[level-membership-for-otolaryngology-category]
CHAPTER 191 Congenital Malformations of the Inner Ear
Development of the inner ear begins early in embryogenesis. By the end of the eighth week, the membranous labyrinth has assumed its characteristic convoluted shape.1 Gradual ossification of the otic capsule develops around the membranous labyrinth and is essentially complete by birth.2 Maturation of the sensory epithelium occurs long after formation of the membranous labyrinth, during the late second and early third trimesters. By weeks 26 to 28 of gestation, hair cell and auditory neural development are largely complete. Thus, the normal human fetus may be able to hear 2.5 to 3 months before birth.
Most inner ear malformations arise when formation of the membranous labyrinth is interrupted during the first trimester of pregnancy.2 This interruption may be either a result of inborn genetic error or a consequence of a teratogenic exposure during the period of inner ear organogenesis between the fourth and eighth weeks of gestation. Genetic errors may be either dominant or recessive and may manifest as sensorineural hearing loss (SNHL) alone or be associated with any of a number of syndromes.4 A partial list of syndromes associated with radiologically detectable inner ear malformation includes Pendred’s, Usher’s, Waardenburg’s, Wildervanck’s (cervico-oculoacoustic), branchio-oto-renal, and Alagille’s sundromes.5–8 Nonsyndromic familial inner ear malformations have also been described.9,10 Teratogenic influences known to affect inner ear organogenesis include in utero viral infection (e.g., rubella, cytomegalovirus infection), chemical teratogens (e.g., thalidomide), and radiation exposure.11 Abnormalities in otic capsule structure and deficiencies in the organ of Corti appear to arise as secondary effects of the earlier error in development of the membranous labyrinth. Derangement of the otic capsule ossification process alone does not appear to be a major mechanism in congenital hearing loss. Ossification of the labyrinthine lumen, however, is a common finding in early acquired deafness, typically arising as a consequence of meningitis.
Developmental damage to cochlear structure and function is not restricted to agents that cause gross structural malformations. Even in doses below those that would be ototoxic in the adult, aminoglycoside antibiotics administered in the animal equivalent of the human first trimester of pregnancy cause severe hearing loss in several species.12 This time frame corresponds to the maturation of the outer hair cells and initiation of the cochlear potentials. Human studies also document this. In 35 of the infants delivered by 72 women who received streptomycin prophylaxis for tuberculosis during the first 4 months of pregnancy, auditory deficits ranging from minor high-frequency threshold elevations to severe bilateral SNHL were noted.13
Incidence
Most children with congenital sensory hearing loss, with the possible exception of auditory neuropathy, would have detectable abnormalities in their inner ears if they could be examined histologically. Because most children with profound bilateral SNHL have radiographically normal inner ears, it can be inferred that malformations limited to the membranous labyrinth predominate. The incidence of malformations reported varies, depending on the hearing level of the study population, the sophistication of imaging used, and the definition of abnormality used by the observer. As a rule of thumb, approximately 20% of cases of congenital SNHL will demonstrate inner ear malformation with modern imaging technology. In a series of 234 children who had SNHL of varying degrees of severity, Reilly14 found cochlear anomalies in only 4% of those evaluated by high-resolution computed tomography (CT). With improvements in CT imaging technology and greater awareness of inner ear deformities, Antonelli and colleagues found anomalies in 31% of 157 children with SNHL of variable degree.15 In another CT study, anomalies were found in 17% of 185 ears of children with SNHL and none in the ears of 309 children without SNHL.16 A large Korean study found 22% incidence of anomalies on radiologic imaging in 590 ears with profound SNHL.17
The incidence of deformities of the semicircular canals (SCCs) and inner ear aqueducts has been less well studied than that of cochlear deformities. In a series of patients with radiographically detectable malformations of the inner ear, the cochlea was involved in 76%, the semicircular canals were involved in 39%, and the vestibular aqueduct (VA) was affected in 32% of ears.3 The total is more than 100%, because many cases demonstrate abnormalities of more than one portion of the inner ear. In recent years, a heightened awareness of VA enlargement, combined with the greater sensitivity of axial CT scans in demonstrating this deformity, has led to a substantial increase in its detection. The rapid accrual of cases by clinicians interested in inner ear malformations suggests that enlargement of the VA will ultimately prove to be the most common radiographically detectable inner ear anomaly.6
Among deaf children with radiographically normal inner ears, histopathologic studies indicate that cochleosaccular dysplasia (Scheibe’s dysplasia) is by far the most common deformity.18 Because of the paucity of pathologic specimens available for examination, it is impossible to estimate the relative frequency of the various membranous malformations.
Classification
The traditional nomenclature used to describe congenital anomalies of the inner ear involves a confusing array of eponyms that stem from the first reports of the various morphologic patterns, usually by 18th or 19th century authors. In this chapter, a descriptive classification system is used, along with the traditional eponymous terminology, to make this topic more logical, easier to learn, and more clinically relevant (Box 191-1). In membranous malformations, this classification is based on histopathologic changes in the inner ear; in combined osseous-membranous deformities, radiographic appearance is used to distinguish among the various entities.3 Correct use of the terminology of pathoembryology is important, although these terms frequently are applied imprecisely in the earlier literature. Key terms are aplasia (complete lack of development), hypoplasia (incomplete development), and dysplasia (aberration in development). The classification scheme used in this chapter has proved to be of practical clinical use and has been employed in most published studies in recent years. Other classification schema that categorize observed anomalies have been proposed.19
Malformations Limited to the Membranous Labyrinth
In malformations limited to the membranous labyrinth, which account for more than 90% of cases of congenital deafness, the bony labyrinth is normal.20–23 In its most severe form, membranous dysplasia involves the entire labyrinth, including the cochlea, semicircular canals (SCCs), utricle, and saccule. A limited form of membranous labyrinthine dysplasia, involving only a portion of the inner ear, also has been described.
Complete Membranous Labyrinthine Dysplasia
Complete membranous labyrinthine dysplasia was first described by Siebenmann and Bing24 and is extremely rare. It has been reported in association with cardioauditory (Jervell and Lange-Nielsen) and Usher syndromes.25
Limited Membranous Labyrinthine Dysplasia
Cochleosaccular Dysplasia (Scheibe’s Dysplasia)
Incomplete development of the pars inferior is the most frequent histopathologic finding in congenital deafness. It was first described by Scheibe26 and commonly is known as cochleosaccular dysplasia. The spectrum of pathologic findings in this anomaly, which is confined to the cochlea and the saccule, has been well described.22,27–30 The organ of Corti is either partially or completely missing. The cochlear duct usually is collapsed, with Reissner’s membrane adherent to the limbus. Less commonly, the duct is distended, presumably as a result of endolymphatic hydrops. The stria vascularis typically is degenerated and may contain colloidal inclusions. Schuknecht31 described characteristic strial changes consisting of aplasia alternating with regions of hyperplasia and gross deformity. Cochlear changes may be severe in the base turn and gradually lessen in intensity toward the apex, or they may be severe throughout. The saccule usually is collapsed and has degenerated sensory epithelium. In cochleosaccular dysplasia, the SCCs and utricle are normal. Auditory neuronal survival is variable but may remain normal into adulthood, at least in some cases. Cochleosaccular dysplasia also has been demonstrated in a number of animal species, including the deaf white cat, Dalmatian dogs, and various mouse mutants.32
Malformations of the Membranous and Osseous Labyrinth
Congenital anomalies of the inner ear that deform the otic capsule are of special interest to the clinician because they may be recognized and differentiated during life through radiographic imaging. As discussed previously, only approximately 20% of congenitally deaf people demonstrate radiographically anomalous inner ears. The clinical manifestations and natural history of these deformities are highly variable. Although some patients are deaf from birth, most maintain some residual hearing into adulthood. Slowly progressive deterioration of hearing during childhood, with eventual stabilization, is common. Sudden decrements in hearing are frequent and may appear to be spontaneous or may be triggered by head trauma, even minor in nature. Presumably, most of these cases are due either to internal fistulization secondary to membrane rupture within the cochlea, with admixture of perilymph and endolymph, or to external fistulization to the middle ear. Fluctuant hearing loss is unusual in these patients, and endolymphatic hydrops is an atypical finding. In some patients, hearing may be best preserved in very high frequencies (greater than 8000 Hz), which are not measured by conventional audiometry.33 Residual ultra-audiometric hearing should be suspected in hearing-impaired children who manifest substantially better auditory function than would be predicted by pure tone results in the speech frequencies.34 Occasionally, malformation of the inner ears may be associated with normal hearing.35,36 This is especially the case for semicircular canal anomalies.37 Vestibular symptoms, which occasionally are severe, are present in approximately 20% of patients.3 Retardation of motor development has been documented in some children with malformed inner ears, particularly those in whom semicircular canals are absent.38 A few patients experience vertigo when exposed to loud sounds—the so-called Tullio phenomenon.39
A wide variety of morphologic patterns of inner ear malformation has been observed radiographically and may involve the cochlea, SCCs, or vestibular aqueduct3,40–45 (Fig. 191-1). Similar diversity has been observed on histologic analysis.21,46–50 A majority of combined osseous and membranous malformations appears to arise from a premature arrest in the development of one or more components of the inner ear (Fig. 191-2). The strongest evidence for this theory comes from the resemblance of most malformed inner ears to the appearance of the inner ear during embryogenesis, particularly between the fourth and eighth weeks of gestation. As a general rule, the earlier the developmental arrest, the more severe the deformity and the worse the hearing will be.
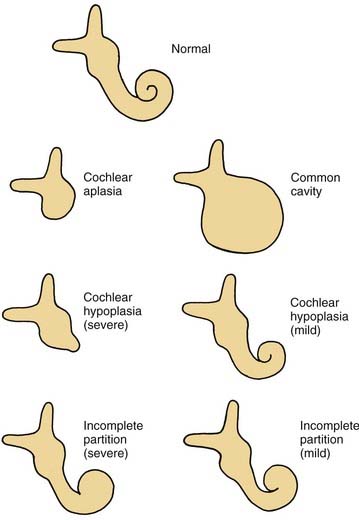
Figure 191-1. Cochlear malformations. Drawings were made from coronal computed tomography scans.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97[Suppl 40]:2.)
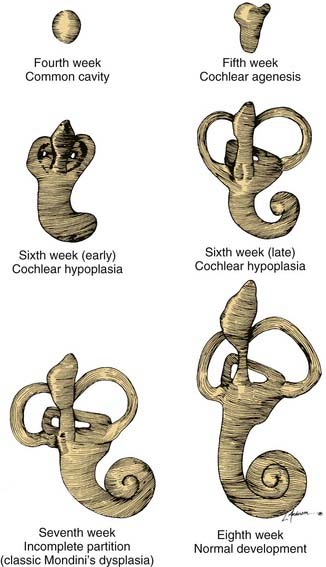
Figure 191-2. Embryogenesis of cochlear malformations.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97(Suppl 40):2.)
Other anomalies cannot be explained by a premature arrest in development alone and appear to arise from an aberrant embryologic process. An example of this type of anomaly is a cochlea of normal length but abnormal size or coiling geometry.46 In humans, the inner ear is of adult size at birth and shows strikingly little variation in size among individual patients. Pappas and colleagues51 suggested that some children with congenital SNHL and apparently normal inner ear morphology on CT possess subtle abnormalities in the dimensions of inner ear structures. These investigators propose that such dimensional variations arise from a teratogenic insult during the second or third trimester, after the membranous labyrinth has formed but before it has reached adult size. Further study is needed to determine the clinical relevance of these observations.
Whereas some inner ear malformations involve only one portion of the inner ear, many patients have a combination of anomalies involving more than one component. Between the fourth and fifth weeks of development, the spheric otocyst develops three buds that ultimately form the cochlea, SCCs, and VA (see Fig. 191-2). An inner ear malformation may be limited to one of these anlages, may involve a combination of two, or may even affect all three.
The frequent coexistence of deformities involving the cochlea, SCCs, and VA has several possible explanations: (1) the anomaly is genetically predetermined; (2) an insult to the embryo occurred before the fifth week; or (3) each of the buds was susceptible to some teratogenic influence at a later stage of development. A majority of inner ear malformations are bilateral and symmetrical. In cases in which radiographs detect an anomaly on only one side, the opposite “normal” inner ear has a hearing loss in approximately 50% of cases.3
Before the evolution of high-resolution imaging technology, clinicians and histopathologists alike tended to lump these malformations together under the term Mondini’s dysplasia, after the first report by Carlo Mondini. We are indebted to the late Dr. Peter Phelps and Latin scholar Gordon Hartley for an English translation of Mondini’s original article.52 Before the Academy of Sciences of the University of Bologna, Mondini described the inner ear findings in a deaf 8-year-old boy who was struck on the foot by a wagon and later died of gangrene.53 The cochlea possessed only 1.5 turns and had a hollow apical cavity. An enlarged vestibule and VA also were noted. This deformity is the most common form of cochlear anomaly (Table 191-1). Although numerous other distinct anatomic patterns of inner ear malformation are discernible radiographically and histologically, many workers continue to use the term Mondini’s dysplasia to describe them all. To avoid a confusing and overly broad nomenclature system, this designation is best reserved for the particular subtype of cochlear malformation first described by Mondini, whether or not it is associated with other inner ear malformations.
| Malformation | Incidence (%) |
|---|---|
| Incomplete partition (Mondini’s dysplasia) | 55 |
| Common cavity | 26 |
| Cochlear hypoplasia | 15 |
| Cochlear aplasia | 3 |
| Complete labyrinthine aplasia (Michel’s aplasia) | 1 |
Complete Labyrinthine Aplasia (Michel’s Aplasia)
The most severe deformity of the membranous and osseous labyrinth, complete labyrinthine aplasia, was first described by Michel.54 This malformation is exceedingly rare. Presumably, a developmental arrest occurs before the formation of an otic vesicle, resulting in a complete absence of inner ear structures. Complete labyrinthine aplasia has been reported in association with anencephaly and thalidomide exposure.21,55 An association with external ear abnormalities also has been reported.56 A purported case of Michel’s aplasia actually described a cystic inner ear of the common cavity type.57 This is but one example of the inaccurate use of traditional eponyms—a frequent occurrence in the literature. The incidence of complete labyrinthine aplasia is overestimated in the radiographic literature because it is confused with labyrinthine ossification. In the latter condition, which usually is acquired during life, a sizable and dense otic capsule is evident radiographically. In complete labyrinthine aplasia, the otic capsule is entirely absent58 (Figs. 191-3 to 191-5). Such ears are, of course, uniformly deaf.
