Chapter 4 Connective Tissues of the Subendothelium
Varieties of Blood Vessels and Their Connective Tissue
The evolutionary transition from an open to a closed circulatory system is clearly reflected in the architecture of the blood vessels.1,2 The size and anatomical organization of individual vessels vary with their specific locations and functions in the body. The major vessels that carry blood directly from the heart are capable of storing and releasing large amounts of energy during the cardiac cycle. As a result, the walls of large arteries are relatively thick and more elastic to allow their expansion and contraction in response to the systolic and diastolic cycles of the heart. Without such elasticity, the intense surge in pressure as blood is ejected from the heart would inhibit its emptying, and the pressure in the vessels would fall too low for the heart to refill. The elasticity of large arteries enables them to store a portion of the stroke volume with each systole and discharge that volume with diastole. Thus, the unique structure of large arteries allows the flow of blood from the heart to be continuous, smooth, and efficient.
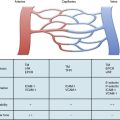
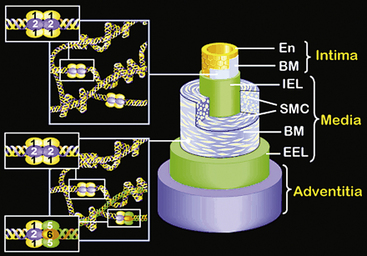
The walls of the large arteries contain three identifiable layers. The luminal surface of arteries contains a single layer of polygonal ECs connected by gap junctions. This cell layer rests on a basement membrane, which in turn is supported by a network of elastic fibers in a fenestrated plate called the internal elastic lamina. This region of the wall is called the tunica intima. The middle layer, called the tunica media, represents the bulk of the vessel wall, contains few elastic fibers but has a large number of VSMCs, with their long axes perpendicular to the lumen axis.3 Smooth muscle cells residing in the tunica media synthesize the major components of ECM that ultimately define the mechanical properties of the vessel. The extracellular space contains a variable mixture of collagen fibers in a continuous sheath adjacent to the elastic fibers. The external elastic lamina separates the medial and adventitial layers of the vessel wall. The outermost layer of the vessel wall, the tunica adventitia, consists primarily of collagen-rich ECM and the vasa vasorum, a network of vessels that supplies nutrients and O2 to the outer portion of arterial walls. Although the unique anatomy and high collagen content of the tunica adventitia help prevent arterial rupture at extremely high pressures, the adventitia is highly susceptible to vascular inflammation.
The walls of smaller arteries are intermediate in size. The tunica intima is relatively thin, as is the medial layer. The tunica adventitia of small arteries usually contains more densely packed collagen fibers arranged longitudinally along the vessel axis. Arterioles have simpler walls; their EC layer is surrounded by VSMCs, and the adventitia is smaller and more pliable compared with those of larger arteries.1,3 Capillaries adjoining the arterioles are surrounded by a few SMCs that control the amount of blood passing through them. The walls of arterial and venous capillaries are lined with flat ECs surrounded by a basement membrane; a discontinuous sheath of pericytes and a fibrous reticulum, made primarily of type III collagen, are attached to the basement membrane. The walls of venules also contain a reticular network of collagen fibers derived from type III collagen, along with smaller quantities of type I collagen fibers.
Vascular Morphogenesis and Extracellular Matrix
The preceding overview underscores the striking structural and phenotypic diversity of different branches of the vascular tree. Therefore it is not surprising that the vascular ECM displays similar complexity depending on its location in the vasculature.2–5 This caveat notwithstanding, all vascular ECM is composed of fibrillar and nonfibrillar components. The fibrillar component of the vascular connective tissue is mainly collagen, and a diversity of proteins and proteoglycans (PGs) make up the rest. What follows is an overview of the structural and functional properties of the major macromolecules that characterize the vascular ECM. For a more detailed discussion of the individual classes of ECM macromolecules, astute readers will need to consult specialized reviews and critical commentaries, a number of which are cited in the chapter.
Collagens
Twenty-eight genetically distinct types of collagen comprising 43 unique α chains have been identified in vertebrates (Table 4-1). The vast majority of these collagens exist in humans.6–9 Based on their domain organization and other structural features (Fig. 4-1), collagens may be categorized as (1) fibril-forming collagens represented by types I, II, III, V, XI, XXIV, and XXVII; (2) fibril-associated collagens with interrupted triple helices (FACIT; e.g., IX, XII, XIV, XVI, XIX, XX, XXI, XXII, and XXVI collagens); (3) collagens capable of forming hexagonal network (e.g., VIII, X); (4) basement membrane collagens represented by IV collagen; (5) collagens that assemble into beaded filaments (e.g., type VI); (6) anchoring fiber-forming collagens (e.g., VII); (7) plasma membrane-spanning types XIII, XVII, XXIII, and XXV collagens; and (8) collagens with unique domain organization, represented by types XV and XVIII.
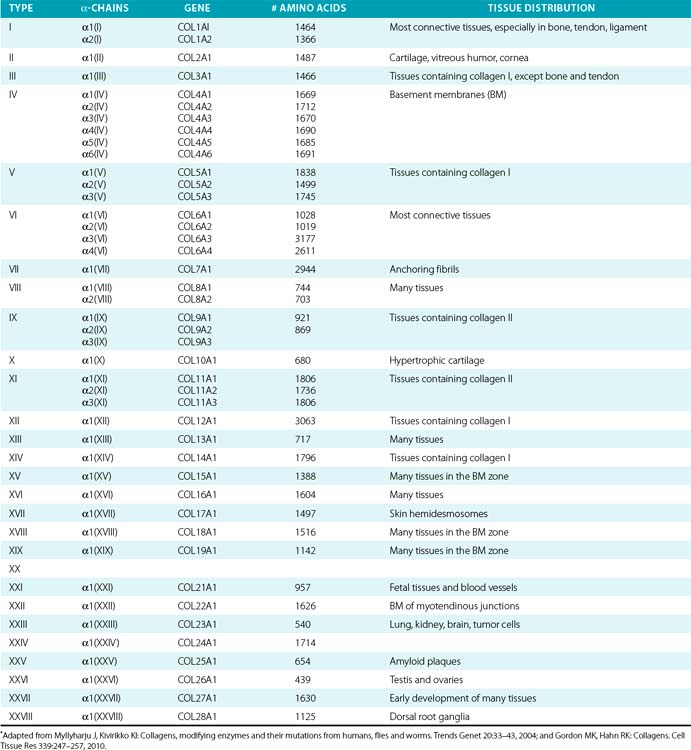
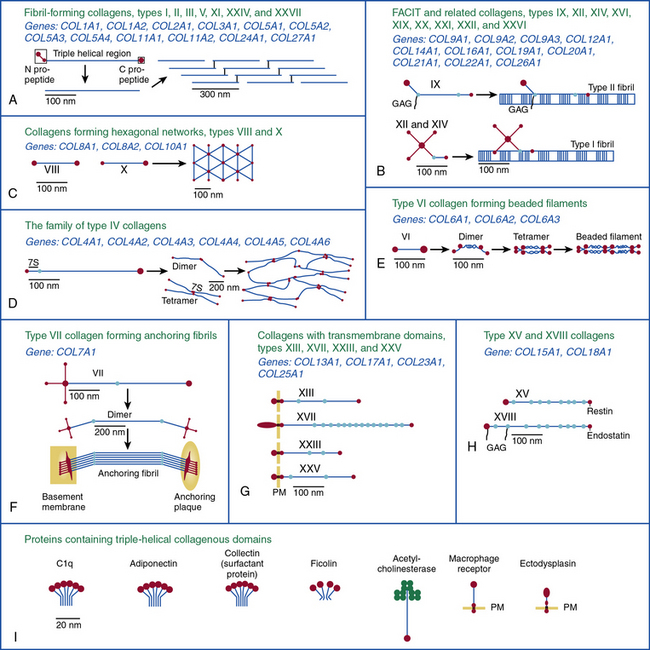
Figure 4-1 Classification of superfamily of vertebrate collagens.
(From Myllyharju J, Kivirikko KI: Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 20:33–43, 2004.)
We should note that the nomenclature of proteins as collagens and their classification into different types is somewhat arbitrary, since collagen fibrils invariably consist of more than one type of collagen. For instance, type I collagen fibrils contain small amounts of type III, V, and XII; similarly, type II collagen fibrils contain significant amounts of collagen types IX and XI. Even more strikingly, types V and IX collagen are known to form hybrid fibrils. The discovery of collagens that have extensive non-triple-helical domains and several proteins that contain triple-helical domains, such as C1q, adiponectin, acetyl cholinesterase, and ectodysplasin (see Fig. 4-1), further challenge the notion of what constitutes a “true collagen” and how it should be classified. Although several collagen types are found in the vasculature, collagen types I and III are the dominant constituents of the blood vessel wall.6,7,9 Collagen types II and X are excluded from our discussion because they are not relevant to the ECM of the vascular endothelium.
Fibrillar Collagens
The collagen molecule, the basic unit of collagen fibers, has an asymmetrical, rodlike structure composed of three polypeptide chains called α chains. Because of the Gly-X-Y repeating units and their stereochemistry, each α chain forms a minor helix (Fig. 4-2). Three α chains wind around a common axis to form a right-handed triple helix. In some collagens, all three α chains are identical, while in others two or three unique α chains form the triple-helical molecule. Type I and type III collagens are the most abundant collagens in the blood vessel and together form the striated fibrils. With the exception of types XXV and XXVII, fibrillar collagens form an uninterrupted triple-helical domain of approximately 300 nm. The type I collagen α chains contain 338 Gly-X-Y repeats, and there are 341 such triplets in type III α chains. At both the NH2 and COOH ends of each α chain are short segments of nonhelical sequences of approximately 15 to 20 amino acid residues, referred to as telopeptides.
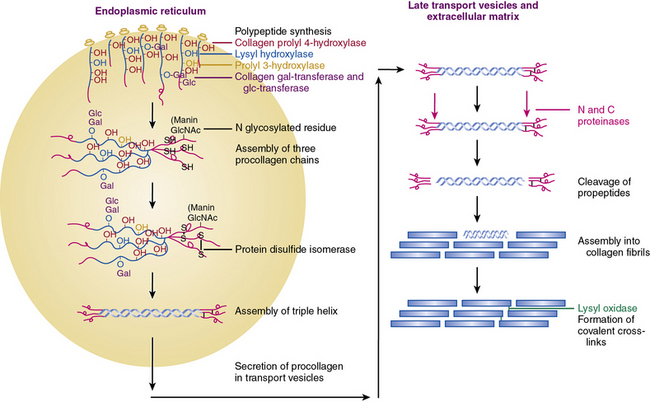
Figure 4-2 An overview of the main steps involved in the synthesis of fibril-forming collagens.
(From Myllyharju J, Kivirikko KI: Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 20:33–43, 2004.)
The collagen triple helix is further stabilized by interchain hydrogen bonds contributed by hydroxyproline residues. Thus, the collagen molecule is a long cylindrical rod with dimensions of 1.5 nm × 300 nm. Under physiological conditions of ionic strength, pH, and temperature, collagen molecules spontaneously aggregate into striated fibrils. Fibril formation occurs by lateral aggregation of collagen molecules, in which each neighboring row of molecules is displaced along its long axis by a distance of 68 nm. In addition, within the same row, there is a gap of approximately 40 nm between the end of one molecule and the beginning of the next (see Figs. 4-1 and 4-2). The short nonhelical telopeptides at the NH2 and COOH ends of each α chain are located in the gap or hole zone of the fibril and are therefore accessible to enzymes that regulate collagen cross-linking.
Network-Forming Collagens
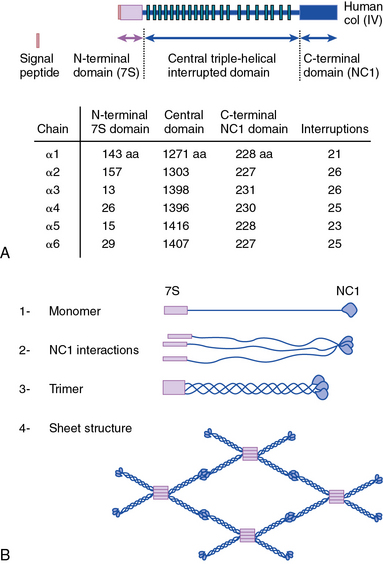
As shown in Figure 4-1, collagen types IV (α1-α6 chains), VI (α1-α5 chains), VIII (α1-α2 chains) and X are known to form networks in the ECM of basement membranes. The supramolecular organization and function of type IV collagen has been extensively characterized. Six different α polypeptide chains of collagen IV are each encoded by an evolutionary conserved gene. The amino and carboxyl propeptides of type IV collagen remain as integral parts of the molecules when they are deposited in the basement membrane. As a result, rather than forming a quarter-stagger, side-by-side alignment of individual molecules, as seen in types I, II, and III collagens, type IV collagen α chains form chicken-wire structures by end-to-end associations stabilized by lysine-derived cross-linking and interchain disulfide bonds (Fig. 4-3). The α1(IV) and α2(IV) collagen chains are more closely related to each other than to α3(IV)1, α4(IV), α5(IV), and α6(VI); the latter share a high degree of sequence homology with each other. The amino terminal domains of α1(IV) and α2(IV) collagen chains are 143 and 167 amino acids, respectively; the NH2-termini of the other four α chains are much smaller (ranging in size from 13 to 19 amino acids). Theoretically, all six α chains of type IV collagen may combine randomly to generate 56 unique triple-helical permutations. However, as shown in Figure 4-4, in vascular basement membranes the most common composition of triple-helical fibrils is [α1(IV)1]2 α2(IV). The [α3(IV)1]2 α4(IV) and [α5(IV)1]2 α6(IV) are also present in basement membrane.9,10
Type VI collagen, another network-forming molecule, is represented by six distinct α chains in the mouse and five α chains in humans; the gene encoding the putative α4(VI) collagen chain is not functional in humans. Heterotrimers of different α chains, encoded by unique genes, form the basic unit of type VI collagen. Alternate splicing of messenger ribonucleic acids (mRNAs) generates additional variants of α2 (VI) and α3 (VI) chains.7,9 The Gly-X-Y domains of α chains of type VI collagen microfibrils are rather short (about 330 amino acid residues) and are flanked by a number of von Willebrand factor (vWF) A domains.
Type VI collagen forms relatively unusual aggregates by a stepwise assembly into the triple-helical monomeric units that form dimers in an antiparallel fashion. The dimers in turn form tetramers, held together by disulfide bonds, to create scissors-like structures. The supramolecular assemblies of type VI collagen, formed by end-to-end associations of tetramers, appear as beads on a string, as revealed by electron microscopy.9 These characteristic structures have been observed in vascular subendothelium and skeletal muscle basement membranes. Type VI collagen microfibrils exhibit unique adhesive properties to other ECM components, such as other collagens, heparin, and vWF, and may be involved in the adhesion of platelets and SMCs. In the medial layer, type VI collagen facilitates interaction between SMCs and elastin by bridging the elastin fibers and cells.11
As illustrated in Figure 4-5 (see discussion in “Metalloproteinases”), types VIII and X collagens comprise a unique subfamily of collagens that form hexagonal networks. These relatively short collagens, containing noncollagenous domains on their NH2 and COOH termini, are collectively known as the multiplexin family of collagens. Type VIII collagen is expressed in many tissues, especially in the endothelium, while type X is exclusively associated with hypertrophic chondrocytes during cartilage and bone development. The preponderance of evidence to date indicates that the two α chains of collagen VIII, encoded by COL8A1 and COL8A2, assemble into homotrimers of α1(VIII) and α2(VIII) (Fig. 4-6). Hexagonal aggregates of type VIII collagen have been observed both in vivo (e. g., Descemet’s membrane of the cornea) and in vitro with purified protein. It is believed that type VIII collagen is capable of assuming other forms of macromolecular aggregates, since hexagonal lattices have yet to be observed in the subendothelial ECM.9
Fibril-Associated Collagens with Interrupted Triple Helices
As the name suggests, the FACIT collagens (types IX, XII, XIV, XVI, XIX, XX, XXI, XXII, and XXVI) do not form fibrils themselves but associate with other fibril-forming collagens.9 Type IX collagen, the prototype of this group, is cross-linked to the surface of type II collagen fibrils in cartilage (see Fig. 4-1); type XII and type XIV collagens are found in both cartilage and noncartilaginous tissues, where they are involved in controlling the diameter of collagen fibrils (see Fig. 4-1). The other FACIT-like collagens (e.g., types XVI, XIX, and XXII) are localized in specialized basement membranes. For instance, XVI collagen is associated with fibrillin 1 near the epidermal basement membrane. Collagen types XXI and XXII are closely related to each other in structure and are involved in formation of supramolecular aggregates in the basement membranes of myotendinous junctions.12,13 As a key constituent of cutaneous basement membranes, anchoring fibrils of type VII collagen form a structural continuum between the dermis and epidermis of normal human skin. The vWF A–like domain in collagen VII binds to fibrils of type I collagen in vitro.14
Minor Collagen Types with Unique Structures
As illustrated in Figure 4-1, the transmembrane collagens (types XIII, XVII, XXIII, and XXV) contain a cytoplasmic domain, a membrane-spanning hydrophobic domain, and extracellular triple-helical domains interspersed with noncollagenous domains; these collagens may also exist in a soluble form. Type XVII collagen is a unique member of this group that is expressed on the basal surface of keratinocytes that bind to laminin found in the basement membrane; compared with the other three members of this group, type XVII has a rather large intracellular domain whose function remains unknown. Collagen types XIII, XXIII, and XXV are similar to each other in their primary structure, but the patterns of their expression appear to be unique. Type XXV collagen is enriched in the senile plaques of Alzheimer’s disease brains.9,12,13,15 High expression of full-length collagen XXIII is found in the lungs, whereas its shed form is enriched in brain, suggesting that shedding of XXIII collagen occurs in a tissue-specific manner.
