[level-membership-for-surgery-category]
Chapter 5 Congenital Malformation of the External Auditory Canal and Middle Ear
 Videos corresponding to this chapter are available online at www.expertconsult.com.
Videos corresponding to this chapter are available online at www.expertconsult.com.
Congenital aural atresia is characterized by aplasia or hypoplasia of the external auditory canal (EAC), often associated with absence or deformity of the auricle (microtia) and the middle ear, with occasional inner ear abnormalities. Aural atresia occurs in 1 in 10,000 to 20,000 live births1–5; unilateral atresia is three times more common than bilateral atresia. This disorder occurs more commonly in males and on the right side.1 EAC atresia is more often bony rather than membranous, and bony atresia is regularly accompanied by malformation of the middle ear cavity and structures of the middle ear.6–9 More severe forms of congenital microtia are usually associated with EAC atresia; rarely, canal atresia may be seen in patients with a normal pinna.10 Generally, a more severe external deformity implies a more severe middle ear abnormality.11,12
Kiesselbach, in 1883, is often credited with the first deep operation attempting to correct this malformation.8 Lascaratos and Assimakopoulos13 noted that the Byzantine physician Paul of Aegina performed a surgical treatment of congenital aural atresia.14 The procedure done by Kiesselbach resulted in facial paralysis. Because of the lack of middle ear microsurgery and the high complication rate, congenital aural atresia surgery was considered dangerous and to be avoided for the most part. In 1914, Page15 reported hearing improvement after surgery in five of eight patients. This report was followed in 1917 by Dean and Gittens,16 who reported an excellent hearing result in a patient and reviewed the various types of operations that had been tried by other surgeons. The prevailing attitude toward surgical correction in these cases remained generally pessimistic, however, despite these and other occasional reports of successful operations, until 1947. That year, Ombredanne17 in France and Pattee18 in the United States each reported a series of patients successfully operated on to improve hearing. Pattee’s technique included removal of the incus to “mobilize” the stapes18; Ombredanne17 added fenestration of the lateral semicircular canal.
With the advent of tympanoplasty techniques in the 1950s, interest in atresiaplasty increased as the teachings of Wullstein and Zollner carried over into surgery of the congenital ear.8 Larger series with greater success rates were reported as surgeons attempted to improve their results, using ossiculoplasty, mastoidectomy, differing degrees of bone removal, and different types and techniques of graft placement.2,4,17,19–26 Ombredanne27 went on to report on more than 600 aplasia cases by 1971 and 1600 cases with major and minor malformations by 1976.8 Gill’s report of 83 cases in 19692 and Jahrsdoerfer’s 1978 article8 are considered landmarks. Crabtree,28 Jahrsdoerfer,29,30 Marquet,31,32 and De la Cruz6,33 and their colleagues all reported on large surgical series, with modifications of classification and operative techniques.
Although techniques of canalplasty, meatoplasty, tympanoplasty, and ossiculoplasty have improved considerably, surgical correction of congenital aural atresia remains one of the most challenging operations performed by otologists. This is a complex surgical problem, requiring application of all tympanoplasty techniques and a thorough knowledge of the surgical anatomy of the facial nerve, oval window, and inner ear, and their congenital variants.1,6,8,17,21,26,27,29-31,33-40 The temporomandibular joint is displaced posteriorly by the lack of development of the EAC, narrowing the distance between the glenoid fossa and the anterior wall of the mastoid tip.19,41 Fusion of the incus and malleus is common, but because of its dual origin, the stapes footplate is usually normal.4,42
The surgical repair of aural atresia is recommended at age 6 years.6 The timing of repair must take into account any planned auricular reconstruction procedures. Criteria for patient selection must be stringent when attempting to achieve closure of the air-bone gap to within 20 to 30 dB. Preoperative counseling and several postoperative visits are essential for optimal results. In this chapter, we discuss these issues and provide guidelines for patient evaluation and selection, surgical techniques, and postoperative management.
EMBRYOLOGY
A review of the normal embryologic development of the ear aids in understanding the myriad of possible combinations of malformations encountered in congenital aural atresia. The inner ear, middle ear, and external ear develop independently and in such a way that deformity of one does not presuppose deformity of another.9,43 Most frequently, abnormalities of the outer and middle ear are encountered in combination with a normal inner ear.44,45
Microtia is a result of first and second branchial arch anomalies. Growth of mesenchymal tissue from the first and second branchial arches forms six hillocks around the primitive meatus that fuse to form the auricle (Table 5-1). By the end of the third month, the primitive auricle has been completed. The external auditory meatus develops from the first branchial groove. During the second month, a solid core of epithelium migrates inward from the rudimentary pinna toward the first branchial pouch. This core, the precursor of the EAC, starts to hollow out and take shape in the sixth month. It canalizes in the seventh month, causing the developing mastoid to become separated from the mandible. Its subsequent posterior and inferior development carries the middle ear and facial nerve to their normal positions.1,43,46–48 Some of the literature supports the notion that microtia grade can indicate the status of middle ear development in aural atresia. The better developed the external ear is, the better developed is the middle ear.
