[level-membership-for-orthopaedics-category]
Chapter 3. Connective tissue inflammation, repair and remodelling
CHAPTER CONTENTS
SUMMARY
This chapter examines the different phases of healing and explores the process of scar tissue formation and its implication in restoring or preventing pain-free function. General principles of treatment are applied to each phase, aiming to facilitate healing, and the factors which promote or impede healing are considered.
The different phases of connective tissue healing are not separate and each is overlapped by the other, with one response signalling another until the wound is bridged by scar tissue (Fig. 3.1). After an initial, relatively short bleeding phase, the inflammatory phase prepares the area for healing, the repair phase rebuilds the structure and the remodelling phase provides the final form of the tissue (Hardy 1989, Broughton et al 2006, Watson 2009).
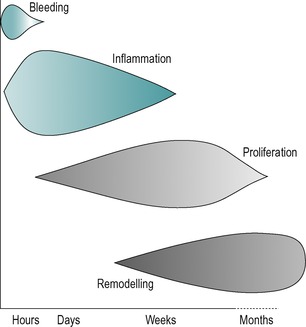 |
| Figure 3.1
Tissue repair phases and timescale.
(Online. Available: http://www.electrotherapy.org. Reprinted by permission of Professor Tim Watson.)
|
The degree of inflammation in response to injury depends on the degree of trauma. A minor injury causes a minimal response whereas a major injury will produce a significant inflammatory response which will pass through acute, subacute and chronic phases.
Lesions encountered in orthopaedic medicine include acute and chronic muscle belly lesions, ligamentous lesions, overuse tendinopathy, tenosynovitis, arthritis, bursitis and mechanical joint displacements.
Acute inflammation is significant and the patient can usually recall the precise time and mode of onset. Following injury the inflammatory response is rapid with noticeable pain and swelling, which can last for hours or days. With chronic inflammation, the patient cannot usually recollect the onset and the reaction is low-grade with less noticeable pain and swelling. Chronic inflammation may occur as a progression from acute inflammation or as a result of overuse, and can last for weeks, months or even years.
As mentioned in Chapter 2, the inflammatory model in chronic tendon lesions has been challenged and the pathological process may involve degenerative rather than inflammatory changes (Cook et al 2000, Khan & Cook 2000). A short discussion on the aetiology and pathology associated with chronic tendon lesions follows here before the phases of healing are described.
The degenerative changes in chronic tendon lesions include collagen fibre breakdown, increased ground substance, neovascularization, increased number of nerve filaments and increased immunoreactivity of substance P and calcitonin; there are no signs of chemical inflammation in chronically painful tendons (Khan & Cook 2000,Cook et al 2000, Wang et al 2006).
Alfredson et al (2002) concur that pathology is linked to degradation of collagen and hypercellularity. Tendons experiencing tendinosis contain no inflammatory cells but exhibit changes in the collagen fibre ultrastructure (Alfredson et al 2002, Tasto et al 2003, Richards et al 2005) with irregular fibre arrangement and a high concentration of glycosaminoglycans (Alfredson et al 2002). Local hypoxia, repetitive microtrauma or impaired wound healing may also contribute to tendinopathy (Richards et al 2005).
Tendinopathy has been adopted as a more appropriate term since it does not commit to pathology. The terms ‘tendinosis’, ‘paratendinitis’ and ‘tendinitis’ are reserved as histopathological labels that need to be confirmed by histopathological studies.
Tenosynovitis is distinct from tendinopathy; it occurs in ensheathed tendons and involves the synovium rather than the tendon itself. Inflammation of the synovium can produce adhesions between the two layers of the sheath which may produce a palpable crepitus (Cyriax 1982).
Characteristics of tendinopathy include a combination of pain, swelling and impaired performance.
The aetiology of tendinopathy appears to be multifactorial and the pathogenesis is unclear. It has increased in the general population and is found in both those who participate in recreational sport and those with a more sedentary lifestyle, as well as in more elite athletes. Other contributing factors will be discussed for specific tendon lesions, as well as their management, in the relevant chapters that follow.
Repetitive strain can reduce the ability of the tendon to endure further tension, disrupting its microscopic and macroscopic structure, but Rees et al (2006) propose that underuse could also be a factor. Inflammation (i.e. tendinitis or peritendinitis) may be the initial finding in tendon overuse and if this progresses the ensuing focal degeneration (i.e. tendinosis) can lead to partial and complete tears (Józsa & Kannus 1997). Other factors such as vascular supply, age and genetics can play a part in the pathogenesis of tendinopathy (Wang et al 2006). More research is needed to understand the mechanisms of tendinopathy at the tissue, cellular and molecular levels to be able to develop more effective evidence-based treatment protocols for tendinopathy.
Where the pain comes from in tendon lesions is also still unclear and Rees et al (2006) put forward mechanical, vascular and neural theories, suggesting that the pain arises from a combination of factors. Alfredson et al (2002) had suggested previously that neovascularization might be involved.
Structural damage noted in tendinopathy may include partial tearing of the collagen fibres, and an element of rest, especially from the aggravating activity, is appropriate to allow time for healing. The low metabolic activity in tendon causes an extended healing period, (Wang et al 2006). A controlled, graded programme of strengthening, stretching and eccentric exercises can be introduced guided by pain and function (Brukner & Khan 2007); ballistic stretching also has an important role in rehabilitation (Witvrouw et al 2007).
INFLAMMATORY PHASE
The initial inflammatory reaction involves vascular and cellular changes. Injury is rapidly followed by transient vasoconstriction, lasting for 5–10 min, and is succeeded by vasodilatation which may result in haemorrhage. If the lesion is still bleeding at the time of treatment this will necessitate careful management.
Figure 3.2 illustrates the components of the acute and chronic inflammatory responses, which will be referred to below. As described in Chapter 2, connective tissue comprises cells embedded in an extracellular matrix of fibres, with an interfibrillar amorphous ground substance that is assumed within the background spaces in the diagram.
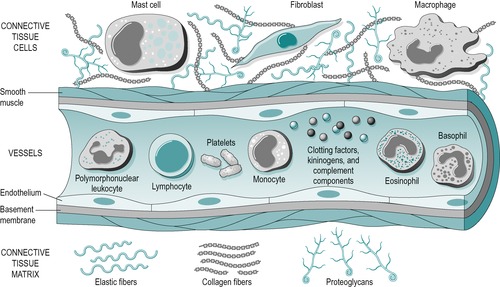 |
| Figure 3.2
The components of acute and chronic inflammatory process, circulating cells and proteins, cells of blood vessels, and cells and proteins of the extracellular matrix.