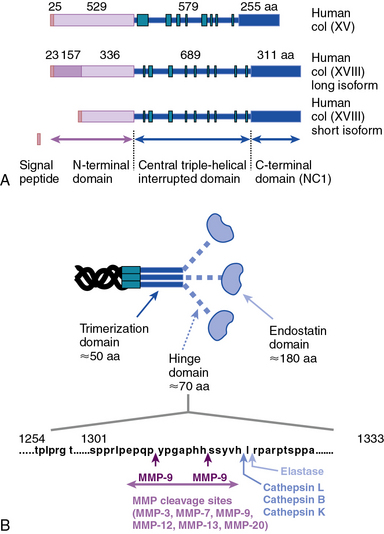
Collagen types XV and XVIII are highly pertinent to the EC biology in several ways.16,17 The full-length types XV and XVIII collagen are basement membrane components; their triple-helical domains share a high degree of homology. Collagen types XV and XVIII were initially identified as PG core proteins containing chondroitin sulfate and heparan sulfate (HS) side chains, respectively. The COOH-terminal domains of XV and XVIII collagens can be cleaved to generate biologically active peptides, endostatin and restin, respectively; these peptides inhibit migration of ECs and thus potently block angiogenesis. In vitro, recombinant collagen XV binds to fibronectin (FN), laminin, and vitronectin (VN) but not to fibrillar collagens, fibril-associated collagens, or decorin.18
Finally, collagens XXVI and XXVIII are newly discovered collagens that are unique both with regard to their structures and tissue-specific distributions. The triple-helical domain of type XXVI is rather small, with only 146 Gly-X-Y repeats. Expression of type XXVI collagen occurs predominantly in testis and ovary. The von Willebrand factor–A domains flank the triple-helical structure of type XXVIII collagen that is almost exclusively expressed in the peripheral nerves.19,20
Regulation of Collagen Biosynthesis
Collagen chains are synthesized as prepro-α chains from which the hydrophobic leader sequence is removed prior to secretion, and the pro-α chains are secreted into the extracellular space (see Fig. 4-2). The pro-α1(I) chain contains an NH2 propeptide (N-peptide) and a COOH propeptide (C-peptide). The N-peptide consists of a 139-residue sequence that precedes a 17-residue sequence of nonhelical telopeptide. This is followed by a 1014 amino acids-long Gly-X-Y helical sequence attached sequentially to a 26 residues-long COOH telopeptide and a 262-residues-long nonhelical C-peptide. The domain organization of pro-α2(I) and pro-α1(III) chains are similar except for minor variations in the number of amino acid residues.6,7,21
The genomic organization and chromosomal locations for genes that encode collagens have been studied. In humans, the genes encoding 43 distinct α chains are dispersed on at least 15 chromosomes. Unlike majority of the collagen-encoding genes, the six homologous α-chains of type IV collagen are encoded by genes that are located in pairs with head-to-head orientation on chromosomes 13 (COL4A1 and COL4A2), 2 (COL4A3 and COL4A4), and the X chromosome (COL4A5 and COL4A6). Interestingly, the promoters of these pairs of type IV collagens overlap, suggesting a coordinate regulation of the gene pairs. The precise molecular mechanisms of this regulation, however, remain incompletely known.6,7,9,21,22
The cis-acting elements of COLA1 and COLA2 genes are modularly organized on either side of the transcription start point (TSP). The regulatory elements are distributed over a distance of 100 to 150 kb of genomic DNA, depending on the specific gene and the assays used to study their transcriptional and posttranscriptional regulation. The tissue-specific and inducible activation of collagen genes involves complex interactions among the cis-acting modules of their promoters and enhancers. Promoters of COLA1 and COLA2 genes contain TATA boxes located 25 to 35 bp upstream of the TSP. Existence of a number of enhancer and repressor cis-elements around the TSP and in the first intron of COLA1 gene has been demonstrated. A key role for CAAT-binding factor, Sp1, Sp3, Ap1, nuclear factor (NF)-κB, and SMADs has been reported for several collagen genes; a number of orientation-dependent enhancer-like elements have also been documented.23,24
Fibrillar and nonfibrillar collagens found in subendothelial ECM are regulated by many cytokines and growth factors; collagen gene expression in response to cytokines (e.g., transforming growth factor [TGF]-β, tumor necrosis factor [TNF]-α, interleukins [ILs]), glucocorticoids, estrogen, androgen, and retinoids has been reported. The signaling cascades initiated by intrinsic and exogenous regulators impinge on a distinct set of cis-acting elements that bind to constitutive and inducible transcription factors. The emerging theme from these studies is that various cis– and trans-acting factors interact to recruit selective transcriptional coactivators and co-repressors in response to specific stimuli.23,24 However, the precise mechanisms that determine combinatorial interactions under physiological and inflammatory conditions remain to be elucidated.
Following translation, pre-procollagen α chains are chaperoned from the endoplasmic reticulum (ER) to the Golgi. It has been reported that the heat shock protein-47 (Hsp47) functions as a collagen-specific chaperone; thus, hsp47 is presumed to provide a quality control mechanism needed for proper maturation of newly synthesized procollagen chains. To demonstrate a role of hsp47 in vivo Nagai and coworkers25 inactivated Hsp47 gene by homologous recombination. The mutant embryos died in utero before 11.5 days of postcoitus development as a result of severely reduced levels of mature type I collagen in their tissues.
As shown in Figure 4-2, fibrillar and nonfibrillar collagens also undergo a number of posttranslational modifications for proper maturation; these include proteolysis of signal peptides, hydroxylation of key proline and lysine residues, glycosylation, and formation of interchain and intrachain disulfide bridges.6,7,21 Thus, optimal biosynthesis and assembly of collagens depends on a number of key enzymes. These include three hydroxylases, two collagen-specific glycosyl transferases, two unique proteinases that cleave the NH2– and COOH-termini, and a collagen-specific oxidase that is needed for cross-link formation. The posttranslational processing of the procollagen molecules also needs a peptidyl proline cis-trans isomerase and a protein disulfide isomerase (PDI).
Vitamin C–dependent 4-prolyl hydroxylase, an α2β2-tetramer located in the ER, plays a central role in collagen synthesis because 4-proline hydroxylation is obligatory for cross-link formation. In humans, there are three known isozymes of 4-prolyl hydroxylases, each with a distinct α subunit, but all contain PDI as their β subunit. Hydroxylation of lysine is carried out by lysyl hydroxylase, which also uses the same cofactors as prolyl hydroxylase and reacts only with a lysine residue in the Y position of the Gly-X-Y triplets. There are three known isozymes of lysyl hydroxylase in humans. The under-hydroxylation of procollagen leads to reduced secretion and rapid degradation. Deficiency of lysine hydroxylase is associated with skeletal deformities, tissue fragility, and vascular malformations.6,7,21
Assembly of procollagen chains into triple-helical molecules is directed by the COOH-terminal propeptide, with formation of interchain disulfide bonds (see Fig. 4-2). There is a high degree of structural conservation within the propeptide of fibrillar collagens across species. Following its triple-helical assembly, the procollagen molecule is secreted into the extracellular space. Once secreted, however, the NH2 and COOH propeptides are removed by the actions of N- and C-specific peptidases to yield the collagen molecule. The two proteinases that remove the NH2 and COOH propeptides from the newly synthesized collagen are represented by three isozymes each. The C-specific peptidases, members of the tolloid family, also cleave a number of other ECM proteins, and fragments of the propeptides can inhibit procollagen synthesis by a feedback mechanism.26,27
Extracellular Maturation of Collagens
During collagen fibril formation, lysyl oxidase catalyzes the oxidative deamination of specific lysine or hydroxylysine residues in the NH2– or COOH-terminal telopeptides to yield allysine and hydroxyallysine, respectively.28 These reactive aldehydes, being located in the hole zone of the fibril, are free to react with the ε-amino group of lysine or hydroxylysine residues on adjacent chains to form a Schiff base, which undergoes Amadori rearrangement to form ketoimine. With time, two ketoimine structures condense to form a trivalent cross-link, 4-hydroxy-pyridinium. All three types of cross-link may coexist in different fibrils.
A second type of cross-link seen in collagen originates from the condensation of two aldehydes in allysine or hydroxyallysine on adjacent chains. The resulting aldol condensate has a free aldehyde that reacts with other ε-amino groups of lysine or histidine, thus potentially linking three or four collagen chains.29 Once the aldehydes of allysine and hydroxyallysine are formed, subsequent aldamine and aldol condensation reactions proceed spontaneously. Thus, inter- and intramolecular cross-linking of fibrillar collagens results in formation of insoluble macromolecular aggregates that possess high tensile strength.
Metalloproteinases
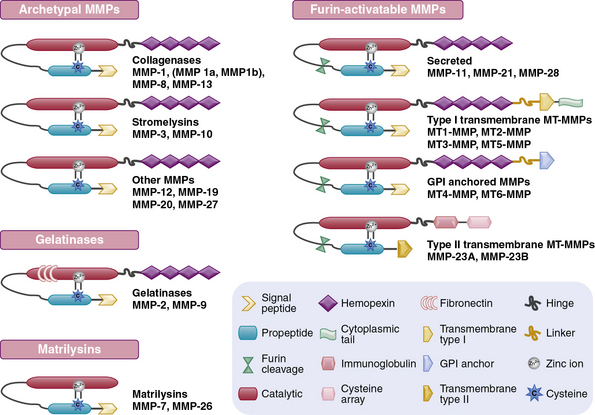
The structural and functional diversity of MMPs rivals that of the superfamily of collagens. The MMPs belong to a large family of zinc-dependent endopeptidases, the first of which was described nearly a half century ago. To date, the presence of 23 distinct MMPs has been reported in human tissues. Based on their cellular localization, these enzymes can be broadly subdivided into secreted and membrane-bound MMPs. However, a more detailed analysis of their structural organization and substrate specificities indicates that MMPs may be better classified as collagenases, gelatinases, stromelysins, metrilysins, and membrane-type MMPs.30–33
The architectural blueprint of a prototype MMP consists of three subdomains: the Pro-domain, the catalytic domain, and the hemopexin-like C-domain, connected to the catalytic domain via a short linker region (see Fig. 4-5). The catalytic domain of MMPs contains a Zn++ ion-binding amino acid sequence motif and a substrate-specific site. The prototypic MMP is synthesized as a pre-proenzyme and is maintained in latent conformation by the Pro-domain via interaction between a cysteine (located in the cysteine switch region of the Pro-domain) and Zn++ ion in the catalytic domain. Only when this interaction is disrupted, either by proteolysis of the Pro-domain or by a chemical modification of the cysteine, MMP becomes activated.32 A number of intracellular and extracellular proteinases, including other MMPs, are known to specifically degrade the Pro-domain to activate MMPs in vivo.
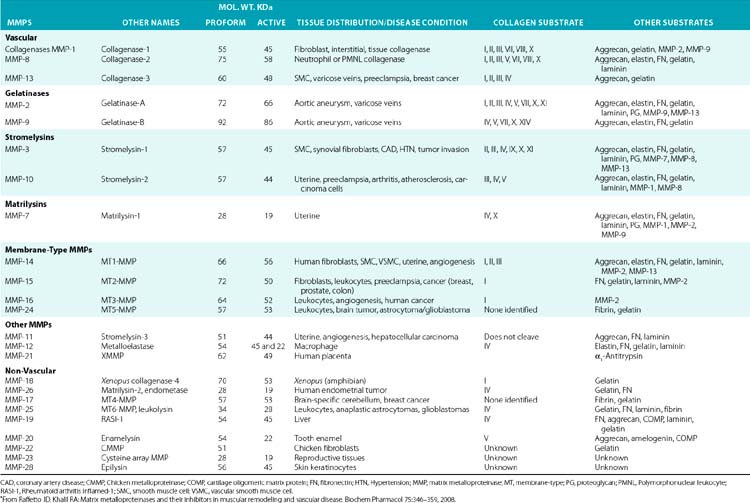
Although in vitro studies have identified numerous substrates for various MMPs (Table 4-2), the precise identities of their in vivo targets remain largely elusive. A number of macromolecules associated with ECM of the endothelium are potential in vivo targets of MMPs. For example, MMP-1 (collagenase 1) readily degrades collagen types I, II, and III, whereas MMP-8 (collagenase 2) digests types I, III, IV, V, VII, X, and XI collagen. Similarly MMP-2 (gelatinase A) degrades types I, III, IV, V, VII, X, and XI collagens, whereas gelatinase B (MMP-9) can degrade collagen types IV, V, XI, and XIV preferentially. MMP-13 (collagenase 3) is also capable of degrading collagens that are prevalent in subendothelial ECM (types I, III, VI, IX, and XIV). Many collagenous and noncollagenous ECM components are readily degraded by stromelysin-1 (MMP-3) and stromelysin-2 (MMP-10), whereas stromelysin-3 (MMP-11) does not degrade known collagens but readily breaks down laminin. Matrix metalloproteinases are also capable of digesting a number of other constituents of ECM, such as FN and elastin, and a variety of other cell- and ECM-associated molecules (see Table 4-2). The actions of some MMPs are likely to mediate highly regulated processing of ECM-bound pro-TGF-β and pro-IL-1.
Table 4-2 Members of the Matrix Metalloproteinase Family in Representative Vascular and Nonvascular Tissues*
Numerous studies have been undertaken to elucidate the molecular mechanisms by which the actions of MMPs are regulated in the tissues under physiological and pathological conditions.32 Two major mechanistic themes have emerged from these studies to explain the exquisite specificity of various MMPs. First, synthesis and localization of various pro-MMPs and their highly tissue-specific inhibitors (TIMPs) are regulated by autocrine and paracrine factors. Thus, cytokines such as IL-1 and TNF-α and a number of other circulating factors regulate expression of various MMPs at the transcriptional and posttranscriptional levels.
The second type of regulation of MMPs is exerted via the unique organization of their functional domains. As outlined earlier, the Pro-domain plays a critical role in maintaining the MMPs in a latent state that is altered by a number of physiological and pathological stimuli. Similarly, the presence of three cysteine-rich repeats, akin to those found in FN (see later discussion) in gelatinase A and gelatinase B, determines their affinities for elastin and collagen. The domain organization of MMPs allows them to be regulated by TIMPs; these inhibitors reversibly bind to MMPs in a 1:1 stoichiometry and inhibit enzymatic activity.34 Tissue inhibitors of MMPs, represented by four homologous proteins (TIMP1 to 4), preferentially inhibit various MMPs.35,36 For example, whereas TIMP3 potently inhibits MMP-9, both TIMP2 and TIMP3 inhibit membrane-type 1 (MT1)-MMP. In contrast, TIMP1 is a very poor inhibitor of MT-3-MMP but a potent inhibitor of MMP-3.34
Concerted actions of various MMPs and their TIMPs regulate key events in the formation of blood vessels in the developing embryo, and the processes of neovasculogenesis and angiogenesis in the adult in response to injury and regeneration (Table 4-3). Formation of new blood vessels from existing vessels is dependent on extensive turnover of subendothelial ECM. This process enables migration of blood vessel–associated cells, liberation of angiogenic factors sequestered in the ECM, and exposure of cryptic cell-regulatory domains found in the intact fibrillar and nonfibrillar components of connective tissue. Therefore, a crucial balance between MMPs and TIMPs is essential for maturation of newly formed blood vessels and ongoing maintenance of their structural integrity. These processes are known to play a critical role during embryogenesis; the formation of solid tumors and their acquisition of invasive, metastatic phenotype is also vitally dependent on the emergence of new blood vessels.37 MMP-2 binds to the αvβ3 integrin and promotes angiogenesis and tumor growth.38 In contrast, the transmembrane MMP, MT1-MMP, cleaves αvβ3 integrin and enhances its affinity for its ligands containing arginine-glycine-aspartic acid (RGD) sequences.