| First Branchial Arch | Second Branchial Arch |
|---|---|
| First hillock—tragus | Fourth hillock—antihelix |
| Second hillock—helical crus | Fifth hillock—antitragus |
| Third hillock—helix | Sixth hillock—lobule and lower helix |
The first branchial pouch grows outward to form the middle ear cleft. The plaque of tissue where this cleft meets the epithelium of the EAC forms the tympanic membrane. While the pouch is forming the eustachian tube, tympanic cavity, and mastoid air cells, Meckel’s cartilage (first branchial arch) is forming the neck and head of the malleus and incus body. Reichert’s cartilage (second branchial arch) forms the remainder of the first two ossicles’ long processes and the stapes superstructure. The footplate has a dual origin from the second arch and the otic capsule. The ossicles attain their final shape by the fourth month. By the end of the seventh to eighth month, the expanding middle ear cleft surrounds the ossicles and covers them with a mucous membrane.1,45,49
The facial nerve is the nerve of the second branchial arch. At 4.5 weeks, this developing nerve divides the blastema, which is the condensation of the second arch mesenchymal cells, into the stapes, the interhyale (stapedius muscle precursor), and the laterohyale (precursor of the posterior wall of the middle ear). The nerve’s intraosseous course is dependent on this bony expansion.45,47 The membranous portion of the inner ear develops during the third to sixth week from an auditory placode on the lateral surface of the hindbrain. The surrounding mesenchyme transforms into the bony otic capsule.50
Congenital aural atresia can range in severity from a thin membranous canal atresia to complete lack of tympanic bone, depending on the time of arrest of intrauterine development.1,51,52 The common finding of a normal inner ear is explained because the inner ear is formed by the time of external/middle ear development arrest. Facial nerve course abnormalities are often seen.
CLASSIFICATION SYSTEMS
Of historical significance is a classification in congenital aural atresia developed in 1955 by Altmann.51 In this system, atresias are categorized into three groups, as follows:
The De la Cruz classification includes surgical feasibility guidelines using only high-resolution computed tomography (CT), taking into consideration mastoid pneumatization, inner ear normality, and facial nerve and footplate relationship.6 The malformations are divided into minor and major malformations (Table 5-2). The clinical importance of this classification is that surgery in cases of minor malformations has a good possibility of yielding serviceable hearing, whereas cases of major malformations are frequently inoperable, but treatable with the bone-anchored hearing aid (BAHA) system.3
TABLE 5-2 De la Cruz Classification of Congenital Aural Atresia
| Minor Malformations |
| 1. Normal mastoid pneumatization |
| 2. Normal oval window/footplate |
| 3. Good facial nerve–footplate relationship |
| 4. Normal inner ear |
| Major Malformations |
| 1. Poor pneumatization |
| 2. Abnormal or absent oval window/footplate |
| 3. Abnormal course of facial nerve |
| 4. Abnormalities of inner ear |
From De la Cruz A, Linthicum FH Jr, Luxford WM: Congenital atresia of the external auditory canal. Laryngoscope 95:421-427, 1985.
Jahrsdoerfer and colleagues53 developed a widely used point grading system to guide surgeons in preoperative assessment of the best candidates for hearing improvement (Table 5-3). This system takes into account the parameters of mastoid pneumatization, presence of the oval and round windows, facial nerve course, status of the ossicles, and external appearance. Point allocation is based primarily on the findings on high-resolution CT. Jahrsdoerfer and colleagues53 proposed that when the preoperative evaluation of the patient is itemized into this grading system, the best results (>80% success) are achieved with a score of 8 or better. A score of 7 indicates the patient is a fair candidate, a score of 6 indicates the patient is a marginal candidate, and with a score less than 5 the patient becomes a poor candidate.
TABLE 5-3 Jahrsdoerfer’s Grading System of Candidacy for Surgery of Congenital Aural Atresia
| Parameter | Points |
|---|---|
| Stapes present | 2 |
| Oval window open | 1 |
| Middle ear space | 1 |
| Facial nerve normal | 1 |
| Malleus-incus complex present | 1 |
| Mastoid well pneumatized | 1 |
| Incus-stapes connection | 1 |
| Round window normal | 1 |
| Appearance of external ear | 1 |
| Total available points | 10 |
| Rating | Type of Candidate |
| 10 | Excellent |
| 9 | Very good |
| 8 | Good |
| 7 | Fair |
| 6 | Marginal |
| ≤5 | Poor |
From Jahrsdoerfer RA, Yeakley JW, Aguilar EA, et al: Grading system for the selection of patients with congenital aural atresia. Am J Otol 13:6-12, 1992.
Ishimoto and associates54 evaluated the relationship between hearing level and temporal bone abnormalities in patients with microtia, using Jahrsdoerfer’s CT scoring system and high-resolution CT scans of the temporal bone. They found that the hearing level in microtic ears correlated with the formation of oval/round windows and ossicular development, but not with the degree of middle ear aeration, facial nerve aberration, or severity of microtia.
Schuknecht’s55 system of classification of congenital aural atresia is based on a combination of clinical and surgical observations. Type A (meatal) atresia is limited to the fibrocartilaginous part of the EAC. Meatoplasty is the surgical procedure of choice, and when performed in a timely fashion prevents formation of canal cholesteatoma and conductive hearing loss. In type B (partial) atresia, there is narrowing of the fibrocartilaginous and bony EAC, but a patent dermal tract allows partial inspection of the tympanic membrane. The tympanic membrane is small and partly replaced by a bony septum. Minor ossicular malformations exist, and hearing loss may be mild to severe. Type C (total) atresia includes all cases with a totally atretic ear canal, but a well-pneumatized tympanic cavity. There is a partial or total bony atretic plate, the tympanic membrane is absent, the heads of the ossicles are fused, there may be no connection to a possibly malformed stapes, and the facial nerve is more likely to have an aberrant course over the oval window. Type D (hypopneumatic total) atresia is a total atresia with poor pneumatization, common in dysplasias such as Treacher Collins syndrome. There are abnormalities of the facial nerve canal and the bony labyrinth. These patients are poor candidates for hearing improvement surgery.
Chiossone’s56 classification is based primarily on the location of the glenoid fossa. In type I, the fossa is in the normal position; in type II, it is moderately displaced; in type III, the fossa overlaps the middle ear; and in type IV, in addition to the fossa overlapping the middle ear, there is lack of mastoid pneumatization. Patients with types I and II are ideal surgical candidates. Type III cases have a tendency toward graft lateralization. Patients with type IV are not surgical candidates. In atresiaplasty surgery, a classification scheme is useful for surgical planning, patient counseling, and comparison of outcomes.
