudest at the mid- to lower left sternal border (Fig. 51e-2) and radiates widely. A thrill is present at the site of maximal intensity in the majority of patients. There is no change in the intensity of the murmur with inspiration. The intensity of the murmur varies as a function of the anatomic size of the defect. Small, restrictive VSDs, as exemplified by the maladie de Roger, create a very loud murmur due to the significant and sustained systolic pressure gradient between the left and right ventricles. With large defects, the ventricular pressures tend to equalize, shunt flow is balanced, and a murmur is not appreciated. The distinction between post-MI ventricular septal rupture and MR has been reviewed previously.
DIASTOLIC HEART MURMURS
Early Diastolic Murmurs (Fig. 51e-1E) Chronic AR results in a high-pitched, blowing, decrescendo, early to mid-diastolic murmur that begins after the aortic component of S2 (A2) and is best heard at the second right interspace (Fig. 51e-6). The murmur may be soft and difficult to hear unless auscultation is performed with the patient leaning forward at end expiration. This maneuver brings the aortic root closer to the anterior chest wall. Radiation of the murmur may provide a clue to the cause of the AR. With primary valve disease, such as that due to congenital bicuspid disease, prolapse, or endocarditis, the diastolic murmur tends to radiate along the left sternal border, where it is often louder than appreciated in the second right interspace. When AR is caused by aortic root disease, the diastolic murmur may radiate along the right sternal border. Diseases of the aortic root cause dilation or distortion of the aortic annulus and failure of leaflet coaptation. Causes include Marfan syndrome with aneurysm formation, annuloaortic ectasia, ankylosing spondylitis, and aortic dissection.
FIGURE 51e-6 Diastolic filling murmur (rumble) in mitral stenosis. In mild mitral stenosis, the diastolic gradient across the valve is limited to the phases of rapid ventricular filling in early diastole and presystole. The rumble may occur during either or both periods. As the stenotic process becomes severe, a large pressure gradient exists across the valve during the entire diastolic filling period, and the rumble persists throughout diastole. As the left atrial pressure becomes greater, the interval between A2 (or P2) and the opening snap (O.S.) shortens. In severe mitral stenosis, secondary pulmonary hypertension develops and results in a loud P2 and the splitting interval usually narrows. ECG, electrocardiogram. (From JA Shaver, JJ Leonard, DF Leon: Examination of the Heart, Part IV, Auscultation of the Heart. Dallas, American Heart Association, 1990, p 55. Copyright, American Heart Association.)
Chronic, severe AR also may produce a lower-pitched mid to late, grade 1 or 2 diastolic murmur at the apex (Austin Flint murmur), which is thought to reflect turbulence at the mitral inflow area from the admixture of regurgitant (aortic) and forward (mitral) blood flow (Fig. 51e-1G). This lower-pitched, apical diastolic murmur can be distinguished from that due to MS by the absence of an opening snap and the response of the murmur to a vasodilator challenge. Lowering afterload with an agent such as amyl nitrite will decrease the duration and magnitude of the aortic–left ventricular diastolic pressure gradient, and thus, the Austin Flint murmur of severe AR will become shorter and softer. The intensity of the diastolic murmur of mitral stenosis (Fig. 51e-6) may either remain constant or increase with afterload reduction because of the reflex increase in cardiac output and mitral valve flow.
Although AS and AR may coexist, a grade 2 or 3 crescendo-decrescendo mid-systolic murmur frequently is heard at the base of the heart in patients with isolated, severe AR and is due to an increased volume and rate of systolic flow. Accurate bedside identification of coexistent AS can be difficult unless the carotid pulse examination is abnormal or the mid-systolic murmur is of grade 4 or greater intensity. In the absence of heart failure, chronic severe AR is accompanied by several peripheral signs of significant diastolic run-off, including a wide pulse pressure, a “water-hammer” carotid upstroke (Corrigan’s pulse), and Quincke’s pulsations of the nail beds. The diastolic murmur of acute, severe AR is notably shorter in duration and lower pitched than the murmur of chronic AR. It can be very difficult to appreciate in the presence of a rapid heart rate. These attributes reflect the abrupt rate of rise of diastolic pressure within the unprepared and noncompliant left ventricle and the correspondingly rapid decline in the aortic–left ventricular diastolic pressure gradient. Left ventricular diastolic pressure may increase sufficiently to result in premature closure of the mitral valve and a soft first heart sound. Peripheral signs of significant diastolic run-off are not present.
Pulmonic regurgitation (PR) results in a decrescendo, early to mid-diastolic murmur (Graham Steell murmur) that begins after the pulmonic component of S2 (P2), is best heard at the second left interspace, and radiates along the left sternal border. The intensity of the murmur may increase with inspiration. PR is most commonly due to dilation of the valve annulus from chronic elevation of the pulmonary artery pressure. Signs of pulmonary hypertension, including a right ventricular lift and a loud, single or narrowly split S2, are present. These features also help distinguish PR from AR as the cause of a decrescendo diastolic murmur heard along the left sternal border. PR in the absence of pulmonary hypertension can occur with endocarditis or a congenitally deformed valve. It is usually present after repair of tetralogy of Fallot in childhood. When pulmonary hypertension is not present, the diastolic murmur is softer and lower pitched than the classic Graham Steell murmur, and the severity of the PR can be difficult to appreciate.
TTE is indicated for the further evaluation of a patient with an early to mid-diastolic murmur. Longitudinal assessment of the severity of the valve lesion and ventricular size and systolic function help guide a potential decision for surgical management. TTE also can provide anatomic information regarding the root and proximal ascending aorta, although computed tomographic or magnetic resonance angiography may be indicated for more precise characterization (Chap. 270e).
Mid-Diastolic Murmurs (Figs. 51e-1G and 51e-1H) Mid-diastolic murmurs result from obstruction and/or augmented flow at the level of the mitral or tricuspid valve. Rheumatic fever is the most common cause of MS (Fig. 51e-6). In younger patients with pliable valves, S1 is loud and the murmur begins after an opening snap, which is a high-pitched sound that occurs shortly after S2. The interval between the pulmonic component of the second heart sound (P2) and the opening snap is inversely related to the magnitude of the left atrial–left ventricular pressure gradient. The murmur of MS is low-pitched and thus is best heard with the bell of the stethoscope. It is loudest at the left ventricular apex and often is appreciated only when the patient is turned in the left lateral decubitus position. It is usually of grade 1 or 2 intensity but may be absent when the cardiac output is severely reduced despite significant obstruction. The intensity of the murmur increases during maneuvers that increase cardiac output and mitral valve flow, such as exercise. The duration of the murmur reflects the length of time over which left atrial pressure exceeds left ventricular diastolic pressure. An increase in the intensity of the murmur just before S1, a phenomenon known as presystolic accentuation (Figs. 51e-1A and 51e-6), occurs in patients in sinus rhythm and is due to a late increase in transmitral flow with atrial contraction. Presystolic accentuation does not occur in patients with atrial fibrillation.
The mid-diastolic murmur associated with tricuspid stenosis is best heard at the lower left sternal border and increases in intensity with inspiration. A prolonged y descent may be visible in the jugular venous waveform. This murmur is very difficult to hear and often is obscured by left-sided acoustical events.