Table 4-3 The Effect of Matrix Turnover on Vascular Pathologies*
| Model | Effects | |
|---|---|---|
| Aneurysm | MMP-3−/− / ApoE−/− | ↓ Aneurysm |
| MMP-9−/− | ↓ Aneurysm | |
| MMP-12−/− | ↔ Aneurysm | |
| Broad-range MMP inhibitor LDLR−/− | ↓ Aneurysm | |
| TIMP-1−/− / ApoE−/− | ↑Aneurysm | |
| TIMP-1 ↑ rat | ↓ Aneurysm | |
| Neointima formation | MMP-9 ↑rat | ↑SMC migration ↓ matrix content ↑Luminal diameter |
| Broad-range MMP inhibitor | ↓ Early and ↔ late neointima formation | |
| LDLR−/− Doxycycline, MMP inhibition rat | ↓ Neointima formation | |
| TIMP-1 ↑human vein | ↓ Neointima formation | |
| TIMP-2 ↑human vein | ↓ Neointima formation | |
| TIMP-3 ↑human and pig veins | ↓ Neointima formation | |
| MMP-9−/−, mouse carotid ligation | ↓ Intimal hyperplasia,↑ collagen content | |
| Remodeling | MMP-12 ↑ | ↓ Luminal diameter |
| MMP inhibitor pig | ↓ Constrictive remodeling | |
| Atherosclerosis | MMP-1 ↑/ ApoE−/− | ↓ Plaque size ↓ collagen content |
| MMP-3−/− / ApoE−/− | ↑ Plaque size ↑ collagen content | |
| MMP-3 ↓ human, promoter polymorphism | ↑Plaque progression | |
| MMP-9 ↑ human, promoter polymorphism | ↑Triple-vessel disease | |
| MMP-9 ↑ human promoter polymorphism | ↔ Coronary artery stenosis | |
| Broad-range MMP inhibitor LDL−/− | ↔ Plaque size | |
| TIMP-1−/− / ApoE−/− | ↓ Plaque size ↑ lipid core content | |
| TIMP-1−/− / ApoE−/− | ↔ Plaque size, medial rupture, micro aneurysms | |
| TIMP-1 ↑/ ApoE−/− | ↓ Plaque size ↑ collagen content | |
| TGF-β inhibition ApoE−/− | ↑ Plaque vulnerability, intraplaque hemorrhage |
MMP, matrix metalloproteinase; Apo, apolipoprotein; LDLR, LDL receptor; TIMP, tissue inhibitor of matrix metalloproteinase; SMC, smooth muscle cell; TGF, transforming growth factor; +/+, transgenic overexpressing mice; −/−, knock-out or homozygous deficient mice; ↑, upregulation or increased; ↓, downregulated or decreased.
*Adapted from Heeneman S, Cleutjens JP, Faber BC, et al: The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. J Pathol 2003:516, 2003.
Elastin
Blood vessels are endowed with a high degree of elasticity, and subendothelial elastic fibers are responsible for the resilience of the vasculature to cycles of deformity and passive recoil during diastole and systole, respectively. The elastic fiber consists of an insoluble core of polymerized tropoelastin surrounded by a mantle of microfibrils. A schematic representation of the modular organization of human tropoelastin is shown in Figure 4-7. The primary structure of tropoelastin consists of hydrophilic and hydrophobic domains; these may be further divided into subdomains based on the composition of their amino acid sequences (see Fig. 4-7). The mechanical properties of the elastic fiber are similar to rubber (i.e., the degree of elongation without irreversible changes per unit force applied to unit cross-sectional areas is high).
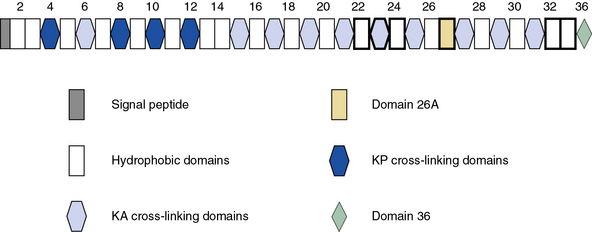
Organization of the elastic fibers has been studied by electron microscopic, biochemical, and genetic approaches, and a number of key insights have been gathered in recent years.3 Elastin is a major constituent of the elastic fiber and may contribute as much as 50% of the dry mass of large arteries.39 The elastic fibers begin to form at mid-gestation by deposition of tropoelastin, the soluble precursor of the cross-linked mature elastin, on a template of fibrillin-rich microfibers. The cross-linked elastin contained in the elastic fibers produced during late fetal and postnatal development generally lasts a lifetime.
Elastin has an amorphous appearance in the electron microscope; microfibrils appear as 10- to 15-nm diameter filaments. The assembly of elastic fibers occurs via a stepwise process that includes formation of a scaffold of microfibrils that facilitate deposition of tropoelastin monomers (Fig. 4-8), followed by extensive cross-linking to form the functional polymer.3,40,41 Tropoelastin is the soluble monomer of elastin that is one of the most apolar and insoluble proteins in nature. Although the glycine and proline content of elastin is similar to fibrillar collagens, elastin contains no hydroxyproline or hydroxylysine, and very small amounts of polar amino acids. Elucidation of the molecular organization of elastin has been difficult because of the technical problems in obtaining large quantities of tropoelastin. Therefore, scientists have relied mainly on the structure of fragments of hydrolyzed soluble elastin and recombinant tropoelastin produced in bacteria.
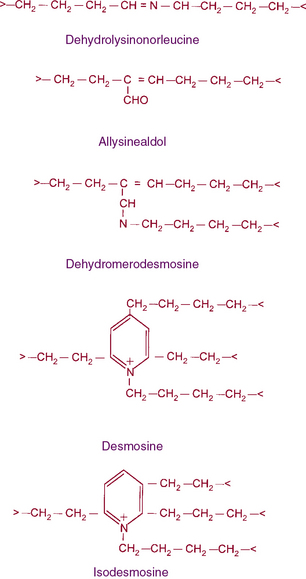
Figure 4-8 The structures of cross-links found in elastin.
Desmosine and isodesmosine represent final products of lysine-derived cross-links.
As illustrated in Figure 4-7, human tropoelastin is encoded as a 72-kD polypeptide that is characterized by a series of tandem repeats. The tropoelastin amino acid sequence is divided into hydrophobic domains that are rich in nonpolar amino acids (glycine, valine, and proline) that typically occur as repeating units; these sequences alternate with hydrophilic domains that are enriched in lysine and alanine. In vitro, elastin undergoes a process of ordered self-aggregation called coacervation (aligning and concentrating the protein in unit spheres) prior to cross-linking. Tropoelastin binds to cell surface glycosaminoglycans as well as to αvβ3 integrins.42 Although the sequential interactions of tropoelastin with fibrillins and its associated molecules are poorly defined, it is thought that the process of elastic fiber assembly is initiated on the cell surface.43,44 This is caused by specific interactions of the individual hydrophobic domains of tropoelastin, since it has an intrinsic ability to organize into polymeric structures.39
In vivo, tropoelastin probably interacts with microfibrils prior to aggregation and becomes cross-linked by lysyl oxidase.40,41,45 Soluble precursors of elastin are not found in extracts of normal tissues. This provides a clue as to the rapid formation of mature, highly cross-linked elastin fibers and the low rate of tropoelastin synthesis. In experimental conditions such as copper deficiency or lathyrism induced by β-aminopropionitrile, which inhibits lysyl oxidase activity and thus cross-link formation, a soluble 72 kD tropoelastin can be extracted from the aorta. Like collagens, newly synthesized tropoelastin undergoes posttranslational modifications before its assembly into elastic fibers; in fact, the same lysyl oxidase reacts with both collagens and elastin.46 In contrast to collagen, however, reduction of double bonds in the elastin cross-link occurs spontaneously, and the quantity of lysine involved in cross-linking is much larger in elastin than in collagen (see Fig. 4-8). Oxidative deamination of lysine residues, followed by subsequent condensation reactions, creates the unusual cross-links found in elastin. All of the cross-links in elastin are derived from lysyl residues through allysine (Fig. 4-9). However, the precise molecular reactions needed to form desmosine remain to be elucidated. Cross-linking in elastin occurs frequently, not only between peptide chains but also within the same polypeptide chain, producing intrapolypeptide links. The cross-linking process is highly efficient, and it is unclear how the cross-linking sites in the monomer get aligned.
Genomic organization of the tropoelastin gene indicates that functionally distinct cross-linking and hydrophobic domains of tropoelastin may be encoded by distinct exons. Short segments rich in alanine and lysine are clustered to apparently delimit the cross-linked region. These amino acids are clustered in the α-helical configuration of tropoelastin, where each begins with tyrosine followed by Ala-Ala-Lys or Ala-Ala-Ala-Lys. In humans, several distinct tropoelastin polypeptides may be generated by alternative splicing (see Fig. 4-7). Space-filling atomic models indicate that lysines separated by two or three alanyl residues in α-helical conformation protrude on the same side of the helix. Hence, the sequence Lys-Ala-Ala-Lys allows formation of dehydrolysinonorleucine, whereas the sequence Lys-Ala-Ala-Ala-Lys accommodates either aldol condensation or dehydrolysinonorleucine formation. Condensation of the two intrachain cross-links could result in the formation of the interchain desmosine cross-links. The alanine- and lysine-rich cross-linking segments are separated by large hydrophobic segments of 6 to 8 kD, which are in a β-spiral structure with elastomeric properties. Within the hydrophobic segments, a repeating pentapeptide (Pro-Gly-Val-Gly-Val) is present. A collagen-like sequence (Gly-Val-Pro-Gly) occurs quite frequently, which would explain the limited susceptibility of tropoelastin to bacterial collagenase (Pro-Gly-X-Y). The sequence Gly-X-Pro-Gly is recognized by the prolyl hydroxylase involved in the cross-linking of collagens (see earlier discussion).
Elastin Metabolism and Vascular Homeostasis
After deposition, tropoelastin production is strikingly reduced; the half-life of elastin in normal humans has been estimated in years. In the event of injury, production of elastin can be quickly initiated. A number of growth factors and cytokines induce biosynthesis of tropoelastin. Under these conditions, a very specific set of proteinases named elastases are responsible for elastin remodeling. Elastin fibers may be degraded by a number of MMPs, particularly MMP-2, -3, -9 and -12, that are present as latent enzymes under physiological conditions but are activated following vessel wall injury.47 The MMPs from neutrophils or macrophages are believed to degrade the elastin-rich ECM found in inflamed tissues. A hereditary defect in circulating elastase inhibitors is associated with a progressive destruction of the elastin-rich alveolar wall, resulting in premature emphysema. Furthermore, experimental instillation of elastase into the lungs of animals causes destruction of the lung similar to that seen in patients with α1-proteinase inhibitor deficiency.
Mice with a disrupted elastin gene have provided important insights into the function of elastin protein. Heterozygous (elastin+/− mice) had decreased arterial compliance and were hypertensive. The homozygous elastin-null mice died young due to arterial obstruction caused by uncontrolled proliferation of smooth muscle cells (SMCs).39,48 A direct link between occlusive vascular diseases and perturbation in the organization of the elastic fibers in the vessels has also been established.49 Mutations in the elastin gene are associated with supravalvular aortic stenosis (SVAS) and Williams-Beuren’s syndrome (WBS), pediatric disorders characterized by hemodynamic stress and loss of elasticity.49 Furthermore, haploinsufficiency of elastin resulting from aberrant degradation of mutated protein in humans or ablation of the elastin gene in transgenic mice caused intimal hyperplasia and thickened arteries.40,45,50–52 Apparently, VSMCs, the primary producers of elastin, organized more cell layers to compensate for lost elasticity and biomechanical support in developing blood vessels of elastin haploinsufficient patients and transgenic mice.
Vascular smooth muscle cells from SVAS patients, WBS patients, and elastin−/− mice show increased rates of proliferation and chemotactic migration, and reduced rates of elastin synthesis in vitro.50–52 Exogenous supplementation of recombinant tropoelastin and α-elastin to these cultures reversed their phenotype. The elastin-rich ECM serves as an autocrine regulator of VSMC; Karnik et al.53 inserted elastin-coated stents in a porcine coronary injury model of restenosis and found that intimal thickness and arterial stenosis were significantly reduced. Although the identities of the specific receptors mediating elastin VSMC interactions and the signaling mechanisms underlying vascular remodeling remain obscure,54 restoring elastin to an injured arterial wall is known to reduce obstructive vascular pathology.55,56 Inhibitors of MMPs have been shown to prevent degradation of elastic fibers after vascular injury and ameliorate neointimal thickening.47
Fibrillins and Other Microfibril-Associated Proteins
Fibrillins, the major constituent of microfibers, are large glycoproteins that form loosely packed bundles in the tissues. The fibrillin superfamily also includes the structurally related latent TGF-β-binding proteins (LTBP1, 2, 3, and 4) and fibulins.40,45,57 Fibrillins are represented by three homologous proteins: fibrillin-1, fibrillin-2, and fibrillin-3. All three fibrillins are approximately 350-kD glycoproteins that display similar modular organization (see Fig. 4-9) that consists of 46/47 epidermal growth factor (EGF)-like domains (42/43 of these are calcium-binding type; cbEGF) interspersed with seven 8-cysteine-containing TGF-β-binding (TB) modules found in LTBPs. Additionally, fibrillins contain two hybrid domains composed of TB/8Cys and cbEGF-like sequences and NH2– and COOH-termini with sequence homologies with respective segments of LTBPs and fibulins. The structural versatility of elastic fibers (e.g., concentric rings in arterial walls vs. parallel bundles in the ocular ligament that anchors the lens to the ciliary body) most probably reflects a selective use of different fibrillins in different locations. Importantly, the function of fibrillin-3 remains to be established, and thus the following narrative is restricted to fibrillin-1 and fibrillin-2.
Fibrillins are thought to organize into microfibrils in which individual molecules are organized in a head-to-tail arrangement as well as sideways. The precise molecular architecture of fibrillins within the microfiber and how its elasticity is regulated are incompletely understood. The developmental role of fibrillins has become evident from studies in transgenic mice. Thus, fibrillin-1-deficient mice display frequent dissecting aneurysm and die soon after birth.58,59 This is in contrast to the vessels of fibrillin-2−/− mice that appear to be structurally and functionally normal. However, mice with haploinsufficiency of both fibrillin-1 and fibrillin-2 elicit variable phenotypes, although many die in utero. These studies indicate that fibrillin-1 and -2 play somewhat unique context-dependent instructive and mechanical roles in the developing vasculature. The four known LTBPs with multiple EGF-repeats of fibrillin-1 and fibrillin-2 and their associated ligands (e.g., perlecan, elastin, fibulin) are mechanistically involved in the developmental actions of these versatile ECM proteins.40,45,57
Fibrillin-rich microfibrils play a vital role in extracellular regulation of TGF-βs and bone morphogenetic proteins (BMPs) by modulating their storage, release, and activation in response to various stimuli.3,40,41,45,49,57 Apparently, LTBP1, 3, and 4 elicit functional redundancy and target the latent TGF-β to elastin-rich microfibrils; LTBP2 does not bind TGF-β but is highly expressed in response to arterial injury. Fibrillins appear to play a direct role in TGF-β signaling, as revealed by fibrillin-1 knockout mice that were born with impaired lungs and emphysema, without measurable signs of inflammation. A detailed analysis of these animals revealed that aberrant TGF-β (Smad2/3) signaling in the developing lungs was responsible for the observed pulmonary phenotype.58,59 More recently, a role of fibrillin-1 mutations in the development of mitral valve prolapse and aortic aneurysm was also reported.
The microfibril-associated glycoproteins MAGP-1 and MAGP-2 are also believed to impart structural integrity to microfibrils.60,61 The expression profile of MAGP-1 in the aorta resembles that of fibrillin-2; both are thought to be critical for embryonic and fetal development of the aorta. Additionally, PGs (e.g., biglycan, decorin, versican) are also associated with microfibrils and are believed to facilitate their incorporation into surrounding ECM.3,40,41,45,49,57
Fibulins represent a family of ECM proteins with cbEGF-like domains and a distinctive COOH-terminal module.57 Seven fibulins have been identified since the discovery of the prototype, fibulin-1. The unique distribution of various fibulins suggests that their contribution to the organization of various types of the elastic fibers may be tissue specific. Based on their length and domain organization, fibulins are classified into two groups. The short fibulins (fibulin-3, -4, -5, and -7) are elastogenic and contain tandem repeats of cbEGF. How various fibulins modify endothelial ECM has been investigated in vitro and in transgenic mice. Whereas fibulin-1 is located in the elastin core, fibulin-2 and -4 are found at the interface between the central elastic core and the mantle of microfibrils. Fibulin-1 knockout mice have dysfunctional vasculature and die of spontaneous bleeding. Mice that lack a functional fibulin-4 gene are also born with severe vascular defects.