INITIAL EVALUATION AND PATIENT SELECTION
When aural atresia is noted in a newborn, several issues must be addressed. Where one congenital abnormality is found, others must be sought. A high-risk registry for hearing loss is helpful in this regard.57 After the degree of aural deformity is assessed by physical examination, evaluation of auditory function in unilateral and bilateral atresia should be performed using auditory brainstem response audiometry within the first few days of life. An 11% to 47% incidence of inner ear abnormality is associated with congenital aural atresia.12 Occasionally, in unilateral cases, there is a total sensorineural hearing loss (SNHL) on the side of the normal-appearing ear, which might otherwise be missed.6,33
In bilateral cases, a bone-conduction hearing aid should be applied as soon as possible, ideally in the third or fourth week of life. In unilateral cases in which the opposite ear hears normally, a hearing aid is unnecessary. A child with aural atresia and associated cephalic abnormalities (e.g., hemifacial microsomia and Treacher Collins, Crouzon, or Pierre Robin syndrome) should be recognized.57–61 In this subset, surgical correction has poor results,55 and a long-term bone-conduction hearing aid or BAHA in these nonoperable bilateral congenital aural atresia situations is beneficial (see later).3
A patient with congenital aural atresia may present with an infected or draining ear or acute facial palsy; 14% have congenital cholesteatoma.6 The priority in these cases is removal of the cholesteatoma and resolution of the infection. Preoperative audiometry and high-resolution CT scanning may be necessary at an earlier age in children with repeated infections in the atretic ear.6,55,62
There are two requirements for planning surgery in congenital aural atresia: radiographic three-dimensional evaluation of the temporal bone and audiometric evidence of cochlear function.8,63 Other conditions mandating prompt surgical intervention are congenital cholesteatoma, a draining postoperative atretic ear, and acute facial palsy. The CT scan should always be reviewed for cholesteatoma, which necessitates surgery at any age.55,62,64 It is not included in any of the grading systems because these are used only for predicting hearing results in elective atresiaplasty surgery.
TIMING OF AURICULAR RECONSTRUCTION AND ATRESIAPLASTY
In bilateral or unilateral atresia, auricular reconstruction and atresiaplasty are recommended at 6 years of age. Before this age, there may be a tendency to form exostosis-like bony growth that may occlude the EAC, and there is less patient cooperation. By age 6, the costal cartilage has developed sufficiently to allow for reconstruction of the auricle, and the mastoid has become as pneumatized as possible. The microtia repair should be done first because the complex flaps and use of autologous rib graft demand excellent blood supply.65,66 Many surgeons do not perform cartilage microtia reconstruction on an ear with previous reconstruction attempts.64 The hearing restoration surgery is performed 2 months after the last step of the microtia repair. Rehabilitation of auricular defects can be done using an osseointegrated percutaneous mastoid implant prosthesis, with and without bone-conduction aids, such as BAHA.3,48,67–69 Alloplastic materials have been used for microtia repair in the past, but there is a high risk of extrusion associated with their use. Newer materials, such as Medpor, seem to be very well tolerated, however.70–72 As noted earlier, Medpor reconstruction can be done before or after atresiaplasty because intact blood supply is not indispensable.
In unilateral cases, atresiaplasty surgery may be indicated in a patient with “minor” unilateral atresia with normal middle ear, ossicles, and facial nerve, and excellent pneumatization. In such patients, atresiaplasty may be offered in childhood with the parents’ consent.33 We often see older adults with unilateral atresia who request surgery when their normal ear begins having high-frequency hearing loss (presbycusis).
Bone-Anchored Hearing Appliances
For patients with bilateral hearing loss, BICROS (Bilateral Contralateral Routing of Signal) hearing aid is an additional microphone that can be used as an accessory to an implanted BAHA fixture and sound processor to overcome the head shadow effect. The accessory microphone is placed in the contralateral ear to the BAHA fixture. The signal is routed from the accessory microphone to the BAHA sound processor via a wire worn behind the neck. It is intended to improve hearing by eliminating the head shadow effect, but it does so with little success and is not well accepted by patients.73
We do not generally recommend implantable hearing aids in children because the surgical scars may preclude any future microtia repair. However, the BAHA titanium implant is ideally placed 5 to 6 cm behind and 3 cm above the ear canal in a hair-bearing area. This placement seems to allow for the possibility of transplanting costal cartilage to an area with unscarred skin for future microtia repair. On the other hand the titanium implants for a bone-anchored ear epithesis are ideally placed 18 to 20 mm behind the (future) ear canal; this interferes with the skin of a future auricle, if reconstruction is contemplated.73
Numerous studies have reviewed results with BAHA in atresia patients.3,74–77 In one series, 45 atresiaplasty surgeries were compared with 39 BAHA surgeries.3 Of 44 ears followed for more than 2 years after atresiaplasty, hearing gain was less than 10 dB in 55% and 10 to 30 dB in 43%. Surgical outcome correlated with Altmann classification: the worse the Altmann classification stage, the worse the surgical outcome. Within 5 years, 24 ears were reoperated on. Of the 39 patients supplied with BAHA, 16 had had prior atresiaplasty work. All BAHA patients in the series considered the implant to be superior to conventional bone-conduction hearing aids, and superior to hearing improvement obtained surgically, for the difficult cases for whom atresiaplasty had been unsuccessful. Van der Pouw and colleagues74 described experience with bilateral BAHA in four patients with bilateral inoperable congenital aural atresia, three of whom had Treacher Collins syndrome. Bilateral BAHA application resulted in improved performance in all tested audiologic parameters, including sound localization, speech recognition in quiet, speech recognition in noise, and a cued listening task.
Kunst and associates75 reported on the audiologic outcome of BAHA in 20 patients with congenital unilateral conductive hearing impairment, with a mean air-bone gap of 50 dB. Small, although not statistically significant, improvements in localization scores were observed in favor of BAHA. Some patients with congenital unilateral conductive hearing impairment had such good directional hearing and speech-in-noise scores in the unaided situation that no overall significant improvement occurred after BAHA fitting. Compliance with BAHA use was remarkably high, however, suggesting patient perceived benefit.
