Control of Ventilation
After reading this chapter, you will be able to:
• Explain how the medullary respiratory center generates the basic breathing pattern
• Describe how the medullary respiratory neurons and pontine centers interact
• Explain how various reflexes and receptors affect ventilation, including the Hering-Breuer inflation reflex, J-receptors, proprioceptors, and muscle spindles
• Show how carbon dioxide (CO2) indirectly stimulates the medullary chemoreceptors
• Explain why high arterial carbon dioxide pressure (PaCO2) more readily stimulates the central chemoreceptors than high arterial levels of metabolically produced fixed acid
• Explain why PaCO2 is a more appropriate controller of ventilation than arterial oxygen pressure (PaO2)
• Explain why hypoxemia plays a more important role in regulating ventilation in patients with chronically high PaCO2 than patients with normal PaCO2
• Differentiate between the immediate and chronic effects of high altitude on ventilation and explain why they occur
• Describe two mechanisms whereby oxygen administration might induce hypercapnia in patients who have severe chronic obstructive pulmonary disease
• Explain how PaCO2 affects the cerebral circulation and intracranial pressure
Medullary Respiratory Center
Rhythmic neural impulses responsible for ventilation originate in the medulla oblongata of the brainstem (Figure 11-1). Animal experiments show that transection of the brainstem just below the medulla (see level IV, Figure 11-1) stops all ventilatory activity. However, rhythmic breathing continues after the brainstem is transected just above the pons (see level I, Figure 11-1).
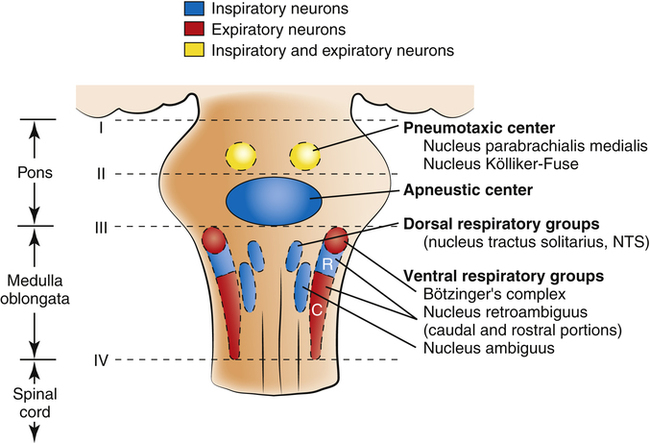
Physiologists previously thought that separate inspiratory and expiratory neuron “centers” in the medulla were responsible for the cyclical pattern of breathing. It was believed that inspiratory and expiratory neurons fired by self-excitation and that they mutually inhibited one another. It is now known that inspiratory and expiratory neurons are anatomically intermingled and do not necessarily inhibit one another.1 These neurons are widely dispersed in the medulla; the dorsal respiratory groups (DRG) contain mainly inspiratory neurons, whereas the ventral respiratory groups (VRG) contain intermingled inspiratory and expiratory neurons.
Dorsal Respiratory Groups
As shown in Figure 11-1, the DRG consist of mainly inspiratory neurons located bilaterally in an area called the nucleus of the tractus solitarius. These neurons send impulses to the phrenic and external intercostal motor nerves in the spinal cord, providing the main stimulus for inspiration.1 Many DRG nerves extend into the VRG, but few VRG fibers extend into the DRG. Thus, reciprocal inhibition is an unlikely explanation for rhythmic, spontaneous breathing.1 The DRG consists of two neuron populations, one of which is inhibited by deep lung inflation (causing cessation of inspiratory effort), whereas the other is excited by lung inflation (causing continued inspiratory effort).1 These neurons are involved in the Hering-Breuer and Head’s reflexes described later in this chapter.
Ventral Respiratory Groups
VRG neurons are located bilaterally in the medulla in two separate nuclei that contain both inspiratory and expiratory neurons (see Figure 11-1). Some inspiratory VRG neurons send motor impulses through the vagus nerve to the laryngeal and pharyngeal muscles, abducting the vocal cords and increasing the diameter of the glottis. Other VRG inspiratory neurons transmit impulses to the diaphragm and external intercostal muscles. Still other VRG neurons have mostly expiratory discharge patterns and send impulses to the internal intercostal and abdominal expiratory muscles. The most rostral (toward the head) part of the VRG, Bötzinger’s complex (see Figure 11-1), contains the only expiratory neurons known to inhibit the inspiratory VRG and DRG impulses.2
Respiratory Rhythm Generation
The exact origin of the basic rhythmic pattern of breathing is unknown; no single group of pacemaker cells have been identified. The Bötzinger complex and a structure located between it and the VRG, the pre-Bötzinger complex, are thought to be responsible for rhythmic breathing.2 Two predominant theories of rhythm generation are the pacemaker hypothesis and the network hypothesis. The pacemaker hypothesis holds that certain medullary cells have intrinsic pacemaker properties—that is, rhythmic self-exciting characteristics—and that these cells drive other medullary neurons. The network hypothesis suggests that rhythmic breathing is the result of a particular pattern of interconnections between neurons dispersed throughout the rostral VRG, the pre-Bötzinger complex, and the Bötzinger complex. This hypothesis assumes that certain populations of inspiratory and expiratory neurons inhibit one another and that one of the neuron types fires in a self-limiting way, such that it becomes less responsive the longer it fires. There is no definitive proof of either hypothesis; the precise origin of respiratory rhythm generation remains elusive.
Inspiratory Ramp Signal
The dorsal and ventral inspiratory neurons do not send an abrupt burst of impulses to the inspiratory muscles; instead, their firing rate increases gradually after expiration ceases, creating a smoothly increasing ramp signal (Figure 11-2). This ramp signal leads to a progressively stronger contraction of inspiratory muscles, smoothly and gradually inflating the lungs instead of filling them in an abrupt inspiratory gasp. During exercise, various peripheral reflexes and receptors influence the medullary neurons, steepening the inspiratory ramp signal and filling the lungs more rapidly.