The preceding description of microfibrils underscores the notion that the elastic fiber and its associated ECM are molecular integrators of extrinsic and intrinsic mechanical signals that impinge on TGF-β and BMP as focal points of tissue homeostasis. Therefore, it is mechanistically probable that diverse assemblies of fibrillin-associated molecules are involved in translating environmental inputs into physiological and pathological responses of the endothelium. Suffice to say, however, that the molecular interactions that regulate the putative extracellular inputs, as well as their corresponding responses both in time and space that mediate remodeling the vasculature during embryogenesis and in the adult, remain to be elucidated.62
Fibronectin
Fibronectin is one of the best characterized molecules of the vascular ECM.63,64 Evolutionary emergence of FN correlates with the appearance of EC lined vasculature in vertebrates.65 There is high degree of interspecies homology and conservation of domain organization of the FN gene across species.65,66 Fibronectin dynamically partners with multiple macromolecules to promote adhesion and spreading of cells, trigger chemotaxis of leukocytes towards injured tissue, and facilitate nonimmune opsonization and phagocytosis of bacteria. Some biologically active modules of FN are normally cryptic and are only exposed under special circumstances. Crosstalk between FN and growth factor/cytokine-mediated signals modulates tissue repair and regeneration and is involved in anchorage-independent growth of cancer cells. Fibronectin is especially abundant in the ECM of the embryo, where it plays a crucial role in phenotypic differentiation of vascular and nonvascular tissues. A functional FN gene is obligatory for development of the cardiovascular system.
Fibronectin Structure
In blood plasma, FN exists in a soluble state, synthesized and secreted by the liver, and is converted into an insoluble supramolecular complex in the ECM. The soluble FN is made of two disulfide-linked monomers of similar or identical mass (220-255 kD). As shown in Figure 4-10, each FN monomer is a mosaic of repeating modules termed type I, II, and III repeats that are 40, 60, and 90 amino acids long, respectively.67,68 A cluster of 15 to 17 type III repeats (depending on alternative splicing) located in the middle of the molecule represents 90% of the FN monomer. In addition, there are 12 type I and 2 type II repeats in each monomer of FN. The type III repeats fold into nearly identical shapes despite having only 20 to 40 amino acid sequence identity (see Fig. 4-10). The striking modular organization of the repeated peptide sequences in FN is reflected in the organization of its gene. The FN gene consists of 47 exons spanning nearly 100 kb in the human genome and generates multiple alternatively spliced mRNAs.69–72 A single gene thus generates about 20 variants of FN protein that may be preferentially synthesized under various physiological and pathological situations.68 Fibronectin monomers containing or lacking the extra domain A (EDA) or B (EDB) are particularly significant with regard to their biological functions. Plasma FN (soluble) lacks both EDA and EDB domains; in contrast, FN assembled into ECM contains variable mixtures of cellular FN with or without EDA and EDB domains (see Fig. 4-10).
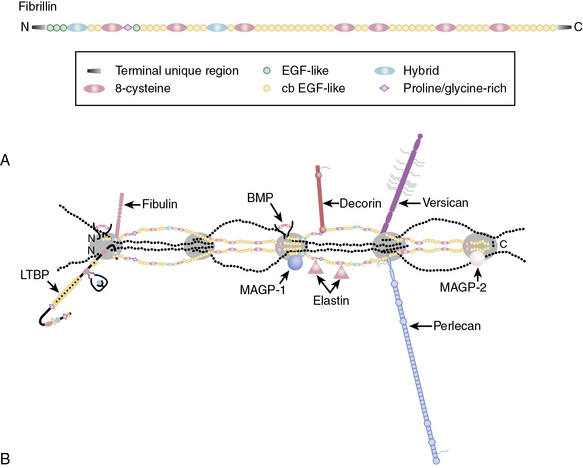
Figure 4-9 A, Domain organization of fibrillin.
(Adapted from Ramirez F, Sakai LY: Biogenesis and function of fibrillin assemblies. Cell Tissue Res 339:71–82, 2010.)
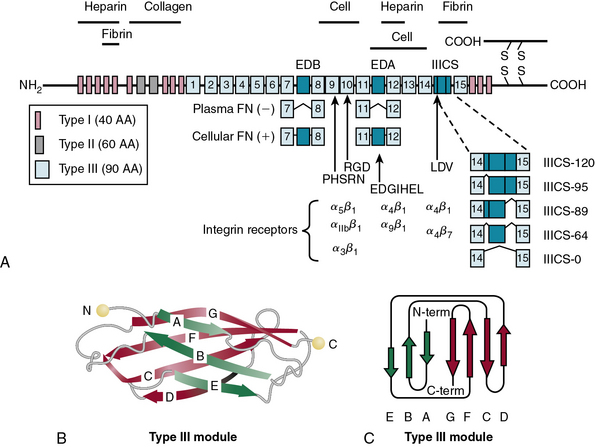
Functional Domains of Fibronectin
Fibronectin is a multifunctional molecule with a series of specialized modules.67,68 Proteolysis of FN generates a number of fragments that bind to specific ligands. The NH2-terminal 70-kD fragment of FN binds to a surprisingly large number of ECM ligands that include collagen, gelatin, fibrin, and heparin. The 70-kD FN also binds to some gram-positive bacteria (e.g., Staphylococcus aureus, Streptococcus pyogenes, Streptococcus pneumoniae) via the so-called microbial surface components recognizing adhesive matrix molecules (MSCRAMM). Thus lipoteichoic acid, M proteins, and several other bacterial adhesins anchored in the cell wall bind to FN and enhance opsonization and phagocytosis of bacteria.73 Gram-negative bacteria do not bind to FN.
The collagen-binding domain
The collagen-binding domain of FN includes type I repeats 6 to 9 and type II repeats 1 and 2.74,75 The first component of complement C1q, which contains a collagen-like structure, also binds FN.76 Denatured collagen (gelatin) has a much greater affinity for FN. Several FN binding sites exist along the collagen α chain, including a high-affinity site in type I collagen in the amino acid sequence targeted for cleavage by MMP-1 and MMP-2. It has been posited that the gelatin-binding domain of FN facilitates clearance of denatured collagen from circulating plasma. However, since triple-helical domains of fibrillar collagens may be partially unwound at body temperature, such local unraveling of the triple helix facilitates FN binding to native collagen and modulates its interactions with other molecules.31,33
The cell-binding domain
Fibronectin binds to cell surfaces via specific heterodimeric receptors called integrins that initiate intracellular signal transduction.68,77,78 Although FN binds to a number of integrins (e.g., α4β1, α5β1,αvβ3, αvβ1), most FN functions in vascular development may be mediated via α5β1 integrin.67 Studies in transgenic mice have demonstrated that specific ablation of α5 integrin causes the most severe defects in vessel formation. The mechanistic relationship between α5 integrin and FN is further highlighted by the observation that the vascular phenotypes of α5β1 integrin knockout and FN-ablated mice are extremely similar.79–81 The RGD site, located in the tenth type III repeat of FN, binds to α5β1 integrin, and this interaction is obligatory for intracellular signaling. The alternatively spliced variants of FN bind to other integrins. For example, a segment of EDA binds to α4β1 integrin, whereas a peptide located in the IIICS segment can bind to both α5β1 and α4β7 integrins.
Based on a number of in vitro assays, subdomains of FN, particularly EDA and EDB, have been ascribed many functions that include cellular adhesion, mitogenic signal transduction, dimer formation, matrix assembly, and regulation of cytokine-dependent secretion of MMPs.68,77,78 However, these studies must be interpreted with caution and need in vivo corroboration. This caveat is highlighted by the observation that mice engineered to express FN without either EDA- or EDB-encoding exons develop normally. Conversely, deletion of both EDA and EDB exons leads to severe cardiovascular anomalies and premature death.82,83
Incorporation of FN into insoluble ECM is a cell-mediated process that is obligatory for vasculogenesis. The NH2-terminal domain of soluble FN binds to the cell surface and is converted into disulfide-linked polymers. Polymerization of FN occurs at specialized surfaces of many cells, including SMCs and fibroblasts, and is coordinated by integrins.84 Since integrins α5β1, aIIbβ3, or αvβ3 can polymerize FN and incorporate it into larger ECM aggregates, different integrins appear to be functionally redundant.64
The heparin-binding domain
The heparin-binding domain of FN is located at the NH2 terminus and overlaps the fibrin-binding site (see later discussion). Several polyanionic molecules (e.g., heparin, heparan sulfate, dextran sulfate, DNA) bind to FN; this binding is specific, since other polyanionic molecules (e.g., chondroitin sulfates, dermatan sulfate [DS]) do not. Some of these macromolecules bind to FN in a cooperative fashion. For example, the presence of heparan sulfate or hyaluronic acid enhances the association between FN and gelatin. Similarly, FN causes precipitation of type I or type III collagens, but only in the presence of heparin. Such cooperative binding of diverse ligands to various modules of FN is likely to facilitate its incorporation into a tissue-selective ECM in vivo.68
The fibrin-binding domains and clotting
The highly organized architecture of blood vessels and their cellular elements are perturbed by persistent hypertension, atherosclerosis, and other vascular pathologies. At these putative sites of injury, thrombus formation is invariably initiated by platelets. Fibronectin participates in blood coagulation and thrombosis.85–87 The human FN monomer contains a fibrin-binding domain at its carboxyl end, and the complex of FN and fibrin is cross-linked by factor XIIIa. The integrin αIIbβ3 found on platelets is recognized by fibrin and FN. Under static conditions, FN binds to αIIbβ3, αvβ3, and α5β1 integrins on platelets; glycoprotein Ib on platelets also binds FN in vitro. The known interactions of various types of FN (e.g., plasma, cellular, basement membrane, α granule stored FN in platelets) with platelets and ECM macromolecules suggest that FN might engage in thrombus formation by both direct and indirect mechanisms.85–87
Clot formation serves a dual function of restoration of vascular integrity and assembly of provisional ECM needed in the initial phase of remodeling and regeneration of injured tissues.88 The provisional ECM assembled in a clot that is also enriched in growth promoting factors facilitates phenotypic transformation of fibroblasts into myofibroblasts. This is followed by deposition of a more permanent ECM that is primarily laid down by myofibroblasts. Thus, an optimal wound healing and repair is dependent on sequential maturation of the ECM. Somewhat similar cell-ECM interactions are believed to occur in atherosclerotic lesions, where VSMCs acquire a proliferative and highly synthetic phenotype not unlike that of myofibroblasts. Increased expression of EDA and EDB domain–containing FN is often associated with a phenotypic transformation of VSMC and transdifferentiation of fibroblasts into myofibroblasts. It is interesting to note that α9β1 and α4β3 integrins that specifically recognize the EDA domain are present on ECs but absent on the surface of platelets.
Laminin
Laminins belong to an ancient family of glycoproteins that polymerize into cruciform structures that form the structural scaffold of all vascular basement membranes. Each laminin molecule is a trimer that consists of one α, one β, and one γ laminin chain. Individual polypeptide chains are joined via a long coiled coil to produce a molecule with one long arm and up to three short arms.89–91 The basement membranes of Hydra contain primordial laminin-like proteins, and there are at least four genes that encode laminins in Caenorhabditis elegans and Drosophila melanogaster. As shown in Figure 4-11, in mammals, there are five distinct α, three β, and three γ chains of laminin that are encoded by LAMA1-5, LAMB1-3, and LAMC1-3 genes, respectively.92,93 Thus, it is theoretically possible to generate more than 45 different laminin trimers; at least 18 distinct isoforms of mammalian laminins have been described to date.90 It is likely that additional isoforms of laminins remain to be discovered.
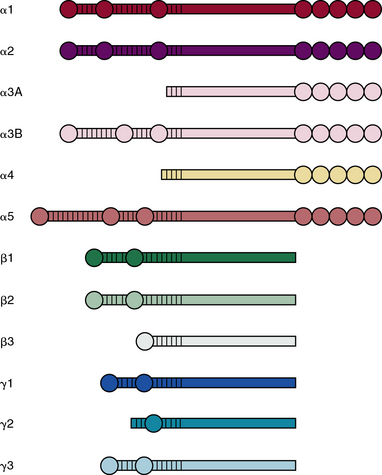
Figure 4-11 Structural motifs found in various laminin subunits.
(Adapted from Durbeej M: Laminins. Cell Tissue Res 339:259–268, 2010.)
The process of trimer formation is not random and is likely to be tissue- and cell-specific. According to a recently adopted system of nomenclature,89 laminin isoforms are named according to their chain composition; for instance, LM-111 consists of α1, β1, and γ1 chains, whereas laminin-511 is made of α5, β1, and γ1 chains. A single type of chain may be incorporated into more than one laminin isoform; for example, laminin-411, laminin-421, and laminin-423 laminins all contain α4 chain.
Various laminin α-chains show tissue-specific expression and are involved in formation of unique basement membranes at different locations in the body.94–96 Laminin-111 (α1β1γ1), the most abundant and best studied laminin, is highly expressed during embryogenesis; laminin-332 (α3β3γ2) and laminin-311 (α3β1γ1) are found preferentially in the basement membranes underlying stratified epithelia of the skin. Basement membranes of the vascular endothelium are enriched in laminins containing α4 or α5 chains.97 The basement membranes of most vessels contain laminin-411 (α4β1γ1) and laminin-421 (α4β2γ1). The laminin α5 chain is expressed by endothelium of the capillaries and venules that contain laminin-511 and laminin-521 in their basement membranes.
All laminin chains share a common structure, with a tandem array of globular and rodlike domains that invariably fold into a cruciform shape, as seen in the prototype, laminin-111 (Fig. 4-12). The NH2-termini of all laminin chains contain globular domains (LG) separated by laminin EGF-like (LEa, LEb, LEc) motifs. The α chain also contains two additional globular domains named L4a and L4b. Similarly, the β and γ chains contain unique LF and L4 domains, respectively. The shaft of the cross is a helical coiled coil formed by one α, one β, and one γ chain of laminin (see Fig. 4-12). The five LG domains of the laminin α chain are attached at the base of the cross. The polymerization of laminin and its incorporation into the supramolecular scaffolds of basement membranes is a cell surface receptor–mediated process, a situation reminiscent of the supramolecular assembly of FN.
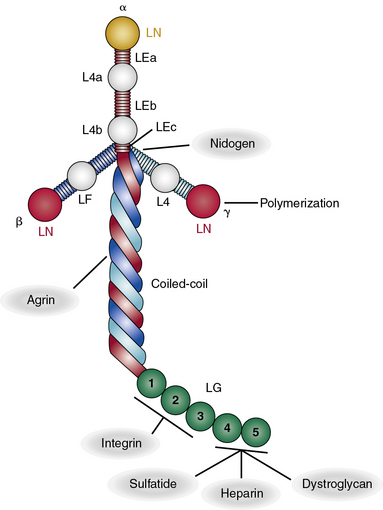
As illustrated in Figure 4-12, various domains of laminin possess unique functional properties that include promotion of architectural scaffolding, binding to cell surfaces via sulfated carbohydrates, or interacting with specific integrins and α-dystroglycan.90,91 Thus, the LN domain, which is present in the α1, α2, α3B, α5, β1, β2, β3, γ1, and γ3 chains, is needed for self-assembly of laminins into trimers that further aggregate into a large network to which additional ECM molecules bind. At least eight unique integrins (α1β1, α2β2, α3β1, α6β1, α6β4, α7β1, α9β1, and αvβ3) and four different types of syndecans bind to laminin.90,91 Similarly, the Lutheran blood group glycoprotein binds to laminins containing the α5 chain, whereas nidogen-1 and -2 bind to a specific region in the laminin γ1 and γ3 chains. Finally, the HS PG agrin binds the central region of the coiled coil domain of laminin (see Fig. 4-12).
Intact laminin is specifically cleaved by a number of proteases (e.g., furin, MT1-MMP, MMP-2, BMP1, plasmin). Controlled proteolysis may uniquely fragment laminin to release its functional domains as well as uncover additional biologically active domains that remain cryptic in the intact molecule. Intact laminin or its proteolytic fragments can modulate adhesion, migration, and phenotypic differentiation of many cell types, hence affecting disparate physiological and pathological events.98,99 These actions of laminins are mediated via their ability to ligate cell surface receptors that trigger intracellular signaling pathways (see later discussion).
The biological functions of laminin isoforms have been deduced from in vitro studies as well as from transgenic mice in which one or both alleles of a particular laminin chain are mutated or deleted.96,100 Congenital ablation of laminin-111 and laminin-511 leads to embryonal death in mice.90 Mice containing dysfunctional α4 or γ1 genes have defective kidneys, placenta, brain, lungs, and limbs. A number of mutations in the human LAMB2 gene are associated with Pierson’s syndrome, an autosomal recessive disorder characterized by ocular and renal defects.101 A deletion of the Lamb2 gene in mice resulted in renal and ocular defects reminiscent of the Pierson’s syndrome.102 Even more significantly, the mutant phenotype in the transgenic mice could be rescued by exogenous expression of the laminin β2 chain.
