Congenital Stem Cell Deficiency
Stem cells are present in all self-renewing tissues.1 Multiple studies have demonstrated that the stem cells for the corneal surface are located at the limbus. Davanger and Evensen2 were the first to speculate that the cells involved in the process of normal corneal epithelial renewal are located at the limbus when they observed that pigmented cells from the area of the limbus were moving centrally in the cornea. The pattern of movement they observed from the peripheral to the central cornea suggested a centripetal migration of corneal epithelial cells; we now know that the process of corneal epithelial cell renewal is very elaborate and involves a migration of epithelial cells from the periphery of the cornea to the central cornea where the older cells are then shed.3 Around the same time, a separate study observed that the basal cells of the limbal area are less differentiated than those cells that are found in other areas of the cornea epithelium. This observation was made by following the patterns of expression of a cornea-specific 64K keratin protein present in all corneal epithelial cells, except the limbal basal cells.4 The absence of expression of this protein in the limbal basal cells led Schermer and coworkers to speculate that corneal epithelial stem cells must be located in the less differentiated limbus.4 Additional studies demonstrated that the cells in the less differentiated limbus display another property in that they exhibit a long cell cycle consistent with the long cell cycles characteristic of other stem cell populations.5 The incorporation of tritiated thymidine for long time intervals into the limbal basal cells demonstrated this concept.5 Finally, Ebato and associates6 reported that human ocular limbal epithelial cells grew better in culture and had a higher rate of mitotic activity than peripheral corneal epithelial cells.
The most common causes of limbal stem cell deficiency (LSCD) are secondary in nature and include chemical and thermal burns, Stevens–Johnson syndrome and ocular cicatricial pemphigoid, multiple surgeries, topical medications, including antimetabolite use, neoplasias, and contact-lens induced LSCD.7–10 Congenital stem cell deficiency is less common and includes such conditions as congenital aniridia, some ectodermal dysplasias and sclerocornea.
Aniridia
Classic congenital aniridia is a bilateral, panocular disorder presenting with decreased best-corrected visual acuity, foveal hypoplasia, and nystagmus.11 Abnormalities in the cornea, anterior chamber angle, iris, lens, optic nerve, macula, and retina have all been reported.11 Glaucoma and cataract may occur. The prevalence is between 1 : 64 000 and 1 : 100 000.11 About two-thirds of cases of aniridia arise via familial inheritance and one-third of cases arise sporadically.11 A PAX6 gene mutation has been identified as the underlying cause of aniridia in the vast majority of patients, although some genetic heterogeneity has recently been identified.12
Aniridic Keratopathy
While ‘aniridia’ was traditionally named for the near complete absence of the iris, foveal hypoplasia is most often responsible for the initial poor visual acuity in affected patients. However, many of these patients are susceptible to further visual loss from a progressive aniridic keratopathy (AK).13 This keratopathy, due to a congenital deficiency of the limbal stem cells, progresses to involve the total cornea, and eventually may lead to severe corneal stromal scarring and complete conjunctivalization of the ocular surface.13
Deficiency of the limbal stem cells in aniridia has been proven. Tseng14 proposed an initial model of LSCD in AK, and this model was further elucidated in a separate study by Nishida et al.15 through the process of impression cytology. Aniridic patients in the Nishida study demonstrated a clinical absence of the limbal palisades of Vogt, and the presence of conjunctival goblet cells on the cornea, implying either a primary absence of the limbal stem cells or abnormal limbal stem cells in aniridia.
Although the term ‘aniridic keratopathy’ was not coined at the time, clinicians began to recognize corneal changes in their aniridic patients well over a hundred years ago. As early as 1893, corneal opacities were mentioned in aniridia, and casual references were made to corneal haze, peripheral dystrophy, and corneal clouding.16 In 1979, Mackman and colleagues16 grouped the corneal changes in AK into four stages. In their stage 0, the aniridic cornea is clinically normal. In their stage 1A, the cornea displays abnormal epithelium and vascularization in the 6- and 12-o’clock meridians, which progresses to 360-degree corneal involvement in stage 1B. In stage 2 AK, there is central corneal involvement with stromal scarring as well. However, the changes seen in AK present in more clinically distinct stages and it may be more helpful to the clinician to separate these out even further.
