[level-membership-for-pediatrics-category]Chapter 76
Conotruncal Anomalies
Conotruncal anomalies are a group of congenital heart defects involving the outflow tract of the heart and great vessels. The conotruncal anomalies include tetralogy of Fallot (TOF), transposition of the great arteries (TGA), double-outlet ventricles, and truncus arteriosus. Interrupted aortic arch type B also is a conotruncal anomaly that will be discussed with other aortic arch anomalies (see Chapter 75).
Conotruncal abnormalities are the result of abnormal division or rotation of the primitive truncus during embryologic development.1,2 The common outlet of the embryonic univentricular heart normally undergoes a complex sequence of events to separate into the right ventricular outflow tract (RVOT) and left ventricular outflow tract (LVOT), the aorta, and the main pulmonary artery.3 Control by numerous genes and migration of the mesenchymal cells from the embryonic neural crest are required for this development.1 Mutations in a number of genes have been associated with conotruncal anomalies in humans and animal models.4
Tetralogy of Fallot
Overview: TOF is the most common cyanotic congenital heart defect. The median described incidence of TOF is 356 per 1 million live births in the United States.5 The classic manifestations include RVOT obstruction, ventricular septal defect (VSD), overriding of the aortic root above the VSD, and right ventricular (RV) hypertrophy (Fig. 76-1, A). These findings are a result of underdevelopment of the subpulmonary infundibulum,6 which is associated with anterior deviation of the conal septum. Rather than sitting between the anterior and posterior limbs of the trabecula septomarginalis (a Y-shaped bundle of muscle along the right side of the ventricular septum), the conal septum in TOF typically is fused with the anterior limb, bringing the aorta over the ventricular septum and leading to the malalignment VSD.7,8 The septal malalignment and hypertrophy of the trabeculations of the infundibular free wall result in RVOT obstruction (Fig. 76-1, B). The VSD in TOF is located between the malaligned conal septum superiorly and the muscular septum inferiorly (i.e., a conoventricular septal defect),9 and it typically is large, nonrestrictive, and subaortic. For further discussion of the trabecula septomarginalis, see the section of this chapter on double-outlet RV.
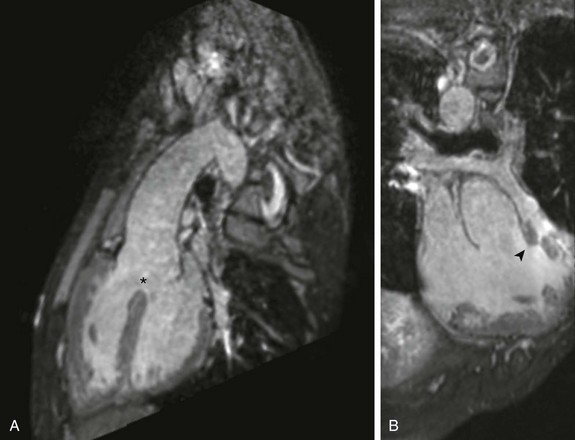
Figure 76-1 Three-dimensional steady-state free-precession imaging in an 18-year-old man with unrepaired tetralogy of Fallot.
A, Sagittal oblique projection demonstrates the aorta overriding an anterior malalignment ventricular septal defect (asterisk). B, Coronal oblique projection demonstrates the deviation of the conal septum (arrowhead), leading to subvalvar and valvar pulmonary stenosis.
The anatomic appearance of TOF varies, including TOF with pulmonary atresia and TOF with dysplastic (absent) pulmonary valve syndrome. The extent of RVOT obstruction is variable, ranging from minimal obstruction to pulmonary atresia.10 The pulmonary valve often is thickened or fused with doming leaflets and a variably hypoplastic annulus causing valvular stenosis. The size of the main and branch pulmonary arteries also varies. Patients with pulmonary valve atresia have no antegrade flow supplying the pulmonary arteries; instead, pulmonary blood flow is supplied by a patent ductus arteriosus, aortopulmonary collateral arteries, or both (e-Fig. 76-2). The central pulmonary arteries can be absent, discontinuous, or diminutive. In persons with TOF and a dysplastic pulmonary valve, congenital severe pulmonary regurgitation occurs, which often is associated with severe dilatation of the central pulmonary arteries and resultant airway compression (e-Fig. 76-3, A and B).11
e-Figure 76-2 A 2-month-old girl with tetralogy of Fallot and pulmonary atresia.
A coronal-oblique maximum intensity projection from a contrast-enhanced magnetic resonance angiogram demonstrates aortopulmonary collaterals (arrowheads). This patient also had a right aortic arch with aberrant left subclavian artery.
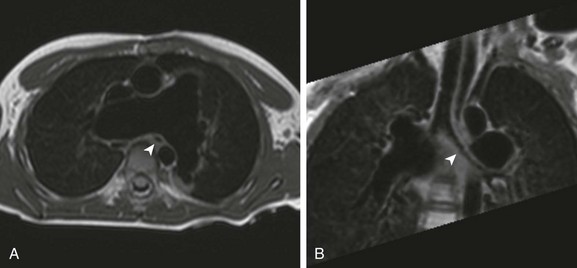
e-Figure 76-3 Turbo spin echo black blood imaging in a 5-year-old boy with tetralogy of Fallot and an absent pulmonary valve after repair with a transannular patch.
A, An axial image demonstrating markedly dilated branch pulmonary arteries. Note the caliber and position of the left mainstem bronchus (arrowhead) relative to the right pulmonary artery. B, Coronal oblique minimum intensity projection demonstrates a narrowed left mainstem bronchus (arrowhead) as it passes posterior to the proximal right pulmonary artery.
Etiology, Pathophysiology, and Clinical Presentation: Genetic abnormalities such as chromosome 22q11 deletion, which also leads to DiGeorge or velocardiofacial syndrome, may play an important role in some patients with this disease.12 Many cases are sporadic, without any specific genetic abnormality identified.
Clinical manifestations are variable. Most patients have adequate pulmonary blood flow at birth, and increasing cyanosis develops early in life.13 If RVOT obstruction is severe, right-to-left shunting occurs, resulting in cyanosis. When the obstruction is less severe, the shunting is predominantly left-to-right (so-called pink TOF); patients with this condition can present with congestive heart failure as a result of the large VSD. Patients who have TOF with pulmonary atresia are dependent on the patent ductus arteriosus or aortopulmonary collaterals for pulmonary artery blood flow. If they are dependent on the patent ductus arteriosus, an infusion of prostaglandin E1 is necessary to maintain ductal patency until a more stable supply of pulmonary blood flow can be established. In patients who have TOF with dysplastic pulmonary valve syndrome, presentation primarily may be with tracheobronchomalacia and air trapping, as well as cyanosis.
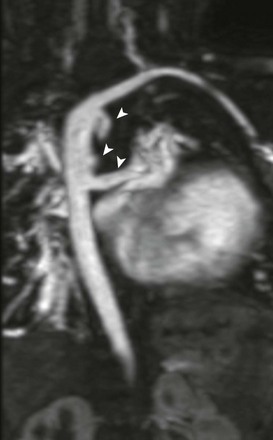
Other congenital heart anomalies can accompany TOF. These anomalies include right aortic arch (25%) and coronary artery anomalies such as abnormal origin of the left anterior descending (LAD) artery arising from the right coronary artery (5% to 6%) or dual LAD coronary arteries.14 When the LAD artery arises from the right, it passes over the RVOT before supplying its usual territory (e-Fig. 76-4).
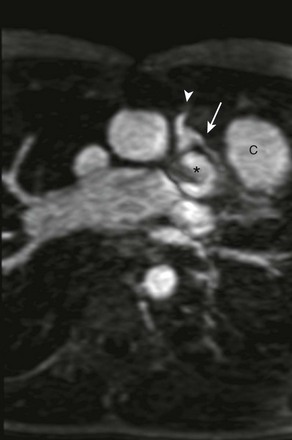
e-Figure 76-4 A 14-year-old boy with tetralogy of Fallot.
Axial oblique maximum intensity projection from a contrast-enhanced magnetic resonance angiogram shows, with a left anterior descending (LAD) coronary artery (arrow) arising from the right coronary artery (arrowhead) and traveling anterior to the right ventricular outflow tract (asterisk), preventing use of a transannular patch. A right ventricle to pulmonary artery conduit (C) was placed instead, anterior to the LAD coronary artery.
Imaging: RV hypertrophy causes uplifting of the cardiac apex. Concavity is present at the location of the main pulmonary artery because of underdevelopment, causing a “wooden shoe “or “boot shape” appearance of the heart (in French, Coeur en sabot) on frontal chest radiographs, a classic sign for TOF (Fig. 76-5). The shadow of the main pulmonary artery is absent, and pulmonary vascularity is decreased.15 Rarely, dilatation of the pulmonary artery occurs as a result of an aneurysm or asymmetric pulmonary vascularity is present as a result of differential pulmonary artery stenosis and collateralization. In the absence of a thymus, the possibility of DiGeorge syndrome should be considered. A right-sided aortic arch also can be seen on the frontal chest radiograph (see Fig. 76-5).
