[level-membership-for-anesthesiology-category]
6 Coronary Physiology and Atherosclerosis
Anatomy and Physiology of Blood Vessels
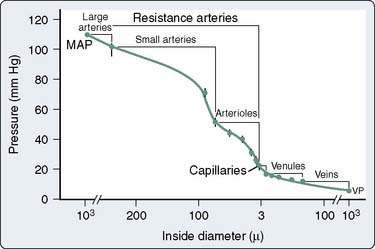
The coronary vasculature has been traditionally divided into three functional groups: large conductance vessels visible on coronary angiography, which offer little resistance to blood flow; small resistance vessels ranging in size from about 10 to 250 μm in diameter; and veins. Although it has been taught that arterioles (precapillary vessels < 50 μm) account for most of the coronary resistance, recent studies indicate that, under resting conditions, 45% to 50% of total coronary vascular resistance resides in vessels larger than 100 μm in diameter1–3 (Figure 6-1). This may be due, in part, to the relatively great length of the small arteries. During intense pharmacologic dilation, the proportion of total coronary vascular resistance because of larger arteries and veins is even greater.2 The regulation of tone in coronary arteries larger than 100 μm in diameter plays an important role in delivering adequate myocardial perfusion.4 One of the early changes in CAD is a diminished ability of the endothelium of epicardial coronary arteries to dilate in response to increased flow (see Endothelium-Derived Relaxing Factors later in this chapter). Advances in technology have enabled measurement, in the beating heart, of diameters of coronary vessels as small as 15 μm. It is becoming evident that, in response to a given intervention, different size classes of coronary vessels can change diameter with different intensity or even in opposite directions.5,6 This heterogeneity of response according to vessel size would be an important consideration in predicting the effects of vasoactive agents on myocardial perfusion. For example, a drug that dilated large vessels and collaterals but not arterioles would be beneficial to patients with CAD (see Coronary Steal later in this chapter).

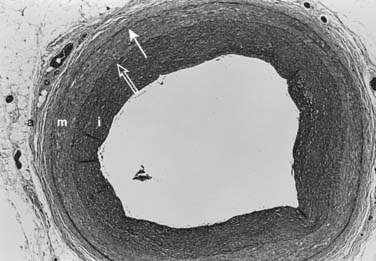
Normal Artery Wall
The arterial lumen is lined by a monolayer of endothelial cells that overlies smooth muscle cells (Figure 6-2). The inner layer of smooth muscle cells, known as the intima, is circumscribed by the internal elastic lamina. Between the internal elastic lamina and external elastic lamina is another layer of smooth muscle cells, the media. Outside the external elastic lamina is an adventitia that is sparsely populated by cells and microvessels of the vasa vasorum.

Intima
Traditionally, the intima has been considered the most important layer of the artery wall.7 The intima can vary from a single endothelial layer to a more complex structure of an endothelium overlying a patchwork of extracellular matrix and vascular smooth muscle cells. As part of the normal development of many large arteries, smooth muscle cells populate this space and form a neointima. This diffuse form of intimal thickening consists of layers of smooth muscle cells and connective tissue, the thickness of which may vary considerably. For convenience, the intima/media ratio is often measured, and the reference range is 0.1 to 1.0. How this benign intima forms is not well understood. Presumably, the intima represents a physiologic adaptation to changes in arterial flow and wall tension. The intima is made up of two distinct layers.8 As seen by electron microscopy, the inner layer subjacent to the luminal endothelium contains an abundance of proteoglycan ground substance. Smooth muscle cells found in this layer are usually distributed as isolated cells in a sea of matrix, rather than in contiguous layers. A few macrophages also may be found in this layer underneath the endothelial monolayer. The outer, musculoelastic layer of the intima is adjacent to the internal elastic lamina and contains smooth muscle cells and elastic fibers.

Media
In normal adult arteries, several smooth muscle cell subpopulations with distinct lineages exist within the media.9 These diverse cell populations likely fulfill different functions to maintain homeostasis in the artery wall. For example, in response to pressure elevations, increases in smooth muscle cell mass and extracellular matrix may be required. Alternatively, for arteries to be able to stretch both longitudinally and circumferentially, smooth muscle cells with variable orientations of cytoskeletal fibers must be present. These distinct cell types may be important not only in health but in disease. In certain experimental models of neointimal formation, proliferation and inward migration of subpopulations of medial smooth muscle cells occur.10 The biologic determinants of medial smooth muscle cell diversity are unknown.11

Adventitia
The adventitia, the outermost layer of the artery wall, normally consists of a sparse collection of fibroblasts, microvessels (vasa vasorum), nerves, and few inflammatory cells. The majority of the vasa vasorum that nourish the inner layers of the artery wall originate in the adventitia. Traditionally, the adventitia has been ignored and is not thought to play a role in vascular lesion formation. However, more recent studies have elucidated the role of the adventitia as not only a source of inflammatory cells in the development of atherosclerosis, but a hub for paracrine signaling that can maintain vascular homeostasis in a variety of vascular diseases. 12

Transmembrane and Transcellular Communication
Figure 6-3 illustrates examples of pathways of transmembrane signaling. Up to five components can be involved: receptor, G protein, effector producing a second messenger, phosphorylation of regulator protein, and the consequent change in cell behavior. G proteins (guanine nucleotide-binding regulatory proteins) are made up of three subunits (α, β, γ) and float in the cell membrane. On contact with a ligand-receptor complex, guanosine diphosphate (GDP) on the α subunit is replaced by guanosine triphosphate (GTP). The activated α subunit then dissociates from the beta-gamma complex and can interact with several membrane targets (see Figure 6-3B). For example, β-receptor activation results in the activation of Gs (s = stimulate), which will stimulate the synthesis of cAMP by adenylyl cyclase. Muscarinic receptor activation activates a Gi (i = inhibit) protein that inhibits adenylyl cyclase. A single G protein can interact with more than one effector. In this way, the G protein can be a branch point for the regulation of multiple effectors in response to a single signal. These proteins already have been implicated in human disease; cholera toxin covalently modifies Gs so that it becomes persistently active in stimulating adenylyl cyclase in intestinal epithelial cells, likely causing the severe diarrhea of cholera.
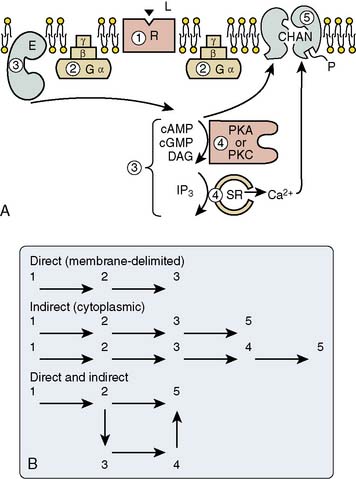
Figure 6-3 Steps in the process whereby hormone-receptor binding results in a change in cell behavior.
(A, B, From Brown AM, Birnbaumer L: Ionic channels and their regulation by G-protein subunits. Annu Rev Physiol 52:197, 1990.)
Several second-messenger systems have been characterized. Gs can directly enhance conductance through calcium channels, with the increased intracellular calcium acting as second messenger. The cyclic nucleotides, cAMP and guanosine monophosphate (GMP), act as second messengers. Their intracellular action is terminated when they are cleaved by phosphodiesterase enzymes, which, in turn, are regulated by stimuli and second messengers. The breakdown products of membrane phosphoinositide constitute another, more recently recognized set of second messengers.13 In response to agonists such as vasopressin, G protein is activated, leading to activation of the membrane-associated enzyme phospholipase C. This enzyme cleaves phosphatidylinositol 4,5-biphosphate on the inner leaflet of the plasma membrane, producing inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). Both are second messengers. IP3 diffuses through the cytoplasm and mobilizes calcium from intracellular stores. DAG remains within the plasma membrane and activates protein kinase C, which modulates cellular activity by phosphorylating intracellular proteins. In many cell types, activation of the same receptors that control phosphoinositide breakdown also results in the liberation of arachidonate and/or eicosanoids (prostaglandins, leukotrienes, and thromboxanes). The resultant change in cell behavior can be the opening of an ion channel, contraction or relaxation of smooth muscle, secretory activity, or initiation of cell division (see Chapter 7).

Endothelium
Although the vascular endothelium was once thought of as an inert lining for blood vessels, it is more accurately characterized as a very active, distributed organ with many biologic functions. It has synthetic (Table 6-1) and metabolic (Table 6-2) capabilities, and contains receptors for a variety of vasoactive substances (Table 6-3). Functions of the endothelium that may play an important role in the pathophysiology of ischemic heart disease are discussed.
| Antithrombotic Substances | Procoagulants |
|---|---|
| Prostacyclin | von Willebrand factor |
| Antithrombin III | Collagen |
| Plasminogen activator | Fibronectin |
| Protein C | Thromboplastin |
| α2-Macroglobulin | Thrombospondin |
| Glycosaminoglycans (heparin) | Plasminogen inhibitors |
| Platelet-activating factor | |
| Thromboxane A2 |
From Bassenge E, Busse R: Endothelial modulation of coronary tone. Prog Cardiovasc Dis 30:349, 1988.
TABLE 6-2 Vasoactive Substances Processed by Vascular Endothelium
| Uptake and Metabolism | Enzymatic Conversion or Degradation |
|---|---|
| Norepinephrine | Angiotensin I to angiotensin II (ACE) |
| Serotonin | Angiotensin II to angiotensin III (angiotensinase) |
| Prostaglandins (E1, E2, E2α) | Bradykinin degradation (ACE) |
| Leukotrienes | Substance P degradation |
| Adenosine |
ACE, angiotensin-converting enzyme.
From Bassenge E, Busse R: Endothelial modulation of coronary tone. Prog Cardiovasc Dis 30:349, 1988.
TABLE 6-3 Stimulators of Endothelium-Mediated Vasodilation
| Transmitters | Adenosine diphosphate (ADP) |
| Acetylcholine | Adenosine |
| Norepinephrine | Serotonin |
| Peptides | Thrombin |
| Angiotensin | Trypsin |
| Bradykinin | Local Hormones |
| Vasopressin | Histamine |
| Oxytocin | Platelet-activating factor |
| Substance P | Physicochemical Stimuli |
| Vasoactive intestinal peptide | Shear stress (flow) |
| Calcitonin gene–related peptide | Mechanical stress (pulsatility) |
| Platelet or Blood Components | Hypoxia |
| Adenosine triphosphate (ATP) |
Modified from Bassenge E, Busse R: Endothelial modulation of coronary tone. Prog Cardiovasc Dis 30:349, 1988.
Endothelium-Derived Relaxing Factors
The first vasoactive endothelial substance to be discovered was prostacyclin (PGI2), a product of the cyclooxygenase pathway of arachidonic acid metabolism (Figure 6-4; Box 6-1).14 The production of PGI2 is activated by shear stress, pulsatility of flow, hypoxia, and a variety of vasoactive mediators. On production it leaves the endothelial cell and acts in the local environment to cause relaxation of the underlying smooth muscle or to inhibit platelet aggregation. Both actions are mediated by the stimulation of adenylyl cyclase in the target cell to produce cAMP.
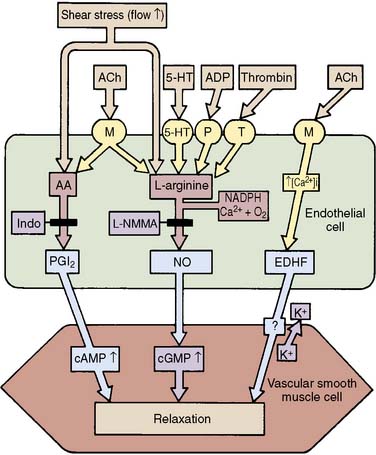
Figure 6-4 The production of endothelium-derived vasodilator substances.
(From Rubanyi GM: Endothelium, platelets, and coronary vasospasm. Coron Artery Dis 1:645, 1990.)
In 1980, Furchgott and Zawadzki15 observed that the presence of an intact endothelium was necessary for acetylcholine-induced vasodilation. Since that time it has been shown that many physiologic stimuli cause vasodilation by stimulating the release of a labile, diffusible, nonprostanoid molecule termed endothelium-derived relaxing factor (EDRF; see Figure 6-4), now known to be nitric oxide (NO). NO is the basis of a widespread paracrine signal transduction mechanism whereby one cell type can modulate the behavior of adjacent cells of different type.16,17 NO is a very small lipophilic molecule that can readily diffuse across biologic membranes and into the cytosol of nearby cells. The half-life of the molecule is less than 5 seconds, so that only the local environment can be affected. NO is synthesized from the amino acid l-arginine by nitric oxide synthase (NOS). In vascular endothelium, the enzyme (endothelial NOS or NOS3) is always present (constitutive) and resides in the cytoplasm. Its function depends on the presence of Ca++ and calmodulin, as well as tetrahydrobiopterin. Serine phosphorylation is important for prolonged activity. The enzyme is activated in response to receptor occupancy or physical stimulation (see Table 6-3). When NO diffuses into the cytosol of the target cell, it binds with the heme group of soluble guanylate cyclase, resulting in a 50- to 200-fold increase in production of cyclic GMP, its second messenger. If the target cells are vascular smooth muscle cells, vasodilation occurs; if the target cells are platelets, adhesion and aggregation are inhibited. In vascular smooth muscle, cyclic GMP leads to activation of protein kinase G, which phosphorylates various intracellular target proteins, including the myosin light-chain regulatory subunit and proteins that control intracellular calcium.18
It is likely that NO is the final common effector molecule of nitrovasodilators (including sodium nitroprusside and organic nitrates such as nitroglycerin). The cardiovascular system is in a constant state of active vasodilation that is dependent on the generation of NO. The molecule is more important in controlling vascular tone in veins and arteries compared with arterioles. When the microcirculation dilates in response to metabolic myocardial demand (e.g., exercise), increased flow through epicardial coronary arteries increases shear stress at the endothelium. This leads to release of NO, which causes vascular smooth muscle relaxation and dilation of the conductance vessels, thereby facilitating the increase in flow. The importance of the loss of this mechanism in atherosclerosis is underlined by the fact that, in this situation, more than 50% of the resistance to flow in the coronary circulation resides in vessels larger than 100 μm in diameter (see Figure 6-1). Abnormalities in the ability of the endothelium to produce NO likely plays a role in diseases such as diabetes, atherosclerosis, and hypertension.19,20 The venous circulation of humans appears to have a lower basal release of NO and an increased sensitivity to nitrovasodilators when compared with the arterial side of the circulation.21
Many agents, such as acetylcholine and norepinephrine, can cause contraction when applied directly to the vascular smooth muscle membrane instead of relaxation, which occurs when it is applied to the intact endothelium (Figure 6-5). The net effect of neural or humoral stimuli depends on a combination of direct effects mediated by binding to vascular smooth muscle receptors and indirect effects because of the ligand binding to endothelial receptors causing NO release from the endothelium. In the presence of healthy endothelium, vasodilation usually predominates. When the endothelium is absent (injured vessel) or diseased (atherosclerosis), vasoconstriction may be the net effect. NO has important roles in neurohumoral regulation of vascular tone, in preventing intravascular platelet aggregation, and in the structural adaptation of blood vessels to the demands of blood flow and pressure. Knowledge of its role in inflammation and atherosclerosis is rapidly expanding.
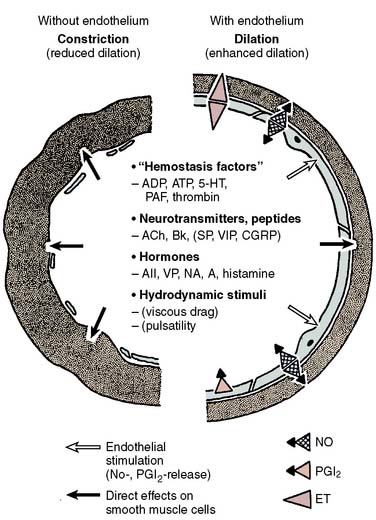
Figure 6-5 Role of endothelium in the control of coronary tone.
(From Bassenge E, Heusch G: Endothelial and neurohumoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol 116:77, 1990.)
In addition to PGI2 and NO, another less-well-understood pathway for receptor-mediated or mechanically induced endothelium-derived vasodilation exists that is associated with smooth muscle hyperpolarization. Both epoxyeicosatrienoic acid (a metabolite of cytochrome P450) and H2O2 have been suggested as possible endothelium-derived hyperpolarizing factors (EDHFs).22,23 Smooth muscle relaxation is a result of hyperpolarization of the myocyte, which leads to decreased intracellular calcium concentration. EDHF-mediated vasodilation can be blocked by inhibition of calcium-dependent potassium channels. EDHF may have an important vasodilator role in the human coronary microcirculation.24
Endothelium-Derived Contracting Factors
Contracting factors produced by the endothelium include prostaglandin H2, thromboxane A2 (TxA2; via cyclooxygenase), and the peptide endothelin (ET). ET is a potent vasoconstrictor peptide (100-fold more potent than norepinephrine)25 with remarkable similarities to the toxin of the burrowing asp. Both have potent coronary constrictor activity to which the strong cardiac toxicity and lethality of the toxin are attributed.26 Three closely related 21 amino acid peptides have been identified: endothelin-1 (ET-1), ET-2, and ET-3. The primary product of vascular endothelium is ET-1, which is synthesized from prepro-ET-1 within vascular endothelial cells by the action of ET-converting enzyme. It is not stored but rapidly synthesized in response to stimuli such as ischemia, hypoxia, and shear stress, and released predominantly abluminally (toward the underlying smooth muscle).27 In vascular smooth muscle cells, ET-1 binds to specific membrane receptors (ETA) and, via phospholipase C, induces an increase in intracellular calcium resulting in long-lasting contractions.28 It is also linked via a Gi protein to voltage-operated calcium channels. This peptide has greater vasoconstricting potency than any other cardiovascular hormone, and in pharmacologic doses can abolish coronary flow, leading to ventricular fibrillation and death.29 Another receptor subtype, ETB, is expressed by both smooth muscle and endothelium and binds ET-1 and ET-3 equally well (Figure 6-6). When isolated vessels are perfused with ET-1, there is an initial NO-mediated vasodilation because of binding with ETB receptors on the endothelial cells, followed by contraction because of binding of ET-1 to ETA receptors on the vascular smooth muscle membrane. Studies utilizing bosentan, a combined ETA– and ETB-receptor antagonist, have demonstrated that ET exerts a basal coronary vasoconstrictor tone in humans.30 There is evidence that ET may play a role in the pathophysiology of pulmonary and arterial hypertension, atherosclerosis, myocardial ischemic syndromes, and heart failure.31 Clinical trials of bosentan in patients with congestive heart failure32 and hypertension have shown promise, but hepatic side effects have limited the dose to less than 500 mg daily, with the primary indication being severe pulmonary hypertension.33
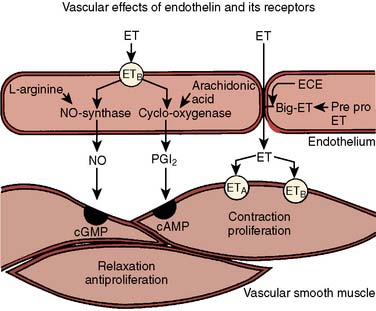
Endothelial Inhibition of Platelets
A primary function of endothelium is to maintain the fluidity of blood. This is achieved by the synthesis and release of anticoagulant (e.g., thrombomodulin, protein C), fibrinolytic (e.g., tissue-type plasminogen activator), and platelet inhibitory (e.g., PGI2, NO) substances (Box 6-2).34 Mediators released from aggregating platelets stimulate the release of NO and PGI2 from intact endothelium, which act together to increase blood flow and decrease platelet adhesion and aggregation (Figure 6-7), thereby flushing away microthrombi and maintaining the patency of the vessel.
BOX 6-2 Endothelial Inhibition Of Platelets
Healthy endothelial cells have a role in maintaining the fluidity of blood by producing:
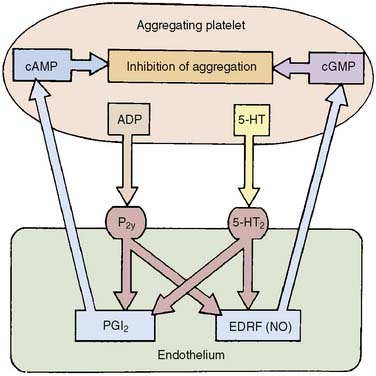
Figure 6-7 Inhibition of platelet adhesion and aggregation by intact endothelium.
(From Rubanyi GM: Endothelium, platelets, and coronary vasospasm. Coron Artery Dis 1:645, 1990.)
With vital roles in modulating the tone of vascular smooth muscle, inhibiting platelets, and processing circulating chemicals, it seems clear that endothelial cell dysfunction would cause or contribute to ischemic syndromes. There is evidence of endothelial dysfunction in atherosclerosis, hyperlipidemia, diabetes, and hypertension.35 Procedures such as coronary artery surgery and angioplasty disrupt the endothelium. The role of endothelium in the pathophysiology of myocardial ischemia is discussed later (see Dynamic Stenosis section).
Determinants of Coronary Blood Flow

Perfusion Pressure and Myocardial Compression
Coronary blood flow is proportional to the pressure gradient across the coronary circulation (Box 6-3). This gradient is calculated by subtracting downstream coronary pressure from the pressure in the root of the aorta. The determination of downstream pressure is complicated because the intramural coronary vessels are compressed with each heartbeat.
During systole, the heart throttles its own blood supply. The force of systolic myocardial compression is greatest in the subendocardial layers, where it approximates intraventricular pressure. Resistance caused by extravascular compression increases with blood pressure, heart rate, contractility, and preload. Because it is difficult to measure intramyocardial pressure, the relative importance of these factors is controversial.36,37 Flow is impeded both by direct compression and by shear caused by twisting of vessels as the heart contracts. Myocardial extravascular compression is less in the right ventricle, where pressures are lower and coronary perfusion persists during systole (Figure 6-8). In pathologic conditions associated with pulmonary hypertension, right coronary flow assumes a phasic pattern similar to left coronary flow. Under normal conditions, extravascular compression contributes only a small component (10% to 25%) to total coronary vascular resistance. When the coronary vessels are dilated by pharmacologic agents such as dipyridamole or during ischemia, the effects of extravascular compression on myocardial perfusion become more important (see Transmural Blood Flow section later in this chapter).
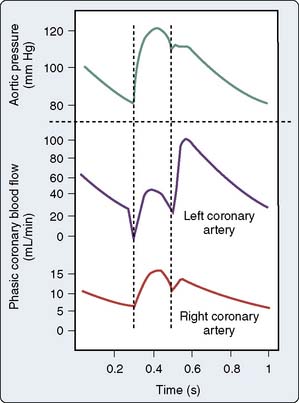
Figure 6-8 Blood flow in the left and right coronary arteries.
(From Berne RM, Levy MN: Special circulations. In Berne RM, Levy MN [eds]: Physiology. St. Louis: CV Mosby, 1988, pp 540–560.)
With each contraction, the intramural vessels are squeezed and blood is expelled forward into the coronary sinus and retrograde into the epicardial arteries. The large coronary arteries on the epicardial surface act as capacitors, charging with blood during systole and expelling blood into the coronary circulation during diastole.38 Coronary capacitance likely explains the findings of Bellamy,39 who reported that flow in the proximal left anterior descending coronary artery of the dog ceased when arterial pressure decreased to less than 45 mm Hg. It was suggested that flow throughout the coronary circulation stopped at pressures far in excess of the pressure at the coronary sinus. This pressure at which flow stopped was termed critical closing pressure or zero-flow pressure (Pzf). This had important implications in the calculation of coronary resistance because the effective downstream pressure would be Pzf and not the much lower coronary venous pressure. This is analogous to a stream with a waterfall, where flow rate over the waterfall depends on the drop from the source to the waterfall edge and is unaffected by the distance to the bottom of the falls. It was later suggested that flow through the intramural coronary vessels continues after coronary inflow near the ostia (measured by Bellamy) has ceased.40,41 There is evidence that antegrade movement of red blood cells in 20-μm arterioles continues until coronary pressure is only a few millimeters of mercury greater than coronary sinus pressure.42 The concept of a critical closing pressure greatly in excess of coronary sinus pressure is probably not valid in the coronary circulation.
Although the true downstream pressure of the coronary circulation is likely close to the coronary sinus pressure, other choices may be more appropriate in clinical circumstances. In patients with CAD, the subendocardial layers of the left ventricle are at greatest risk for ischemia and necrosis (see Transmural Blood Flow section later in this chapter). Because this layer is perfused mostly when the aortic valve is closed, the most appropriate measure of the driving pressure for flow here is the average pressure in the aortic root during diastole. This can be approximated by aortic diastolic or mean pressure. Pressures monitored in peripheral arteries by routine methods in clinical settings can differ from central aortic readings. This is due to distortion of the pressure waveform as it is propagated through the arterial tree and inaccuracies associated with the hydraulic and electronic components of the monitoring system. Under these conditions, the mean arterial pressure may be the most reliable measure of coronary driving pressure. The true downstream pressure of the left ventricular subendocardium is the left ventricular diastolic pressure, which can be estimated by pulmonary artery occlusion pressure. When the right ventricle is at risk for ischemia (e.g., severe pulmonary hypertension), right ventricular diastolic pressure or central venous pressure may be more appropriate choices for downstream pressure.