House and Kutz76 reported the incidence of complications associated with implantation of BAHA in 149 patients and the management of these complications. There were no intraoperative or perioperative complications. Significant postoperative complications requiring intervention occurred in 12.8% of patients. Skin overgrowing the abutment occurred in 7.4%, and all but one of these patients required revision in the operating room. Skin overgrowth was a late complication, occurring an average of 12 months (range 3 months to 2 years) after the initial procedure. Implant extrusion from failure to osseointegrate occurred in 3.4%. Two patients had local wound infections requiring oral antibiotics.
One study evaluated hearing results in pediatric patients managed with EAC reconstruction or BAHA.77 The investigators also evaluated the medical cost-effectiveness of each procedure. They reviewed 36 ears that underwent surgical canal reconstruction and 6 patients who underwent BAHA placement. Most (93%) of the patients undergoing EAC reconstruction required some form of amplification postoperatively. The investigators concluded that BAHA can achieve acceptable hearing (<15 dB) in school-aged children with normal bone curves, and it can match the bone curves for children with SNHL. The two-staged BAHA system placement may be provided at almost one third the cost to the medical system of surgical EAC reconstruction, on a decibel-for-decibel basis, with single-stage placement yielding even greater cost savings. Implantable hearing appliances seem to offer a good alternative for patients with inoperable atresia and for patients in whom the operative prognosis is poor.
PREOPERATIVE EVALUATION AND PATIENT COUNSELING
Early diagnosis with auditory brainstem response and auditory enhancement with bone-conduction hearing aids in bilateral cases should be done in the first weeks of life.78 Corrective surgery begins at 6 years of age. Imaging is deferred until age 6. Microtia repair is performed before atresiaplasty. The size of the mastoid on physical examination can be estimated by palpation of the mastoid tip, suprameatal spine of Henle (if present), condyle, and zygomatic arch. This is a useful measure because the new ear canal is constructed at the expense of the mastoid air-cell system.
The preoperative imaging study is a high-resolution CT scan of the temporal bone in coronal and axial planes (Fig. 5-1).5,12,63,79 The CT scan must be examined carefully by the otologic surgeon together with the otoradiologist.41,80 The four important imaging elements that are most helpful to the surgeon planning reconstruction of a congenitally malformed ear are (1) the degree of pneumatization of the temporal bone; (2) the course of the facial nerve, including the relationship of the horizontal portion to the footplate, and the location of the vertical segment; (3) the presence of the oval window and stapes footplate; and (4) the status of the inner ear.5,6,63 CT also provides information on thickness and form of the atretic bone, size and status of the middle ear cavity, and soft tissue contribution to the atresia,6 but these are less critical for the repair.
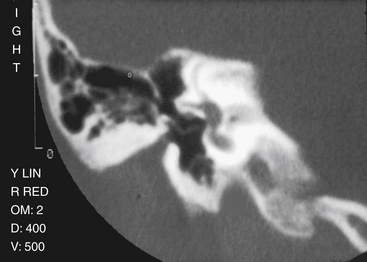
Lack of pneumatization is the major cause of inoperability in congenital aural atresia.6,33 Normal pneumatization is present in most cases. The facial nerve over the oval window may prevent ossiculoplasty and hearing improvement. If the oval window or footplate are absent, surgery with fenestration of the lateral semicircular canal or placement of a hearing aid is indicated. There is potential for facial nerve injury in atresia surgery.81 The nerve may describe a more acute angle rather than its usual 90 to 120 degrees at the mastoid genu, and often lies more lateral than usual (Fig. 5-2).82 On high-resolution CT, it is important not to “identify” mistakenly the vertical lie of the facial nerve in the marrow bone leading to the styloid process and the hypoplastic mastoid process (Fig. 5-3).83 Even in atretic ears in which the facial nerve does not have an abnormal course, a significantly reduced distance is found between the facial canal and the temporomandibular joint,41 and the facial canal and the posterior wall of the cavum tympani.47
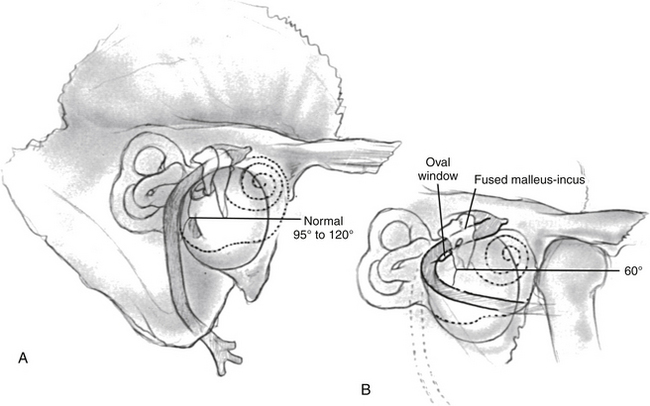
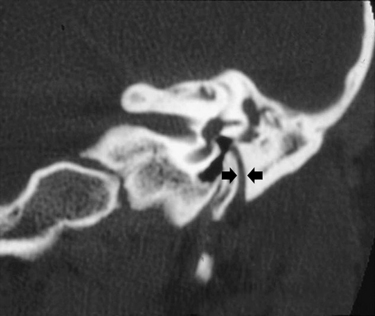
To be considered a surgical candidate, the patient must have sufficient cochlear function as determined by auditory brainstem response or routine audiometry; a normal-appearing inner ear on CT scan; and, preferably, a well-developed mastoid, and good oval window/footplate and facial nerve relationship. The patient and parents are counseled regarding the success of atresiaplasty repair and the chances of successful hearing improvement. Patients with a degree of malformation equivalent to an 8 or better on the Jahrsdoerfer grading scale are given a greater than 80% chance of hearing improvement. The risk to the facial nerve is small, made more so by the use of the facial nerve monitor intraoperatively.33,82 Patients are informed that a split-thickness skin graft (STSG) from the hypogastrium is used to line the new EAC. Initially, frequent postoperative visits are necessary. The possibilities of a facial nerve paralysis (<1%), profound SNHL (approximately 7%, no dead ears), postoperative canal stenosis (<4%), tympanic membrane graft lateralization (<4%), and ossicular chain refixation (<4%) are noted.84,85
SURGICAL APPROACH
Early atresiaplasty operations failed because of poor tympanoplasty techniques, large mastoidectomies, and faulty skin grafting techniques.18 Advances in meatoplasty added to the success rate of atresiaplasty surgery, however.19,86 Although fenestration remains a rare option in bilateral congenital absence of the oval window, modern methods of ossiculoplasty are used preferentially and yield superior results.6,8,19,23,26,28,31,34-36,87-89
A postauricular temporo-occipital incision is made. In cases in which microtia repair has been done, care is taken not to expose the costal cartilage graft or alloplastic material used in the auricular reconstruction. Subcutaneous tissue is elevated anteriorly to the temporomandibular joint. An incision is made in the periosteum, which is elevated forward exposing the mastoid cortex and, anteriorly, the temporomandibular joint space (Fig. 5-4). Care is taken to avoid injury to an anomalous facial nerve exiting the temporal bone in this area. Temporalis fascia is harvested, trimmed of excess soft tissue if needed, and placed aside to dry. The temporomandibular joint space is explored to verify that the facial nerve or tympanic bone is not lying within it.