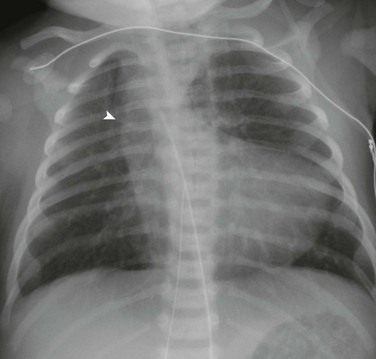
Figure 76-5 A 1-day-old girl who has tetralogy of Fallot with pulmonary atresia.
Anteroposterior chest radiograph demonstrates the upturned apex and concavity in the region of the pulmonary artery (a boot-shaped heart). Note the shadow to the right of the trachea (arrowhead), which is indicative of a right aortic arch.
Complete anatomic diagnosis in a neonate with TOF usually is made by echocardiography, with an infrequent need for cross-sectional imaging. CT or MRI typically is requested to determine pulmonary artery anatomy and sources of pulmonary blood flow, including the central pulmonary arteries, patent ductus arteriosus, and aortopulmonary collaterals. CT angiography is an effective modality for delineating pulmonary artery and collateral anatomy in these patients, but it has the disadvantage of using ionizing radiation.16 MRI also can accurately describe these anatomic details but without the risks of ionizing radiation. Turbo spin echo techniques can display vessels clearly, with the added advantage of demonstrating airway anatomy. The mainstay of MRI for these anatomic questions is three-dimensional, gadolinium contrast-enhanced magnetic resonance (MR) angiography, which is highly accurate compared with diagnostic catheterization.17
Treatment and Follow-up: Current management of TOF in most large centers is early single-stage reconstructive surgery, typically performed at 3 to 6 months of age.18 Staged reconstruction can be required if significant hypoplasia of the central pulmonary arteries is present; a palliative shunt is placed from the systemic to the pulmonary circulation to provide stable pulmonary blood flow. When pulmonary supply is from multiple aortopulmonary collaterals, a staged approach of unifocalization of collaterals to either a shunt or the central pulmonary arteries is utilized, eventually bringing the major vessels into continuity with the RV. VSD closure often is not tolerated until later in life in this subgroup of patients.18
The goal of surgical repair of TOF is to close the VSD and relieve the RVOT obstruction, thus providing unobstructed flow to the pulmonary vessels from the RV. The approach depends on the anatomy, including the degree of pulmonary valve annulus hypoplasia and anatomy of the pulmonary arteries. The entire repair can be performed transatrially, including VSD closure and division of muscle bundles within the RVOT to relieve obstruction, with no right ventriculotomy. If the pulmonary valve annulus is hypoplastic, a limited transannular patch may be needed to relieve the obstruction.18 This procedure inevitably results in severe pulmonary regurgitation. In the past this operation was performed with a large right ventriculotomy, which now usually is avoided. In patients who have TOF with pulmonary atresia, a limited transannular patch may be sufficient to relieve RVOT obstruction. With a longer segment atresia, an RV to pulmonary artery conduit may be required. In patients with an LAD coronary artery arising from the right coronary artery and crossing the RVOT, an RV to pulmonary artery conduit occasionally is necessary to avoid damaging the vessel (see e-Fig. 76-4).11
In patients with a transannular patch, a pulmonary valve replacement may be indicated later in life to remove the volume load of pulmonary regurgitation from the RV (Video 76-1). The appropriate timing of this valve replacement is the subject of intense interest.19 In patients who require an RV to pulmonary artery conduit, a conduit replacement will be needed in time because of the somatic growth of the patient. More recently, a percutaneous pulmonary valve has become available for placement within a conduit to relieve both stenosis and regurgitation (Video 76-2).
Repaired TOF is a frequent referral diagnosis for cardiac MRI. Chronic, severe pulmonary regurgitation can lead to RV pathology. Cardiac MRI is widely considered the gold standard for assessment of RV size and function, making it particularly useful in this patient population (Video 76-3). Other questions in this patient population include anatomy of the RVOT (Video 76-4), quantification of pulmonary regurgitation (Video 76-5 and Fig. 76-6, A), assessment of branch and segmental pulmonary artery anatomy, measurement of branch pulmonary artery flow, anatomy of aortopulmonary collaterals, assessment of the left ventricle (LV), and aortic valve and root pathology.
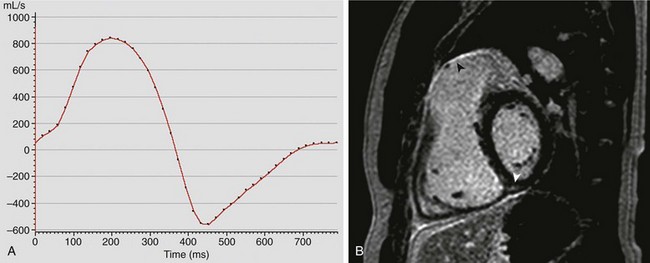
Figure 76-6 Cardiovascular magnetic resonance evaluation of a 31-year-old man with tetralogy of Fallot after he underwent repair of the lesion.
A, Graphical representation (flux vs. time) of antegrade and regurgitant flow in the main pulmonary artery. This patient had severe pulmonary insufficiency, with a regurgitant fraction of 57%. B, Late gadolinium enhancement imaging in the short axis of the ventricles demonstrates enhancement (arrowheads) along the right ventricular outflow tract and at the inferior insertion point of the interventricular septum. See Videos 76-3, 76-4, and 76-5.
These studies typically are performed with steady-state free-precession imaging in the vertical and horizontal long-axis planes and short axis of the ventricles, as well as parallel to the RVOT. Gadolinium-enhanced three-dimensional MR angiography offers high-resolution assessment of the distal pulmonary arteries and can evaluate for aortopulmonary collaterals. Velocity-encoded phase-contrast imaging assesses the ratio of pulmonary to systemic flow, differential pulmonary blood flow, and valve regurgitation. Delayed enhancement imaging reveals scarring or fibrosis in the heart (Fig. 76-6, B).1,11
Transposition of the Great Arteries
Overview: TGA is defined by discordant ventriculoarterial relations; the aorta is connected to the RV and the pulmonary artery is connected to the LV. The most common type of TGA, defined by the segmental anatomy of the heart, is {S, D, D} transposition—that is, visceral and atrial situs solitus (S), ventricular D loop (D), and dextroposition of the aortic valve (D) (see Chapter 63). The aortic valve is side by side or anterior and rightward of the pulmonary valve (Fig. 76-7, A) and usually is separated from the tricuspid valve by conal tissue. “D-looped TGA” also is acceptable terminology.1
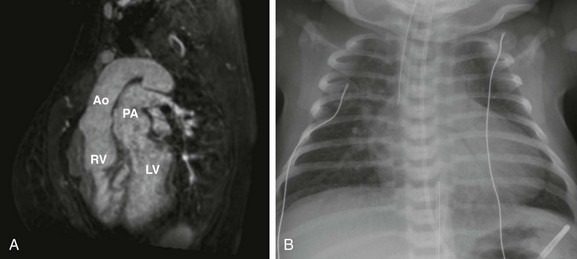
Figure 76-7 Imaging appearance of {S,D,D} transposition of the great arteries.
A, Sagittal oblique projection from three-dimensional steady-state free-precession imaging in a 29-year-old woman with {S,D,D} transposition of the great arteries, after an atrial switch procedure, demonstrates parallel outflow tracts, with the aorta (Ao) arising from the right ventricle (RV) and the pulmonary artery (PA) arising from the left ventricle (LV). B, A chest radiograph of a neonate with {S,D,D} TGA prior to repair. As a result of the parallel, anterior-posterior relationship of the great arteries, patients with {S,D,D} transposition of the great arteries have a narrow mediastinum, which, combined with cardiomegaly and increased vascular flow, produces the classic “egg on a string” appearance.
{S, D, D} TGA is the second most common cyanotic congenital heart disease.11 The median described incidence of {S, D, D} TGA is 303 per 1 million live births.5
Etiology, Pathophysiology, and Clinical Presentation: TGA usually is not associated with extracardiac anomalies or syndromes.15 It is associated with VSD in approximately 40% to 45% of cases. Other anomalies that can be seen in patients with TGA are LVOT obstruction, aortic coarctation or interrupted aortic arch, tricuspid valve abnormalities, or, less commonly, mitral valve abnormalities, leftward juxtaposition of the atrial appendages, and RV hypoplasia.20
Imaging: The radiographic appearance is variable. The classic finding of TGA by chest radiography is the “egg on a string” sign (Fig. 76-7, B), which is caused by a narrow mediastinum and the cardiac shadow. The narrow mediastinum is a result of stress-related thymic atrophy and the parallel position of the great vessels, with the pulmonary artery obscured by the aorta. The heart size varies from normal to enlarged.15
Treatment and Follow-up: In newborn infants with {S, D, D} TGA, adequacy of the communications between the pulmonary and systemic circulations is critical. Infusion of prostaglandin E1 maintains ductal patency. If the atrial septum is restrictive, urgent cardiac catheterization for balloon atrial septostomy often is required.21 In this procedure, a balloon septostomy catheter is passed across the atrial septum into the left atrium. The balloon is inflated and the catheter is sharply pulled back, fracturing the septum and enlarging the opening, allowing for greater mixing of oxygenated and deoxygenated blood.
In the early days of surgical repair of TGA, the atrial switch was used (i.e., “Mustard” or “Senning” procedures). With the atrial switch procedure, an intraatrial baffle is created with use of pericardium or native atrial tissue, which directs systemic venous return to the morphologic LV and pulmonary artery, and pulmonary venous return to the morphologic RV and aorta (Fig. 76-8, A and B).