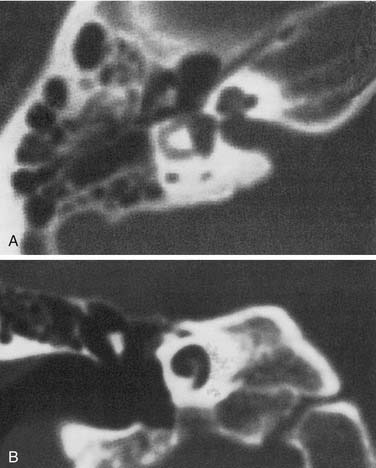
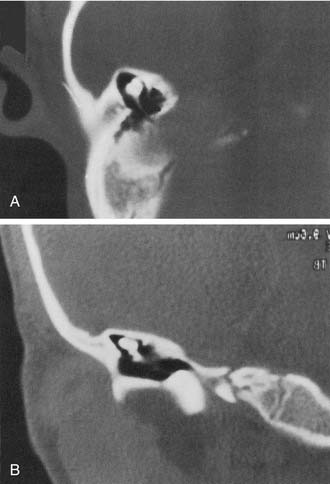
Cochlear Anomalies
Cochlear Aplasia
In cochlear aplasia the cochlea is completely absent, presumably as a result of an arrest in the development of the cochlear bud at the fifth week of gestation (see Fig. 191-1). This morphologic pattern is rare. Radiographically, only a vestibule and SCCs (usually deformed) are present. To differentiate this anomaly from labyrinthine ossification, it is necessary to assess the amount of otic capsule bone anterior to the internal auditory canal (IAC). In cochlear aplasia, the otic capsule is absent, whereas in osseous obliteration, it is dense and of normal dimensions. Ears with cochlear aplasia are devoid of auditory function.
Cochlear Hypoplasia
An arrest during the sixth week of gestation results in a hypoplastic cochlea consisting of a single turn or less. This deformity comprises approximately 15% of all cochlear anomalies. Radiographically, a small bud of variable length (usually 1 to 3 mm) protrudes from the vestibule (Fig. 191-6). The vestibule frequently is enlarged, with accompanying semicircular malformations in approximately one half of the cases. Small cochleas lacking a modiolus or other internal architecture have been described histologically.27,50,59 Hearing is variable in these ears and may be remarkably good in view of the minute size of the cochlea. The variability of hearing presumably is accounted for by the degree of membranous labyrinthine development within the truncated cochlear lumen.
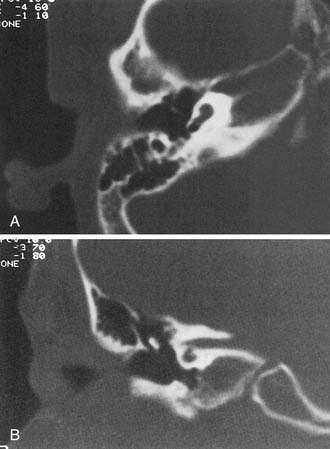
Incomplete Partition (Mondini)
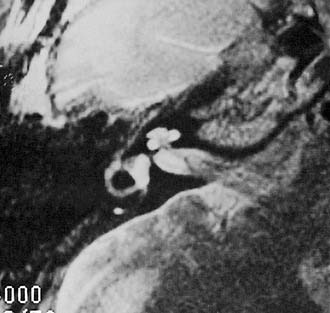
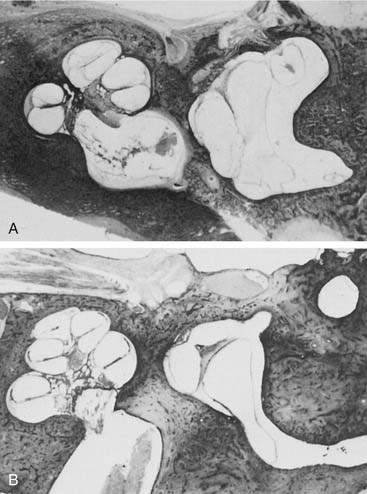
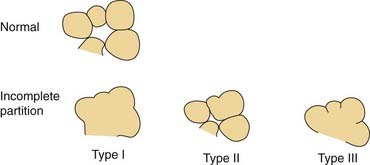
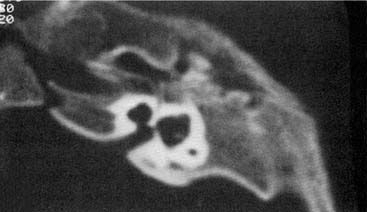
Arrest at the seventh week of gestation yields a cochlea that has only 1.5 turns. This is the most common type of cochlear malformation, accounting for more than 50% of all such deformities. Radiographically, the cochlea is smaller than normal and partially or completely lacks an interscalar septum (Figs. 191-7 and 191-8). Although the cochlea usually measures 8 to 10 mm vertically, it is typically in the 5- to 6-mm range in incomplete partition deformity. Care must be exercised in counting the number of cochlear turns radiographically because this may be difficult to determine even using high-resolution CT. The radiographic diagnosis depends more on cochlear size and the absence of a scalar septum than on the number of cochlear turns perceived. Histologically, incomplete partition appears to be the radiographic correlate of classical Mondini’s dysplasia (Fig. 191-9). In numerous reported cases, a small cochlea with 1.5 turns possessing an apical scala communis secondary to deficiency in the osseous spiral lamina has been described.27,46,48 Sennaroglu and Saatci have subtyped the incomplete partition deformity into three variants60 (Fig. 191-10). Type I lacks the entire modiolus and interscalar septa and demonstrates a cystic appearance. Type II has a normal base turn but a cystic apex (“Mondini type”). Recently Sennaroglu has proposed a type III variant with deficient modiolus and partial interscalar septation at the cochlea’s periphery (L. Sennaroglu, personal communication, 2007). Organ of Corti development is variable, as is the auditory neural population. As might be expected, auditory function also is variable, ranging from normal to profound SNHL. The mean hearing threshold (three-tone average) in a group of 41 ears with incomplete partition was 75 dB.3 SCC deformities accompany incomplete partition of the cochlea in approximately 20% of cases.
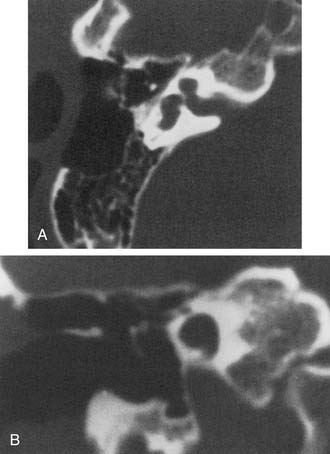
Common Cavity
A deformed inner ear in which the cochlea and vestibule are confluent, forming an ovoid cystic space without internal architecture, may be explained by an arrest at the week 4 otocyst stage. Alternatively, this deformity may result from aberrant development at a later stage. An empty ovoid space typically longer in its horizontal dimension is seen radiographically. Although the size of the cyst may vary, it averages 7 mm vertically and 10 mm horizontally. It is quite easy to misdiagnose a dysplastic lateral SCC as a common cavity deformity. The key to differentiating between the two is that a common cavity cochlea lies predominantly anterior to the IAC on axial-plane CT, and a dysplastic vestibular system lies posterior to it. Histologically, an ovoid or spherical smooth-walled cystic cavity containing primordia of the membranous labyrinth has been described.48,50,55 Sensory and supporting cells may be differentiated into recognizable organs of Corti that are scattered peripherally around the walls of the cyst. Neural population usually is sparse or absent. Hearing is usually, but not invariably, poor.
Labyrinthine Anomalies
Semicircular Canal Dysplasia
Dysplasia of the lateral SCC is a common type of inner ear malformation (Fig. 191-11). Approximately 40% of ears with a malformed cochlea will have an accompanying dysplasia of the lateral SCC.3 Occasionally, dysplasia of the lateral SCC exists as the sole inner ear malformation. During the sixth week of development, the budding SCC normally forms a semicircular evagination from the vestibular anlage. The central portion of the pocket-shaped protrusion adheres, leaving a peripheral semicircular tube. When this central adhesion fails to occur, SCC dysplasia results (Fig. 191-12; see also Fig. 191-2). SCC dysplasia occasionally takes the form of a small bud, rather than the more common half-disk shape, presumably because of a slightly earlier timing of the developmental insult. The lateral SCC is deformed more often than the posterior or superior SCC, apparently because it forms earlier in embryogenesis. The typical radiographic appearance of SCC dysplasia is that of a short, broad cystic space confluent with the vestibule (Fig. 191-13).
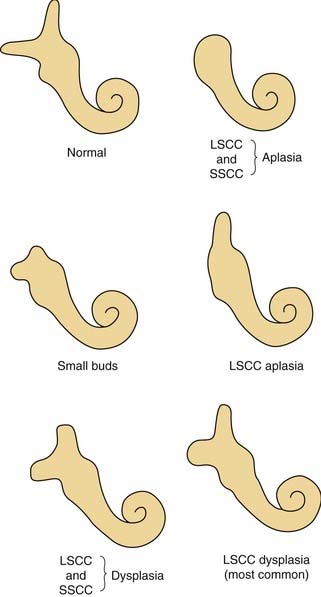
Figure 191-11. Semicircular canal malformations. LSCC, lateral semicircular canal; SSCC, superior semicircular canal.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97[Suppl 40]:2.)
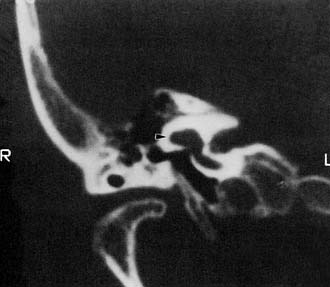
Numerous histologic descriptions of SCC dysplasia exist in the literature.46–50 The half-disk shaped cavity may contain a rudimentary crista ampullaris. The utricle and saccule may be distended, collapsed, or entirely absent. Caloric responses in SCC dysplasia are functionally absent or reduced in most cases, but a few may have normal responsiveness.3 Ears with malformations limited to the vestibular system often have normal or near-normal hearing. When the cochlea also is abnormal, sensory hearing levels tend to be impaired, to a variable degree.62 SCC dysplasia appears to have an association with conductive hearing loss, presumably because of inner ear micromechanical factors rather than stapes fixation.63
Semicircular Canal Aplasia
SCC aplasia is only one-fourth as common as SCC dysplasia.3 It is usually associated with cochlear anomalies.29 Presumably, SCC aplasia arises from a failure in the development of the vestibular anlage before the sixth week of gestation. Most cases are syndromic, with a predominance of the CHARGE association (coloboma, heart defects, atresia of the choanae, retardation of growth and development, genital, and ear abnormalities or hearing loss).64,65
Malformations of the Vestibular and Cochlear Aqueducts
Enlargement of the Vestibular Aqueduct
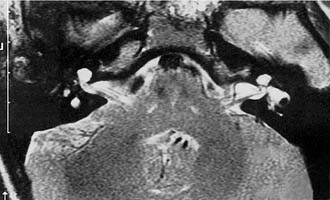
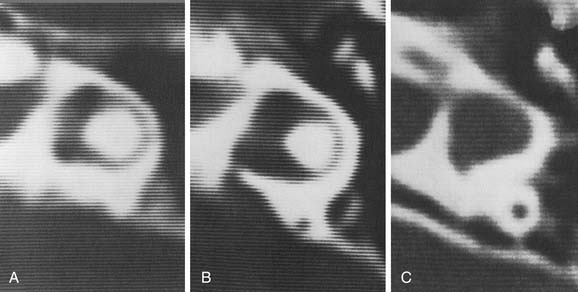
Experience suggests that enlargement of the VA is the most common radiographically detectable malformation of the inner ear.57,66–71 In earlier literature its incidence was underestimated, partly because of a lack of awareness but mostly because enlargement of the VA could be visualized only by lateral polytomography at a time when most studies were confined to the anteroposterior plane. The advent of high-resolution CT in the axial plane has made assessment of the VA much easier. The diameter of a normal VA, when measured halfway between the common crus and its external aperture, is between 0.4 and 1 mm. Enlargement of the VA is diagnosed when its diameter exceeds 2 mm, although enlarged VAs may exceed 6 mm in width. Whereas the VA is well visualized on axial CT (Fig. 191-14), the dilated endolymphatic sac is better seen with T2-weighted magnetic resonance imaging (MRI)72,73 (Fig. 191-15). The sac often is seen to be enormously enlarged on MRI, sometimes measuring 2 cm or more in diameter.74 In many cases, VA enlargement accompanies malformation of the cochlea or SCC. It also may be the sole radiographically detectable abnormality of the inner ear in a child with hearing loss. This condition is commonly referred to as the large VA syndrome, after Valvassori and Clemis’s first descriptions.75,76
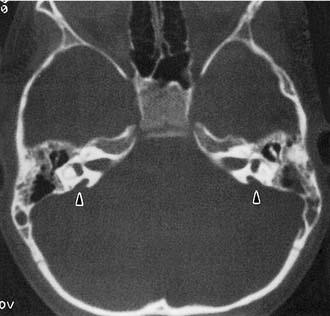
Figure 191-14. Bilateral enlargement of the vestibular aqueducts (arrowheads) as seen on an axial computed tomography scan.
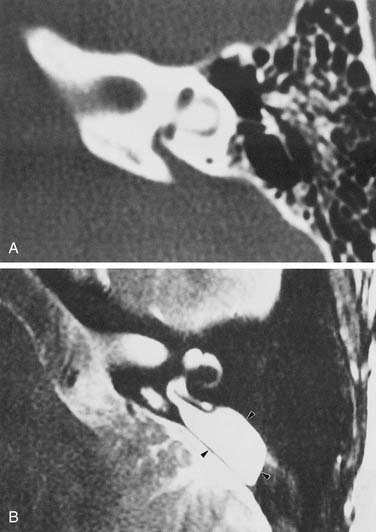
The VA derives from a diverticulum formed in the wall of the otocyst during the fifth week of gestation. It begins as a short, broad pouch but gradually elongates and thins until it achieves its characteristic J shape of adulthood (see Fig. 191-2). A premature arrest in development would be expected to produce a VA that is abnormally short and broad. However, an increasing body of evidence suggests that VA enlargement does not stem from an early arrest of sac development, but rather is an acquired deformity. Histologically, the sac and aqueduct are thin-walled and lack both vascularity and the rugose features thought to be important for physiologic function. Three observations suggest that a large VA stems from an abnormal communication between the subarachnoid space and the fluid chambers of the inner ear. The bone surrounding the VA may show signs of erosion, a finding that is inconsistent with a stable congenital deformity.77 Serial MR images show variability in both the size and signal characteristics of the enlarged sac.78 Presence of cerebrospinal fluid (CSF) under pressure, with consequent “gusher,” within the inner ear has been observed in ears with large VAs during both cochlear implantation and stapedectomy.31,79 Furthermore, the existence of abnormal CSF in the inner ear pathway frequently has been observed with CT or MRI. This anomaly typically consists of a defect of the cochlear modiolus at the distal end of the IAC.12,73 For CSF to become confluent with the endolymphatic space, the CSF fistula (subarachnoid space to perilymph compartment) must be accompanied by a second fistula joining the endolymph and perilymph spaces.