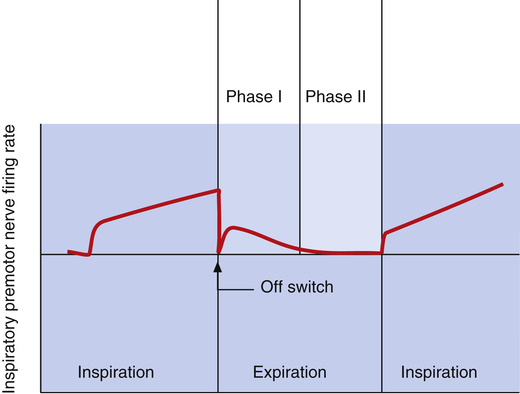
As the inspiratory ramp signal strengthens, inhibitory neurons begin to fire with increasing frequency.2 After approximately 2 seconds, these restraining impulses become strong enough to switch off the inspiratory signal abruptly. Expiration proceeds for about 3 seconds.3 As expiration begins, inspiratory neurons fire briefly, “braking” the early phase of expiration by maintaining some inspiratory muscle tone (see Figure 11-2). Inspiratory neuronal activity completely stops in the last phase of expiration. The inhibitory neurons that switch off the inspiratory ramp arise from the pneumotaxic center and pulmonary stretch receptors, which are discussed in the next section.
Pontine Centers
If the brainstem is transected above the medulla (see Figure 11-1, level III), spontaneous respiration continues, although in a more irregular pattern; thus, the pons promotes rhythmic breathing by modifying the output of the medullary centers. Figure 11-1 shows two groups of neurons in the pons: (1) the apneustic center and (2) the pneumotaxic center.
Apneustic and Pneumotaxic Centers
The two respiratory centers in the pons (apneustic and pneumotaxic) have been identified through animal brain transection experiments. If the brainstem is severed at the midpons level (see Figure 11-1, level II), and the vagus nerves connecting this area with the lower parts of the brain and spinal cord are also cut, a breathing pattern called apneusis results (Figure 11-3). Apneusis consists of prolonged inspiratory gasps interrupted by occasional expirations. The apneustic center is thought to be in a region in the lower pons, but it has never been anatomically identified with certainty.2 Its existence and function can be demonstrated only if its connections to the higher pneumotaxic center are severed and the vagus nerves are cut. Under these circumstances, the area in the lower pons (i.e., the apneustic center) sends signals to the DRG neurons, preventing the inspiratory ramp signal from being switched off.3 Apparently, vagal and pneumotaxic center impulses hold the apneustic center’s stimulatory effect on DRG neurons in check because apneusis does not occur if the vagus is left intact (see Figure 11-3).
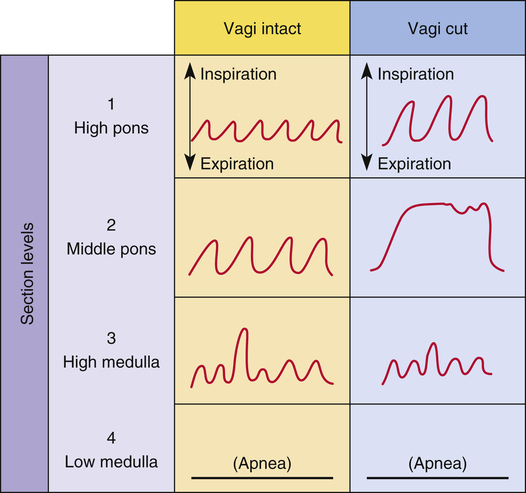
The pneumotaxic centers are bilateral groups of neurons in the upper pons (see Figure 11-1). These centers control the off-switch point of the DRG’s inspiratory ramp signal, sending inhibitory impulses to medullary inspiratory neurons; thus, pneumotaxic signals control the length of inspiration. Strong pneumotaxic signals shorten the inspiratory time and increase the respiratory rate. Weak signals produce prolonged inspiration and large tidal volumes. The primary function of the pneumotaxic center is to limit inspiration and hold apneustic center impulses in check. The exact nature of the interaction between the apneustic and pneumotaxic centers remains poorly understood; they apparently work together to control the depth of inspiration.3 The effect of the pontine centers on the lower medullary centers seems to be fine-tuning the respiratory pattern because the transection of the brainstem between the pons and medulla (see Figures 11-1, level III, and 11-3) results in an irregular breathing pattern.1
Reflex Control of Breathing
Hering-Breuer Inflation Reflex
The Hering-Breuer inflation reflex, described by Hering and Breuer in 1868, is generated by stretch receptors located in smooth muscle of large and small airways. These receptors are called slowly adapting receptors because their activity continues as long as the stimulus persists (see Chapter 2). When stretched, these receptors send inhibitory impulses through the vagus nerve to the DRG neurons, stopping further inspiration. Thus, the Hering-Breuer reflex has an effect similar to that of the pneumotaxic center.3 The Hering-Breuer reflex in adults is activated only at large tidal volumes (≥800 to 1000 mL) and is apparently not an important control mechanism in quiet breathing. However, this reflex is important in regulating the respiratory rate and depth during moderate to strenuous exercise.2
Diseases associated with low lung compliance (i.e., strong elastic retractive forces) may stimulate the inflation stretch receptors by increasing the mechanical stress acting on the airways during inspiratory efforts.2 This stimulation tends to increase the respiratory rate because it sends an early inhibitory signal, shortening the inspiratory phase.