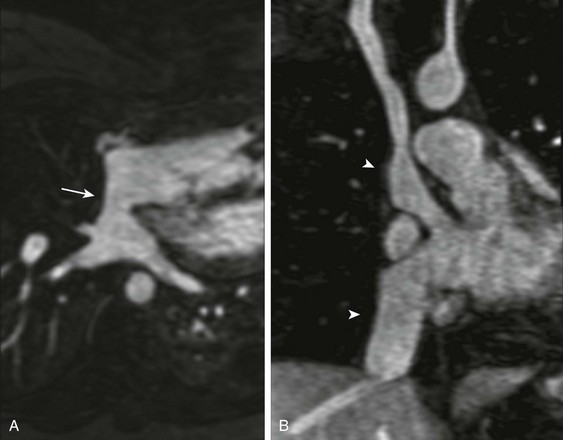
Figure 76-8 A contrast-enhanced magnetic resonance angiogram in a 29-year-old woman with {S,D,D} transposition of the great arteries, after an atrial switch procedure.
A, Transverse oblique projection demonstrating an unobstructed pulmonary venous baffle (arrow) to the tricuspid valve. B, A coronal oblique projection demonstrating unobstructed pathways from the superior and inferior vena cavae (arrowheads) to the mitral valve. See Video 76-6.
The atrial switch has been replaced by the arterial switch operation, which has been used widely in the United States since the mid to late 1980s. In this operation the ascending aorta and pulmonary artery are transected at the sinotubular junction and anastomosed to the concordant ventricle, the aorta to the left and the pulmonary artery to the right, after the pulmonary artery is relocated anterior to the aorta (the “Lecompte maneuver”) (Fig. 76-9, A). The coronary arteries are relocated from the native aorta to the neoaorta (Fig. 76-9, B).22
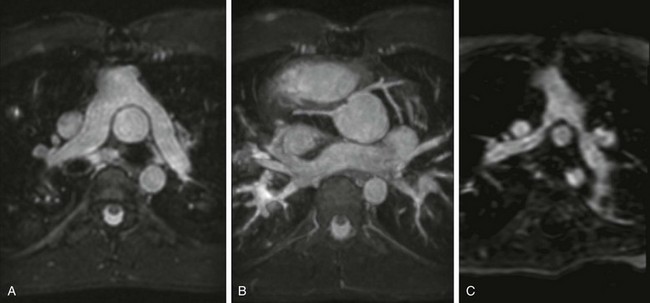
Figure 76-9 Cardiovascular magnetic resonance evaluation after arterial switch operation.
A, Transverse projection demonstrating the pulmonary artery bifurcation anterior to the ascending aorta, with no narrowing of the proximal branch pulmonary arteries. B, Maximum intensity projection in the transverse oblique plane from three-dimensional steady-state free precession in a 16-year-old boy with {S,D,D} transposition of the great arteries, after an arterial switch operation, demonstrates unobstructed proximal coronary arteries that have been translocated to the facing sinuses of the native pulmonary (neoaortic) root. C, Transverse oblique projection of a contrast-enhanced magnetic resonance angiogram in a 19-month boy with {S,D,D} transposition of the great arteries, after an arterial switch operation, demonstrates proximal narrowing of both left and right branch pulmonary arteries.
In the presence of a VSD and LVOT obstruction, a Rastelli repair can be performed,23 with baffling of the LV through the VSD to the native aortic valve, along with placement of an RV to the pulmonary artery conduit (e-Fig. 76-10).
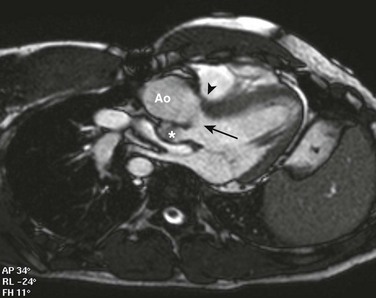
e-Figure 76-10 A 22-year-old man with {S,D,D} transposition of the great arteries, a ventricular septal defect, and pulmonary stenosis after undergoing the Rastelli procedure.
Steady-state free-precession imaging in a transverse oblique plane shows a baffle (arrowhead) redirects flow from the left ventricle via the ventricular septal defect (arrow) to the aorta (Ao). Note the hypoplastic native pulmonary valve (asterisk), which ends in a blind pouch. A right ventricle to pulmonary artery conduit was placed to reconstitute pulmonary blood flow (not shown in this image).
For the atrial switch procedure, the following early and mid-term complications may occur:
1. Systemic baffle obstruction, most commonly affecting the superior limb of the baffle at the junction of the right atrium with the superior vena cava24
2. Baffle leaks (in approximately 20% of patients) (e-Fig. 76-11)25
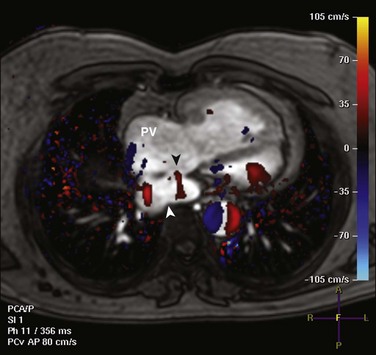
e-Figure 76-11 A 40-year-old man with {S,D,D} transposition of the great artereries after undergoing the Mustard procedure.
Axial phase contrast imaging with in-plane velocity encoding demonstrates a baffle leak, with flow from the pulmonary venous pathway to the systemic venous pathway (black arrowhead). Red denotes flow from anterior to posterior from the pulmonary venous pathway (PV) into the inferior vena cava portion of the systemic venous pathway (white arrowhead).
3. Pulmonary venous obstruction (less common) (e-Fig. 76-12)
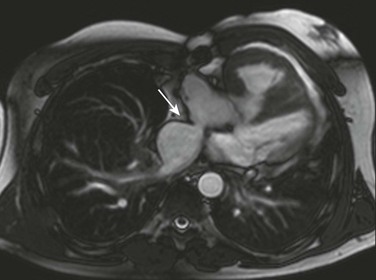
The most important complication after the atrial switch procedure, early or late, is failure of the systemic RV and tricuspid regurgitation. Less common complications include conduction and rhythm disturbances, which might require mechanical pacing or even cause sudden death.11
MRI is frequently used for assessment after the atrial switch procedure is performed. The MRI protocol is tailored for assessment of the aforementioned complications.1,11 Specifically, MRI is useful for the following assessments:
1. Evaluation of the size and function of the ventricles (Video 76-6)
2. Evaluation of the systemic and pulmonary venous pathways for obstruction and/or baffle leak(s) (e-Fig. 76-13)
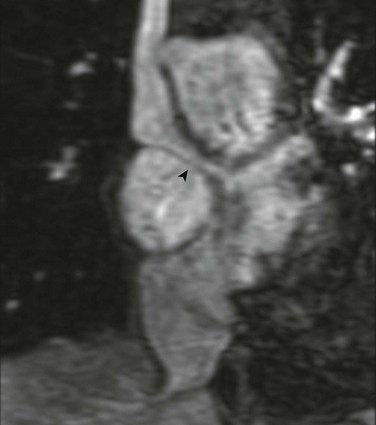
e-Figure 76-13 A 42-year-old man with {S,D,D} transposition of the great arteries after undergoing the atrial switch procedure.
Coronal oblique projection from a contrast-enhanced magnetic resonance angiogram demonstrates the typical location of narrowing of the superior vena cava pathway (arrowhead) to the mitral valve.
3. Assessment of tricuspid valve regurgitation
4. Evaluation of the LVOT and RVOT for obstruction
5. Late gadolinium enhancement for evaluation of fibrosis in the RV26
2. Stenosis at the arterial anastomotic sites, most commonly pulmonary stenosis
3. Branch pulmonary artery obstruction (see Fig. 76-9, C)
MRI also is used frequently for assessment of patients who have undergone an arterial switch procedure, with clinical questions dictated by the aforementioned potential complications1,11:
1. RV and LV size and function
2. Evaluation of the RVOT and LVOT for obstruction and for valvular regurgitation
1. LV and RV size and systolic function
2. Anatomy and potential obstruction of the LV to aorta pathway
3. Stenosis and regurgitation of the RV to pulmonary artery conduit
CT is used postoperatively in selected cases to answer anatomic questions, particularly when MRI is contraindicated. CT is particularly useful for assessment of extracardiac anatomy such as repaired aortic coarctation, arterial anastomotic sites, and branch pulmonary arteries after an arterial switch procedure is performed (e-Fig. 76-14) and venous baffles after an atrial switch procedure is performed. Functional evaluation of the heart is limited compared with MRI.
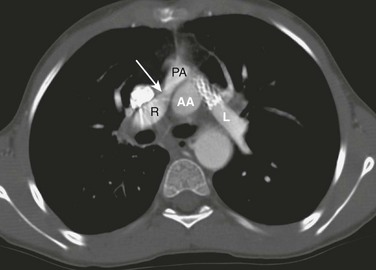
e-Figure 76-14 An axial computed tomography scan in a patient after the arterial switch procedure was performed complicated by pulmonary stenoses.
Mild right pulmonary artery (R) narrowing (arrow) developed in this patient in between the superior vena cava and the ascending aorta (AA). Previous left pulmonary artery (L) narrowing has been alleviated with a stent. PA, Pulmonary artery.