Myocardial Metabolism
Myocardial blood flow, like flow in the brain and skeletal muscle, is primarily under metabolic control. Even when the heart is cut off from external control mechanisms (neural and humoral factors), its ability to match blood flow to its metabolic requirements is almost unaffected.35 Because coronary venous oxygen tension is normally 15 to 20 mm Hg, there is only a small amount of oxygen available through increased extraction. A major increase in cardiac oxygen consumption (Mvo2) can occur only if oxygen delivery is increased by augmentation of coronary blood flow. Normally, flow and metabolism are closely matched so that over a wide range of oxygen consumption, coronary sinus oxygen saturation changes little.43
Despite intensive research over the last several decades, the mediator or mediators linking myocardial metabolism so effectively to myocardial blood flow are still unknown. Hypotheses of metabolic control propose that vascular tone is linked either to a substrate that is depleted, such as oxygen or adenosine triphosphate (ATP), or to the accumulation of a metabolite such as CO2 or hydrogen ion (Box 6-4). Adenosine has been proposed in both categories. Feigl43 has proposed six criteria for a chemical transmitter between the cardiac myocyte and the coronary vascular smooth muscle cell:
Many potential mediators of metabolic regulation have been proposed.44 Although NO has a role in many coronary vasoregulatory pathways, it does not fulfill the role of metabolic regulator because blockade of NOS does not alter the increase in myocardial blood flow associated with an increase in myocardial oxygen demand.45 The arguments for oxygen, carbon dioxide, and adenosine are briefly examined.
Oxygen
The coronary smooth muscle would have to be more sensitive to lack of oxygen than the working cardiocytes for oxygen to regulate coronary flow through a direct vascular action. Coronary microvessels in vitro do not relax until Po2 is less than 5 mm Hg, a level well below the average Po2 of 20 mm Hg in cardiac muscle cytosol.46,47 With myocardial oxygen consumption (Mvo2) held constant, increases in arterial oxygen content cause coronary flow to decrease, whereas decreases in arterial oxygen content cause flow to increase. These changes could explain only 40% of the increase in flow observed with tachycardia.48 It is undecided whether the constancy of myocardial oxygen tension is the cause or the consequence of the excellent match between myocardial metabolism and myocardial blood flow.49
Carbon Dioxide
The end product of substrate oxidation is CO2, the formation of which is directly related to the level of cardiac work. Carbon dioxide is highly diffusible and can easily reach coronary smooth muscle cells. Unfortunately, it is difficult to separate the effects on coronary tone of increasing CO2 from concomitant increases in other metabolites. Broten et al48 pump-perfused the left main coronary artery of dogs and used an oxygenator in the perfusion circuit to alter coronary arterial Pco2 and Po2 at a constant level of myocardial metabolism. Increases in arterial and coronary sinus Pco2 caused increases in coronary blood flow in the absence of changes in Mvo2. Interestingly, there was a synergistic action of Pco2 and Po2: The increase in flow with elevation of Pco2 was much greater at low Po2 and vice versa. The effect of increasing CO2, however, could not completely account for flow changes associated with an increase in Mvo2.
Adenosine
Adenosine is a powerful coronary vasodilator via its activation of receptors on vascular endothelium and smooth muscle. In 1963, both Berne50 and Gerlach51 independently demonstrated the production of adenosine in ischemic heart muscle. They hypothesized that the release of adenosine may serve as a feedback signal inducing coronary vasodilation and augmenting coronary blood flow in proportion to myocardial metabolic needs. Initially, it was suggested that adenosine formation was coupled to myocardial oxygen tension.50 A substrate theory has been proposed whereby adenosine production is linked to the cardiac energy state by the regulation of cytosolic AMP concentration to explain metabolic regulation by adenosine under both normoxic and ischemic conditions.52 According to this theory, increases in cardiac work lead to a decline in ATP potential, which results in a quantitatively appropriate change in cytosolic AMP concentration, leading to increased adenosine release. In this way, the rate of adenosine production is determined by the myocardial oxygen supply/demand ratio. It is likely that adenosine causes coronary arteriolar dilation through stimulation of A1 receptors directly coupled to ATP-sensitive K+ (K+ATP) channels and A2 receptor–mediated elevation of cAMP/protein kinase A, which lead to vasodilation, in part, by opening of K+ ATP channels.53,54
Evidence against the adenosine hypothesis is accumulating. Adenosine deaminase is an enzyme that, when introduced in sufficient quantity into the myocardium, can significantly reduce the interstitial concentration of adenosine. Aminophylline and theophylline interfere with the coronary dilating effects of adenosine by acting on the receptor on vascular smooth muscle. Experiments using these agents to inhibit adenosine effect have shown that resting coronary blood flow, exercise-induced coronary dilation, autoregulation, and reactive hyperemia are largely unrelated to adenosine.55–58 Measuring coronary microvessel diameters in beating hearts in situ, Kanatsuka et al5 found that when Mvo2 was doubled by pacing, vessels between 40 and 380 μm dilated, whereas when a similar increase in flow was induced by the infusion of adenosine or dipyridamole at constant Mvo2, only vessels smaller than 150 μm dilated. Although adenosine does not seem to have an important role in metabolic regulation in the normal heart, adenosine blockade has been shown to cause a decrease in blood flow to hypoperfused myocardium sufficient to decrease systolic segment shortening.59 Adenosine may have other important roles in ischemia, in which there is evidence of a cardioprotective action.60,61

Neural and Humoral Control
Coronary Innervation
The heart is supplied with branches of the sympathetic and parasympathetic divisions of the autonomic nervous system. Thicker vagal fibers end in the adventitia of coronary vessels, whereas fine nonmedullated sympathetic fibers end on vascular smooth muscle cells.62 Large and small coronary arteries, as well as veins, are richly innervated. The sympathetic nerves to the heart and coronary vessels arise from the superior, middle, and inferior cervical sympathetic ganglia, as well as the first four thoracic ganglia. The stellate ganglion (formed when the inferior cervical and first thoracic ganglia merge) is a major source of cardiac sympathetic innervation. The vagi supply the heart with efferent cholinergic nerves.
Parasympathetic Control
Vagal stimulation causes bradycardia, decreased contractility, and lower blood pressure. The resultant decline in Mvo2 causes a metabolically mediated coronary vasoconstriction. When myocardial metabolism is held constant, however, cholinergic coronary dilation is consistently observed in response to exogenous acetylcholine, electrical vagal stimulation, and reflex activation through baroreceptors, chemoreceptors, and ventricular receptors.35,43,63 These effects can be abolished by atropine.
In patients with angiographically normal coronary arteries, the response to intracoronary acetylcholine injection is predominantly dilation, whereas in atherosclerotic segments of epicardial arteries, constriction is observed.64–66 Acetylcholine injected intraluminally binds to muscarinic receptors on the endothelium and stimulates the release of NO, which causes smooth muscle dilation. Acetylcholine is not normally found circulating in the blood but is released from vagal fibers and reaches the coronary smooth muscle from the adventitial side. Surprisingly, activation of muscarinic receptors on vascular smooth muscle cells causes constriction. Parasympathetic stimulation normally causes coronary vasodilation. This response depends on the ability of the coronary endothelium to elaborate NO and perhaps also EDHF (see earlier).67,68 Parasympathetic control has not been shown to be important in the initiation of myocardial ischemia.
β-Adrenergic Coronary Dilation
β-Receptor activation causes dilation of both large and small coronary vessels even in the absence of changes in blood flow.35,43 Studies in animals indicate that both β1 and β2 receptors are present throughout the coronary circulation, but β1 receptors predominate in the conductance vessels, whereas β2 receptors predominate in the resistance vessels. Mature canine coronary collaterals respond similarly to the conductance vessels.69,70 β-Adrenergic coronary dilation may improve the speed and accuracy of coronary blood flow regulation during exercise.71
α-Adrenergic Coronary Constriction
Activation of the sympathetic nerves to the heart results in increases in heart rate, contractility, and blood pressure, which lead to a marked, metabolically mediated increase in coronary blood flow (Box 6-5). This suggested to early investigators that the effect of sympathetic coronary innervation is vasodilation. More recent investigation has demonstrated that the direct effect of sympathetic stimulation is coronary vasoconstriction, which is in competition with the metabolically mediated dilation of exercise or excitement. Whether adrenergic coronary constriction is powerful enough to further diminish blood flow in ischemic myocardium or whether it can have some beneficial effect in the distribution of myocardial blood flow is controversial.
Classification
α-Adrenergic receptors can be classified anatomically as presynaptic or postsynaptic and also according to their pharmacologic properties as α1 and α2 (Table 6-4). The receptors can be further divided into subtypes according to their signal transduction mechanism (G-protein subtype) and second messenger (adenylyl cyclase, phospholipase C, etc.).72
TABLE 6-4 Classification of α-Adrenergic Receptor Subtypes in the Heart
| Selective Agonists | Selective Antagonists | Effects of Activation |
|---|---|---|
| α1 | ||
| Phenylephrine | Prazosin | Presynaptic: feedback inhibition of norepinephrine release |
| Methoxamine | Postsynaptic: coronary vasoconstriction, increase in myocardial arrhythmias | |
| Inotropism | ||
| α2 | ||
| Clonidine | Yohimbine | Presynaptic: feedback inhibition of norepinephrine release |
| Azepexole | Rauwolscine | Postsynaptic: coronary vasoconstriction, arrhythmias (?) |
| BHT 920 | Idazoxan | |
| UK 14, 304 |
Norepinephrine is a nonselective agonist. Phentolamine and phenoxybenzamine are nonselective antagonists. Phenylephrine also causes β-receptor activation.
Modified from Heusch G: Alpha-adrenergic mechanisms in myocardial ischemia. Circulation 81:1, 1990.
Presynaptic α Receptors
α receptors on cardiac sympathetic nerve terminals mediate feedback inhibition of neuronal norepinephrine release. Both α1 and α2 receptors appear to be involved because exercise-induced increases in heart rate and contractility can be potentiated by either idazoxan (α2-blockade) or prazosin (α1-blockade).73
Cardiac Muscle Cells
Activation of myocardial α1 receptors results in a positive inotropic effect that, in contrast with β-receptor activation, is associated with prolongation of contraction. Although normally of minor functional importance, this effect may serve as an inotropic reserve mechanism when β-receptor–mediated inotropy is impaired (e.g., hypothyroidism, cardiac failure, chronic propranolol treatment).74 The importance of this mechanism in humans is uncertain. An increase in inotropy caused by stimulation of myocardial α receptors would result in increased Mvo2 and a metabolically mediated coronary dilation.
Coronary Endothelium
Binding of norepinephrine to α2 receptors on vascular endothelium stimulates the release of NO, which acts to relax vascular smooth muscle. The endothelium can also act to limit the effect of norepinephrine by metabolizing it. In these ways, the endothelium modulates the direct constrictive effects of α-adrenergic activation. Abnormal endothelial function in atherosclerosis may predispose to excessive α-adrenergic constriction and is implicated in the pathogenesis of myocardial ischemia (see Dynamic Stenosis later in this chapter).
Coronary Resistance
The magnitude of α-adrenergic vasoconstriction that occurs in the coronary bed is small compared with that which occurs in the skin and skeletal muscle. In the presence of β-blockade, intense sympathetic stimulation results in only a 20% to 30% increase in coronary resistance.75 Mohrman and Feigl76 examined the effect of sympathetic activation on coronary flow in the absence of β-blockade. The net effect of α-receptor vasoconstriction was to restrict the metabolically related flow increase by 30%, thereby increasing oxygen extraction and decreasing coronary sinus oxygen content.
Epicardial coronary diameter changes little during sympathetic stimulation.77 α1-Adrenergic and α2-adrenergic receptors are found throughout the coronary circulation; however, α1 receptors appear to be more important in the large epicardial vessels, whereas α2 predominate in small coronary vessels less than 100 μm in diameter.78 Studies of mature coronary collateral vessels in dogs have generally failed to provide evidence of α-receptor–mediated vasoconstriction.79 After heart transplant, patients demonstrated a lesser increase in myocardial blood flow after a cold pressor test in denervated regions of the heart.80 The authors argue that this was not due to increased myocardial metabolism secondary to myocardial β-receptor activation. They suggest that sympathetic innervation has an important role in coronary vessel dilation during stress.
Exercise
α-Adrenergic coronary constrictor tone during exercise is exerted predominantly by circulating catecholamines.81 Numerous studies indicate that myocardial blood flow during exercise is limited by α vasoconstriction.35 In a study of exercising dogs, Huang and Feigl82 found that despite an increase in total coronary flow in an α-blocked region of myocardium, flow to the inner, subendocardial layer was diminished. These results suggest a beneficial effect of α-adrenergic coronary constriction on the distribution of blood flow within the myocardium.
Myocardial Ischemia
Buffington and Feigl83 demonstrated the persistence of α-adrenergic coronary vasoconstriction distal to a moderate coronary stenosis during norepinephrine infusion. Investigations in dogs have demonstrated that, as coronary reserve is depleted by increasing stenosis severity, the response to sympathetic stimulation shifts from a metabolically induced coronary dilation to coronary constriction.84,85 These observations suggest that sympathetic coronary vasoconstriction limits coronary blood flow even during myocardial ischemia, when autoregulatory reserve is exhausted (see Coronary Reserve later in this chapter). There is no consensus as to the importance of α1 vs α2 receptors in ischemic myocardium.35 Using constant flow coronary perfusion in anesthetized dogs, Nathan and Feigl86 compared the transmural distribution of myocardial blood flow in α-blocked and intact regions of myocardium during hypoperfusion. Surprisingly, α-blockade diverted blood flow from the subendocardium to the subepicardium. This suggests that α vasoconstriction had limited flow more in the subepicardium, thereby producing an antisteal effect, and improved perfusion of the more vulnerable inner layers of the left ventricle. Chilian and Ackell87 found similar results in exercising dogs with an artificial coronary stenosis. In contrast, work from Heusch and colleagues88,89 demonstrated improved subendocardial perfusion distal to a severe coronary stenosis with α2-receptor blockade. This controversy is unresolved.90 α-Receptor blockers have not been shown to have a role in the treatment of myocardial ischemia in patients with CAD.
Studies in Humans
Studies indicate that there is little α-adrenoceptor–mediated tone in resting humans.91 Clinical studies have failed to provide convincing evidence that α-adrenergic coronary constriction plays an important role in Prinzmetal’s variant angina (angina with ST-segment elevation at rest).92 During sympathetic activation, however, there is evidence that α vasoconstriction can precipitate myocardial ischemia by further narrowing diseased coronary arteries. This has been shown during isometric exercise, dynamic exercise, and with the cold pressor test93–98 (see Dynamic Stenosis later in this chapter).
Humoral Control
A complete understanding of the effects of circulating substances on the coronary vessels would require determining their effects on large versus small coronary vessels, while separating direct effects on coronary vessels from changes in tone mediated by changes in myocardial metabolism. This is further complicated by the critical role of an intact vascular endothelium in modulating these responses (see Endothelium earlier in this chapter). Some of the better studied agents are discussed briefly later.
The peptide hormones include vasopressin (arginine vasopressin [AVP] or antidiuretic hormone [ADH]), atrial natriuretic peptide (ANP), vasoactive intestinal peptide, neuropeptide Y, and calcitonin gene–related peptide.44 Of these, AVP and ANP have been the most studied. It has been demonstrated in dogs that AVP, in concentrations 3 to 30 times those found in stressed patients, can cause vasoconstriction sufficient to produce myocardial ischemia.99 In large coronary arteries, the dilator response (via NO) likely exceeded the constrictor response.35 This was due to constriction of the small-resistance vessels. In physiologic concentrations, AVP acts primarily as an ADH with little effect on the coronary circulation. ANP can cause endothelium-dependent coronary dilation but is not known to have significant vascular effects in physiologic concentrations.100
Angiotensin-converting enzyme (ACE) is present on vascular endothelium and converts angiotensin I to angiotensin II (AII), which causes coronary vasoconstriction. AII also facilitates release of norepinephrine from presynaptic adrenergic nerve terminals. ACE inactivates bradykinin, which can attenuate vasoconstriction via NO stimulation. Thus, ACE inhibition can reduce coronary tone by suppressing AII formation and degrading bradykinin, and perhaps also by decreasing norepinephrine release. Despite these theoretical considerations, ACE inhibition has not been shown to be of benefit in human myocardial ischemia other than through control of afterload.101
PGI2 and TxA2 are synthesized from arachidonic acid in a reaction catalyzed by cyclooxygenase. PGI2 is synthesized in the vascular endothelium and, in addition to inhibiting platelet aggregation, induces vasodilation (see Endothelium-Derived Relaxing Factors section earlier in this chapter). TxA2 is mainly synthesized in platelets and causes platelet aggregation and vasoconstriction in the presence of damaged vascular endothelium. In response to TxA2, the intact endothelium releases NO, causing both vasodilation and platelet disaggregation, mechanisms to maintain patency of normal vessels (see Endothelial Inhibition of Platelets section earlier in this chapter). Unlike platelets, the vascular endothelium can synthesize proteins de novo, and thus cyclooxygenase acetylation by aspirin administration has a lesser effect in reducing vascular PGI2 than platelet TxA2. Other than in platelet-vessel interactions and inflammation, prostaglandins are not known to have an important role in the regulation of coronary blood flow.35 Serotonin (5-HT) is another platelet product that can cause endothelium-dependent dilation of coronary arterial vessels smaller than 100 μm, but causes constriction of larger epicardial coronary arteries.102
Histamine receptors are present in the coronary vessels. H1 receptors are located on vascular smooth muscle cells of large and small coronary arteries, and mediate vasoconstriction. H2 receptors are located on smooth muscle cells of arterioles and mediate vasodilation. H1 receptors also are located on vascular endothelium and can mediate vasodilation via stimulation of NO release. In patients with vasospastic angina and endothelial dysfunction, administration of exogenous histamine can cause vasospasm.103
Coronary Pressure-Flow Relations

Autoregulation
Autoregulation is the tendency for organ blood flow to remain constant despite changes in arterial perfusion pressure.104 Autoregulation can maintain flow to myocardium served by stenotic coronary arteries despite low perfusion pressure distal to the obstruction. This is a local mechanism of control and can be observed in isolated, denervated hearts. If Mvo2 is fixed, coronary blood flow will remain relatively constant between mean arterial pressures of 60 to 140 mm Hg. Figure 6-9 illustrates that, at a given cardiac workload, the level of flow (determined by metabolic regulation) is maintained constant over a broad range of pressure by autoregulation.
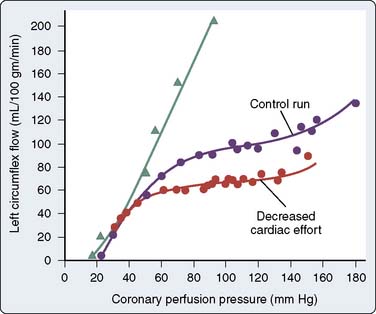
Figure 6-9 Autoregulation at two levels of myocardial oxygen consumption.
(From Mosher P, Ross J Jr, McFate PA, Shaw RF: Control of coronary blood flow by an autoregulatory mechanism. Circ Res 14:250, 1964.)
Coronary perfusion pressure must be varied while holding Mvo2 constant to study autoregulation. This is difficult in the heart because changing aortic pressure changes both the perfusion pressure for the coronary arteries and the afterload of the left ventricle. Thus, changes in aortic pressure inevitably change Mvo2. This problem is overcome by cannulating the coronary arteries and perfusing them with a pump. However, even when heart rate and aortic pressure are held constant, Mvo2 changes with changing coronary pressure. This is because myocardial contractility and metabolism increase when coronary pressure is increased to more than the normal autoperfused level. This phenomenon is known as the Gregg effect and may be explained by the “garden hose” hypothesis of Lochner, whereby engorgement of the coronary vasculature elongates the myocardial sarcomere length during diastole and contractile strength is increased because of the Frank–Starling mechanism (for a detailed review, see Feigl43 and Gregg105).
In addition to the Gregg effect, two other issues complicate studies of autoregulation: collateral flow and myocardial oxygen extraction. If pressure is lowered in the left coronary artery and not in the right, there will be a pressure gradient for flow from the right to left coronary artery via collateral vessels. Flow measured proximally in the left coronary artery will then underestimate flow reaching the myocardium. Normal coronary sinus oxygen tension (CSo2) is less than 20 mm Hg. Dole106 observed that autoregulation was effective when CSo2 was less than 25 mm Hg, but was completely lost when CSo2 exceeded 32 mm Hg. Autoregulation can be intensified by vasoconstriction (increased oxygen extraction) and attenuated by vasodilation (decreased oxygen extraction).107 The degradation of autoregulation with α-receptor blockade suggests a benefit of adrenergic coronary vasoconstriction.108
Early reports indicated that autoregulation is less effective in the right ventricle than the left. More recently, it has been suggested that increases in right coronary pressures may produce large changes in Mvo2, perhaps because of an exaggerated Gregg effect. When changes in myocardial metabolism are taken into account, autoregulation in the right and left ventricle is similar.109,110
Quantitation of the degree of autoregulation must involve a comparison of the observed change in vascular resistance to the change in resistance that would have occurred in the absence of flow autoregulation. Some degree of autoregulation exists when the relative change in flow (ΔF/F) is less than the relative change in pressure (ΔP/P). From these definitions, Dole107 has derived an autoregulation index that can be used to quantify the effects of different agents on coronary autoregulation.111
Three theories have been proposed to explain coronary autoregulation: the tissue pressure theory, the myogenic theory, and the metabolic theory.112 The tissue pressure hypothesis proposes that changes in perfusion pressure result in directionally similar changes in capillary filtration and, therefore, tissue pressure. In this way, extravascular resistance would oppose changes in flow with changes in perfusion pressure. Experimental evidence has shown, however, that there is no relation between the degree of autoregulation and the magnitude of change in tissue pressure. Arterial smooth muscle contracts in response to augmented intraluminal pressure; this is known as the myogenic response. Recently, this response has been demonstrated in coronary arterioles in the presence and absence of functioning endothelium.113 The argument for myogenic regulation of coronary flow is that myocardial metabolic changes are not rapid enough to explain large decreases in resistance after coronary occlusions for one or two heartbeats. However, myocardial metabolic events have been shown to occur during the course of a single cardiac contraction.114 The metabolic theory of autoregulation proposes that coronary arteriolar tone is determined by the balance of myocardial oxygen supply and demand. An increase in flow above the requirements of metabolism would wash out metabolites or cause accumulation of substrates, and this would be the signal for an appropriate change in coronary tone. Although metabolic regulation and autoregulation are separate phenomena, they may, therefore, have a common underlying mechanism. Metabolic regulation is discussed earlier (see Myocardial Metabolism). For an instructive, three-dimensional, graphic analysis of the interrelations among coronary artery pressure, myocardial metabolism, and coronary blood flow, see Feigl et al.115