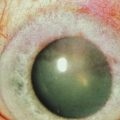
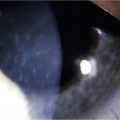
Holland has proposed the following classification of AK, which more accurately details the cornea changes (unpublished results). AK occurs in five stages, with each stage progressing to the next unless the LSCD is corrected (Figs 32.1–32.7). In stage 1, a peripheral corneal epitheliopathy exists. Patients may display increased fluorescein uptake in the area of the peripheral cornea and exhibit a characteristic of LSCD known as late-staining. Clinically abnormal epithelium is evident in the periphery of the cornea. Stage 2 AK occurs when the peripheral epitheliopathy moves centrally, involving the pericentral cornea. In stage 3 AK, the epitheliopathy involves the central cornea. At any stage, neovascularization may be seen and is non-specific for AK and not helpful in identifying the stage of AK a patient may be exhibiting. In stage 4 AK, the cornea displays complete epitheliopathy and subepithelial fibrosis. Total epitheliopathy is required before the process of subepithelial fibrosis (SEF) can occur. Without further treatment, stage 5 AK ensues, and in this stage the cornea exhibits total epitheliopathy and deeper scarring now involving the stroma. Clinically, it is important to recognize and diagnose AK before SEF and deep scarring of the cornea ensues (prior to stage 4), as the patient will require both correction of the LSCD and a cornea transplant beyond this point.
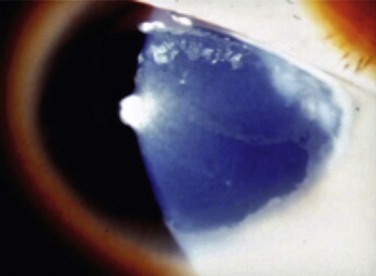
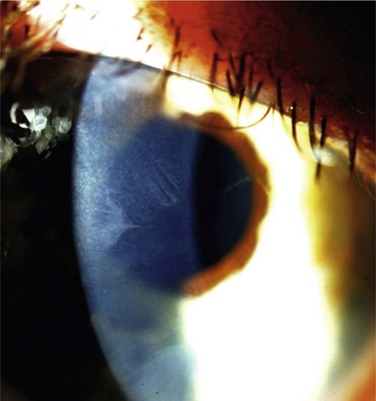
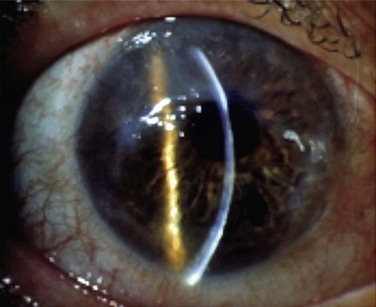
Figure 32.3 Stage 2 AK. Note the peripheral epitheliopathy.
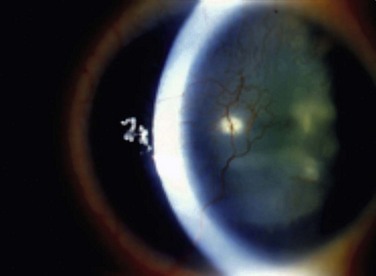
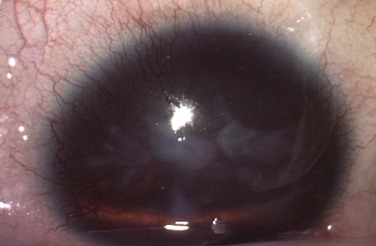
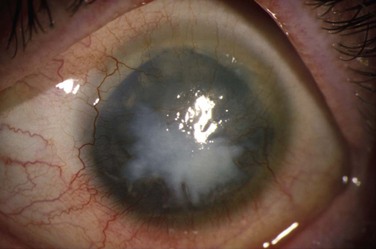
Figure 32.6 Stage 5 AK. There is total epitheliopathy along with subepithelial and stromal scar tissue.
The PAX6 gene plays an important role for the ocular surface in all individuals, not just aniridics. Studies have demonstrated PAX6 expression in the limbal stem cells.17 PAX6 has also been demonstrated in embryonic, as well as mature corneal and conjunctival tissues, and it may play a role in the maintenance and proliferation of the limbal stem cells.17 As mentioned, the underlying genetic defect in aniridia involves a mutation in the PAX6 gene, although other genes are now being identified. In aniridia, one copy of the PAX6 gene expresses the mutation while the second copy of the gene is normal. Patients who are born with two mutated copies of the PAX6 gene do not live.18
The mutated PAX6 gene has implications for the aniridic patient. The normal centripetal migration of corneal epithelial cells from the area of the limbus to the central cornea is impaired in aniridia secondary to the mutated PAX6 gene.11 PAX6 also regulates the expression of cytokeratins 3 and 12, which are critical for normal cell-to-cell binding, and in anchoring cells to their underlying basal lamina. In aniridia, the epithelium is more fragmented secondary to the reduced expression of cytokeratins as a result of the mutated PAX6 gene.11 Furthermore, the expression of adhesion molecules desmogleine, B-catenin, and gamma-catenin, is decreased in the aniridic because these too are controlled by the PAX6 gene.11 The absence of these adhesion molecules gives rise to spaces between the corneal epithelial cells.11 Finally, PAX6 regulates the development of cell surface proteins that are required for the normal migration of cells in response to injury, and in aniridia these proteins are absent.11 Cells cannot migrate in response to injury. The PAX6 gene also controls the production of matrix metalloproteinase 9 (MMP 9), a protein required for wound healing which is as a result, abnormal in aniridia. The end result is that aniridics have a fragile corneal surface that is not able to properly heal itself, and patients experience repeated epithelial defects and ulcerations.