Physiologically Corrected Transposition of the Great Arteries
Overview: Physiologically corrected or {S,L,L} TGA is rare, with an estimated incidence of 30 per 1 million live births.28 The segmental anatomy of this lesion is visceral and atrial situs solitus (S), L-ventricular loop (L), and levo-malposition of the aortic valve (L) (see Chapter 63). In L-looped TGA, atrioventricular and ventriculoarterial discordance occurs. The morphologic RV receives the pulmonary venous blood via the left atrium and connects to the aorta (Fig. 76-15, A, and Video 76-7). The LV receives systemic venous blood via the right atrium and connects to the pulmonary artery. The outflow tracts are parallel and the aorta is leftward and anterior to the pulmonary artery.
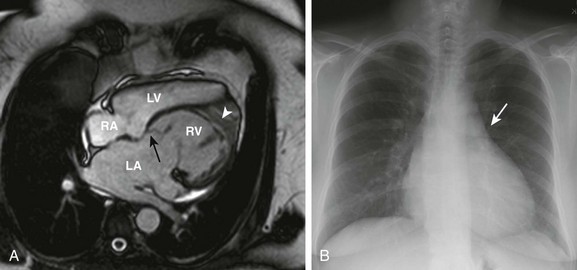
Figure 76-15 Imaging characteristics of {S,L,L} congenitally corrected transposition of the great arteries in a 56-year-old woman with no previous intervention.
A, Steady-state free-precession imaging in the four-chamber view demonstrates apical displacement of the septal insertion of the left-sided atrioventricular valve (black arrow) and a moderator band (white arrowhead) and coarse trabeculation of the left-sided ventricle, consistent with a left-sided morphologic right ventricle (RV). Note the bowing of the septum into the right-sided, morphologic left (subpulmonary) ventricle (LV). B, A posterior-anterior chest radiograph demonstrates a straightened upper left heart border (arrow) as a result of leftward malposition of the aorta. LA, Left atrium; RA, right atrium. See Video 76-7.
Etiology, Pathophysiology, and Clinical Presentation: Physiologically corrected TGA is attributed to abnormal cardiac looping during embryologic development. It can be characterized as ventricular inversion, with the systemic and pulmonary circulations in series. Most patients have associated heart lesions, but if no associated lesions are present, the patient is not cyanotic and typically is asymptomatic in early life. Patients with this lesion can present for the first time in adulthood, although this presentation is not typical.
More than 90% of patients with physiologically corrected TGA have additional anatomic abnormalities. Associated anomalies include dextrocardia, tricuspid valve abnormalities such as Ebstein anomaly, VSD (e-Fig. 76-16), RV hypoplasia, subvalvar and valvar pulmonary stenosis (Video 76-8), and conduction abnormalities, including complete heart block.1
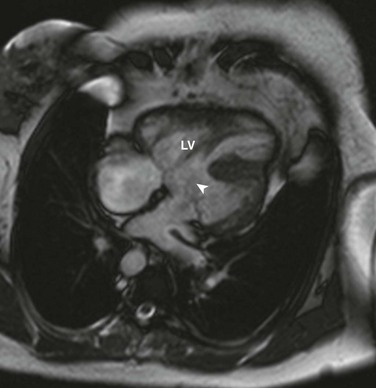
e-Figure 76-16 Steady-state free-precession imaging in a 52-year-old woman with {S,L,L} congenitally corrected transposition of the great arteries, with ventricular septal defect and pulmonary stenosis, after prior subpulmonic resection.
A four-chamber view demonstrates ventricular inversion, with a right-sided morphologic left ventricle (LV) and a large inlet ventricular septal defect (arrowhead). See Video 76-8.
Imaging: The ascending aorta is not visible on the right and the descending aorta and pulmonary artery may not be visible on the left. The vascular pedicle looks narrow, and straightening of the left heart border occurs as a result of the anterior and leftward position of the aorta. The cardiac silhouette has a “humped” appearance and is more vertical than usual (see Fig. 76-15, B).29 In the presence of systemic atrioventricular valve regurgitation and ventricular dysfunction, the heart may be enlarged. Dextrocardia or mesocardia also occurs with {S,L,L} TGA; if the chest radiograph reveals the gastric bubble on the left (abdominal situs solitus) and the apex of the heart on the right, this lesion should be suspected. Findings seen with associated cardiac lesions such as VSD and pulmonary atresia also can be detected.
Treatment and Follow-up: The early management of patients with physiologically corrected TGA who have no associated structural anomalies is controversial. Because this lesion is noncyanotic and usually asymptomatic, it can be managed medically.11,30 However, some physicians believe that earlier surgery should be performed, particularly in the presence of tricuspid valve (systemic) regurgitation.31
Until the early to mid 1990s, repair of physiologically corrected TGA was intended only to repair the pertinent associated anomalies, including VSD, pulmonary stenosis or atresia, and tricuspid valve abnormalities,22 leaving the RV as the systemic ventricle. However, in many patients with physiologically corrected transposition, systemic (RV) ventricular failure develops over time.32 Tricuspid (systemic atrioventricular) valve regurgitation also is associated with RV dysfunction and heart failure. In addition to RV failure and tricuspid regurgitation, complete heart block develops in many patients. These complications become more prevalent with increasing patient age and have led to greater interest in performing early surgery for an anatomic correction with a systemic LV. Depending on the individual anatomy, this correction can be accomplished with a combination of an atrial switch with an arterial switch (a “double switch”), or, in the presence of LVOT obstruction and a VSD, with a Rastelli procedure.22 In the absence of significant LVOT obstruction, the LV is not prepared to function at systemic pressure. A pulmonary artery band is first placed to “train” the LV to pump at higher pressures. After approximately 6 months, the complete repair is undertaken.
Double-Outlet Right Ventricle
Overview: According to the Congenital Heart Surgery Nomenclature and Database Project, double-outlet right ventricle (DORV) is “a type of ventriculoarterial connection in which both great vessels arise entirely or predominantly from the right ventricle.”33 DORV has been defined by some investigators as the degree of aortic override, with diagnosis if the aorta is more than 50% over the RV.34 However, this definition can be difficult to apply practically, because the degree of override may appear different from various anatomic planes, which can lead to lack of clarity, for example, in differentiating DORV from TOF. The absence of fibrous continuity between the aortic and mitral valve also has been referred to as a criterion for diagnosis of DORV; however, according to the Congenital Heart Surgery Nomenclature and Database Project, this finding should not be used as an absolute prerequisite for the diagnosis of DORV.33,35 Although the definition may be problematic, the median described incidence of DORV is 127 per 1 million live births.5
The anatomy and consequent physiology of a person with DORV is differentiated by the position of the VSD, the conal morphology, and the relationship of the great vessels. An understanding of these anatomic-physiologic variants is important to the surgical approach and management of the patients.1 The VSD usually is found between the limbs of the trabecula septomarginalis (Fig. 76-17). The aorta can be located rightward and posterior, directly rightward, anterior and rightward, directly anterior, or anterior and leftward of the pulmonary artery (Fig. 76-18, A). Because both vessels arise from one ventricle, abnormal positioning of the great arteries is termed “malposition” rather than “transposition.”36
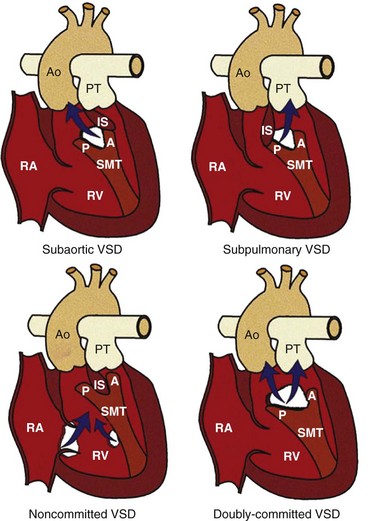
The relationship of the VSD to the conal septum and great arteries has been used as the basis for one of the most widely used clinical classifications, in which DORV is classified into four anatomic subtypes (see Fig. 76-17). This clinical classification was first described by Lev et al. in 197235,37:
1. DORV with subaortic VSD (most common): the conal septum (outlet septum) is attached to the anterior limbus of the trabecula septomarginalis
2. DORV with subpulmonary VSD: the conal septum is attached to the posterior limbus of the trabecula septomarginalis
3. DORV with doubly committed VSD: the conal septum is hypoplastic or absent
4. DORV with noncommitted VSD: the defect is located at the inlet or at the trabecular zone of the interventricular septum, usually as an atrioventricular canal–type or apical muscular defect
Etiology, Pathophysiology, and Clinical Presentation: In one review, chromosomal abnormalities, including trisomies 13 and 18 and deletion of chromosome 22q11, were the most commonly associated cytogenetic lesions.35
DORV also can be classified by physiologic subtypes, which dictate clinical presentation1:
1. VSD physiology: Subaortic VSD and no pulmonary stenosis
2. TOF physiology: Subaortic VSD and pulmonary stenosis
3. TGA physiology: Subpulmonary VSD, with or without systemic outflow obstruction
4. Single-ventricle physiology: DORV with mitral atresia, unbalanced atrioventricular canal, or severe hypoplasia of one of the ventricular sinuses (often in association with heterotaxy syndrome)
DORV is associated with several cardiac anomalies. VSD is almost ubiquitous. Obstruction to either pulmonary or systemic outflow is common. Coarctation of the aorta is common in patients with a subpulmonary VSD, particularly when aortic outflow is obstructed. Coronary artery anomalies often follow the physiology. For example, in DORV with subaortic VSD and pulmonary stenosis (TOF-type), supply of the LAD coronary artery from the right coronary artery can occur, crossing the subpulmonary outflow tract. In DORV with subpulmonary VSD (TGA-type), anomalies can vary, such as the circumflex coronary artery supplied from the right coronary artery.11
Imaging: Given the many anatomic and physiologic variants of DORV, no specific pattern is described in the chest radiograph of patients with DORV. Chest radiograph findings follow the anatomic-physiologic type of DORV, such as an enlarged heart and increased pulmonary vascular markings in persons with a VSD, decreased pulmonary vascularity, a concave main pulmonary artery shadow and upward lifted heart apex in persons with TOF, or varied findings in persons with TGA, sometimes including cardiomegaly and increased pulmonary vascularity.