There are several other causes of mid-diastolic murmurs. Large left atrial myxomas may prolapse across the mitral valve and cause variable degrees of obstruction to left ventricular inflow (Chap. 289e). The murmur associated with an atrial myxoma may change in duration and intensity with changes in body position. An opening snap is not present, and there is no presystolic accentuation. Augmented mitral diastolic flow can occur with isolated severe MR or with a large left-to-right shunt at the ventricular or great vessel level and produce a soft, rapid filling sound (S3) followed by a short, low-pitched mid-diastolic apical murmur. The Austin Flint murmur of severe, chronic AR has already been described.
A short, mid-diastolic murmur is rarely heard during an episode of acute rheumatic fever (Carey-Coombs murmur) and probably is due to flow through an edematous mitral valve. An opening snap is not present in the acute phase, and the murmur dissipates with resolution of the acute attack. Complete heart block with dyssynchronous atrial and ventricular activation may be associated with intermittent mid- to late diastolic murmurs if atrial contraction occurs when the mitral valve is partially closed. Mid-diastolic murmurs indicative of increased tricuspid valve flow can occur with severe, isolated TR and with large ASDs and significant left-to-right shunting. Other signs of an ASD are present (Chap. 282), including fixed splitting of S2 and a mid-systolic murmur at the mid- to upper left sternal border. TTE is indicated for evaluation of a patient with a mid- to late diastolic murmur. Findings specific to the diseases discussed above will help guide management.
CONTINUOUS MURMURS
(Figs. 51e-1H and 51e-7) Continuous murmurs begin in systole, peak near the second heart sound, and continue into all or part of diastole. Their presence throughout the cardiac cycle implies a pressure gradient between two chambers or vessels during both systole and diastole. The continuous murmur associated with a patent ductus arteriosus is best heard at the upper left sternal border. Large, uncorrected shunts may lead to pulmonary hypertension, attenuation or obliteration of the diastolic component of the murmur, reversal of shunt flow, and differential cyanosis of the lower extremities. A ruptured sinus of Valsalva aneurysm creates a continuous murmur of abrupt onset at the upper right sternal border. Rupture typically occurs into a right heart chamber, and the murmur is indicative of a continuous pressure difference between the aorta and either the right ventricle or the right atrium. A continuous murmur also may be audible along the left sternal border with a coronary arteriovenous fistula and at the site of an arteriovenous fistula used for hemodialysis access. Enhanced flow through enlarged intercostal collateral arteries in patients with aortic coarctation may produce a continuous murmur along the course of one or more ribs. A cervical bruit with both systolic and diastolic components (a to-fro murmur, Fig. 51e-7) usually indicates a high-grade carotid artery stenosis.
FIGURE 51e-7 Comparison of the continuous murmur and the to-fro murmur. During abnormal communication between high-pressure and low-pressure systems, a large pressure gradient exists throughout the cardiac cycle, producing a continuous murmur. A classic example is patent ductus arteriosus. At times, this type of murmur can be confused with a to-fro murmur, which is a combination of systolic ejection murmur and a murmur of semilunar valve incompetence. A classic example of a to-fro murmur is aortic stenosis and regurgitation. A continuous murmur crescendos to near the second heart sound (S2), whereas a to-fro murmur has two components. The mid-systolic ejection component decrescendos and disappears as it approaches S2. (From JA Shaver, JJ Leonard, DF Leon: Examination of the Heart, Part IV, Auscultation of the Heart. Dallas, American Heart Association, 1990, p 55. Copyright, American Heart Association.)
Not all continuous murmurs are pathologic. A continuous venous hum can be heard in healthy children and young adults, especially during pregnancy; it is best appreciated in the right supraclavicular fossa and can be obliterated by pressure over the right internal jugular vein or by having the patient turn his or her head toward the examiner. The continuous mammary souffle of pregnancy is created by enhanced arterial flow through engorged breasts and usually appears during the late third trimester or early puerperium. The murmur is louder in systole. Firm pressure with the diaphragm of the stethoscope can eliminate the diastolic portion of the murmur.
DYNAMIC AUSCULTATION
(Table 51e-2; see Table 267-1) Careful attention to the behavior of heart murmurs during simple maneuvers that alter cardiac hemodynamics can provide important clues to their cause and significance.
|
DYNAMIC AUSCULTATION: BEDSIDE MANEUVERS THAT CAN BE USED TO CHANGE THE INTENSITY OF CARDIAC MURMURS (SEE TEXT) |
Respiration Auscultation should be performed during quiet respiration or with a modest increase in inspiratory effort, as more forceful movement of the chest tends to obscure the heart sounds. Left-sided murmurs may be best heard at end expiration, when lung volumes are minimized and the heart and great vessels are brought closer to the chest wall. This phenomenon is characteristic of the murmur of AR. Murmurs of right-sided origin, such as tricuspid or pulmonic regurgitation, increase in intensity during inspiration. The intensity of left-sided murmurs either remains constant or decreases with inspiration.
Bedside assessment also should evaluate the behavior of S2 with respiration and the dynamic relationship between the aortic and pulmonic components (Fig. 51e-8). Reversed splitting can be a feature of severe AS, HOCM, left bundle branch block, right ventricular pacing, or acute myocardial ischemia. Fixed splitting of S2 in the presence of a grade 2 or 3 mid-systolic murmur at the mid- or upper left sternal border indicates an ASD. Physiologic but wide splitting during the respiratory cycle implies either premature aortic valve closure, as can occur with severe MR, or delayed pulmonic valve closure due to PS or right bundle branch block.
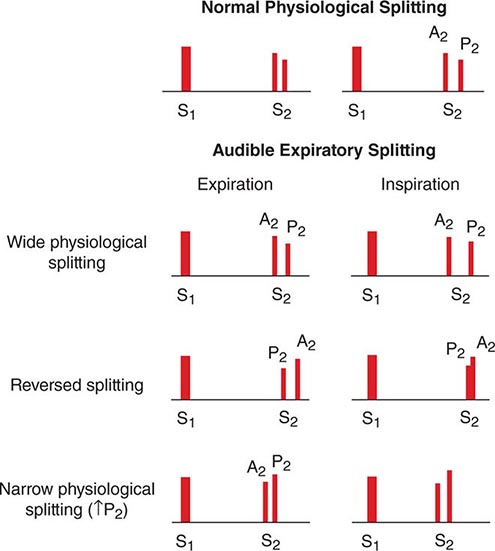
FIGURE 51e-8 Top. Normal physiologic splitting. During expiration, the aortic (A2) and pulmonic (P2) components of the second heart sound are separated by <30 ms and are appreciated as a single sound. During inspiration, the splitting interval widens, and A2 and P2 are clearly separated into two distinct sounds. Bottom. Audible expiratory splitting. Wide physiologic splitting is caused by a delay in P2. Reversed splitting is caused by a delay in A2, resulting in paradoxical movement; i.e., with inspiration P2 moves toward A2, and the splitting interval narrows. Narrow physiologic splitting occurs in pulmonary hypertension, and both A2 and P2 are heard during expiration at a narrow splitting interval because of the increased intensity and high-frequency composition of P2. (From JA Shaver, JJ Leonard, DF Leon: Examination of the Heart, Part IV, Auscultation of the Heart. Dallas, American Heart Association, 1990, p 17. Copyright, American Heart Association.)
Alterations of Systemic Vascular Resistance Murmurs can change characteristics after maneuvers that alter systemic vascular resistance and left ventricular afterload. The systolic murmurs of MR and VSD become louder during sustained handgrip, simultaneous inflation of blood pressure cuffs on both upper extremities to pressures 20–40 mmHg above systolic pressure for 20 s, or infusion of a vasopressor agent. The murmurs associated with AS or HOCM will become softer or remain unchanged with these maneuvers. The diastolic murmur of AR becomes louder in response to interventions that raise systemic vascular resistance.