Various combinations of respiratory rate and tidal volume can produce the level of ventilation needed to meet metabolic gas exchange demands, but there is always a specific combination that results in the least work. The information stretch receptors send to the medullary centers, which can be consciously sensed, probably helps to establish this optimal combination. The sensation of dyspnea is thought to be produced when the medullary centers demand a level of ventilation greater than the actual ventilation achieved.2
Hering-Breuer Deflation Reflex
Hering and Breuer also observed that sudden collapse of the lung stimulates strong inspiratory efforts and increases the respiratory rate. This deflation reflex may be the result of decreased stretch receptor activity, or it may be caused by the stimulation of other receptors, such as the irritant receptors and J-receptors (discussed later). Although it is unclear which receptors are involved, it is clear that the vagus nerve is the pathway (as it is for the Hering-Breuer reflex) and that the effect is hyperpnea.1 The deflation reflex is probably responsible for the hyperpnea observed with pneumothorax.
Head’s Paradoxical Reflex
In 1889, Head observed that if the Hering-Breuer reflex is partially blocked by cooling the vagus nerve, lung hyperinflation causes a further increase in inspiratory effort, opposite of the Hering-Breuer reflex. The receptors for this paradoxical reflex are called rapidly adapting receptors because they stop firing promptly after a volume change occurs. Head’s reflex may help maintain large tidal volumes during exercise and may be involved in periodic deep sighs during quiet breathing. Periodic sighs help prevent alveolar collapse or atelectasis. This reflex also may be involved in stimulating the first breaths of a newborn infant.1
J-Receptors
C-fibers in the lung parenchyma near pulmonary capillaries are called juxtacapillary receptors, or J-receptors (see Chapter 1). They are stimulated by alveolar inflammatory processes (pneumonia), pulmonary vascular congestion (congestive heart failure), and edema. J-receptor stimulation causes rapid, shallow breathing; a sensation of dyspnea; and expiratory narrowing of the glottis. Expiratory narrowing of the glottis causes grunting on expiration, especially in infants.
Peripheral Proprioceptors
Proprioceptors (positional sensors) in muscles, tendons, and joints, and pain receptors in muscles and skin send stimulatory impulses to the medullary centers, increasing inspiratory activity and hyperpnea.2 For this reason, moving the limbs, slapping the skin, and other painful stimuli stimulate ventilation in patients who have respiratory depression. Splashing cold water on the skin has a similar effect.
Proprioceptors in joints and tendons may be important in initiating and maintaining increased ventilation at the beginning of exercise. Passive limb movement around a joint increases the breathing rate in anesthetized animals and unanesthetized humans.3
Muscle Spindles
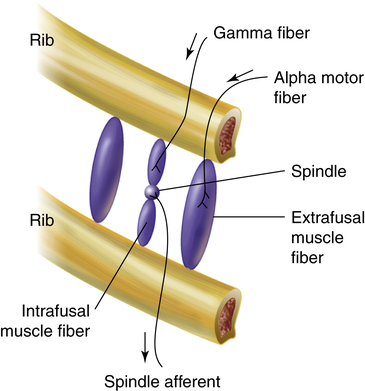
Muscle spindles in the diaphragm and intercostal muscles are part of a reflex arc that helps the muscles adjust to an increased load. Muscle spindles are stretch-sensing elements located on intrafusal muscle fibers, which are arranged in parallel with the main extrafusal muscle fibers (Figure 11-4). The extrafusal skeletal muscle fibers that elevate the ribs (see Figure 11-4) are innervated by different motor fibers (alpha motor fibers) than the intrafusal spindle fibers (gamma motor fibers). As the main extrafusal muscle fiber and the intrafusal fibers contract together in parallel, the spindle-sensing element stretches and sends impulses over spindle afferent nerves directly to the spinal cord (see Figure 11-4). The number of impulses sent is proportional to the spindle’s stretch. The spindle’s afferent (sensory) nerve synapses directly with the same alpha motor neuron in the spinal cord that sends motor impulses back to the main extrafusal muscle fiber; this creates a single synapse reflex arc. When stimulated by the spindle’s afferent nerve signal, the alpha motor neuron sends signals to only the main extrafusal muscle fibers, causing them to contract with greater force, which unloads and shortens the adjacent intrafusal muscle fibers. As a result, the stretch-sensitive spindle is unloaded, and its impulses cease. In this way, inspiratory muscle force automatically adjusts to the load imposed by decreased lung compliance or increased airway resistance.
Chemical Control of Ventilation
the medulla respond to H+, which normally arises from the reaction between dissolved CO2 and H2O in the cerebrospinal fluid (CSF). Peripherally located chemoreceptors in the fork of the common carotid arteries and the aortic arch are also sensitive to H+ and thus are indirectly sensitive to CO2. In addition, peripheral chemoreceptors are indirectly sensitive to hypoxemia because hypoxemia increases their sensitivity to hydrogen ions.2
Central (Medullary) Chemoreceptors
Hydrogen ions, not CO2 molecules, stimulate highly responsive chemosensitive nerve cells, located bilaterally in the medulla. Nevertheless, these central chemoreceptors are extremely responsive to CO2 because the [H+] surrounding them is dependent on the reaction between CO2 and H2O in their local environment. The chemoreceptors are not in direct contact with arterial blood. Instead, they are bathed in the cerebral spinal fluid, separated from the blood by a semipermeable membrane called the blood-brain barrier. This membrane is almost impermeable to H+ and HCO3−, but it is freely permeable to CO2. When the PaCO2 rises, CO2 diffuses rapidly through the blood-brain barrier into the CSF. In the CSF, CO2 reacts with H2O to form hydrogen ions (Figure 11-5); this stimulates the central chemoreceptors, which in turn stimulate the medullary inspiratory neurons. Because CSF contains no protein buffers, CO2 diffusion from the blood into the CSF increases [H+] almost instantly, exciting the central chemoreceptors within seconds. Through this mechanism, alveolar ventilation increases by approximately 2 to 3 L/min for each millimeter of mercury increase in PaCO2.4 In this indirect fashion, PaCO2 is the principal minute-to-minute stimulus for ventilation mediated through the central chemoreceptors.