Echocardiography is adequate for diagnosis and surgical planning in most newborns or infants with DORV. Cardiac MRI or CT is helpful in the evaluation of complex anomalies of the aortic arch, pulmonary arteries, aortopulmonary collaterals, and systemic or pulmonary venous anomalies that are not completely delineated by echocardiography. Considerations for CT versus MRI have been discussed previously. Assessment of the relationship between the VSD and the position of the great vessels in relation to the conal septum also can be answered by cardiac MRI (Fig. 76-18, B)1 and can be helpful in planning complex surgical baffling of the LV to the aorta. In addition, MRI can accurately assess the size of the RV when its adequacy for a two-ventricle repair is in question.
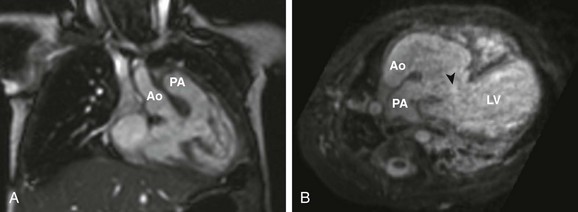
Figure 76-18 Examples of variations of double-outlet right ventricle (DORV).
A, A coronal oblique steady-state free-precession image in a 5-month old girl with DORV, side-by-side great arteries (aorta to the right), with no subaortic or subpulmonary stenosis, requiring previous pulmonary artery banding. B, Transverse oblique projection from three-dimensional steady-state free-precession imaging in a 4-month-old boy with DORV, anterior-posterior great arteries (aorta anterior to the pulmonary artery), subpulmonary ventricular septal defect (VSD) (arrowhead), and subvalvar and valvar pulmonary stenosis. This patient subsequently underwent a Rastelli procedure, with complicated baffle of the VSD to the anterior aortic valve and a right ventricle to the pulmonary artery conduit. Ao, aorta; PA, pulmonary artery; LV, left ventricle.
Treatment and Follow-up: The goal of surgical treatment of DORV is the connection of the LV to the systemic circulation and the RV to the pulmonary circulation. The preferred approach is to use an intraventricular tunnel repair, but such a repair may not be possible because of anatomic barriers, which may preclude a two-ventricle circulation. Alternative surgical procedures have been applied in these situations.33
In patients with a subaortic VSD, an intraventricular patch directing LV outflow to the aorta is the usual approach. Resection of RVOT obstruction, with or without an outflow patch, may be necessary, analogous to TOF repair. In persons with subpulmonary VSD, an intraventricular patch can be used to direct LV outflow to the pulmonary artery, which is accompanied by an arterial switch operation. Alternatively, the pulmonary artery can be closed proximally, the LV baffled through the VSD to the aorta, and a valved conduit placed between the RV and distal pulmonary artery (Rastelli procedure).36 More complex forms of DORV with heterotaxy syndrome, severe hypoplasia or absence of one of the ventricular sinuses, major straddling of an AV valve, or mitral atresia are palliated as a single ventricle.1
MRI is an important modality in the assessment of patients with repaired DORV. Questions for MRI depend on the anatomic-physiologic subtype of DORV and follow the descriptions in the previous sections for MRI of TOF and {S,D,D} TGA. In addition, assessment of the LV to aortic pathway for potential obstruction is crucial, similar to the aforementioned description for a Rastelli repair. Imaging for patients with single ventricle physiology can be found under the section regarding Fontan imaging in Chapter 74.
Truncus Arteriosus
Overview: Truncus arteriosus is defined as a single vessel arising from the heart that has a single semilunar valve, giving rise to the coronary arteries, aorta, and at least one branch of the pulmonary artery. Truncus arteriosus is uncommon, with a reported incidence of 94 per 1 million live births.5
The classification of truncus arteriosus is based on the branching pattern of the pulmonary arteries. Van Praagh and Van Praagh modified the original classification of Collett and Edwards1,38:
• Type I: The branch pulmonary arteries arise from a short main pulmonary artery
• Type II: The branch pulmonary arteries arise directly from the arterial trunk through separate orifices (Fig. 76-19, A and B)
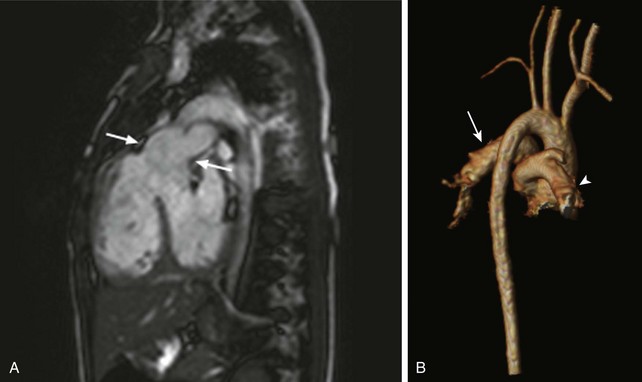
Figure 76-19 Cardiac magnetic resonance imaging of a 3-year-old girl with unrepaired truncus arteriosus, type II.
A, A sagittal oblique steady-state free-precession image demonstrates a truncal root (arrows) overriding a conoventricular ventricular septal defect, with the origin of the pulmonary arteries from the truncal root. B, A three-dimensional reconstruction (viewed posteriorly) from a contrast-enhanced magnetic resonance angiogram demonstrates the origin of the left (arrow) and right (arrowhead) branch pulmonary arteries from the truncal root.
• Type III: One branch pulmonary artery arises from the ascending segment of the trunk; collateral vessels usually supply the contralateral lung
• Type IV: Truncus arteriosus with aortic arch hypoplasia, coarctation, or interruption (usually type B, between the common carotid and subclavian arteries) (see Chapter 75).
Etiology, Pathophysiology, and Clinical Presentation: Truncus arteriosus results from failure of septation of the conotruncus into a separate aorta and pulmonary artery. It is associated with DiGeorge syndrome and chromosome 22q11 deletion.39
The semilunar valve of the truncus, that is, the truncal valve, is most often tricommissural. A bicommissural or quadricommissural valve occurs less frequently. The truncal valve often is thickened with deformed leaflets, causing stenosis or insufficiency. The conal septum is usually absent, and a malalignment VSD is present in almost all patients (>80%). An interrupted aortic arch is a common associated lesion (occurring in 11% to 14% of patients). Abnormalities of the mitral valve, coronary arteries, and pulmonary venous connections also are among the associated cardiac abnormalities.11,38
Neonates with truncus arteriosus generally are asymptomatic, while pulmonary vascular resistance remains elevated. As pulmonary vascular resistance decreases in early life, excessive pulmonary blood flow and symptoms of congestive heart failure develop. These children are at high risk for pulmonary vascular disease if they are not treated in the first 3 to 6 months of life.18
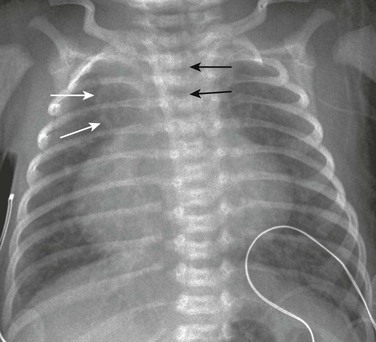
Imaging: Cardiomegaly, increased pulmonary vascularity, and right-sided aortic arch (in 30% of cases) are consistent with truncus arteriosus (e-Fig. 76-20). A depressed diaphragm and thymic atrophy also are noted.
Treatment and Follow-up: Surgical repair is undertaken within the first few weeks of life. In 1968, McGoon et al.40 described the first surgical repair of truncus arteriosus. In this description, the pulmonary arteries were separated from the aorta, and a valved homograft was placed from the RV to the pulmonary arteries. The VSD was closed with a patch to align the truncal valve with the LV. This technique, which is similar to the Rastelli procedure, remains the predominant method of repair of truncus arteriosus.18 Aortic arch interruption or coarctation is repaired at the same time. A severe valve abnormality can be addressed at the time of repair as well.41
In the absence of associated lesions, repair can be accomplished with very good survival rates in the neonatal and early infancy period.42 Newborns with truncus arteriosus and associated interrupted aortic arch have a poorer prognosis.43
Important complications after truncal repair are RV–pulmonary artery conduit stenosis or regurgitation, branch pulmonary artery stenosis, neoaortic valve (truncal) insufficiency or stenosis, residual VSD, and aortic arch obstruction. Conduits require replacement over time because of somatic growth.18,43 More recently, a percutaneous pulmonary valve is available for placement via cardiac catheterization for relief of conduit obstruction and regurgitation.