Opposite changes in systolic and diastolic murmurs may occur with the use of pharmacologic agents that lower systemic vascular resistance. Inhaled amyl nitrite is now rarely used for this purpose but can help distinguish the murmur of AS or HOCM from that of either MR or VSD, if necessary. The former two murmurs increase in intensity, whereas the latter two become softer after exposure to amyl nitrite. As noted previously, the Austin Flint murmur of severe AR becomes softer, but the mid-diastolic rumble of MS becomes louder, in response to the abrupt lowering of systemic vascular resistance with amyl nitrite.
Changes in Venous Return The Valsalva maneuver results in an increase in intrathoracic pressure, followed by a decrease in venous return, ventricular filling, and cardiac output. The majority of murmurs decrease in intensity during the strain phase of the maneuver. Two notable exceptions are the murmurs associated with MVP and obstructive HOCM, both of which become louder during the Valsalva maneuver. The murmur of MVP may also become longer as leaflet prolapse occurs earlier in systole at smaller ventricular volumes. These murmurs behave in a similar and parallel fashion with standing. Both the click and the murmur of MVP move closer in timing to S1 on rapid standing from a squatting position (Fig. 51e-3). The increase in the intensity of the murmur of HOCM is predicated on the augmentation of the dynamic left ventricular outflow tract gradient that occurs with reduced ventricular filling. Squatting results in abrupt increases in both venous return (preload) and left ventricular afterload that increase ventricular volume, changes that predictably cause a decrease in the intensity and duration of the murmurs associated with MVP and HOCM; the click and murmur of MVP move away from S1 with squatting. Passive leg raising can be used to increase venous return in patients who are unable to squat and stand. This maneuver may lead to a decrease in the intensity of the murmur associated with HOCM but has less effect in patients with MVP.
Post-Premature Ventricular Contraction A change in the intensity of a systolic murmur in the first beat after a premature beat, or in the beat after a long cycle length in patients with atrial fibrillation, can help distinguish AS from MR, particularly in an older patient in whom the murmur of AS is well transmitted to the apex. Systolic murmurs due to left ventricular outflow obstruction, including that due to AS, increase in intensity in the beat after a premature beat because of the combined effects of enhanced left ventricular filling and post-extrasystolic potentiation of contractile function. Forward flow accelerates, causing an increase in the gradient and a louder murmur. The intensity of the murmur of MR does not change in the post-premature beat as there is relatively little further increase in mitral valve flow or change in the left ventricular–left atrial gradient.
THE CLINICAL CONTEXT
Additional clues to the etiology and importance of a heart murmur can be gleaned from the history and other physical examination findings. Symptoms suggestive of cardiovascular, neurologic, or pulmonary disease help focus the differential diagnosis, as do findings relevant to the jugular venous pressure and waveforms, the arterial pulses, other heart sounds, the lungs, the abdomen, the skin, and the extremities. In many instances, laboratory studies, an ECG, and/or a chest x-ray may have been obtained earlier and may contain valuable information. A patient with suspected infective endocarditis, for example, may have a murmur in the setting of fever, chills, anorexia, fatigue, dyspnea, splenomegaly, petechiae, and positive blood cultures. A new systolic murmur in a patient with a marked fall in blood pressure after a recent MI suggests myocardial rupture. By contrast, an isolated grade 1 or 2 mid-systolic murmur at the left sternal border in a healthy, active, and asymptomatic young adult is most likely a benign finding for which no further evaluation is indicated. The context in which the murmur is appreciated often dictates the need for further testing.
ECHOCARDIOGRAPHY
(Fig. 51e–9; Chaps. 267 and 270e) Echocardiography with color flow and spectral Doppler is a valuable tool for the assessment of cardiac murmurs. Information regarding valve structure and function, chamber size, wall thickness, ventricular function, estimated pulmonary artery pressures, intracardiac shunt flow, pulmonary and hepatic vein flow, and aortic flow can be ascertained readily. It is important to note that Doppler signals of trace or mild valvular regurgitation of no clinical consequence can be detected with structurally normal tricuspid, pulmonic, and mitral valves. Such signals are not likely to generate enough turbulence to create an audible murmur.
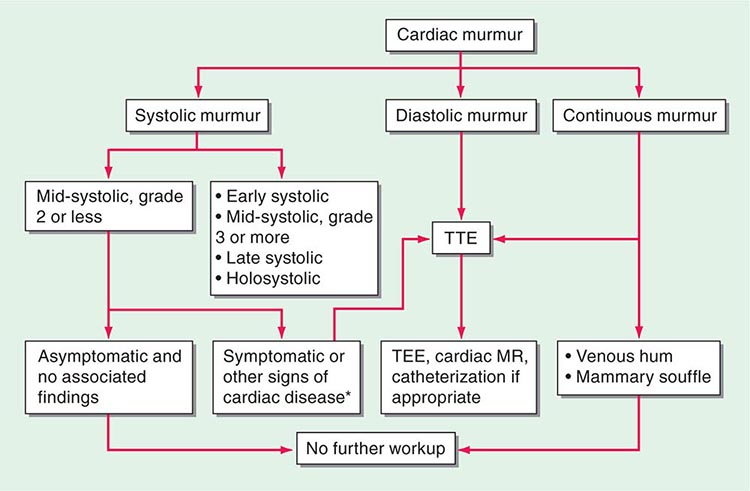
FIGURE 51e-9 Strategy for evaluating heart murmurs. *If an electrocardiogram or chest x-ray has been obtained and is abnormal, echocardiography is indicated. TTE, transthoracic echocardiography; TEE, transesophageal echocardiography; MR, magnetic resonance. (Adapted from RO Bonow et al: J Am Coll Cardiol 32:1486, 1998.)
Echocardiography is indicated for the evaluation of patients with early, late, or holosystolic murmurs and patients with grade 3 or louder mid-systolic murmurs. Patients with grade 1 or 2 mid-systolic murmurs but other symptoms or signs of cardiovascular disease, including those from ECG or chest x-ray, should also undergo echocardiography. Echocardiography is also indicated for the evaluation of any patient with a diastolic murmur and for patients with continuous murmurs not due to a venous hum or mammary souffle. Echocardiography also should be considered when there is a clinical need to verify normal cardiac structure and function in a patient whose symptoms and signs are probably noncardiac in origin. The performance of serial echocardiography to follow the course of asymptomatic individuals with valvular heart disease is a central feature of their longitudinal assessment and provides valuable information that may have an impact on decisions regarding the timing of surgery. Routine echocardiography is not recommended for asymptomatic patients with a grade 1 or 2 mid-systolic murmur without other signs of heart disease. For this category of patients, referral to a cardiovascular specialist should be considered if there is doubt about the significance of the murmur after the initial examination.
The selective use of echocardiography outlined above has not been subjected to rigorous analysis of its cost-effectiveness. For some clinicians, handheld or miniaturized cardiac ultrasound devices have replaced the stethoscope. Although several reports attest to the improved sensitivity of such devices for the detection of valvular heart disease, accuracy is highly operator-dependent, and incremental cost considerations and outcomes have not been addressed adequately. The use of electronic or digital stethoscopes with spectral display capabilities has also been proposed as a method to improve the characterization of heart murmurs and the mentored teaching of cardiac auscultation.