While other genes are being discovered, the vast majority of patients with classic congenital aniridia are found to be heterozygous for a mutation in the PAX6 gene. Interestingly, only two of six patients with a congenital aniridia variant (variant aniridia; see below) in a study by Skeens et al.18 had definitive PAX6 mutations and one had a potentially disease-causing variant. Although the precise reason for this is unclear, one possibility is that patients with a milder, variant aniridic keratopathy phenotype may have sequence changes in regulatory or intronic regions of the PAX6 gene. In fact, distinct cis-acting DNA modules have been shown to specifically direct PAX6 expression in ocular surface tissues and lens during development.19Alternatively, there may be genetic heterogeneity for this phenotype. Although, the molecular techniques in the aforementioned study were not designed to identify large genomic deletions or rearrangements of the PAX6 gene, most patients with such deletions have a classic congenital aniridia phenotype, and the authors of this study felt that this was unlikely to be a cause of disease in their patients. Also of note is that, as mentioned, while the vast majority of aniridia is caused by a PAX6 gene mutation, there are isolated patients who may have mutations in other genes leading to the development of aniridia.
Variant Aniridia
Early reports of classic congenital aniridia emphasized the severe abnormalities of the iris on clinical examination as essential in making the diagnosis of this condition.20 Because of this terminology, many ophthalmologists now think to diagnose aniridia only in patients who display a near complete absence of the iris (Fig. 32.8). The term ‘aniridia’ is actually a misnomer. In 1994, Pearce first reported on the variability that may occur in the structural changes in the iris in aniridia.20 He cited the case reports of other authors who had not described complete absence of the iris, but instead reported atypical colobomas, stromal hypoplasia, full-thickness iris holes, and radial stromal defects in patients who had the additional ophthalmic findings consistent with what we define as classic congenital aniridia (poor visual acuity, fovea hypoplasia, nystagmus, cataract, and glaucoma).20 Hence to be diagnosed with ‘aniridia,’ a patient may have not only a near complete absence of the iris, but may present with any of these structural defects of the iris instead (Fig. 32.9).20 In fact, the abnormalities in each ocular structure affected in classic congenital aniridia may present along a wide spectrum. For example, not all patients develop cataracts and glaucoma, and patients may present with varying degrees of foveal hypoplasia, visual acuity, and nystagmus. While the process is bilateral, it can be asymmetric in its severity and progression. As previously discussed, Skeens et al. described a subset of patients referred for undiagnosed cornea pathology who presented with signs and symptoms of aniridic keratopathy.18 Because iris findings were generally mild and because nystagmus or other findings of classic aniridia, including foveal hypoplasia, were mild or not present, the diagnosis of aniridic keratopathy was not previously entertained by the referring physicians. Nonetheless, it was demonstrated that a subset of these patients had defined mutations in PAX6 (see above) and all patients responded well to keratolimbal allograft.
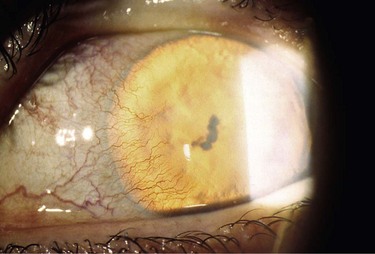
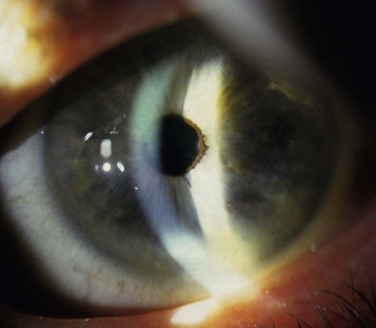
Figure 32.9 This patient has variant aniridia. Note the presence of ectropion uveae along with the cornea nodule.