Coronary Reserve
Myocardial ischemia causes intense coronary vasodilation. After a 10- to 30-second coronary occlusion, restoration of perfusion pressure is accompanied by a marked increase in coronary flow. This large increase in flow, which can be five or six times resting flow in the dog, is termed reactive hyperemia. Figure 6-10 illustrates that the repayment volume is greater than the debt volume. There is, however, no overpayment of the oxygen debt because oxygen extraction declines during the hyperemia.116 The presence of high coronary flows when coronary venous oxygen content is high suggests that mediators other than oxygen are responsible for this metabolically induced vasodilation.43 The difference between resting coronary blood flow and peak flow during reactive hyperemia represents the autoregulatory coronary flow reserve: the further capacity of the arteriolar bed to dilate in response to ischemia. In Figure 6-9, the flow reserve is the vertical distance from the autoregulating pressure-flow curve (purple or red circles) to the nonautoregulating curve (triangles). The reserve is greater at higher perfusing pressure and lower Mvo2. Unlike cannula-perfused preparations in which these data are obtained, in the clinical setting, increases in pressure increase both perfusing pressure and Mvo2. Reactive hyperemia responses have been used in animals and humans to estimate coronary reserve in conditions such as obstructive coronary disease, aortic stenosis, and left ventricular hypertrophy117–119. The myocardial fractional flow reserve (FFR) is calculated by dividing the pressure in a coronary vessel distal to a stenosis during maximal pharmacologic dilation by the aortic root pressure. This ratio (FFR) easily can be measured in the angiography suite and has been recommended as a useful index of the functional severity of coronary stenoses of intermediate morphologic severity on angiography, as well as a measure of residual obstruction after interventions.120 Indeed, the relevance of a reduction in the FFR is highlighted in a recent randomized, controlled study that demonstrated improved clinical outcomes in FFR-guided percutaneous coronary interventions as opposed to angiography alone.121
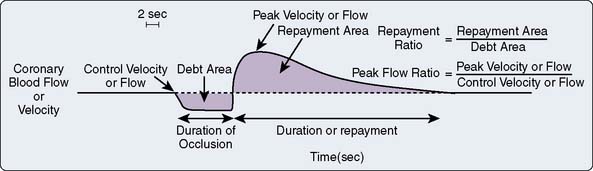
Figure 6-10 Schematic diagram of the reactive hyperemic response to a 10-second coronary occlusion.
(From Marcus ML: Metabolic regulation of coronary blood flow. In Marcus ML [ed]: The coronary circulation in health and disease. New York: McGraw-Hill, 1983, pp 65–92. Reproduced by permission of McGraw-Hill Companies.)
It has been generally accepted that the coronary resistance vessels are maximally dilated when coronary perfusion pressure is reduced sufficiently to cause myocardial ischemia. In fact, agents such as adenosine, carbochromene, and dipyridamole can cause further increases in coronary flow in the presence of intense ischemia, when autoregulatory reserve is believed to be exhausted. This pharmacologic vasodilator reserve is greater than the autoregulatory vasodilator reserve. If flow to ischemic myocardium can be increased by pharmacologic dilation of resistance vessels, the use of these agents should reverse ischemic dysfunction and metabolism. Arteriolar dilators have, in general, not been found to be beneficial during myocardial ischemia. Coronary blood flow in the different layers of the ventricle must be reviewed to understand why (Box 6-6).
BOX 6-6 Transmural Blood Flow

Transmural Blood Flow
It is well-known that, when coronary perfusion pressure is inadequate, the inner one third to one fourth of the left ventricular wall is the first region to become ischemic or necrotic.122 This increased vulnerability of the subendocardium may be caused by an increased demand for perfusion or a decreased supply, compared with the outer layers. There has been extensive study of the transmural distribution of oxygen consumption, use of oxidizable substrates, activity of glycolytic and mitochondrial enzymes, tissue contents of endogenous substrates, high-energy phosphates, lactate, isoforms of contractile proteins, and fiber stress and fiber shortening. In general, these studies indicate that if such differences exist between the layers of the left ventricle, they are unlikely to exceed 10% to 20%.43,123 It is likely that preferential underperfusion of the subendocardium is the primary determinant of its increased vulnerability.
Regional blood flow in the myocardium is usually determined using radioactive microspheres. These plastic beads, labeled with a radioisotope, are injected into the bloodstream. The assumption is that they will mix uniformly with blood and be distributed in proportion to blood flow, as if they were red blood cells.124 Because they are rigid and larger than red cells (9- or 15-μm diameters are usually chosen), they are trapped in the microcirculation. At the end of an experiment, the heart can be divided into small blocks and the amount of radioactivity in each piece measured in a gamma counter. The blood flow to each block of tissue will be proportional to the number of microspheres in each piece, which can be determined from its radioactivity. By using different radioisotopes as labels, several sets of microspheres can be injected during an experiment, giving “snapshots” of what flow was at the time of each injection. It is difficult to reduce the variability of the technique below 10%.43,125 Subendocardial blood flow is found using this technique to be about 10% greater than subepicardial blood flow under normal circumstances. This gives a normal subendocardial/subepicardial or inner/outer (I/O) blood flow ratio of 1.10. This ratio is maintained at normal perfusing pressures even at heart rates greater than 200 beats/min.
If coronary pressure is gradually reduced, autoregulation is exhausted and flow decreases in the inner layers of the left ventricle before it begins to decrease in the outer layers (Figure 6-11). This indicates that there is less flow reserve in the subendocardium than in the subepicardium. The limits of autoregulation will depend on the level of cardiac work (see Autoregulation section earlier in this chapter) and on the experimental conditions. In conscious dogs, the mean coronary pressure at which evidence of subendocardial ischemia appeared was 38 mm Hg at a heart rate of 100 beats/min, and increased to 61 mm Hg at 200 beats/min. Subepicardial flow during tachycardia did not decline even at pressures as low as 33 mm Hg.126 Because subepicardial flow is rarely inadequate, a subendocardial/subepicardial blood flow ratio close to 1.0 indicates adequate subendocardial flow and an appropriate matching of myocardial oxygen supply to oxygen demand. For this reason, the I/O ratio is often used as a measure of the adequacy of myocardial blood flow.
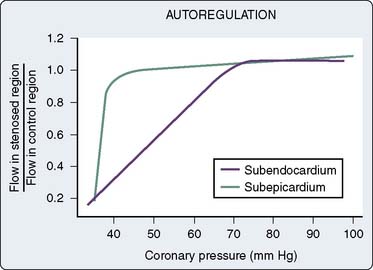
Three mechanisms have been proposed to explain the decreased coronary reserve in the subendocardium: differential systolic intramyocardial pressure, differential diastolic intramyocardial pressure, and interactions between systole and diastole. Because the force of systolic myocardial compression is greatest in the inner layers of the ventricle and is low at the subepicardium, it was believed that the outer layers of the heart were perfused throughout the cardiac cycle, whereas the subendocardium was perfused only during diastole. The subendocardium would have to obtain its entire flow during only a portion of the cycle and, therefore, would have to have a lower resistance. Recent studies, suggesting that there may be little systolic flow even to the outer layers, argue against this explanation.127 The second mechanism is based on the high coronary pressures observed when coronary flow has ceased during a long diastole, Pzf (see Perfusion Pressure and Myocardial Compression earlier in this chapter).39 The shape of the pressure-flow relation during a long diastole suggests that Pzf is higher in the subendocardium. This would mean that perfusion pressure for the subendocardium is lower in diastole compared with the outer layers of myocardium. Available evidence suggests that Pzf is not high in any layer and is unlikely to be more than 2 to 3 mm Hg greater in the subendocardium than in the subepicardium.127 Hoffman109,127 proposed an interaction between systole and diastole as the explanation for the increased vulnerability of the subendocardium to ischemia. During systole, intramyocardial pressure is high enough throughout most of the ventricular wall to squeeze blood out of the intramural vessels and into the extramural coronary veins and arteries. Because the compressive force is greatest in the subendocardium, vessels here are the narrowest at end systole. At the beginning of diastole, blood will be directed first to vessels with the lowest resistance, the larger vessels in the subepicardium, and last to the most narrowed vessels in the subendocardium. In this way, should the duration of diastole or the diastolic perfusion pressure be reduced, the subendocardial muscle would receive the least flow. Spaan36 presents an interesting analysis of the interaction between arterial pressure and force of contraction as an intramyocardial pump. Although this theory is compatible with existing evidence, support for it will remain indirect until it becomes possible to measure phasic pressures and flows in separate layers of myocardium.
When the left ventricle hypertrophies in response to a pressure load (aortic stenosis, hypertension), myofibrillar growth outstrips the capillary network, resulting in decreased capillary density and increased diffusion distances. The net effect is to reduce coronary autoregulatory reserve.128 The transmural gradient of reserve is exaggerated as well, so the subendocardium is at increased risk for ischemia in the hypertrophied heart compared with normal.129
In addition to the transmural gradient of coronary reserve from outer to inner layer of the left ventricle, there is also marked variation of reserve between small regions of myocardium within a layer.130 This heterogeneity of flow reserve may explain why pharmacologic reserve exceeds autoregulatory reserve (see Coronary Reserve section earlier in this chapter). During hypoperfusion, regional myocardial blood flow is decreased, but in all layers some small pieces of muscle will have no flow reserve left, whereas adjacent pieces can have substantial reserve. Fewer pieces will retain reserve in the subendocardium than in the subepicardium. The increase in flow in response to an infusion of adenosine is due to increased flow in the small regions with reserve, with no change in the adjacent fully dilated regions.131–133 These findings suggest that ischemia causes maximal coronary vasodilation, and that increases in flow with adenosine or dipyridamole are due to dilation of vessels in nonischemic regions. Contrasting evidence is provided by Duncker and Bache,54 who used a balloon occluder to simulate a coronary stenosis in exercising dogs. The occluder was adjusted to maintain distal coronary pressure constant at 43 mm Hg. During exercise, an intracoronary infusion of adenosine increased blood flow to all myocardial layers and improved regional systolic segment shortening. Although this is evidence of vasodilator reserve in ischemic myocardium, the constant distal pressure preparation does not faithfully mimic a coronary stenosis because it makes transmural steal impossible (see Coronary Steal later in this chapter). In general, pharmacologic dilation of resistance vessels has the potential to worsen ischemia by producing coronary steal. Dilation of larger penetrating vessels (50 to 500 μm in diameter) with nitrovasodilators could preferentially decrease resistance to blood flow to the subendocardium, and this may, in addition to favorable effects on the systemic circulation, explain the usefulness of nitrates in the treatment of angina.54
Atherosclerosis
The atherosclerotic lesion consists of an excessive accumulation of smooth muscle cells in the intima, with quantitative and qualitative changes in the noncellular connective tissue components of the artery wall, and intracellular and extracellular deposition of lipoproteins and mineral components (e.g., calcium; Box 6-7). By definition, atherosclerosis is a combination of atherosis and sclerosis. The latter term, sclerosis, refers to the hard, collagenous material that accumulates in lesions and is usually more voluminous than the pultaceous “gruel” of the atheroma (Figure 6-12).
BOX 6-7 Atherosclerosis
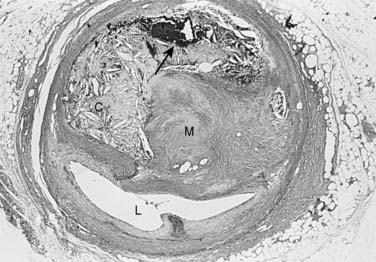
Stary and colleagues134 note that the earliest detectable change in the evolution of coronary atherosclerosis in young people was the accumulation of intracellular lipid in the subendothelial region, creating lipid-filled macrophages or “foam cells.” Grossly, a collection of foam cells may give the artery wall the appearance of a “fatty streak.” In general, fatty streaks are covered by a layer of intact endothelium and are not characterized by excessive smooth muscle cell accumulation. At later stages of atherogenesis, extracellular lipoproteins accumulate in the musculoelastic layer of the intima, eventually forming an avascular core of lipid-rich debris that is separated from the central arterial lumen by a fibrous cap of collagenous material. Foam cells are not usually seen deep within the atheromatous core, but are frequently found at the periphery of the lipid core.

Atherogenesis
Certain human arteries are more prone to develop atherosclerosis than others. For example, the coronary, renal, and internal carotid arteries, as well as some areas of the aorta, are known to be common sites for lesion formation.134 In the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study, the aorta and right coronary arteries of 1378 young people aged 15 to 34 who died as a result of trauma were studied.135 Two-dimensional maps of lipid-laden fatty streaks, as well as fibrous plaques, were made for each vessel. Although atherosclerosis is usually clinically silent until middle age or later, these investigators found that the disease process begins in adolescence or childhood. Moreover, fatty streaks and fibrous plaques do not occur randomly in the circulation but follow a well-defined distribution pattern. For example, in the right coronary artery, fatty streaks were found with the highest probability in the proximal 2 cm of this vessel, which closely parallels the distribution of raised fibrous lesions. However, in the abdominal aorta, where aortic lesions are commonly found, the high prevalence of fatty streaks did not always correlate with the prevalence of raised fibrous lesions. Therefore, at least in the aorta, the role of childhood fatty streaks in the development of adult fibrous lesions is uncertain.
Historically, there are two classical theories of atherogenesis. According to von Rokitansky’s136 thrombogenic (or encrustation) theory, fibrin is the initiating factor in lesion development. Later, Duguid137 expanded on this theory by suggesting that atherosclerosis is the result of altered fibrinolysis, whereas more recent studies have documented the overexpression of prothrombotic factors, such as plasminogen activator inhibitor-1, in atherosclerotic plaques.138 Alternatively, in 1856, Virchow’s imbibition (or insudation) theory proposed that atherosclerotic lesions were the result of altered vessel wall permeability.139 Variations of this theory have been suggested by others, and all support the concept that the accumulation of various plasma components, including lipoproteins, may be important during lesion formation. For example, Ross and Glomset140 blended the concepts of these original hypotheses into the “response-to-injury” hypothesis in which both lipid infiltration and thrombus formation play important roles in atherogenesis. Similarly, Schwartz and colleagues141 compared arterial narrowing in atherosclerotic arteries with the process of wound healing. This perspective has advantages, as it allows a multifactorial process such as atherosclerosis to be broken down into components of a more completely understood process such as the biology of a skin wound. For example, wound healing of any form begins with the formation of a clot (fibrin- and fibronectin-containing gel) that fills the wound and provides a provisional matrix for inflammatory cells, fibroblasts, and newly formed microvessels.142,143 This is followed by the proliferation and migration of fibroblasts into the wound.144 By day 7 after injury, microvessels grow into the base of the wound and form granulation tissue. As the wound matures and undergoes contracture, these blood vessels regress and fibroblasts disappear. After resorption of microvessels, tissue hypoxia develops and likely plays a role in the completion of the final scarring process.145 As discussed later, there is now ample evidence to suggest that many similar events take place during arterial wound healing; however, because atherosclerosis is a chronic process, it is likely that vascular lesion formation involves indolent levels of inflammation with ongoing cycles of injury and repair over many years.142,146,147

Arterial Wall Inflammation
A number of studies have demonstrated the presence of monocytes/macrophages and T lymphocytes in the arteries of not only advanced lesions but early atherosclerotic lesions of young adults.148,149 Moreover, in experimental atherosclerosis, leukocyte infiltration into the vascular wall is known to precede smooth muscle cell hyperplasia.150 Once inside the artery wall, mononuclear cells may play several important roles in lesion development. For example, monocytes may transform into macrophages and become involved in the local oxidation of low-density lipoproteins (LDLs) and accumulation of oxidized LDLs. Alternatively, macrophages in the artery wall may act as a rich source of factors that, for example, promote cell proliferation, migration, or the breakdown of local tissue barriers. The latter process of local tissue degradation may be important for the initiation of acute coronary artery syndromes because loss of arterial wall integrity may lead to plaque fissuring or rupture.151
Normally, the endothelium exhibits a low affinity for circulating leukocytes. Therefore, the transmigration of leukocytes into the artery wall must occur as a facilitated process. The release of proinflammatory cytokines such as interleukin-1 may promote the expression of leukocyte adhesive molecules.152 For simplicity, the interaction between leukocytes and the endothelium can be considered to involve three steps.153 First, leukocytes in the bloodstream must loosely associate and roll along the endothelium—a process that is mediated by selectins expressed on endothelial cells.152 Second, firm adhesion of these leukocytes to endothelial cells occurs via the interaction between integrins, such as α4β1 (also known as very late antigen-4 [VLA-4]), expressed on leukocytes, and counter-receptors, such as vascular cell adhesion molecule-1 (VCAM-1), on endothelial cells.154 Finally, the transmigration of leukocytes into the subendothelial space is mediated by various migration-inducing factors, such as monocyte chemoattractant protein-1 (MCP-1).155
Dysfunction, discontinuity, or injury of the endothelial cell monolayer has been postulated to play a significant role in facilitating the transmigration of leukocytes into the intima and the development of intimal hyperplasia. However, the premise that regrowth of a healthy endothelium will limit neointimal accumulation is inconsistent with the results of several independent lines of investigation. For example, in experimental models, smooth muscle cell proliferation is not increased in arterial regions devoid of an endothelium.156 Moreover, restoration of the endothelium, as might be achieved by seeding endothelial cells back into a denuded artery, does not decrease neointimal accumulation after vascular interventions.157 Therefore, the presence of an endothelium in the central lumen of an artery and resistance to intimal growth do not appear to be inextricably linked. Finally, it is important to note that the endothelium is not restricted to the central lumen, as the artery wall is also invested with a rich supply of microvessels (i.e., vasa vasorum).158–161 The vasa vasorum are likely another portal of entry for inflammatory cells into the artery wall, particularly because the expression of certain adhesion molecules is more abundant in the endothelium lining these microvessels than that of the central arterial lumen.162

Role of Lipoproteins in Lesion Formation
Much of the pioneering work in understanding cholesterol metabolism is based on seminal observations by Brown and Goldstein.163 The work of these two investigators focused on LDL, the so-called bad form of cholesterol, and the absence (or deficient forms) of the LDL receptor that are seen in familial hypercholesterolemia (FH). Patients with FH have high levels of LDL cholesterol and suffer from accelerated forms of atherosclerosis as cholesterol moieties enter the cell via an alternate route. In the absence of a functional LDL receptor, LDL cholesterol is oxidized and taken up by scavenger receptors of monocytes and macrophages resident within the artery wall. Steinberg164 and others have integrated these data into a theory of atherogenesis that highlights the central role of LDL oxidation and the formation of lipid-laden monocytes in fatty streaks.
One of the major consequences of cholesterol accumulation in the artery wall is thought to be the impairment of endothelial function. The endothelium is more than a physical barrier between the bloodstream and the artery wall. Under normal conditions, the endothelium is capable of modulating vascular tone (e.g., via nitrous oxide), thrombogenicity, fibrinolysis, platelet function, and inflammation. In the presence of traditional risk factors, particularly dyslipidemias, these protective endothelial functions are reduced or lost. Notably, the loss of these endothelial-derived functions may occur in the presence or absence of an underlying atherosclerotic plaque and may simply imply that atherogenesis has begun. Aggressive attempts to normalize atherosclerotic risk factors (e.g., diet and lipid-lowering therapies) may markedly attenuate endothelial dysfunction—even in the presence of extensive atherosclerosis. A number of clinical studies now demonstrate dramatic improvements in endothelial function, as well as cardiovascular morbidity and mortality, with the use of inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HGM-CoA) reductase, or “statins.”165–167 Future studies may help clarify the exact mechanisms by which dyslipidemias (and other risk factors) alter endothelial function.