OTHER CARDIAC TESTING
(Chap. 270e, Fig. 51e-9) In relatively few patients, clinical assessment and TTE do not adequately characterize the origin and significance of a heart murmur. Transesophageal echocardiography (TEE) can be considered for further evaluation, especially when the TTE windows are limited by body size, chest configuration, or intrathoracic pathology. TEE offers enhanced sensitivity for the detection of a wide range of structural cardiac disorders. Electrocardiographically gated cardiac magnetic resonance (CMR) imaging, although limited in its ability to display valvular morphology, can provide quantitative information regarding valvular function, stenosis severity, regurgitant fraction, regurgitant volume, shunt flow, chamber and great vessel size, ventricular function, and myocardial perfusion. CMR has largely supplanted the need for cardiac catheterization and invasive hemodynamic assessment when there is a discrepancy between the clinical and echocardiographic findings. Invasive coronary angiography is performed routinely in most adult patients before valve surgery, especially when there is a suspicion of coronary artery disease predicated on symptoms, risk factors, and/or age. The use of computed tomography coronary angiography (CCTA) to exclude coronary artery disease in patients with a low pretest probability of disease before valve surgery is gaining wider acceptance.
INTEGRATED APPROACH
The accurate identification of a heart murmur begins with a systematic approach to cardiac auscultation. Characterization of its major attributes, as reviewed above, allows the examiner to construct a preliminary differential diagnosis, which is then refined by integration of information available from the history, associated cardiac findings, the general physical examination, and the clinical context. The need for and urgency of further testing follow sequentially. Correlation of the findings on auscultation with the noninvasive data provides an educational feedback loop and an opportunity for improving physical examination skills. Cost constraints mandate that noninvasive imaging be justified on the basis of its incremental contribution to diagnosis, treatment, and outcome. Additional study is required to assess the cost-effective application of newer imaging technology in patients with heart murmurs.
52 |
Palpitations |
Palpitations are extremely common among patients who present to their internists and can best be defined as an intermittent “thumping,” “pounding,” or “fluttering” sensation in the chest. This sensation can be either intermittent or sustained and either regular or irregular. Most patients interpret palpitations as an unusual awareness of the heartbeat and become especially concerned when they sense that they have had “skipped” or “missing” heartbeats. Palpitations are often noted when the patient is quietly resting, during which time other stimuli are minimal. Palpitations that are positional generally reflect a structural process within (e.g., atrial myxoma) or adjacent to (e.g., mediastinal mass) the heart.
Palpitations are brought about by cardiac (43%), psychiatric (31%), miscellaneous (10%), and unknown (16%) causes, according to one large series. Among the cardiovascular causes are premature atrial and ventricular contractions, supraventricular and ventricular arrhythmias, mitral valve prolapse (with or without associated arrhythmias), aortic insufficiency, atrial myxoma, and pulmonary embolism. Intermittent palpitations are commonly caused by premature atrial or ventricular contractions: the post-extrasystolic beat is sensed by the patient owing to the increase in ventricular end-diastolic dimension following the pause in the cardiac cycle and the increased strength of contraction (post-extrasystolic potentiation) of that beat. Regular, sustained palpitations can be caused by regular supraventricular and ventricular tachycardias. Irregular, sustained palpitations can be caused by atrial fibrillation. It is important to note that most arrhythmias are not associated with palpitations. In those that are, it is often useful either to ask the patient to “tap out” the rhythm of the palpitations or to take his/her pulse during palpitations. In general, hyperdynamic cardiovascular states caused by catecholaminergic stimulation from exercise, stress, or pheochromocytoma can lead to palpitations. Palpitations are common among athletes, especially older endurance athletes. In addition, the enlarged ventricle of aortic regurgitation and accompanying hyperdynamic precordium frequently lead to the sensation of palpitations. Other factors that enhance the strength of myocardial contraction, including tobacco, caffeine, aminophylline, atropine, thyroxine, cocaine, and amphetamines, can cause palpitations.
Psychiatric causes of palpitations include panic attacks or disorders, anxiety states, and somatization, alone or in combination. Patients with psychiatric causes for palpitations more commonly report a longer duration of the sensation (>15 min) and other accompanying symptoms than do patients with other causes. Among the miscellaneous causes of palpitations are thyrotoxicosis, drugs (see above) and ethanol, spontaneous skeletal muscle contractions of the chest wall, pheochromocytoma, and systemic mastocytosis.
SECTION 6 |
ALTERATIONS IN GASTROINTESTINAL FUNCTION |
53 |
Dysphagia |
Dysphagia—difficulty with swallowing—refers to problems with the transit of food or liquid from the mouth to the hypopharynx or through the esophagus. Severe dysphagia can compromise nutrition, cause aspiration, and reduce quality of life. Additional terminology pertaining to swallowing dysfunction is as follows. Aphagia (inability to swallow) typically denotes complete esophageal obstruction, most commonly encountered in the acute setting of a food bolus or foreign body impaction. Odynophagia refers to painful swallowing, typically resulting from mucosal ulceration within the oropharynx or esophagus. It commonly is accompanied by dysphagia, but the converse is not true. Globus pharyngeus is a foreign body sensation localized in the neck that does not interfere with swallowing and sometimes is relieved by swallowing. Transfer dysphagia frequently results in nasal regurgitation and pulmonary aspiration during swallowing and is characteristic of oropharyngeal dysphagia. Phagophobia (fear of swallowing) and refusal to swallow may be psychogenic or related to anticipatory anxiety about food bolus obstruction, odynophagia, or aspiration.
PHYSIOLOGY OF SWALLOWING
Swallowing begins with a voluntary (oral) phase that includes preparation during which food is masticated and mixed with saliva. This is followed by a transfer phase during which the bolus is pushed into the pharynx by the tongue. Bolus entry into the hypopharynx initiates the pharyngeal swallow response, which is centrally mediated and involves a complex series of actions, the net result of which is to propel food through the pharynx into the esophagus while preventing its entry into the airway. To accomplish this, the larynx is elevated and pulled forward, actions that also facilitate upper esophageal sphincter (UES) opening. Tongue pulsion then propels the bolus through the UES, followed by a peristaltic contraction that clears residue from the pharynx and through the esophagus. The lower esophageal sphincter (LES) relaxes as the food enters the esophagus and remains relaxed until the peristaltic contraction has delivered the bolus into the stomach. Peristaltic contractions elicited in response to a swallow are called primary peristalsis and involve sequenced inhibition followed by contraction of the musculature along the entire length of the esophagus. The inhibition that precedes the peristaltic contraction is called deglutitive inhibition. Local distention of the esophagus anywhere along its length, as may occur with gastroesophageal reflux, activates secondary peristalsis that begins at the point of distention and proceeds distally. Tertiary esophageal contractions are nonperistaltic, disordered esophageal contractions that may be observed to occur spontaneously during fluoroscopic observation.
The musculature of the oral cavity, pharynx, UES, and cervical esophagus is striated and directly innervated by lower motor neurons carried in cranial nerves (Fig. 53-1). Oral cavity muscles are innervated by the fifth (trigeminal) and seventh (facial) cranial nerves; the tongue, by the twelfth (hypoglossal) cranial nerve. Pharyngeal muscles are innervated by the ninth (glossopharyngeal) and tenth (vagus) cranial nerves.