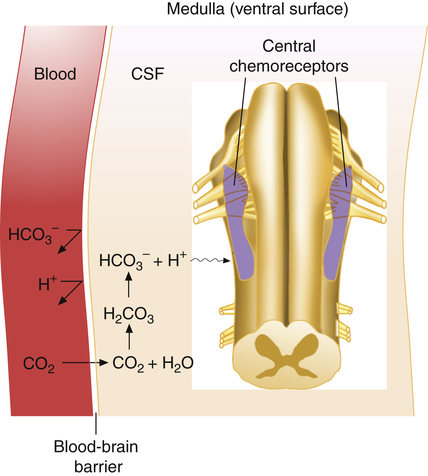
Figure 11-6 compares the ventilatory response to increased PaCO2 with the ventilatory response to increased arterial [H+] arising from fixed acid accumulation (i.e., acidemia from a nonrespiratory cause). A great change in ventilation occurs between PaCO2 values of 40 mm Hg and 80 mm Hg compared with a relatively small change in ventilation caused by a fixed acid–induced fall in arterial pH from 7.40 to 7.00 (see Figure 11-6).
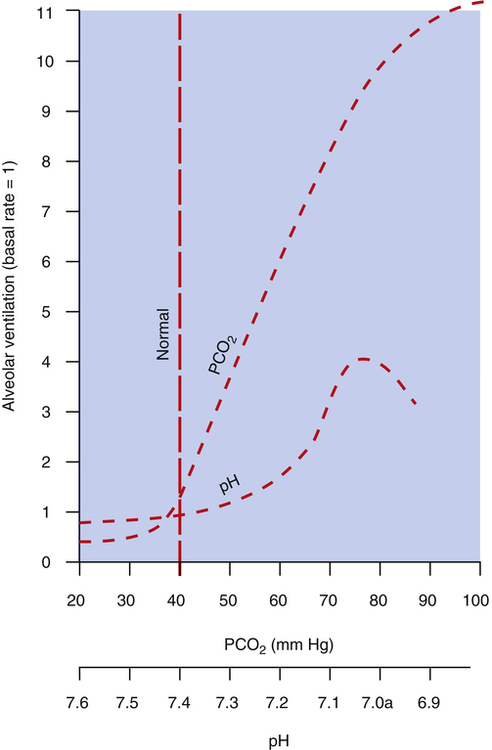
The stimulatory effect of CO2 on the central chemoreceptors gradually declines over 1 to 2 days because of renal compensatory responses. The kidneys increase blood bicarbonate ion [HCO3−] concentration in response to respiratory acidosis (see Chapter 10), which returns blood pH to the normal range. The increased number of bicarbonate ions in the blood eventually diffuses across the blood-brain barrier into the CSF where they buffer H+ and bring the CSF pH back to normal; this removes the stimulus to the chemoreceptors, and ventilation decreases. Although an increase in PaCO2 has an extremely powerful effect on ventilation, its effect is greatly weakened after 1 or 2 days of adaptation.
Unimportance of Oxygen as a Primary Controller of Ventilation
Although it would seem that oxygen should play an important role in controlling ventilation, oxygen molecules have no effect on the medullary chemoreceptors. Ventilation does not control arterial PO2 or hemoglobin oxygen saturation with any degree of precision. If an individual’s normal resting ventilation is cut in half, PaO2 decreases from 100 mm Hg to about 60 mm Hg, which still produces an oxygen hemoglobin saturation of about 90% (see Chapter 8). If ventilation then increases, and a normal PaO2 of 100 mm Hg is restored, oxyhemoglobin saturation increases to about 98%; in other words, an increase in ventilation that raises the PaO2 from 60 mm Hg to 100 mm Hg adds only marginally more oxygen to the blood. Even maximal hyperventilation cannot raise the PaO2 higher than 125 to 130 mm Hg (refer to the alveolar air equation in Chapter 7). This degree of hyperventilation (which increases the PaO2 from 100 mm Hg to 130 mm Hg) increases the hemoglobin saturation by only 1.5%. Thus, in healthy individuals under normal conditions, ventilation ranging from about half normal to many times normal produces very little change in arterial oxygen content; this is explained by the fact that the oxyhemoglobin equilibrium curve is nearly flat from 60 to 125 mm Hg. In contrast, the CO2 equilibrium curve is practically linear in the physiological range, which means that doubling the alveolar ventilation decreases the PaCO2 to half of normal, whereas cutting alveolar ventilation to half normal doubles the PaCO2. Such changes in PaCO2 drastically affect the blood pH. Thus, it makes sense that carbon dioxide (not oxygen) is the main controller of ventilation.