Smooth Muscle Cell Proliferation, Migration, and Arterial Remodeling
The dominant cell type in atherosclerotic lesions is the smooth muscle cell, and as lesions progress, the number of smooth muscle cells in the artery wall tends to increase. Therefore, smooth muscle replication must occur at some time during atherogenesis. Perhaps the first line of evidence that cell replication occurs in human arteries is from the observation that atherosclerotic plaques contain monoclonal cell populations. Elegant studies by Benditt and Benditt168 demonstrated that groups (or clones) of cells that arise from a single progenitor cell are present in tissue from atherosclerotic coronary arteries of women who were deficient in glucose-6-phosphate dehydrogenase (G-6-PD). Because G-6-PD is an X-chromosome–linked enzyme that has two isoforms, cells would express only one isoform, with the other isoform being suppressed on the inactivated X chromosome. Therefore, groups of cells in an atherosclerotic plaque that contain only one isoform of G-6-PD are likely the result of proliferation of a single progenitor cell. More recently, Murry and colleagues169 studied the monoclonality of atherosclerotic plaques using X-chromosome inactivation patterns. Using the polymerase chain reaction, they examined the monoclonality of plaques according to the methylation pattern of the human androgen receptor gene, a highly polymorphic locus on the X chromosome for which 90% of women are heterozygous. These investigators note that diseased and normal arteries contain monoclonal populations (or patches) of cells. Therefore, they speculate that the monoclonality of plaques might be caused by expansion of a preexisting monoclonal patch of cells, rather than mutation or selection of individual cells in the artery wall.
Little is known about when and why cells proliferate in the artery wall. However, it is known that early in life there is a rapid expansion in neointimal smooth muscle cell mass. Sims and Gavin170,171 describe the accumulation of intimal smooth muscle cells in the left anterior descending coronary artery of neonates. Using electron microscopy, these investigators demonstrate interruptions in the internal elastic lamina in coronary arteries where a neointima had formed.171 These interruptions in the internal elastic lamina are not present in all human arteries. Indeed, the internal mammary artery, which typically is devoid of atherosclerosis, has an intact internal elastic lamina. Therefore, it has been suggested that medial smooth muscle cells migrate inward through breaks in the internal elastic lamina to expand and form a neointima. The frequency and degree of smooth muscle cell replication in adult coronary arteries have been examined by various investigators. The majority of these studies have demonstrated very low replication rates in tissue from both normal and diseased arteries.172–175 Whether these low cell replication rates are sufficient to gradually result in advanced lesions, or whether sporadic bursts of replication occur in response to injury, is unknown. Finally, it is recognized that programmed cell death, or apoptosis, occurs in the artery wall.176 Therefore, the accumulation of cells in the artery wall is a function of not only cell proliferation, but also apoptosis.
The role of smooth muscle cell migration in adult CAD is poorly understood. It has been suggested, however, that like fibroblasts that migrate into the base of a wound, arterial wall smooth muscle cells migrate inward to expand plaque mass. Smooth muscle cell migration into the intima has been studied in various animal models of neointimal formation (e.g., rat carotid artery model).11 The majority of these models demonstrate the inward migration of medial smooth muscle cells after normal arteries are subjected to balloon injury. A number of growth factors (e.g., platelet-derived growth factor) have been shown to play an important role in facilitating smooth muscle cell migration in these models.177–179 Unfortunately, the clinical relevance of these experimental observations remains to be clarified, as the milieu for cell migration in complex human lesions appears to be very different from that of normal animal arteries that are subjected to injury. More information is required regarding the factors that regulate smooth muscle cell migration, as well as why smooth muscle cells differ in their propensity to migrate after injury.
Finally, it is important to point out that the buildup of atherosclerotic plaque does not always translate into the formation of arterial obstructions.142 For example, Glagov180 notes that human vessels can accumulate massive amounts of atherosclerotic plaque without encroaching on the central arterial lumen. Instead, abluminal expansion of the artery wall may occur until 40% of the area encompassed by the internal elastic lamina is occupied by plaque. Thereafter no further enlargement may occur, and luminal narrowing may ensue. Although this form of compensatory enlargement is referred to as “remodeling,” the term is confusing because it holds different meaning in different contexts (Figures 6-13 and 6-14).180–182 For example, remodeling has also been used to describe the arterial response to changes in blood flow (e.g., during pregnancy or in the neonatal period) or pressure (e.g., hypertension).183 In addition, remodeling has been invoked as a key component of the response to arterial injury—however, with quite a different meaning.184 In animal models of arterial injury, as well as studies of human coronary arteries that have undergone angioplasty (or percutaneous transluminal coronary angioplasty [PTCA]), “shrinkage” or constrictive remodeling of the artery wall is a major determinant of lumenal narrowing, whereas neointimal formation plays a minor role in this process.185–192
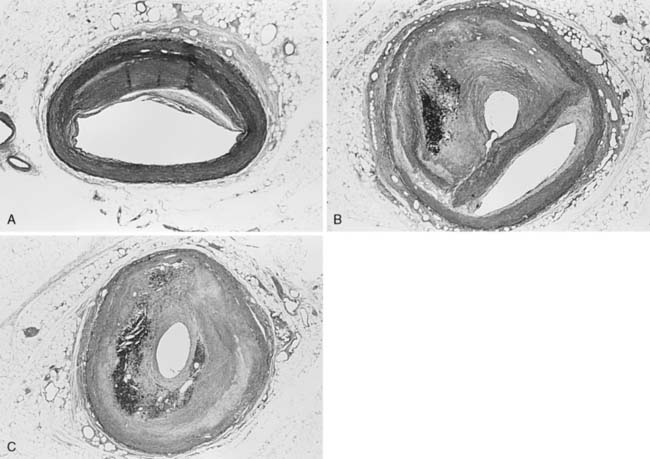
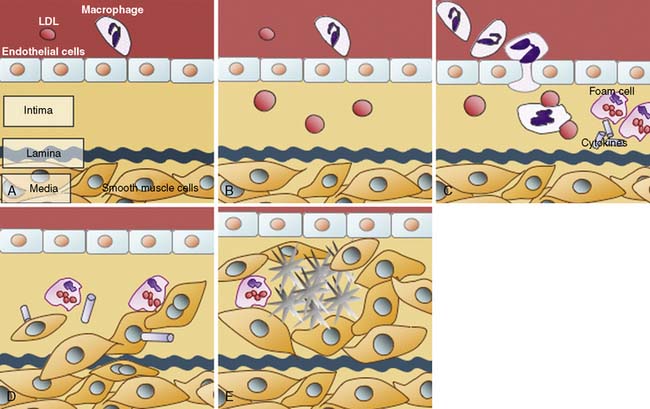
How arterial wall constriction is accomplished or why some, but not all, arteries undergo compensatory dilatation to preserve lumen area is incompletely understood.193–195 Blood flow and shear stress are known to play a critical role in remodeling. The response of arteries to chronic alterations in blood flow are endothelium dependent.196,197 For example, Langille and O’Donnell196 demonstrated in rabbit carotid arteries that decreased blood flow results in narrowing of the vessel diameter that is unchanged with papaverine and likely caused by structural changes in the artery wall. However, when the endothelium is removed from these vessels, the response to reduced blood flow is abolished. In atherosclerotic arteries that contain a rich network of endothelial cell-lined microvessels or vasa vasorum, the role of the endothelium in regulating remodeling may be important.198–200

Assessment of Atherosclerosis by Intravascular Ultrasonography
A detailed description of both diagnostic and therapeutic procedures performed in the cardiac catheterization laboratory is provided in Chapter 3. However, given that an integral understanding of the anatomy of the coronary artery and the atherosclerotic lesion is necessary for the appropriate interpretation and use of these technologies, a brief review of new developments in invasive assessment of atherosclerosis by intravascular ultrasonography (IVUS) is included here.
Standard coronary angiography gives operators a two-dimensional representation of the lumen. By examining arteries in multiple views, the operator estimates coronary stenoses by comparison of the lumen diameter at the point of maximal narrowing to adjacent disease-free segments. However, as discussed earlier, development of the atherosclerotic plaque results not only in luminal encroachment but arterial remodeling,201 meaning significant disease may be overlooked on traditional angiography. Thus, technologies, such as IVUS, are becoming more prevalent in the assessment of CAD.
IVUS of coronary arteries was first popularized in the 1990s, 202 with subsequent refinement of catheter delivery systems and commercialization making it now commonplace in the modern catheterization laboratory. Compatible with most guiding catheters, IVUS probes are delivered to the coronary arteries via standard angiography techniques, and both manual and mechanical pullback of the IVUS probe allows operators to assess real-time cross-sectional images. Complementary software can then allow users to generate either longitudinal or three-dimensional reconstructions of the interrogated vessel.
IVUS images demonstrate remarkable fidelity to cross-sectional histologic specimens and permit accurate visualization and measurement of the intima, media, and, in some instances, the adventitia (Figure 6-15). Arterial remodeling with significant intimal hyperplasia but relatively intact lumen diameter can thus identify occult disease not otherwise appreciated on standard angiography. One landmark article has noted that even if only minor luminal irregularities exist, atherosclerotic disease can be demonstrated throughout most other vessels in the coronary circulation, suggesting luminograms may be simply the tip of the atherosclerotic iceberg. 203 Indeed, IVUS is now commonly used to more accurately quantify lesions in cases of intermediate severity lesions,204 or in regions that are otherwise difficult to assess on standard angiography, such as left main disease. As well, IVUS allows operators to assess arteries after percutaneous intervention for not only adequate stent deployment but also for complications, such as arterial dissection, which can be missed on standard angiography.205 However, IVUS is not limited to simply documenting and quantifying atherosclerotic burden. Plaque composition also can be assessed qualitatively and classified based on acoustic impedance, allowing differentiation among fibromuscular “soft” lesions, dense “fibrous” lesions, and “calcified” hyperechoic lesions.206 Although not yet reliably predictable, ongoing studies are aimed at identifying which plaques are “vulnerable” or susceptible to rupture, thus causing acute vessel closure and myocardial infarction.207,208
Pathophysiology of Coronary Blood Flow

Coronary Artery Stenoses and Plaque Rupture
Coronary atherosclerosis is a chronic disease that develops over decades, remaining clinically silent for prolonged periods (Box 6-8). Clinical manifestations of CAD occur when the atherosclerotic plaque mass encroaches on the vessel lumen and obstructs coronary blood flow, causing angina. Alternatively, cracks or fissures may develop in the atherosclerotic lesions and result in acute thromboses that cause unstable angina or myocardial infarction.
On angiography, patients with stable angina typically have lesions with smooth borders. Only a minority of coronary lesions are concentric, most having a complex geometry varying in shape over their length. Eccentric stenoses, with a remaining pliable, musculoelastic arc of normal wall, can vary in diameter and resistance in response to changes in vasomotor tone or intraluminal pressure. Most human coronary stenoses are compliant.209 The intima of the normal portion of the vessel wall is often thickened, making endothelial dysfunction probable (see Dynamic Stenosis section later in this chapter). In contrast, patients with unstable angina usually have lesions characterized by overhanging edges, scalloped or irregular borders, or multiple irregularities. These complicated stenoses likely represent ruptured plaque or partially occlusive thrombus, or both.210 On angiography, these lesions may appear segmental, confined to a short segment of an otherwise normal proximal coronary artery. At autopsy, however, the most common pathologic finding is diffuse vessel involvement with superimposed segmental obstruction of greater severity.211 In a diffusely narrowed vessel, even modest progression of luminal narrowing can be significant. In such a patient, rating the significance of the obstruction by the percentage of diameter reduction relative to adjacent vessel segments will underestimate its physiologic importance.212,213 Therefore, understanding the characteristics of atherosclerotic plaques is of central importance to the management of acute coronary artery syndromes.
The intuitive notion that the severity of coronary artery stenoses should correlate with the risk for complications from CAD has been disproved by several key investigations. Ambrose and colleagues214 reviewed the coronary angiograms of 38 patients who had had a Q-wave myocardial infarct in the interval between serial studies. On the preinfarct angiograms, the mean percentage stenosis at the coronary segment that was later responsible for infarction was only 34%. Similarly, Little and colleagues215 reviewed the coronary angiograms of 42 patients who also had this procedure performed at an interval before and after myocardial infarction. Total occlusion of a previously patent artery was observed in 29 patients; yet, for 19 of these occluded arteries, the degree of stenosis was less than 50% on the initial angiogram. Therefore, although the revascularization of arteries with critical stenoses in target lesions is appropriately indicated to reduce symptoms and myocardial ischemia, the risk for further cardiac events remains because atherosclerosis is a diffuse process and mild or modest angiographic stenoses are more likely to result in subsequent myocardial infarction than are severe stenoses.
With this background comes the question of predicting which arterial segments with minimal angiographic disease will later develop new critical stenoses. Clues to the answer for this question are emerging from careful pathologic studies of lesions by Davies and Thomas.216 Superficial intimal injury (plaque erosions) and intimal tears of variable depth (plaque fissures) with overlying microscopic mural thrombosis are commonly found in atherosclerotic plaques. In the absence of obstructive luminal thrombosis, these intimal injuries do not cause clinical events. However, disruption of the fibrous cap, or plaque rupture, is a more serious event that typically results in the formation of clinically significant arterial thromboses. From autopsy studies, it is known that rupture-prone plaques tend to have a thin, friable fibrous cap.217 The site of plaque rupture is thought to be the shoulder of the plaque, where substantial numbers of mononuclear inflammatory cells are commonly found.218 The mechanisms responsible for the local accumulation of these cells at this location in the plaque are unknown; presumably, monocyte chemotactic factors, the expression of leukocyte cell adhesion molecules, and specific cytokines are involved.162,219 Moreover, macrophages in plaques have been shown to express factors such as stromelysin, which promote the breakdown of the extracellular matrix, and thereby weaken the structural integrity of the plaque.151 Currently, no effective strategies have been designed to limit the possibility of plaque rupture; however, as discussed later, aggressive lipid-lowering therapy may be a helpful preventative measure.

Hemodynamics
If accurate angiographic assessment of the geometry of a coronary stenosis is made, hydrodynamic principles can be used to estimate the physiologic significance of the obstruction.220 Energy is lost when blood flows through a stenosis because of entrance effects, frictional losses in the stenotic segment, and separation losses caused by turbulence as blood exits the stenosis (Figure 6-16). The equation relating stenosis geometry to hemodynamic severity is:
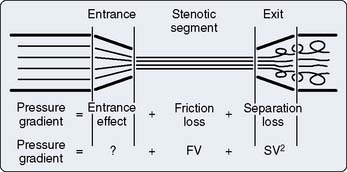
Figure 6-16 Sources of energy loss across a stenosis.
(From Marcus ML: The physiologic effects of a coronary stenosis. In Marcus ML [ed]: The coronary circulation in health and disease, New York: McGraw-Hill, 1983, pp 242–269. Reproduced by permission of McGraw-Hill Companies.)
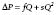
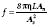
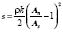
where ρ is blood density, and k is an experimentally determined coefficient. Thus, frictional losses are directly proportional to the first power of stenosis length but are inversely proportional to the square of the area (or fourth power of diameter). Separation losses are particularly prominent because they increase with the square of flow. Even at resting flows, more than 75% of energy loss is due to this turbulence when blood exits the stenosis. Except for very long stenoses, the frictional term can be neglected.209 Thus, the amount of energy loss or pressure decline across the obstruction increases exponentially as flow rate increases. For this reason, exercise, anemia, and arteriolar vasodilator drugs (e.g., dipyridamole) are poorly tolerated in the presence of a severe stenosis. Figure 6-17 illustrates that, although resting flow is unaffected until coronary diameter is reduced by more than 80%, maximal flow begins to decline when diameter is reduced by 50%.
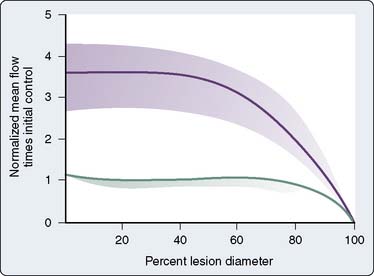
Figure 6-17 Effect of increasing stenosis severity at resting and maximal coronary flows.
(From Gould KL, Lipscomb K: Effect of coronary stenoses on coronary flow reserve and resistance. Am J Cardiol 34:48, 1974.)
Resting flow in Figure 6-17 remains constant as lumen diameter decreases because the coronary arterioles progressively dilate, thereby reducing the resistance of the distal coronary bed sufficiently to compensate for the resistance of the stenosis. As the severity of the stenosis increases further, the arteriolar bed can no longer compensate and flow begins to fall. This is an example of autoregulation: As stenosis severity increases, distal perfusion pressure decreases, arterioles dilate to maintain flow until autoregulation is exhausted (in the subendocardium first) and flow becomes pressure dependent. As illustrated in Figure 6-9, the distal pressure (or stenosis diameter) at which flow becomes pressure dependent is lower at low levels of myocardial metabolism (Mvo2). The interpretation of normal resting flow can be difficult in the presence of CAD. A coronary artery supplying blood through collaterals to a large mass of myocardium will require high resting flow rates and even a mild stenosis may be flow limiting.
The term critical stenosis is frequently used. This is usually defined as a coronary constriction sufficient to prevent an increase in flow over resting values in response to increased myocardial oxygen demands.221 This is a greater degree of obstruction than an angiographically significant stenosis, which is usually defined as a reduction in cross-sectional area of 75%, which is equivalent to a 50% decrease in the diameter of a concentric stenosis.212 A critical stenosis is demonstrated experimentally by blunting or abolishing reactive hyperemia (see Autoregulation section earlier in this chapter). This is evidence that autoregulation has been exhausted in at least the inner layer of myocardium (see Transmural Blood Flow section earlier in this chapter). Notably, the critical nature of the stenosis is relative to the resting Mvo2. If oxygen demand decreases, some coronary autoregulatory reserve will be recovered and the stenosis will no longer be critical. The failure to recognize this fact has led to misinterpretation of studies designed to demonstrate coronary steal (see later).

Coronary Collaterals
Coronary collaterals are anastomotic connections, without an intervening capillary bed, between different coronary arteries or between branches of the same artery. In the normal human heart, these vessels are small and have little or no functional role. In patients with CAD, well-developed coronary collateral vessels may play a critical role in preventing death and myocardial infarction. Individual differences in the capability of developing a sufficient collateral circulation is a determinant of the vulnerability of the myocardium to coronary occlusive disease.222 There is great interspecies variation in the ability of the collateral circulation to support myocardial perfusion after acute coronary occlusion; pigs and rats have little collateral circulation and infarct almost all the area at risk, whereas dogs and cats with better collateralization will infarct less than 75% of the area at risk.223 In the guinea pig, collaterals are so well developed that coronary occlusion does not even decrease myocardial blood flow. There are also differences in the location of collateral vessels; in dogs, collaterals develop in a narrow subepicardial zone, at the border of the potentially ischemic region, whereas in pigs, a dense subendocardial plexus develops in response to coronary occlusion. In the presence of coronary disease, humans exhibit a small number of large epicardial collateral vessels and numerous small subendocardial vessels.
In response to coronary occlusion, native coronary collateral vessels do not passively stretch but undergo an active growth process that within 8 weeks, in the dog, can restore perfusion sufficient to support normal myocardial function, even during exercise. Human collaterals have a tortuous corkscrew-like pattern visible on angiography. This may be because of an embryonal pattern of vascular development in which longitudinal growth of smooth muscle cells occurs at the same time as radial growth. In the nongrowing adult heart, this increase in length results in tortuosity.224 There is much interest in discovering the factors that control collateral vessel growth in the hope of providing therapy for patients who cannot be revascularized otherwise. Arteriogenesis refers to the transformation of preexisting collateral arterioles into functional arteries with a thick muscular coat and the acquisition of viscoelastic and vasomotor properties.225 Fujita and Tambara226 provide an overview of the process: A high-grade coronary stenosis decreases distal intra-arterial pressure, resulting in an increased pressure gradient across the preexisting collateral network. Increased collateral blood flow results in increased shear stress at the endothelium, which upregulates cell adhesion molecules. This leads to adherence of monocytes, which transform into macrophage, and the production and release of growth factors such as granulocyte macrophage colony-stimulating factor (GM-CSF), MCP-1, and basic fibroblast growth factor (bFGF). Angiogenesis is not directly related to collateral vessel development but refers to the proliferation, migration, and tube formation of capillaries in the central area of ischemic regions.227 The development of a treatment to promote collateral growth in patients with intractable CAD is currently a subject of intense investigation.
Evidence in dogs suggests that mature coronary collaterals respond differently to neurohumoral stimulation than normal coronary arteries. Collaterals do not constrict in response to α-receptor activation, but do dilate in response to β1– or β2-agonists. They constrict in response to prostaglandin F2α (PGF2α) and AII, but less so than normal vessels. Remarkably, collateral vessels constrict in response to vasopressin to a much greater extent than normal vessels. In vivo studies in dogs indicate that levels of vasopressin present during stress (hemorrhage, cardiopulmonary bypass) can diminish flow to collateral-dependent myocardium. This is likely due to constriction of collateral vessels, as well as enhanced vasoconstriction of the resistance vessels in the collateral-dependent myocardium.79 It is possible that the endothelial cells of both types of vessel are dysfunctional.228 Relaxation in response to nitroglycerin was enhanced. This is a further mechanism for the beneficial effects of nitroglycerin in CAD. The deleterious effects of coronary arteriolar dilators such as adenosine and dipyridamole are discussed later (see Coronary Steal).
It has been estimated that, in humans, perfusion via collaterals can equal perfusion via a vessel with a 90% diameter obstruction.229 Although coronary collateral flow can be sufficient to preserve structure and resting myocardial function, muscle dependent on collateral flow usually becomes ischemic when oxygen demand increases above resting levels.230 It is possible that evidence from patients with angina underestimates collateral function of the population of all patients with CAD. Perhaps individuals with coronary obstructions but excellent collateralization remain asymptomatic and are not studied.

Pathogenesis of Myocardial Ischemia
Ischemia is the condition of oxygen deprivation accompanied by inadequate removal of metabolites consequent to reduced perfusion.231 Clinically, myocardial ischemia is a decline in the blood flow supply/demand ratio resulting in impaired function. There is no universally accepted “gold standard” for the presence of myocardial ischemia. In practice, symptoms, anatomic findings, and evidence of myocardial dysfunction must be combined before concluding that myocardial ischemia is present.232 Conclusive evidence of anaerobic metabolism in the setting of reduced coronary blood flow (relative to demand) would be convincing. Such evidence is extremely difficult to obtain, even in experimental preparations.