The large VA syndrome typically is bilateral. Affected children usually are born with normal or mildly impaired hearing that gradually deteriorates through childhood into adolescence and early adulthood. Hearing levels are variable, although at least 40% eventually develop profound SNHL.80 As with other inner ear malformations, there is a tendency to suffer sudden decrements of hearing, particularly after head trauma. Conductive hearing loss may be present but is likely to be from intracochlear micromechanical disturbances rather than from impairment of ossicular mobility. Stapes surgery is best avoided because of the risk of CSF otorrhea.79 Vestibular complaints in these patients have frequently been reported.81,82 Large VA syndrome has been observed to occur in familial clusters.10
Surgical manipulation of the endolymphatic sac has been attempted in patients with large VAs. In a sizable series of shunt procedures, there was a high incidence of postoperative hearing loss.67 Furthermore, a comparison of the operated and contralateral unoperated ears revealed no evidence of hearing stabilization after surgery. Recently, surgical obliteration of the enlarged endolymphatic sac with muscle and fascia has been advocated.83 Results in a multicenter study indicated that the procedure not only lacks efficacy but actually injures one half of the operated ears.84 Cochlear implantation has been quite successful in both adults and children with large VAs.85,86
Enlargement of the Cochlear Aqueduct
Enlargement of the cochlear aqueduct (CA) frequently is mentioned in the otologic literature because of its purported association with stapedectomy gusher and transotic CSF leak. Despite an active interest in the field and an extensive review of published radiographic images, I have never seen a radiograph that convincingly demonstrated CA enlargement.87 Most cases presented as enlargement of the CA are misinterpretations of the wide internal funnel that opens into the posterior fossa.88,89 In healthy subjects, the radiographic diameter of this aperture averages 3 to 4 mm but ranges from radiographically invisible to more than 10 mm46,90–92 (Figs. 191-16 and 191-17). For enlargement of the CA to be diagnosed radiographically, the intraosseous portion coursing toward the vestibule must be enlarged beyond 1 mm, the practical resolution limit of contemporary CT scanners. If criteria analogous to those for enlargement of the VA are used, then an enlarged CA must have a diameter exceeding 2 mm throughout its course between the inner ear and the posterior fossa.
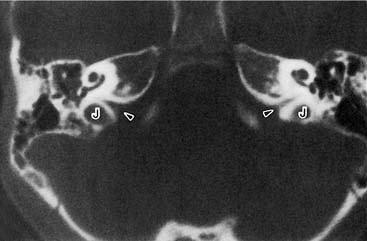
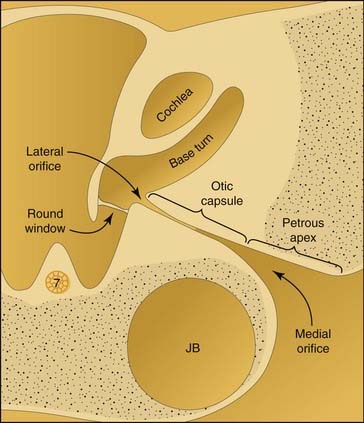
The preponderance of evidence suggests that the human CA usually is functionally patent.92–94 In most persons, however, it can neither transmit sudden large pressure changes to the inner ear nor allow free flow of spinal fluid in any significant quantity (e.g., with outflow of perilymphatic fluid from perilymphatic fistula or an open vestibule). This observation reflects two anatomic features of the aqueduct: a narrow diameter of the bony channel and the presence of a fibrous tissue meshwork occupying and baffling the lumen. Schuknecht and Reisser95 evaluated more than 1400 temporal bones in the Massachusetts Eye and Ear Infirmary collection and found no CA that exceeded 0.2 mm at its narrowest point. This series included 29 congenitally malformed inner ears. Of note, the CA was absent or nonpatent in 21 of these dysmorphic ears.
Some persons have less than normal or no fibrous tissue within the lumen, and the bony diameter is wider than normal. An opportunity may then exist for free flow of CSF from the oval window through the CA. According to Poiseuille’s law, fluid flow through a tube varies with the fourth power of the radius, suggesting the possibility of free flow in an individual with a large-diameter unimpeded channel, as discussed by Allen.96 Several histologic studies of congenital inner ear malformations have detected slightly larger CAs, particularly at their lateral orifice at the vestibule.24,49 Even in these malformed inner ears, however, the aqueduct diameter remained well in the submillimeter range. From a clinical standpoint, a CA less than 1 mm in diameter is undetectable radiographically. Despite the theoretical possibility that a widely patent CA may cause a stapedectomy gusher, abnormal connections between the IAC and the vestibule appear to be etiologic in a majority of cases.9,89,97,98
Developmental Anomalies of the Internal Auditory Canal
Wide Internal Auditory Canal
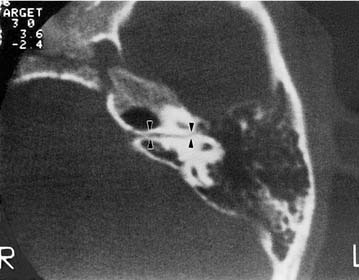
Unlike congenital narrowness of the IAC, a congenitally large canal may be an incidental finding in healthy individuals (Fig. 191-18). When a large IAC (larger than 10 mm in diameter) accompanies a malformation of the inner ear, it does not, as an independent variable, correlate with the level of hearing.3 One report suggests that a wide IAC may be correlated with hearing loss.99 The primary importance of detecting enlargement of the IAC is its association with spontaneous CSF leak and the occurrence of gusher during stapes surgery.97 Because hearing after stapedectomy complicated by CSF gusher (often erroneously called “perilymph gusher”) frequently is poor, a CT scan should be obtained before stapedectomy to address congenital fixation is undertaken. Dilation of the IAC, especially when the partition between the lateral end of the canal and inner ear appears deficient, should contraindicate stapedectomy.
Narrow Internal Auditory Canal
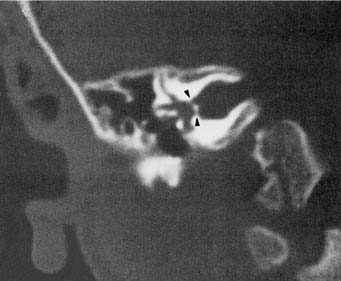
A narrow IAC may indicate a failure of eighth cranial nerve development. When a patient has normal facial function and an IAC less than 3 mm in diameter, it is likely that the bony canal transmits only the facial nerve (Fig. 191-19). A narrow IAC may accompany inner ear malformations or may be the sole radiographically detectable anomaly in a deaf child. A narrow IAC has been considered a relative contraindication to cochlear implantation, because it suggests that the eighth nerve may be insufficiently developed to conduct an auditory signal.50 After cochlear implantation, some patients with narrow IACs have experienced facial pain and twitching without useful auditory sensation.
Anomalies of the Eighth Nerve
Hypoplasia and aplasia of the eighth nerve are often, but not inevitably, associated with congenital narrowness or even absence of the IAC. Similarly, although eighth nerve maldevelopment frequently accompanies malformation of the inner ear, the presence of a normal cochlea and semicircular canals does not guarantee normal development of the audiovestibular nerve.100 High-resolution, thin-section MRI with T2-weighted sequences currently is the best means of assessing the fine anatomy of the eighth nerve in the IAC.101,102 Such a study is warranted before cochlear implantation when the bony IAC is narrow on the CT scan, with severe types of inner ear malformation, and in syndromes known to be associated with maldevelopment of the eighth nerve (e.g., the CHARGE association, Möbius’s syndrome).103 Unilateral cochlear nerve aplasia, sometimes familial, is increasingly recognized as an important cause of congenital unilateral profound SNHL.104,105 The internal auditory canal frequently is normal.106 Increasingly refined MR techniques may reveal hypoplasia of the auditory nerve as more common than previously recognized.107
Syndromic Correlations and Familial Patterns
In Waardenburg’s syndrome type I, absence of all three semicircular canals (17%) and cochlear hypoplasia (8%) have been reported.108 In Waardenburg’s syndrome type II, maldevelopment of the posterior semicircular canal has been described.109 Semicircular canal agenesis or hypoplasia also has been noted in CHARGE syndrome.110,111 In branchio-oto-renal syndrome, hypoplasia of the cochlea, sometimes accompanied by large VA, has been reported.7,112,113 In Pendred’s syndrome, large VA is a common feature, sometimes accompanied by modiolar deficiency.114,115 Otic capsule anomalies also have been described in trisomy E and Usher’s syndrome.5,8 Down syndrome (trisomy 21) is associated with semicircular canal dysplasia and a variety of other inner ear malformations.116
Nonsyndromic familial patterns of inner ear malformation also have been identified.9,10 An autosomal recessive pattern of Mondini’s dysplasia has been reported.117 In familial Mondini’s dysplasia, genetic analysis has identified a deletion in locus DFN3.118 Inherited complete labyrinthine aplasia (Michel’s aplasia) has been described with a probable autosomal recessive inheritance pattern.119 Familial lateral semicircular canal malformation has been described in association with anomalies of the external and middle ear.120 Nonsyndromic familial occurrence of large VA has been associated with the Pendred gene.121
Cerebrospinal Fluid Leakage and Meningitis
Congenitally malformed inner ears may be a source of CSF otorhinorrhea.122,123 For CSF leakage to occur, two abnormal connections must be present: one between the subarachnoid space and the inner ear and a second between the inner ear and middle ear. The most common pathologic interconnection between the subarachnoid space and the inner ear is through the fundus of the IAC. Histologically, large defects in the modiolar end of the IAC have been shown to exist in 10% of dysplastic ears.95 Leakage may occur less often around the facial nerve at the lateral end of the IAC124–126 (Fig. 191-20). The junction between the IAC and the inner ear may be evaluated with CT (Figs. 191-21 and 191-22). A well-defined bony plate normally partitions the distal IAC from the labyrinthine lumen. In a number of well-documented cases of CSF leakage, radiopaque contrast placed into the cerebellopontine angle cistern could be traced through the IAC into the inner ear.122,124,127–129 The propensity of a dysplastic inner ear to leak CSF depends on its morphologic type. The common cavity deformity appears to be at particularly high risk. By contrast, Phelps and coworkers130,131 maintain that the incomplete partition deformity (Mondini’s dysplasia) is not associated with a heightened risk of CSF leakage. In ears with CSF fistulas, the SCCs usually are severely deformed. Although enlargement of the VA is a sign that CSF pressure is present within the inner ear, it does not necessarily imply the existence of an external leak into the middle ear. Rarely, transotic CSF leakage may be accompanied by a second congenital fistula in the anterior skull base.132
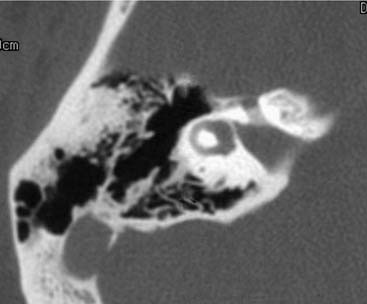
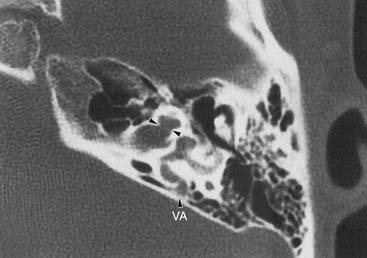
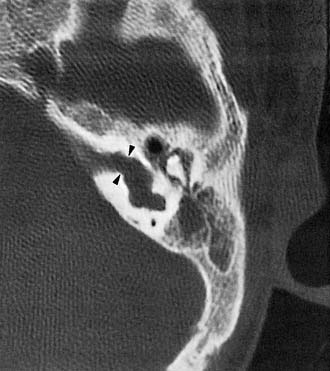
A second potential pathway between the subarachnoid space and the inner ear is the CA. As discussed previously in this chapter, there is little evidence to suggest that this pathway is a source of vigorous CSF leakage. In one published report, a CT metrizamide cisternogram was said to demonstrate a large CSF leak occurring through the CA; however, the published image showed the defect to be far anterior and superior to the region of the CA.122
A direct route between the subarachnoid space and middle ear, bypassing the inner ear, may exist in rare cases. Hyrtl’s fissure, a congenital cleft that runs between the hypotympanum and posterior cranial fossa passing in proximity to the jugular bulb, has been reported to be a source of CSF leakage.125 It is interesting that a diligent search of the historical literature has failed to find a publication on this subject by Hyrtl, despite his long eponymic association with this deformity.133 In the interest of clarity, the descriptive term tympanomeningeal fissure is preferable.
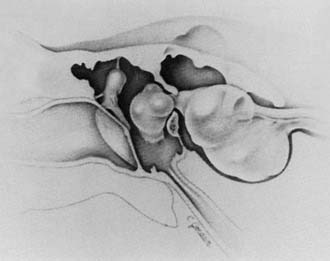
The pathologic interconnection between the inner ear and middle ear most frequently is at the oval window. Numerous reports have described defects in the central portion of the footplate with a protruding membrane, most probably arachnoid123,124,134–137 (Fig. 191-23). In other cases the defect is adjacent to the footplate, particularly just anterior to it. Much less frequently, leakage may occur through the round window or a fissure on the promontory.123 Schuknecht and Reisser95 evaluated the oval and round windows in 29 ears with congenital malformations of the inner ear. On histologic examination, the stapes were absent in 4, fixed in 6, and normal in 15 cases. In 4 cases the footplates were noted to have defects that were bridged by thin membranes only. The round windows were normal in 28 cases and abnormal in only 1 (bony closure). No deficiencies in the round window membranes were noted.
The primary clinical importance of CSF leakage is the risk of meningitis. There have been numerous reports of meningitis complicating congenital malformation of the inner ear.122–124,134,136–138 Recurrent bouts of meningitis are typical, and recognition of the inner ear as the source frequently comes only after several episodes have occurred. Acute otitis media appears to be the bacterial source in most cases. All children with recurrent meningitis without an obvious cause should undergo CT of the inner ear to exclude inner ear malformation.139 Even children with a single episode of meningitis should have a postrecovery audiogram followed by CT if unilateral or bilateral SNHL is discovered. The causative organisms in a review of 24 reported cases have been Streptococcus pneumoniae (in 71% of the cases), Haemophilus influenzae (in 33%), and β-hemolytic streptococci (in 8%). The total exceeds 100% because of the incidence of polymicrobial infection.