Laminins are a key determinant of the structure and function of basement membranes and are therefore essential for optimal performance of the vascular tree.91,101 In addition to contributing to the formation of the blood vessels, laminins (1) impart structural and mechanical stability to mature blood vessels, (2) modulate the barrier function of vessel walls, and (3) act as mechanosensors of shear stress relayed by ECs. In the developing embryo, the cell-attached scaffold of polymerized laminin nucleates the formation of basement membranes that acquire structural maturity, ligand diversity, and functional complexity as other ECM molecules (e.g., IV collagen, PGs) are incorporated into the laminin scaffold.96
The barrier function of blood vessel walls is facilitated by laminins that directly and indirectly regulate movement of charged macromolecules, leukocytes, and tumor cells through subendothelial basement membranes.90,91,101,103 LM-411 was shown to facilitate extravasation of T cells, whereas their transmigration through the vessel wall was restricted by LM-511. Similarly, the growth of primary tumors and metastasis were accelerated in α4−/− mice that elicited defective angiogenesis (e.g., irregular growth of vessel sprouts, dilated vessels). Signaling mechanisms induced by the α4 chain that binds to EC-specific integrins (α6β1 and α3β1) play a vital role in angiogenesis. It is therefore significant that not only intact laminin but also its subfragments may be functionally relevant for angiogenesis under physiological and pathological conditions. The COOH-terminal LG4-LG5 domains of α4 laminin inhibit EC migration and blood vessel sprouting in vitro. Mice lacking α4 are born with widespread defects in their vasculature.104 Expression of endothelial laminins is induced by proinflammatory cytokines that may promote transmigration of circulating leukocytes and tumor cells.
Proteoglycans
Glycosaminoglycan are linear chains of negatively charged polysaccharides that may be divided into (1) sulfated GAGs such as chondroitin-4 and -6 sulfates (CS), DS, HS, heparin, keratan sulfate (KS), and (2) nonsulfated GAGs represented by hyaluranan.105–107 The repeating disaccharide unit of a GAG contains one sugar that is invariably a glucosamine or a galactosamine, and the other is either a glucuronic or an L-iduronic acid; KS contains a galactose in the place of the hexuronic acid. Hexosamine is, as a rule, N-acetylated except in HS, where it may be N-sulfated. The structures of the repeating units of GAGs are presented in Figure 4-13.
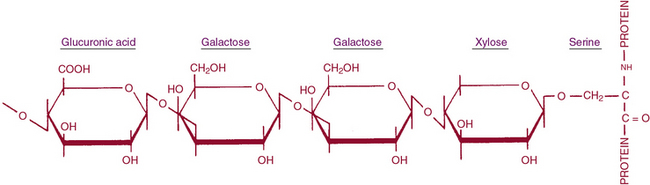
Figure 4-13 Oligosaccharide linkage between glycosaminoglycans (GAGs) and protein core.
(Adapted from Silber JE: Structure and metabolism of proteoglycans and glycosaminoglycans. J Invest Dermatol 79:31, 1982. Copyright by Williams & Wilkins.)
The uronic acid linkage in CS and HA is β1,3, and the analogous linkage in DS is α1,3 because of the presence of L-iduronic acid; the hexosaminidic linkage in all three GAGs is β1,4. The disaccharide unit of KS is β-galactosyl (1,4)-N-acetylglucosamine that is polymerized by a β1,3 glucosaminidic bond. The structure of HS contains some disaccharide units composed of D-glucosamine and D-glucuronic acid and others of D-glucosamine and L-iduronic acid. The uronyl linkage in HS is α1,4 rather than β1,3, and the hexosaminidic linkage is α1,4 rather than β1,4. The glucosamine residues are partly N-sulfated as well as O-sulfated. Heparan sulfate is structurally related to the anticoagulant heparin, which actually has a higher sulfate content than HS. The GAG chains range in molecular weights from several thousand to several million daltons. All hexosamine residues are N-substituted with either acetyl or sulfate groups. In addition, most GAGs have ester O-sulfate on one or both sugars of the disaccharide unit.105–108
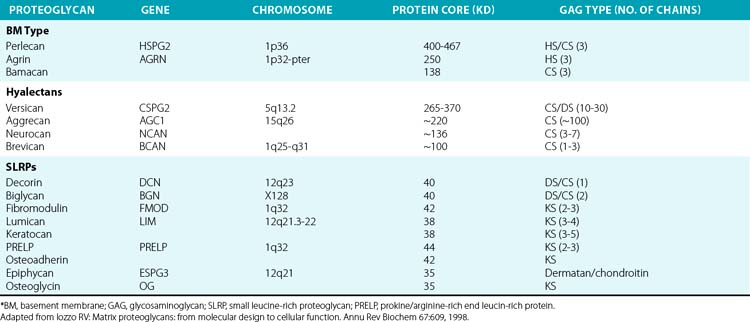
The superfamily of PGs consists of about 30 members that are subdivided into three classes: modular PGs, small leucine-rich PGs (SLRPs) and cell surface PGs. Modular PGs are further subdivided into hyalectans and non-HA-binding PGs (Table 4-4 and Fig. 4-14). A number of recent reviews105–109 may be consulted for more comprehensive structural and functional descriptions of various classes of PGs. Following is a brief outline of the major PGs, with an emphasis on their function in subendothelial ECM.
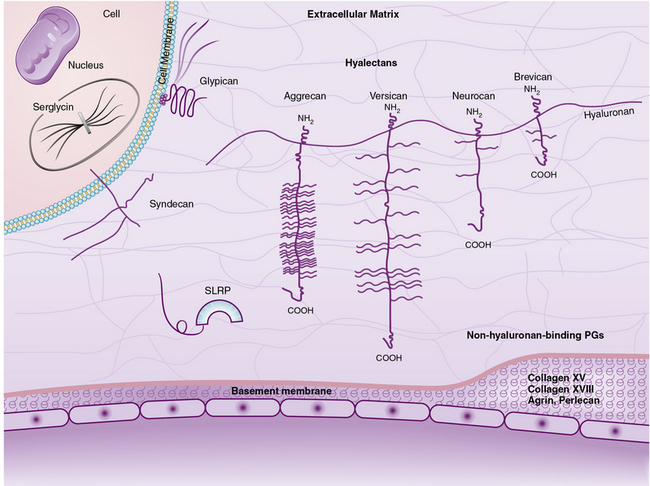
Hyaluronan- and Lectin-Binding Proteoglycans
Four distinct PGs—versican, aggrecan, neurocan, and brevican—constitute the hyalectan family of PGs.105–109 The tripartite organization of their core proteins consists of the NH2– and COOH-terminal domains separated by a distinct central domain where GAGs are attached. The amino terminal domain of these PGs binds to HA and their carboxyl termini contain lectin-like domains, thus the name hyalectans (see Table 4-4). The central domain of hyalectans contains variable numbers of GAG chains, ranging from 3 seen in brevican to around 100 found in aggrecan. Versican, the largest member of the hyalectan family, may be considered a prototype109,110 (see Fig. 4-14 and Table 4-4). Versican and other hyalectans are believed to connect lectin-containing proteins on the cell surface with HA in the intercellular space. Versican has an immunoglobulin (Ig)-like motif and two tandem HA-binding domains near the NH2-terminus; an EGF-like domain and a lectin- like motif are located near the COOH-terminus of versican. Four alternatively spliced versican isoforms (V0, V1, V2, and V3) are preferentially expressed in different tissues. Versican binds to numerous cell-associated and ECM molecules that include type I collagen, tenascin, fibulins, fibrillin-1, FN, selectins, chemokines, CD-44, integrin β1, EGF, and Toll-like receptors.109–113
Aggrecan typically contains around 100 CS-enriched and 30 KS-enriched GAGs that are covalently linked to about a 220-kD core protein (see Table 4-4). As the most abundant constituent of cartilage ECM, aggrecan is found in giant aggregates with link proteins and HA, and occupies a large hydrodynamic volume (2 × 10− 12 cm3) that may be equivalent to a bacterium.108 The lectin modules of both versican and aggrecan can interact with simple sugars found in glycoproteins; this binding is calcium dependent.114 Defective cartilage and shortened limb development have been demonstrated in mice, chickens, and humans that contain mutated aggrecan genes.115,116
Neurocan and brevican, with tripartite organization of core proteins characteristic of hyalectans, are the most abundant PGs of this class in the central nervous system.117 Brevican is synthesized in the brain as a secreted full-length molecule, as well as a truncated form that lacks the COOH-terminal domain. The short form of brevican is attached to the plasma membrane via a GPI anchor. Neurocan and brevican promote neuronal attachment and outgrowth of neurites in developing neurons. Brevican activates EGF receptor (EGFR) signaling that results in enhanced expression of cell adhesion molecules such as FN. Highly aggressive central nervous system tumors elicit accelerated synthesis and proteolysis of brevican and thus may promote tumor metastasis.
Non-Hyaluronan-Binding (Basement Membrane) Proteoglycans
Perlecan, agrin, and bamacan are usually present in the vascular and epithelial basement membranes of mammalian tissues.97,109 Whereas the three GAG chains of perlecan and agrin consist primarily of HS and CS, the three bamacan GAG chains contain only CS. The core protein of the human perlecan is about 470 kD in size and contains five well-defined domains (I through V) that are mosaics of sequences found in other proteins. Thus, domain I of perlecan consists of three serine-glycine-asparagine triplets to which HS side chains are covalently attached, and an SEA module (containing sperm protein, enterokinase, and agrin homology sequence). Domain II contains four low-density lipoprotein (LDL) receptor class-A repeats located next to IgG-like motifs. Three laminin IV globular domains interspersed with nine laminin EGF-like motifs comprise the domain III of perlecan. Domain IV contains 21 IgG-like motifs that share homology with neural cell adhesion molecules (N-CAM). Finally, domain V is made of three laminin G motifs separated by two sets of EGF-like repeats; the 85-kD COOH-terminal fragment of perlecan called endorepellin is a potent antiangiogenic molecule.97,109,118
In addition to imparting structural integrity to the basement membranes, PGs modulate cellular behavior because of their ability to interact with a large number of molecules, as exemplified by perlecan.97,109,110,118 Although perlecan is embedded within the subendothelial basement membranes, it binds to fibroblast growth factor 2 (FGF-2), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), several cell surface molecules, and ECM proteins. Heparan sulfate GAGs of perlecan associate with FGF-2 and serve as its reservoir in blood vessel walls. During aortic morphogenesis, there is an inverse correlation between perlecan expression and smooth muscle proliferation in the rat. Perlecan interacts with α1β2 integrin, an integrin that also binds to fibrillar collagens. Dynamic interactions of perlecan and fibrillar collagens with integrins potentiate atherosclerosis, angiogenesis, and carcinogenesis. Perlecan regulates the motility of ECs and transformed cells and promotes metastasis.119 Perlecan-null mice die in utero or shortly after birth.
Small Leucine-Rich Proteoglycans
Nine known members of this family of PGs, characterized by central leucine-rich domains and DS/KS GAG chains, are currently known.120 Based on their primary structures and evolutionary relationships, SLRPs may be further divided into three subclasses (see Table 4-4). Members of the SLRP family have been121 implicated in diverse functions that include regulation of growth factor accessibility (e.g., TGF-β) and control of collagen fibrillogenesis.119 Presence of decorin, fibromodulin, or lumican retards collagen fibrillogenesis in vitro.122–125 A reciprocal relationship between the amount of decorin and rate of collagen fibril growth in the developing tendon of the chicken was also demonstrated.126 The abnormal collagen fibril formation and reduced tensile strength of the skin seen in decorin knockout mice support a functional role for decorin in proper collagen fibrillogenesis.122
Decorin, a prototype of the SLRP, is organized into four discernible domains.109 Core protein of decorin binds TGF-β1, -2, and -3 with high affinity. Because the TGF-β/decorin complex is incapable of intracellular signaling, decorin is believed to facilitate deposition of inactive TGF-βs at specific tissue locations. Perturbation of this interaction and activation of TGF-βs may occur in response to inflammatory reactions. Decorin itself has been shown to regulate cell proliferation, and ectopic expression of decorin could suppress the growth of cancer cells. Decorin directly interacts with EGFR with a 1:1 stoichiometry; this interaction inactivates EGFR and its downstream signaling.127 Vascular ECs undergoing cord formation that precedes angiogenesis in vitro synthesize decorin, whereas proliferating ECs showed enhanced synthesis of biglycan; these two structurally similar SLRPs appear to regulate EC phenotype in an opposite manner.
Cell Surface Proteoglycans
Two main classes of cell surface PGs are membrane-spanning syndecans and GPI-linked PGs represented by glypicans.109,128,129 There are four members of the syndecan subfamily. Syndecan-4 is distributed ubiquitously, but syndecan-1, -2, and -3 have more restricted tissue- or development-specific expression.128–130 For example, syndecan-1 is highly expressed in the developing embryo, and syndecan-3 is primarily enriched the neural tissue. Syndecans are involved in multiple signaling pathways to regulate cell proliferation, adhesion, motility, and differentiation. The cell surface HS-enriched syndecans serve as co-receptors for FGF and EGF to facilitate their binding and signal transduction. Syndecan-1 is cleaved by MMPs, and the soluble ectodomain of syndecan promotes tumor growth and invasiveness in vitro.121,131,132 Exogenous treatment of microvascular ECs with EGF and FGF leads to shedding of syndecan-2 that in turn affects EC behavior.133
The subfamily of GPI-linked cell surface PGs includes six member glypicans and a splicing variant of brevican that lacks the COOH-terminal lectin binding and EGF motifs. While most tissues express glypican-1, expression of glypican-3, -4, and -5 is restricted to the central nervous system. In contrast, glipican-2 is expressed abundantly in the embryo, whereas glypican-6 is found mainly in the heart, kidney, and intestine. Glypican-3, which inhibits hedgehog (Hh) signaling by competing for its receptor Patched, is up-regulated in neuroblastoma and Wilms tumor. Glypican regulates binding and signaling of a number of other morphogens and growth factors that include Wnts, slit, FGFs, insulin-like growth factors (IGFs), and BMPs. The regulatory actions of glypicans on proliferation and differentiation of cells appear to be context dependent.129,134,135
Biosynthesis of Proteoglycans
Biosynthesis of all PGs involves similar steps, the rate-limiting step being translation of the core protein.119,136,137 Following its synthesis, core protein undergoes covalent modification with GAG chains that begins with the linkage of xylose to a specific serine(s). Proteoglycan synthesis occurs in late ER and the Golgi with attachment of a xylose residue to the OH group of serine in the protein core.138 Linking of galactose to xylose is carried out by galactosyl transferase. A second galactose is then transferred by a distinct galactosyl transferase that is followed by addition of the first glucuronic acid by UDP–glucuronic acid transferase. Growth of the GAG chain then occurs by alternating transfer of hexosamine and uronic acid residues. Thus, the UDP derivatives of N-acetylglucosamine and glucuronic acid are precursors for HA, heparin, and HS; whereas N-acetylgalactosamine and glucuronic acid are precursors for CS and DS. After addition of the first sugar, elongation occurs by the same N-acetyl-hexosaminyltransferase and glucuronosyltransferase, regardless of which chain is being synthesized. The respective chains are variably modified by pathway-specific epimerization and sulfation reactions to yield iduronic acid and sulfation.107,127
Degradation of Proteoglycans
Compared to collagen and elastin, PGs have more rapid rates of turnover, with turnover of 2 to 10 days in younger animals.97,109,110,119 Degradation of the PGs involves proteolysis of the core protein by MMPs, breakdown of the sugar chain, and desulfation of sugars. The dramatic loss of cartilage matrix that results from experimental intravenous injection of papain illustrates the importance of the protein core to the structural integrity of PGs. Fibroblasts, macrophages, and neutrophils produce a variety of enzymes that can degrade PGs at neutral pH. Degradation of sugar chains occurs mainly in lysosomes. Perhaps the best characterized GAG-degrading enzyme is testicular hyaluronidase, which degrades HA, CS, and DS to oligosaccharides. Other glycosidases are required to complete the breakdown of oligosaccharides to monosaccharides. Lysosomes contain glucuronidase and N-acetylhexosaminidases that remove the terminal glucuronic acid and hexosamine residues, respectively. Lysosomes also contain β-xylosidase, β-galactosidase, and α-iduronidase, which complete the breakdown. Lysosomal sulfatases are responsible for removal of sulfate groups from oligosaccharides. Inherited defects in the activity of various GAG-degrading enzymes cause mucopolysaccharidoses, characterized by faulty catabolism of one or another type of GAGs.97,109,110,119
Subendothelial Extracellular Matrix as a Regulator of Cell Signaling
The central role of ECM, to endow blood vessels with the mechanical ability to undergo repeated cycles of extension and passive recoil throughout the life of the organism, has been appreciated for nearly a century. However, in recent years we have also discovered that ECM, beyond providing scaffolding for the vascular walls, has many effects on their cellular inhabitants. Thus ECs, pericytes, and VSMCs dynamically sense their physical (e.g., shear stress) and biochemical microenvironment and adjust cellular behavior accordingly.139–141 Numerous experimental observations indicate that ECM is a key component of this bidirectional communication between cells and their microenvironment.142
Vascular cells actively synthesize and mold their ECM into unique configurations to ensure it optimal stiffness and deformability; molecular constituents of the subendothelial ECM in turn profoundly modulate the adhesion, polarity, motility, survival, proliferation, and differentiation of vascular cells. The fibrillar and nonfibrillar ECM interact with dozens of cell-associated and extracellular molecules that alter the signaling repertoire of ECs, VSMCs, and platelets.139,143,144
The functional diversity of ECM emanates from the architectural complexity of its molecular constituents, which have myriad specifically folded domains, some uniquely juxtaposed in the basement membranes of the vascular tree. Subendothelial ECM, in addition to forming an adhesive scaffold via integrins that are capable of bi-directional intracellular signal transduction, serves as a reservoir of growth factors.78,145 The anchorage-dependent survival and growth of normal epithelial cells, ECs, and VSMCs depends on their adhesive interactions with ECM. When this adhesive normalcy is lost, cells acquire anchorage-independent growth potential, a hallmark of cancerous transformation and metastasis.146 Thus, as highly organized solid-phase signal inducers, ECM molecules can integrate numerous signals in the microenvironment of the blood vessels to regulate their development, maturation, and homeostasis. The following is a brief summary of the mechanisms that underlie the dynamic two-way signaling between cells and their ECM.