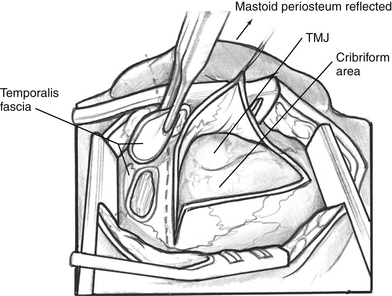
The literature has fostered the belief that there are separate and distinct approaches to the bony work necessary in atresiaplasty: the anterior approach, the transmastoid approach, and modification of the anterior approach.6,8,24,33,35,57,64 We believe that strict distinction between these surgical approaches is unnecessary because each can be used alone or in combination to facilitate the atresiaplasty.
Surgical Technique
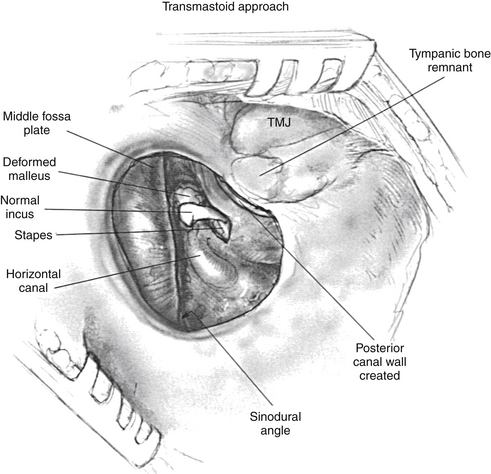
If a remnant of tympanic bone is present, drilling for the new ear canal begins at the cribriform area. If no such remnant exists, drilling of a 12 mm cylindrical-shaped canal begins at the level of the linea temporalis, just posterior to the glenoid fossa. Dissection proceeds anteriorly and medially, keeping in mind the lack of landmarks in atretic bone. The middle fossa plate is identified and followed to the epitympanum, where the fused malleus head/incus body mass is identified (Fig. 5-5). Care is taken to avoid exposure of the temporomandibular joint space or to avoid opening an excessive number of mastoid air cells. A radical mastoidectomy–like approach should be avoided. When the ossicular mass is identified, the atretic bone is removed with diamond microdrills and curettes to expose the ossicles completely (Fig. 5-6A).
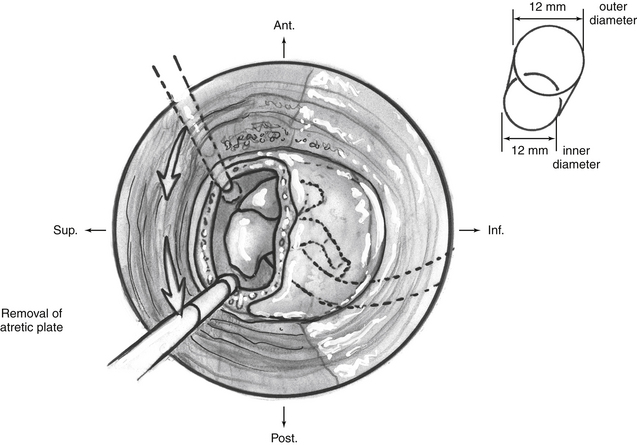
FIGURE 5-5 Removal of atretic plate and adhesions, exposing the ossicles, and drilling for the new ear canal.
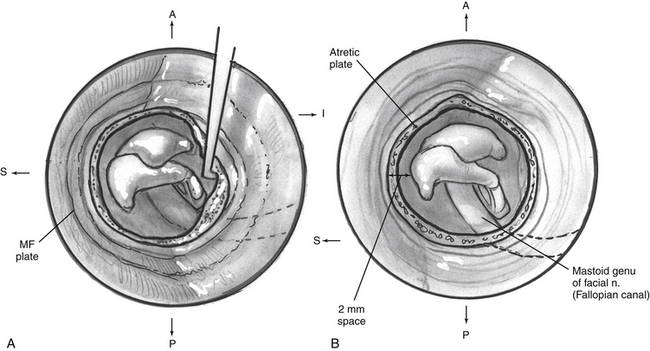
It is important to minimize drilling on the ossicular mass because transmission of high-speed drill energy to the inner ear may result in high-tone SNHL. An argon laser is used to free the malleus-incus complex from its soft tissue attachments to reduce potential drill trauma to the inner ear. The ossicular mass in the epitympanum is meticulously dissected free of the atresia plate and left intact. Although the temporomandibular joint often limits anterior dissection, particular effort is made to try to create a space of 2 mm or greater, anterior to the ossicular mass (Fig. 5-6B). The horizontal facial nerve always lies medial to the ossicular mass. While dissecting the inferior and posterior aspect of the canal, an aberrant facial nerve may be encountered as it passes laterally through the atretic bone. Drilling for the new ear canal continues until it measures about 12 mm. This circumference is most difficult to achieve medially, where the facial nerve, temporomandibular joint capsule, and middle fossa plate limit larger exposure.