Other authors have confirmed the same findings. In 1986, Kivlin et al. coined the term ‘dominantly inherited keratitis’ to represent a group of patients presenting with a dominantly inherited fibrovascular replacement of Bowman’s layer, associated with inflammation and vascularization of the cornea.21 Abnormalities of the iris were not reported, but their patients underwent penetrating keratoplasty of the cornea with a quick recurrence in the grafts of all original findings. Pearce et al. challenged, dominantly inherited keratitis. as a possible ‘aniridia variant’ when they reported the same cornea changes as Kivlin’s in 15 affected family members of a four-generation family who also had associated macular hypoplasia and iris abnormalities that included stromal atrophy and ectropion uveae.22 Some of their patients also underwent penetrating keratoplasty with a recurrence of the cornea findings in the grafts. The authors also noted that the patients had more subtle degrees of macular hypoplasia, with visual acuities as good as 20/25 to 20/30, and iris abnormalities which included radial defects, ectropion uveae, iris coloboma, and absent crypts and collarettes. Pearce thus declared a wider picture of familial aniridia to include those patients with the less severe changes in iris structure, little to no nystagmus, and improved visual acuities.22 He suggested we reconsider the diagnosis of ‘aniridia without aniridia.’22 A PAX6 gene mutation was identified as the underlying cause of ‘autosomal dominant keratitis.’23
Foveal hypoplasia may be identified clinically by the absence of a foveal light reflex, the absence of the normal darker pigmentation seen in the fovea, and/or the presence of vessels crossing the fovea. Foveal hypoplasia has also recently been elucidated using optical coherence tomography (OCT).24
Patients may undergo keratolimbal allograft with success following their clinical diagnosis (Fig. 32.10). All grafts in the study cited by Skeens et al. were successful,18 and the patients in that study currently maintain a stable ocular surface. It is important to identify that the corneal changes in these patients are associated with AK because without limbal stem cell transplantation, routine penetrating keratoplasty is destined to fail.
Treatment of the keratopathy of classic congenital or variant aniridia is of great value in patients whose vision has been reduced by the corneal changes. Cornea transplant (PK or deep anterior lamellar keratoplasty – DALK) alone will fail because without correction of the primary limbal stem cell deficiency, the findings of AK will develop in the transplanted tissue.25 Since the underlying corneal abnormality arises in the limbal stem cells, replacement of the limbal stem cells is required for proper treatment of AK. As previously mentioned, it is also important to treat the LSCD prior to the development of stage 4 and 5 AK, as described by Holland, so that further cornea transplant will not also be required following limbal stem cell replacement.
Treatment of Aniridic Keratopathy
Holland and coauthors25 reported on the success of KLAL in 31 eyes of 23 patients with classic congenital aniridia. Seventy-four percent of patients achieved a stable ocular surface. Visual acuity improved in the patients from 20/1000 to 20/165. Ninety percent of patients in the study who received systemic immunosuppression maintained a stable ocular surface whereas only 40% of patients not receiving systemic immunosuppression maintained a stable ocular surface. KLAL was determined to be an effective treatment for AK with patients receiving systemic immunosuppression more likely to maintain a healthy ocular surface than those who did not.
The safety of systemic immunosuppression in patients receiving ocular surface stem cell transplants has been reviewed. Holland and coauthors26 reported on the effects of systemic immunosuppression in 136 patients who underwent stem cell transplantation for varying diagnosis between 1997 and 2007. The most common systemic immunosuppression regimen consisted of tacrolimus, mycophenolate mofetil, and a short course of oral prednisone. The mean duration of use of systemic immunosuppression was 42.1 months. Only two patients experienced severe adverse events and 19 patients experienced minor adverse events, with 47.6% of those 19 having previous systemic co-morbidities. The authors concluded that the prevention of graft rejection post ocular surface transplant is critical and should be approached with the same rigor as in solid organ transplantation. With proper long-term follow-up by the transplant surgeon and close monitoring of physical health while taking immunosuppression, ocular surface transplant patients experience minimal irreversible toxicity.
Another approach to ocular surface stem cell transplantation in these patients involves the use of HLA-matched living related conjunctival tissue, termed a living related conjunctival limbal allograft (lr-CLAL). The advantage to this approach is that the patient may not need to be systemically immunosuppressed or may require a shorter term of immunosuppression because the tissue is matched for donor compatibility. Scocco et al.27 reported on 39 eyes of 32 patients with bilateral surface disorders and LSCD who underwent HLA-matched lr-CLAL. The donor limbus was obtained from a sibling or a parent after an appropriate class I and II HLA match. Vision improved in 46.2% of patients, ambulatory vision was achieved in 48.7%, and a stable corneal surface was achieved in 84.6% of the eyes. They concluded that HLA-matched lr-CLAL could be an adequate method of treatment for bilateral surface disorders. While none of the patients in their study had aniridia, three had ectodermal dysplasia, another congenital stem cell deficiency disorder discussed later in this chapter.