Surgical closure of transotic CSF leakage may be attempted at four anatomic levels: (1) the posterior cranial fossa; (2) the dysplastic inner ear; (3) the middle ear windows; and (4) the eustachian tube. In an ear with residual hearing, tympanotomy and overlay grafting of the site of leakage with a connective tissue graft is indicated as a first attempt. Unfortunately, recurrent leakage is common. When this technique fails and a useful amount of hearing persists, posterior fossa craniotomy with placement of a muscle plug in the IAC or connective tissue graft over the fistulous tract may prove to be successful and spare hearing.122 When the hearing is poor, a direct approach to the dysplastic inner ear is indicated. After tympanotomy and removal of the footplate, an oversized piece of muscle may be used to obliterate the vestibule.18,25,123,140 If the anatomy is unfavorable for this maneuver, a postauricular translabyrinthine approach to the IAC should prove to be effective. Farrior and Endicott141 proposed a hypotympanic approach to ablation of the CA. However, this aqueduct has never been convincingly demonstrated to cause a high-volume CSF leak. The final option, closure of the eustachian tube, is not a good choice for children. After this procedure, the anomalous CSF pathway is still open to the middle ear—a disadvantageous situation at an age when acute otitis media is common. Subsequent to the repair of any CSF leakage, the patient is placed on bed rest with the head elevated 30 degrees for several days. Acetazolamide (Diamox) is administered to reduce CSF production, and fluids are restricted. Placement of a lumbar drain is a useful adjunctive measure, although it may be difficult to insert and maintain in young children.90
X-Linked Stapes Gusher Syndrome
A form of hereditary mixed conductive and sensory hearing loss associated with CSF gusher during stapes surgery has recently been described.142 Inherited hearing loss follows an X-linked pattern.143 The characteristic inner ear deformity may be seen radiographically.144,145 On CT, the cochlear modiolus is deficient and the lateral portion of the IAC often is bulbous (see Fig. 191-21). These observations provide strong evidence that the abnormal interconnection between the subarachnoid space and the inner ear is by way of the lateral terminus of the IAC. The labyrinthine segment of the fallopian canal may be dilated. In the vestibular system, the posterior semicircular canal often is deformed, and the VA may be enlarged.
The occurrence of stapes fixation in X-linked gusher syndrome is questionable. It has been proposed that the observed air-bone gap may be accounted for by alteration in cochlear micromechanics rather than dysfunction of the conducting system.146 Results of acoustic reflex testing and auditory evoked responses are more consistent with a pure sensory hearing loss than a mixed form. The outcome of stapes surgery in these patients usually is poor, with anacusis in the operated ear a frequent result. When surgery is contemplated for a congenital conductive hearing loss, preoperative imaging is advisable to screen for inner ear malformation.
Perilymphatic Fistula
Much controversy surrounds the diagnosis and management of perilymphatic fistula in children. The candidate group for exploratory tympanotomy consists of children with progressive or sudden SNHL. CT of the inner ear may demonstrate pneumolabyrinth, although this finding is rare (Fig. 191-24). Conservative surgeons have advocated exploration only in children with radiographically abnormal inner ears who have a clear antecedent event of head trauma or barometric pressure change.147 Other workers have espoused the notion of surgical exploration in all children with unexplained SNHL.14,148,149 Great variability exists in the number of fistulas “confirmed” at surgery. Pappas and colleagues147 found only 4 fistulas in 36 ears (11%) explored for progressive SNHL during childhood. Of note, 50% of these ears had radiographically malformed inner ears. By contrast, Parnes and McCabe148 found fistulas in 20 of 26 children’s ears (77%), only 6 of whom had inner ear anomalies radiographically. A high incidence of fistulas among children with sensory hearing loss (64%), both with and without inner ear malformation, has been reported.150,151 Furthermore, one group of investigators claim to have objective verification of the presence of perilymph in the middle ear through evaluation of β2-transferrin levels in surgical aspirates.152 An important point is that considerable doubt exists regarding whether this test is valid. A recent study in which perilymph samples were directly obtained during either stapedectomy or cochlear implant surgery failed to detect β2-transferrin in this fluid.153
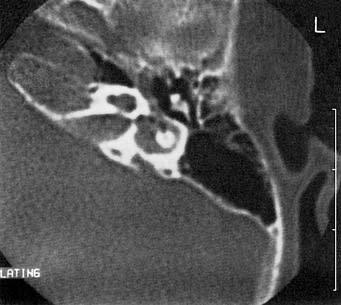
Figure 191-24. Air bubbles in the inner ear (pneumolabyrinth) in a congenitally deaf child with a perilymphatic fistula.
In exploration of a perilymph fistula, it is important to perform provocative maneuvers to render subtle perilymph flows more visible. Such maneuvers include performing a Valsalva maneuver, using the Trendelenburg position, and compressing the jugular veins. Many surgeons are either missing subtle or intermittent perilymphatic fistulas during explorations or overdiagnosing them by misinterpreting middle ear secretions or local anesthetic as perilymph. Attempts to make a practical biochemical test for the presence of perilymph or CSF in the middle ear—through either assay of β2-transferrin or intrathecal fluorescein administration—have not yet proved useful.154,155 Most surgeons patch both oval and round windows with connective tissue whether or not a perilymphatic fistula was identified during exploration.
Rather than dwell on the controversy about how many fistulas are found, the critical observer should judge the effectiveness of the therapeutic intervention by analyzing its outcome relative to the natural history of the disease. In this regard, results of perilymphatic fistula exploration in children have been disappointing. In one series of 36 patients, hearing was better in 3, unchanged in 21, and worse in 12.147 Similar results have been obtained in other childhood perilymphatic fistula series. It would be difficult to argue that these data represent a significant improvement over the natural history of the inner ear disease. Congenital progressive SNHL often is characterized by long periods of stability interspersed with periods of rapid deterioration. Even a relative improvement in hearing may occur after an episode of sudden loss. It is apparent that most children with sudden or progressive SNHL have suffered from cochlear diseases other than perilymphatic fistula. Examples are hereditary progressive loss, viral labyrinthitis (e.g., measles, mumps, cytomegalovirus), autoimmune inner ear disease (e.g., Cogan’s syndrome), and endolymphatic hydrops. Even in the most suggestive clinical situation—sudden loss triggered by head trauma in a child with radiographically malformed inner ears—only a minority of patients demonstrate oval or round window fistulization. Many of these children probably lose hearing as a result of internal fistulization between the endolymphatic and perilymphatic space because of deficiencies of the osseous spiral lamina, rather than because of external fistulization to the middle ear. A minority of children with sudden or progressive SNHL have prominent vestibular symptoms. Results of perilymphatic fistula repair in the relief of vertigo have been quite good, although spontaneous recovery from vestibulopathy is common in this age group.
Inner Ear Malformations in Congenital Aural Atresia
Development of the external and middle ears is embryologically distinct from that of the inner ear. Anomalies of the lateral portion of the temporal bone typically result from derangement of the first or second branchial arches. Despite their embryologic separateness, congenital aural atresia may coexist with malformation of the inner ear.156 In one study of patients with atresia, 12% (8 of 66) had a radiographically detectable inner ear abnormality.157 In this group, SCC abnormalities were most common, including some ears with normal bone-conduction hearing. Cochlear anomalies were present in 5% (3 of 66) of these patients, all of whom had poor bone conduction hearing. It is an embryologic enigma that one patient with unilateral atresia had a malformed inner ear only on the contralateral side. In another polytomographic study of atretic ears, 8% (4 of 48) had SCC anomalies, and 2% (1 of 48) had a deformed cochlea.158 As indicated by a comprehensive review of the relevant literature, approximately 5% of patients with atresia have cochlear deformities, whereas 10% have malformed SCCs. As a general rule, surgical reconstruction of atretic ears should not be attempted when they are associated with malformation of the inner ear. The deformed cochlea is more vulnerable to injury from vibratory trauma, and the risk of inducing a sensory hearing loss is increased. This surgery also carries a heightened risk of CSF leakage.
Imaging
During the 1980s and 1990s, CT was the gold standard imaging modality for investigation of patients suspected of having an inner ear malformation. In the early 21st century, new MRI techniques are capable of producing images superior to those obtained with CT. In contrast to CT, which depicts otic capsule bone, MRI displays inner ear morphology by imaging inner ear fluids. High-resolution, thin-section, T2-weighted sequences (e.g., fast-spin echo) provide exquisite detail of inner ear anatomy (see Fig. 191-4).159 MRI is superior to CT in a number of ways. MRI can distinguish between a cochlea filled with fluid and one occluded by soft tissue. CT is unable to make this distinction—an observation of great importance in assessing scala tympani patency before cochlear implantation. MRI provides more information about large VAs than that provided by CT.72,73,160 It displays not only the dilated osseous canal but also the dimensions of the often impressively expanded endolymphatic sac. Direct visualization of the eighth nerve in the IAC, where it appears as a linear filling defect outlined by CSF, is yet another advantage of MRI. This capability is important in certain severe deformities in which neural agenesis may be found. Three-dimensional renderings of inner ear malformations have been produced using both CT and MRI.161,162
In assessing images for possible inner ear malformation, it is important to measure the size of structures and not merely look for anomalous patterns. Normative data of inner ear dimension have been published to aid in this determination.163 Quantitative measurement has been shown to increase diagnositc yield in cochlear hypoplasia (less than 4.4 mm cochlear height on coronal images) and semicircular canal dysplasia (bony island width on axial projection) even for experienced neuroradiologists.164
When selecting MRI as an imaging study to screen for inner ear malformation, it is important for the physician to be aware that standard MRI protocols designed to screen for central nervous system abnormalities provide only limited information concerning the inner ear. To be useful in screening for inner ear anomalies, specially designed protocols are needed.159 One disadvantage of MRI is the difficulty young children have in tolerating the confined spaces of the magnet. In addition, fast-spin echo MRI requires considerably longer scan time than does CT. Whereas simple sedation usually is sufficient for CT, acquiring high-quality magnetic resonance images may require use of general anesthesia in pediatric patients. Accordingly, despite the numerous advantages of MRI (including the absence of ionizing radiation), CT is the pediatric examination of choice in many centers for this important practical reason.
Evaluation and Management
High-resolution CT or MRI of the inner ears is recommended for all children with suspected congenital malformation of the inner ear, including those with otherwise unexplained SNHL, regardless of whether it has existed from birth or occurred later. Scans usually are obtained when hearing impairment is first recognized. Use of thin slices of 1- or 1.5-mm thickness with 0.5-mm overlap will adequately image the minute inner ear structures.59 If only one plane of view is to be obtained, the axial orientation is preferred because it provides superior images of the VA, a frequently malformed structure that is difficult to visualize on coronal scans. When possible, both axial and coronal views should be obtained, because certain subtle deformities, such as deficiency of the interscalar septum, are best seen in the coronal orientation. It is important to examine thoroughly all portions of the inner ear, because anomalies frequently are multiple (Fig. 191-25).
Few patients with congenitally malformed inner ears have a fluctuating pattern of hearing loss suggestive of endolymphatic hydrops. When fluctuations are encountered, however, a low-salt diet and diuretics are prescribed. Surgical intervention is indicated in a few well-defined circumstances. Obviously, CSF leakage and meningitis require intervention. Exploration for perilymphatic fistula is reserved for patients with radiographically abnormal inner ears who have suffered a sudden hearing deterioration temporally related to a precipitating event such as head trauma, rapid barometric pressure change, vigorous exercise, or sneezing. As discussed previously, even in these patients, only a minority have a fistula, and those who do frequently continue to lose hearing despite repair. House165 and later others advocated endolymphatic sac surgery for congenital malformations of the inner ear166–168; however, a careful analysis of the outcome in a large series revealed no benefit of this surgery in terms of hearing stabilization.169 In fact, nearly 30% of the patients suffered significant postoperative hearing deterioration. Patients with large VAs appear to be at particularly high risk for poor outcome after sac surgery. Surgical obliteration of the endolymphatic sac with muscle and fascia has been advocated in patients with enlargement of the VA.83 Unfortunately, a multicenter study showed that this procedure not only fails to stabilize hearing but is associated with a 50% rate of sensorineural hearing deterioration.84
Preventive measures are important in the management of patients with congenitally malformed inner ears. The risk of CSF leakage may be estimated by CT or MRI of the inner ear. Anatomic features of concern include a wide IAC, deficient partitioning between the IAC and inner ear, and enlargement of the vestibule in the horizontal plane. Parents should be made aware of the risk of meningitis and instructed in recognizing its early symptoms and signs. Because most episodes of meningitis associated with inner ear malformation are pneumococcal, use of the pneumococcal vaccine is recommended.170 Children with malformed inner ears should avoid head trauma and rapid barometric pressure changes, because these risk provoking a sudden hearing loss or even CSF leakage. Activities such as contact sports and scuba diving are discouraged. Finally, establishing a prognosis for future auditory function in a hearing-impaired child serves as a guide for educational and rehabilitative efforts. Recognizing an inner ear malformation on CT may be helpful, because certain morphologic patterns have more favorable prognoses than others.
Cochlear Implantation
Electrical stimulation of the auditory nerve has proved useful in many forms of sensory deafness, including those associated with congenital malformations of the inner ear. The fundamental premise of cochlear implantation is that some of the cochlear neural population survive despite the absence of hair cells. In congenital malformations of the inner ear, the auditory nerve population typically is less than with other forms of sensory deafness. The normal human spiral ganglion cell count usually is 25,000 to 35,000. In eight congenitally dysplastic ears, Schmidt171 reported cell counts ranging from 7677 to 16,110, with an average of only 11,478. By contrast, ears with profound sensory hearing loss from ototoxicity, otosclerosis, or mechanisms of sudden loss have ganglion cell populations in the range of 18,000 to 22,000. This observation suggests that malformed inner ears could be electrically stimulated but that improvements in speech discrimination would fall below average for the deaf population at large. A narrow IAC on pre-implantation CT (less than 3 mm in diameter) is a strongly adverse predictor of neural survival.23
Cochlear implantation in deformities that involve both the membranous and osseous labyrinth raises several special considerations. Cochlear implantation in malformations limited to the membranous labyrinth is equivalent to implantation in the prelingually deaf child (see Chapter 159). Results of implantation with short, single-channel devices in patients with cochlear hypoplasia, common cavity, and incomplete partition deformities appear in the literature.36,172,173 In successful cases, sound detection levels are similar to those in other prelingually deaf children.174,175 Implantation of deformed cochleas with multichannel devices also has proved to be valuable in a majority of cases but is associated with a higher rate of complications.31,176–179 Performance varies widely and appears, on average, to be somewhat below that realized in most other forms of deafness. The design of multichannel electrodes makes certain assumptions about the geometry of the scala tympani that affect the anticipated orientation of stimulating electrodes to neural elements. Although standard electrodes are not optimized for dysplastic cochleae, fabrication of custom electrodes for each malformed cochlea is not feasible with current technology. The degree of electrode insertion varies, in large part because of variability in size and geometry of the deformed cochlea.180 Only partial insertion may be realized, particularly in hypoplastic cochleas, whereas complete insertion is more common with milder deformities such as incomplete partition.
One complication of unpredictable electrode positioning is stimulation of the facial nerve at current levels lower than the threshold for auditory perception. Cross-stimulation of the facial nerve has been reported with both single-channel and multichannel designs and has prevented some patients from using the device. Multichannel systems have a distinct advantage in this regard because the offending electrode pair or pairs may be programmed out of the stimulation map, thereby preserving the ability to provide auditory stimulation. The presence of a dysplastic inner ear may be a warning that the facial nerve is malpositioned.181 An anomalous facial nerve course has been reported to occur in 16% of malformed inner ears.176 In one patient with cochlear hypoplasia, an anomalous facial nerve was injured immediately above the round window niche, a highly vulnerable location during implant surgery.31 Use of electrophysiologic facial nerve monitoring during implantation in patients with malformed inner ears is advisable. It also is possible for a persistent stapedial artery to traverse the round window region, thereby precluding implantation.182
Another complication of implantation in malformed cochleas is facial twitching consequent to inadvertent penetration of the IAC by the electrode.179 When such a malpositioned electrode is activated, vigorous facial twitching may occur at an extremely low stimulation threshold. To avoid this complication, post-implantation CT is advisable before stimulation is undertaken to assess electrode position after implantation in a malformed cochlea.