Peripheral Chemoreceptors
The peripheral chemoreceptors are small, highly vascular tissues known as the carotid and aortic bodies. The carotid bodies are small organs located bilaterally in bifurcations of the common carotid arteries (Figure 11-7). The aortic bodies are found in the arch of the aorta. These neural structures increase their firing rates in response to increased arterial [H+] regardless of its origin—that is, whether from fixed acid accumulation or increased CO2. The carotid bodies contain far more chemoreceptors than the aortic bodies; they send their impulses over the glossopharyngeal nerve to medullary DRG and VRG, whereas the aortic bodies send their impulses over the vagus nerve (see Figure 11-7). The carotid bodies exert much more influence over the respiratory centers than do the aortic bodies, especially with respect to arterial hypoxemia and acidemia.1
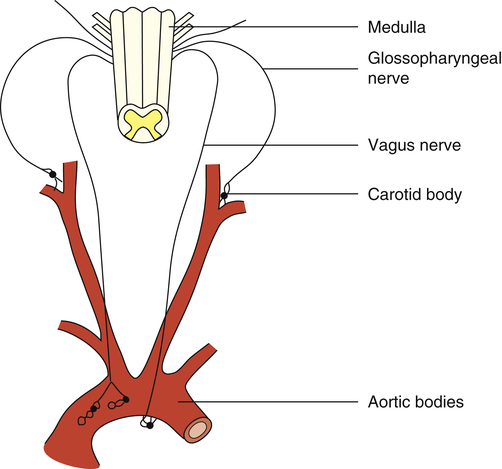
The carotid bodies receive an extremely high blood flow, about 20 times their weight each minute.3 This extremely high flow rate means the carotid bodies have little time to remove oxygen from the blood, so the venous blood leaving the carotid bodies has almost the same oxygen content as the arterial blood entering them. The carotid bodies are exposed at all times to arterial (not venous) blood, and they sense arterial (not venous) [H+].
Response to Decreased Arterial Oxygen
Traditionally, it was believed that the carotid bodies directly sensed decreased PaO2 levels, implying that arterial hypoxemia represents an independent drive to breathe—the so-called hypoxic drive. Although peripheral chemoreceptors fire more frequently in the presence of arterial hypoxemia, they do so only because hypoxemia makes them more sensitive to hydrogen ions.2 That is, in the presence of hypoxemia, carotid body sensitivity to a given [H+] increases; in this way, hypoxia increases ventilation for any given pH. Conversely, a very high PO2 (hyperoxia) decreases carotid body sensitivity to [H+]. Thus, the carotid bodies respond to arterial hypoxemia but only because hypoxia makes them more sensitive to [H+]; this implies that if the arterial [H+] is extremely low, as in severe alkalemia, hypoxemia has little effect on the carotid bodies.2 Simply stated, the ultimate effect of hypoxemia is to increase the neural firing rate of the peripheral chemoreceptors, which increases minute ventilation.
Because carotid body tissues have extremely high blood flow rates, they respond to decreased PaO2 (in the indirect way just described) rather than to an actual decrease in arterial oxygen content. That is, the carotid bodies’ extraction of oxygen from each unit of rapidly flowing blood is so small that their oxygen needs are met entirely by dissolved oxygen in the plasma. This is why conditions associated with low arterial oxygen content but normal PaO2 (e.g., anemia and carbon monoxide poisoning) do not stimulate ventilation.4
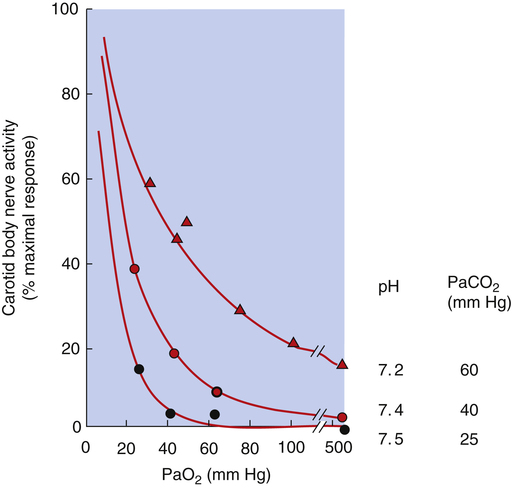
When pH and PaCO2 are normal (i.e., pH = 7.40 and PaCO2 = 40 mm Hg), the carotid bodies’ rate of nerve impulse transmission does not increase significantly until PaO2 decreases to about 60 mm Hg.4 As PaO2 decreases from 60 mm Hg to 30 mm Hg, the rate of impulse transmission increases sharply and linearly because hypoxemia makes the carotid bodies much more sensitive to a pH of 7.40 (Figure 11-8). A decrease in PaO2 from 60 mm Hg to 30 mm Hg corresponds to the sharpest decrease in oxygen content on the oxygen-hemoglobin equilibrium curve—that is, the steepest part of the curve. Arterial hypoxemia does not stimulate ventilation greatly until the PaO2 decreases to values less than 60 mm Hg; this is why ventilation increases as one ascends to high altitudes. Decreased barometric pressure at high altitudes decreases inspired and arterial PO2 values, which increases the sensitivity of peripheral chemoreceptors to hydrogen ions. However, the resulting increase in ventilation is less than expected because hyperventilation increases arterial pH by decreasing PaCO2 and subsequently, blood [H+]. The increase in arterial pH depresses the medullary respiratory center, counteracting the excitatory effect of decreased PaO2 on peripheral chemoreceptors. However, lung mechanics in certain conditions, such as advanced chronic obstructive pulmonary disease, are so deranged that the stimulatory effect of hypoxemia on ventilation fails to decrease PaCO2 regardless of the patient’s effort. In such instances, there is no alkalosis to counteract stimulatory effects of hypoxemia on ventilation.
Response to Increased Arterial Carbon Dioxide Pressure and Hydrogen Ions
Similar to the central chemoreceptors, the peripheral chemoreceptors are directly sensitive to hydrogen ions and indirectly sensitive to PCO2. That is, ventilation controls arterial [H+] by controlling PaCO2 (which generates H+ through the hydration reaction), and in this way, the peripheral chemoreceptors are sensitive to PaCO2. Increased PaCO2 increases blood [H+], directly exciting the carotid bodies and stimulating ventilation. However, this increase in ventilation is far less than the increase produced by the central chemoreceptors when they are stimulated by CO2-generated H+. The peripheral chemoreceptors account for only 20% to 30% of the ventilatory response to hypercapnia.4 However, the peripheral chemoreceptors respond to a fixed acid-induced rise in arterial [H+] about five times more quickly than the central chemoreceptors.3 In contrast to the central chemoreceptors, the carotid bodies are directly exposed to arterial blood. Thus, the initial ventilatory response to fixed acid accumulation is quick, offsetting the fact that hydrogen ions cross the blood-brain barrier with great difficulty.