Determinants of Myocardial Oxygen Supply/Demand Ratio
An increase in myocardial oxygen requirement beyond the capacity of the coronary circulation to deliver oxygen results in myocardial ischemia (Box 6-9). This is the most common mechanism leading to ischemic episodes in chronic stable angina and during exercise testing. Intraoperatively, the anesthesiologist must measure and control the determinants of Mvo2 and protect the patient from “demand” ischemia. The major determinants of Mvo2 are heart rate, myocardial contractility, and wall stress (chamber pressure × radius/wall thickness). Shortening, activation, and basal metabolic requirements are minor determinants of Mvo2 (Figure 6-18).
BOX 6-9 Determinants of Myocardial Oxygen Supply/Demand Ratio
The major determinants of myocardial oxygen consumption are:
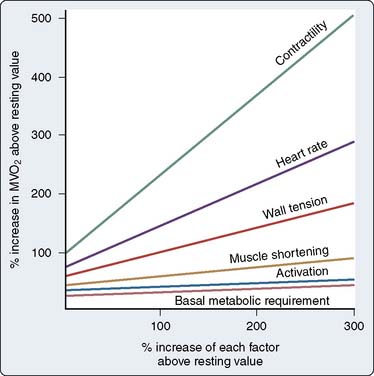
Figure 6-18 Relative importance of variables that determine myocardial oxygen consumption (Mvo2).
(From Marcus ML: Metabolic regulation of coronary blood flow. In Marcus ML [ed]: The coronary circulation in health and disease. New York: McGraw-Hill, 1983, pp 65–92. Reproduced by permission of McGraw-Hill Companies.)
An increase in heart rate can reduce subendocardial perfusion by shortening diastole. Coronary perfusion pressure may decline because of reduced systemic pressure or increased left ventricular end-diastolic pressure. With the onset of ischemia, perfusion may be further compromised by delayed ventricular relaxation (decreased subendocardial perfusion time) and decreased diastolic compliance (increased left ventricular end-diastolic pressure). Anemia and hypoxia also can compromise delivery of oxygen to the myocardium. Several indices of myocardial oxygen supply/demand ratio have been proposed to guide therapy. The rate-pressure product (heart rate × systolic blood pressure) gives a good estimate of Mvo2 but does not correlate well with ischemia. A patient with a systolic pressure of 160 and heart rate of 70 has a much lower likelihood of ischemia than a patient with a pressure of 70 and rate of 160, although both have a rate-pressure product of 11,200. The ratio of the diastolic pressure–time index (DPTI) to the systolic pressure–time index (SPTI) was devised to estimate subendocardial perfusion and takes into account determinants of oxygen delivery (Figure 6-19).233–235 When blood oxygen content was included, the index became a good predictor of endocardial flow in animals with normal coronary arteries. More recently, the ratio of mean arterial pressure/heart rate has been proposed as a correlate of myocardial ischemia.236 In dogs with moderate-to-severe coronary stenoses, systolic shortening was best with high pressures and low heart rate, and worst with low pressure and high heart rate. None of these indices has proved to be reliable in the clinical setting. Their major value is to bring attention to the important variables determining the supply/demand ratio. These variables should be measured (or estimated) and controlled individually.
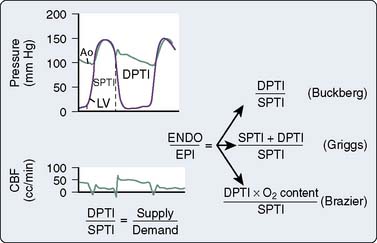

Dynamic Stenosis
Patients with CAD can have variable exercise tolerance during the day and between days. Ambulatory monitoring of the electrocardiogram has demonstrated that ST-segment changes indicative of myocardial ischemia, in the absence of changes in oxygen demand, are common.237 These findings are explained by variations over time in the severity of the obstruction to blood flow imposed by coronary stenoses.
Although the term hardening of the arteries suggests rigid, narrowed vessels, in fact, most stenoses are eccentric and have a remaining arc of compliant tissue (Figure 6-20). A modest amount (10%) of shortening of the muscle in the compliant region of the vessel can cause dramatic changes in lumen caliber.209 This was part of Prinzmetal’s original proposal to explain coronary spasm. Maseri et al238 suggest that the term spasm should be reserved for “situations where coronary constriction is both focal, sufficiently profound to cause transient coronary occlusion, and is responsible for reversible attacks of angina at rest” (i.e., variant angina). Although this syndrome is rare, lesser degrees of obstruction in response to vasoconstrictor stimuli are common among patients with CAD.
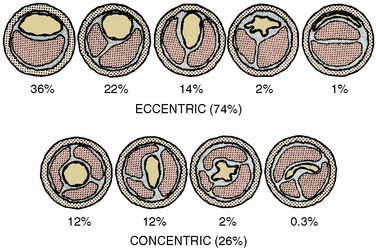
Sympathetic tone can be increased by the cold pressor test (immersing the arm in ice water) or isometric handgrip testing. In response to this maneuver, coronary resistance decreased in normal subjects but increased in patients with coronary disease, some of whom experienced angina.239 This increase in resistance appears to be mediated by α receptors because it can be prevented by phentolamine. Studies using quantitative coronary angiography have documented reductions in caliber in diseased vessels in contrast with dilation of vessels in healthy subjects.98,240 Zeiher et al240 showed that the same vessel segments that constricted with the cold pressor test also constricted in response to an infusion of acetylcholine. Because the normal, dilatory response to acetylcholine is dependent on intact endothelium, these findings suggest that the abnormal response of stenotic coronary arteries is due to endothelial dysfunction.
Animal models of coronary vasospasm demonstrate that enhanced vascular smooth muscle reactivity also may underlie vasospasm.241 Rho, a GTP-binding protein, sensitizes vascular smooth muscle cells to calcium by inhibiting myosin phosphatase activity through an effector protein called Rho-kinase. Upregulation of this pathway may be a mechanism of coronary vasospasm. Interestingly, Rho-kinase inhibitors have been shown to block agonist-induced vasoconstriction of internal thoracic artery segments from patients undergoing coronary artery surgery.242
It also has been noted that, in patients with coronary disease, some of the angiographically normal-appearing segments also respond abnormally.243 It appears likely that, during the development of coronary atherosclerosis, endothelial dysfunction precedes the appearance of visible stenoses. In patients with angiographically smooth coronary arteries, Vita et al244 found that an abnormal response to acetylcholine was correlated with serum cholesterol, male sex, age, and family history of coronary disease. The normal dilation of epicardial coronary arteries in response to increased blood flow (shear stress) has been shown to be absent in atherosclerotic vessels.245 As well, it has been demonstrated that patients with coronary disease respond to 5-HT with coronary vasoconstriction instead of the normal vasodilatory response.246,247 The concentrations of 5-HT used were within the range found in coronary sinus blood of patients with coronary disease. Very high concentrations of 5-HT may be found on the endothelium at the site of aggregating platelets.246 All these findings point to the central role of endothelial dysfunction in the abnormal coronary vasomotion of patients with atherosclerosis248 (see Endothelium section earlier in this chapter).