From Pathologic Basis of Disease 7th edn by V Kumar, N Fausto and A K Abbas. Reprinted by permission of Elsevier Ltd.
|
In the early stage of inflammation the blood vessel walls become more permeable and plasma and leukocytes leak into the surrounding tissues as inflammatory exudate or oedema. Swelling may take a few hours to develop and the amount of swelling is determined by the type of tissue involved in the injury. For example, muscle bellies may produce considerable swelling and bleeding but the structure of tendons prevents the collection of fluid and they do not easily swell. Similarly, ligaments themselves do not usually show dramatic swelling but capsular ligaments (e.g. the medial collateral ligament of the knee) may provoke a traumatic arthritis of the joint causing considerable pain and swelling. Swelling may also be restricted physically by fascial bands and intermuscular septa.
The vascular response is directly due to damage of blood vessel walls and indirectly due to the influence of chemical mediators. These chemical mediators include heparin and histamine, released by the mast cells (Fig. 3.2), bradykinin originating from plasma and plasma proteins, serotonin from platelets and prostaglandins, which are hormone-like compounds, produced by all cells in the body.
Heparin temporarily prevents coagulation of the excess tissue fluid and blood in the area. Bradykinins have multiple effects. They are potent mediators of the inflammatory response, can directly cause pain and vasodilatation, can activate the production of substance P and can enhance prostaglandin release. Substance P promotes vasodilatation, increases vascular permeability and stimulates phagocytosis and mast cell degranulation, with the subsequent release of histamine and serotonin. Prostaglandins may provoke or inhibit the inflammatory response. Histamine and serotonin produce a short-lived vascular effect whereas both bradykinins and prostaglandins promote more long-term vasodilatation (Broughton et al 2006). The overall vascular activity is responsible for the gross signs of inflammation: heat ( calor), redness ( rubor), swelling ( tumor), pain and tenderness ( dolor) and disturbed function ( functio laesa) (Peery & Miller 1971).
Inflammation causes pain and tenderness. Mechanical pain is due to mechanical stress, tissue damage, muscle spasm and the accumulated oedema causing excess pressure on surrounding tissues. Chemical pain arises through chemosensitive nerve receptors which are sensitive to histamine, serotonin, bradykinins and prostaglandins which are released into the tissues during this inflammatory phase. However, most nociceptors (pain receptors) are sensitive to more than one type of stimulus. The inflammatory substances may cause extreme stimulation of nerve fibres without necessarily causing them damage.
The nociceptors become progressively more sensitive the longer the pain stimulus is maintained and pro-inflammatory prostaglandins are believed to sensitize nociceptors leading to a state of hyperalgesia, an increased response to a painful stimulus (Wilkerson 1985, Kloth & Miller 1990).
If the tissue is still haemorrhaging, attempts must be made to stop this as blood is a strong irritant and will cause chemical and mechanical pain, as well as prolonging the inflammatory process (Dingman 1973, Evans 1980). This is particularly true of a haemarthrosis where aspiration should be considered.
In the first few hours of the early inflammatory phase fibronectin, a structural glycoprotein which acts as a tissue ‘glue’, appears in the wound, deposited along strands of fibrin in the clot (Broughton et al 2006, Standring 2009). This fibrin–fibronectin meshwork is associated with immature fibroblasts, which are thought to deposit type III collagen fibres to provide a scaffold for platelet adhesion and anchorage for further invading fibroblasts (Nimni 1980, Lehto et al 1985).
In minor injury, the inflammatory process is short and the scar tissue produced is minimal. The red blood cells break down into cellular debris and haemoglobin pigment, and the platelets (Fig. 3.2) release thrombin, an enzyme which changes fibrinogen into fibrin. The fibrin forms a meshwork of fibres which trap the blood clot and an early scar is formed.
If the injury is significant, the next stage of the inflammatory phase is phagocytic. Circulating monocytes modulate into macrophages (Fowler 1989) (Fig. 3.2) and join the resident macrophage population to clear the debris from the site of injury through phagocytic action (Leibovich & Ross 1974). The macrophages increase in great numbers during the first 3 or 4 days (Dingman 1973). They engulf any matter with which they come into contact, clearing the wound environment and preparing it for subsequent repair. As well as performing a phagocytic role, the macrophage acts as a director cell, directing the repair process by chemically influencing an appropriate number of fibroblast cells activated in the area.
Macrophages also have a role in muscle regeneration, stimulating the production of satellite cells that align themselves to muscle fibres where they are capable of forming completely new muscle fibres or restoring damaged muscle fibres (Grefte et al 2007). Further support for their importance is provided by Shen et al (2008) who observed that a reduced number of macrophages in muscle tissue is associated with reduced muscle regeneration. Fisher & Rathgaber (2006) observed muscle regeneration after acute blunt trauma to the gastrocnemius muscle in rats and noted an initial degenerative process with early peaking of macrophages and fibroblasts in the first 3 days, following which numbers of macrophages declined and satellite cells were noted beneath the basal lamina of muscle fibres. A regenerative process then began and after 6 days numerous irregularly arranged sarcomeres were observed, composed of thick and thin microfilaments and Z bands.
A stage of neovascularization is also reached, with capillaries starting to develop after about 12 h and continuing to develop for a further 2 or 3 days (Daly 1990). The new vessels supply oxygen and nourishment to the injured tissues, but are delicate and easily disrupted. Relative immobilization is necessary at this stage and heat is contraindicated as it will cause increased bleeding from the fragile vessels (Hardy 1989).
Inflammation is a normal response to either trauma or infection and to have no inflammatory response would mean that healing would not occur. Too little inflammation will delay healing and too much inflammation will lead to excessive scarring. The so-called PRICE principle of employing protection, relative rest, ice, compression and elevation in the early stages can help to control heat, swelling and pain and to reduce, but not abolish, inflammation.
The control of swelling is important towards regaining function since the greater the amount of inflammatory exudate, the more fibrin will be found in the area which becomes organized into scar tissue (Evans 1980). Although pain inhibits normal movement, graded mobilization should be encouraged as early as possible to promote healing and to avert adverse scar tissue formation.