As stated earlier, hypoxemia increases the sensitivity of the peripheral chemoreceptors to hydrogen ions and thus indirectly to PaCO2. Conversely, an excessively high PaO2 (hyperoxia) decreases the peripheral chemoreceptors’ PaCO2 sensitivity to almost zero.2 Thus, when PaO2 is increased, the ventilatory response to PaCO2 is mainly mediated by the central chemoreceptors, which are unaffected by hypoxemia.
The fact that the only effect of hypoxia on the peripheral chemoreceptors is to increase their sensitivity to arterial [H+] and subsequently to PaCO2 means that (1) high PO2 renders the peripheral chemoreceptors almost unresponsive to PCO2, and (2) decreased PaCO2 (low arterial [H+]) renders the peripheral chemoreceptors almost unresponsive to hypoxemia.2 Coexisting arterial hypoxemia, acidemia, and high PaCO2 (i.e., asphyxia) maximally stimulate the peripheral chemoreceptors (see Figure 11-8).
Control of Breathing during Chronic Hypercapnia
A sudden increase in PaCO2 causes an immediate increase in ventilation because CO2 rapidly diffuses from the blood into the CSF, increasing [H+] surrounding the central chemoreceptors. On the other hand, if PaCO2 increases gradually over many years, as might occur in the development of severe COPD and steadily worsening lung mechanics, the kidneys compensate by increasing the plasma bicarbonate concentration, keeping the arterial pH within normal limits (see Chapter 10). As plasma bicarbonate levels increase, these ions slowly diffuse across the blood-brain barrier, keeping CSF pH in its normal range. Because the central chemoreceptors respond to [H+], not the CO2 molecule, they sense a normal pH environment, even though the PaCO2 is abnormally high.
Oxygen-Associated Hypercapnia
CLINICAL FOCUS 11-3 Chronic Physiological Effect of Low Inspired Oxygen at High Altitude
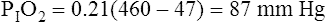
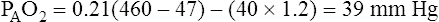
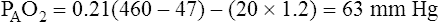
why alternative explanations have been sought is that the reduction in minute ventilation after oxygen breathing in patients with advanced COPD is not always severe enough to account for the increased PaCO2.5,6
Several investigators have suggested that oxygen breathing worsens the lungs’ ventilation-perfusion (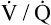 ) relationships, creating more dead space.5,6 Other investigators suggested that oxygen-induced hypercapnia is caused by the combined effects of hypoxic stimulus removal and redistribution of
) relationships, creating more dead space.5,6 Other investigators suggested that oxygen-induced hypercapnia is caused by the combined effects of hypoxic stimulus removal and redistribution of 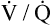 relationships in the lungs.7–9
relationships in the lungs.7–9
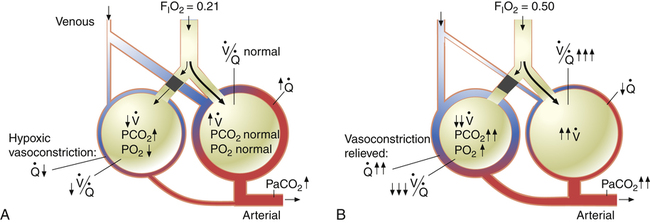
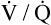 relationships may worsen in the lungs after oxygen administration because improved oxygenation abolishes hypoxic pulmonary vasoconstriction in poorly ventilated lung regions. The resulting reduction in vascular resistance shifts more blood flow to these underventilated areas, drawing blood flow away from well-ventilated regions (Figure 11-9). As poorly ventilated regions receive more blood flow, they become even less ventilated as oxygen-rich inspired gas washes out resident nitrogen gas, making alveoli more subject to absorption atelectasis; that is, oxygen may be absorbed by the pulmonary circulation more rapidly than the slowed ventilation can replenish it (notice the further decreased
relationships may worsen in the lungs after oxygen administration because improved oxygenation abolishes hypoxic pulmonary vasoconstriction in poorly ventilated lung regions. The resulting reduction in vascular resistance shifts more blood flow to these underventilated areas, drawing blood flow away from well-ventilated regions (Figure 11-9). As poorly ventilated regions receive more blood flow, they become even less ventilated as oxygen-rich inspired gas washes out resident nitrogen gas, making alveoli more subject to absorption atelectasis; that is, oxygen may be absorbed by the pulmonary circulation more rapidly than the slowed ventilation can replenish it (notice the further decreased 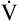 in Figure 11-9, B). As a result, inspired gas flows preferentially to the already well-ventilated alveoli (see Figure 11-9, B), increasing their
in Figure 11-9, B). As a result, inspired gas flows preferentially to the already well-ventilated alveoli (see Figure 11-9, B), increasing their 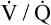 ratios. The increased
ratios. The increased 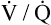 in these alveoli is further exaggerated because they lose some blood flow to the poorly ventilated regions, which now have lower vascular resistance owing to oxygen breathing. In the end, more blood flow is directed to poorly ventilated alveoli, which takes blood flow away from well-ventilated alveoli. The key point is that when already underventilated alveoli receive additional blood flow, arterial blood PCO2 increases further. These events occur without a decline in overall minute ventilation.
in these alveoli is further exaggerated because they lose some blood flow to the poorly ventilated regions, which now have lower vascular resistance owing to oxygen breathing. In the end, more blood flow is directed to poorly ventilated alveoli, which takes blood flow away from well-ventilated alveoli. The key point is that when already underventilated alveoli receive additional blood flow, arterial blood PCO2 increases further. These events occur without a decline in overall minute ventilation.