In congenital stem cell deficiency, the patient will require 360-degree replacement of the limbal tissues. A third alternative in patients with congenital stem cell deficiency involves the combination of lr-CLAL and KLAL. Biber et al.28 have reported on the success of ‘The Cincinnati Procedure.’ In this technique, 50% of the recipient limbus is replaced by HLA-matched lr-CLAL, and the remainder is replaced by non-matched KLAL. Again, the advantage to this technique is a decreased duration of systemic immunosuppression use.
Finally, the Boston keratoprosthesis is a suitable alternative to limbal stem cell transplantation in patients with congenital stem cell deficiency (Figs 32.11, 32.12). Akpek et al.29 evaluated the long-term outcomes of keratoprosthesis as an alternative surgical procedure in the management of AK. Sixteen eyes of 15 patients in their study underwent a Boston type I keratoprosthesis procedure. Device retention rate, intraoperative and postoperative complications, and preoperative and postoperative visual acuity were reviewed. The patients were followed for an average of 17 months. All devices were retained throughout the follow-up period with only one device requiring repair, but not exchange. The visual acuity improved in all patients with the exception of one. The authors concluded that the keratoprosthesis provides significant vision benefits in this group of patients.
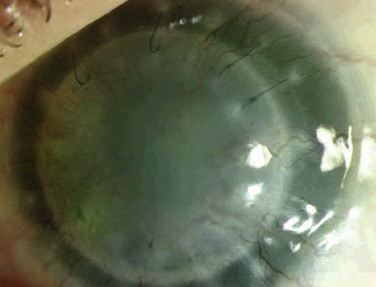
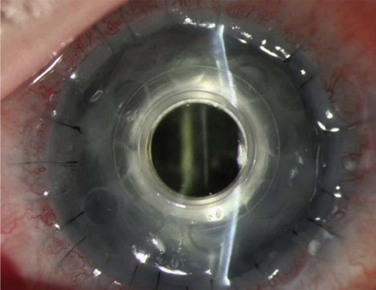
Figure 32.12 The same patient as in Figure 32.11 after undergoing keratoprosthesis placement.
Ectodermal Dysplasia
Over 150 separate forms of ectodermal dysplasia exist.30 Ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC) is one form of ectodermal dysplasia. EEC consists of ectrodactyly (lobster-claw deformity) of the hands and feet, ectodermal dysplasia, and cleft lip and palate. The EEC syndrome, an acronym coined by Rüdiger et al. in 1970,31 was first recognized as a distinct clinical entity by Cockayne in 1936.32 He drew attention to the dacryocystitis that commonly affects these patients, and found that it was associated with atresia of the lacrimal ducts. Since that description in 1936, several other reports have appeared in the literature detailing the ocular findings in the EEC syndrome. Many ocular structures are derived from surface ectoderm, so it is not surprising that the abnormal ocular findings in EEC may consist of absent meibomian glands, lacrimal drainage abnormalities, and a progressive keratopathy.33 Interestingly, the crystalline lens, a surface ectoderm-derived structure, is not affected.33
Limbal stem cell failure is the cause of the progressive keratopathy that develops in the EEC syndrome. Iorio et al.34 reviewed 23 patients affected by the EEC syndrome to assess the pathogenic basis of their visual morbidity. While all patients had ocular involvement, and the commonest involvement was anomaly of the meibomian glands and lacrimal drainage system, the major cause of visual morbidity was progressive LSCD that occurred in 61% of patients. LSCD was related to advancing age and caused a progressive keratopathy resulting in a dense corneal pannus.34 Clinical examination of the limbal palisades of Vogt along with impression cytology by the authors did confirm the presence of LSCD. The authors also confirmed that heterozygous mutations in the p63 gene are responsible for the limbal stem cell failure.34 Approximately 40 different pathogenic p63 mutations have been identified in EEC syndrome.34 Patients develop corneal conjunctivalization, cornea ulceration, and corneal neovascularization as a result of the LSCD that develops secondary to the mutations.34 Mutational analysis of p63 was performed in this study by polymerase chain reaction-based bidirectional Sanger sequencing.34 The p63 gene is essential in epithelial development for the regenerative proliferation of epithelial cells.34 The p63 gene also distinguishes human keratinocyte stem cells from their transient amplifying cell (TAC) progeny, and is expressed by the basal cells of the limbal epithelium but not by the transient amplifying cells covering the corneal surface.34 Briefly, the normal process of a stem cell involves an asymmetric cell division so that one of the daughter cells remains a stem cell while the other becomes a TAC.35 The TAC then differentiates into a postmitotic cell (PMC) and finally into a terminally differentiated cell (TDC).35 The TAC and PMC have no ability to divide.