Pain itself can be reduced further by analgesic drugs. Non-steroidal anti-inflammatory drugs (NSAIDs) modify the inflammatory response, reduce chemical pain and reduce temperature by inhibiting the production of prostaglandins. There are exponents of the so-called ‘NICE’ principle, adding in the prescription of non-steroidal drugs to the usual first-line after-care of injury. However, a counter view is that these should not be prescribed for the first 2 or 3 days after injury since they will tend to delay healing (Boruta et al 1990, Watson 2009). It may be more appropriate to allow the body’s natural inflammatory response to proceed with analgesics such as paracetamol and/or physical measures such as mobilization, massage and electrotherapy as appropriate methods of pain control.
Corticosteroids are contraindicated in the acute inflammatory phase as they inhibit macrophage activity which delays debridement of the wound and scar tissue production by delaying the onset and proliferation of the fibroblasts (Leibovich & Ross 1974, Hardy 1989). Each stage of the inflammatory phase is essential to the repair process and suppression in the early stages will delay healing.
Gentle transverse frictions and controlled mobilization can be initiated early to allow healing in the presence of movement, as guided by the irritability of the tissues. Mobilization is initiated very gently, to start moving the injured connective tissue towards regaining painless function, but without unduly stressing the healing breach. Overstressing the healing tissue would disrupt the early fragile scar and set up a secondary inflammatory response, leading to excess scar tissue formation. Mobilization also has an effect on the mechanoreceptors which is thought to reduce pain.
Gentle frictions together with gentle mobilization will agitate tissue fluid and increase the chance contact of the macrophage with cellular debris, so promoting healing (Evans 1980).
REPAIR PHASE
The repair phase is simultaneous with the inflammatory phase and overlaps the remodelling phase.
Some tissues, e.g. the synovial lining of the joints, bone and skeletal muscle, are capable of direct regeneration. All other connective tissues are incapable of regeneration, and repair of these structures involves a reconstruction process of the damaged tissue by collagen fibre or scar tissue formation. Scar tissue does not have exactly the same properties or tensile strength as the tissue it is rebuilding, but its structure comes to resemble that tissue closely to ensure that normal function is regained (Douglas et al 1969, Hardy 1989, Watson 2009).
Once the wound has been prepared by phagocytosis, the macrophage becomes the director of repair and signals an appropriate number of fibroblasts to the area. As the inflammatory phase subsides the fibroblast becomes the dominant cell in the repair phase and synthesizes the connective tissue matrix, comprising the amorphous ground substance and collagen (Fig. 3.2). Fibroblasts may appear in the wound during the first 24 h after injury, but maximum numbers are not achieved until day 5–10 (Bryant 1977, Chamberlain 1982, Fowler 1989). They do not decrease in number until 3 weeks after the injury (Chamberlain 1982).
Fibroblasts secrete the amorphous ground substance which provides the cross-linking mechanism for the collagen fibres it also synthesizes. This arrangement ‘glues’ the wound together, with cross-links forming at appropriate nodal intersect points (see Ch. 2). Once the fibroblasts are stimulated to produce collagen there is rapid closure of the healing breach. Collagen fibres proper are laid down approximately 5–10 days after injury and the repair process continues as they arrange themselves into larger units or bundles (Stearns 1940b, Chamberlain 1982).
Macrophages continue to stimulate the production of satellite cells, important for the regeneration and repair of muscle tissue. At the proliferation stage the satellite cells become myoblasts that may either fuse to each other to create new myofibres or may fuse to existing damaged myofibres for repair (Grefte et al 2007).
The rate of repair is directly related to the size of the wound (Stearns 1940a, 1940b). A small wound with approximated edges will heal quickly with a minimal inflammatory response and collagen fibres will be laid down early to bind the edges together, provided that the edges remain in apposition. Consider a clean, stitched skin wound, when the stitches are usually safely removed after 7 days and the wound has sufficient tensile strength to withstand movement.
Large, unapproximated wounds are deep as well as wide and healing initially requires the formation of granulation tissue. It may be several days after injury before the fibroblasts initiate fibre formation and several weeks before there is sufficient collagen to provide enough tensile strength for the wound to withstand normal movement. Therefore all timings mentioned above are approximate and the clinician will conduct a thorough assessment. The level of irritability of the tissues is a guide to the time for application of the appropriate graded mobilization.
During the early part of the repair process, a stage of wound contraction occurs assisted by the contractile action of myofibroblasts (Gabbiani et al 1971, Daly 1990). Linear wounds contract rapidly while circular or rectangular wounds contract relatively slowly (Grillo et al 1958, Fowler 1989, Hardy 1989, Daly 1990, Standring 2009). Wound contracture, as distinct from contraction, results from fibrosis or adhesion formation and this will be discussed later in this chapter.
Initial collagen fibre formation is random. The number of collagen fibres and the tensile strength of the wound increase substantially during the first 3 weeks after injury to become approximately 15–20% of the normal strength of the tissue (Hardy 1989, Daly 1990, Hardy & Woodall 1998). However, the tensile strength does not depend entirely on the number of fibres, since after this time the number of collagen fibres stabilizes but the tensile strength of the wound continues to increase. Tensile strength is related to a balance between the synthesis and lysis of collagen (the production and breakdown of collagen, a continuous, dynamic process), the development of collagen cross-links and the orientation of collagen fibres into the existing weave. This process of maturation is known as the remodelling phase (van der Meulen 1982, Standring 2009).
REMODELLING PHASE
The remodelling phase sees the new collagen or scar tissue attempt to take on the physical characteristics of the tissue it is replacing. It begins in earnest approximately 21 days after injury and continues for 6 months or more, possibly even for years. Broughton et al (2006) suggest that the process can actually begin much earlier than the peak of 21 days, from approximately 8 days, and provide support for the remodelling stage continuing for I year. Remodelling is responsible for the final structural orientation and arrangement of the fibres as well as the tensile strength.
Initial, immature scar tissue is weak and the fibres are oriented in all directions through several planes. Remodelling allows these randomly arranged fibres to become rearranged in both a linear and a lateral orientation in a ‘well-mannered network’ (Broughton et al 2006). The orientation of collagen fibres occurs in two ways: through induction and through tension.
Normal tissue adjacent to the wound induces structure in the replacement scar tissue. Thus dense tissue induces dense, highly cross-linked scar tissue while pliable tissue induces loose, less cross-linked scar tissue (Hardy 1989). The final physical weave of the collagen so formed is responsible for the functional behaviour of the wound within the connective tissue it is replacing.