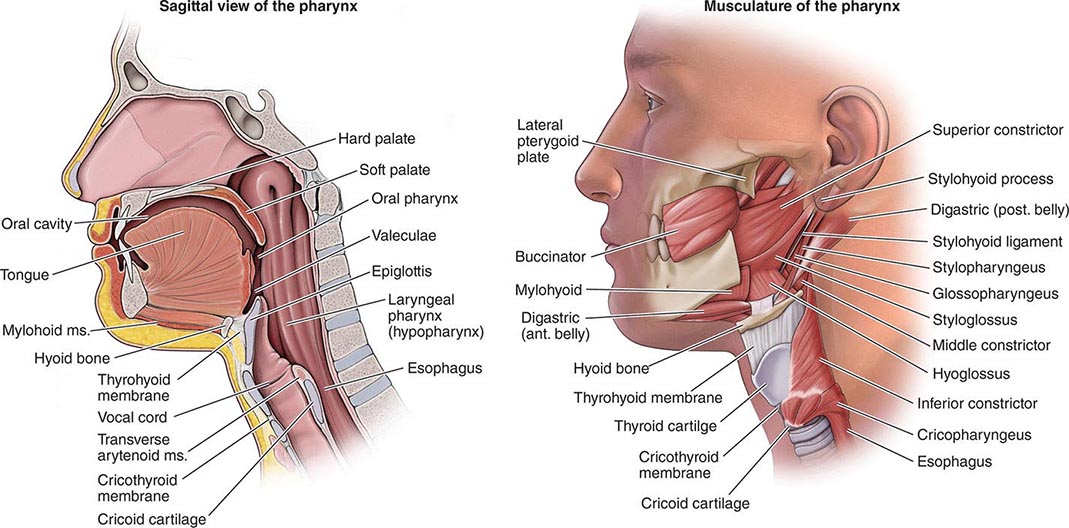
FIGURE 53-1 Sagittal and diagrammatic views of the musculature involved in enacting oropharyngeal swallowing. Note the dominance of the tongue in the sagittal view and the intimate relationship between the entrance to the larynx (airway) and the esophagus. In the resting configuration illustrated, the esophageal inlet is closed. This is transiently reconfigured such that the esophageal inlet is open and the laryngeal inlet closed during swallowing. (Adapted from PJ Kahrilas, in DW Gelfand and JE Richter [eds]: Dysphagia: Diagnosis and Treatment. New York: Igaku-Shoin Medical Publishers, 1989, pp. 11–28.)
Physiologically, the UES consists of the cricopharyngeus muscle, the adjacent inferior pharyngeal constrictor, and the proximal portion of the cervical esophagus. UES innervation is derived from the vagus nerve, whereas the innervation to the musculature acting on the UES to facilitate its opening during swallowing comes from the fifth, seventh, and twelfth cranial nerves. The UES remains closed at rest owing to both its inherent elastic properties and neurogenically mediated contraction of the cricopharyngeus muscle. UES opening during swallowing involves both cessation of vagal excitation to the cricopharyngeus and simultaneous contraction of the suprahyoid and geniohyoid muscles that pull open the UES in conjunction with the upward and forward displacement of the larynx.
The neuromuscular apparatus for peristalsis is distinct in proximal and distal parts of the esophagus. The cervical esophagus, like the pharyngeal musculature, consists of striated muscle and is directly innervated by lower motor neurons of the vagus nerve. Peristalsis in the proximal esophagus is governed by the sequential activation of the vagal motor neurons in the nucleus ambiguus. In contrast, the distal esophagus and LES are composed of smooth muscle and are controlled by excitatory and inhibitory neurons within the esophageal myenteric plexus. Medullary preganglionic neurons from the dorsal motor nucleus of the vagus trigger peristalsis via these ganglionic neurons during primary peristalsis. Neurotransmitters of the excitatory ganglionic neurons are acetylcholine and substance P; those of the inhibitory neurons are vasoactive intestinal peptide and nitric oxide. Peristalsis results from the patterned activation of inhibitory followed by excitatory ganglionic neurons, with progressive dominance of the inhibitory neurons distally. Similarly, LES relaxation occurs with the onset of deglutitive inhibition and persists until the peristaltic sequence is complete. At rest, the LES is contracted because of excitatory ganglionic stimulation and its intrinsic myogenic tone, a property that distinguishes it from the adjacent esophagus. The function of the LES is supplemented by the surrounding muscle of the right diaphragmatic crus, which acts as an external sphincter during inspiration, cough, or abdominal straining.
PATHOPHYSIOLOGY OF DYSPHAGIA
Dysphagia can be subclassified both by location and by the circumstances in which it occurs. With respect to location, distinct considerations apply to oral, pharyngeal, or esophageal dysphagia. Normal transport of an ingested bolus depends on the consistency and size of the bolus, the caliber of the lumen, the integrity of peristaltic contraction, and deglutitive inhibition of both the UES and the LES. Dysphagia caused by an oversized bolus or a narrow lumen is called structural dysphagia, whereas dysphagia due to abnormalities of peristalsis or impaired sphincter relaxation after swallowing is called propulsive or motor dysphagia. More than one mechanism may be operative in a patient with dysphagia. Scleroderma commonly presents with absent peristalsis as well as a weakened LES that predisposes patients to peptic stricture formation. Likewise, radiation therapy for head and neck cancer may compound the functional deficits in the oropharyngeal swallow attributable to the tumor and cause cervical esophageal stenosis.
Oral and Pharyngeal (Oropharyngeal) Dysphagia Oral-phase dysphagia is associated with poor bolus formation and control so that food has prolonged retention within the oral cavity and may seep out of the mouth. Drooling and difficulty in initiating swallowing are other characteristic signs. Poor bolus control also may lead to premature spillage of food into the hypopharynx with resultant aspiration into the trachea or regurgitation into the nasal cavity. Pharyngeal-phase dysphagia is associated with retention of food in the pharynx due to poor tongue or pharyngeal propulsion or obstruction at the UES. Signs and symptoms of concomitant hoarseness or cranial nerve dysfunction may be associated with oropharyngeal dysphagia.
Oropharyngeal dysphagia may be due to neurologic, muscular, structural, iatrogenic, infectious, and metabolic causes. Iatrogenic, neurologic, and structural pathologies are most common. Iatrogenic causes include surgery and radiation, often in the setting of head and neck cancer. Neurogenic dysphagia resulting from cerebrovascular accidents, Parkinson’s disease, and amyotrophic lateral sclerosis is a major source of morbidity related to aspiration and malnutrition. Medullary nuclei directly innervate the oropharynx. Lateralization of pharyngeal dysphagia implies either a structural pharyngeal lesion or a neurologic process that selectively targeted the ipsilateral brainstem nuclei or cranial nerve. Advances in functional brain imaging have elucidated an important role of the cerebral cortex in swallow function and dysphagia. Asymmetry in the cortical representation of the pharynx provides an explanation for the dysphagia that occurs as a consequence of unilateral cortical cerebrovascular accidents.
Oropharyngeal structural lesions causing dysphagia include Zenker’s diverticulum, cricopharyngeal bar, and neoplasia. Zenker’s diverticulum typically is encountered in elderly patients, with an estimated prevalence between 1:1000 and 1:10,000. In addition to dysphagia, patients may present with regurgitation of particulate food debris, aspiration, and halitosis. The pathogenesis is related to stenosis of the cricopharyngeus that causes diminished opening of the UES and results in increased hypopharyngeal pressure during swallowing with development of a pulsion diverticulum immediately above the cricopharyngeus in a region of potential weakness known as Killian’s dehiscence. A cricopharyngeal bar, appearing as a prominent indentation behind the lower third of the cricoid cartilage, is related to Zenker’s diverticulum in that it involves limited distensibility of the cricopharyngeus and can lead to the formation of a Zenker’s diverticulum. However, a cricopharyngeal bar is a common radiographic finding, and most patients with transient cricopharyngeal bars are asymptomatic, making it important to rule out alternative etiologies of dysphagia before treatment. Furthermore, cricopharyngeal bars may be secondary to other neuromuscular disorders.