Saw et al.36 reported on a subgroup of ectodermal dysplasia patients presenting with severe cicatrizing conjunctivitis and sharing clinical and immunopathological features of ocular mucous membrane pemphigoid (MMP). Six patients with ectodermal dysplasia (four with EEC and two with hypohidrotic ectodermal dysplasia) demonstrated circulating and mucosa-deposited antibasement membrane zone antibodies. Clinically severe ocular surface inflammation (OSI) was controlled with systemic immunosuppression in 83% of patients. Patients with MMP develop severe cicatrizing conjunctivitis and LSCD as a result of uncontrolled OSI.37 The need to control OSI in these patients in order to prevent cornea conjunctivalization and vascularization with loss of vision is evident in MMP, as should it be in patients with ectodermal dysplasia presenting with severe inflammation of the ocular surface.
Case reports of penetrating keratoplasty in patients with EEC syndrome and keratopathy cite unsuccessful results, which is not surprising given that limbal stem cell failure has been identified as the cause of the corneal abnormalities. Mader and Stulting38 reported the outcome of PK in two related patients with EEC syndrome who underwent surgery after a spontaneous corneal perforation. One patient developed microbial keratitis postoperatively, and the second developed recurrent vascularization, peripheral scarring, and recurrent epithelial erosions. Baum and Bull39 reported on a 5-year-old child with EEC syndrome who underwent PK, but 1 month postoperatively only 1 to 2 mm of the donor button had re-epithelialized and the corneal graft subsequently opacified and failed. Replacement of the abnormal limbus in these patients with stem cells prior to PK is beneficial. Daya and Ilari40 reported on the success of HLA matched lr-CLAL in two eyes with ectodermal dysplasia and one additional with EEC syndrome. Keratoprosthesis placement could be an alternative to limbal stem cell transplant in these patients; however, no reports exist to date on this treatment.
Not all forms of ectodermal dysplasia display corneal involvement. Keratitis–ichthyosis–deafness, or KID, syndrome, also called Senter’s syndrome, is an ectodermal dysplasia that does, however, have corneal involvement with presumed limbal stem cell deficiency. In 1915, Burns described a patient with progressive corneal inflammatory disease, diffuse hyperkeratotic erythroderma, and neurosensory hearing loss.41 The acronym ‘KID’ was coined by Skinner et al.42 to represent the triad of keratitis, ichthyosis, and deafness. KID syndrome consists of congenital neurosensory hearing loss, ectodermal dysplasia, and progressive corneal vascularization and conjunctivalization.43 Chronic bacterial and mycotic infections, an increased risk of squamous cell carcinoma, liver disease, and mental retardation have all been associated in some patients as well.43 Gicquel et al.44 reported on LSCD as a major pathologic factor in this syndrome.
Hereditary hypohidrotic ectodermal dysplasia may be a variant of the KID syndrome. Patients present with decreased sweating, generalized hypotrichosis, and dental abnormalities, as well as ectodermal dysplasia. Corneal vascularization and conjunctivalization have been reported.45 Bowman’s membrane is replaced with a fibrovascular pannus with inflammatory cells present.45 The corneal epithelium demonstrates acanthosis and dyskeratosis.45
Autoimmune Polyglandular Endocrinopathy–Candidiasis–Ectodermal Dysplasia (APECED)
Keratitis early in life associated with multiple endocrine deficiency (failure of the parathyroid glands, adrenal cortex, gonads, pancreatic beta cells, gastric parietal cells, thyroid gland, and hepatitis), chronic mucocutaneous candidiasis, dystrophy of dental enamel and nails, alopecia, and vitiligo, has been coined the APECED syndrome.46 Also known as autoimmune polyendocrinopathy syndrome type 1, APECED syndrome is a rare autosomal recessive disease, due to a defect in the AIRE (autoimmune regulator) gene.47 Corneal conjunctivalization and vascularization is characteristic.47 Impression cytology of the cornea demonstrates goblet cells consistent with conjunctivalization of the cornea.47 It is uncertain whether this represents a congenital stem cell deficiency or an acquired one. Stem cell grafts from living related donors have been reported in this disorder.48
Sclerocornea
Sclerocornea is congenital and usually bilateral. The cornea is often flat with complete opacification and no appreciable limbal area, or only peripheral corneal opacification may exist. Autosomal dominant inheritances, as well as sporadic occurrence have been reported.49 Sclerocornea is often not an isolated anomaly but is seen in conjunction with Peter’s anomaly, microphthalmos, and aniridia. In peripheral sclerocornea, the changes are usually not progressive, suggesting that stem cells are present to maintain central corneal clarity.50 Demonstration of limbal stem cell deficiency has not been reported, but in the more severe cases with total sclerocornea and no defined limbus, it is likely that a stem cell deficiency is present.