Internal and external stresses apply tension to the wound during the remodelling phase, e.g. muscle tension, joint movement, passive gliding of fascial planes, connective tissue loading and unloading, temperature changes and mobilization (Hardy 1989). It is recognized that both mobilization and immobilization can strongly influence the structural orientation of collagen fibres (Stearns 1940a, 1940b, Akeson et al 1987).
During the maturation phase of scar formation, immature scar tissue is converted to mature scar tissue and the cross-linking system changes from weak hydrogen bonding to strong covalent bonding (Price 1990). While the scar tissue is relatively immature the weak electrostatic bonding forms reducible cross-links which allow scar tissue to be mobilized with a gentle, steady stress. During this stage transverse frictions and mobilization within the limits of pain are appropriate to maintain the mobility of immature scar tissue with graded mobilization promoting the alignment of fibres.
Remodelling involves the reorganization of scar while it matures with fibres being absorbed, replaced and re-oriented. When stresses arising from mobilization are applied to collagen fibres, the resultant piezo-electric effect (generation of small voltages called streaming potentials) is believed to be responsible for the production, maintenance, alignment and absorption of collagen fibrils (Price 1990, Standring 2009).
Cross-linking is responsible for the tensile strength of new, desirable scar tissue, but if the cross-linking becomes excessive it will be responsible for the toughness and lack of resilience of unwanted fibrous adhesions (Kloth & Miller 1990). Immobilization causes loss of the ground substance, which reduces the interfibrillar distance and causes friction between the collagen subunits, facilitating the formation of excessive cross-links (Akeson et al 1967, Akeson 1990).
Collagen fibre formation and orientation conform to lines of stress within connective tissue, and are similar in this respect to osseous alignment (Le Gros Clark 1965, Price 1990, Standring 2009).
Ligaments require strong scar tissue fibres within their parallel wavy weave to be capable of resisting excessive joint stresses as well as being able to relax and fold when the tension is removed. The scar tissue formed between the ligament and bone must mimic the normal weave to allow normal movement of the joint (Hardy 1989). Scar tissue formed within a muscle belly needs to be flexible to allow the muscle fibres to broaden as the muscle contracts and extensible enough to allow the muscle to lengthen when stretched. Scar tissue within tendons must be oriented in a parallel weave along the lines of mechanical stress to ensure maximum tensile strength of the tendon while also maintaining its gliding properties.
To avoid adverse scar tissue formation, gentle transverse frictions and a progressively increasing range of mobilization should be continued until a full pain-free range of movement is restored. This aims to prevent excessive cross-links occurring between individual fibres and encourages fibre alignment. Applying appropriate stress to restore length should avoid the necessity to stretch.
Collagen synthesis and remodelling continue in the 6 months following injury with the tissue returning to its normal state of activity 6–12 months after injury under normal conditions (Daly 1990). However, the increase in tensile strength in fascia and other dense connective tissues is thought to take much longer (Dingman 1973). Fully repaired skin wounds eventually achieve only approximately 70% of their original strength (Douglas et al 1969, Bryant 1977, Daly 1990).
As the scar matures, it becomes dense, tough and less resilient than immature scar tissue. The developing stable cross-links become more prolific and the stronger covalent bonds which form do not yield as readily to applied stresses (Price 1990).
An increasing depth of transverse frictions provides pressure and lateral stretch to the mature scar. Deeper transverse frictions and a greater range of mobilization are required to mobilize mature scar tissue compared with immature scar tissue.
Adhesion formation and contracture
Synthesis and lysis, together with orientation of the collagen fibres (i.e. remodelling), ensure the final form of scar tissue. A balance is needed between collagen synthesis and lysis for an appropriate turnover of collagen and sufficient stresses should be applied to the tissue to stimulate fibre orientation but without disrupting the healing breach. This has implications for the grade of mobilization applied and is discussed in Chapter 4.
Increasing the size of the healing breach would set up secondary inflammatory changes, leading to excessive scar tissue formation and eventual contracture, adhesions and fibrosis. Excessive scar tissue, adhesions or contracture within any soft tissue structure will impede function and cause pain. Pain itself, as a characteristic of inflammation, acts as an inhibitor to normal function and, if a state of chronic inflammation is maintained, the function of the tissue will continue to deteriorate. This self-perpetuating inflammation presents an ongoing chronic functional problem which may be difficult to treat.
An abnormal excessive production of scar tissue may result in hypertrophic or keloid scars (Daly 1990, Price 1990). Hypertrophic scars develop when excessive collagen is deposited within the original wound site while keloid scarring involves excessive collagen deposits in the tissues surrounding the scar. In the connective tissue structures important to orthopaedic medicine, this excessive production of scar tissue may be seen as adhesion formation and contracture either within the healing structure or within the surrounding tissues.
In the treatment of hypertrophic scarring, prolonged pressure has been used to restore the balance between collagen synthesis and lysis (Hardy 1989). In a chronically inflamed wound where excessive scar tissue has been produced, the technique of deep transverse frictions applies pressure to the area of scar tissue as well as providing a lateral stress to mobilize adhesions.
The use of intralesional corticosteroid is said to produce keloid regression through its multiple steroid effects, which include inhibition of fibroblast migration, decreased collagen synthesis and increased collagenase activity (Carrico et al 1984). This effect may be transferred to the use of intralesional steroid for chronic inflammation in chronic connective tissue lesions such as tendinopathy, bursitis and some chronic ligament strains.
FACTORS WHICH MAY AFFECT WOUND HEALING
In considering various factors which can promote, delay or lead to poor repair, assessment of connective tissue lesions should take into account the following factors:
• Time of onset and the time lapse since injury.
• Extent of the lesion.
• Inappropriate or overaggressive mobilization relating to irritability of the lesion.
• Stage reached in the inflammation, repair, remodelling phases.
• Anatomical structures involved directly and indirectly in the lesion.
• Medical conditions which may affect wound healing, e.g. circulatory disorders, clotting disorders, diabetes.
• Age.
• Medications which might affect management and healing, e.g. anticoagulants, analgesics, anti-inflammatory drugs.
Chronic trauma can cause excessive movement or tension on devitalized tissues, promoting unwanted scarring. This may be the mechanism of chronic overuse syndromes in which repetitive trauma disrupts tissue unity.