Since the pharyngeal phase of swallowing occurs in less than a second, rapid-sequence fluoroscopy is necessary to evaluate for functional abnormalities. Adequate fluoroscopic examination requires that the patient be conscious and cooperative. The study incorporates recordings of swallow sequences during ingestion of food and liquids of varying consistencies. The pharynx is examined to detect bolus retention, regurgitation into the nose, or aspiration into the trachea. Timing and integrity of pharyngeal contraction and opening of the UES with a swallow are analyzed to assess both aspiration risk and the potential for swallow therapy. Structural abnormalities of the oropharynx, especially those which may require biopsies, also should be assessed by direct laryngoscopic examination.
Esophageal Dysphagia The adult esophagus measures 18–26 cm in length and is anatomically divided into the cervical esophagus, extending from the pharyngoesophageal junction to the suprasternal notch, and the thoracic esophagus, which continues to the diaphragmatic hiatus. When distended, the esophageal lumen has internal dimensions of about 2 cm in the anteroposterior plane and 3 cm in the lateral plane. Solid food dysphagia becomes common when the lumen is narrowed to <13 mm but also can occur with larger diameters in the setting of poorly masticated food or motor dysfunction. Circumferential lesions are more likely to cause dysphagia than are lesions that involve only a partial circumference of the esophageal wall. The most common structural causes of dysphagia are Schatzki’s rings, eosinophilic esophagitis, and peptic strictures. Dysphagia also occurs in the setting of gastroesophageal reflux disease without a stricture, perhaps on the basis of altered esophageal sensation, distensibility, or motor function.
Propulsive disorders leading to esophageal dysphagia result from abnormalities of peristalsis and/or deglutitive inhibition, potentially affecting the cervical or thoracic esophagus. Since striated muscle pathology usually involves both the oropharynx and the cervical esophagus, the clinical manifestations usually are dominated by oropharyngeal dysphagia. Diseases affecting smooth muscle involve both the thoracic esophagus and the LES. A dominant manifestation of this, absent peristalsis, refers to either the complete absence of swallow-induced contraction or the presence of nonperistaltic, disordered contractions. Absent peristalsis and failure of deglutitive LES relaxation are the defining features of achalasia. In diffuse esophageal spasm (DES), LES function is normal, with the disordered motility restricted to the esophageal body. Absent peristalsis combined with severe weakness of the LES is a nonspecific pattern commonly found in patients with scleroderma.
APPROACH TO THE PATIENT:
Dysphagia
Figure 53-2 shows an algorithm for the approach to a patient with dysphagia.
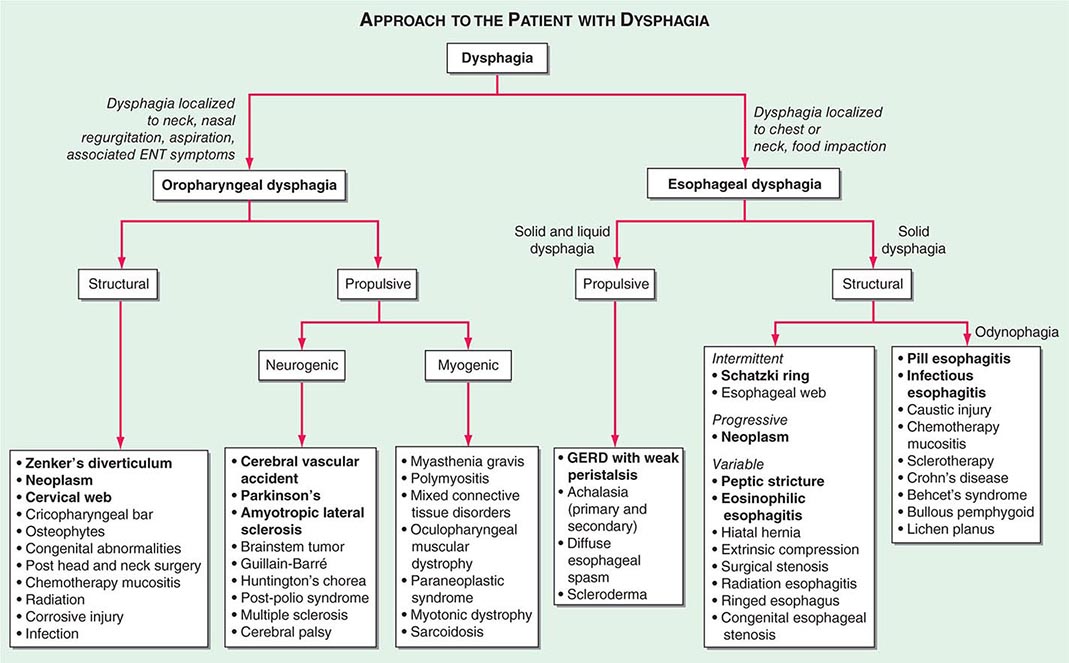
FIGURE 53-2 Approach to the patient with dysphagia. Etiologies in bold print are the most common. ENT, ear, nose, and throat; GERD, gastroesophageal reflux disease.
HISTORY
The patient history is extremely valuable in making a presumptive diagnosis or at least substantially restricting the differential diagnoses in most patients. Key elements of the history are the localization of dysphagia, the circumstances in which dysphagia is experienced, other symptoms associated with dysphagia, and progression. Dysphagia that localizes to the suprasternal notch may indicate either an oropharyngeal or an esophageal etiology as distal dysphagia is referred proximally about 30% of the time. Dysphagia that localizes to the chest is esophageal in origin. Nasal regurgitation and tracheobronchial aspiration manifest by coughing with swallowing are hallmarks of oropharyngeal dysphagia. Severe cough with swallowing may also be a sign of a tracheoesophageal fistula. The presence of hoarseness may be another important diagnostic clue. When hoarseness precedes dysphagia, the primary lesion is usually laryngeal; hoarseness that occurs after the development of dysphagia may result from compromise of the recurrent laryngeal nerve by a malignancy. The type of food causing dysphagia is a crucial detail. Intermittent dysphagia that occurs only with solid food implies structural dysphagia, whereas constant dysphagia with both liquids and solids strongly suggests a motor abnormality. Two caveats to this pattern are that despite having a motor abnormality, patients with scleroderma generally develop mild dysphagia for solids only and, somewhat paradoxically, that patients with oropharyngeal dysphagia often have greater difficulty managing liquids than solids. Dysphagia that is progressive over the course of weeks to months raises concern for neoplasia. Episodic dysphagia to solids that is unchanged over years indicates a benign disease process such as a Schatzki’s ring or eosinophilic esophagitis. Food impaction with a prolonged inability to pass an ingested bolus even with ingestion of liquid is typical of a structural dysphagia. Chest pain frequently accompanies dysphagia whether it is related to motor disorders, structural disorders, or reflux disease. A prolonged history of heartburn preceding the onset of dysphagia is suggestive of peptic stricture and, infrequently, esophageal adenocarcinoma. A history of prolonged nasogastric intubation, esophageal or head and neck surgery, ingestion of caustic agents or pills, previous radiation or chemotherapy, or associated mucocutaneous diseases may help isolate the cause of dysphagia. With accompanying odynophagia, which usually is indicative of ulceration, infectious or pill-induced esophagitis should be suspected. In patients with AIDS or other immunocompromised states, esophagitis due to opportunistic infections such as Candida, herpes simplex virus, or cytomegalovirus and to tumors such as Kaposi’s sarcoma and lymphoma should be considered. A strong history of atopy increases concerns for eosinophilic esophagitis.