CSF leak also has been a frequent complication during implantation of the malformed cochlea, occurring with an incidence of approximately 40%.176 The leak typically is evident immediately on opening the round window and presumably occurs as a result of deficiencies in the partition between the modiolus and the dural envelope at the lateral end of the IAC. Leaks have been successfully handled by packing connective tissue around the electrode, but temporary CSF diversion may be required to obtain a lasting seal.183 Multichannel cochlear implants, especially certain varieties designed to approximate the modiolus, have been associated with an increased long-term incidence of meningitis.184 The incidence may well be higher in patients with malformed inner ears.185
Alexander G. Zur Pathologie und Pathologischen Anatomie der kongenitalen Taubheit. Arch Otor Nas Kohlk Heilk. 1904;61:183.
Anson BJ. The endolymphatic and perilymphatic aqueducts of the human ear: developmental and adult anatomy of their parietes and contents in relation to otological surgery. Acta Otolaryngol (Stockh). 1965;59:140.
Bamiou DE, Worth S, Phelps P, et al. Eighth nerve aplasia and hypoplasia in cochlear implant candidates: the clinical perspective. Otol Neurotol. 2001;22:492.
Bluestone CB. Cochlear malformations, meningitis, and cochlear implants: what have we learned? Otol Neurotol. 2003;24:349.
Ceruti S, Stinckens C, Cremers CW, et al. Temporal bone anomalies in the branchio-oto-renal syndrome: detailed computed tomographic and magnetic resonance imaging findings. Otol Neurotol. 2002;23:200.
Collins WO, Buchman CA. Bilateral semicircular canal aplasia: a characteristic of the CHARGE association. Otol Neurotol. 2002;23:233.
Cremers WR, Bolder C, Admiraal RJ, et al. Progressive sensorineural hearing loss and a widened vestibular aqueduct in Pendred syndrome. Arch Otolaryngol Head Neck Surg. 1998;124:501.
Eisenman DJ, Ashbaugh C, Zwolan TA, et al. Implantation of the malformed cochlea. Otol Neurotol. 2001;22:834.
Govaerts PJ, Casselman J, Daemers K, et al. Audiological findings in large vestibular aqueduct syndrome. Int J Pediatr Otorhinolaryngol. 1999;51:157.
Ishinaga H, Shimizu T, Yuta A, et al. Pendred’s syndrome with goiter and enlarged vestibular aqueducts diagnosed by PDS gene mutation. Head Neck. 2002;24:710.
Ito K, Ishimoto S, Shotaro K. Isolated cochlear nerve hypoplasia with various internal auditory meatus deformities in children. Ann Otol Rhinol Laryngol. 2007;116:520.
Jackler RK, Hwang PH. Enlargement of the cochlear aqueduct: fact or fiction? Otolaryngol Head Neck Surg. 1993;109:14.
Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope.. 1987;97(Suppl 40):2.
Johnson J, Lalwani AK. Sensorineural and conductive hearing loss associated with lateral semicircular canal malformation. Laryngoscope. 2000;110:1673.
Konigsmark BW, Gorlin RJ. Genetic and Metabolic Deafness. Philadelphia: WB Saunders; 1976.
Miyamoto RT, Robbins AJ, Myres WA, et al. Cochlear implantation in the Mondini inner ear malformation. Am J Otol. 1986;7:258.
Mondini C. Anatomia surdi nedi sectio. De Bononiensi Scientiarum et Artium Instituto Arque Academia Commentarii, Bologna. 1791;7:419.
Mondini C. The minor works of Carlo Mondini: the anatomical section of a boy born deaf. Am J Otol. 1997;18:288.
Nadol JB, Young YS, Glynn RJ. Survival of spiral ganglion cells in profound sensorineural deafness: its implications for cochlear implantation. Ann Otol Rhinol Laryngol. 1989;98:411.
Paparella MM. Mondini’s deafness: a review of histopathology. Ann Otol Rhinol Laryngol Suppl. 1980;89:1.
Phelps PD. Imaging for congenital deformities of the ear. Clin Radiol. 1994;49:663.
Phelps PD, Coffey RA, Trembath RC, et al. Radiological malformations of the ear in Pendred syndrome. Clin Radiol. 1998;53:268.
Phelps PD, Proops D, Sellars S, et al. Congenital cerebrospinal fluid fistula through the inner ear and meningitis. J Laryngol Otol. 1993;107:492.
Phelps PD, Reardon W, Pembrey M, et al. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology. 1991;33:326.
Purcell DD, Fischbein N, Patel A, et al. Two temporal bone computed tomography measurements increase recognition of malformations and predict sensorineural hearing loss. Laryngoscope. 2006;116:1439.
Raphael Y, Fein A, Nebel L. Transplacental kanamycin ototoxicity in the guinea pig. Arch Otorhinolaryngol. 1983;238:45.
Rich PM, Graham J, Phelps PD. Hyrtl’s fissure. Otol Neurotol. 2002;23:476.
Satar B, Mukherji SK, Telian SA. Congenital aplasia of the semicircular canals. Otol Neurotol. 2003;24:437.
Schuknecht HF. Pathology of the Ear. Cambridge: Harvard University Press; 1974.
Schuknecht HF, Reisser C. The morphologic basis for perilymphatic gushers and oozers. Adv Otorhinolaryngol. 1988;39:1.
Sennaroglu L, Saatci I. Unpartitioned versus incompletely partitioned cochleae: radiologic differentiation. Otol Neurotol. 2004;25:520.
Sennaroglu L, Saatci I. A new classification for cochleovestibular malformations. Laryngoscope. 2002;112:2230.
Streeter GL. On the development of the membranous labyrinth and the acoustic and facial nerves in the human embryo. Am J Anat. 1906;6:139.
Streeter GL. The histogenesis and growth of the otic capsule and its contained periotic tissue-spaces in the human embryo. Carnegie Contrib Embryol. 1918;7:5.
Usami S, Abe S, Weston MD, et al. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188.
Wilson JT, Leivy SW, Sofferman RA, et al. Mondini dysplasia: spontaneous cerebrospinal fluid otorrhea. New perspectives in management. Pediatr Neurosurg. 1990-1991;16:260.
Wootten CT, Backous DD, Haynes DS. Management of cerebrospinal fluid leakage from cochleostomy during cochlear implant surgery. Laryngoscope. 2006;116:2055.
Zheng Y, Schachern PA, Djalilian HR, et al. Temporal bone histopathology related to cochlear implantation in congenital malformation of the bony cochlea. Otol Neurotol. 2002;23:181.
1. Streeter GL. On the development of the membranous labyrinth and the acoustic and facial nerves in the human embryo. Am J Anat. 1906;6:139.
2. Streeter GL. The histogenesis and growth of the otic capsule and its contained periotic tissue-spaces in the human embryo. Carnegie Contrib Embryol. 1918;7:5.
3. Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97(Suppl 40):2.
4. Gorlin RJ, Toriello HV, Cohen MM. Hereditary Hearing Loss and Its Syndromes. New York: Oxford University Press; 1995.
5. Harada T, Takahara T, Ishii S. Three-dimensional reconstruction of the temporal bone from a case of E trisomy syndrome. Laryngoscope. 1993;103:541.
6. Johnsen T, Larsen C, Friis J, et al. Pendred’s syndrome: acoustic, vestibular, and radiological findings in 17 unrelated patients. J Laryngol Otol. 1987;101:1187.
7. Ostri B, Johnsen T, Bergmann I. Temporal bone findings in a family with branchio-oto-renal syndrome (BOR). Clin Otolaryngol. 1991;16:163.
8. van Aarem A, Cremers WR, Benraad-van Rens MJ. Usher syndrome: a temporal bone report. Arch Otolaryngol Head Neck Surg. 1995;121:916.
9. Chan KH, Eelkema EA, Furman JM, et al. Familial sensorineural hearing loss: a correlative study of audiologic, radiographic, and vestibular findings. Ann Otol Rhinol Laryngol. 1991;100:620.
10. Griffith AJ, Arts A, Downs C, et al. Familial large aqueduct syndrome. Laryngoscope. 1996;106:960.
11. Bauman NM, Kirby-Keyser LJ, Dolan KD, et al. Mondini dysplasia and congenital cytomegalovirus infection. J Pediatr. 1994;124:71.
12. Naganawa S, Ito T, Iwayama E, et al. MR imaging of the cochlear modiolus: area measurement in healthy subjects and in patients with a large endolymphatic duct and sac. Radiology. 1999;213:819.
13. Snider DEJr, Layde PM, Johnson MW, et al. Treatment of tuberculosis during pregnancy. Ann Rev Respir Dis. 1980;122:65.
14. Reilly JS. Congenital perilymphatic fistula: a prospective study in infants and children. Laryngoscope. 1989;99:393.
15. Antonelli PJ, Varela AE, Mancuso AA. Diagnostic yield of high resolution computed tomography for pediatric sensorineural hearing loss. Laryngoscope. 1999;109:1642.
16. McClay JE, Tandy R, Gnundfast K, et al. Major and minor temporal bone abnormalities in children with and without congenital sensorineural hearing loss. Arch Otolaryngol Head Neck Surg. 2002;128:664.
17. Shim HJ, Shin JE, Chung JW, et al. Inner ear anomalies in cochlear implantees: importance of radiologic measurements in the classification. Otol Neurotol. 2006;27:831.
18. Schuknecht HF. Dysmorphogenesis of the inner ear. Birth Defects. 1980;16:47.
19. Zheng Y, Schachern PA, Cureoglu S, et al. The shortened cochlea: its classification and histopathologic features. Int J Pediatr Otorhinolaryngol. 2002;63:29.
20. Altmann F. Malformations, anomalies, and vestigial structures of the inner ear. Arch Otolaryngol Head Neck Surg. 1953;57:591.
21. Lindsay JR. Profound childhood deafness: inner ear pathology. Ann Otol Rhinol Laryngol Suppl. 1973;83:1.
22. Ormerod FC. The pathology of congenital deafness. J Laryngol. 1960;74:919.
23. Shelton C, Luxford WM, Tonokawa LL, et al. The narrow internal auditory canal in children: a contraindication to cochlear implants. Otolaryngol Head Neck Surg. 1989;100:227. 231
24. Siebenmann F, Bing R. Uber den labyrinth and Hirnbefund bei einem an Retinitis pigmentosa erblindeten angeboren taubstummen. Z Ohrenheilk. 1907;54:265.
25. Friedmann I, Fraser GR, Frogan P. Pathology of the inner ear in the cardioauditory syndrome of Servell and Lange-Neilsen. J Laryngol. 1966;80:451.
26. Scheibe A. Ein fall von trubstummheit mit acusticus-atrophie und gildung sangomalien im hautigen labyrinth beiderseits. Z Ohrenh. 1892;22:11.
27. Beal DD, Davey PR, Lindsay JR. Inner ear pathology of congenital deafness. Arch Otolaryngol Head Neck Surg. 1967;85:134.
28. Bergstrom L. Pathology of congenital deafness: present status and future prospects. Ann Otol Rhinol Laryngol Suppl. 1980;89:31.
29. Friedmann I. Pathology of the Ear. London: Blackwell Scientific; 1974.
30. Nomura Y, Kawabata I. Scheibe dysgenesis of the inner ear. J Laryngol Otol. 1980;94:1345.
31. Slattery WH, Luxford WL. Cochlear implantation in congenitally malformed cochlea. Laryngoscope. 1995;105:1184.
32. Steel KP, Bock GR. Hereditary inner ear abnormalities in animals: relationships with human abnormalities. Arch Otolaryngol Head Neck Surg. 1983;109:22.
33. Feghali JG, Leone CA, Linthicum FH. Residual high frequency hearing in a patient with Mondini’s deformity: clinical implications. Am J Otol. 1985;6:336.
34. Berlin CI, Wexler KF, Jerger JF, et al. Superior ultra-audiometric hearing: a new type of hearing loss which correlates highly with unusually good speech in the “profoundly deaf.”. Trans Am Acad Ophthalmol Otolaryngol. 1978;86:111.
35. Komune S, Nogami K, Inoue H, et al. Bilateral Mondini dysplasia with normal hearing. J Otorhinolaryngol Relat Spec. 1993;55:143.
36. Mizuno M, Harada T. Labyrinthine anomalies with normal cochlear function. J Otorhinolaryngol Relat Spec. 1992;54:278.
37. Yukawa K, Horiguchi S, Suzuki M. Congenital inner ear malformations without sensorineural hearing loss. Auris Nasus Larynx. 2008;35:121.
38. Tsuzuku T, Kaga K. The relationship between motor function development and vestibular function tests in four children with inner ear anomaly. Acta Otolaryngol (Stockh). 1991;481:443.
39. Kwee HL. The occurrence of the Tullio phenomenon in congenitally deaf children. J Laryngol Otol. 1976;90:501.
40. Curtin HD. Congenital malformations of the ear. Otolaryngol Clin North Am. 1988;21:317.
41. Mafee MF, Selis JE, Yannias DA, et al. Congenital sensorineural hearing loss. Radiology. 1984;150:427.
42. Murata K, Isono M, Ohta F, et al. Quantitative analysis of inner ear anomalies by high resolution CT scanning of the temporal bone. J Otolaryngol. 1987;16:133.
43. Olson JE, Dorwart RH, Brant WE. Use of high resolution thin section CT scanning of the petrous bone in temporal bone anomalies. Laryngoscope. 1982;92:1274.
44. Phelps PD, Lloyd GAS. Radiological investigation of congenital deafness. In: Phelps PD, Lloyd GAS, editors. Radiology of the Ear. Oxford: Blackwell Scientific, 1983.
45. Phelps PD. Mondini and “pseudo Mondini” [editorial]. Clin Otolaryngol. 1990;15:99.
46. Johnsson L, Hawkins JEJr, Rouse RC, et al. Four variations of the Mondini inner ear malformations as seen in microdissections. Am J Otolaryngol. 1984;5:242.
47. Monsell EM, Jackler RK, Motta G, et al. Congenital malformations of the inner ear: histologic findings in five temporal bones. Laryngoscope. 1987;97(Suppl 40):18.
48. Paparella MM. Mondini’s deafness: a review of histopathology. Ann Otol Rhinol Laryngol Suppl. 1980;89:1.
49. Sando I, Takahara T, Owaga A. Congenital anomalies of the inner ear. Ann Otol Rhinol Laryngol Suppl. 1984;93:110.
50. Schuknecht HF. Mondini dysplasia: a clinical and pathological study. Ann Otol Rhinol Laryngol Suppl. 1980;89:1.
51. Pappas DG, Simpson LC, McKenzie RA, et al. High-resolution computed tomography: determination of the cause of pediatric sensorineural hearing loss. Laryngoscope. 1990;100:564.