Haematoma formation retards the healing process by acting as an irritant, producing a mechanical blockage which separates the torn edges and provides a medium for infection (Dingman 1973).
Infection of the injured tissue presents a serious complication which delays the healing process.
Age, according to Mulder (1990), can delay cell migration and proliferation, wound contracture and collagen remodelling, and decrease the tensile strength of the wound, so increasing the chance of wound dehiscence or splitting. Experimental wounds in young rats showed better mechanical properties in terms of greater strength, elastic stiffness and energy absorption than those in older rats. The fibre organization was more complex and better organized in the young rats and healing was observed to be faster (Holm-Pedersen & Viidik 1971).
Changes in the gel–fibre ratio occurring with age are consistent with the changes occurring with immobilization. Contractures tend to occur more frequently, after less trauma and after shorter periods of time in the relatively immobile joints of the elderly. Chemical changes in the gel–fibre ratio have been noted in such tissues as the skin and the nucleus pulposus of older individuals (Akeson et al 1968).
The following medications, therapies and conditions may also affect wound healing.
While anti-inflammatory medication may not be the most appropriate treatment for acute inflammation, its use in chronic lesions is most appropriate for suppressing inflammation and relieving pain. NSAIDs, either taken orally or topically applied, may be used in conjunction with physical measures (PRICE). NSAIDs do not cause a significant change in collagen synthesis; they inhibit production of histamine, serotonin and prostaglandins (Wilkerson 1985). However, aspirin, in addition to its anti-inflammatory function, inhibits platelet aggregation and may prolong bleeding (Rang et al 2003).
The oral intake of corticosteroids inhibits collagen synthesis, reduces tensile strength and delays wound healing (Dingman 1973, Ahonen et al 1980, Mulder 1990). Corticosteroids administered in the acute inflammatory phase interfere with macrophage migration but if delivered after the macrophage invasion, i.e. after 3 days, their effect on wound healing is much less severe (Fowler 1989).
Anticoagulants, e.g. heparin and warfarin, prolong bleeding and delay wound healing.
The effect of chemotherapy depends on the drugs used and their dosage, but fibroblast proliferation may be affected and subsequently collagen synthesis is delayed (Carrico et al 1984).
Radiotherapy radiation can damage fibroblasts, cause vascular damage and decrease collagen production, but it depends on the dose, frequency and location of the irradiated area in relation to the injury site (Mulder 1990).
Acquired immunedeficiency syndrome (AIDS) patients are in a state of immunosuppression and this will delay the healing process.
Diabetes appears to affect the inflammatory stage rather than collagen synthesis, implying that insulin is important in the early phase of healing (Carrico et al 1984).
Other factors which could affect healing include vitamin A and C deficiency, protein deprivation, low temperature (Watson 2009), systemic vascular disorders and systemic connective tissue disorders.
REFERENCES
Ahonen, J.; Jiborn, H.; Zederfeldt, B., Hormone influence on wound healing, In: (Editor: Hunt, T.K.) Wound Healing and Wound Infection: Theory and Surgical Practice ( 1980)Appleton Century Croft, New York, pp. 95–105.
Akeson, W., The response of ligaments to stress modulation and overview of the ligament healing response, In: (Editors: Daniel, D.; Akeson, W.H.; O’Connor, J.J.) Knee Ligaments: Structure, Function, Injury and Repair ( 1990)Lippincott, Williams & Wilkins, Philadelphia, pp. 315–327.
Akeson, W.; Amiel, D.; La Violette, D., The connective tissue response to immobility: a study of chondroitin-4 and 6-sulfate and dermatan sulfate changes in periarticular connective tissue of control and immobilised knees of dogs, Clin. Orthop. 51 (1967) 183–197.
Akeson, W.; Amiel, D.; LaViolette, D.; et al., The connective tissue response to immobility: an accelerated ageing response?Exp. Gerontol. 3 (1968) 289–301.
Akeson, W.; Amiel, D.; Abel, M.F.; et al., Effects of immobilisation on joints, Clin. Orthop. Relat. Res. 219 (1987) 28–37.
Alfredson, H.; Bjur, D.; Thorsen, K.; et al., High intratendinous lactate levels in painful chronic Achilles tendinosis. An investigation using microdialysis technique, J. Orthop. Res. 20 (5) ( 2002) 934–938.
Boruta, P.M.; Bishop, J.O.; Braly, W.G.; et al., Acute lateral ankle ligament injuries: literature review, Foot Ankle 11 (1990) 107–113.
Broughton, G.; Janis, J.; Attinger, C., The basic science of wound healing, Plast. Reconstr. Surg. 117 (7S) ( 2006) 12S–34S.
Brukner, P.; Khan, K., Clinical Sports Medicine. 3rd edn. ( 2007)McGraw-Hill, Sydney.
Bryant, W., Wound healing, Clin. Symp. 29 (1977) 2–28.
Carrico, T.J.; Mehrhof, A.I.; Cohen, I.K., Biology of wound healing, Surg. Clin. N. Am. 64 (1984) 721–731.
Chamberlain, G.J., Cyriax’s friction massage: a review, J. Orthop. Sports Phys. Ther. 4 (1982) 16–22.
Cook, J.L.; Khan, K.M.; Maffulli, N.; et al., Overuse tendinosis, not tendinopathy, part 2: applying the new approach to patellar tendinopathy, Phys. Sports Med. 28 (6) ( 2000) 31–41.
Cyriax, J., 8th edn.Textbook of Orthopaedic Medicine. vol. 1 ( 1982)Baillière Tindall, London.
Daly, T.J., The repair phase of wound healing – re-epithelialization and contraction, In: (Editors: Kloth, L.C.; McCullock, J.M.; Feedar, J.A.) Wound Healing: Alternatives in Management ( 1990)F A Davis, Philadelphia, pp. 14–29.
Dingman, R.O., Factors of clinical significance affecting wound healing, Laryngoscope 83 (1973) 1540–1554.
Douglas, D.M.; Forrester, J.C.; Ogilvie, R.R., Physical characteristics of collagen in the later stages of wound healing, Br. J. Surg. 56 (1969) 219–222.
Evans, P., The healing process at cellular level: a review, Physiotherapy 66 (1980) 256–259.
Fisher, B.D.; Rathgaber, M., An overview of muscle regeneration following acute injury, J. Phys. Ther. Sci. 18 (1) ( 2006) 57–66.