MRI and CT are useful for postoperative imaging of repaired truncus arteriosus. The anatomic and functional issues in patients who have undergone this repair are similar to those in patients with repaired TOF, specifically those with an RV to pulmonary artery conduit. Neoaortic valve dysfunction and aortic arch obstruction may require additional investigation. The postoperative MRI should address the following questions (e-Fig. 76-21)1:
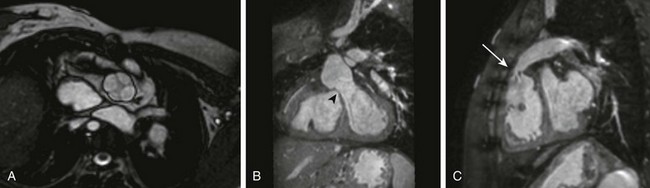
e-Figure 76-21 Cardiac magnetic resonance images demonstrating postoperative anatomy in a 13-year-old girl with truncus arteriosus after undergoing repair of the lesion.
A, A steady-state free-precession image in the short axis of the truncal root demonstrates a quadricommissural valve. B, A coronal oblique projection from three-dimensional steady-state free-precession imaging demonstrates a patch (arrowhead) closing the ventricular septal defect, connecting the left ventricle to the truncal (neoaortic) valve. C, A sagittal oblique steady-state free-precession image demonstrates the right ventricle to pulmonary artery conduit, which is narrowed proximally (arrow).
1. Quantitative assessment of LV and RV volumes, function, and mass
2. Measurements of pulmonary (conduit) and neoaortic valve regurgitation
3. Imaging of the RVOT, conduit, and pulmonary arteries
 What the Clinician Needs to Know
What the Clinician Needs to Know
• Ventricular size and systolic function
• Status of the RVOT and LVOT, including anatomic or functional obstruction, and valve regurgitation
• Presence of obstruction of surgical baffles, such as the venous baffles in an atrial switch procedure or the intraventricular baffle in a Rastelli procedure
Lai WW, Mertens LL, Cohen MS, et al, eds. Echocardiography in pediatric and congenital heart disease: from fetus to adult. Chichester, UK: Wiley-Blackwell, 2009.
Lewin, MB, Salerno, JC. Truncus arteriosus. In: Echocardiography in pediatric and congenital heart disease: from fetus to adult. Chichester, UK: Wiley-Blackwell; 2009.
Lopez, L. Double outlet ventricle. In: Echocardiography in pediatric and congenital heart disease: from fetus to adult. Chichester, UK: Wiley-Blackwell; 2009.
Jonas, RA. Comprehensive surgical management of congenital heart disease. London: Arnold; 2004.
Mertens, LL, Otto Vogt, M, Marek, J, et al. Transposition of the great arteries. In: Echocardiography in pediatric and congenital heart disease: from fetus to adult. Chichester, UK: Wiley-Blackwell; 2009.
Oechslin, E. Physiologically “corrected” transposition of the great arteries. In: Echocardiography in pediatric and congenital heart disease: from fetus to adult. Chichester, UK: Wiley-Blackwell; 2009.
Srivastava, S, Parness, IA. Tetralogy of Fallot. In: Echocardiography in pediatric and congenital heart disease: from fetus to adult. Chichester, UK: Wiley-Blackwell; 2009.
References
1. Dorfman, AL, Geva, T. Magnetic resonance imaging evaluation of congenital heart disease: conotruncal anomalies. J Cardiovasc Magn Reson. 2006;8(4):645–659.
2. Donnelly, LF, Higgins, CB. MR imaging of conotruncal abnormalities. AJR Am J Roentgenol. 1996;166(4):925–928.
3. Restivo, A, Piacentini, G, Placidi, S, et al. Cardiac outflow tract: a review of some embryogenetic aspects of the conotruncal region of the heart. Anat Rec A Discov Mol Cell Evol Biol. 2006;288(9):936–943.
4. De Luca, A, Sarkozy, A, Ferese, R, et al. New mutations in zfpm2/fog2 gene in tetralogy of Fallot and double outlet right ventricle. Clin Genet. 2011;80(2):184–190.
5. Hoffman, JI, Kaplan, S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–1900.
6. Van Praagh, R, Van Praagh, S, Nebesar, RA, et al. Tetralogy of Fallot: underdevelopment of the pulmonary infundibulum and its sequelae. Am J Cardiol. 1970;26(1):25–33.
7. Sommer, RJ, Hijazi, ZM, Rhodes, JF. Pathophysiology of congenital heart disease in the adult: part iii: complex congenital heart disease. Circulation. 2008;117(10):1340–1350.
8. Shinebourne, EA, Babu-Narayan, SV, Carvalho, JS. Tetralogy of Fallot: from fetus to adult. Heart. 2006;92(9):1353–1359.
9. Van Praagh, R, Geva, T, Kreutzer, J. Ventricular septal defects: how shall we describe, name and classify them? J Am Coll Cardiol. 1989;14(5):1298–1299.
10. Fox, D, Devendra, GP, Hart, SA, et al. When ‘blue babies’ grow up: what you need to know about tetralogy of Fallot. Cleve Clin J Med. 2010;77(11):821–828.
11. Frank, L, Dillman, JR, Parish, V, et al. Cardiovascular MR imaging of conotruncal anomalies. Radiographics. 2010;30(4):1069–1094.
12. Lu, JH, Chung, MY, Betau, H, et al. Molecular characterization of tetralogy of Fallot within DiGeorge critical region of the chromosome 22. Pediatr Cardiol. 2001;22(4):279–284.
13. Apitz, C, Webb, GD, Redington, AN. Tetralogy of Fallot. Lancet. 2009;374(9699):1462–1471.
14. Need, LR, Powell, AJ, del Nido, P, et al. Coronary echocardiography in tetralogy of Fallot: Diagnostic accuracy, resource utilization and surgical implications over 13 years. J Am Coll Cardiol. 2000;36(4):1371–1377.
15. Ferguson, EC, Krishnamurthy, R, Oldham, SA. Classic imaging signs of congenital cardiovascular abnormalities. Radiographics. 2007;27(5):1323–1334.
16. Brenner, D, Elliston, C, Hall, E, et al. Estimated risks of radiation-induced fatal cancer from pediatric CT. AJR Am J Roentgenol. 2001;176(2):289–296.
17. Geva, T, Greil, GF, Marshall, AC, et al. Gadolinium-enhanced 3-dimensional magnetic resonance angiography of pulmonary blood supply in patients with complex pulmonary stenosis or atresia: comparison with x-ray angiography. Circulation. 2002;106(4):473–478.
18. Gaca, AM, Jaggers, JJ, Dudley, LT, et al. Repair of congenital heart disease: a primer—part 2. Radiology. 2008;248(1):44–60.
19. Geva, T. Repaired tetralogy of Fallot: the roles of cardiovascular magnetic resonance in evaluating pathophysiology and for pulmonary valve replacement decision support. J Cardiovasc Magn Reson. 2011;13:9.
20. Wernovsky, G. Transposition of the great arteries. In Allen HD, Gutgesell HP, Clark EB, et al, eds.: Moss and Adams’ heart disease in infants, children, and adolescents, 6th ed, Philadelphia: Lippincott, 2001.
21. Fulton, DR, Fyler, DC. D-Transposition of the great arteries. In Keane JF, Lock JE, Fyler DC, eds.: Nadas’ pediatric cardiology, 2nd ed, Philadelphia: Saunders, 2006.
22. Gaca, AM, Jaggers, JJ, Dudley, LT, et al. Repair of congenital heart disease: a primer—part 1. Radiology. 2008;247(3):617–631.
23. Rastelli, GC, Wallace, RB, Ongley, PA. Complete repair of transposition of the great arteries with pulmonary stenosis. A review and report of a case corrected by using a new surgical technique. Circulation. 1969;39(1):83–95.
24. Bottega, NA, Silversides, CK, Oechslin, EN, et al. Stenosis of the superior limb of the systemic venous baffle following a mustard procedure: an under-recognized problem. Int J Cardiol. 2012;154(1):32–37.
25. Chin, AJ, Sanders, SP, Norwood, WI, et al. Two-dimensional echocardiographic localization of residual atrial shunts after the Senning procedure. Am J Cardiol. 1985;55(9):1238–1289.
26. Giardini, A, Lovato, L, Donti, A, et al. Relation between right ventricular structural alterations and markers of adverse clinical outcome in adults with systemic right ventricle and either congenital complete (after Senning operation) or congenitally corrected transposition of the great arteries. Am J Cardiol. 2006;98(9):1277–1282.
27. Warnes, CA, Williams, RG, Bashore, TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults with Congenital Heart Disease). Circulation. 2008;118(23):e714–e833.
28. Wallis, GA, Debich-Spicer, D, Anderson, RH. Congenitally corrected transposition. Orphanet J Rare Dis. 2011;6(1):22.
29. Warnes, CA. Transposition of the great arteries. Circulation. 2006;114(24):2699–2709.
30. Graham, TP, Jr., Bernard, YD, Mellen, BG, et al. Long-term outcome in congenitally corrected transposition of the great arteries: a multi-institutional study. J Am Coll Cardiol. 2000;36(1):255–261.