Coronary Steal
Steal occurs when the perfusion pressure for a vasodilated vascular bed (in which flow is pressure dependent) is lowered by vasodilation in a parallel vascular bed, both beds usually being distal to a stenosis. Two kinds of coronary steal are illustrated: collateral and transmural (Figure 6-21).
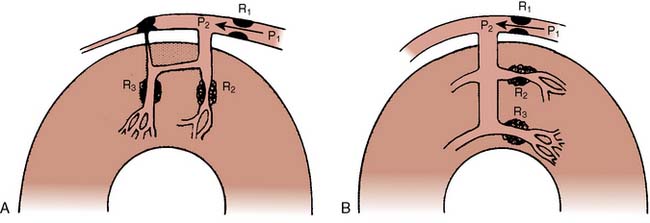
Collateral steal in which one vascular bed (R3), distal to an occluded vessel, is dependent on collateral flow from a vascular bed (R2) supplied by a stenotic artery is shown (see Fig 6-21A). Because collateral resistance is high, the R3 arterioles are dilated to maintain flow in the resting condition (autoregulation). Dilation of the R2 arterioles will increase flow across the stenosis, R1, and decrease pressure, P2. If R3 resistance cannot further decrease sufficiently, flow there will decline, producing or worsening ischemia in the collateral-dependent bed. The values of all the resistances, including collaterals, and the baseline myocardial metabolic state will determine how powerful the vasodilator stimulus must be to produce ischemia in the collateral bed. Failure to recognize this has confounded studies of vasodilator drugs. If collateral vessels are very well developed or Mvo2 is low, sufficient autoregulatory reserve may remain in the collateral-dependent bed to maintain adequate myocardial blood flow even with the administration of a moderately powerful vasodilator.
Transmural steal is also illustrated (see Figure 6-21B). Normally, vasodilator reserve is less in the subendocardium (see Transmural Blood Flow section earlier in this chapter). In the presence of a stenosis, flow may become pressure dependent in the subendocardium, whereas autoregulation is maintained in the subepicardium. This is illustrated in Figure 6-11, where at a perfusion pressure of 50 mm Hg, flow has declined in the subendocardium, whereas the subepicardium retains autoregulatory reserve. Dilation of the subepicardial arterioles, R2, will then increase flow across the stenosis (R1) causing P2 to fall and resulting in decreased flow to the subendocardium as subepicardial flow increases.
Future Directions
Interestingly, clues to the biology of plaque rupture are indirectly emerging from clinical trials with statins, in that these trials show modest reductions in the angiographic degree of vessel stenoses but consistently demonstrate a significant decrease in acute ischemic events well before the effects of LDL cholesterol lowering should appear. For example, the MIRACL (Myocardial Ischemia Reduction with Acute Cholesterol Lowering) trial demonstrated that treating only 38 patients with acute coronary syndromes with high-dose atorvastatin could prevent one recurrent infarction, refractory angina, or death as early as 30 days after the index event. 249 Similarly, the ARMYDA (Atorvastatin for Reduction of Myocardial Damage during Angioplasty),250 ARMYDA-ACS (Atorvastatin for Reduction of Myocardial Damage During Angioplasty–Acute Coronary Syndromes),251 and ARMYDA RECAPTURE (Atorvastatin for Reduction of Myocardial Damage During Angioplasty)252 studies have demonstrated that acute treatment with atorvastatin in patients undergoing percutaneous coronary interventions results in reductions in periprocedural myocardial infarctions when started just days before the procedure. Certainly, given the impressive clinical benefits, there is much interest in exploring the pleiotropic effects of statins that extend beyond lipid lowering and understanding how plaque stabilization is occurring.253,254
Most recently, there has become a renewed focus on the adventitia of the coronary artery and its role in initiation of inflammation in the vessel wall and subsequent development of atherosclerosis.12,255 Although much effort has been devoted to studying the intima and media in development of vascular lesions, the adventitia is just now garnering the focus it may deserve. The adventitia is unique in that it houses the vaso vasorum, which provides nutrients and vasoactive factors and acts as a portal of entry for inflammatory cells into the media and intima of epicardial coronary arteries. As well, the adventitia is unique in that it alone supplies all neural input to the vessel wall—an input that has been implicated in plaque progression and destabilization.256 Supporting this notion of an “outside-in” hypothesis to the development of atherosclerosis is that changes in the adventitial vaso vasorum often precede intimal changes.257 In addition to atherosclerosis, the adventitia also has been implicated as a source of cells in neointimal development after vascular injury such as balloon angioplasty.258 Given the dynamic nature of the adventitia in disease and the unique role it serves in vessel homeostasis, understanding of adventitial biology is lacking.
There also is a growing need to better understand the role of vascular progenitor cells in both arterial repair259 and lesion formation after vascular injury.260 Studies suggest that endothelial and smooth muscle cells appear to be derived from multiple sources such as circulating stem and progenitor cells, as well as tissue-resident progenitor cell populations.259–261 A number of observational clinical studies have inversely correlated endothelial progenitor cell (EPC) number and cardiovascular risk, fueling the hypothesis that impaired progenitor cell–mediated repair of arteries is a risk factor for atherosclerosis and clinical events.262,263 Moreover, reports of the involvement of vascular progenitor cells in the pathogenesis of other vascular lesions are also emerging. For example, observations from organ transplantations highlight the involvement of a blood-borne population of human vascular cells in development of transplant arteriosclerosis.264 Circulating EPC levels also have been found to be lower or have impaired adhesion capacity in patients who develop in-stent restenosis compared with patients with patent stents or an absence of CAD. 265,266 Hence, taken together, the emerging data suggest that vascular progenitor cells may play a critical role, not only in maintaining the artery wall but in ensuring that appropriate repair mechanisms occur in the face of injury. Therapeutic strategies that target EPC mobilization, homing, and differentiation may prove beneficial. Already, early clinical trials have suggested potential benefit of progenitor cell transplantation after myocardial infarction in improving left ventricular ejection fraction,267 and a combined clinical end point of death, recurrent myocardial infarction, or revascularization.268 Although these early clinical studies are promising, significant progress in our understanding the role vascular progenitors play in the pathogenesis of a wide variety of vascular lesions remains hampered by the absence of a clear definition of what constitutes a true vascular stem cell.269
1 Zweifach B.W., Lipowsky H.H., Pressure-flow relations in blood and lymph microcirculation, Berne R., Sperelakis N., editors, Handbook of Physiology, Section 2: The Cardiovascular System; vol III; 1984. William & Wilkins. Baltimore; 251-307; Microcirculation
2 Marcus M.L., Chilian W.M., Kanatsuka, et al. Understanding the coronary circulation through studies at the microvascular level. Circulation. 1990;82(1):1.
3 Mulvany M.J., Aalkjaer C. Structure and function of small arteries. Physiol Rev. 1990;70:921.
4 Merkus D., Chilian W.M., Stepp D.W. Functional characteristics of the coronary microcirculation. Herz. 1999;24:7.
5 Kanatsuka H., Lamping K.G., Eastham C.L., et al. Comparison of the effects of increased myocardial oxygen consumption and adenosine on the coronary microvascular resistance. Circ Res. 1989;65:1296.
6 Chilian W.M., Layne S.M., Eastham C.L., et al. Heterogeneous microvascular coronary alpha-adrenergic vasoconstriction. Circ Res. 1989;64:376.
7 Schwartz S.M., deBlois D., O’Brien E.R. The intima: Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445.
8 Stary H.C., Blankenhorn D.H., Chandler A.B., et al. A definition of the intima of human arteries and of its atherosclerosis-prone regions. Circulation. 1992;85:391.
9 Frid M.G., Moiseeva E.P., Stenmark K.R. Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo. Circ Res. 1994;75:669.
10 Clowes A.W., Clowes M.M., Reidy M.A. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle growth in the absence of endothelium. Lab Invest. 1983;49:327.
11 Clowes A.W., Schwartz S.M. Significance of quiescent smooth muscle migration in the injured rat carotid artery. Circ Res. 1985;56:139.
12 Maiellaro K., Taylor W.R. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:4.
13 Rana R.S., Hokin L.E. Role of phosphoinositides in transmembrane signaling. Physiol Rev. 1990;70:115.
14 Moncada S., Higgs E.A., Vane J.R. Human arterial and venous tissues generate prostacyclin (prostaglandin X), a potent inhibitor of platelet aggregation. Lancet. 1977;I:18.
15 Furchgott R.F., Zawadzki J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373.
16 Ignarro L.J. Nitric oxide. A novel signal transduction mechanism for transcellular communication. Hypertension. 1990;16:477.
17 Ignarro L.J. Biological actions and properties of endothelium-derived nitric oxide formed and released from artery and vein. Circ Res. 1989;65:1.
18 Lincoln T.M., Dey N., Sellak H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J Appl Physiol. 2001;91:3.
19 Moncada S., Palmer R.MJ., Higgs E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109.
20 Harrison D.G., Cai H. Endothelial control of vasomotion and nitric oxide production. Cardiol Clin. 2003;21:3.
21 Vallance P., Collier J., Moncada S. Nitric oxide synthesised from l-arginine mediates endothelium-dependent dilatation in human veins. Cardiovasc Res. 1989;23:1053.
22 Miura H., Bosnjak J.J., Ning G., et al. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:2.
23 Busse R., Edwards G., Feletou M., et al. EDHF: Bringing the concepts together. Trends Pharmacol Sci. 2002;23:8.
24 Miura H., Wachtel R.E., Liu Y., et al. Flow-induced dilation of human coronary arterioles: Important role of Ca(2+)-activated K(+) channels. Circulation. 2001;103:15.
25 Miyauchi T., Tomobe Y., Shiba R., et al. Involvement of endothelin in the regulation of human vascular tonus. Potent vasoconstrictor effect and existence in endothelial cells. Circulation. 1990;81:6.
26 Lee S.Y., Lee C.Y., Chen Y.M., et al. Coronary vasospasm as the primary cause of death due to the venom of the burrowing asp Atractaspis engaddensis. Toxicon. 1986;24:285.
27 Goodwin A.T., Yacoub M.H. Role of endogenous endothelin on coronary flow in health and disease. Coron Artery Dis. 2001;12:6.
28 Hirata Y., Yoshimi H., Takata S. Cellular mechanism of action by a novel vasoconstrictor endothelin in cultured rat vascular smooth muscle cells. Biochem Biophys Res Commun. 1988;154:868.
29 Clozel J.-P., Clozel M. Effects of endothelin on the coronary vascular bed in open-chest dogs. Circ Res. 1989;65:1193.
30 Wenzel R.R., Fleisch M., Shaw S., et al. Hemodynamic and coronary effects of the endothelin antagonist bosentan in patients with coronary artery disease. Circulation. 1998;98:21.
31 Goodwin A.T., Yacoub M.H. Role of endogenous endothelin on coronary flow in health and disease. Coron Artery Dis. 2001;12:6.
32 Mylona P., Cleland J.G. Update of REACH-1 and MERIT-HF clinical trials in heart failure. Cardionet Editorial Team. Eur J Heart Fail. 1999;1:2.
33 Humbert M., Sitbon O., Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:14.
34 Hoak J.C. The endothelium, platelets, and coronary vasospasm. Adv Intern Med. 1989;34:353.
35 Bassenge E., Heusch G. Endothelial and neuro-humoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol. 1990;116:77.
36 Spaan J.A. Mechanical determinants of myocardial perfusion. Basic Res Cardiol. 1995;90:2.
37 Westerhof N. Physiological hypotheses—intramyocardial pressure. A new concept, suggestions for measurement. Basic Res Cardiol. 1990;85:105.
38 Mates R.E., Klocke F.J., Canty J.M.Jr. Coronary capacitance. Prog Cardiovasc Dis. 1988;31(1):1.
39 Bellamy R.F. Diastolic coronary artery pressure-flow relations in the dog. Circ Res. 1978;43:92.
40 Eng C., Jentzer J.H., Kirk E.S. The effects of coronary capacitance on the interpretation of diastolic pressure-flow relationships. Circ Res. 1982;50:334.
41 Messina L.M., Hanley F.L., Uhlig P.N., et al. Effects of pressure gradients between branches of the left coronary artery on the pressure axis intercept and the shape of steady state circumflex pressure-flow relations in dogs. Circ Res. 1985;56:11.
42 Kanatsuka H., Ashikawa K., Suzuki T., et al. Diameter change and pressure-red blood cell velocity relations in coronary microvessels during long diastoles in the canine left ventricle. Circ Res. 1990;66:503.
43 Feigl E.O. Coronary physiology. Physiol Rev. 1983;63:1.
44 Hoffman J.I., Piedimonte G., Maxwell A.J., et al. Aspects of coronary vasomotor regulation. Adv Exp Med Biol. 1995;381:135.
45 Tune J.D., Richmond K.N., Gorman M.W., et al. Control of coronary blood flow during exercise. Exp Biol Med (Maywood). 2002;227:4.
46 Gellai M., Norton J.M., Detar R. Evidence for direct control of coronary vascular tone by oxygen. Circ Res. 1973;32:279.
47 Weiss H.R., Neubauer J.A., Lipp J.A. Quantitative determination of regional oxygen consumption in the dog heart. Circ Res. 1978;42:394.
48 Broten T.P., Romson J.L., Fullerton D.A., et al. Synergistic action of myocardial oxygen and carbon dioxide in controlling coronary blood flow. Circ Res. 1991;68:531.
49 Spaan J.A., Dankelman J. Theoretical analysis of coronary blood flow and tissue oxygen pressure-control. Adv Exp Med Biol. 1993;346:189.
50 Berne R.M. Cardiac nucleotides in hypoxia: Possible role in regulation of coronary blood flow. Am J Physiol. 1963;204:317.
51 Gerlach E., Deuticke B., Dreisbach R.H. Der Nucleotid-Abbau im Herzmuskel bei Sauerstoffmangel und seine mogliche Bedeutung fur die Coronardurchblutung. Naturwissenschaften. 1963;6:228.
52 Olsson R.A., Bunger R. Metabolic control of coronary blood flow. Prog Cardiovasc Dis. 1987;29:369.
53 Standen N.B., Quayle J.M. K+ channel modulation in arterial smooth muscle. Acta Physiol Scand. 1998;164:4.
54 Duncker D.J., Bache R.J. Regulation of coronary vasomotor tone under normal conditions and during acute myocardial hypoperfusion. Pharmacol Ther. 2000;86:1.
55 Kroll K., Feigl E.O. Adenosine is unimportant in controlling coronary blood flow in unstressed dog hearts. Am J Physiol. 1985;249:H1176.
56 Bache R.J., Dai X., Schwarts J.S., et al. Role of adenosine in coronary vasodilation during exercise. Circ Res. 1988;62:846.
57 Dole W.P., Yamada N., Bishop V.S., et al. Role of adenosine in coronary blood flow regulation after reductions in perfusion pressure. Circ Res. 1985;56:517.
58 Saito D., Steinhard C.R., Nixon D.G., et al. Intracoronary adenosine deaminase reduces canine myocardial reactive hyperemia. Circ Res. 1981;49:1262.
59 Laxson D.D., Homans D.C., Bache R.J. Inhibition of adenosine-mediated coronary vasodilation exacerbates myocardial ischemia during exercise. Am J Physiol. 1993;5(Pt 2):265.
60 Baxter G.F. Role of adenosine in delayed preconditioning of myocardium. Cardiovasc Res. 2002;55:3.
61 Zaugg M., Lucchinetti E., Uecker M., et al. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth. 2003;91:4.
62 Woollard H.H. The innervation of the heart. J Anat Physiol. 1926;60:345.
63 Van Winkle D.M., Feigl E.O. Acetylcholine causes coronary vasodilation in dogs and baboons. Circ Res. 1989;65:6.
64 Horio Y., Yasue H., Okumura K.T., et al. Effects of intracoronary injection of acetylcholine on coronary arterial hemodynamics and diameter. Am J Cardiol. 1988;62:13.
65 Hodgson J.M., Marshall J.J. Direct vasoconstriction and endothelium-dependent vasodilation. Mechanisms of acetylcholine effects on coronary flow and arterial diameter in patients with nonstenotic coronary arteries. Circulation. 1989;79:5.
66 Ludmer P.L., Selwyn A.P., Shook T.L., et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046.
67 Feigl E.O. EDRF—a protective factor? Nature. 1988;331:490.
68 Feigl E.O. Neural control of coronary blood flow. J Vasc Res. 1998;35:2.
69 Feldman R.D., Christy J.P., Paul S.T., et al. β-adrenergic receptors on canine coronary collateral vessels: Characterization and function. Am J Physiol. 1989;257:H1634.
70 Mosher P., Ross J.Jr, McFate P.A., et al. Control of coronary blood flow by an autoregulatory mechanism. Circ Res. 1964;14:250.
71 Gorman M.W., Tune J.D., Richmond K.N., et al. Feedforward sympathetic coronary vasodilation in exercising dogs. J Appl Physiol. 2000;89:5.
72 Holtz J. Alpha-adrenoceptor subtypes in the coronary circulation. Basic Res Cardiol. 1990;85(Suppl 1):81.
73 Guth B.D., Thaulow E., Heusch G., et al. Myocardial effects of selective alpha-adrenoceptor blockade during exercise in dogs. Circ Res. 1990;66:1703.
74 Heusch G. Alpha-adrenergic mechanisms in myocardial ischemia. Circulation. 1990;81:1.
75 Kelley K.O., Feigl E.O. Segmental alpha-receptor-mediated vasoconstriction in the canine coronary circulation. Circ Res. 1978;43:908.
76 Mohrman D.E., Feigl E.O. Competition between sympathetic vasoconstriction and metabolic vasodilation in the canine coronary circulation. Circ Res. 1978;42:79.
77 Heusch G., Deussen A. Nifedipine prevents sympathetic vasoconstriction distal to severe coronary stenoses. J Cardiovasc Pharmacol. 1984;6:378.
78 Chilian W.M. Adrenergic vasomotion in the coronary microcirculation. Basic Res Cardiol. 1990;85(Suppl 1):111.
79 Harrison D.G., Sellke F.W., Quillen J.E. Neurohumoral regulation of coronary collateral vasomotor tone. Basic Res Cardiol. 1990;85(Suppl 1):121.
80 DiCarli M.F., Tobes M.C., Mangner T., et al. Effects of cardiac sympathetic innervation on coronary blood flow. N Engl J Med. 1997;336:1208.
81 Chilian W.M., Harrison D.G., Haws C.W., et al. Adrenergic coronary tone during submaximal exercise in the dog is produced by circulating catecholamines. Evidence for adrenergic denervation supersensitivity in the myocardium but not in coronary vessels. Circ Res. 1986;58:68.
82 Huang A.H., Feigl E.O. Adrenergic coronary vasoconstriction helps maintain uniform transmural blood flow distribution during exercise. Circ Res. 1988;62:286.
83 Buffington C.W., Feigl E.O. Adrenergic coronary vasoconstriction in the presence of coronary stenosis in the dog. Circ Res. 1981;48:416.
84 Heusch G., Deussen A. The effects of cardiac sympathetic nerve stimulation on the perfusion of stenotic coronary arteries in the dog. Circ Res. 1983;53:8.
85 Bassenge E., Walter P., Doutheil U. Wirkungsmumkehr der adrenergischen coronargefassreakton in abhangigkeit vom coronargefasstonus. Pflugers Arch. 1967;297:146.
86 Nathan H.J., Feigl E.O. Adrenergic coronary vasoconstriction lessens transmural steal during coronary hypoperfusion. Am J Physiol. 1986;250:H645.
87 Chilian W.M., Ackell P.H. Transmural differences in sympathetic coronary constriction during exercise in the presence of coronary stenosis. Circ Res. 1988;62:216.
88 Seitelberger R., Guth B.D., Heusch G., et al. Intracoronary alpha 2-adrenergic receptor blockade attenuates ischemia in conscious dogs during exercise. Circ Res. 1988;62:436.
89 Baumgart D., Heusch G. Neuronal control of coronary blood flow. Basic Res Cardiol. 1995;90:2.
90 Feigl E.O. Adrenergic control of transmural coronary blood flow. Basic Res Cardiol. 1990;85(Suppl 1):167.
91 Hodgson J.MB., Cohen M.D., Szentpetery S., et al. Effects of regional alpha- and beta-blockade on resting and hyperemic coronary blood flow in conscious, unstressed humans. Circulation. 1989;79:797.
92 Chierchia S., Davies G., Berkenboom G., et al. Alpha-adrenergic receptors and coronary spasm: An elusive link. Circulation. 1984;69:8.
93 Brown B.G., Lee A.B., Bolson E.L., et al. Reflex constriction of significant coronary stenosis as a mechanism contributing to ischemic left ventricular dysfunction during isometric exercise. Circulation. 1984;70:18.
94 Berkenboom G., Unger P. Alpha-adrenergic coronary constriction in effort angina. Basic Res Cardiol. 1990;85(Suppl 1):359.
95 Berkenboom G.M., Abramowicz M., Vandermoten P., et al. Role of alpha-adrenergic coronary tone in exercise induced angina pectoris. Am J Cardiol. 1986;57:195.
96 Gage J.E., Hess O.M., Murakami T., et al. Vasoconstriction of stenotic coronary arteries during dynamic exercise in patients with classic angina pectoris: Reversibility by nitroglycerin. Circulation. 1986;73:865.
97 Chilian W.M., Layne S.M. Coronary microvascular responses to reductions in perfusion pressure. Evidence for persistent arteriolar vasomotor tone during coronary hypoperfusion. Circ Res. 1990;66:1227.
98 Nabel E.G., Ganz P., Gordon J.B., et al. Dilation of normal and constriction of atherosclerotic coronary arteries caused by the cold pressor test. Circulation. 1988;77:43.
99 Maturi M.F., Martin S.E., Markle D., et al. Production of myocardial ischemia in dogs by constriction of nondiseased small vessels. Circulation. 1991;83:2111.
100 Chu A., Morris K., Kuehl W., et al. Effects of atrial natriuretic peptide on the coronary arterial vasculature in humans. Circulation. 1989;80:1627.
101 Linder C., Heusch G. ACE-inhibitors for the treatment of myocardial ischemia? Cardiovasc Drugs Ther. 1990;4:1375.
102 Lamping K.G., Kanatsuka H., Eastham C.L., et al. Nonuniform vasomotor responses of the coronary microcirculation to serotonin and vasopressin. Circ Res. 1989;65:2.
103 Ginsburg R., Bristow M.R., Kantrowitz N., et al. Histamine provocation of clinical coronary artery spasm: Implications concerning pathogenesis of variant angina pectoris. Am Heart J. 1981;102:5.
104 Johnson P.C. Autoregulation of blood flow. Circ Res. 1986;59:483.
105 Gregg D.E. Effect of coronary perfusion pressure or coronary flow on oxygen usage of the myocardium. Circ Res. 1963;13:497.
106 Dole W.P., Nuno D.W. Myocardial oxygen tension determines the degree and pressure range of coronary autoregulation. Circ Res. 1986;59:202.
107 Dole W.P. Autoregulation of the coronary circulation. Prog Cardiovasc Dis. 1987;29:293.
108 Jones C.E., Liang I.YS., Gwirtz P.A. Effects of alpha-adrenergic blockade on coronary autoregulation in dogs. Am J Physiol. 1987;253:H365.
109 Hoffman J.IE., Spaan J.A. Pressure-flow relations in coronary circulation. Physiol Rev. 1990;70(2):331.
110 Feigl E.O. Coronary autoregulation. J Hypertens. 1989;7(Suppl):S55.
111 Hickey R.F., Sybert P.E., Verrier E.D., et al. Effects of halothane, enflurane, and isoflurane on coronary blood flow, autoregulation, and coronary vascular reserve in the canine heart. Anesthesiology. 1988;68:21.
112 Sestier F.J., Mildenberger R.R., Klassen G.A. Role of autoregulation in spatial and temporal perfusion heterogeneity of canine myocardium. Am J Physiol. 1978;235(1):H64.
113 Kuo L., Chilian W.M., Davis M.J. Coronary arteriolar myogenic response is independent of endothelium. Circ Res. 1990;66:860.
114 Fossel E.T., Morgan H.E., Ingwall J.S. Measurement of changes in high-energy phosphates in the cardiac cycle by using 31P nuclear magnetic resonance. Proc Natl Acad Sci U S A. 1980;77:3654.
115 Feigl E.O., Neat G.W., Huang A.H. Interrelations between coronary artery pressure, myocardial metabolism and coronary blood flow. J Mol Cell Cardiol. 1990;22:375.
116 Ruiter J.H., Spaan J.AE., Laird J.D. Transient oxygen uptake during myocardial reactive hyperemia in the dog. Am J Physiol. 1978;235:H87.
117 Marcus M.L., Metabolic regulation of coronary blood flow. In Marcus ML (eds): The Coronary Circulation in Health and Disease. New York, McGraw-Hill;1983:65-92
118 Bourdarias J.P. Coronary reserve: Concept and physiological variations. Eur Heart J. 1995;16:2.
119 Nitenberg A., Antony I. Coronary vascular reserve in humans: A critical review of methods of evaluation and of interpretation of the results. Eur Heart J. 1995;16:7.
120 Aude Y.W., Garza L. How to prevent unnecessary coronary interventions: Identifying lesions responsible for ischemia in the cath lab. Curr Opin Cardiol. 2003;18:5.
121 Tonino P.A., De B.B., Pijls N.H., et al. Fractional flow reserve versus angiography for guiding percutaneous coronary intervention. N Engl J Med. 2009;360:3.
122 Hoffman J.IE. Transmural myocardial perfusion. Prog Cardiovasc Dis. 1987;29:429.
123 van der Vusse T., Arts, Glatz J.F., et al. Transmural differences in energy metabolism of the left ventricular myocardium: Fact or fiction. J Mol Cell Cardiol. 1990;22:1.
124 Bassingthwaighte J.B., Malone M.A., Moffett T.C., et al. Validity of microsphere depositions for regional myocardial flows. Am J Physiol. 1987;253:H184.
125 Buckberg G.D., Luck J.C., Payne D.B., et al. Some sources of error in measuring regional blood flow with radioactive microspheres. J Appl Physiol. 1971;31:4.
126 Canty J.M.Jr, Giglia J., Kandath D. Effect of tachycardia on regional function and transmural myocardial perfusion during graded coronary pressure reduction in conscious dogs. Circulation. 1990;82:1815.
127 Hoffman J.IE. Transmural myocardial perfusion. Prog Cardiovasc Dis. 1990;29:429-464.
128 Harrison D.G., Florentine M.S., Brooks L.A., et al. The effect of hypertension and left ventricular hypertrophy on the lower range of coronary autoregulation. Circulation. 1988;77:1108.
129 Tomanek R.J. Response of the coronary vasculature to myocardial hypertrophy. J Am Coll Cardiol. 1990;15:528.
130 Hoffman J.I. Heterogeneity of myocardial blood flow. Basic Res Cardiol. 1995;90:2.
131 Coggins D.L., Flynn A.E., Austin R.E., et al. Nonuniform loss of regional flow reserve during myocardial ischemia in dogs. Circ Res. 1990;67:253.
132 Pantely G.A., Bristow J.D., Swenson L.J., et al. Incomplete coronary vasodilation during myocardial ischemia in swine. Am J Physiol. 1985;249:638.
133 Aversano T., Becker L.C. Persistence of coronary vasodilator reserve despite functionally significant flow reduction. Am J Physiol. 1985;248:H403.
134 Stary H.C. Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis. 1989;9(Suppl 1):I-19.
135 PDAY Research Group. Relationship of atherosclerosis in young men to serum lipoprotein cholesterol concentrations and smoking: A preliminary report from the Pathobiological Determinants of Atherosclerosis in Youth. JAMA. 1990;264:3018.
136 von Rokitansky C., A manual of pathological anatomy. London, The Sydenham Society; 1852.
137 Duguid J.B. Thrombosis as a factor in atherogenesis. J Pathol Bacteriol. 1946;58:207.
138 Schneiderman J., Sawdey M.S., Keeton M.R., et al. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci U S A. 1992;89:6998.
139 Virchow R., Gesammelte abhandlungen zur wissenschaftlichen medicin, phlogose und thrombose im gefassystem. Berlin, Meidinger Sohn and Co; 1856;458-463
140 Ross R., Glomset J.A. The pathogenesis of atherosclerosis. N Engl J Med. 1976;295:369.
141 Schwartz S.M., Murry C.E., O’Brien E.R. Vessel wall response to injury. Sci Med. 1996;3:2.
142 Dvorak H.F., Harvey V.S., Estrella P., et al. Fibrin containing gels induce angiogenesis. Implications for tumor stroma generation and wound healing. Lab Invest. 1987;57:673.
143 Hunt T.K., Knighton D.R., Thakral K.K., et al. Studies on inflammation and wound healing: Angiogenesis and collagen synthesis stimulated in vivo by resident and activated wound macrophage. Surgery. 1984;96(1):48.
144 Gabbiani G. The biology of the myofibroblast. Kidney Int. 1996;41:530.
145 Dvorak H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:26.
146 Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature. 1993;362:801.
147 Munro J.M., Cotran R.S. The pathogenesis of atherosclerosis: Atherogenesis and inflammation. Lab Invest. 1988;58:3.
148 Jonasson L., Holm J., Skalli O., et al. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:2.
149 Katsuda S., Boyd H.C., Fligner C., et al. Human atherosclerosis: Immunocytochemical analysis of the cell composition of lesions of young adults. Am J Pathol. 1992;140:907.
150 Prescott M.F., McBride C.K., Court M. Development of intimal lesions after leukocyte migration into the vascular wall. Am J Pathol. 1989;135:5.
151 Henney A.M., Wakeley P.R., Davies M.J., et al. Localization of stromelysin gene expression in atherosclerotic plaques by in situ hybridization. Proc Natl Acad Sci U S A. 1991;88(18):8154.
152 Bevilacqua M.P., Slengelin S., Gimbrone M.AJ., et al. Endothelial leukocyte adhesion molecule 1: An inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160.
153 Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell. 1994;76:301.
154 Osborn L., Hession C., Tizard R., et al. Direct expression cloning of vascular cell adhesion molecules 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. 1989;59:1203.
155 Nelken N.A., Coughlin S.R., Gordon D., et al. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88:1121.
156 Reidy M.A., Silver M. Endothelial regeneration. Lack of intimal proliferation after defined injury to rat aorta. Am J Pathol. 1985;118:2.
157 Conte M.S., Choudhry R.P., Shirakowa M., et al. Endothelial cell seeding fails to attenuate intimal thickening in balloon-injured rabbit arteries. J Vasc Surg. 1995;21:413.
158 Winternitz M.C,., Thomas R.M., LeCompte P.M., The biology of arteriosclerosis. Springfield, IL, Charles C. Thomas; 1938;
159 Barger A.C., Beeuwkes R., Lainey L.L., et al. Hypothesis: Vasa vasorum and neovascularization of human coronary arteries. N Engl J Med. 1984;310:175.
160 O’Brien E.R., Garvin M.R., Dev R., et al. Angiogenesis in human coronary atherosclerotic plaques. Am J Pathol. 1994;145(4):883.
161 Pels K., Labinaz M., O’Brien E.R. Arterial wall neovascularization: Potential role in atherosclerosis and restenosis. Japan Circ J. 1997;35:241.
162 O’Brien K.D., McDonald T.O., Chait A., et al. Neovascular expression of E-selection intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation. 1996;93:672.
163 Brown M.S., Goldstein J.L. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34.
164 Steinberg D. Antioxidant vitamins and coronary heart disease. N Engl J Med. 1993;328(20):1444.
165 The Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344:8934.
166 Treasure C.B., Klein J.L., Weintraub W.S. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med. 1995;332:481.
167 Anderson T.J., Meredith I.T., Yeung A.C., et al. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med. 1995;332:488.