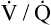
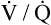
Although some investigators believe the mechanisms just described explain why oxygen administration induces hypercapnia in patients with COPD, other studies support an equally important role for oxygen suppression of the hypoxic ventilatory stimulus.7–9 These studies demonstrated that in exacerbated, chronically hypercapnic patients with COPD, oxygen administration significantly reduced their ventilation8,9 and elevated the PaCO2 level required to stimulate ventilation via the medullary chemoreceptors.7
Medullary Response to Acute Carbon Dioxide Increase in Chronic Hypercapnia
The resulting ventilatory response is depressed for both chemical and mechanical reasons: (1) the blood’s increased buffering capacity (high HCO3− level) in chronic hypercapnia prevents arterial pH from decreasing as sharply as it would in normal conditions, and (2) abnormal breathing mechanics impair the lungs’ ability to increase ventilation appropriately. To illustrate the blood’s changed buffering capacity, compare a healthy person (pH = 7.40; PaCO2 = 40 mm Hg; HCO3− = 24 mEq/L) with a chronically hypercapnic patient (pH = 7.38; PaCO2 = 60 mm Hg; HCO3− = 34 mEq/L). If PaCO2 suddenly increases by 30 mm Hg in both individuals, the healthy person’s arterial pH decreases to 7.21, and the hypercapnic patient’s pH decreases to only 7.24. (These values are calculated using the Henderson-Hasselbalch equation, assuming a 1-mEq/L increase in plasma HCO3− concentration for each acute 10-mm Hg increase in PaCO2.) Thus, the chronically hypercapnic patient’s central chemoreceptors experience less stimulation than the central chemoreceptors of the healthy person for the same increase in PaCO2. Several investigators have confirmed the reduced ventilatory response to CO2 in patients with chronic hypercapnia.10,11
Ventilatory Response to Exercise
During strenuous exercise, CO2 production and oxygen consumption may increase 20-fold.3 These increases are extremely well matched by ventilation and cardiac output. Ventilation keeps pace with CO2 production, keeping PaCO2, PaO2, and arterial pH remarkably constant. What drives the greatly increased ventilation that accompanies exercise if arterial blood gases do not change?
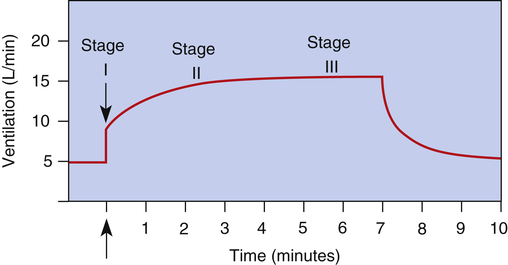
The exact mechanisms responsible for increased ventilation during exercise are not well understood. Figure 11-10 illustrates the three stages of exercise: I, onset; II, period of adjustment; III, steady state. Especially mysterious is the abrupt increase in ventilation at the onset of exercise (stage I), long before any chemical or humoral changes can occur in the body. The two predominating theories are as follows: (1) when the cerebral motor cortex sends impulses to exercising muscles, it apparently sends collateral excitatory impulses to the medullary respiratory centers, and (2) exercising limbs moving around their joints stimulate proprioceptors, which transmit excitatory impulses to the medullary centers.3 Evidence also suggests that the sudden increase in ventilation at the onset of exercise is a learned response.3 In other words, with repeated experience, the brain learns to anticipate the proper amount of ventilation required to maintain normal blood gases during exercise.
Abnormal Breathing Patterns
CLINICAL FOCUS 11-4 Oxygen-Induced Hypoventilation in Chronic Hypercapnia
What is the explanation for these blood gas results?
Given this patient’s history, chronic hypoxemia probably increases peripheral chemoreceptor sensitivity enough (via mechanisms explained in this chapter) to have an effect on his normal ventilation. His initial PaO2 of 50 mm Hg is low enough to increase significantly peripheral chemoreceptor sensitivity to H+. It stands to reason that the relatively high fractional concentration of oxygen in inspired gas (FIO2) of 0.40 played a role in decreasing peripheral chemoreceptor sensitivity because the PaO2 increased to 95 mm Hg. Whatever the mechanism might be (whether it is removal of the hypoxic stimulus, worsening of the 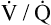 ratio, or a combination of both), this patient hypoventilated in response to oxygen therapy as is shown by the increase in PaCO2 to 80 mm Hg. This acute increase in CO2 and the accompanying acidemia depress the central nervous system, causing lethargy, which can ultimately lead to a coma. Aside from the causative mechanism, the clinically important fact is that uncontrolled oxygen therapy can lead to hypoventilation and acute acidemia in patients who are chronically hypercapnic. For this reason, spontaneously breathing, hypoxemic, chronically hypercapnic patients with COPD who are experiencing exacerbations are generally given low concentrations of oxygen (24% to 28%). Devices that produce a fixed FIO2 regardless of the patient’s ventilatory pattern should be used. Initial oxygen therapy is monitored closely through arterial blood gases to detect acute CO2 retention and acidemia. Low levels of inspired oxygen often increase the arterial oxygen content quite effectively in these patients because their blood PO2 is on the steep part of the oxygen-hemoglobin equilibrium curve (see Chapter 8). In other words, small increases in PaO2 produce relatively large increases in oxygen content. However, concern about inducing hypoventilation, hypercapnia, and acidemia is never a justifiable reason for withholding oxygen from a patient with tissue hypoxia; tissue hypoxia has a far greater potential to be life threatening than hypercapnia and acidemia. The clinician must be prepared to mechanically ventilate these patients as necessary.