52. Mondini C. Minor works of Carlo Mondini: the anatomical section of a boy born deaf. Am J Otol. 1997;18:288.
53. Phelps PD. Ear dysplasia after Mondini. J Laryngol Otol. 1994;108:461.
54. Michel EM. Memoires sur les anomalies congenitales de l’oreille interne. Gaz Med Strasbourg. 1863;3:55.
55. Jorgensen MB, Kristensen HK, Buch MH. Thalidomide-induced aplasia of the inner ear. J Laryngol Otol. 1964;78:1095.
56. Hersh JH, Ganzel TM, Fellows RA. Michel’s anomaly, type I microtia and microdontia. Ear Nose Throat J. 1991;70:155.
57. Kavanagh KT, Magill HL. Michel dysplasia: common cavity inner ear deformity. Pediatr Radiol. 1989;19:343.
58. Marsot-Dupuch K, Dominguez-Brito A, Ghasli K, et al. CT and MR findings of Michel anomaly: inner ear aplasia. AJNR Am J Neuroradiol. 1999;20:281.
59. Jackler RK, Dillon WP. Computed tomography and magnetic resonance imaging of the inner ear. Otolaryngol Head Neck Surg. 1988;99:494.
60. Sennaroglu L, Saatci I. Unpartitioned versus incompletely partitioned cochleae: radiologic differentiation. Otol Neurotol. 2004;25:520.
61. Tian Q, Linthicum FH, Fayad JN. Human cochleae with three turns: an unreported malformation. Laryngoscope. 2006;116:800.
62. Yu K. Molecular genetic advances in semicircular canal abnormalities and sensorineural hearing loss: a report of 16 cases. Otolaryngol Head Neck Surg. 2003;129:637.
63. Johnson J, Lalwani AK. Sensorineural and conductive hearing loss associated with lateral semicircular canal malformation. Laryngoscope. 2000;110:1673.
64. Satar B, Mukherji SK, Telian SA. Congenital aplasia of the semicircular canals. Otol Neurotol. 2003;24:437.
65. Morimoto AK, Wiggins RH, Hudgins PA, et al. Absent semicircular canals in CHARGE syndrome: radiologic spectrum of findings. AJNR Am J Neuroradiol. 2006;27:1663.
66. Arcand P, Desrosiers M, Dubé J, et al. The large vestibular aqueduct syndrome and sensorineural hearing loss in the pediatric population. J Otolaryngol. 1991;20:247.
67. Jackler RK, De La Cruz A. The large vestibular aqueduct syndrome. Laryngoscope. 1989;99:1238.
68. Levenson MJ, Parisier SC, Jacobs M, et al. The large vestibular aqueduct in children. Arch Otolaryngol Head Neck Surg. 1989;115:54.
69. Mafee MF, Charletta D, Kumar A, et al. Large vestibular aqueduct and congenital sensorineural hearing loss. Am J Neuroradiol. 1992;13:805.
70. Okumura T, Takahashi H, Honjo I, et al. Sensorineural hearing loss in patients with large vestibular aqueduct. Laryngoscope. 1995;105:289.
71. Zalzal GH, Tomaski SM, Vezina LG, et al. Enlarged vestibular aqueduct and sensorineural hearing loss in childhood. Arch Otolaryngol Head Neck Surg. 1995;121:23.
72. Harnsberger HR, Dahlen RT, Shelton C, et al. Advanced techniques in magnetic resonance imaging in the evaluation of the large endolymphatic duct and sac syndrome. Laryngoscope. 1995;105:1037.
73. Hirsch BE, Weissman JL, Curtin HD, et al. Magnetic resonance imaging of the large vestibular aqueduct. Arch Otolaryngol Head Neck Surg. 1992;118:1124.
74. Reussner LA, Dutcher PO, House WF. Large vestibular aqueduct syndrome with massive endolymphatic sacs. Otolaryngol Head Neck Surg. 1995;113:606.
75. Valvassori GE, Clemis JD. Abnormal vestibular aqueduct in cochleovestibular disorders. Adv Otorhinolaryngol. 1978;24:100.
76. Valvassori GE. The large vestibular aqueduct and associated anomalies of the inner ear. Otolaryngol Clin North Am. 1983;16:95.
77. Gussen R. The endolymphatic sac in Mondini disorder. Arch Otorhinol Laryngol. 1985;242:71.
78. Naganawa S, Koshikawa T, Fukatsu H, et al. Serial MR imaging studies in enlarged endolymphatic duct and sac syndrome. Eur Radiol. 2002;12(Suppl 3):S114.
79. Shirazi A, Fenton JE, Fagan PA. Large vestibular aqueduct syndrome and stapes fixation. J Laryngol Otol. 1994;108:989.
80. Govaerts PJ, Casselman J, Daemers K, et al. Audiological findings in large vestibular aqueduct syndrome. Int J Pediatr Otorhinolaryngol. 1999;51:157.
81. Schessel DA, Nedzelski JM. Presentation of large vestibular aqueduct syndrome to a dizziness unit. J Otolaryngol. 1992;21:265.
82. Yetiser S, Kertmen M, Ozkaptan Y. Vestibular disturbance in patients with large vestibular aqueduct syndrome (LVAS). Acta Otolaryngol. 1999;119:641.
83. Wilson DF, Hodgson RS, Talbot JM. Endolymphatic sac obliteration for large vestibular aqueduct. Am J Otol. 1997;18:101.
84. Welling DB, Slater PW, Martyn MD, et al. Sensorineural hearing loss after occlusion of the enlarged vestibular aqueduct. Am J Otol. 1999;20:338.
85. Harker LA, Vanderheiden S, Veazey D, et al. Multichannel cochlear implantation in children with large vestibular aqueduct syndrome. Ann Otol Rhinol Laryngol Suppl. 1999;177:39.
86. Miyamoto RT, Bichey BG, Wynne MK, et al. Cochlear implantation with large vestibular aqueduct syndrome. Laryngoscope. 2002;112:1178.
87. Jackler RK, Hwang PH. Enlargement of the cochlear aqueduct: fact or fiction? Otolaryngol Head Neck Surg. 1993;109:14.
88. Narcy P, Viala P, Sellier N, et al. [The cochlear aqueduct and congenital perilymphatic fistula. An initial report.]. Ann Otolaryngol Chir Cervicofac. 1989;106:57.
89. Phelps PD. Congenital cerebrospinal fluid fistulae of the petrous temporal bone. Clin Otolaryngol. 1986;11:79.
90. Bhimani S, Virapongse C, Sarwar M. High resolution computed tomographic appearance of the normal human cochlear aqueduct. Am J Neuroradiol. 1984;5:715.
91. Muren C, Wilbrand H. Anatomic variations of the human cochlear aqueduct: a radioanatomic investigation. Acta Radiol Diagn. 1986;27:11.
92. Rask-Anderson H, Stahle J, Wilbrand H. Human cochlear aqueduct and its accessory canals. Ann Otol Rhinol Laryngol Suppl. 1977;86:1.
93. Bachor E, Byahatti S, Karmody CS. New aspects in the histopathology of the cochlear aqueduct in children. Am J Otol. 1999;20:612.
94. Palva T, Dammerr K. Human cochlear aqueduct. Acta Otolaryngol Suppl (Stockh). 1969;246:1.
95. Schuknecht HF, Reisser C. The morphologic basis for perilymphatic gushers and oozers. Adv Otorhinolaryngol. 1988;39:1.
96. Allen G. Fluid flow in the cochlear aqueduct and cochlea—hydrodynamic considerations in perilymph fistula, stapes gusher, and secondary endolymphatic hydrops. Am J Otol. 1987;8:319.
97. Flood LM, Kernink JL, Karnish JM. Pneumocephalus following treatment of a stapes gusher. Am J Otol. 1985;6:508.
98. Glasscock ME. The stapes gusher. Arch Otolaryngol Head Neck Surg. 1973;98:82.
99. Birman CS, Gibson WPR. Hearing loss associated with large internal auditory meatus: a report of five paediatric cases. J Laryngol Otol. 1999;113:1015.
100. Bamiou DE, Worth S, Phelps P, et al. Eighth nerve aplasia and hypoplasia in cochlear implant candidates: the clinical perspective. Otol Neurotol. 2001;22:492.
101. Casselman JW, Offeciers FE, Govaerts PJ, et al. Aplasia and hypoplasia of the vestibulocochlear nerve: diagnosis with MR imaging. Radiology. 1997;202:773.
102. Glastonbury CM, Davidson HC, Harnsberger HR, et al. Imaging findings of cochlear nerve deficiency. AJNR Am J Neuroradiol. 2002;23:635.
103. Gray RF, Ray J, Baguley DM, et al. Cochlear implant failure due to unexpected absence of the eighth nerve: a cautionary tale. J Laryngol Otol. 1998;112:646.
104. Patel N, Oghalai JS. Familial unilateral cochlear nerve aplasia. Otol Neurotol. 2006;27:443.
105. Ito K, Endo A, Monobe H, et al. Nonsyndromic isolated unilateral cochlear nerve aplasia without narrow internal auditory meatus: a previously overlooked cause of unilateral profound deafness in childhood. Ann Otology Rhinol Laryngol. 2005;114:859.
106. Adunka OF, Roush PA, Teagle HF, et al. Internal auditory canal morphology in children with cochlear nerve deficiency. Otol Neurotol. 2006;27:793.
107. Ito K, Ishimoto S, Shotaro K. Isolated cochlear nerve hypoplasia with various internal auditory meatus deformities in children. Ann Otol Rhinol Laryngol. 2007;116:520.
108. Oysu C, Oysu A, Aslan I, et al. Temporal bone imaging findings in Waardenburg’s syndrome. Int J Pediatr Otorhinolaryngol. 2001;58:215.
109. Higashi K, Matsuki C, Sarashina N. Aplasia of posterior semicircular canal in Waardenburg syndrome type II. J Otolaryngol. 1992;21:262.
110. Amiel J, Attieé-Bitach T, Marianowski R, et al. Temporal bone anomaly proposed as a major criteria for diagnosis of CHARGE syndrome. Am J Med Genet. 2001;99:124.
111. Collins WO, Buchman CA. Bilateral semicircular canal aplasia: a characteristic of the CHARGE association. Otol Neurotol. 2002;23:233.
112. Ceruti S, Stinckens C, Cremers CW, et al. Temporal bone anomalies in the branchio-oto-renal syndrome: detailed computed tomographic and magnetic resonance imaging findings. Otol Neurotol. 2002;23:200.
113. Kemperman MH, Stinckens C, Kumar S, et al. Progressive fluctuant hearing loss, enlarged vestibular aqueduct, and cochlear hypoplasia in branchio-oto-renal syndrome. Otol Neurotol. 2001;22:637.
114. Cremers WR, Bolder C, Admiraal RJ, et al. Progressive sensorineural hearing loss and a widened vestibular aqueduct in Pendred syndrome. Arch Otolaryngol Head Neck Surg. 1998;124:501.
115. Ishinaga H, Shimizu T, Yuta A, et al. Pendred’s syndrome with goiter and enlarged vestibular aqueducts diagnosed by PDS gene mutation. Head Neck. 2002;24:710.
116. Blaser S, Propst EJ, Martin D, et al. Inner ear dysplasia is common in children with Down syndrome (trisomy 21). Laryngoscope. 2006;16:2113.
117. Griffith AJ, Telian SA, Downs C, et al. Familial Mondini dysplasia. Laryngoscope. 1998;108:1368.
118. Arellano B, Ramírez Camacho R, García Berrocal JR, et al. Sensorineural hearing loss and Mondini dysplasia caused by a deletion at locus DFN3. Arch Otolaryngol Head Neck Surg. 2000;126:1065.
119. Daneshi A, Farhadi M, Asghari A, et al. Three familial cases of Michel’s aplasia. Otol Neurotol. 2002;23:346.
120. Matsunaga T, Hirota E. Familial lateral semicircular canal malformation with external and middle ear abnormalities. Am J Med Genet. 2003;116A:360.
121. Usami S, Abe S, Weston MD, et al. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188.
122. Park TS, Hoffman HJ, Humphreys RP, et al. Spontaneous cerebrospinal fluid otorrhea in association with a congenital defect of the cochlear aqueduct and Mondini dysplasia. Neurosurgery. 1982;11:356.
123. Quiney RE, Mitchell DB, Djazeri B, et al. Recurrent meningitis in children due to inner ear abnormalities. J Laryngol Otol. 1989;103:473.
124. Barcz DV, Wood RP2nd, Stears J, et al. Subarachnoid space: middle ear pathways and recurrent meningitis. Am J Otol. 1985;6:157.
125. Gacek RR, Leipzig B. Congenital cerebrospinal otorrhea. Ann Otol Rhinol Laryngol. 1979;88:358.
126. Romo LV, Curtin HD. Anomalous facial nerve canal with cochlear malformations. AJNR Am J Neuroradiol. 2001;22:838.
127. Burton EM, Keith JW, Linden BE, et al. CSF fistula in a patient with Mondini deformity: demonstration by CT cisternography. AJNR Am J Neuroradiol. 1990;11:205.
128. Kaufman B, Jordan VM, Pratt LL. Positive contrast demonstration of a cerebrospinal fluid fistula through the fundus of the internal auditory meatus. Acta Radiol. 1969;9:83.
129. Rockett FX, Wittenborg MH, Shillito JJr, et al. Pantopaque visualization of a congenital dural defect of the internal auditory meatus causing rhinorrhea. AJR Am J Roentgenol. 1964;91:640.
130. Phelps PD, Proops D, Sellars S, et al. Congenital cerebrospinal fluid fistula through the inner ear and meningitis. J Laryngol Otol. 1993;107:492.
131. Phelps PD, King A, Michaels L. Cochlear dysplasia and meningitis. Am J Otol. 1994;15:551.
132. Clarós P, Guirado C, Clarós A, et al. Association of spontaneous anterior fossa CSF rhinorrhea and congenital perilymphatic fistula in a patient with recurrent meningitis. Int J Pediatr Otorhinolaryngol. 1993;27:65.
133. Rich PM, Graham J, Phelps PD. Hyrtl’s fissure. Otol Neurotol. 2002;23:476.
134. Herther C, Schindler RA. Mondini’s dysplasia with recurrent meningitis. Laryngoscope. 1985;95:655.
135. Luntz M, Frank I, Yurovitzki I, et al. Large perilymph fistulas. Am J Otol. 1986;7:282.
136. Ohlms LA, Edwards MS, Mason EO, et al. Recurrent meningitis and Mondini dysplasia. Arch Otolaryngol Head Neck Surg. 1990;116:608.
137. Parisier SC, Birken EA. Recurrent meningitis secondary to idiopathic oval window CSF leak. Laryngoscope. 1977;86:1503.
138. MacRae DL, Ruby RR. Recurrent meningitis secondary to perilymph fistula in young children. J Otolaryngol. 1990;19:222.