Reconstruction with the patient’s own ossicular chain is performed in almost all cases, even if the ossicular chain is deformed. If the ossicular chain is intact, it is left in place and used for ossicular reconstruction. When this is impossible, a total or partial ossicular reconstruction prosthesis to either a mobile footplate or the stapes head is used for reconstruction. The prosthesis is covered with cartilage before grafting the new drum. Occasionally, the stapes footplate may not be seen well because of anomalous facial nerve anatomy, making placement of an ossicular prosthesis difficult or dangerous. In these instances, gentle transposition of the facial nerve has been described, but more commonly hearing restoration can be accomplished with lateral semicircular canal fenestration or the patient can be counseled to use a hearing aid only.4,8,17,26,31,36,42
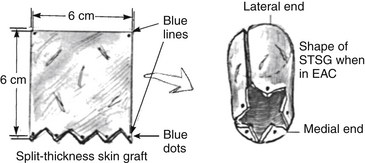
A dermatome is used to obtain a 0.008-inch thick, 6 × 6 cm STSG from the hypogastrium. Pressure with a gauze sponge soaked in 1% lidocaine with epinephrine 1:100,000 and thrombin solution aids hemostasis of the donor site. When dry, the donor site is dressed with a sterile Tegaderm dressing.90 This dressing reduces most of the pain associated with other methods of donor site care. One edge of the skin graft is cut in a zigzag fashion to create four or five triangular points (Fig. 5-7). To assist inspection of the skin graft in the final stages of the procedure, the tips of each point and the two corners on the opposite edge are colored with a skin marker.
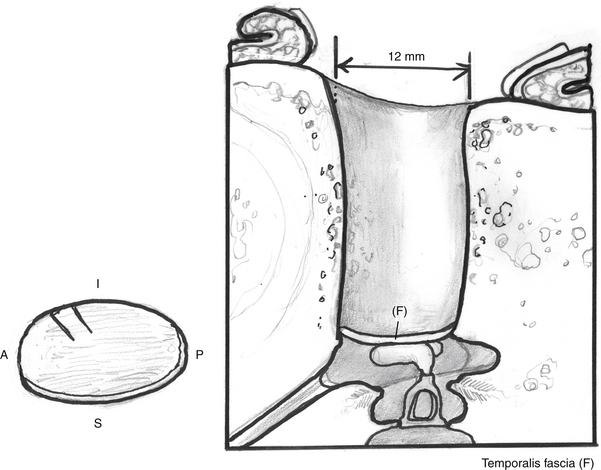
The dried temporalis fascia is trimmed to size, ideally a 20 × 15 mm oval, and a small 4 mm “tab” is cut into the anterior-inferior aspect of the graft in an attempt to prevent lateralization.91 Nitrous oxide is discontinued 30 minutes before grafting begins. The fascia is placed over the ossicular chain medial to the malleus, if present, or over the cartilage that covers the prosthesis (Fig. 5-8). The tab of the graft is placed medially into the protympanum to help prevent lateralization of the new eardrum.
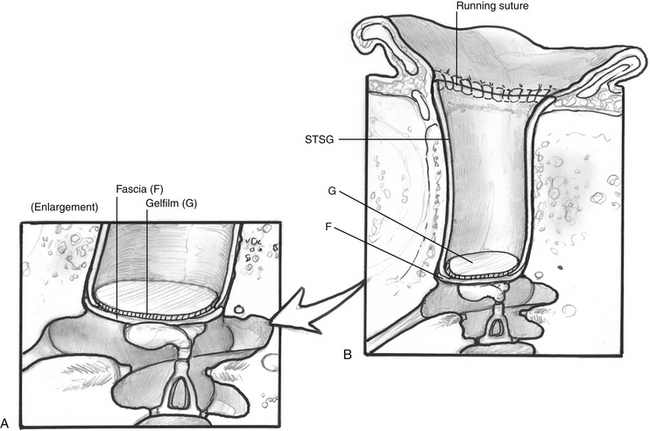
Next, the new ear canal is circumferentially lined with the STSG so that all the bone is completely covered (Fig. 5-9). The triangular corners of the skin graft are placed medially and partially overlap the fascia. The colored points help ensure that no skin lies folded on itself, and that the entire width of the graft is used. A single layer of antibiotic-soaked absorbable gelatin sponge (Gelfoam) holds the fascia and skin graft in place. To reproduce the anterior tympanomeatal angle, a disk of absorbable gelatin film (Gelfilm) is placed over the fascia and skin graft. Lateralization is the most common delayed cause of a poor hearing outcome, usually occurring in the first 12 months after surgery. When the graft starts lateralizing, it pulls away from the ossicles, and the original good hearing results slowly deteriorate. After the fascia and skin graft are in proper position, three 0.020 inch silicone elastomer (Silastic) strips are placed to line the skin (Fig. 5-10). The Silastic strips help prevent adhesions between the skin graft and the ear canal packing, and ensure good contact between the skin graft and ear canal bone. A 9 × 15 mm Merocel wick is placed in the bony canal.