31. Bacha, E. Patients with congenitally corrected transposition of the great arteries and systemic tricuspid valve regurgitation should be referred for surgical consultation as soon as the diagnosis of regurgitation is made. J Am Coll Cardiol. 2011;57(20):2018–2019.
32. Silversides, CK, Salehian, O, Oechslin, E, et al. Canadian Cardiovascular Society 2009 Consensus conference on the management of adults with congenital heart disease: complex congenital cardiac lesions. Can J Cardiol. 2009;26(3):e98–e117.
33. Walters, HL, 3rd., Mavroudis, C, Tchervenkov, CI, et al. Congenital heart surgery nomenclature and database project: double outlet right ventricle. Ann Thorac Surg. 2000;69(4 suppl):S249–S263.
34. Kirklin, JW, Pacifico, AD, Bargeron, LM, Jr., et al. Cardiac repair in anatomically corrected malposition of the great arteries. Circulation. 1973;48(1):153–159.
35. Obler, D, Juraszek, AL, Smoot, LB, et al. Double outlet right ventricle: Aetiologies and associations. J Med Genet. 2008;45(8):481–497.
36. Weir, EK, Joffe, HS, Barnard, CN, et al. Double outlet right ventricle: Clinical and anatomical spectrum. Thorax. 1978;33(3):283–289.
37. Lev, M, Bharati, S, Meng, CC, et al. A concept of double-outlet right ventricle. J Thorac Cardiovasc Surg. 1972;64(2):271–281.
38. Van Praagh, R, Van Praagh, S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. A study of 57 necropsy cases. Am J Cardiol. 1965;16(3):406–425.
39. Goldmuntz, E, Clark, BJ, Mitchell, LE, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32(2):492–498.
40. McGoon, DC, Rastelli, GC, Ongley, PA. An operation for the correction of truncus arteriosus. JAMA. 1968;205(2):69–73.
41. Mavroudis, C, Backer, CL. Surgical management of severe truncal insufficiency: experience with truncal valve remodeling techniques. Ann Thorac Surg. 2001;72(2):396–400.
42. Brown, JW, Ruzmetov, M, Okada, Y, et al. Truncus arteriosus repair: outcomes, risk factors, reoperation and management. Eur J Cardiothorac Surg. 2001;20(2):221–227.
43. Sinzobahamvya, N, Boscheinen, M, Blaschczok, HC, et al. Survival and reintervention after neonatal repair of truncus arteriosus with valved conduit. Eur J Cardiothorac Surg. 2008;34(4):732–737.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 76
Conotruncal Anomalies
Conotruncal anomalies are a group of congenital heart defects involving the outflow tract of the heart and great vessels. The conotruncal anomalies include tetralogy of Fallot (TOF), transposition of the great arteries (TGA), double-outlet ventricles, and truncus arteriosus. Interrupted aortic arch type B also is a conotruncal anomaly that will be discussed with other aortic arch anomalies (see Chapter 75).
Conotruncal abnormalities are the result of abnormal division or rotation of the primitive truncus during embryologic development.1,2 The common outlet of the embryonic univentricular heart normally undergoes a complex sequence of events to separate into the right ventricular outflow tract (RVOT) and left ventricular outflow tract (LVOT), the aorta, and the main pulmonary artery.3 Control by numerous genes and migration of the mesenchymal cells from the embryonic neural crest are required for this development.1 Mutations in a number of genes have been associated with conotruncal anomalies in humans and animal models.4
Tetralogy of Fallot
Overview: TOF is the most common cyanotic congenital heart defect. The median described incidence of TOF is 356 per 1 million live births in the United States.5 The classic manifestations include RVOT obstruction, ventricular septal defect (VSD), overriding of the aortic root above the VSD, and right ventricular (RV) hypertrophy (Fig. 76-1, A). These findings are a result of underdevelopment of the subpulmonary infundibulum,6 which is associated with anterior deviation of the conal septum. Rather than sitting between the anterior and posterior limbs of the trabecula septomarginalis (a Y-shaped bundle of muscle along the right side of the ventricular septum), the conal septum in TOF typically is fused with the anterior limb, bringing the aorta over the ventricular septum and leading to the malalignment VSD.7,8 The septal malalignment and hypertrophy of the trabeculations of the infundibular free wall result in RVOT obstruction (Fig. 76-1, B). The VSD in TOF is located between the malaligned conal septum superiorly and the muscular septum inferiorly (i.e., a conoventricular septal defect),9 and it typically is large, nonrestrictive, and subaortic. For further discussion of the trabecula septomarginalis, see the section of this chapter on double-outlet RV.
Figure 76-1 Three-dimensional steady-state free-precession imaging in an 18-year-old man with unrepaired tetralogy of Fallot.
A, Sagittal oblique projection demonstrates the aorta overriding an anterior malalignment ventricular septal defect (asterisk). B, Coronal oblique projection demonstrates the deviation of the conal septum (arrowhead), leading to subvalvar and valvar pulmonary stenosis.
The anatomic appearance of TOF varies, including TOF with pulmonary atresia and TOF with dysplastic (absent) pulmonary valve syndrome. The extent of RVOT obstruction is variable, ranging from minimal obstruction to pulmonary atresia.10 The pulmonary valve often is thickened or fused with doming leaflets and a variably hypoplastic annulus causing valvular stenosis. The size of the main and branch pulmonary arteries also varies. Patients with pulmonary valve atresia have no antegrade flow supplying the pulmonary arteries; instead, pulmonary blood flow is supplied by a patent ductus arteriosus, aortopulmonary collateral arteries, or both (e-Fig. 76-2). The central pulmonary arteries can be absent, discontinuous, or diminutive. In persons with TOF and a dysplastic pulmonary valve, congenital severe pulmonary regurgitation occurs, which often is associated with severe dilatation of the central pulmonary arteries and resultant airway compression (e-Fig. 76-3, A and B).11
e-Figure 76-2 A 2-month-old girl with tetralogy of Fallot and pulmonary atresia.
A coronal-oblique maximum intensity projection from a contrast-enhanced magnetic resonance angiogram demonstrates aortopulmonary collaterals (arrowheads). This patient also had a right aortic arch with aberrant left subclavian artery.
e-Figure 76-3 Turbo spin echo black blood imaging in a 5-year-old boy with tetralogy of Fallot and an absent pulmonary valve after repair with a transannular patch.
A, An axial image demonstrating markedly dilated branch pulmonary arteries. Note the caliber and position of the left mainstem bronchus (arrowhead) relative to the right pulmonary artery. B, Coronal oblique minimum intensity projection demonstrates a narrowed left mainstem bronchus (arrowhead) as it passes posterior to the proximal right pulmonary artery.
Etiology, Pathophysiology, and Clinical Presentation: Genetic abnormalities such as chromosome 22q11 deletion, which also leads to DiGeorge or velocardiofacial syndrome, may play an important role in some patients with this disease.12 Many cases are sporadic, without any specific genetic abnormality identified.
Clinical manifestations are variable. Most patients have adequate pulmonary blood flow at birth, and increasing cyanosis develops early in life.13 If RVOT obstruction is severe, right-to-left shunting occurs, resulting in cyanosis. When the obstruction is less severe, the shunting is predominantly left-to-right (so-called pink TOF); patients with this condition can present with congestive heart failure as a result of the large VSD. Patients who have TOF with pulmonary atresia are dependent on the patent ductus arteriosus or aortopulmonary collaterals for pulmonary artery blood flow. If they are dependent on the patent ductus arteriosus, an infusion of prostaglandin E1 is necessary to maintain ductal patency until a more stable supply of pulmonary blood flow can be established. In patients who have TOF with dysplastic pulmonary valve syndrome, presentation primarily may be with tracheobronchomalacia and air trapping, as well as cyanosis.
Other congenital heart anomalies can accompany TOF. These anomalies include right aortic arch (25%) and coronary artery anomalies such as abnormal origin of the left anterior descending (LAD) artery arising from the right coronary artery (5% to 6%) or dual LAD coronary arteries.14 When the LAD artery arises from the right, it passes over the RVOT before supplying its usual territory (e-Fig. 76-4).
e-Figure 76-4 A 14-year-old boy with tetralogy of Fallot.
Axial oblique maximum intensity projection from a contrast-enhanced magnetic resonance angiogram shows, with a left anterior descending (LAD) coronary artery (arrow) arising from the right coronary artery (arrowhead) and traveling anterior to the right ventricular outflow tract (asterisk), preventing use of a transannular patch. A right ventricle to pulmonary artery conduit (C) was placed instead, anterior to the LAD coronary artery.
Imaging: RV hypertrophy causes uplifting of the cardiac apex. Concavity is present at the location of the main pulmonary artery because of underdevelopment, causing a “wooden shoe “or “boot shape” appearance of the heart (in French, Coeur en sabot) on frontal chest radiographs, a classic sign for TOF (Fig. 76-5). The shadow of the main pulmonary artery is absent, and pulmonary vascularity is decreased.15 Rarely, dilatation of the pulmonary artery occurs as a result of an aneurysm or asymmetric pulmonary vascularity is present as a result of differential pulmonary artery stenosis and collateralization. In the absence of a thymus, the possibility of DiGeorge syndrome should be considered. A right-sided aortic arch also can be seen on the frontal chest radiograph (see Fig. 76-5).