Extracellular Matrix–Integrin Bi-Directional Signaling
Each of the many molecules found in the vascular ECM recognizes a variety of cell surface proteins predominated by integrins, a superfamily of heterodimeric transmembrane receptors (Fig. 4-15). Integrins are assembled by selective pairings of 18 individual α and 8 unique β chains. Twenty-four integrin receptors with distinct ligand selectivity, cell-specific expression, and signaling properties have been described in mammals.147,148 The extracellular segment of the α subunit of integrin consists of a β-propeller domain, an I-domain, and three Ig-like domains. The ectodomain of the integrin β subunit also has a modular organization, with two tandem I-domains, an Ig hybrid motif, a plexin-semaphorin-integrin (PSI) domain, and four EGF-like domains. Integrins specifically bind to several ECM molecules, their subfragments, and divalent cations. With respect to vasculature, ECs express integrins that specifically bind to collagen (α1β1, α2β1, α10β1, and α11β1), FN (α4β1 and α4β1), and laminin (α3β1, α6β1, and α6β4). Based on a number of in vitro and in vivo observations, at least eight integrins (α117, α2β1, α3β1, α4β1, α5β1, α6β1, αvβ3, and αvβ5) have been implicated in the process of angiogenesis.149,150 In contrast, leukocytes express a number of unique integrins on their surface that include α4β7, αLβ2, and αMβ2 integrins. We should note that the ligand selectivity of integrins is far from absolute, as exemplified by αvβ3, which binds to several RGD sequence–containing proteins and peptides.147,148 The following discussion summarizes numerous observations that underscore the fact that integrins lie at a unique crossroads of extracellular microenvironment, cytoskeleton mechanics, and intracellular signaling networks to alter the behavior of vascular walls in health and disease (see Fig. 4-15).
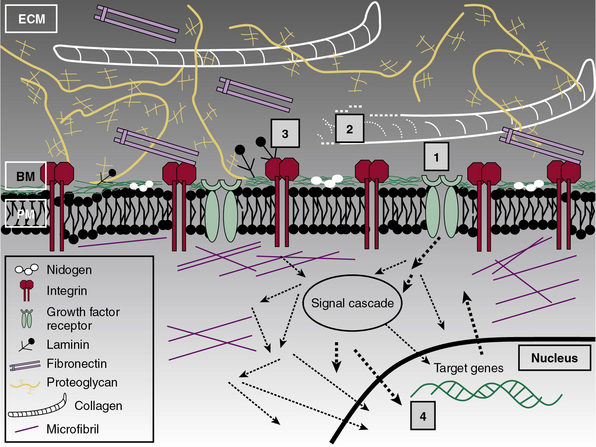
Integrins are unusual proteins among the transmembrane receptors, with an ability to relay signals in both directions.147,148,151 The intracellular changes induced by ECM-liganded integrins are referred to as outside-in signaling. Conversely, inside-out signaling occurs when intracellular biochemical changes trigger reorganization of the cytoskeleton, which alters the shape of the ectodomain of integrin and its affinity for the ligand. The ECM-liganded integrins are clustered as dot-like foci that sequentially evolve into focal adhesions, fibrillar adhesions, and finally into supramolecular three-dimensional adhesions. Such mass clustering of integrins into focal adhesions results in summation of numerous weak-affinity interactions of individual integrins into an adhesive unit with high affinity and high avidity.
Clustering and activation of integrins induce a number of characteristic biochemical and physical changes in the cells that are collectively referred to as outside-in signal transduction.143,144,152 Since integrins themselves lack catalytic activity, they signal indirectly via a host of accessory proteins that assemble multi-protein platforms to recruit bona fide signaling catalysts into the focal adhesions. The assembly of bi-directional signaling complexes depends on interactions among a large number of integrin-binding proteins (e.g., talin), adapter molecules (e.g., vinculin, paxillin), and signaling enzymes (e.g., FAK, RhoA-kinase [ROCK], myosin phosphatase). It has been estimated that the “integrin adhesome” consists of more than 150 unique proteins; therefore it is conceivable that recruitment of unique sets of adapter and signaling molecules to focal adhesions might be different under differing physiological and pathological conditions.153,154
The inside-out signaling of integrins is best exemplified by their own activation, particularly on the surface of leukocytes, where integrins are normally present as inactive receptors.143,144,152 This mechanism enables the immune cells and platelets to circulate through the bloodstream without undesirable adherence to the vessels or causing premature thrombosis, respectively. The activation of T cells, neutrophils, and platelets can occur by integrin-independent pathways (i.e., occupancy of the T-cell receptor by MHC-loaded antigenic peptides, ligation of selectins on the surface of neutrophils, and interaction between platelet glycoprotein IV and collagen. Such activation initiates a series of intracellular reactions that lead to binding of kindlin and talin to intracellular tails of β-integrin and its conformation-dependent activation. As a consequence, the affinity of integrin for its ligands is greatly enhanced, as is its signaling strength.
Integrin signaling is mainly propagated by kinases and phosphatases that, by dynamic phosphorylation and dephosphorylation, alter protein-protein interactions of their substrates and catalytic activities of signaling enzymes in focal adhesions.143,144,152,155 The major enzymes involved in this process include tyrosine kinase, FAK, protein kinase C (PKC; a serine/threonine kinase), a lipid kinase (PI3-kinase), and the receptor protein tyrosine phosphatase α (PTPα). Activation of integrin immediately initiates tyrosine phosphorylation of specific substrates that include the integrin β tails. Concomitantly, there is a surge in intracellular concentration of lipid second messengers, phosphatidylinositol-4,5-bisphosphates and phosphatidylinositol-3,4,5-trisphosphates, and a reorganization of the cytoskeleton.
As shown in Figure 4-16, the characteristic physical link between integrins and cytoskeleton is initiated and maintained by integrin-bound proteins that also bind to actin (e.g., talin, filamin) in collaboration with other proteins that regulate the structure of cytoskeleton indirectly (e.g., paxillin, FAK, kindlin). Additionally, the integrin-cytoskeleton linkage and signal propagation is critically dependent on actin-binding proteins (e.g., vinculin) and several signaling adapters (e.g., RhoA, Rac1, cdc42). Numerous biochemical and biophysical analyses have revealed that talin, vinculin, α-actinin, and integrin-linked kinase (ILK) are indispensable for bidirectional signaling of focal adhesions.143,144,152,155
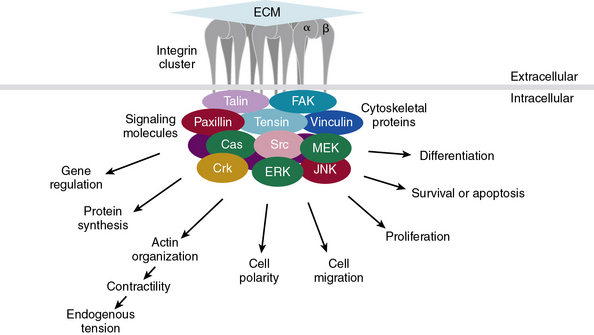
Figure 4-16 General model of cell–extracellular matrix (ECM) adhesions and downstream signaling pathways.
Cell-ECM adhesions containing clusters of integrins recruit cytoplasmic proteins (e.g., talin, paxillin, vinculin) and kinases (focal adhesion kinase [FAK], Src, and c-jun NH 2-terminal kinase [JNK]), which in cooperation with other cell surface receptors (growth factor receptors; see Fig. 4-17) control diverse cellular processes, functions, and phenotypes. Details of these interactions are described in text. ERK, extracellular signal regulated kinase; MEK, mitogen extracellular signal regulated kinase.
(Adapted from Berrier AL, Yamada KM: Cell-matrix adhesion. J Cell Physiol 213:565–573, 2007.)
The unique molecular composition of focal adhesions at different locations in the vasculature is likely to affect signal strength and quality.77,78,154 The striking diversity of signal transduction cascades documented in various cell types also involves crosstalk among the canonical and alternative signaling pathways (see Fig. 4-16). Finally, the growth factor microenvironment (see later discussion) greatly influences the mechanisms that control attachment, polarity, and directional motility of vascular cells. Aberration of these signals leads to dramatic changes in the ability of cells to attach, polarize, and migrate, as well as their growth factor– and anchorage-independent proliferation.
Signaling Triad of Extracellular Matrix, Integrin, and Growth Factors
Although the ECM-integrin signaling axis has been the focus of most investigations, it has become evident in recent years that subendothelial connective tissues make additional contributions to the bidirectional signaling of vascular and nonvascular cells. A number of growth factors and developmental morphogens are sequestered in the fibrillar and nonfibrillar constituents of ECM. Presumably, such ECM-bound factors have the potential to be released in a highly regulated manner in response to developmental cues or tissue injury and inflammation.78,145,146 Furthermore, a number of ECM molecules serve as coreceptors for growth factors, as is the case for FGF and VEGF; both bind to heparin and HS chains of PGs that are needed for optimal ligand presentation and signal transduction. Proteoglycans modulate TGF-β signaling in a number of ways. Transforming growth factor beta binds to decorin in the ECM and in this state cannot bind to its cell surface receptors. Additionally, the binding of TGF-β to its signaling receptors is dependent on a plasma membrane-bound PG (β-glycan) and the cell surface glycoprotein endoglin, both of which serve as co-receptors. Finally, some growth factors can independently activate a number of downstream effectors of integrin signaling (FAK, Src, PI3-K, MAPK); this is exemplified by synergistic modulation of ECM-integrin signaling by IGF-1 and PDGF (Fig. 4-17).
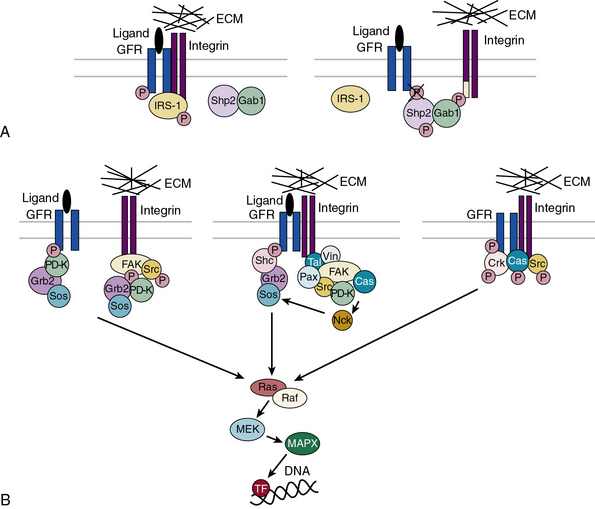
Figure 4-17 Regulation of growth factor receptor (GFR) signaling by integrins.
(Adapted from Alam N, Goel HL, Zarif MJ, et al: The integrin–growth factor receptor duet. J Cell Physiol 213:649–653, 2007.)
Comodulation of integrin and growth factor signaling has been documented for EGF, hepatocyte growth factor (HGF), IGF-1, and PDGF, both during early development of model organisms and in cell lines.156–162 Evidently, a trimolecular interaction among the BMP homolog DPP, its cell surface receptor, and type IV collagen is crucial for optimal relay of signals involved in morphogenesis of the dorso-ventral axis in Drosophila. A similar mechanistic interaction of HGF with two other proteins has been reported. Apparently, fibronectin- or VN-bound HGF forms a trimolecular complex that contains (FN or VN)-HGF, c-Met (HGF receptor), and α3β4 integrin. A functional role of these interactions was corroborated by showing that ligand-independent activation of c-Met by cellular adhesion plays a crucial role in the growth and invasive potential of epithelial cells in response to HGF. Finally, it has been reported that the downstream signaling evoked by the tyrosine kinase transmembrane glycoprotein EGFR was complemented by integrins; thus, EGFR was shown to laterally coaggregate with αvβ3, α2β1, and α6β4 integrins.
As reviewed in a number of excellent publications,78,144 the crosstalk between growth factors and integrins involves a number of mechanisms (see Fig. 4-17). First, it involves integrin’s ability to concentrate signaling adapters in close proximity of growth factor receptors to enhance their intracellular signaling ability. An additional mechanism of crosstalk may be dependent on lateral mass aggregation of integrins, a process posited to alter the location and/or concentration of growth factor receptors on the cell surface. The observation that α1β2 integrin and EGFRs were coaggregated at the foci of cell-cell contact bolsters such a mechanism. Furthermore, integrin-mediated adhesive interactions may alter the rate of internalization and degradation of some receptors (e.g., PDGFR). Conversely, integrin activation may lead to enhanced expression of some growth factors; thus, it was shown that increased biosynthesis of VEGF and IGF-2 occurred via the actions of α6β4 and α1β1A integrins, respectively. These observations and many others are consistent with the notion that bidirectional crosstalk between integrins and growth factors is highly relevant to cellular behavior and phenotype. However, the relative contribution of these two processes to bidirectional signaling remains controversial. Similarly, the hierarchical relationship between integrin and growth factor signaling pathways remains to be sorted out.78,143,145
Vascular Extracellular Matrix and Platelet Interaction
Circulating platelets act as sentinels of vascular integrity and do not attach to vessel walls under normal conditions. However, upon injury of blood vessel walls, platelets rapidly adhere to ECM of the subendothelium and neointima (formed after repeated damage to vessel wall) and aggregate with each other at the site of injury. The adhesive interactions between subendothelial ECM and platelets and thrombus formation have been extensively studied.86,163–165 Binding and activation of platelets by the subendothelial ECM is modulated by hemodynamic stress, composition of the vascular tissue, and extent and depth of the lesion.
The fibrillar collagens, of which types I, III, IV, V, and VI are widely distributed in the vascular ECM, are key initiators of thrombosis in injured vessels.7,9,166 The hemostatic cascade begins when platelets adhere to the exposed subendothelial collagens via vWF, a large glycoprotein that is constitutively synthesized by ECs.86,87 The vWF both circulates in plasma as a soluble molecule and is stored in the α granules of platelets. Endothelial or plasma vWF, which forms multimeric aggregates, binds to collagen. It is believed that high hemodynamic stress causes local uncoiling of multimeric vWF, further facilitating its binding to platelet GPIbα and collagen.167,168 Additional interactions of collagen with α2β1 integrin and glycoprotein VI (GPVI) synergize the binding of platelets to vascular ECM. Although a pivotal role of collagens in thrombosis is borne out by numerous observations, the relative contributions of individual fibrillar collagens to this process appears to be highly context dependent.86,87,169 This view is supported by the observation that platelets adhere to and are activated by types I-V collagens under relatively high and moderate hemodynamic stress. In contrast, binding and activation of platelets by type V-VIII collagens occurs at much lower shear stress, and type V collagen binds to platelets only under static conditions.
Over the years, many platelet-associated proteins have been claimed as candidate collagen receptors.170–178 However, a careful review of the literature would suggest that some of these claims reflect vagaries of different experimental models (in vivo vs. ex vivo), while others may have emanated from a lack of technical or interpretational rigor.87,166,169 Historically, the search for collagen receptors on platelets began with studies using purified type I and III collagens or their cyanogen bromide (CB) subfragments that were tested for their ability to bind and activate platelets. These studies led to the discovery that purified native type I/III collagens and some of their CB fragments interacted with platelet integrin α2β1. More recently, a series of overlapping triple-helical peptides spanning human collagen types I and III have been systematically tested for their ability to bind and activate platelets.179–181 These elegant studies have unraveled discrete sequence motifs on types I and III collagen that interact with receptors on the surface of platelets and vWF. Based on these analyses, it has been surmised that type I collagen contains four α2β1 integrin-binding sites of varying affinities; apparently, similar sequence motifs are also conserved in type III collagen. The molecular interactions between high-affinity type I collagen triple-helical peptide and α2β1 integrin have been elucidated with precision. These and related studies have shown that fibrillar collagens also interact with αIIbβ3, albeit indirectly. Finally, the use of well-defined triple-helical collagen peptides has led to the discovery of amino acid sequence motifs in types I and III collagen that specifically bind to the platelet surface GPVI, as well as to vWF.86,87,180
How collagen-platelet interactions culminate in the formation of a thrombus has been investigated in considerable detail.86,87,180 Under high shear stress, the obligatory first step is co-engagement of α2β1 integrin and vWF by collagens. Platelet proteins GPVI and GPIV (CD36) strengthen the initial encounter with collagen. Glycoprotein VI has a modular organization that is composed of 2 Ig domains, an O-linked glycosylated stalk, an intramembrane domain, and an intracellular tail. In platelets, GPVI exists in association with the FcR γ-chain, a transmembrane protein containing an immunoreceptor tyrosine activation motif (ITAM) motif. Interaction of platelets with collagen triggers phosphorylation of the FcR γ-chain and signaling that mimics signaling cascades associated with T-cell activation. The intracellular signaling is mainly driven by Ca++ mobilization that also occurs in response to other platelet-activating stimuli (e.g., thrombin, adenosine diphosphate [ADP]). The role of CD36 in thrombosis is somewhat debatable in light of the observation that nearly 5% of Japanese lack functional CD36 protein in their platelets without overt complications of hemostasis.