168 Benditt E.P., Benditt J.M. Evidence for a monoclonal origin of human atherosclerotic plaques. Proc Natl Acad Sci U S A. 1973;70:1753.
169 Murry C.E., Gipaya C.T., Bartosek T., et al. Monoclonality of smooth muscle cells in human atherosclerosis. Am J Pathol. 1997;151:3.
170 Sims F.H., Gavin J.B., Vanderwee M.A. The intima of human coronary arteries. Am Heart J. 1989;118:32.
171 Sims F.H., Gavin J.B. The early development of intimal thickening of human coronary arteries. Coron Artery Dis. 1990;1:205.
172 Gordon D., Reidy M.A., Benditt E.P., et al. Cell proliferation in human coronary arteries. Proc Natl Acad Sci U S A. 1990;87:4600.
173 O’Brien E.R., Alpers C.E., Stewart D.K., et al. Proliferation in primary and restenotic coronary atherectomy tissue: Implications for anti-proliferative therapy. Circ Res. 1993;73(2):223.
174 Villaschi S., Spagnoli L.G. Autoradiographic and ultrastructural studies on the human fibroatheromatous plaque. Atherosclerosis. 1983;48:95.
175 Spagnoli L.G., Villaschi S., Neri L., et al. Autoradiographic studies of the smooth muscle cells in human arteries. Artery Wall. 1981;7:107.
176 Bennett M.R., Evan G.I., Schwartz S.M. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest. 1995;95:2266.
177 Lindner V., Olson N.E., Clowes A.W., et al. Heparin inhibits smooth muscle cell proliferation in injured rat arteries by displacing basic fibroblast growth factor. J Clin Invest. 1992;90:2044.
178 Jackson C.L., Raines E.W., Ross R., et al. Role of endogenous platelet-derived growth factor in arterial smooth muscle cell migration after balloon catheter injury. Arterioscler Thromb. 1993;13(8):1218.
179 Reidy M.A., Irvin C., Lindner V. Migration of arterial wall cells. Expression of plasminogen activators and inhibitors in injured rat arteries. Circ Res. 1996;78:3.
180 Glagov S., Weisenberg E., Zarins C.K., et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371.
181 Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1982;62:347.
182 Jamal A., Bendeck M., Langille B.L. Structural changes and recovery of function after arterial injury. Arterioscler Thromb. 1992;12:307.
183 Folkow B. “Structural factor” in primary and secondary hypertension. Hypertension. 1990;16:89.
184 Isner J.M. Vascular remodeling: Honey, I think I shrunk the artery. Circulation. 1994;89(6):2937.
185 Lafont A., Guzman L.A., Whitlow P.L., et al. Restenosis after experimental angioplasty. Intimal, medial, and adventitial changes associated with constrictive remodeling. Circ Res. 1995;76:996.
186 Kakuta T., Currier J.W., Haudenschild C.C., et al. Differences in compensatory vessel enlargement, not intimal formation, account for restenosis following angioplasty in the hypercholesterolemic rabbit model. Circulation. 1994;89:2809.
187 Post M.J., Borst C., Kuntz R.E. The relative importance of arterial remodeling compared with intimal hyperplasia in lumen renarrowing after balloon angioplasty. A study in the normal rabbit and the hypercholesterolemic Yucatan micropig. Circulation. 1994;89:2816.
188 Pasterkamp G., Borst C., Post M.J., et al. Atherosclerotic arterial remodeling in the superficial femoral artery. Individual variation in local compensatory enlargement response. Circulation. 1996;93:1818.
189 Kimura T., Kaburagi S., Tamura T., et al. Remodeling of human coronary arteries undergoing coronary angioplasty or atherectomy. Circulation. 1999;96:475.
190 Luo H., Nishioka T., Eigler N.L., et al. Coronary artery restenosis after balloon angioplasty in humans is associated with circumferential coronary artery constriction. Arterioscler Thromb Vasc Biol. 1999;16:1393.
191 Mintz G.S., Popma J.J., Pichard A.D., et al. Arterial remodeling after coronary angioplasty. A serial intravascular ultrasound study. Circulation. 1996;94:35.
192 Pasterkamp G., de Kleijn D., Borst C. Arterial remodeling in atherosclerosis, restenosis and after alteration of blood flow: Potential mechanisms and clinical implications. Cardiovasc Res. 2000;45:843.
193 Schwartz S.M., Murry C.E., O’Brien E.R. Vessel wall response to injury. Sci Med. 1996;3:2.
194 Shi Y., Pieniek M., Fard A., et al. Adventitial remodeling after coronary artery injury. Circulation. 1996;93:340.
195 Bryant S.R., Bjercke R.J., Erichsen D.A., et al. Vascular remodeling in response to altered blood flow is mediated by fibroblast growth factor-2. Circ Res. 1999;84:323.
196 Langille B.L., O’Donnell F. Reductions in arterial diameter produced by chronic diseases in blood flow are endothelium-dependent. Science. 1986;231:405.
197 Jamal A., Bendeck M., Langille B.L. Structural changes and recovery of function after arterial injury. Arterioscler Thromb. 1992;12:307.
198 Koester W. Endarteriitis and arteriitis. Berl Klin Wochenschr. 1876;13:454.
199 Geiringer E. Intimal vascularization and atherosclerosis. J Pathol Bacteriol. 1951;63:201.
200 Heistad D.H., Armstrong M.L. Blood flow through vasa vasorum of coronary arteries in atherosclerotic monkeys. Arteriosclerosis. 1986;6:326.
201 Glagov S., Weisenberg E., Zarins C.K., et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371.
202 Nissen S.E., Gurley J.C., Grines C.L., et al. Intravascular ultrasound assessment of lumen size and wall morphology in normal subjects and patients with coronary artery disease. Circulation. 1991;84:3.
203 Topol E.J., Nissen S.E. Our preoccupation with coronary luminology. The dissociation between clinical and angiographic findings in ischemic heart disease. Circulation. 1995;92:8.
204 Tobis J., Azarbal B., Slavin L. Assessment of intermediate severity coronary lesions in the catheterization laboratory. J Am Coll Cardiol. 2007;49:8.
205 Escolar E., Weigold G., Fuisz A., et al. New imaging techniques for diagnosing coronary artery disease. CMAJ. 2006;174:4.
206 Nissen S.E. Application of intravascular ultrasound to characterize coronary artery disease and assess the progression or regression of atherosclerosis. Am J Cardiol. 2002;4A:89.
207 Virmani R., Burke A.P., Kolodgie F.D., et al. Pathology of the thin-cap fibroatheroma: A type of vulnerable plaque. J Interv Cardiol. 2003;16:3.
208 Ambrose J.A. In search of the “vulnerable plaque”: Can it be localized and will focal regional therapy ever be an option for cardiac prevention? J Am Coll Cardiol. 2008;51:16.
209 Brown B.G., Bolson E.L., Dodge H.T. Dynamic mechanisms in human coronary stenosis. Circulation. 1984;70:917.
210 Levin D.C., Gardiner G.A., Coronary arteriography. In Braunwald E (ed): Heart disease, 3rd ed. Philadelphia, WB Saunders; 1988:268-310
211 Arnett E.N., Isner J.M., Redwood D.R., et al. Coronary artery narrowing in coronary heart disease: Comparison of cineangiographic and necropsy findings. Ann Intern Med. 1979;91:350.
212 Wilson R.F. Assessing the severity of coronary artery stenoses. N Engl J Med. 1996;334:1735.
213 Marcus M.L., The physiologic effects of a coronary stenosis. In Marcus ML (ed): The coronary circulation in health and disease. New York, McGraw Hill; 1983:242-269
214 Ambrose J.A., Tannenbaum M.A., Alexopoulos D., et al. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12(1):56.
215 Little W.C., Constantinescu M., Applegate R.J., et al. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild to moderate coronary artery disease? Circulation. 1988;78:1157.
216 Davies M.J., Thomas A.C. Plaque fissuring—the cause of acute myocardial infarction, sudden ischemic death, and crescendo angina. Br Heart J. 1985;53(4):363.
217 Richardson P.D., Davies M.J., Born G.V. Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet. 1989;2(8669):941.
218 Van der Wal A.C., Becker A.E., van der Loos C.M., et al. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36.
219 Libby P., Hansson G.K. Involvement of the immune system in human atherogenesis: Current knowledge and unanswered questions. Lab Invest. 1991;64:5.
220 Demer L., Gould K.L., Kirkeeide R. Assessing stenosis severity; coronary flow reserve, collateral function, quantitative coronary arteriography, positron imaging, and digital subtraction angiography. A review and analysis. Prog Cardiovasc Dis. 1988;30(5):307.
221 Gould K.L., Lipscomb K., Hamilton G.W. Physiologic basis for assessing critical coronary stenosis. Am J Cardiol. 1974;33:87.
222 Koerselman J., van der Graaf Y., De Jaegere P.P., Grobbee D.E. Coronary collaterals: An important and underexposed aspect of coronary artery disease. Circulation. 2003;107:19.
223 Schaper W., Gorge G., Winkler B., et al. The collateral circulation of the heart. Prog Cardiovasc Dis. 1988;1:57.
224 Schaper W., Biological and molecular biological aspects of angiogenesis in coronary collateral development. In Nakamwa M, Vanhoutte PM (eds): Coronary circulation in physiological and pathophysiological states. Tokyo, Springer-Verlag,; 1991:21-27
225 Conway E.M., Collen D., Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:3.
226 Fujita M., Tambara K. Recent insights into human coronary collateral development. Heart. 2004;90:3.
227 Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:4.
228 Sellke F.W., Quillen J.E., Brooks L.A., et al. Endothelial modulation of the coronary vasculature in vessels perfused via mature collaterals. Circulation. 1990;81:1938.
229 Verani M.S. The functional significance of coronary collateral vessels: Anecdote confronts science. Cathet Cardiovasc Diagn. 1983;9:333.
230 Marcus M.L., The coronary collateral circulation, Marcus ML., editor, The coronary circulation in health and disease; 1983. McGraw Hill: New York; 221-241
231 Braunwald E., Sobel B.E., Coronary blood flow and myocardial ischemia. 3rd ed. Braunwald E., editor. Heart disease; 1988. Philadelphia. WB Saunders; 1191-1221
232 Hlatky M.A., Mark D.B., Califf R.M., et al. Angina, myocardial ischemia and coronary disease: Gold standards, operational definitions and correlations. J Clin Epidemiol. 1989;42(5):381.
233 Buckberg G.D., Fixler D.E., Archie J.P., et al. Experimental subendocardial ischemia in dogs with normal coronary arteries. Circ Res. 1972;30:67.
234 Griggs D.M., Nakamura Y. Effects of coronary constriction on myocardial distribution of Iodoantipyrine-I-131. Am J Physiol. 1968;215:1082.
235 Brazier J., Cooper N., Buckberg G.D. The adequacy of subendocardial oxygen delivery: The interaction of determinants of flow, arterial oxygen content and myocardial oxygen need. Circulation. 1974;49:968.
236 Buffington C.W. Hemodynamic determinants of ischemic myocardial dysfunction in the presence of coronary stenosis in dogs. Anesthesiology. 1985;63:651.
237 Stone P.H. Mechanisms of silent myocardial ischemia: Implications for selection of optimal therapy. Adv Cardiol. 1990;37:328.
238 Maseri A., Newman C., Davies G. Coronary vasomotor tone: A heterogeneous entity. Eur Heart J. 1989;10(Suppl F):2.
239 Malacoff R.F., Mudge G.H., Holman B.L., et al. Effect of cold pressor test on regional myocardial blood flow in patients with coronary artery disease. Am Heart J. 1983;106:78.
240 Zeiher A.M., Drexler H., Wollschlaeger H., et al. Coronary vasomotion in response to sympathetic stimulation in humans: Importance of the functional integrity of the endothelium. J Am Coll Cardiol. 1989;14:1181.
241 Konidala S., Gutterman D.D. Coronary vasospasm and the regulation of coronary blood flow. Prog Cardiovasc Dis. 2004;46:4.
242 Batchelor T.J., Sadaba J.R., Ishola A., et al. Rho-kinase inhibitors prevent agonist-induced vasospasm in human internal mammary artery. Br J Pharmacol. 2001;132:1.
243 Werns S.W., Walton J.A., Hsia H.H., et al. Evidence of endothelial dysfunction in angiographically normal coronary arteries of patients with coronary artery disease. Circulation. 1989;79:2.
244 Vita J.A., Treasure C.B., Nabel E.G., et al. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation. 1990;81:491.
245 Nabel E.G., Selwyn A.P., Ganz P. Large coronary arteries in humans are responsive to changing blood flow: An endothelium-dependent mechanism that fails in patients with atherosclerosis. J Am Coll Cardiol. 1990;16:349.
246 Golino P., Piscione F., Willerson J.T., et al. Divergent effects of serotonin on coronary artery dimensions and blood flow in patients with coronary atherosclerosis and control patients. N Engl J Med. 1991;324:641.
247 McFadden E.P., Clarke J.G., Davies G.J., et al. Effect of intracoronary serotonin on coronary vessels in patients with stable angina and patients with variant angina. N Engl J Med. 1991;324:648.
248 Golino P., Maseri A. Serotonin receptors in human coronary arteries. Circulation. 1994;90:3.
249 Schwartz G.G., Olsson A.G., Ezekowitz M.D., et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: The MIRACL study: A randomized controlled trial. JAMA. 2001;285:13.
250 Pasceri V., Patti G., Nusca A., et al. Randomized trial of atorvastatin for reduction of myocardial damage during coronary intervention: Results from the ARMYDA (Atorvastatin for Reduction of MYocardial Damage during Angioplasty) study. Circulation. 2004;110:6.
251 Patti G., Pasceri V., Colonna G., et al. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: Results of the ARMYDA-ACS randomized trial. J Am Coll Cardiol. 2007;49:12.
252 Di S.G., Patti G., Pasceri V., et al. Efficacy of atorvastatin reload in patients on chronic statin therapy undergoing percutaneous coronary intervention: Results of the ARMYDA-RECAPTURE (Atorvastatin for Reduction of Myocardial Damage During Angioplasty) Randomized Trial. J Am Coll Cardiol. 2009;54:6.
253 Schonbeck U., Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: Statins as antiinflammatory agents? Circulation. 2004;109(Suppl 1):21.
254 Halcox J.PJ., Deanfield J.E. Beyond the laboratory: Clinical implications for statin pleiotropy. Circulation. 2004;109(Suppl 1):21.
255 Pagano P.J., Gutterman D.D. The adventitia: The outs and ins of vascular disease. Cardiovasc Res. 2007;75:4.
256 Bot I., de Jager S.C., Bot M., et al. The neuropeptide substance P mediates adventitial mast cell activation and induces intraplaque hemorrhage in advanced atherosclerosis. Circ Res. 2010;106:1.
257 Herrmann J., Lerman L.O., Rodriguez-Porcel M., et al. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc Res. 2001;51:4.
258 Shi Y., O’Brien J., Fard A., et al. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation. 1996;94(7):1655.
259 Zampetaki A., Kirton J.P., Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008;78:3.
260 Hibbert B., Olsen S., O’Brien E.R. Involvement of progenitor cells in vascular repair. Trends Cardiovasc Med. 2003;13:8.
261 Ma X., Hibbert B., White D., et al. Contribution of recipient-derived cells in allograft neointima formation and the response to stent implantation. PLoS ONE. 2008;3:3.
262 Hill J.M., Zalos G., Halcox J.P., et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:7.
263 Werner N., Kosiol S., Schiegl T., et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:10.
264 Quaini F., Urbanek K., Beltrami A.P., et al. Chimerism of the transplanted heart. N Engl J Med. 2002;346:1.
265 George J., Herz I., Goldstein E., et al. Number and adhesive properties of circulating endothelial progenitor cells in patients with in-stent restenosis. Arterioscler Thromb Vasc Biol. 2003;23:12.
266 Hibbert B., Chen Y.-X., O’Brien E.R. The opposing roles of c-kit immunopositive vascular progenitor cells and endothelial progenitor cells in human coronary in-stent restenosis. Am J Physiol (Heart Circ Physiol). 2004;287:H518.
267 Assmus B., Honold J., Schachinger V., et al. Transcoronary transplantation of progenitor cells after myocardial infarction. N Engl J Med. 2006;355:12.
268 Schachinger V., Erbs S., Elsasser A., et al. Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006;355:12.
269 Hirschi K.K., Ingram D.A., Yoder M.C. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:9.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
6 Coronary Physiology and Atherosclerosis
Anatomy and Physiology of Blood Vessels
The coronary vasculature has been traditionally divided into three functional groups: large conductance vessels visible on coronary angiography, which offer little resistance to blood flow; small resistance vessels ranging in size from about 10 to 250 μm in diameter; and veins. Although it has been taught that arterioles (precapillary vessels < 50 μm) account for most of the coronary resistance, recent studies indicate that, under resting conditions, 45% to 50% of total coronary vascular resistance resides in vessels larger than 100 μm in diameter1–3 (Figure 6-1). This may be due, in part, to the relatively great length of the small arteries. During intense pharmacologic dilation, the proportion of total coronary vascular resistance because of larger arteries and veins is even greater.2 The regulation of tone in coronary arteries larger than 100 μm in diameter plays an important role in delivering adequate myocardial perfusion.4 One of the early changes in CAD is a diminished ability of the endothelium of epicardial coronary arteries to dilate in response to increased flow (see Endothelium-Derived Relaxing Factors later in this chapter). Advances in technology have enabled measurement, in the beating heart, of diameters of coronary vessels as small as 15 μm. It is becoming evident that, in response to a given intervention, different size classes of coronary vessels can change diameter with different intensity or even in opposite directions.5,6 This heterogeneity of response according to vessel size would be an important consideration in predicting the effects of vasoactive agents on myocardial perfusion. For example, a drug that dilated large vessels and collaterals but not arterioles would be beneficial to patients with CAD (see Coronary Steal later in this chapter).
Normal Artery Wall
The arterial lumen is lined by a monolayer of endothelial cells that overlies smooth muscle cells (Figure 6-2). The inner layer of smooth muscle cells, known as the intima, is circumscribed by the internal elastic lamina. Between the internal elastic lamina and external elastic lamina is another layer of smooth muscle cells, the media. Outside the external elastic lamina is an adventitia that is sparsely populated by cells and microvessels of the vasa vasorum.
Intima
Traditionally, the intima has been considered the most important layer of the artery wall.7 The intima can vary from a single endothelial layer to a more complex structure of an endothelium overlying a patchwork of extracellular matrix and vascular smooth muscle cells. As part of the normal development of many large arteries, smooth muscle cells populate this space and form a neointima. This diffuse form of intimal thickening consists of layers of smooth muscle cells and connective tissue, the thickness of which may vary considerably. For convenience, the intima/media ratio is often measured, and the reference range is 0.1 to 1.0. How this benign intima forms is not well understood. Presumably, the intima represents a physiologic adaptation to changes in arterial flow and wall tension. The intima is made up of two distinct layers.8 As seen by electron microscopy, the inner layer subjacent to the luminal endothelium contains an abundance of proteoglycan ground substance. Smooth muscle cells found in this layer are usually distributed as isolated cells in a sea of matrix, rather than in contiguous layers. A few macrophages also may be found in this layer underneath the endothelial monolayer. The outer, musculoelastic layer of the intima is adjacent to the internal elastic lamina and contains smooth muscle cells and elastic fibers.
Media
In normal adult arteries, several smooth muscle cell subpopulations with distinct lineages exist within the media.9 These diverse cell populations likely fulfill different functions to maintain homeostasis in the artery wall. For example, in response to pressure elevations, increases in smooth muscle cell mass and extracellular matrix may be required. Alternatively, for arteries to be able to stretch both longitudinally and circumferentially, smooth muscle cells with variable orientations of cytoskeletal fibers must be present. These distinct cell types may be important not only in health but in disease. In certain experimental models of neointimal formation, proliferation and inward migration of subpopulations of medial smooth muscle cells occur.10 The biologic determinants of medial smooth muscle cell diversity are unknown.11
Adventitia
The adventitia, the outermost layer of the artery wall, normally consists of a sparse collection of fibroblasts, microvessels (vasa vasorum), nerves, and few inflammatory cells. The majority of the vasa vasorum that nourish the inner layers of the artery wall originate in the adventitia. Traditionally, the adventitia has been ignored and is not thought to play a role in vascular lesion formation. However, more recent studies have elucidated the role of the adventitia as not only a source of inflammatory cells in the development of atherosclerosis, but a hub for paracrine signaling that can maintain vascular homeostasis in a variety of vascular diseases. 12
Transmembrane and Transcellular Communication
Figure 6-3 illustrates examples of pathways of transmembrane signaling. Up to five components can be involved: receptor, G protein, effector producing a second messenger, phosphorylation of regulator protein, and the consequent change in cell behavior. G proteins (guanine nucleotide-binding regulatory proteins) are made up of three subunits (α, β, γ) and float in the cell membrane. On contact with a ligand-receptor complex, guanosine diphosphate (GDP) on the α subunit is replaced by guanosine triphosphate (GTP). The activated α subunit then dissociates from the beta-gamma complex and can interact with several membrane targets (see Figure 6-3B). For example, β-receptor activation results in the activation of Gs (s = stimulate), which will stimulate the synthesis of cAMP by adenylyl cyclase. Muscarinic receptor activation activates a Gi (i = inhibit) protein that inhibits adenylyl cyclase. A single G protein can interact with more than one effector. In this way, the G protein can be a branch point for the regulation of multiple effectors in response to a single signal. These proteins already have been implicated in human disease; cholera toxin covalently modifies Gs so that it becomes persistently active in stimulating adenylyl cyclase in intestinal epithelial cells, likely causing the severe diarrhea of cholera.
Figure 6-3 Steps in the process whereby hormone-receptor binding results in a change in cell behavior.
(A, B, From Brown AM, Birnbaumer L: Ionic channels and their regulation by G-protein subunits. Annu Rev Physiol 52:197, 1990.)
Several second-messenger systems have been characterized. Gs can directly enhance conductance through calcium channels, with the increased intracellular calcium acting as second messenger. The cyclic nucleotides, cAMP and guanosine monophosphate (GMP), act as second messengers. Their intracellular action is terminated when they are cleaved by phosphodiesterase enzymes, which, in turn, are regulated by stimuli and second messengers. The breakdown products of membrane phosphoinositide constitute another, more recently recognized set of second messengers.13 In response to agonists such as vasopressin, G protein is activated, leading to activation of the membrane-associated enzyme phospholipase C. This enzyme cleaves phosphatidylinositol 4,5-biphosphate on the inner leaflet of the plasma membrane, producing inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). Both are second messengers. IP3 diffuses through the cytoplasm and mobilizes calcium from intracellular stores. DAG remains within the plasma membrane and activates protein kinase C, which modulates cellular activity by phosphorylating intracellular proteins. In many cell types, activation of the same receptors that control phosphoinositide breakdown also results in the liberation of arachidonate and/or eicosanoids (prostaglandins, leukotrienes, and thromboxanes). The resultant change in cell behavior can be the opening of an ion channel, contraction or relaxation of smooth muscle, secretory activity, or initiation of cell division (see Chapter 7).
Endothelium
Although the vascular endothelium was once thought of as an inert lining for blood vessels, it is more accurately characterized as a very active, distributed organ with many biologic functions. It has synthetic (Table 6-1) and metabolic (Table 6-2) capabilities, and contains receptors for a variety of vasoactive substances (Table 6-3). Functions of the endothelium that may play an important role in the pathophysiology of ischemic heart disease are discussed.
| Antithrombotic Substances | Procoagulants |
|---|---|
| Prostacyclin | von Willebrand factor |
| Antithrombin III | Collagen |
| Plasminogen activator | Fibronectin |
| Protein C | Thromboplastin |
| α2-Macroglobulin | Thrombospondin |
| Glycosaminoglycans (heparin) | Plasminogen inhibitors |
| Platelet-activating factor | |
| Thromboxane A2 |
From Bassenge E, Busse R: Endothelial modulation of coronary tone. Prog Cardiovasc Dis 30:349, 1988.
TABLE 6-2 Vasoactive Substances Processed by Vascular Endothelium
| Uptake and Metabolism | Enzymatic Conversion or Degradation |
|---|---|
| Norepinephrine | Angiotensin I to angiotensin II (ACE) |
| Serotonin | Angiotensin II to angiotensin III (angiotensinase) |
| Prostaglandins (E1, E2, E2α) | Bradykinin degradation (ACE) |
| Leukotrienes | Substance P degradation |
| Adenosine |
ACE, angiotensin-converting enzyme.
From Bassenge E, Busse R: Endothelial modulation of coronary tone. Prog Cardiovasc Dis 30:349, 1988.
TABLE 6-3 Stimulators of Endothelium-Mediated Vasodilation
| Transmitters | Adenosine diphosphate (ADP) |
| Acetylcholine | Adenosine |
| Norepinephrine | Serotonin |
| Peptides | Thrombin |
| Angiotensin | Trypsin |
| Bradykinin | Local Hormones |
| Vasopressin | Histamine |
| Oxytocin | Platelet-activating factor |
| Substance P | Physicochemical Stimuli |
| Vasoactive intestinal peptide | Shear stress (flow) |
| Calcitonin gene–related peptide | Mechanical stress (pulsatility) |
| Platelet or Blood Components | Hypoxia |
| Adenosine triphosphate (ATP) |
Modified from Bassenge E, Busse R: Endothelial modulation of coronary tone. Prog Cardiovasc Dis 30:349, 1988.
Endothelium-Derived Relaxing Factors
The first vasoactive endothelial substance to be discovered was prostacyclin (PGI2), a product of the cyclooxygenase pathway of arachidonic acid metabolism (Figure 6-4; Box 6-1).14 The production of PGI2 is activated by shear stress, pulsatility of flow, hypoxia, and a variety of vasoactive mediators. On production it leaves the endothelial cell and acts in the local environment to cause relaxation of the underlying smooth muscle or to inhibit platelet aggregation. Both actions are mediated by the stimulation of adenylyl cyclase in the target cell to produce cAMP.
Figure 6-4 The production of endothelium-derived vasodilator substances.
(From Rubanyi GM: Endothelium, platelets, and coronary vasospasm. Coron Artery Dis 1:645, 1990.)
In 1980, Furchgott and Zawadzki15 observed that the presence of an intact endothelium was necessary for acetylcholine-induced vasodilation. Since that time it has been shown that many physiologic stimuli cause vasodilation by stimulating the release of a labile, diffusible, nonprostanoid molecule termed endothelium-derived relaxing factor (EDRF; see Figure 6-4), now known to be nitric oxide (NO). NO is the basis of a widespread paracrine signal transduction mechanism whereby one cell type can modulate the behavior of adjacent cells of different type.16,17 NO is a very small lipophilic molecule that can readily diffuse across biologic membranes and into the cytosol of nearby cells. The half-life of the molecule is less than 5 seconds, so that only the local environment can be affected. NO is synthesized from the amino acid l-arginine by nitric oxide synthase (NOS). In vascular endothelium, the enzyme (endothelial NOS or NOS3) is always present (constitutive) and resides in the cytoplasm. Its function depends on the presence of Ca++ and calmodulin, as well as tetrahydrobiopterin. Serine phosphorylation is important for prolonged activity. The enzyme is activated in response to receptor occupancy or physical stimulation (see Table 6-3). When NO diffuses into the cytosol of the target cell, it binds with the heme group of soluble guanylate cyclase, resulting in a 50- to 200-fold increase in production of cyclic GMP, its second messenger. If the target cells are vascular smooth muscle cells, vasodilation occurs; if the target cells are platelets, adhesion and aggregation are inhibited. In vascular smooth muscle, cyclic GMP leads to activation of protein kinase G, which phosphorylates various intracellular target proteins, including the myosin light-chain regulatory subunit and proteins that control intracellular calcium.18
It is likely that NO is the final common effector molecule of nitrovasodilators (including sodium nitroprusside and organic nitrates such as nitroglycerin). The cardiovascular system is in a constant state of active vasodilation that is dependent on the generation of NO. The molecule is more important in controlling vascular tone in veins and arteries compared with arterioles. When the microcirculation dilates in response to metabolic myocardial demand (e.g., exercise), increased flow through epicardial coronary arteries increases shear stress at the endothelium. This leads to release of NO, which causes vascular smooth muscle relaxation and dilation of the conductance vessels, thereby facilitating the increase in flow. The importance of the loss of this mechanism in atherosclerosis is underlined by the fact that, in this situation, more than 50% of the resistance to flow in the coronary circulation resides in vessels larger than 100 μm in diameter (see Figure 6-1). Abnormalities in the ability of the endothelium to produce NO likely plays a role in diseases such as diabetes, atherosclerosis, and hypertension.19,20 The venous circulation of humans appears to have a lower basal release of NO and an increased sensitivity to nitrovasodilators when compared with the arterial side of the circulation.21