Figure 76-5 A 1-day-old girl who has tetralogy of Fallot with pulmonary atresia.
Anteroposterior chest radiograph demonstrates the upturned apex and concavity in the region of the pulmonary artery (a boot-shaped heart). Note the shadow to the right of the trachea (arrowhead), which is indicative of a right aortic arch.
Complete anatomic diagnosis in a neonate with TOF usually is made by echocardiography, with an infrequent need for cross-sectional imaging. CT or MRI typically is requested to determine pulmonary artery anatomy and sources of pulmonary blood flow, including the central pulmonary arteries, patent ductus arteriosus, and aortopulmonary collaterals. CT angiography is an effective modality for delineating pulmonary artery and collateral anatomy in these patients, but it has the disadvantage of using ionizing radiation.16 MRI also can accurately describe these anatomic details but without the risks of ionizing radiation. Turbo spin echo techniques can display vessels clearly, with the added advantage of demonstrating airway anatomy. The mainstay of MRI for these anatomic questions is three-dimensional, gadolinium contrast-enhanced magnetic resonance (MR) angiography, which is highly accurate compared with diagnostic catheterization.17
Treatment and Follow-up: Current management of TOF in most large centers is early single-stage reconstructive surgery, typically performed at 3 to 6 months of age.18 Staged reconstruction can be required if significant hypoplasia of the central pulmonary arteries is present; a palliative shunt is placed from the systemic to the pulmonary circulation to provide stable pulmonary blood flow. When pulmonary supply is from multiple aortopulmonary collaterals, a staged approach of unifocalization of collaterals to either a shunt or the central pulmonary arteries is utilized, eventually bringing the major vessels into continuity with the RV. VSD closure often is not tolerated until later in life in this subgroup of patients.18
The goal of surgical repair of TOF is to close the VSD and relieve the RVOT obstruction, thus providing unobstructed flow to the pulmonary vessels from the RV. The approach depends on the anatomy, including the degree of pulmonary valve annulus hypoplasia and anatomy of the pulmonary arteries. The entire repair can be performed transatrially, including VSD closure and division of muscle bundles within the RVOT to relieve obstruction, with no right ventriculotomy. If the pulmonary valve annulus is hypoplastic, a limited transannular patch may be needed to relieve the obstruction.18 This procedure inevitably results in severe pulmonary regurgitation. In the past this operation was performed with a large right ventriculotomy, which now usually is avoided. In patients who have TOF with pulmonary atresia, a limited transannular patch may be sufficient to relieve RVOT obstruction. With a longer segment atresia, an RV to pulmonary artery conduit may be required. In patients with an LAD coronary artery arising from the right coronary artery and crossing the RVOT, an RV to pulmonary artery conduit occasionally is necessary to avoid damaging the vessel (see e-Fig. 76-4).11
In patients with a transannular patch, a pulmonary valve replacement may be indicated later in life to remove the volume load of pulmonary regurgitation from the RV (Video 76-1). The appropriate timing of this valve replacement is the subject of intense interest.19 In patients who require an RV to pulmonary artery conduit, a conduit replacement will be needed in time because of the somatic growth of the patient. More recently, a percutaneous pulmonary valve has become available for placement within a conduit to relieve both stenosis and regurgitation (Video 76-2).
Repaired TOF is a frequent referral diagnosis for cardiac MRI. Chronic, severe pulmonary regurgitation can lead to RV pathology. Cardiac MRI is widely considered the gold standard for assessment of RV size and function, making it particularly useful in this patient population (Video 76-3). Other questions in this patient population include anatomy of the RVOT (Video 76-4), quantification of pulmonary regurgitation (Video 76-5 and Fig. 76-6, A), assessment of branch and segmental pulmonary artery anatomy, measurement of branch pulmonary artery flow, anatomy of aortopulmonary collaterals, assessment of the left ventricle (LV), and aortic valve and root pathology.
Figure 76-6 Cardiovascular magnetic resonance evaluation of a 31-year-old man with tetralogy of Fallot after he underwent repair of the lesion.
A, Graphical representation (flux vs. time) of antegrade and regurgitant flow in the main pulmonary artery. This patient had severe pulmonary insufficiency, with a regurgitant fraction of 57%. B, Late gadolinium enhancement imaging in the short axis of the ventricles demonstrates enhancement (arrowheads) along the right ventricular outflow tract and at the inferior insertion point of the interventricular septum. See Videos 76-3, 76-4, and 76-5.
These studies typically are performed with steady-state free-precession imaging in the vertical and horizontal long-axis planes and short axis of the ventricles, as well as parallel to the RVOT. Gadolinium-enhanced three-dimensional MR angiography offers high-resolution assessment of the distal pulmonary arteries and can evaluate for aortopulmonary collaterals. Velocity-encoded phase-contrast imaging assesses the ratio of pulmonary to systemic flow, differential pulmonary blood flow, and valve regurgitation. Delayed enhancement imaging reveals scarring or fibrosis in the heart (Fig. 76-6, B).1,11
Transposition of the Great Arteries
Overview: TGA is defined by discordant ventriculoarterial relations; the aorta is connected to the RV and the pulmonary artery is connected to the LV. The most common type of TGA, defined by the segmental anatomy of the heart, is {S, D, D} transposition—that is, visceral and atrial situs solitus (S), ventricular D loop (D), and dextroposition of the aortic valve (D) (see Chapter 63). The aortic valve is side by side or anterior and rightward of the pulmonary valve (Fig. 76-7, A) and usually is separated from the tricuspid valve by conal tissue. “D-looped TGA” also is acceptable terminology.1
Figure 76-7 Imaging appearance of {S,D,D} transposition of the great arteries.
A, Sagittal oblique projection from three-dimensional steady-state free-precession imaging in a 29-year-old woman with {S,D,D} transposition of the great arteries, after an atrial switch procedure, demonstrates parallel outflow tracts, with the aorta (Ao) arising from the right ventricle (RV) and the pulmonary artery (PA) arising from the left ventricle (LV). B, A chest radiograph of a neonate with {S,D,D} TGA prior to repair. As a result of the parallel, anterior-posterior relationship of the great arteries, patients with {S,D,D} transposition of the great arteries have a narrow mediastinum, which, combined with cardiomegaly and increased vascular flow, produces the classic “egg on a string” appearance.
{S, D, D} TGA is the second most common cyanotic congenital heart disease.11 The median described incidence of {S, D, D} TGA is 303 per 1 million live births.5
Etiology, Pathophysiology, and Clinical Presentation: TGA usually is not associated with extracardiac anomalies or syndromes.15 It is associated with VSD in approximately 40% to 45% of cases. Other anomalies that can be seen in patients with TGA are LVOT obstruction, aortic coarctation or interrupted aortic arch, tricuspid valve abnormalities, or, less commonly, mitral valve abnormalities, leftward juxtaposition of the atrial appendages, and RV hypoplasia.20
Imaging: The radiographic appearance is variable. The classic finding of TGA by chest radiography is the “egg on a string” sign (Fig. 76-7, B), which is caused by a narrow mediastinum and the cardiac shadow. The narrow mediastinum is a result of stress-related thymic atrophy and the parallel position of the great vessels, with the pulmonary artery obscured by the aorta. The heart size varies from normal to enlarged.15
Treatment and Follow-up: In newborn infants with {S, D, D} TGA, adequacy of the communications between the pulmonary and systemic circulations is critical. Infusion of prostaglandin E1 maintains ductal patency. If the atrial septum is restrictive, urgent cardiac catheterization for balloon atrial septostomy often is required.21 In this procedure, a balloon septostomy catheter is passed across the atrial septum into the left atrium. The balloon is inflated and the catheter is sharply pulled back, fracturing the septum and enlarging the opening, allowing for greater mixing of oxygenated and deoxygenated blood.
In the early days of surgical repair of TGA, the atrial switch was used (i.e., “Mustard” or “Senning” procedures). With the atrial switch procedure, an intraatrial baffle is created with use of pericardium or native atrial tissue, which directs systemic venous return to the morphologic LV and pulmonary artery, and pulmonary venous return to the morphologic RV and aorta (Fig. 76-8, A and B).
Figure 76-8 A contrast-enhanced magnetic resonance angiogram in a 29-year-old woman with {S,D,D} transposition of the great arteries, after an atrial switch procedure.
A, Transverse oblique projection demonstrating an unobstructed pulmonary venous baffle (arrow) to the tricuspid valve. B, A coronal oblique projection demonstrating unobstructed pathways from the superior and inferior vena cavae (arrowheads) to the mitral valve. See Video 76-6.
The atrial switch has been replaced by the arterial switch operation, which has been used widely in the United States since the mid to late 1980s. In this operation the ascending aorta and pulmonary artery are transected at the sinotubular junction and anastomosed to the concordant ventricle, the aorta to the left and the pulmonary artery to the right, after the pulmonary artery is relocated anterior to the aorta (the “Lecompte maneuver”) (Fig. 76-9, A). The coronary arteries are relocated from the native aorta to the neoaorta (Fig. 76-9, B).22
Figure 76-9 Cardiovascular magnetic resonance evaluation after arterial switch operation.
A, Transverse projection demonstrating the pulmonary artery bifurcation anterior to the ascending aorta, with no narrowing of the proximal branch pulmonary arteries. B,
[/not-level-membership-for-pediatrics-category]