Platelets also adhere to other components of ECM, particularly to FN and laminin, both of which are essential ECM components of the subendothelial connective tissues.86,87,182,183 Since the original 1978 report of Hynes et al. supporting a role of FN in platelet adhesion, a number of apparently conflicting data have muddled this issue.86,87,182,183 There is little doubt that platelets adhere to FN; platelets possess two integrin receptors that bind FN (i.e., α5β1, αIIbβ3). Depletion of FN from plasma reduced the ability of platelets for thrombus formation on fibrillar substratum or subendothelium, and if FN was restored, interplatelet aggregation and activation was restored. The NH2-terminal 70-kD fragment of FN could be incorporated in the clot and was cross-linked to fibrin. Further bolstering a positive role of FN in thrombosis is the phenotype of mice with various gene knockouts. Thus, FN−/− mice were shown to elicit defective thrombus formation. Similarly, mice lacking both fibrinogen and vWF elicited thrombosis primarily driven by FN and other proteins at the sites of blood vessel injury. However, it was noted that FN-platelet interactions were apparently too weak to withstand the normal shear stress. Based on these observations, it has been posited that FN plays a complementary role in thrombus formation in the presence of fibrinogen and vWF.184,185
These paradoxical observations on the role of FN in platelet activation may be reconciled in a model that suggests that the ultimate response of platelets may be determined by their initial encounter with different sets of ECM molecules in the endothelium. It is known that platelets can assemble a fibrillar network of FN from soluble FN. However, when platelets attach to vWF, it suppresses their ability to deposit FN. Under these conditions, aggregation and activation of platelets may be driven mainly by fibrinogen-αIIbβ3 integrin interaction. In contrast, if fibrin or collagen initiates adhesion of platelets, they acquire a greater ability to convert plasma FN into supramolecular FN that gets incorporated it into thrombi. It follows then that aggregation and clustering of platelets via fibrinogen or assembled FN may be regulated in a mutually exclusive manner.86,87
In the injured vessel wall, platelets are exposed to various forms of laminin; thus, laminin-411 and laminin-511 are present not only in the vascular ECM but also in platelets that contain laminin-522. Laminins bind to platelets and thus may play a role in their adhesion and activation. Despite the presence of laminin-binding integrin receptors on the platelet surface, the relative role of laminin-mediated adhesion in thrombus formation remains to be more precisely delineated. Analogous to more nuanced regulation of FN-platelet interaction, where interplay among fibrinogen, vWF, and VN negatively regulate FN assembly, interactions among laminin, fibrin, and collagen elicit an opposite effect on FN. Thus, it is not unreasonable to conclude that the precise outcome of platelets’ encounter with the endothelium is highly context dependent.186,187
Finally, we should note that nonfibrillar constituents of the subendothelium might also impinge on the function of platelets in a number of ways. For instance, heparin binds to serine protease inhibitors (SERPINS), antithrombin III, and heparin cofactor II and thus accelerates formation of the thrombin-antithrombin complex. Thrombin is a pivotal serine protease of the coagulation pathway, and it also exerts a positive feedback effect on its own biogenesis by activating factors V and VIII. Inhibition of thrombin by GAGs is a key mechanism in the regulation of blood clotting.188–191 Platelets contain platelet factor 4 (PF4) in α granules, where it is stored in a complex with chondroitin-4-sulfate PG. When released, PF4 has the ability to neutralize the anticoagulant effects of heparin and heparan. The PG-PF4 complex is dissociated by conditions of high ionic strength and by GAGs. Based on these interactions, it is conceivable that any HS– and DS–containing PGs present in blood vessel walls could serve to control deposition of PF4 locally by competitively dissociating the PG-PF4 complex.
Perspectives
Consistent with their role in imparting mechanical integrity and tensile strength to blood vessels, a spectrum of recessive mutations in genes that encode fibrillar collagens and elastic fiber-associated proteins has been reported. Thus, haploinsufficiency of types I and III collagen have been associated with arterial aneurysms and Ehlers Danlos syndrome (EDS) type IV in patients. Ablation of type I collagen expression in mice led to death due to rupture of large vessels between day 12 and 14 of gestation. Although mutations in type I collagen gene were mainly associated with osteogenesis imperfecta (OI), a number of OI patients were predisposed to dissection and rupturing of their aorta. Finally, Biswas et al. reported that a point mutation in the COL8A2 gene (encodes transmembrane collagen type VIII) was associated with two different types of endothelial dystrophy in the cornea.192 It is noteworthy that aberrant turnover of ECM as a result of mutations in MMPs or TIMPs also yielded vascular phenotypes in transgenic mice. Mice deficient in either MMP-2 or MMP-9 are resistant to CaCl2-induced formation of aneurysm; conversely, disruption of TIMP-1, an inhibitor of MMP-9, leads to enhanced propensity of mice to develop aneurysms.
1 Burton A.C. Relation of structure to function of the tissues of the wall of blood vessels. Physiol Rev. 1954;34(4):619–642.
2 Eble J.A., Niland S. The extracellular matrix of blood vessels. Curr Pharm Des. 2009;15(12):1385–1400.
3 Wagenseil J.E., Mecham R.P. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89(3):957–989.
4 Davis G.E. The development of the vasculature and its extracellular matrix: a gradual process defined by sequential cellular and matrix remodeling events. Am J Physiol Heart Circ Physiol. 2010;299(2):H245–H247.
5 Davis G.E., Senger D.R. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97(11):1093–1107.
6 Myllyharju J., Kivirikko K.I. Collagens and collagen-related diseases. Ann Med. 2001;33(1):7–21.
7 Myllyharju J., Kivirikko K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20(1):33–43.
8 Kadler K.E., Baldock C., Bella J., et al. Collagens at a glance. J Cell Sci. 2007;120(Pt 12):1955–1958.
9 Gordon M.K., Hahn R.A. Collagens. Cell Tissue Res. 2010;339(1):247–257.
10 Khoshnoodi J., Pedchenko V., Hudson B.G. Mammalian collagen IV. Microsc Res Tech. 2008;71(5):357–370.
11 Murakami M., Simons M. Regulation of vascular integrity. J Mol Med. 2009;87(6):571–582.
12 Koch M., Schulze J., Hansen U., et al. A novel marker of tissue junctions, collagen XXII. J Biol Chem. 2004;279(21):22514–22521.
13 Koch M., Veit G., Stricker S., et al. Expression of type XXIII collagen mRNA and protein. J Biol Chem. 2006;281(30):21546–21557.
14 Villone D., Fritsch A., Koch M., et al. Supramolecular interactions in the dermo-epidermal junction zone: anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J Biol Chem. 2008;283(36):24506–24513.
15 Hashimoto T., Wakabayashi T., Watanabe A., et al. CLAC: a novel Alzheimer amyloid plaque component derived from a transmembrane precursor, CLAC-P/collagen type XXV. EMBO J. 2002;21(7):1524–1534.
16 Sasaki T., Larsson H., Tisi D., et al. Endostatins derived from collagens XV and XVIII differ in structural and binding properties, tissue distribution and anti-angiogenic activity. J Mol Biol. 2000;301(5):1179–1190.
17 Li D., Clark C.C., Myers J.C. Basement membrane zone type XV collagen is a disulfide-bonded chondroitin sulfate proteoglycan in human tissues and cultured cells. J Biol Chem. 2000;275(29):22339–22347.
18 Hurskainen M, Ruggiero F, Hagg P, et al: Recombinant human collagen XV regulates cell adhesion and migration, J Biol Chem 285(8):5258–5265.
19 Sato K., Yomogida K., Wada T., et al. Type XXVI collagen, a new member of the collagen family, is specifically expressed in the testis and ovary. J Biol Chem. 2002;277(40):37678–37684.
20 Grimal S, Puech S, Wagener R, et al: Collagen XXVIII is a distinctive component of the peripheral nervous system nodes of Ranvier and surrounds nonmyelinating glial cells, Glia 58(16):1977–1987.
21 Prockop D.J., Kivirikko K.I. Collagens: molecular biology, diseases, and potentials for therapy. Annu Rev Biochem. 1995;64:403–434.
22 Plenz G.A., Deng M.C., Robenek H., et al. Vascular collagens: spotlight on the role of type VIII collagen in atherogenesis. Atherosclerosis. 2003;166(1):1–11.
23 Ghosh A.K. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood). 2002;227(5):301–314.
24 Ghosh A.K., Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J Cell Physiol. 2007;213(3):663–671.
25 Nagai N., Hosokawa M., Itohara S., et al. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol. 2000;150(6):1499–1506.
26 Aycock R.S., Raghow R., Stricklin G.P., et al. Post-transcriptional inhibition of collagen and fibronectin synthesis by a synthetic homolog of a portion of the carboxyl-terminal propeptide of human type I collagen. J Biol Chem. 1986;261(30):14355–14360.
27 Bornstein P. Regulation of expression of the alpha 1 (I) collagen gene: a critical appraisal of the role of the first intron. Matrix Biol. 1996;15(1):3–10.
28 Fujii K., Tanzer M.L., Cooke P.H. Collagen fibrogenesis and the formation of complex cross-links. J Mol Biol. 1976;106(1):223–227.
29 Bernstein P.H., Mechanic G.L. A natural histidine-based imminium cross-link in collagen and its location. J Biol Chem. 1980;255(21):10414–10422.
30 Bourboulia D, Stetler-Stevenson WG: Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion, Semin Cancer Biol 20(3):161–168.
31 Kessenbrock K, Plaks V, Werb Z: Matrix metalloproteinases: regulators of the tumor microenvironment, Cell 141(1):52–67.
32 Fanjul-Fernandez M., Folgueras A.R., Cabrera S., et al. Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta. 1803;1:3–19.
33 Raffetto J.D., Khalil R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75(2):346–359.
34 Brew K., Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 1803;1:55–71.
35 Sternlicht M.D., Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516.
36 Woessner J., Nagase H. matrix metalloproteinases and TIMPs. New York: Oxford University Press; 2002.
37 Ruoslahti E. Specialization of tumour vasculature. Nat Rev Cancer. 2002;2(2):83–90.
38 Silletti S., Kessler T., Goldberg J., et al. Disruption of matrix metalloproteinase 2 binding to integrin alpha vbeta 3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc Natl Acad Sci U S A. 2001;98(1):119–124.
39 Wise S.G., Weiss A.S. Tropoelastin. Int J Biochem Cell Biol. 2009;41(3):494–497.
40 Ramirez F., Dietz H.C. Fibrillin-rich microfibrils: structural determinants of morphogenetic and homeostatic events. J Cell Physiol. 2007;213(2):326–330.
41 Royce P.M., Steinmann B., Kielty C.M., et al. The collagen family: structure, assembly, and organization in the extracellular matrix. In: Royce P.M., Steinmann B. Connective tissue and its heritable disorders: molecular, genetic, and medical aspects. ed 2. New York: Wiley & Sons; 2002:159.
42 Broekelmann T.J., Kozel B.A., Ishibashi H., et al. Tropoelastin interacts with cell-surface glycosaminoglycans via its COOH-terminal domain. J Biol Chem. 2005;280(49):40939–40947.
43 Czirok A., Zach J., Kozel B.A., et al. Elastic fiber macro-assembly is a hierarchical, cell motion-mediated process. J Cell Physiol. 2006;207(1):97–106.
44 Kozel B.A., Rongish B.J., Czirok A., et al. Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol. 2006;207(1):87–96.
45 Ramirez F., Sakai L.Y. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010;339(1):71–82.
46 Sato F., Wachi H., Ishida M., et al. Distinct steps of cross-linking, self-association, and maturation of tropoelastin are necessary for elastic fiber formation. J Mol Biol. 2007;369(3):841–851.
47 Galis Z.S., Khatri J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90(3):251–262.
48 Pezet M., Jacob M.P., Escoubet B., et al. Elastin haploinsufficiency induces alternative aging processes in the aorta. Rejuvenation Res. 2008;11(1):97–112.
49 Brooke B.S., Bayes-Genis A., Li D.Y. New insights into elastin and vascular disease. Trends Cardiovasc Med. 2003;13(5):176–181.
50 Li D.Y., Brooke B., Davis E.C., et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393(6682):276–280.
51 Li D.Y., Faury G., Taylor D.G., et al. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102(10):1783–1787.
52 Urban Z., Riazi S., Seidl T.L., et al. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet. 2002;71(1):30–44.
53 Karnik S.K., Brooke B.S., Bayes-Genis A., et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130(2):411–423.
54 Raines E.W. The extracellular matrix can regulate vascular cell migration, proliferation, and survival: relationships to vascular disease. Int J Exp Pathol. 2000;81(3):173–182.
55 Waugh J.M., Li-Hawkins J., Yuksel E., et al. Therapeutic elastase inhibition by alpha-1-antitrypsin gene transfer limits neointima formation in normal rabbits. J Vasc Interv Radiol. 2001;12(10):1203–1209.
56 Zaidi S.H., You X.M., Ciura S., et al. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105(4):516–521.
57 Yanagisawa H, Davis EC: Unraveling the mechanism of elastic fiber assembly: the roles of short fibulins, Int J Biochem Cell Biol 42(7):1084–1093.
58 Arteaga-Solis E., Gayraud B., Lee S.Y., et al. Regulation of limb patterning by extracellular microfibrils. J Cell Biol. 2001;154(2):275–281.
59 Carta L., Smaldone S., Zilberberg L., et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 2009;284(9):5630–5636.
60 Trask B.C., Trask T.M., Broekelmann T., et al. The microfibrillar proteins MAGP-1 and fibrillin-1 form a ternary complex with the chondroitin sulfate proteoglycan decorin. Mol Biol Cell. 2000;11(5):1499–1507.
61 Trask T.M., Trask B.C., Ritty T.M., et al. Interaction of tropoelastin with the amino-terminal domains of fibrillin-1 and fibrillin-2 suggests a role for the fibrillins in elastic fiber assembly. J Biol Chem. 2000;275(32):24400–24406.
62 Ramirez F., Dietz H.C. Extracellular microfibrils in vertebrate development and disease processes. J Biol Chem. 2009;284(22):14677–14681.
63 Hynes R.O. Fibronectins. New York: Springer; 1990.
64 Magnusson M.K., Mosher D.F. Fibronectin: structure, assembly, and cardiovascular implications. Arterioscler Thromb Vasc Biol. 1998;18(9):1363–1370.
65 Hynes R.O., Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150(2):F89–F96.
66 Whittaker C.A., Bergeron K.F., Whittle J., et al. The echinoderm adhesome. Dev Biol. 2006;300(1):252–266.
67 Pankov R., Yamada K.M. Fibronectin at a glance. J Cell Sci. 2002;115(Pt 20):3861–3863.
68 White E.S., Baralle F.E., Muro A.F. New insights into form and function of fibronectin splice variants. J Pathol. 2008;216(1):1–14.
69 Gutman A., Kornblihtt A.R. Identification of a third region of cell-specific alternative splicing in human fibronectin mRNA. Proc Natl Acad Sci U S A. 1987;84(20):7179–7182.
70 Kornblihtt A.R., Vibe-Pedersen K., Baralle F.E. Human fibronectin: molecular cloning evidence for two mRNA species differing by an internal segment coding for a structural domain. EMBO J. 1984;3(1):221–226.
71 Schwarzbauer J.E., Patel R.S., Fonda D., et al. Multiple sites of alternative splicing of the rat fibronectin gene transcript. EMBO J. 1987;6(9):2573–2580.
72 Schwarzbauer J.E., Tamkun J.W., Lemischka I.R., et al. Three different fibronectin mRNAs arise by alternative splicing within the coding region. Cell. 1983;35(2 Pt 1):421–431.
73 Bisno A.L., Brito M.O., Collins C.M. Molecular basis of group A streptococcal virulence. Lancet Infect Dis. 2003;3(4):191–200.
74 Klebe R.J. Isolation of a collagen-dependent cell attachment factor. Nature. 1974;250(463):248–251.
75 Kleinman H.K., Klebe R.J., Martin G.R. Role of collagenous matrices in the adhesion and growth of cells. J Cell Biol. 1981;88(3):473–485.
76 Engvall E., Ruoslahti E., Miller E.J. Affinity of fibronectin to collagens of different genetic types and to fibrinogen. J Exp Med. 1978;147(6):1584–1595.