Fowler, J.D., Wound healing: an overview, Semin. Vet. Med. Surg. 4 (1989) 256–262.
Gabbiani, G.; Ryan, G.B.; Majno, G., Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction, Experientia 27 (1971) 549–550.
Grefte, S.; Kuijpers-Jagtman, A.M.; Torensma, R.; et al., Skeletal muscle development and regeneration, Stem Cells Dev. 16 (2007) 857–868.
Grillo, H.C.; Watts, G.T.; Gross, J., Studies in wound healing: I. contraction and the wound contents, Ann. Surg. 148 (1958) 145–152.
Hardy, M.A., The biology of scar formation, Phys. Ther. 69 (1989) 1014–1023.
Hardy, M.; Woodall, W., Therapeutic effects of heat, cold and stretch on connective tissue, J. Hand Ther. 11 (2) ( 1998) 148–152.
Holm-Pedersen, P.; Viidik, A., Tensile properties and morphology of healing wounds in young and old rats, Scand. J. Plast. Reconstr. Surg. 6 (1971) 24–35.
Józsa, L.; Kannus, P., Human Tendons: Anatomy, Physiology and Pathology. ( 1997)Human Kinetics, Champaign, Illinois.
Khan, K.M.; Cook, J.L., Overuse tendon injuries: where does the pain come from?Sports Med. Arthrosc. Rev. 8 (2000) 17–31.
Kloth, L.C.; Miller, K.H., The inflammatory response to wounding, In: (Editors: Kloth, L.C.; McCullock, J.M.; Feedar, J.A.) Wound Healing: Alternatives in Management ( 1990)F A Davis, Philadelphia, pp. 14–29.
Kumar, V.; Fausto, N.; Abbas, A.K., Pathologic Basis of Disease. 7th edn. ( 2006)Saunders, Philadelphia.
Le Gros Clark, W.E., The Tissues of the Body. 5th edn. ( 1965)Oxford University Press, Oxford.
Lehto, M.; Duance, V.C.; Restall, D., Collagen and fibronectin in a healing skeletal muscle injury, J. Bone Joint Surg. 67B (1985) 820–827.
Leibovich, S.J.; Ross, R., The role of the macrophage in wound repair, Am. J. Pathol. 78 (1974) 71–91.
Mulder, G.D., Factors complicating wound repair, In: (Editors: Kloth, L.C.; McCullock, J.M.; Feedar, J.) Wound Healing: Alternatives in Management ( 1990)F A Davis, Philadelphia, pp. 43–51.
Nimni, M.E., The molecular organisation of collagen and its role in determining the biophysical properties of the connective tissues, Biorheology 17 (1980) 51–82.
Peery, T.M.; Miller, F.N., Pathology: A Dynamic Introduction to Medicine and Surgery. 2nd edn. ( 1971)Little, Brown, Boston.
Price, H., Connective tissue in wound healing, In: (Editors: Kloth, L.C.; McCullock, J.M.; Feedar, J.) Wound Healing: Alternatives in Management ( 1990)F A Davis, Philadelphia, pp. 31–41.
Rang, H.P.; Dale, M.M.; Ritter, J.M.; et al., Pharmacology. 5th edn. ( 2003)Churchill Livingstone, Edinburgh.
Rees, J.; Wilson, A.; Wolman, R., Current concepts in the management of tendon disorders, Rheumatology 45 (2006) 508–521.
Richards, P.; Win, T.; Jones, P., The distribution of microvascular response in Achilles tendinopathy assessed by colour and power Doppler, Skeletal Radiol. 34 (6) ( 2005) 336–342.
Shen, W.; Li, Y.; Zhu, J.; et al., Interaction between macro-phages. TGF-B1, and the COX-2 pathway during the inflammatory phase of skeletal muscle healing after injury, J. Cell. Physiol. 214 (2008) 405–412.
Standring, S., Gray’s Anatomy: The Anatomical Basis of Clinical Practice. 40th edn. ( 2009)Churchill Livingstone, Edinburgh.
Stearns, M.L., Studies on the development of connective tissue in transparent chambers in the rabbit’s ear, I, Am. J. Anat. 66 (1940) 133–176.
Stearns, M.L., Studies on the development of connective tissue in transparent chambers in the rabbit’s ear, II, Am. J. Anat. 67 (1940) 55–97.
Tasto, J.P.; Cummings, J.; Medlock, V.; et al., The tendon treatment center: new horizons in the treatment of tendinosis, Arthroscopy 19 (Suppl. 1) ( 2003) 213–223.
van der Meulen, J.H.C., Present state of knowledge on processes of healing in collagen structures, Int. J. Sports Med. 3 (1982) 4–8.
Wang, J.; Losifidus, M.; Fu, F., Biomechanical basis for tendinopathy, Clin. Orthop. Relat. Res. 443 (2006) 320–332.
Watson, T. (2009). Tissue repair phases and timescale. Online. Available: < http://www.electrotherapy.org>.
Wilkerson, G.B., Inflammation in connective tissue: etiology and management, Athl. Train. Winter (1985) 298–301.
Witvrouw, E.; Mahieu, N.; Roosen, P.; et al., The role of stretching in tendon injuries, Br. J. Sports Med. 41 (4) ( 2007) 224–226.
[/level-membership-for-orthopaedics-category][not-level-membership-for-orthopaedics-category]
Chapter 3. Connective tissue inflammation, repair and remodelling
CHAPTER CONTENTS
SUMMARY
This chapter examines the different phases of healing and explores the process of scar tissue formation and its implication in restoring or preventing pain-free function. General principles of treatment are applied to each phase, aiming to facilitate healing, and the factors which promote or impede healing are considered.
The different phases of connective tissue healing are not separate and each is overlapped by the other, with one response signalling another until the wound is bridged by scar tissue (Fig. 3.1). After an initial, relatively short bleeding phase, the inflammatory phase prepares the area for healing, the repair phase rebuilds the structure and the remodelling phase provides the final form of the tissue (Hardy 1989, Broughton et al 2006, Watson 2009).
|
|
| Figure 3.1
Tissue repair phases and timescale.
(Online. Available: http://www.electrotherapy.org. Reprinted by permission of Professor Tim Watson.)
|
The degree of inflammation in response to injury depends on the degree of trauma. A minor injury causes a minimal response whereas a major injury will produce a significant inflammatory response which will pass through acute, subacute and chronic phases.