ratio, or a combination of both), this patient hypoventilated in response to oxygen therapy as is shown by the increase in PaCO2 to 80 mm Hg. This acute increase in CO2 and the accompanying acidemia depress the central nervous system, causing lethargy, which can ultimately lead to a coma. Aside from the causative mechanism, the clinically important fact is that uncontrolled oxygen therapy can lead to hypoventilation and acute acidemia in patients who are chronically hypercapnic. For this reason, spontaneously breathing, hypoxemic, chronically hypercapnic patients with COPD who are experiencing exacerbations are generally given low concentrations of oxygen (24% to 28%). Devices that produce a fixed FIO2 regardless of the patient’s ventilatory pattern should be used. Initial oxygen therapy is monitored closely through arterial blood gases to detect acute CO2 retention and acidemia. Low levels of inspired oxygen often increase the arterial oxygen content quite effectively in these patients because their blood PO2 is on the steep part of the oxygen-hemoglobin equilibrium curve (see Chapter 8). In other words, small increases in PaO2 produce relatively large increases in oxygen content. However, concern about inducing hypoventilation, hypercapnia, and acidemia is never a justifiable reason for withholding oxygen from a patient with tissue hypoxia; tissue hypoxia has a far greater potential to be life threatening than hypercapnia and acidemia. The clinician must be prepared to mechanically ventilate these patients as necessary.
Cheyne-Stokes breathing, the respiratory rate and tidal volume gradually increase and then gradually decrease to complete apnea (absence of ventilation), which may last several seconds. The tidal volume and breathing frequency gradually increase again, repeating the cycle (Figure 11-11). This pattern occurs when cardiac output is low, as in congestive heart failure, which delays the blood transit time between the lungs and the brain.3 As shown in Figure 11-11, changes in respiratory center PCO2 lag behind changes in PaCO2. When increasing PaCO2 reaches the medullary neurons, ventilation is stimulated, decreasing the arterial blood PCO2. When this blood, now low in PaCO2, eventually reaches the medulla, it inhibits ventilation, but it is so slow to reach the medullary neurons that hyperventilation persists for an inappropriately long time. When the blood from the lung
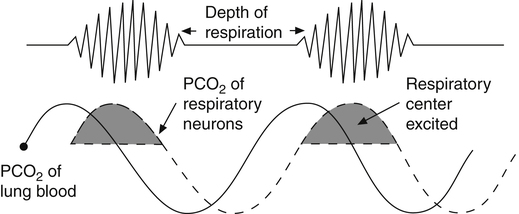
finally reaches the medullary centers, its decreased PaCO2 greatly depresses ventilation to the point of apnea. PaCO2 then increases, but an increase in medullary center PCO2 is delayed because of the low blood flow rate. The brain eventually receives the high PaCO2 signal, and the cycle is repeated. Cheyne-Stokes breathing also may be caused by brain injuries in which the respiratory centers overrespond to changes in PCO2. In this situation, slight increases in PCO2 cause exaggerated increases in ventilation, and low PCO2 may completely turn off the medullary centers.3
Biot’s breathing is similar to Cheyne-Stokes breathing except tidal volumes have essentially the same depth. The mechanism for this pattern is unclear; it occurs in patients with lesions of the pons.13
Apneustic breathing (discussed previously) also indicates damage to the pons.13 Central reflex hyperpnea (formerly known as central neurogenic hyperventilation) is continuous deep breathing driven by abnormal neural stimuli. It is related to midbrain and upper pons damage that may occur with head trauma, severe brain hypoxia, or lack of blood flow to the brain.13 Conversely, in central reflex hypopnea (formerly known as central neurogenic hypoventilation), the respiratory centers do not respond appropriately to ventilatory stimuli such as CO2. Central reflex hypopnea is associated with head trauma, brain hypoxia, and narcotic suppression of the respiratory center.2
Carbon Dioxide and Cerebral Blood Flow
CO2 plays an important role in regulating cerebral blood flow (CBF). Its effect is mediated through the formation of hydrogen ions by CO2.12 Increased PCO2 dilates cerebral vessels, increasing CBF, whereas decreased PCO2 constricts cerebral vessels and reduces CBF. In patients with traumatic brain injury (TBI), the brain swells acutely; this increases the intracranial pressure (ICP) in the rigid skull to such high levels that blood supply to the brain might be cut off, causing cerebral hypoxia (ischemia)—increased ICP may exceed cerebral arterial pressure and stop blood flow. Normal ICP is less than 10 mm Hg; a pressure of 20 mm Hg places the brain at risk for ischemia and requires active treatment to reduce the ICP.12 The brain has a high oxygen requirement; it accounts for about 20% of the entire body’s resting oxygen consumption. To meet this oxygen demand, the body maintains a constant CBF of about 50 to 60 mL per 100 g of brain tissue per minute; CBF less than 18 mL/100 g/min is the threshold for cerebral ischemia.18 To sustain the CBF, cerebral perfusion pressure (CPP) of about 60 mm Hg must be maintained. CPP is the difference between mean arterial pressure and ICP; cerebral ischemia is likely to occur if CPP decreases to less than 55 mm Hg.12
Mechanical hyperventilation has been used for many years in TBI to reduce PaCO2 and reduce CBF and ICP. In patients with TBI, a cerebral blood volume reduction of only 0.5 to 0.7 mL decreases ICP by 1 mm Hg; on the other hand, for every 1-mm Hg acute reduction in PaCO2 (between 20 mm Hg and 60 mm Hg), there is a 3% reduction in CBF.12 Thus, although an acute reduction in PaCO2 reduces ICP, it also reduces CBF and potentially causes cerebral ischemia. In any event, hypoventilation in a patient with head trauma and a preexisting elevated ICP is especially dangerous because the resulting hypercapnia dilates cerebral vessels and elevates ICP even more.