77 Hynes R.O. Cell-matrix adhesion in vascular development. J Thromb Haemost. 2007;5(Suppl 1):32–40.
78 Hynes R.O. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219.
79 George E.L., Georges-Labouesse E.N., Patel-King R.S., et al. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119(4):1079–1091.
80 Astrof S., Kirby A., Lindblad-Toh K., et al. Heart development in fibronectin-null mice is governed by a genetic modifier on chromosome four. Mech Dev. 2007;124(7–8):551–558.
81 Francis S.E., Goh K.L., Hodivala-Dilke K., et al. Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler Thromb Vasc Biol. 2002;22(6):927–933.
82 Astrof S., Crowley D., Hynes R.O. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev Biol. 2007;311(1):11–24.
83 Astrof S., Crowley D., George E.L., et al. Direct test of potential roles of EIIIA and EIIIB alternatively spliced segments of fibronectin in physiological and tumor angiogenesis. Mol Cell Biol. 2004;24(19):8662–8670.
84 Wierzbicka-Patynowski I., Schwarzbauer J.E. The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003;116(Pt 16):3269–3276.
85 Cho J., Mosher D.F. Role of fibronectin assembly in platelet thrombus formation. J Thromb Haemost. 2006;4(7):1461–1469.
86 Lowenberg E.C., Meijers J.C., Levi M. Platelet-vessel wall interaction in health and disease. Neth J Med. 2010;68(6):242–251.
87 Ruggeri Z.M., Mendolicchio G.L. Adhesion mechanisms in platelet function. Circ Res. 2007;100(12):1673–1685.
88 Raghow R. The role of extracellular matrix in postinflammatory wound healing and fibrosis. FASEB J. 1994;8(11):823–831.
89 Aumailley M., Bruckner-Tuderman L., Carter W.G., et al. A simplified laminin nomenclature. Matrix Biol. 2005;24(5):326–332.
90 Durbeej M. Laminins. Cell Tissue Res. 2010;339(1):259–268.
91 Miner J.H., Yurchenco P.D. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284.
92 Aumailley M., Pesch M., Tunggal L., et al. Altered synthesis of laminin 1 and absence of basement membrane component deposition in (beta)1 integrin-deficient embryoid bodies. J Cell Sci. 2000;113(Pt 2):259–268.
93 Colognato H., Yurchenco P.D. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218(2):213–234.
94 Scheele S., Falk M., Franzen A., et al. Laminin alpha1 globular domains 4–5 induce fetal development but are not vital for embryonic basement membrane assembly. Proc Natl Acad Sci U S A. 2005;102(5):1502–1506.
95 Scheele S., Nystrom A., Durbeej M., et al. Laminin isoforms in development and disease. J Mol Med. 2007;85(8):825–836.
96 Tzu J., Marinkovich M.P. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol. 2008;40(2):199–214.
97 Van Agtmael T., Bruckner-Tuderman L. Basement membranes and human disease. Cell Tissue Res. 2010;339(1):167–188.
98 Engbring J.A., Kleinman H.K. The basement membrane matrix in malignancy. J Pathol. 2003;200(4):465–470.
99 Kleinman H.K., Koblinski J., Lee S., et al. Role of basement membrane in tumor growth and metastasis. Surg Oncol Clin N Am. 2001;10(2):329–338. ix
100 Miner J.H. Laminins and their roles in mammals. Microsc Res Tech. 2008;71(5):349–356.
101 Gubler M.C. Inherited diseases of the glomerular basement membrane. Nat Clin Pract Nephrol. 2008;4(1):24–37.
102 Patton B.L., Wang B., Tarumi Y.S., et al. A single point mutation in the LN domain of LAMA2 causes muscular dystrophy and peripheral amyelination. J Cell Sci. 2008;121(Pt 10):1593–1604.
103 Timpl R., Tisi D., Talts J.F., et al. Structure and function of laminin LG modules. Matrix Biol. 2000;19(4):309–317.
104 Thyboll J., Kortesmaa J., Cao R., et al. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol. 2002;22(4):1194–1202.
105 Esko J.D., Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108(2):169–173.
106 Gandhi N.S., Mancera R.L. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72(6):455–482.
107 Bulow H.E., Hobert O. The molecular diversity of glycosaminoglycans shapes animal development. Annu Rev Cell Dev Biol. 2006;22:375–407.
108 Roughley P.J. The structure and function of cartilage proteoglycans. Eur Cell Mater. 2006;12:92–101.
109 Schaefer L., Schaefer R.M. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339(1):237–246.
110 Theocharis A.D., Skandalis S.S., Tzanakakis G.N., et al. Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 2010;277(19):3904–3923.
111 Wu Y.J., La Pierre D.P., Wu J., et al. The interaction of versican with its binding partners. Cell Res. 2005;15(7):483–494.
112 Kim S., Takahashi H., Lin W.W., et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457(7225):102–106.
113 Iozzo R.V., Zoeller J.J., Nystrom A. Basement membrane proteoglycans: modulators par excellence of cancer growth and angiogenesis. Mol Cells. 2009;27(5):503–513.
114 Drickamer K. A conserved disulphide bond in sialyltransferases. Glycobiology. 1993;3(1):2–3.
115 Li H., Schwartz N.B., Vertel B.M. cDNA cloning of chick cartilage chondroitin sulfate (aggrecan) core protein and identification of a stop codon in the aggrecan gene associated with the chondrodystrophy, nanomelia. J Biol Chem. 1993;268(31):23504–23511.
116 Watanabe H., Kimata K., Line S., et al. Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nat Genet. 1994;7(2):154–157.
117 Kwok J.C., Afshari F., Garcia-Alias G., et al. Proteoglycans in the central nervous system: plasticity, regeneration and their stimulation with chondroitinase ABC. Restor Neurol Neurosci. 2008;26(2–3):131–145.
118 Iozzo R.V. Basement membrane proteoglycans: from cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6(8):646–656.
119 Iozzo R.V. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–652.
120 Iozzo R.V., Schaefer L. Proteoglycans in health and disease: novel regulatory signaling mechanisms evoked by the small leucine-rich proteoglycans. FEBS J. 2010;277(19):3864–3875.
121 Brule S., Charnaux N., Sutton A., et al. The shedding of syndecan-4 and syndecan-1 from HeLa cells and human primary macrophages is accelerated by SDF-1/CXCL12 and mediated by the matrix metalloproteinase-9. Glycobiology. 2006;16(6):488–501.
122 Danielson K.G., Baribault H., Holmes D.F., et al. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136(3):729–743.
123 Hedbom E., Heinegard D. Binding of fibromodulin and decorin to separate sites on fibrillar collagens. J Biol Chem. 1993;268(36):27307–27312.
124 Nurminskaya M.V., Birk D.E. Differential expression of fibromodulin mRNA associated with tendon fibril growth: isolation and characterization of a chicken fibromodulin cDNA. Biochem J. 1996;317(Pt 3):785–789.
125 Vogel K.G., Paulsson M., Heinegard D. Specific inhibition of type I and type II collagen fibrillogenesis by the small proteoglycan of tendon. Biochem J. 1984;223(3):587–597.
126 Birk D.E., Nurminskaya M.V., Zycband E.I. Collagen fibrillogenesis in situ: fibril segments undergo post-depositional modifications resulting in linear and lateral growth during matrix development. Dev Dyn. 1995;202(3):229–243.
127 Goldoni S., Iozzo R.V. Tumor microenvironment: modulation by decorin and related molecules harboring leucine-rich tandem motifs. Int J Cancer. 2008;123(11):2473–2479.
128 Heinegard D. Proteoglycans and more–from molecules to biology. Int J Exp Pathol. 2009;90(6):575–586.
129 Filmus J., Capurro M., Rast J. Glypicans. Genome Biol. 2008;9(5):224.
130 Alexopoulou A.N., Multhaupt H.A., Couchman J.R. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol. 2007;39(3):505–528.
131 Li Q., Park P.W., Wilson C.L., et al. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111(5):635–646.
132 Endo K., Takino T., Miyamori H., et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278(42):40764–40770.
133 Manon-Jensen T, Itoh Y, Couchman JR: Proteoglycans in health and disease: the multiple roles of syndecan shedding, FEBS J 277(19):3876–3889.
134 Rodgers K.D., San Antonio J.D., Jacenko O. Heparan sulfate proteoglycans: a GAGgle of skeletal-hematopoietic regulators. Dev Dyn. 2008;237(10):2622–2642.
135 Beckett K., Franch-Marro X., Vincent J.P. Glypican-mediated endocytosis of hedgehog has opposite effects in flies and mice. Trends Cell Biol. 2008;18(8):360–363.
136 Kjellen L., Lindahl U. Proteoglycans: structures and interactions. Annu Rev Biochem. 1991;60:443–475.
137 Lindahl U., Kusche-Gullberg M., Kjellen L. Regulated diversity of heparan sulfate. J Biol Chem. 1998;273(39):24979–24982.
138 Ruoslahti E. Structure and biology of proteoglycans. Annu Rev Cell Biol. 1988;4:229–255.
139 Geiger B., Spatz J.P., Bershadsky A.D. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10(1):21–33.
140 Lock J.G., Wehrle-Haller B., Stromblad S. Cell-matrix adhesion complexes: master control machinery of cell migration. Semin Cancer Biol. 2008;18(1):65–76.
141 Delon I., Brown N.H. Integrins and the actin cytoskeleton. Curr Opin Cell Biol. 2007;19(1):43–50.
142 Tilghman R.W., Parsons J.T. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. Semin Cancer Biol. 2008;18(1):45–52.
143 Berrier A.L., Yamada K.M. Cell-matrix adhesion. J Cell Physiol. 2007;213(3):565–573.
144 Legate K.R., Wickstrom S.A., Fassler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23(4):397–418.
145 Alam N., Goel H.L., Zarif M.J., et al. The integrin-growth factor receptor duet. J Cell Physiol. 2007;213(3):649–653.
146 Xu R., Boudreau A., Bissell M.J. Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis Rev. 2009;28(1–2):167–176.
147 Hynes R.O. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687.
148 Humphries J.D., Byron A., Humphries M.J. Integrin ligands at a glance. J Cell Sci. 2006;119(Pt 19):3901–3903.
149 Astrof S., Hynes R.O. Fibronectins in vascular morphogenesis. Angiogenesis. 2009;12(2):165–175.
150 Gui P., Chao J.T., Wu X., et al. Coordinated regulation of vascular Ca2 + and K+ channels by integrin signaling. Adv Exp Med Biol. 2010;674:69–79.
151 Luo B.H., Carman C.V., Springer T.A. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647.
152 Jarvelainen H., Sainio A., Koulu M., et al. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61(2):198–223.
153 Zaidel-Bar R., Itzkovitz S., Ma’ayan A., et al. Functional atlas of the integrin adhesome. Nat Cell Biol. 2007;9(8):858–867.
154 Lo S.H. Focal adhesions: what’s new inside. Dev Biol. 2006;294(2):280–291.
155 Li S., Guan J.L., Chien S. Biochemistry and biomechanics of cell motility. Annu Rev Biomed Eng. 2005;7:105–150.
156 Goel H.L., Breen M., Zhang J., et al. Beta1A integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res. 2005;65(15):6692–6700.
157 Goel H.L., Moro L., King M., et al. Beta1 integrins modulate cell adhesion by regulating insulin-like growth factor-II levels in the microenvironment. Cancer Res. 2006;66(1):331–342.
158 Sridhar S.C., Miranti C.K. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene. 2006;25(16):2367–2378.
159 Baron V., Schwartz M. Cell adhesion regulates ubiquitin-mediated degradation of the platelet-derived growth factor receptor beta. J Biol Chem. 2000;275(50):39318–39323.
160 Cabodi S., Moro L., Bergatto E., et al. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem Soc Trans. 2004;32(Pt3):438–442.
161 Bill H.M., Knudsen B., Moores S.L., et al. Epidermal growth factor receptor-dependent regulation of integrin-mediated signaling and cell cycle entry in epithelial cells. Mol Cell Biol. 2004;24(19):8586–8599.
162 Chen X., Abair T.D., Ibanez M.R., et al. Integrin alpha1beta1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol Cell Biol. 2007;27(9):3313–3326.
163 Hawiger. Adhesive interactions of blood cells and vascular wall in hemostasis and thrombosis. In: Coleman R., Hirsch J., Marder V., et al. Hemostasis and thrombosis: basic principles and clinical practices. ed 3. Philadelphia: JB Lippincott; 1994:639–653.
164 Ruggeri Z.M. Platelets in atherothrombosis. Nat Med. 2002;8(11):1227–1234.
165 Gawaz M., Langer H., May A.E. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115(12):3378–3384.
166 Nieswandt B., Watson S.P. Platelet-collagen interaction: is GPVI the central receptor? Blood. 2003;102(2):449–461.
167 Wagner D.D. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–246.
168 McGrath RT, McRae E, Smith OP, et al: Platelet von Willebrand factor–structure, function and biological importance, Br J Haematol 148(6):834–843.
169 Farndale R.W., Sixma J.J., Barnes M.J., et al. The role of collagen in thrombosis and hemostasis. J Thromb Haemost. 2004;2(4):561–573.
170 Asch A.S., Liu I., Briccetti F.M., et al. Analysis of CD36 binding domains: ligand specificity controlled by dephosphorylation of an ectodomain. Science. 1993;262(5138):1436–1440.
171 Chiang T.M., Rinaldy A., Kang A.H. Cloning, characterization, and functional studies of a nonintegrin platelet receptor for type I collagen. J Clin Invest. 1997;100(3):514–521.
172 Kato K., Kanaji T., Russell S., et al. The contribution of glycoprotein VI to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102(5):1701–1707.
173 Kunicki T.J., Nugent D.J., Staats S.J., et al. The human fibroblast class II extracellular matrix receptor mediates platelet adhesion to collagen and is identical to the platelet glycoprotein Ia-IIa complex. J Biol Chem. 1988;263(10):4516–4519.
174 Moroi M., Jung S.M., Okuma M., et al. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. 1989;84(5):1440–1445.
175 Moroi M., Jung S.M., Shinmyozu K., et al. Analysis of platelet adhesion to a collagen-coated surface under flow conditions: the involvement of glycoprotein VI in the platelet adhesion. Blood. 1996;88(6):2081–2092.
176 Nieswandt B., Brakebusch C., Bergmeier W., et al. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001;20(9):2120–2130.
177 Nieuwenhuis H.K., Akkerman J.W., Houdijk W.P., et al. Human blood platelets showing no response to collagen fail to express surface glycoprotein Ia. Nature. 1985;318(6045):470–472.
178 Santoro S.A. Identification of a 160,000 dalton platelet membrane protein that mediates the initial divalent cation-dependent adhesion of platelets to collagen. Cell. 1986;46(6):913–920.
179 Smethurst P.A., Onley D.J., Jarvis G.E., et al. Structural basis for the platelet-collagen interaction: the smallest motif within collagen that recognizes and activates platelet glycoprotein VI contains two glycine-proline-hydroxyproline triplets. J Biol Chem. 2007;282(2):1296–1304.
180 Farndale R.W., Lisman T., Bihan D., et al. Cell-collagen interactions: the use of peptide toolkits to investigate collagen-receptor interactions. Biochem Soc Trans. 2008;36(Pt 2):241–250.
181 Herr A.B., Farndale R.W. Structural insights into the interactions between platelet receptors and fibrillar collagen. J Biol Chem. 2009;284(30):19781–19785.
182 Gardner J.M., Hynes R.O. Interaction of fibronectin with its receptor on platelets. Cell. 1985;42(2):439–448.
183 Ni H., Yuen P.S., Papalia J.M., et al. Plasma fibronectin promotes thrombus growth and stability in injured arterioles. Proc Natl Acad Sci U S A. 2003;100(5):2415–2419.
184 Hynes R.O. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8(9):918–921.
185 Ginsberg M.H., Partridge A., Shattil S.J. Integrin regulation. Curr Opin Cell Biol. 2005;17(5):509–516.
186 White-Adams TC, Berny MA, Patel IA, et al: Laminin promotes coagulation and thrombus formation in a factor XII-dependent manner, J Thromb Haemost 8(6):1295–1301.
187 Bennett J.S., Berger B.W., Billings P.C. The structure and function of platelet integrins. J Thromb Haemost. 2009;7(Suppl 1):200–205.
188 Carrell R.W., Huntington J.A., Mushunje A., et al. The conformational basis of thrombosis. Thromb Haemost. 2001;86(1):14–22.
189 Casu B. Structural features and binding properties of chondroitin sulfates, dermatan sulfate, and heparan sulfate. Semin Thromb Hemost. 1991;17(Suppl 1):9–14.
190 Furie B., Furie B.C. Molecular and cellular biology of blood coagulation. N Engl J Med. 1992;326(12):800–806.
191 Warkentin T.E., Greinacher A. Heparin-induced thrombocytopenia and cardiac surgery. Ann Thorac Surg. 2003;76(2):638–648.
192 Biswas S., Munier F.L., Yardley J., et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10(21):2415–2423.