References
1. Holland, EJ. Epithelial transplantation for the management of severe ocular surface disease. Trans Am Ophthalmol Soc. 1996;94:677–743.
2. Davanger, M, Evensen, A. Role of the pericorneal papillary structure in renewal of corneal epithelium. Nature. 1971;229:560–561.
3. Lim, M, Goldstein, MH, Tuli, S, et al. Growth factor, cytokine, and protease interactions during cornea wound healing. Ocul Surf. 2003;1:53–65.
4. Schermer, S, Galvin, S, Sun, TT. Differentiation-related expression of a major 64K corneal keratin in vivo and in culture suggests limbal location of corneal epithelial stem cells. J Cell Biol. 1986;103:49–62.
5. Cotsarelis, G, Dong, G, Sun, TT, et al, Differential response of limbal and corneal epithelial to phorbol myristate acetate (TPA). Invest Ophthalmol Vis Sci. 1987;28(suppl. 1:1.
6. Ebato, B, Friend, J, Thoft, RA. Comparison of limbal and peripheral human corneal epithelium in tissue culture. Invest Ophthalmol Vis Sci. 1988;29:1533–1537.
7. Margo, CE. Congenital aniridia: a histopahologic study of the anterior segment in children. J Pediatr Ophahalmol. Strabismus. 1983;20:192–198.
8. Nelson, LB, Spaeth, GL, Nowinski, T, et al. Aniridia: a review. Surv Ophthalmol. 1984;28:621–642.
9. Tugal-Tutkun, I, Akova, YA, Foster, CS. Penetrating keratoplasty in cicatrizing conjunctival diseases. Ophthalmology. 1995;102:576–585.
10. Jenkins, C, Tuft, S, Liu, C, et al. Limbal transplantation in the management of chronic contact-lens associated epitheliopathy. Eye. 1993;7:629–633.
11. Lee, H, Khan, R, O’Keefe, M. Aniridia: Current pathology and management. Acta Ophthalmologica. 2008;86:708–715.
12. Ito, Y, Footz, T, Berry, F, et al. Severe molecular defects of a novel FOXC1 W152G mutation result in aniridia. Invest Ophthalmol Vis Sci. 2009;50:3573–3579.
13. Mayer, KL, Nordlund, ML, Schwartz, GS, et al. Keratopathy in congenital aniridia. Ocul Surf. 2003;1:74–79.
14. Tseng, SCG. Concept and application of limbal stem cells. Eye. 1989;3:141–157.
15. Nishida, K, Kinoshita, S, Ohashi, Y, et al. Ocular surface abnormalities in aniridia. Am J Ophthalmol. 1995;120:368–375.
16. Mackman, G, Brightbill, FS, Optiz, JM. Corneal changes in aniridia. Am J Ophthalmol. 1979;87:497–502.
17. Karoma, BM, Yang, JM, Sundin, OH. The Pax-6 homeobox gene is expressed throughout the corneal and conjunctival epithelia. Invest Ophthalmol Vis Sci. 1997;38:108–120.
18. Skeens, HM, Brooks, BP, Holland, EJ. Congenital aniridia variant: minimally abnormal irides with severe limbal stem cell deficiency. Ophthalmology. 2011;118:1260–1264.
19. Kammandel, B, Chowdhury, K, Stoykova, A, et al. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev Biol. 1999;205:79–97.
20. Pearce, WG. Variability of iris defects in autosomal dominant aniridia. Can J Ophthalmol. 1994;29:25–29.
21. Kivlin, JD, Apple, DJ, Olson, RJ, et al. Dominant inherited keratitis. Arch Ophthalmol. 1986;104:1621–1623.
22. Pearce, WG, Mielke, BW, Hassard, DT, et al. Autosomal dominant keratitis: a possible aniridia variant. Can J Ophthalmol. 1995;30:131–137.
23. Mirzayans, F, Pearce, WG, MacDonald, IM, et al. Mutation of the PAX6 gene in patients with autosomal dominant keratitis. Am J Hum Genet. 1995;57:539–548.
24. Holstrom, G, Eriksson, U, Hellgren, K, et al. Optical coherence tomography is helpful in the diagnosis of foveal hypoplasia. Acta Ophthalmol. 2010;88:439–442.
25. Holland, EJ, Djalilian, AR, Schwartz, GS. Management of aniridic keratopathy with keratolimbal allograft: a limbal stem cell transplantation technique. Ophthalmology. 2003;110:125–130.