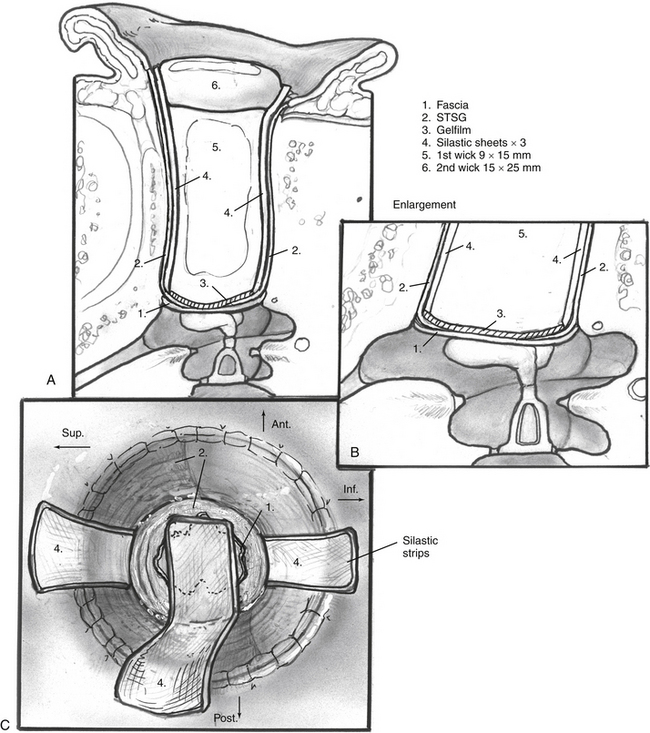
A 12 mm meatus is created, anticipating that a third of its diameter will reduce because of normal healing. Before starting the meatoplasty, the ear is turned back, and the periosteum is brought back in place with one absorbable suture at the superior level of the new ear canal; this stabilizes the pinna and helps ensure that the meatus is at the same level as the new ear canal. Skin, subcutaneous tissue, and cartilage are removed in a 12 mm diameter core over the new meatus. An attempt is made to avoid exposing the grafted cartilage or other materials used for auricular reconstruction. The lateral edge of the STSG is brought through the meatoplasty, and the lateral portion of the new ear canal and the meatus are packed with a large 15 × 25 mm Ambrus wick (Ambrus Merocel, Medtronic Xomed Surgical Products, Jackonsiville, Florida) this applies diffuse pressure over the entire lateral skin graft and widely packs the meatoplasty. Five tacking sutures of 5-0 braided polyester (Ticron) attach the lateral edge of the skin graft circumferentially to the meatal skin. Finally, 6-0 fast-absorbing plain gut is used in a running manner between each Ticron suture (Fig. 5-11).
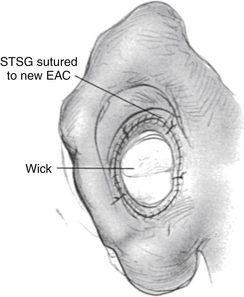
FIGURE 5-11 Skin graft sutured into place at meatus. EAC, external auditory canal; STSG, split-thickness skin graft.
Silastic sheets and Merocel wicks have helped decrease the incidence of tympanic membrane lateralization by maintaining the tympanic membrane graft in position and the incidence of postoperative EAC stenosis by covering all exposed areas of bone that may cause granulation tissue formation.85 The Merocel wicks, which are now available in many sizes for canal stenting, help prevent postoperative infection and granulation tissue by allowing administration of topical antibiotics to the entire EAC.
Transmastoid Approach (Canal Wall Down Technique)
The transmastoid approach is of historical interest only (Fig. 5-12). It has not been used since 1975. In this approach, the drilling starts as posterior as possible from the temporomandibular joint. It starts at the linea temporalis and follows the sinodural angle to the ossicular mass. Mastoid air cell exenteration and lowering of the facial ridge to the facial nerve allows the creation of an EAC with a canal wall down technique. Bone pâté and soft tissue obliteration of the large mastoid cells is performed before undertaking meatoplasty and canal skin grafting. The transmastoid approach resulted in a large mastoid cavity with frequent postoperative drainage, and patients were not allowed to swim.
POSTOPERATIVE CARE
The mastoid dressing is removed on the first postoperative day. The Steri-Strips are left in place over the postauricular incision for 7 days. The patient is counseled to keep the operative site dry and to change the cotton ball over the canal/meatus packing four times daily. The Tegaderm over the skin graft donor site is left on for at least 3 weeks and requires no special attention. Epithelialization occurs under the plastic, and the typical pain associated with older methods of donor site dressing is absent.90 The patient is given a 5-day course of oral antibiotics.
RESULTS
Numerous publications have described atresiaplasty results in more than 500 patients from our institution over time.6,33,84,85,92 Our most recent reviews were published in 2003 and 2004 and included 116 atretic ears operated at the House Ear Clinic.84,85 Closure of the air-bone gap to 30 dB at short-term (<6 months) follow-up occurred in 58.5% of primary surgeries and 56% of revisions.84 The long-term (≥6 months) postoperative air-bone gap was 30 dB or less in 50.8% and 39.1% respectively. There was no significant change in air-bone gap from short-term to long-term follow-up for either primary or revision cases. Soft tissue stenosis occurred in 8% of primary surgeries and 3.4% of revisions, and tympanic membrane lateralization occurred in 3.4% and 3.4% respectively, an improvement from the previous series reported in 1995.
By 1994, all the modifications used in current practice (use of argon laser, thinner STSG, Silastic sheets in the EAC, and Merocel wicks) were routinely being employed. On this basis, the patients were divided into two groups: old cases performed before the changes in surgical technique (n = 36), and new cases that included surgeries performed after these changes were introduced (n = 80).85 We compared complication rates and hearing results before and after the introduction of the new techniques. Overall, the new group had better hearing results and lower rates of complications. Closure of the air-bone gap to 30 dB or less at short-term follow-up occurred in 63.1% of surgeries performed after modifications in the surgical technique and 44.5% of surgeries performed before the modifications. Soft tissue stenosis and bony growth of the EAC were seen in 3.8% of surgeries performed after and 13.9% of surgeries performed before the surgical technique changes. Tympanic membrane lateralization occurred in none of the cases performed after and in 11.1% of the surgeries performed before the surgical technique changes. Ossicular chain refixation occurred in 3.8% of surgeries performed after and 25% of surgeries performed before the modifications. There were no dead ears and no facial palsies in either group.