PHYSICAL EXAMINATION
Physical examination is important in the evaluation of oral and pharyngeal dysphagia because dysphagia is usually only one of many manifestations of a more global disease process. Signs of bulbar or pseudobulbar palsy, including dysarthria, dysphonia, ptosis, tongue atrophy, and hyperactive jaw jerk, in addition to evidence of generalized neuromuscular disease, should be elicited. The neck should be examined for thyromegaly. A careful inspection of the mouth and pharynx should disclose lesions that may interfere with passage of food. Missing dentition can interfere with mastication and exacerbate an existing cause of dysphagia. Physical examination is less helpful in the evaluation of esophageal dysphagia as most relevant pathology is restricted to the esophagus. The notable exception is skin disease. Changes in the skin may suggest a diagnosis of scleroderma or mucocutaneous diseases such as pemphigoid, lichen planus and epidermolysis bullosa, all of which can involve the esophagus.
DIAGNOSTIC PROCEDURES
Although most instances of dysphagia are attributable to benign disease processes, dysphagia is also a cardinal symptom of several malignancies, making it an important symptom to evaluate. Cancer may result in dysphagia due to intraluminal obstruction (esophageal or proximal gastric cancer, metastatic deposits), extrinsic compression (lymphoma, lung cancer), or paraneoplastic syndromes. Even when not attributable to malignancy, dysphagia is usually a manifestation of an identifiable and treatable disease entity, making its evaluation beneficial to the patient and gratifying to the practitioner. The specific diagnostic algorithm to pursue is guided by the details of the history (Fig. 53-2). If oral or pharyngeal dysphagia is suspected, a fluoroscopic swallow study, usually done by a swallow therapist, is the procedure of choice. Otolaryngoscopic and neurologic evaluation also can be important, depending on the circumstances. For suspected esophageal dysphagia, upper endoscopy is the single most useful test. Endoscopy allows better visualization of mucosal lesions than does barium radiography and also allows one to obtain mucosal biopsies. Endoscopic or histologic abnormalities are evident in the leading causes of esophageal dysphagia: Schatzki ring, gastroesophageal reflux disease and eosinophilic esophagitis. Furthermore, therapeutic intervention with esophageal dilation can be done as part of the procedure if it is deemed necessary. The emergence of eosinophilic esophagitis as a leading cause of dysphagia in both children and adults has led to the recommendation that esophageal mucosal biopsies be obtained routinely in the evaluation of unexplained dysphagia even if endoscopically identified esophageal mucosal lesions are absent. For cases of suspected esophageal motility disorders, endoscopy is still the appropriate initial evaluation as neoplastic and inflammatory conditions can secondarily produce patterns of either achalasia or esophageal spasm. Esophageal manometry is done if dysphagia is not adequately explained by endoscopy or to confirm the diagnosis of a suspected esophageal motor disorder. Barium radiography can provide useful adjunctive information in cases of subtle or complex esophageal strictures, prior esophageal surgery, esophageal diverticula, or paraesophageal herniation. In specific cases, computed tomography (CT) examination and endoscopic ultrasonography may be useful.
Treatment of dysphagia depends on both the locus and the specific etiology. Oropharyngeal dysphagia most commonly results from functional deficits caused by neurologic disorders. In such circumstances, the treatment focuses on utilizing postures or maneuvers devised to reduce pharyngeal residue and enhance airway protection learned under the direction of a trained swallow therapist. Aspiration risk may be reduced by altering the consistency of ingested food and liquid. Dysphagia resulting from a cerebrovascular accident usually, but not always, spontaneously improves within the first few weeks after the event. More severe and persistent cases may require gastrostomy and enteral feeding. Patients with myasthenia gravis (Chap. 461) and polymyositis (Chap. 388) may respond to medical treatment of the primary neuromuscular disease. Surgical intervention with cricopharyngeal myotomy is usually not helpful, with the exception of specific disorders such as the idiopathic cricopharyngeal bar, Zenker’s diverticulum, and oculopharyngeal muscular dystrophy. Chronic neurologic disorders such as Parkinson’s disease and amyotrophic lateral sclerosis may manifest with severe oropharyngeal dysphagia. Feeding by a nasogastric tube or an endoscopically placed gastrostomy tube may be considered for nutritional support; however, these maneuvers do not provide protection against aspiration of salivary secretions or refluxed gastric contents.
Treatment of esophageal dysphagia is covered in detail in Chap. 347. The majority of causes of esophageal dysphagia are effectively managed by means of esophageal dilatation using bougie or balloon dilators. Cancer and achalasia are often managed surgically, although endoscopic techniques are available for both palliation and primary therapy, respectively. Infectious etiologies respond to antimicrobial medications or treatment of the underlying immunosuppressive state. Finally, eosinophilic esophagitis has emerged as an important cause of dysphagia that is amenable to treatment by elimination of dietary allergens or administration of swallowed, topically acting glucocorticoids.
54 |
Nausea, Vomiting, and Indigestion |
Nausea is the subjective feeling of a need to vomit. Vomiting (emesis) is the oral expulsion of gastrointestinal contents due to contractions of gut and thoracoabdominal wall musculature. Vomiting is contrasted with regurgitation, the effortless passage of gastric contents into the mouth. Rumination is the repeated regurgitation of food residue, which may be rechewed and reswallowed. In contrast to emesis, these phenomena may exhibit volitional control. Indigestion is a term encompassing a range of complaints including nausea, vomiting, heartburn, regurgitation, and dyspepsia (the presence of symptoms thought to originate in the gastroduodenal region). Some individuals with dyspepsia report predominantly epigastric burning, gnawing, or pain. Others experience postprandial fullness, early satiety (an inability to complete a meal due to premature fullness), bloating, eructation (belching), and anorexia.
NAUSEA AND VOMITING
MECHANISMS
Vomiting is coordinated by the brainstem and is effected by responses in the gut, pharynx, and somatic musculature. Mechanisms underlying nausea are poorly understood but likely involve the cerebral cortex, as nausea requires conscious perception. This is supported by functional brain imaging studies showing activation of a range of cerebral cortical regions during nausea.
Coordination of Emesis Brainstem nuclei—including the nucleus tractus solitarius; dorsal vagal and phrenic nuclei; medullary nuclei regulating respiration; and nuclei that control pharyngeal, facial, and tongue movements—coordinate initiation of emesis. Neurokinin NK1, serotonin 5-HT3, and vasopressin pathways participate in this coordination.
Somatic and visceral muscles respond stereotypically during emesis. Inspiratory thoracic and abdominal wall muscles contract, producing high intrathoracic and intraabdominal pressures that evacuate the stomach. The gastric cardia herniates above the diaphragm, and the larynx moves upward to propel the vomitus. Distally migrating gut contractions are normally regulated by an electrical phenomenon, the slow wave, which cycles at 3 cycles/min in the stomach and 11 cycles/min in the duodenum. During emesis, the slow wave is abolished and is replaced by orally propagating spikes that evoke retrograde contractions that assist in expulsion of gut contents.