26. Holland, EJ, Mogilishetty, G, Skeens, HM, et al. Systemic immunosuppression in ocular surface stem cell transplantation: results of a 10-year experience. Cornea. 2012;31:655–661.
27. Scocco, C, Kwitko, S, Rymer, S, et al. HLA-matched living-related conjunctival limbal allograft for bilateral ocular surface disorders: long-term results. Arq Bras Oftalmol. 2008;71:781–787.
28. Biber, JM, Skeens, HM, Neff, KD, et al. The Cincinnati procedure: technique and outcomes of combined living-related conjunctival limbal allografts and keratolimbal allografts in severe ocular surface failure. Cornea. 2011;30:765–771.
29. Arpek, EK, Harissi-Dagher, M, Petrarca, R, et al. Outcomes of Boston keratoprosthesis in aniridia: a retrospective multicenter study. Am J Ophthalmol. 2007;144:227–231.
30. Freire-Maia, N, Pinheiro, M. Ectodermal dysplasias: a review of the conditions described after 1984 with an overall analysis of all the conditions belonging to this nosologic group. Rev Brasil Genet. 1988;10:403–414.
31. Rüdiger, RA, Haase, W, Passarge, E. Association of ectrodactyly, ectodermal dysplasia, and cleft lip-palate. Am J Dis Child. 1970;120:160–163.
32. Cockayne, EA. Cleft palate–lip, hare lip, dacryocystitis and cleft hand and foot. Biometrika. 1936;28:60–63.
33. Mcnab, A, Potts, M, Welham, R. The EEC syndrome and its ocular manifestations. Br J Ophthalmol. 1989;73:261–264.
34. Di Iorio, E, Kaye, S, Ponzin, D, et al. Limbal stem cell deficiency and ocular phenotype in ectrodactyly-ectodermal dysplasia-clefting syndrome caused by p63 mutations. Ophthalmology. 2010;119:74–83.
35. Leblond, CP. The life history of cells in renewing systems. Am J Anat. 1981;160:114–158.
36. Saw, VP, Dart, JK, Sitaru, C, et al. Cicatrising conjunctivitis with anti-basement membrane autoantibodies in ectodermal dysplasia. Br J Ophthalmol. 2008;92:1403–1410.
37. Thorne, JE, Anhalt, G, Jabs, D. Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology. 2004;111:45–52.
38. Mader, TH, Stulting, RD. Penetrating keratoplasty in ectodermal dysplasia. Am J Ophthalmol. 1990;110:319–320.
39. Baum, JL, Bull, MJ. Ocular manifestations of the ectrodactyly, ectodermal dysplasia, cleft lip-palate syndrome. Am J Ophthalmol. 1974;78:211–216.
40. Daya, SM, Ilari, L. Living related conjunctival limbal allograft for the treatment of stem cell deficiency. Ophthalmology. 2001;108:126–134.
41. Burns, FS. A case of generalized congenital keratoderma, with unusual involvement of the eyes, ears, and nasal and buccal mucous membranes. J Cutan Dis. 1915;33:255–260.
42. Skinner, BA, Greist, MC, Norins, AL. The keratitis, ichthyosis, and deafness (KID) syndrome. Arch Dermatol. 1981;117:285–289.
43. Messmer, EM, Kenyon, KR, Rittinger, O, et al. Ocular manifestations of Keratitis-Ichthyosis-Deafness (KID) syndrome. Ophthalmology. 2005;112:e1–e6.
44. Gicquel, JJ, Lami, MC, Catier, A, et al. Limbal stem cell deficiency associated with KID syndrome, about a case [in French]. J Fr Ophtalmol. 2002;25:1061–1064.
45. Wilson, FM, Grayson, M, Pieroni, D. Corneal changes in ectodermal dysplasia. Am J Ophthalmol. 1973;75:17–27.
46. Ahonen, P, Myllarniemi, S, Sipila, I, et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829–1836.
47. Rajendram, R, Deane, JA, Barnes, M, et al. Rapid onset childhood cataracts leading to the diagnosis of Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Am J Ophthalmol. 2003;136:951–952.
48. Tseng, SCG, Meller, D, Pires, RTF, et al. Corneal surface reconstruction by limbal epithelial cells ex vivo expanded in amniotic membrane. Investigative Ophthalmol Vis Sci. 2000;41:S756. [Abst No 4016].
49. Elliott, JH, Feman, SS, O’Day, DM, et al. Hereditary sclerocornea. Arch Ophthalmol. 1985;103:676–679.
50. Waizenegger, UR, Kohnen, T, Weidle, EG, et al. Kongenitale familiar cornea plana mit Ptosis, peripherer Sklerokornea und Bindehaut-Xerose. Klin Monatsbl Augenheilk. 1995;206:111–116.