Lesions encountered in orthopaedic medicine include acute and chronic muscle belly lesions, ligamentous lesions, overuse tendinopathy, tenosynovitis, arthritis, bursitis and mechanical joint displacements.
Acute inflammation is significant and the patient can usually recall the precise time and mode of onset. Following injury the inflammatory response is rapid with noticeable pain and swelling, which can last for hours or days. With chronic inflammation, the patient cannot usually recollect the onset and the reaction is low-grade with less noticeable pain and swelling. Chronic inflammation may occur as a progression from acute inflammation or as a result of overuse, and can last for weeks, months or even years.
As mentioned in Chapter 2, the inflammatory model in chronic tendon lesions has been challenged and the pathological process may involve degenerative rather than inflammatory changes (Cook et al 2000, Khan & Cook 2000). A short discussion on the aetiology and pathology associated with chronic tendon lesions follows here before the phases of healing are described.
The degenerative changes in chronic tendon lesions include collagen fibre breakdown, increased ground substance, neovascularization, increased number of nerve filaments and increased immunoreactivity of substance P and calcitonin; there are no signs of chemical inflammation in chronically painful tendons (Khan & Cook 2000,Cook et al 2000, Wang et al 2006).
Alfredson et al (2002) concur that pathology is linked to degradation of collagen and hypercellularity. Tendons experiencing tendinosis contain no inflammatory cells but exhibit changes in the collagen fibre ultrastructure (Alfredson et al 2002, Tasto et al 2003, Richards et al 2005) with irregular fibre arrangement and a high concentration of glycosaminoglycans (Alfredson et al 2002). Local hypoxia, repetitive microtrauma or impaired wound healing may also contribute to tendinopathy (Richards et al 2005).
Tendinopathy has been adopted as a more appropriate term since it does not commit to pathology. The terms ‘tendinosis’, ‘paratendinitis’ and ‘tendinitis’ are reserved as histopathological labels that need to be confirmed by histopathological studies.
Tenosynovitis is distinct from tendinopathy; it occurs in ensheathed tendons and involves the synovium rather than the tendon itself. Inflammation of the synovium can produce adhesions between the two layers of the sheath which may produce a palpable crepitus (Cyriax 1982).
Characteristics of tendinopathy include a combination of pain, swelling and impaired performance.
The aetiology of tendinopathy appears to be multifactorial and the pathogenesis is unclear. It has increased in the general population and is found in both those who participate in recreational sport and those with a more sedentary lifestyle, as well as in more elite athletes. Other contributing factors will be discussed for specific tendon lesions, as well as their management, in the relevant chapters that follow.
Repetitive strain can reduce the ability of the tendon to endure further tension, disrupting its microscopic and macroscopic structure, but Rees et al (2006) propose that underuse could also be a factor. Inflammation (i.e. tendinitis or peritendinitis) may be the initial finding in tendon overuse and if this progresses the ensuing focal degeneration (i.e. tendinosis) can lead to partial and complete tears (Józsa & Kannus 1997). Other factors such as vascular supply, age and genetics can play a part in the pathogenesis of tendinopathy (Wang et al 2006). More research is needed to understand the mechanisms of tendinopathy at the tissue, cellular and molecular levels to be able to develop more effective evidence-based treatment protocols for tendinopathy.
Where the pain comes from in tendon lesions is also still unclear and Rees et al (2006) put forward mechanical, vascular and neural theories, suggesting that the pain arises from a combination of factors. Alfredson et al (2002) had suggested previously that neovascularization might be involved.
Structural damage noted in tendinopathy may include partial tearing of the collagen fibres, and an element of rest, especially from the aggravating activity, is appropriate to allow time for healing. The low metabolic activity in tendon causes an extended healing period, (Wang et al 2006). A controlled, graded programme of strengthening, stretching and eccentric exercises can be introduced guided by pain and function (Brukner & Khan 2007); ballistic stretching also has an important role in rehabilitation (Witvrouw et al 2007).
INFLAMMATORY PHASE
The initial inflammatory reaction involves vascular and cellular changes. Injury is rapidly followed by transient vasoconstriction, lasting for 5–10 min, and is succeeded by vasodilatation which may result in haemorrhage. If the lesion is still bleeding at the time of treatment this will necessitate careful management.
Figure 3.2 illustrates the components of the acute and chronic inflammatory responses, which will be referred to below. As described in Chapter 2, connective tissue comprises cells embedded in an extracellular matrix of fibres, with an interfibrillar amorphous ground substance that is assumed within the background spaces in the diagram.
|
|
| Figure 3.2
The components of acute and chronic inflammatory process, circulating cells and proteins, cells of blood vessels, and cells and proteins of the extracellular matrix.
From Pathologic Basis of Disease 7th edn by V Kumar, N Fausto and A K Abbas. Reprinted by permission of Elsevier Ltd.
|
In the early stage of inflammation the blood vessel walls become more permeable and plasma and leukocytes leak into the surrounding tissues as inflammatory exudate or oedema. Swelling may take a few hours to develop and the amount of swelling is determined by the type of tissue involved in the injury. For example, muscle bellies may produce considerable swelling and bleeding but the structure of tendons prevents the collection of fluid and they do not easily swell. Similarly, ligaments themselves do not usually show dramatic swelling but capsular ligaments (e.g. the medial collateral ligament of the knee) may provoke a traumatic arthritis of the joint causing considerable pain and swelling. Swelling may also be restricted physically by fascial bands and intermuscular septa.
The vascular response is directly due to damage of blood vessel walls and indirectly due to the influence of chemical mediators. These chemical mediators include heparin and histamine, released by the mast cells (Fig. 3.2), bradykinin originating from plasma and plasma proteins, serotonin from platelets and prostaglandins, which are hormone-like compounds, produced by all cells in the body.
Heparin temporarily prevents coagulation of the excess tissue fluid and blood in the area. Bradykinins have multiple effects. They are potent mediators of the inflammatory response, can directly cause pain and vasodilatation, can activate the production of substance P and can enhance prostaglandin release. Substance P promotes vasodilatation, increases vascular permeability and stimulates phagocytosis and mast cell degranulation, with the subsequent release of histamine and serotonin. Prostaglandins may provoke or inhibit the inflammatory response. Histamine and serotonin produce a short-lived vascular effect whereas both bradykinins and prostaglandins promote more long-term vasodilatation (Broughton et al 2006). The overall vascular activity is responsible for the gross signs of inflammation: heat ( calor), redness ( rubor), swelling ( tumor), pain and tenderness ( dolor) and disturbed function ( functio laesa) (Peery & Miller 1971).