139. Drummond DS, de Jong AL, Giannoni C, et al. Recurrent meningitis in the pediatric patient-the otolaryngologist’s role. Int J Pediatr Otorhinolaryngol. 1999;48:199.
140. Stevenson DS, Proops DW, Phelps PD. Severe cochlear dysplasia causing recurrent meningitis: a surgical lesson. J Laryngol Otol. 1993;107:726.
141. Farrior JB, Endicott JN. Congenital mixed deafness: cerebrospinal fluid otorrhea. Ablation of the aqueduct of the cochlea. Laryngoscope. 1971;81:684.
142. Cremers CW, Hombergen GC, Scaf JJ, et al. X-linked progressive mixed deafness with perilymphatic gusher during stapes surgery. Arch Otolaryngol Head Neck Surg. 1985;111:249.
143. Phelps PD, Reardon W, Pembrey M, et al. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology. 1991;33:326.
144. Piussan C, Hanauer A, Dahl N, et al. X-linked progressive mixed deafness: a new micro deletion that involves a more proximal region in Xq21. Am J Hum Genet. 1995;56:224.
145. Talbot JM, Wilson DF. Computed tomographic diagnosis of X-linked congenital mixed deafness, fixation of the stapedial footplate, and perilymphatic gusher. Am J Otol. 1994;15:177.
146. Snik A, Hombergen GC, Mylanus EA, et al. Air-bone gap in patients with X-linked stapes gusher syndrome. Am J Otol. 1995;16:241.
147. Pappas DG, Simpson LC, Godwin GH. Perilymphatic fistula in children with pre-existing sensorineural hearing loss. Laryngoscope. 1988;98:507.
148. Parnes LS, McCabe BF. Perilymph fistula: an important cause of deafness and dizziness in children. Pediatrics. 1987;80:524.
149. Supance JS, Bluestone CD. Perilymph fistulas in infants and children. Otolaryngol Head Neck Surg. 1982;91:663.
150. Weber PC, Perez BA, Bluestone CD. Congenital perilymphatic fistula and associated middle ear abnormalities. Laryngoscope. 1993;103:160.
151. Weissman JL, Weber PC, Bluestone CD. Congenital perilymphatic fistula: computed tomography appearance of middle and inner ear anomalies. Otolaryngol Head Neck Surg. 1994;111:243.
152. Weber PC, Bluestone CD, Kenna MA, et al. Correlation of beta-2 transferrin and middle ear abnormalities in congenital perilymph fistula. Am J Otol. 1995;16:277.
153. Levenson MJ, Desloge RB, Parisier SC. Beta-2 transferrin: limitations of use as a clinical marker for perilymph. Laryngoscope. 1996;106:159.
154. Buchman CA, Luxford WM, Hirsch BE, et al. Beta-2 transferrin assay in the identification of perilymph. Am J Otol. 1999;20:174.
155. Gehrking E, Wisst F, Remmert S, et al. Intraoperative assessment of perilymphatic fistulas with intrathecal administration of fluorescein. Laryngoscope. 2002;112:1614.
156. Jahrsdoerfer RA. Congenital atresia of the ear. Laryngoscope. 1978;88(Suppl 13):1.
157. Naunton RF, Valvassori GE. Inner ear anomalies: their association with atresia. Laryngoscope. 1968;78:1041.
158. Potter GD. Inner ear abnormalities in association with congenital atresia of the external auditory canal. Ann Otol Rhinol Laryngol. 1969;78:598.
159. Phelps PD. Fast spin echo MRI in otology. J Laryngol Otol. 1994;108:383.
160. Papadopoulos A, Vlahos L, Xenelis J, et al. Value of Gd-DTPA-enhanced MR imaging of the labyrinth in patients with sudden hearing loss. Magn Reson Imaging. 1995;13:387.
161. Isono M, Murata K, Aiba K, et al. Minute findings of inner ear anomalies by three-dimensional CT scanning. Int J Pediatr Otorhinolaryngol. 1997;42:41.
162. Miyashita H, Isono M, Murata K, et al. Three-dimensional magnetic resonance imaging findings of inner ear anomaly. Acta Otolaryngol Suppl. 2000;542:67.
163. Purcell D, Johnson J, Fischbein N, et al. Establishment of normative cochlear and vestibular measurements to aid the diagnosis of inner ear malformations. Otolaryngol Head Neck Surg. 2003;128:78.
164. Purcell DD, Fischbein N, Patel A, et al. Two temporal bone computed tomography measurements increase recognition of malformations and predict sensorineural hearing loss. Laryngoscope. 2006;116:1439.
165. House WF, House JW. Mondini deformity: surgical treatment. Paper presented at the IVth Pan-Pacific Surgical Association Congress, Honolulu, Hawaii, April 1978.
166. Goin DW, Rasband RW, Mischke RE. Endolymphatic sac surgery in Mondini’s dysplasia: a report of 16 cases. Laryngoscope. 1984;94:343.
167. Mangabeira-Albernaz PL, Fukuka Y, Chammas F, et al. The Mondini dysplasia—a clinical study. J Otorhinolaryngol Relat Spec. 1981;43:131.
168. Mitchell DP, Rubin AM. Mondini dysplasia: late complications. J Otolaryngol. 1985;14:265.
169. Jackler RK, Luxford WM, Brackmann DE, et al. Endolymphatic sac surgery in congenital malformations of the inner ear. Laryngoscope. 1988;98:698.
170. Bluestone CB. Cochlear malformations, meningitis, and cochlear implants: what have we learned? Otol Neurotol. 2003;24:349.
171. Schmidt JM. Cochlear neuronal populations in development defects of the inner ear: implications for cochlear implantation. Acta Otolaryngol (Stockh). 1985;99:14.
172. Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: sound detection with cochlear implant in five ears of four children with congenital malformations of the cochlea. Laryngoscope Suppl. 1987;97:15.
173. Weber BP, Dillo W, Dietrich B, et al. Pediatric cochlear implantation in cochlear malformations. Am J Otol. 1998;19:747.
174. Gijs KA, Van Wermeskerken GKA, Dunnebier EA, et al. Audiological performance after cochlear implantation: a 2-year follow-up in children with inner ear malformations. Acta Otolaryngol. 2007;127:252.
175. Eisenman DJ, Ashbaugh C, Zwolan TA, et al. Implantation of the malformed cochlea. Otol Neurotol. 2001;22:834.
176. Hoffman RA, Downey LL, Waltzman SB, et al. Cochlear implantation in pediatric patients with Mondini deformities. Am J Otol. 1997;18:184.
177. Molter DW, Pate BRJr, McElveen JTJr. Cochlear implantation in the congenitally malformed ear. Otolaryngol Head Neck Surg. 1993;108:174.
178. Silverstein H, Smouha E, Morgan N. Multichannel cochlear implantation in a patient with bilateral Mondini deformities. Am J Otol. 1988;9:451.
179. Tucci DL, Telian SA, Zimmerman-Phillips S, et al. Cochlear implantation in patients with cochlear malformations. Arch Otolaryngol Head Neck Surg. 1995;121:833.
180. Zheng Y, Schachern PA, Djalilian HR, et al. Temporal bone histopathology related to cochlear implantation in congenital malformation of the bony cochlea. Otol Neurotol. 2002;23:181.
181. Raine CH, Hussain SS, Khan S, et al. Anomaly of the facial nerve and cochlear implantation. Ann Otol Rhinol Laryngol Suppl. 1995;166:430.
182. Wardrop P, Kerr AI, Moussa SA. Persistent stapedial artery preventing successful cochlear implantation: a case report. Ann Otol Rhinol Laryngol Suppl. 1995;166:443.
183. Wootten CT, Backous DD, Haynes DS. Management of cerebrospinal fluid leakage from cochleostomy during cochlear implant surgery. Laryngoscope. 2006;116:2055.
184. O’Donoghue G, Balkany T, Cohen N, et al. Meningitis and cochlear implantation. Otol Neurotol. 2002;23:823.
185. Page EL, Eby TL. Meningitis after cochlear implantation in Mondini malformation. Otolaryngol Head Neck Surg. 1997;116:104.
[/level-membership-for-otolaryngology-category][not-level-membership-for-otolaryngology-category]
CHAPTER 191 Congenital Malformations of the Inner Ear
Development of the inner ear begins early in embryogenesis. By the end of the eighth week, the membranous labyrinth has assumed its characteristic convoluted shape.1 Gradual ossification of the otic capsule develops around the membranous labyrinth and is essentially complete by birth.2 Maturation of the sensory epithelium occurs long after formation of the membranous labyrinth, during the late second and early third trimesters. By weeks 26 to 28 of gestation, hair cell and auditory neural development are largely complete. Thus, the normal human fetus may be able to hear 2.5 to 3 months before birth.
Most inner ear malformations arise when formation of the membranous labyrinth is interrupted during the first trimester of pregnancy.2 This interruption may be either a result of inborn genetic error or a consequence of a teratogenic exposure during the period of inner ear organogenesis between the fourth and eighth weeks of gestation. Genetic errors may be either dominant or recessive and may manifest as sensorineural hearing loss (SNHL) alone or be associated with any of a number of syndromes.4 A partial list of syndromes associated with radiologically detectable inner ear malformation includes Pendred’s, Usher’s, Waardenburg’s, Wildervanck’s (cervico-oculoacoustic), branchio-oto-renal, and Alagille’s sundromes.5–8 Nonsyndromic familial inner ear malformations have also been described.9,10 Teratogenic influences known to affect inner ear organogenesis include in utero viral infection (e.g., rubella, cytomegalovirus infection), chemical teratogens (e.g., thalidomide), and radiation exposure.11 Abnormalities in otic capsule structure and deficiencies in the organ of Corti appear to arise as secondary effects of the earlier error in development of the membranous labyrinth. Derangement of the otic capsule ossification process alone does not appear to be a major mechanism in congenital hearing loss. Ossification of the labyrinthine lumen, however, is a common finding in early acquired deafness, typically arising as a consequence of meningitis.
Developmental damage to cochlear structure and function is not restricted to agents that cause gross structural malformations. Even in doses below those that would be ototoxic in the adult, aminoglycoside antibiotics administered in the animal equivalent of the human first trimester of pregnancy cause severe hearing loss in several species.12 This time frame corresponds to the maturation of the outer hair cells and initiation of the cochlear potentials. Human studies also document this. In 35 of the infants delivered by 72 women who received streptomycin prophylaxis for tuberculosis during the first 4 months of pregnancy, auditory deficits ranging from minor high-frequency threshold elevations to severe bilateral SNHL were noted.13
Incidence
Most children with congenital sensory hearing loss, with the possible exception of auditory neuropathy, would have detectable abnormalities in their inner ears if they could be examined histologically. Because most children with profound bilateral SNHL have radiographically normal inner ears, it can be inferred that malformations limited to the membranous labyrinth predominate. The incidence of malformations reported varies, depending on the hearing level of the study population, the sophistication of imaging used, and the definition of abnormality used by the observer. As a rule of thumb, approximately 20% of cases of congenital SNHL will demonstrate inner ear malformation with modern imaging technology. In a series of 234 children who had SNHL of varying degrees of severity, Reilly14 found cochlear anomalies in only 4% of those evaluated by high-resolution computed tomography (CT). With improvements in CT imaging technology and greater awareness of inner ear deformities, Antonelli and colleagues found anomalies in 31% of 157 children with SNHL of variable degree.15 In another CT study, anomalies were found in 17% of 185 ears of children with SNHL and none in the ears of 309 children without SNHL.16 A large Korean study found 22% incidence of anomalies on radiologic imaging in 590 ears with profound SNHL.17
The incidence of deformities of the semicircular canals (SCCs) and inner ear aqueducts has been less well studied than that of cochlear deformities. In a series of patients with radiographically detectable malformations of the inner ear, the cochlea was involved in 76%, the semicircular canals were involved in 39%, and the vestibular aqueduct (VA) was affected in 32% of ears.3 The total is more than 100%, because many cases demonstrate abnormalities of more than one portion of the inner ear. In recent years, a heightened awareness of VA enlargement, combined with the greater sensitivity of axial CT scans in demonstrating this deformity, has led to a substantial increase in its detection. The rapid accrual of cases by clinicians interested in inner ear malformations suggests that enlargement of the VA will ultimately prove to be the most common radiographically detectable inner ear anomaly.6
Among deaf children with radiographically normal inner ears, histopathologic studies indicate that cochleosaccular dysplasia (Scheibe’s dysplasia) is by far the most common deformity.18 Because of the paucity of pathologic specimens available for examination, it is impossible to estimate the relative frequency of the various membranous malformations.
Classification
The traditional nomenclature used to describe congenital anomalies of the inner ear involves a confusing array of eponyms that stem from the first reports of the various morphologic patterns, usually by 18th or 19th century authors. In this chapter, a descriptive classification system is used, along with the traditional eponymous terminology, to make this topic more logical, easier to learn, and more clinically relevant (Box 191-1). In membranous malformations, this classification is based on histopathologic changes in the inner ear; in combined osseous-membranous deformities, radiographic appearance is used to distinguish among the various entities.3 Correct use of the terminology of pathoembryology is important, although these terms frequently are applied imprecisely in the earlier literature. Key terms are aplasia (complete lack of development), hypoplasia (incomplete development), and dysplasia (aberration in development). The classification scheme used in this chapter has proved to be of practical clinical use and has been employed in most published studies in recent years. Other classification schema that categorize observed anomalies have been proposed.19
Malformations Limited to the Membranous Labyrinth
In malformations limited to the membranous labyrinth, which account for more than 90% of cases of congenital deafness, the bony labyrinth is normal.20–23 In its most severe form, membranous dysplasia involves the entire labyrinth, including the cochlea, semicircular canals (SCCs), utricle, and saccule. A limited form of membranous labyrinthine dysplasia, involving only a portion of the inner ear, also has been described.