Activators of Emesis Emetic stimuli act at several sites. Emesis evoked by unpleasant thoughts or smells originates in the brain, whereas cranial nerves mediate vomiting after gag reflex activation. Motion sickness and inner ear disorders act on the labyrinthine system. Gastric irritants and cytotoxic agents like cisplatin stimulate gastroduodenal vagal afferent nerves. Nongastric afferents are activated by intestinal and colonic obstruction and mesenteric ischemia. The area postrema, in the medulla, responds to bloodborne stimuli (emetogenic drugs, bacterial toxins, uremia, hypoxia, ketoacidosis) and is termed the chemoreceptor trigger zone.
Neurotransmitters mediating vomiting are selective for different sites. Labyrinthine disorders stimulate vestibular muscarinic M1 and histaminergic H1 receptors. Vagal afferent stimuli activate serotonin 5-HT3 receptors. The area postrema is served by nerves acting on 5-HT3, M1, H1, and dopamine D2 subtypes. Cannabinoid CB1 pathways may participate in the cerebral cortex. Optimal pharmacologic therapy of vomiting requires understanding of these pathways.
DIFFERENTIAL DIAGNOSIS
Nausea and vomiting are caused by conditions within and outside the gut as well as by drugs and circulating toxins (Table 54-1).
|
CAUSES OF NAUSEA AND VOMITING |
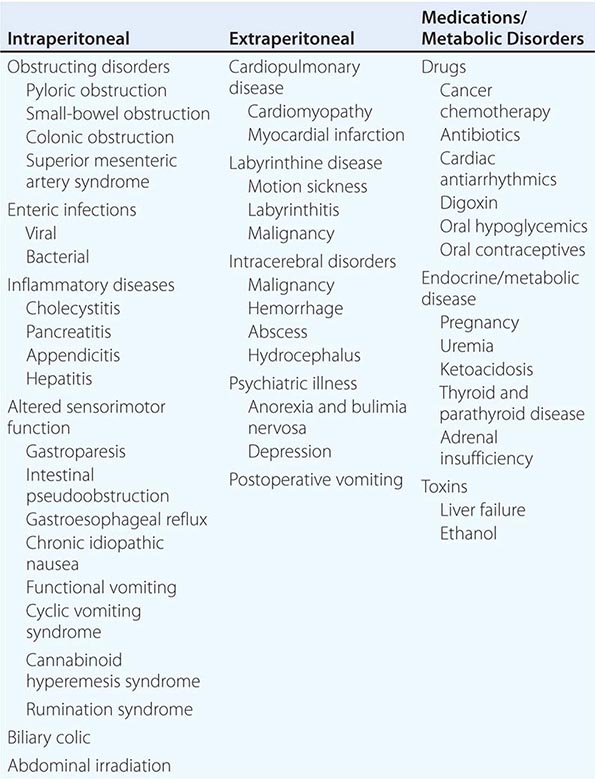
Intraperitoneal Disorders Visceral obstruction and inflammation of hollow and solid viscera may elicit vomiting. Gastric obstruction results from ulcers and malignancy, whereas small-bowel and colon blockage occur because of adhesions, benign or malignant tumors, volvulus, intussusception, or inflammatory diseases like Crohn’s disease. The superior mesenteric artery syndrome, occurring after weight loss or prolonged bed rest, results when the duodenum is compressed by the overlying superior mesenteric artery. Abdominal irradiation impairs intestinal motor function and induces strictures. Biliary colic causes nausea by acting on local afferent nerves. Vomiting with pancreatitis, cholecystitis, and appendicitis is due to visceral irritation and induction of ileus. Enteric infections with viruses like norovirus or rotavirus or bacteria such as Staphylococcus aureus and Bacillus cereus often cause vomiting, especially in children. Opportunistic infections like cytomegalovirus or herpes simplex virus induce emesis in immunocompromised individuals.
Gut sensorimotor dysfunction often causes nausea and vomiting. Gastroparesis presents with symptoms of gastric retention with evidence of delayed gastric emptying and occurs after vagotomy or with pancreatic carcinoma, mesenteric vascular insufficiency, or organic diseases like diabetes, scleroderma, and amyloidosis. Idiopathic gastroparesis is the most common etiology. It occurs in the absence of systemic illness and may follow a viral illness, suggesting an infectious trigger. Intestinal pseudoobstruction is characterized by disrupted intestinal and colonic motor activity with retention of food residue and secretions; bacterial overgrowth; nutrient malabsorption; and symptoms of nausea, vomiting, bloating, pain, and altered defecation. Intestinal pseudoobstruction may be idiopathic, inherited as a familial visceral myopathy or neuropathy, result from systemic disease, or occur as a paraneoplastic consequence of malignancy (e.g., small-cell lung carcinoma). Patients with gastroesophageal reflux may report nausea and vomiting, as do some with irritable bowel syndrome (IBS) or chronic constipation.
Other functional gastroduodenal disorders without organic abnormalities have been characterized in adults. Chronic idiopathic nausea is defined as nausea without vomiting occurring several times a week. Functional vomiting is defined as one or more vomiting episodes weekly in the absence of an eating disorder or psychiatric disease. Cyclic vomiting syndrome presents with periodic discrete episodes of relentless nausea and vomiting in children and adults and shows an association with migraine headaches, suggesting that some cases may be migraine variants. Some adult cases have been described in association with rapid gastric emptying. A related condition, cannabinoid hyperemesis syndrome, presents with cyclical vomiting with intervening well periods in individuals (mostly men) who use large quantities of cannabis over many years and resolves with its discontinuation. Pathologic behaviors such as taking prolonged hot baths or showers are associated with the syndrome. Rumination syndrome, characterized by repetitive regurgitation of recently ingested food, is often misdiagnosed as refractory vomiting.
Extraperitoneal Disorders Myocardial infarction and congestive heart failure may cause nausea and vomiting. Postoperative emesis occurs after 25% of surgeries, most commonly laparotomy and orthopedic surgery. Increased intracranial pressure from tumors, bleeding, abscess, or blockage of cerebrospinal fluid outflow produces vomiting with or without nausea. Patients with psychiatric illnesses including anorexia nervosa, bulimia nervosa, anxiety, and depression often report significant nausea that may be associated with delayed gastric emptying.
Medications and Metabolic Disorders Drugs evoke vomiting by action on the stomach (analgesics, erythromycin) or area postrema (opiates, anti-parkinsonian drugs). Other emetogenic agents include antibiotics, cardiac antiarrhythmics, antihypertensives, oral hypoglycemics, antidepressants (selective serotonin and serotonin norepinephrine reuptake inhibitors), smoking cessation drugs (varenicline, nicotine), and contraceptives. Cancer chemotherapy causes vomiting that is acute (within hours of administration), delayed (after 1 or more days), or anticipatory. Acute emesis from highly emetogenic agents (e.g., cisplatin) is mediated by 5-HT3 pathways, whereas delayed emesis is less dependent on 5-HT3 mechanisms. Anticipatory nausea may respond to anxiolytic therapy rather than antiemetics.
Metabolic disorders elicit nausea and vomiting. Pregnancy is the most prevalent endocrinologic cause, and nausea affects 70% of women in the first trimester. Hyperemesis gravidarum is a severe form of nausea of pregnancy that produces significant fluid loss and electrolyte disturbances. Uremia, ketoacidosis, adrenal insufficiency, and parathyroid and thyroid disease are other metabolic etiologies.
Circulating toxins evoke emesis via effects on the area postrema. Endogenous toxins are generated in fulminant liver failure, whereas exogenous enterotoxins may be produced by enteric bacterial infection. Ethanol intoxication is a common toxic etiology of nausea and vomiting.