Inflammation causes pain and tenderness. Mechanical pain is due to mechanical stress, tissue damage, muscle spasm and the accumulated oedema causing excess pressure on surrounding tissues. Chemical pain arises through chemosensitive nerve receptors which are sensitive to histamine, serotonin, bradykinins and prostaglandins which are released into the tissues during this inflammatory phase. However, most nociceptors (pain receptors) are sensitive to more than one type of stimulus. The inflammatory substances may cause extreme stimulation of nerve fibres without necessarily causing them damage.
The nociceptors become progressively more sensitive the longer the pain stimulus is maintained and pro-inflammatory prostaglandins are believed to sensitize nociceptors leading to a state of hyperalgesia, an increased response to a painful stimulus (Wilkerson 1985, Kloth & Miller 1990).
If the tissue is still haemorrhaging, attempts must be made to stop this as blood is a strong irritant and will cause chemical and mechanical pain, as well as prolonging the inflammatory process (Dingman 1973, Evans 1980). This is particularly true of a haemarthrosis where aspiration should be considered.
In the first few hours of the early inflammatory phase fibronectin, a structural glycoprotein which acts as a tissue ‘glue’, appears in the wound, deposited along strands of fibrin in the clot (Broughton et al 2006, Standring 2009). This fibrin–fibronectin meshwork is associated with immature fibroblasts, which are thought to deposit type III collagen fibres to provide a scaffold for platelet adhesion and anchorage for further invading fibroblasts (Nimni 1980, Lehto et al 1985).
In minor injury, the inflammatory process is short and the scar tissue produced is minimal. The red blood cells break down into cellular debris and haemoglobin pigment, and the platelets (Fig. 3.2) release thrombin, an enzyme which changes fibrinogen into fibrin. The fibrin forms a meshwork of fibres which trap the blood clot and an early scar is formed.
If the injury is significant, the next stage of the inflammatory phase is phagocytic. Circulating monocytes modulate into macrophages (Fowler 1989) (Fig. 3.2) and join the resident macrophage population to clear the debris from the site of injury through phagocytic action (Leibovich & Ross 1974). The macrophages increase in great numbers during the first 3 or 4 days (Dingman 1973). They engulf any matter with which they come into contact, clearing the wound environment and preparing it for subsequent repair. As well as performing a phagocytic role, the macrophage acts as a director cell, directing the repair process by chemically influencing an appropriate number of fibroblast cells activated in the area.
Macrophages also have a role in muscle regeneration, stimulating the production of satellite cells that align themselves to muscle fibres where they are capable of forming completely new muscle fibres or restoring damaged muscle fibres (Grefte et al 2007). Further support for their importance is provided by Shen et al (2008) who observed that a reduced number of macrophages in muscle tissue is associated with reduced muscle regeneration. Fisher & Rathgaber (2006) observed muscle regeneration after acute blunt trauma to the gastrocnemius muscle in rats and noted an initial degenerative process with early peaking of macrophages and fibroblasts in the first 3 days, following which numbers of macrophages declined and satellite cells were noted beneath the basal lamina of muscle fibres. A regenerative process then began and after 6 days numerous irregularly arranged sarcomeres were observed, composed of thick and thin microfilaments and Z bands.
A stage of neovascularization is also reached, with capillaries starting to develop after about 12 h and continuing to develop for a further 2 or 3 days (Daly 1990). The new vessels supply oxygen and nourishment to the injured tissues, but are delicate and easily disrupted. Relative immobilization is necessary at this stage and heat is contraindicated as it will cause increased bleeding from the fragile vessels (Hardy 1989).
Inflammation is a normal response to either trauma or infection and to have no inflammatory response would mean that healing would not occur. Too little inflammation will delay healing and too much inflammation will lead to excessive scarring. The so-called PRICE principle of employing protection, relative rest, ice, compression and elevation in the early stages can help to control heat, swelling and pain and to reduce, but not abolish, inflammation.
The control of swelling is important towards regaining function since the greater the amount of inflammatory exudate, the more fibrin will be found in the area which becomes organized into scar tissue (Evans 1980). Although pain inhibits normal movement, graded mobilization should be encouraged as early as possible to promote healing and to avert adverse scar tissue formation.
Pain itself can be reduced further by analgesic drugs. Non-steroidal anti-inflammatory drugs (NSAIDs) modify the inflammatory response, reduce chemical pain and reduce temperature by inhibiting the production of prostaglandins. There are exponents of the so-called ‘NICE’ principle, adding in the prescription of non-steroidal drugs to the usual first-line after-care of injury. However, a counter view is that these should not be prescribed for the first 2 or 3 days after injury since they will tend to delay healing (Boruta et al 1990, Watson 2009). It may be more appropriate to allow the body’s natural inflammatory response to proceed with analgesics such as paracetamol and/or physical measures such as mobilization, massage and electrotherapy as appropriate methods of pain control.
Corticosteroids are contraindicated in the acute inflammatory phase as they inhibit macrophage activity which delays debridement of the wound and scar tissue production by delaying the onset and proliferation of the fibroblasts (Leibovich & Ross 1974, Hardy 1989). Each stage of the inflammatory phase is essential to the repair process and suppression in the early stages will delay healing.
Gentle transverse frictions and controlled mobilization can be initiated early to allow healing in the presence of movement, as guided by the irritability of the tissues. Mobilization is initiated very gently, to start moving the injured connective tissue towards regaining painless function, but without unduly stressing the healing breach. Overstressing the healing tissue would disrupt the early fragile scar and set up a secondary inflammatory response, leading to excess scar tissue formation. Mobilization also has an effect on the mechanoreceptors which is thought to reduce pain.
Gentle frictions together with gentle mobilization will agitate tissue fluid and increase the chance contact of the macrophage with cellular debris, so promoting healing (Evans 1980).
REPAIR PHASE
The repair phase is simultaneous with the inflammatory phase and overlaps the remodelling phase.
Buy Membership for Orthopaedics Category to continue reading. Learn more here
[/not-level-membership-for-orthopaedics-category]