Complete Membranous Labyrinthine Dysplasia
Complete membranous labyrinthine dysplasia was first described by Siebenmann and Bing24 and is extremely rare. It has been reported in association with cardioauditory (Jervell and Lange-Nielsen) and Usher syndromes.25
Limited Membranous Labyrinthine Dysplasia
Cochleosaccular Dysplasia (Scheibe’s Dysplasia)
Incomplete development of the pars inferior is the most frequent histopathologic finding in congenital deafness. It was first described by Scheibe26 and commonly is known as cochleosaccular dysplasia. The spectrum of pathologic findings in this anomaly, which is confined to the cochlea and the saccule, has been well described.22,27–30 The organ of Corti is either partially or completely missing. The cochlear duct usually is collapsed, with Reissner’s membrane adherent to the limbus. Less commonly, the duct is distended, presumably as a result of endolymphatic hydrops. The stria vascularis typically is degenerated and may contain colloidal inclusions. Schuknecht31 described characteristic strial changes consisting of aplasia alternating with regions of hyperplasia and gross deformity. Cochlear changes may be severe in the base turn and gradually lessen in intensity toward the apex, or they may be severe throughout. The saccule usually is collapsed and has degenerated sensory epithelium. In cochleosaccular dysplasia, the SCCs and utricle are normal. Auditory neuronal survival is variable but may remain normal into adulthood, at least in some cases. Cochleosaccular dysplasia also has been demonstrated in a number of animal species, including the deaf white cat, Dalmatian dogs, and various mouse mutants.32
Malformations of the Membranous and Osseous Labyrinth
Congenital anomalies of the inner ear that deform the otic capsule are of special interest to the clinician because they may be recognized and differentiated during life through radiographic imaging. As discussed previously, only approximately 20% of congenitally deaf people demonstrate radiographically anomalous inner ears. The clinical manifestations and natural history of these deformities are highly variable. Although some patients are deaf from birth, most maintain some residual hearing into adulthood. Slowly progressive deterioration of hearing during childhood, with eventual stabilization, is common. Sudden decrements in hearing are frequent and may appear to be spontaneous or may be triggered by head trauma, even minor in nature. Presumably, most of these cases are due either to internal fistulization secondary to membrane rupture within the cochlea, with admixture of perilymph and endolymph, or to external fistulization to the middle ear. Fluctuant hearing loss is unusual in these patients, and endolymphatic hydrops is an atypical finding. In some patients, hearing may be best preserved in very high frequencies (greater than 8000 Hz), which are not measured by conventional audiometry.33 Residual ultra-audiometric hearing should be suspected in hearing-impaired children who manifest substantially better auditory function than would be predicted by pure tone results in the speech frequencies.34 Occasionally, malformation of the inner ears may be associated with normal hearing.35,36 This is especially the case for semicircular canal anomalies.37 Vestibular symptoms, which occasionally are severe, are present in approximately 20% of patients.3 Retardation of motor development has been documented in some children with malformed inner ears, particularly those in whom semicircular canals are absent.38 A few patients experience vertigo when exposed to loud sounds—the so-called Tullio phenomenon.39
A wide variety of morphologic patterns of inner ear malformation has been observed radiographically and may involve the cochlea, SCCs, or vestibular aqueduct3,40–45 (Fig. 191-1). Similar diversity has been observed on histologic analysis.21,46–50 A majority of combined osseous and membranous malformations appears to arise from a premature arrest in the development of one or more components of the inner ear (Fig. 191-2). The strongest evidence for this theory comes from the resemblance of most malformed inner ears to the appearance of the inner ear during embryogenesis, particularly between the fourth and eighth weeks of gestation. As a general rule, the earlier the developmental arrest, the more severe the deformity and the worse the hearing will be.
Figure 191-1. Cochlear malformations. Drawings were made from coronal computed tomography scans.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97[Suppl 40]:2.)
Figure 191-2. Embryogenesis of cochlear malformations.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97(Suppl 40):2.)
Other anomalies cannot be explained by a premature arrest in development alone and appear to arise from an aberrant embryologic process. An example of this type of anomaly is a cochlea of normal length but abnormal size or coiling geometry.46 In humans, the inner ear is of adult size at birth and shows strikingly little variation in size among individual patients. Pappas and colleagues51 suggested that some children with congenital SNHL and apparently normal inner ear morphology on CT possess subtle abnormalities in the dimensions of inner ear structures. These investigators propose that such dimensional variations arise from a teratogenic insult during the second or third trimester, after the membranous labyrinth has formed but before it has reached adult size. Further study is needed to determine the clinical relevance of these observations.
Whereas some inner ear malformations involve only one portion of the inner ear, many patients have a combination of anomalies involving more than one component. Between the fourth and fifth weeks of development, the spheric otocyst develops three buds that ultimately form the cochlea, SCCs, and VA (see Fig. 191-2). An inner ear malformation may be limited to one of these anlages, may involve a combination of two, or may even affect all three.
The frequent coexistence of deformities involving the cochlea, SCCs, and VA has several possible explanations: (1) the anomaly is genetically predetermined; (2) an insult to the embryo occurred before the fifth week; or (3) each of the buds was susceptible to some teratogenic influence at a later stage of development. A majority of inner ear malformations are bilateral and symmetrical. In cases in which radiographs detect an anomaly on only one side, the opposite “normal” inner ear has a hearing loss in approximately 50% of cases.3
Before the evolution of high-resolution imaging technology, clinicians and histopathologists alike tended to lump these malformations together under the term Mondini’s dysplasia, after the first report by Carlo Mondini. We are indebted to the late Dr. Peter Phelps and Latin scholar Gordon Hartley for an English translation of Mondini’s original article.52 Before the Academy of Sciences of the University of Bologna, Mondini described the inner ear findings in a deaf 8-year-old boy who was struck on the foot by a wagon and later died of gangrene.53 The cochlea possessed only 1.5 turns and had a hollow apical cavity. An enlarged vestibule and VA also were noted. This deformity is the most common form of cochlear anomaly (Table 191-1). Although numerous other distinct anatomic patterns of inner ear malformation are discernible radiographically and histologically, many workers continue to use the term Mondini’s dysplasia to describe them all. To avoid a confusing and overly broad nomenclature system, this designation is best reserved for the particular subtype of cochlear malformation first described by Mondini, whether or not it is associated with other inner ear malformations.
| Malformation | Incidence (%) |
|---|---|
| Incomplete partition (Mondini’s dysplasia) | 55 |
| Common cavity | 26 |
| Cochlear hypoplasia | 15 |
| Cochlear aplasia | 3 |
| Complete labyrinthine aplasia (Michel’s aplasia) | 1 |
Complete Labyrinthine Aplasia (Michel’s Aplasia)
The most severe deformity of the membranous and osseous labyrinth, complete labyrinthine aplasia, was first described by Michel.54 This malformation is exceedingly rare. Presumably, a developmental arrest occurs before the formation of an otic vesicle, resulting in a complete absence of inner ear structures. Complete labyrinthine aplasia has been reported in association with anencephaly and thalidomide exposure.21,55 An association with external ear abnormalities also has been reported.56 A purported case of Michel’s aplasia actually described a cystic inner ear of the common cavity type.57 This is but one example of the inaccurate use of traditional eponyms—a frequent occurrence in the literature. The incidence of complete labyrinthine aplasia is overestimated in the radiographic literature because it is confused with labyrinthine ossification. In the latter condition, which usually is acquired during life, a sizable and dense otic capsule is evident radiographically. In complete labyrinthine aplasia, the otic capsule is entirely absent58 (Figs. 191-3 to 191-5). Such ears are, of course, uniformly deaf.
Cochlear Anomalies
Cochlear Aplasia
In cochlear aplasia the cochlea is completely absent, presumably as a result of an arrest in the development of the cochlear bud at the fifth week of gestation (see Fig. 191-1). This morphologic pattern is rare. Radiographically, only a vestibule and SCCs (usually deformed) are present. To differentiate this anomaly from labyrinthine ossification, it is necessary to assess the amount of otic capsule bone anterior to the internal auditory canal (IAC). In cochlear aplasia, the otic capsule is absent, whereas in osseous obliteration, it is dense and of normal dimensions. Ears with cochlear aplasia are devoid of auditory function.
Cochlear Hypoplasia
An arrest during the sixth week of gestation results in a hypoplastic cochlea consisting of a single turn or less. This deformity comprises approximately 15% of all cochlear anomalies. Radiographically, a small bud of variable length (usually 1 to 3 mm) protrudes from the vestibule (Fig. 191-6). The vestibule frequently is enlarged, with accompanying semicircular malformations in approximately one half of the cases. Small cochleas lacking a modiolus or other internal architecture have been described histologically.27,50,59 Hearing is variable in these ears and may be remarkably good in view of the minute size of the cochlea. The variability of hearing presumably is accounted for by the degree of membranous labyrinthine development within the truncated cochlear lumen.
Incomplete Partition (Mondini)
Arrest at the seventh week of gestation yields a cochlea that has only 1.5 turns. This is the most common type of cochlear malformation, accounting for more than 50% of all such deformities. Radiographically, the cochlea is smaller than normal and partially or completely lacks an interscalar septum (Figs. 191-7 and 191-8). Although the cochlea usually measures 8 to 10 mm vertically, it is typically in the 5- to 6-mm range in incomplete partition deformity. Care must be exercised in counting the number of cochlear turns radiographically because this may be difficult to determine even using high-resolution CT. The radiographic diagnosis depends more on cochlear size and the absence of a scalar septum than on the number of cochlear turns perceived. Histologically, incomplete partition appears to be the radiographic correlate of classical Mondini’s dysplasia (Fig. 191-9). In numerous reported cases, a small cochlea with 1.5 turns possessing an apical scala communis secondary to deficiency in the osseous spiral lamina has been described.27,46,48 Sennaroglu and Saatci have subtyped the incomplete partition deformity into three variants60 (Fig. 191-10). Type I lacks the entire modiolus and interscalar septa and demonstrates a cystic appearance. Type II has a normal base turn but a cystic apex (“Mondini type”). Recently Sennaroglu has proposed a type III variant with deficient modiolus and partial interscalar septation at the cochlea’s periphery (L. Sennaroglu, personal communication, 2007). Organ of Corti development is variable, as is the auditory neural population. As might be expected, auditory function also is variable, ranging from normal to profound SNHL. The mean hearing threshold (three-tone average) in a group of 41 ears with incomplete partition was 75 dB.3 SCC deformities accompany incomplete partition of the cochlea in approximately 20% of cases.
Common Cavity
A deformed inner ear in which the cochlea and vestibule are confluent, forming an ovoid cystic space without internal architecture, may be explained by an arrest at the week 4 otocyst stage. Alternatively, this deformity may result from aberrant development at a later stage. An empty ovoid space typically longer in its horizontal dimension is seen radiographically. Although the size of the cyst may vary, it averages 7 mm vertically and 10 mm horizontally. It is quite easy to misdiagnose a dysplastic lateral SCC as a common cavity deformity. The key to differentiating between the two is that a common cavity cochlea lies predominantly anterior to the IAC on axial-plane CT, and a dysplastic vestibular system lies posterior to it. Histologically, an ovoid or spherical smooth-walled cystic cavity containing primordia of the membranous labyrinth has been described.48,50,55 Sensory and supporting cells may be differentiated into recognizable organs of Corti that are scattered peripherally around the walls of the cyst. Neural population usually is sparse or absent. Hearing is usually, but not invariably, poor.
Labyrinthine Anomalies
Semicircular Canal Dysplasia
Dysplasia of the lateral SCC is a common type of inner ear malformation (Fig. 191-11). Approximately 40% of ears with a malformed cochlea will have an accompanying dysplasia of the lateral SCC.3 Occasionally, dysplasia of the lateral SCC exists as the sole inner ear malformation. During the sixth week of development, the budding SCC normally forms a semicircular evagination from the vestibular anlage. The central portion of the pocket-shaped protrusion adheres, leaving a peripheral semicircular tube. When this central adhesion fails to occur, SCC dysplasia results (Fig. 191-12; see also Fig. 191-2). SCC dysplasia occasionally takes the form of a small bud, rather than the more common half-disk shape, presumably because of a slightly earlier timing of the developmental insult. The lateral SCC is deformed more often than the posterior or superior SCC, apparently because it forms earlier in embryogenesis. The typical radiographic appearance of SCC dysplasia is that of a short, broad cystic space confluent with the vestibule (Fig. 191-13).
Figure 191-11. Semicircular canal malformations. LSCC, lateral semicircular canal; SSCC, superior semicircular canal.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97[Suppl 40]:2.)
Numerous histologic descriptions of SCC dysplasia exist in the literature.46–50 The half-disk shaped cavity may contain a rudimentary crista ampullaris. The utricle and saccule may be distended, collapsed, or entirely absent. Caloric responses in SCC dysplasia are functionally absent or reduced in most cases, but a few may have normal responsiveness.3 Ears with malformations limited to the vestibular system often have normal or near-normal hearing. When the cochlea also is abnormal, sensory hearing levels tend to be impaired, to a variable degree.62 SCC dysplasia appears to have an association with conductive hearing loss, presumably because of inner ear micromechanical factors rather than stapes fixation.63
Semicircular Canal Aplasia
SCC aplasia is only one-fourth as common as SCC dysplasia.3 It is usually associated with cochlear anomalies.29 Presumably, SCC aplasia arises from a failure in the development of the vestibular anlage before the sixth week of gestation. Most cases are syndromic, with a predominance of the CHARGE association (coloboma, heart defects, atresia of the choanae, retardation of growth and development, genital, and ear abnormalities or hearing loss).64,65
Malformations of the Vestibular and Cochlear Aqueducts
Enlargement of the Vestibular Aqueduct
Experience suggests that enlargement of the VA is the most common radiographically detectable malformation of the inner ear.57,66–71 In earlier literature its incidence was underestimated, partly because of a lack of awareness but mostly because enlargement of the VA could be visualized only by lateral polytomography at a time when most studies were confined to the anteroposterior plane. The advent of high-resolution CT in the axial plane has made assessment of the VA much easier. The diameter of a normal VA, when measured halfway between the common crus and its external aperture, is between 0.4 and 1 mm. Enlargement of the VA is diagnosed when its diameter exceeds 2 mm, although enlarged VAs may exceed 6 mm in width. Whereas the VA is well visualized on axial CT (Fig. 191-14), the dilated endolymphatic sac is better seen with T2-weighted magnetic resonance imaging (MRI)72,73 (Fig. 191-15). The sac often is seen to be enormously enlarged on MRI, sometimes measuring 2 cm or more in diameter.74 In many cases, VA enlargement accompanies malformation of the cochlea or SCC. It also may be the sole radiographically detectable abnormality of the inner ear in a child with hearing loss. This condition is commonly referred to as the large VA syndrome, after Valvassori and Clemis’s first descriptions.75,76
Figure 191-14. Bilateral enlargement of the vestibular aqueducts (arrowheads) as seen on an axial computed tomography scan.
The VA derives from a diverticulum formed in the wall of the otocyst during the fifth week of gestation. It begins as a short, broad pouch but gradually elongates and thins until it achieves its characteristic J shape of adulthood (see Fig. 191-2). A premature arrest in development would be expected to produce a VA that is abnormally short and broad. However, an increasing body of evidence suggests that VA enlargement does not stem from an early arrest of sac development, but rather is an acquired deformity. Histologically, the sac and aqueduct are thin-walled and lack both vascularity and the rugose features thought to be important for physiologic function. Three observations suggest that a large VA stems from an abnormal communication between the subarachnoid space and the fluid chambers of the inner ear. The bone surrounding the VA may show signs of erosion, a finding that is inconsistent with a stable congenital deformity.77 Serial MR images show variability in both the size and signal characteristics of the enlarged sac.78 Presence of cerebrospinal fluid (CSF) under pressure, with consequent “gusher,” within the inner ear has been observed in ears with large VAs during both cochlear implantation and stapedectomy.31,79 Furthermore, the existence of abnormal CSF in the inner ear pathway frequently has been observed with CT or MRI. This anomaly typically consists of a defect of the cochlear modiolus at the distal end of the IAC.12,73
[/not-level-membership-for-otolaryngology-category]