Many agents, such as acetylcholine and norepinephrine, can cause contraction when applied directly to the vascular smooth muscle membrane instead of relaxation, which occurs when it is applied to the intact endothelium (Figure 6-5). The net effect of neural or humoral stimuli depends on a combination of direct effects mediated by binding to vascular smooth muscle receptors and indirect effects because of the ligand binding to endothelial receptors causing NO release from the endothelium. In the presence of healthy endothelium, vasodilation usually predominates. When the endothelium is absent (injured vessel) or diseased (atherosclerosis), vasoconstriction may be the net effect. NO has important roles in neurohumoral regulation of vascular tone, in preventing intravascular platelet aggregation, and in the structural adaptation of blood vessels to the demands of blood flow and pressure. Knowledge of its role in inflammation and atherosclerosis is rapidly expanding.
Figure 6-5 Role of endothelium in the control of coronary tone.
(From Bassenge E, Heusch G: Endothelial and neurohumoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol 116:77, 1990.)
In addition to PGI2 and NO, another less-well-understood pathway for receptor-mediated or mechanically induced endothelium-derived vasodilation exists that is associated with smooth muscle hyperpolarization. Both epoxyeicosatrienoic acid (a metabolite of cytochrome P450) and H2O2 have been suggested as possible endothelium-derived hyperpolarizing factors (EDHFs).22,23 Smooth muscle relaxation is a result of hyperpolarization of the myocyte, which leads to decreased intracellular calcium concentration. EDHF-mediated vasodilation can be blocked by inhibition of calcium-dependent potassium channels. EDHF may have an important vasodilator role in the human coronary microcirculation.24
Endothelium-Derived Contracting Factors
Contracting factors produced by the endothelium include prostaglandin H2, thromboxane A2 (TxA2; via cyclooxygenase), and the peptide endothelin (ET). ET is a potent vasoconstrictor peptide (100-fold more potent than norepinephrine)25 with remarkable similarities to the toxin of the burrowing asp. Both have potent coronary constrictor activity to which the strong cardiac toxicity and lethality of the toxin are attributed.26 Three closely related 21 amino acid peptides have been identified: endothelin-1 (ET-1), ET-2, and ET-3. The primary product of vascular endothelium is ET-1, which is synthesized from prepro-ET-1 within vascular endothelial cells by the action of ET-converting enzyme. It is not stored but rapidly synthesized in response to stimuli such as ischemia, hypoxia, and shear stress, and released predominantly abluminally (toward the underlying smooth muscle).27 In vascular smooth muscle cells, ET-1 binds to specific membrane receptors (ETA) and, via phospholipase C, induces an increase in intracellular calcium resulting in long-lasting contractions.28 It is also linked via a Gi protein to voltage-operated calcium channels. This peptide has greater vasoconstricting potency than any other cardiovascular hormone, and in pharmacologic doses can abolish coronary flow, leading to ventricular fibrillation and death.29 Another receptor subtype, ETB, is expressed by both smooth muscle and endothelium and binds ET-1 and ET-3 equally well (Figure 6-6). When isolated vessels are perfused with ET-1, there is an initial NO-mediated vasodilation because of binding with ETB receptors on the endothelial cells, followed by contraction because of binding of ET-1 to ETA receptors on the vascular smooth muscle membrane. Studies utilizing bosentan, a combined ETA– and ETB-receptor antagonist, have demonstrated that ET exerts a basal coronary vasoconstrictor tone in humans.30 There is evidence that ET may play a role in the pathophysiology of pulmonary and arterial hypertension, atherosclerosis, myocardial ischemic syndromes, and heart failure.31 Clinical trials of bosentan in patients with congestive heart failure32 and hypertension have shown promise, but hepatic side effects have limited the dose to less than 500 mg daily, with the primary indication being severe pulmonary hypertension.33
Endothelial Inhibition of Platelets
A primary function of endothelium is to maintain the fluidity of blood. This is achieved by the synthesis and release of anticoagulant (e.g., thrombomodulin, protein C), fibrinolytic (e.g., tissue-type plasminogen activator), and platelet inhibitory (e.g., PGI2, NO) substances (Box 6-2).34 Mediators released from aggregating platelets stimulate the release of NO and PGI2 from intact endothelium, which act together to increase blood flow and decrease platelet adhesion and aggregation (Figure 6-7), thereby flushing away microthrombi and maintaining the patency of the vessel.
BOX 6-2 Endothelial Inhibition Of Platelets
Healthy endothelial cells have a role in maintaining the fluidity of blood by producing:
Figure 6-7 Inhibition of platelet adhesion and aggregation by intact endothelium.
(From Rubanyi GM: Endothelium, platelets, and coronary vasospasm. Coron Artery Dis 1:645, 1990.)
With vital roles in modulating the tone of vascular smooth muscle, inhibiting platelets, and processing circulating chemicals, it seems clear that endothelial cell dysfunction would cause or contribute to ischemic syndromes. There is evidence of endothelial dysfunction in atherosclerosis, hyperlipidemia, diabetes, and hypertension.35 Procedures such as coronary artery surgery and angioplasty disrupt the endothelium. The role of endothelium in the pathophysiology of myocardial ischemia is discussed later (see Dynamic Stenosis section).
Determinants of Coronary Blood Flow
Perfusion Pressure and Myocardial Compression
Coronary blood flow is proportional to the pressure gradient across the coronary circulation (Box 6-3). This gradient is calculated by subtracting downstream coronary pressure from the pressure in the root of the aorta. The determination of downstream pressure is complicated because the intramural coronary vessels are compressed with each heartbeat.
During systole, the heart throttles its own blood supply. The force of systolic myocardial compression is greatest in the subendocardial layers, where it approximates intraventricular pressure. Resistance caused by extravascular compression increases with blood pressure, heart rate, contractility, and preload. Because it is difficult to measure intramyocardial pressure, the relative importance of these factors is controversial.36,37 Flow is impeded both by direct compression and by shear caused by twisting of vessels as the heart contracts. Myocardial extravascular compression is less in the right ventricle, where pressures are lower and coronary perfusion persists during systole (Figure 6-8). In pathologic conditions associated with pulmonary hypertension, right coronary flow assumes a phasic pattern similar to left coronary flow. Under normal conditions, extravascular compression contributes only a small component (10% to 25%) to total coronary vascular resistance. When the coronary vessels are dilated by pharmacologic agents such as dipyridamole or during ischemia, the effects of extravascular compression on myocardial perfusion become more important (see Transmural Blood Flow section later in this chapter).
Figure 6-8 Blood flow in the left and right coronary arteries.
(From Berne RM, Levy MN: Special circulations. In Berne RM, Levy MN [eds]: Physiology. St. Louis: CV Mosby, 1988, pp 540–560.)
With each contraction, the intramural vessels are squeezed and blood is expelled forward into the coronary sinus and retrograde into the epicardial arteries. The large coronary arteries on the epicardial surface act as capacitors, charging with blood during systole and expelling blood into the coronary circulation during diastole.38 Coronary capacitance likely explains the findings of Bellamy,39 who reported that flow in the proximal left anterior descending coronary artery of the dog ceased when arterial pressure decreased to less than 45 mm Hg. It was suggested that flow throughout the coronary circulation stopped at pressures far in excess of the pressure at the coronary sinus. This pressure at which flow stopped was termed critical closing pressure or zero-flow pressure (Pzf). This had important implications in the calculation of coronary resistance because the effective downstream pressure would be Pzf and not the much lower coronary venous pressure. This is analogous to a stream with a waterfall, where flow rate over the waterfall depends on the drop from the source to the waterfall edge and is unaffected by the distance to the bottom of the falls. It was later suggested that flow through the intramural coronary vessels continues after coronary inflow near the ostia (measured by Bellamy) has ceased.40,41 There is evidence that antegrade movement of red blood cells in 20-μm arterioles continues until coronary pressure is only a few millimeters of mercury greater than coronary sinus pressure.42 The concept of a critical closing pressure greatly in excess of coronary sinus pressure is probably not valid in the coronary circulation.
Although the true downstream pressure of the coronary circulation is likely close to the coronary sinus pressure, other choices may be more appropriate in clinical circumstances. In patients with CAD, the subendocardial layers of the left ventricle are at greatest risk for ischemia and necrosis (see Transmural Blood Flow section later in this chapter). Because this layer is perfused mostly when the aortic valve is closed, the most appropriate measure of the driving pressure for flow here is the average pressure in the aortic root during diastole. This can be approximated by aortic diastolic or mean pressure. Pressures monitored in peripheral arteries by routine methods in clinical settings can differ from central aortic readings. This is due to distortion of the pressure waveform as it is propagated through the arterial tree and inaccuracies associated with the hydraulic and electronic components of the monitoring system. Under these conditions, the mean arterial pressure may be the most reliable measure of coronary driving pressure. The true downstream pressure of the left ventricular subendocardium is the left ventricular diastolic pressure, which can be estimated by pulmonary artery occlusion pressure. When the right ventricle is at risk for ischemia (e.g., severe pulmonary hypertension), right ventricular diastolic pressure or central venous pressure may be more appropriate choices for downstream pressure.
Myocardial Metabolism
Myocardial blood flow, like flow in the brain and skeletal muscle, is primarily under metabolic control. Even when the heart is cut off from external control mechanisms (neural and humoral factors), its ability to match blood flow to its metabolic requirements is almost unaffected.35 Because coronary venous oxygen tension is normally 15 to 20 mm Hg, there is only a small amount of oxygen available through increased extraction. A major increase in cardiac oxygen consumption (Mvo2) can occur only if oxygen delivery is increased by augmentation of coronary blood flow. Normally, flow and metabolism are closely matched so that over a wide range of oxygen consumption, coronary sinus oxygen saturation changes little.43
Despite intensive research over the last several decades, the mediator or mediators linking myocardial metabolism so effectively to myocardial blood flow are still unknown. Hypotheses of metabolic control propose that vascular tone is linked either to a substrate that is depleted, such as oxygen or adenosine triphosphate (ATP), or to the accumulation of a metabolite such as CO2 or hydrogen ion (Box 6-4). Adenosine has been proposed in both categories. Feigl43 has proposed six criteria for a chemical transmitter between the cardiac myocyte and the coronary vascular smooth muscle cell:
Many potential mediators of metabolic regulation have been proposed.44 Although NO has a role in many coronary vasoregulatory pathways, it does not fulfill the role of metabolic regulator because blockade of NOS does not alter the increase in myocardial blood flow associated with an increase in myocardial oxygen demand.45 The arguments for oxygen, carbon dioxide, and adenosine are briefly examined.
Oxygen
The coronary smooth muscle would have to be more sensitive to lack of oxygen than the working cardiocytes for oxygen to regulate coronary flow through a direct vascular action. Coronary microvessels in vitro do not relax until Po2 is less than 5 mm Hg, a level well below the average Po2 of 20 mm Hg in cardiac muscle cytosol.46,47 With myocardial oxygen consumption (Mvo2) held constant, increases in arterial oxygen content cause coronary flow to decrease, whereas decreases in arterial oxygen content cause flow to increase. These changes could explain only 40% of the increase in flow observed with tachycardia.48 It is undecided whether the constancy of myocardial oxygen tension is the cause or the consequence of the excellent match between myocardial metabolism and myocardial blood flow.49
Carbon Dioxide
The end product of substrate oxidation is CO2, the formation of which is directly related to the level of cardiac work. Carbon dioxide is highly diffusible and can easily reach coronary smooth muscle cells. Unfortunately, it is difficult to separate the effects on coronary tone of increasing CO2 from concomitant increases in other metabolites. Broten et al48 pump-perfused the left main coronary artery of dogs and used an oxygenator in the perfusion circuit to alter coronary arterial Pco2 and Po2 at a constant level of myocardial metabolism. Increases in arterial and coronary sinus Pco2 caused increases in coronary blood flow in the absence of changes in Mvo2. Interestingly, there was a synergistic action of Pco2 and Po2: The increase in flow with elevation of Pco2 was much greater at low Po2 and vice versa. The effect of increasing CO2, however, could not completely account for flow changes associated with an increase in Mvo2.
Adenosine
Adenosine is a powerful coronary vasodilator via its activation of receptors on vascular endothelium and smooth muscle. In 1963, both Berne50 and Gerlach51 independently demonstrated the production of adenosine in ischemic heart muscle. They hypothesized that the release of adenosine may serve as a feedback signal inducing coronary vasodilation and augmenting coronary blood flow in proportion to myocardial metabolic needs. Initially, it was suggested that adenosine formation was coupled to myocardial oxygen tension.50 A substrate theory has been proposed whereby adenosine production is linked to the cardiac energy state by the regulation of cytosolic AMP concentration to explain metabolic regulation by adenosine under both normoxic and ischemic conditions.52 According to this theory, increases in cardiac work lead to a decline in ATP potential, which results in a quantitatively appropriate change in cytosolic AMP concentration, leading to increased adenosine release. In this way, the rate of adenosine production is determined by the myocardial oxygen supply/demand ratio. It is likely that adenosine causes coronary arteriolar dilation through stimulation of A1 receptors directly coupled to ATP-sensitive K+ (K+ATP) channels and A2 receptor–mediated elevation of cAMP/protein kinase A, which lead to vasodilation, in part, by opening of K+ ATP channels.53,54
Evidence against the adenosine hypothesis is accumulating. Adenosine deaminase is an enzyme that, when introduced in sufficient quantity into the myocardium, can significantly reduce the interstitial concentration of adenosine. Aminophylline and theophylline interfere with the coronary dilating effects of adenosine by acting on the receptor on vascular smooth muscle. Experiments using these agents to inhibit adenosine effect have shown that resting coronary blood flow, exercise-induced coronary dilation, autoregulation, and reactive hyperemia are largely unrelated to adenosine.55–58 Measuring coronary microvessel diameters in beating hearts in situ, Kanatsuka et al5 found that when Mvo2 was doubled by pacing, vessels between 40 and 380 μm dilated, whereas when a similar increase in flow was induced by the infusion of adenosine or dipyridamole at constant Mvo2, only vessels smaller than 150 μm dilated. Although adenosine does not seem to have an important role in metabolic regulation in the normal heart, adenosine blockade has been shown to cause a decrease in blood flow to hypoperfused myocardium sufficient to decrease systolic segment shortening.59 Adenosine may have other important roles in ischemia, in which there is evidence of a cardioprotective action.60,61
Neural and Humoral Control
Coronary Innervation
The heart is supplied with branches of the sympathetic and parasympathetic divisions of the autonomic nervous system. Thicker vagal fibers end in the adventitia of coronary vessels, whereas fine nonmedullated sympathetic fibers end on vascular smooth muscle cells.62 Large and small coronary arteries, as well as veins, are richly innervated. The sympathetic nerves to the heart and coronary vessels arise from the superior, middle, and inferior cervical sympathetic ganglia, as well as the first four thoracic ganglia. The stellate ganglion (formed when the inferior cervical and first thoracic ganglia merge) is a major source of cardiac sympathetic innervation. The vagi supply the heart with efferent cholinergic nerves.
Parasympathetic Control
Vagal stimulation causes bradycardia, decreased contractility, and lower blood pressure. The resultant decline in Mvo2 causes a metabolically mediated coronary vasoconstriction. When myocardial metabolism is held constant, however, cholinergic coronary dilation is consistently observed in response to exogenous acetylcholine, electrical vagal stimulation, and reflex activation through baroreceptors, chemoreceptors, and ventricular receptors.35,43,63 These effects can be abolished by atropine.
In patients with angiographically normal coronary arteries, the response to intracoronary acetylcholine injection is predominantly dilation, whereas in atherosclerotic segments of epicardial arteries, constriction is observed.64–66 Acetylcholine injected intraluminally binds to muscarinic receptors on the endothelium and stimulates the release of NO, which causes smooth muscle dilation. Acetylcholine is not normally found circulating in the blood but is released from vagal fibers and reaches the coronary smooth muscle from the adventitial side. Surprisingly, activation of muscarinic receptors on vascular smooth muscle cells causes constriction. Parasympathetic stimulation normally causes coronary vasodilation. This response depends on the ability of the coronary endothelium to elaborate NO and perhaps also EDHF (see earlier).67,68 Parasympathetic control has not been shown to be important in the initiation of myocardial ischemia.
β-Adrenergic Coronary Dilation
β-Receptor activation causes dilation of both large and small coronary vessels even in the absence of changes in blood flow.35,43 Studies in animals indicate that both β1 and β2 receptors are present throughout the coronary circulation, but β1 receptors predominate in the conductance vessels, whereas β2 receptors predominate in the resistance vessels. Mature canine coronary collaterals respond similarly to the conductance vessels.69,70 β-Adrenergic coronary dilation may improve the speed and accuracy of coronary blood flow regulation during exercise.71
α-Adrenergic Coronary Constriction
Activation of the sympathetic nerves to the heart results in increases in heart rate, contractility, and blood pressure, which lead to a marked, metabolically mediated increase in coronary blood flow (Box 6-5). This suggested to early investigators that the effect of sympathetic coronary innervation is vasodilation. More recent investigation has demonstrated that the direct effect of sympathetic stimulation is coronary vasoconstriction, which is in competition with the metabolically mediated dilation of exercise or excitement. Whether adrenergic coronary constriction is powerful enough to further diminish blood flow in ischemic myocardium or whether it can have some beneficial effect in the distribution of myocardial blood flow is controversial.
Classification
α-Adrenergic receptors can be classified anatomically as presynaptic or postsynaptic and also according to their pharmacologic properties as α1 and α2 (Table 6-4). The receptors can be further divided into subtypes according to their signal transduction mechanism (G-protein subtype) and second messenger (adenylyl cyclase, phospholipase C, etc.).72
TABLE 6-4 Classification of α-Adrenergic Receptor Subtypes in the Heart
| Selective Agonists | Selective Antagonists | Effects of Activation |
|---|---|---|
| α1 | ||
| Phenylephrine | Prazosin | Presynaptic: feedback inhibition of norepinephrine release |
| Methoxamine | Postsynaptic: coronary vasoconstriction, increase in myocardial arrhythmias | |
| Inotropism | ||
| α2 | ||
| Clonidine | Yohimbine | Presynaptic: feedback inhibition of norepinephrine release |
| Azepexole | Rauwolscine | Postsynaptic: coronary vasoconstriction, arrhythmias (?) |
| BHT 920 | Idazoxan | |
| UK 14, 304 |
Norepinephrine is a nonselective agonist. Phentolamine and phenoxybenzamine are nonselective antagonists. Phenylephrine also causes β-receptor activation.
Modified from Heusch G: Alpha-adrenergic mechanisms in myocardial ischemia. Circulation 81:1, 1990.
Presynaptic α Receptors
α receptors on cardiac sympathetic nerve terminals mediate feedback inhibition of neuronal norepinephrine release. Both α1 and α2 receptors appear to be involved because exercise-induced increases in heart rate and contractility can be potentiated by either idazoxan (α2-blockade) or prazosin (α1-blockade).73
Cardiac Muscle Cells
Activation of myocardial α1 receptors results in a positive inotropic effect that, in contrast with β-receptor activation, is associated with prolongation of contraction. Although normally of minor functional importance, this effect may serve as an inotropic reserve mechanism when β-receptor–mediated inotropy is impaired (e.g., hypothyroidism, cardiac failure, chronic propranolol treatment).74 The importance of this mechanism in humans is uncertain. An increase in inotropy caused by stimulation of myocardial α receptors would result in increased Mvo2 and a metabolically mediated coronary dilation.
Coronary Endothelium
Binding of norepinephrine to α2 receptors on vascular endothelium stimulates the release of NO, which acts to relax vascular smooth muscle. The endothelium can also act to limit the effect of norepinephrine by metabolizing it. In these ways, the endothelium modulates the direct constrictive effects of α-adrenergic activation. Abnormal endothelial function in atherosclerosis may predispose to excessive α-adrenergic constriction and is implicated in the pathogenesis of myocardial ischemia (see Dynamic Stenosis later in this chapter).
Coronary Resistance
The magnitude of α-adrenergic vasoconstriction that occurs in the coronary bed is small compared with that which occurs in the skin and skeletal muscle. In the presence of β-blockade, intense sympathetic stimulation results in only a 20% to 30% increase in coronary resistance.75 Mohrman and Feigl76 examined the effect of sympathetic activation on coronary flow in the absence of β-blockade. The net effect of α-receptor vasoconstriction was to restrict the metabolically related flow increase by 30%, thereby increasing oxygen extraction and decreasing coronary sinus oxygen content.
Epicardial coronary diameter changes little during sympathetic stimulation.77 α1-Adrenergic and α2-adrenergic receptors are found throughout the coronary circulation; however, α1 receptors appear to be more important in the large epicardial vessels, whereas α2 predominate in small coronary vessels less than 100 μm in diameter.78 Studies of mature coronary collateral vessels in dogs have generally failed to provide evidence of α-receptor–mediated vasoconstriction.79 After heart transplant, patients demonstrated a lesser increase in myocardial blood flow after a cold pressor test in denervated regions of the heart.80 The authors argue that this was not due to increased myocardial metabolism secondary to myocardial β-receptor activation. They suggest that sympathetic innervation has an important role in coronary vessel dilation during stress.
Exercise
α-Adrenergic coronary constrictor tone during exercise is exerted predominantly by circulating catecholamines.81 Numerous studies indicate that myocardial blood flow during exercise is limited by α vasoconstriction.35 In a study of exercising dogs, Huang and Feigl82 found that despite an increase in total coronary flow in an α-blocked region of myocardium, flow to the inner, subendocardial layer was diminished. These results suggest a beneficial effect of α-adrenergic coronary constriction on the distribution of blood flow within the myocardium.
Myocardial Ischemia
Buffington and Feigl83 demonstrated the persistence of α-adrenergic coronary vasoconstriction distal to a moderate coronary stenosis during norepinephrine infusion. Investigations in dogs have demonstrated that, as coronary reserve is depleted by increasing stenosis severity, the response to sympathetic stimulation shifts from a metabolically induced coronary dilation to coronary constriction.84,85 These observations suggest that sympathetic coronary vasoconstriction limits coronary blood flow even during myocardial ischemia, when autoregulatory reserve is exhausted (see Coronary Reserve later in this chapter). There is no consensus as to the importance of α1 vs α2 receptors in ischemic myocardium.35 Using constant flow coronary perfusion in anesthetized dogs, Nathan and Feigl86 compared the transmural distribution of myocardial blood flow in α-blocked and intact regions of myocardium during hypoperfusion. Surprisingly, α-blockade diverted blood flow from the subendocardium to the subepicardium. This suggests that α vasoconstriction had limited flow more in the subepicardium, thereby producing an antisteal effect, and improved perfusion of the more vulnerable inner layers of the left ventricle. Chilian and Ackell87 found similar results in exercising dogs with an artificial coronary stenosis. In contrast, work from Heusch and colleagues88,89 demonstrated improved subendocardial perfusion distal to a severe coronary stenosis with α2-receptor blockade. This controversy is unresolved.90 α-Receptor blockers have not been shown to have a role in the treatment of myocardial ischemia in patients with CAD.
Studies in Humans
Studies indicate that there is little α-adrenoceptor–mediated tone in resting humans.91 Clinical studies have failed to provide convincing evidence that α-adrenergic coronary constriction plays an important role in Prinzmetal’s variant angina (angina with ST-segment elevation at rest).92 During sympathetic activation, however, there is evidence that α vasoconstriction can precipitate myocardial ischemia by further narrowing diseased coronary arteries. This has been shown during isometric exercise, dynamic exercise, and with the cold pressor test93–98 (see Dynamic Stenosis later in this chapter).
Humoral Control
A complete understanding of the effects of circulating substances on the coronary vessels would require determining their effects on large versus small coronary vessels, while separating direct effects on coronary vessels from changes in tone mediated by changes in myocardial metabolism. This is further complicated by the critical role of an intact vascular endothelium in modulating these responses (see Endothelium earlier in this chapter). Some of the better studied agents are discussed briefly later.
The peptide hormones include vasopressin (arginine vasopressin [AVP] or antidiuretic hormone [ADH]), atrial natriuretic peptide (ANP), vasoactive intestinal peptide, neuropeptide Y, and calcitonin gene–related peptide.44 Of these, AVP and ANP have been the most studied. It has been demonstrated in dogs that AVP, in concentrations 3 to 30 times those found in stressed patients, can cause vasoconstriction sufficient to produce myocardial ischemia.99 In large coronary arteries, the dilator response (via NO) likely exceeded the constrictor response.35 This was due to constriction of the small-resistance vessels. In physiologic concentrations, AVP acts primarily as an ADH with little effect on the coronary circulation. ANP can cause endothelium-dependent coronary dilation but is not known to have significant vascular effects in physiologic concentrations.100
Angiotensin-converting enzyme (ACE) is present on vascular endothelium and converts angiotensin I to angiotensin II (AII), which causes coronary vasoconstriction. AII also facilitates release of norepinephrine from presynaptic adrenergic nerve terminals. ACE inactivates bradykinin, which can attenuate vasoconstriction via NO stimulation. Thus, ACE inhibition can reduce coronary tone by suppressing AII formation and degrading bradykinin, and perhaps also by decreasing norepinephrine release. Despite these theoretical considerations, ACE inhibition has not been shown to be of benefit in human myocardial ischemia other than through control of afterload.101
PGI2 and TxA2 are synthesized from arachidonic acid in a reaction catalyzed by cyclooxygenase. PGI2 is synthesized in the vascular endothelium and, in addition to inhibiting platelet aggregation, induces vasodilation (see Endothelium-Derived Relaxing Factors section earlier in this chapter). TxA2 is mainly synthesized in platelets and causes platelet aggregation and vasoconstriction in the presence of damaged vascular endothelium. In response to TxA2, the intact endothelium releases NO, causing both vasodilation and platelet disaggregation, mechanisms to maintain patency of normal vessels (see Endothelial Inhibition of Platelets section earlier in this chapter). Unlike platelets, the vascular endothelium can synthesize proteins de novo, and thus cyclooxygenase acetylation by aspirin administration has a lesser effect in reducing vascular PGI2 than platelet TxA2
[/not-level-membership-for-anesthesiology-category]