Shih and Crabtree93 reviewed long-term surgical results for 39 ears. Hearing averages of 25 dB, 40 dB, and 46 dB were achieved for mild, moderate, and severe atresia. Restenosis occurred in 33%, and 31% had recurrent cavity/canal skin infections. This rate was reduced with the use of STSG.
Digoy and Cueva94 reviewed their short-term (<1 year) and long-term (>1 year) surgical and hearing results for congenital aural atresia in 45 patients (54 ears). Approximately 50% of their patients achieved a speech reception threshold of 30 dB or better in the short term and long term. The average improvement in air-bone gap was 22 dB. Short-term and long-term outcomes were not significantly different. Patients with an intact ossicular chain did not seem to have a significant advantage in hearing compared with patients with a reconstruction prosthesis. They reported meatal stenosis in 7% and tympanic membrane lateralization in 18% of the cases.
COMPLICATIONS AND THEIR MANAGEMENT
Complications of atresiaplasty at our institution for cases operated with the most current technique include lateralization of the tympanic membrane in 3.4%, stenosis of the meatus in 3.8%, high-tone SNHL in 7.5%, and facial nerve palsy in less than 1%.84,85 Other authors have reported meatal stenosis in 7% and tympanic membrane lateralization in 18% of their cases.94
The incidence of stenosis has been greatly reduced with the use of one-piece STSG covering all exposed bone, and Silastic sheets and Merocel wicks in the EAC.85 Local care and prevention of infection avoid graft failure and early restenosis. Careful inspection of the meatus and early restenting with large Merocel wicks can obviate reoperation.
Bony EAC stenosis with a “bumpy” exostosis-like appearance tends to occur more frequently in young patients84; this is probably due to active bone growth at this age, resulting in an exuberant bone regrowth in the newly created EAC. This problem seems to be greatly underreported in the literature.
UNILATERAL ATRETIC EAR
Controversy remains over whether children with unilateral atresia should undergo surgery. Jahrsdoerfer8 and De la Cruz and coworkers6 have argued for atresia surgery in selected patients with unilateral atresia. Other authors support this position.95 In the past, Schuknecht,96 Crabtree,28 and Bellucci52 recommended against operating on children with unilateral atresia. These authors argued that the benefit to be gained is minimal in the presence of a contralateral normal-hearing ear. Hearing results at that time were unpredictable and often did not approach the 20 dB air-bone gap needed for useful hearing in the atretic ear. Risks of surgery, including facial nerve injury, also precluded operating on the unilateral atretic ear. In a review of more than 1000 operations for aural atresia with and without cholesteatoma, however, Jahrsdoerfer and Lambert97 showed that the risk was minimal (1%).
The incidence of major complications (total SNHL and facial nerve injury) and of other complications (tympanic membrane graft lateralization and restenosis) has decreased over the years, but these are still potential complications. The decision to operate on the unilateral atretic ear must weigh these potential complications along with the possibility of a draining ear. Nevertheless, with excellent preoperative imaging, improved surgical techniques, and advances in technology, we believe the results of atresia surgery are now more predictable. Closure of the air-bone gap to within 30 dB in a properly selected patient can be consistently achieved, and hearing remains stable over the length of follow-up.84 A review examining long-term stability of hearing results in patients operated on for aural atresia does show some drop-off in hearing thresholds (speech reception threshold) over time, however.84,85,98 In the hands of an experienced otologic surgeon with an anatomically favorable patient who (with the parents) understands the risks of potential complications and the need for postoperative care, atresiaplasty in the patient with unilateral atresia is a rewarding operation for the surgeon and the patient. Surgical correction of unilateral atresia offers the benefits of a clean, dry ear with binaural hearing, including sound localization and improved hearing in noise.
1. Federspil P., Delb W. Treatment of congenital malformations of the external and middle ear. In: Ars B., editor. Congenital External and Middle Ear Malformations: Management. Amsterdam: Kugler Publications; 1992:47-70.
2. Gill N.W. Congenital atresia of the ear: A review of the surgical findings in 83 cases. J Laryngol Otol. 1969;83:551-587.
3. Granstrom G., Bergstrom K., Tjellstrom A. The bone-anchored hearing aid and bone-anchored epithesis for congenital ear malformations. Otolaryngol Head Neck Surg. 1993;109:46-53.
4. House H.P. Management of congenital ear canal atresia. Laryngoscope. 1953;63:916-946.
5. Mehra Y.N., Dubey S.P., Mann S.B.S., Suri S. Correlation between high resolution computed tomography and surgical findings in congenital aural atresia. Arch Otolaryngol Head Neck Surg. 1988;114:137-141.
6. De la Cruz A., Linthicum F.H.Jr., Luxford W.M. Congenital atresia of the external auditory canal. Laryngoscope. 1985;95:421-427.
7. Hiraide F., Nomura Y., Nakamura K. Histopathology of atresia auris congenita. J Laryngol Otol. 1974;88:1249-1256.
8. Jahrsdoerfer R.A. Congenital atresia of the ear. Laryngoscope. 1978;88(Suppl 13):1-48.
9. Kelemen G.D. Aural participation in congenital malformations of the organism. Acta Otolaryngol Suppl. 1974;321:1-35.
10. Grundfast K.M., Camilon F. External auditory canal stenosis and partial atresia without associated anomalies. Ann Otol Rhinol Laryngol. 1986;95:505-509.
11. Harada O., Ishii H. The condition of the auditory ossicles in microtia: Findings in 57 middle ear operations. Plast Reconstr Surg. 1972;50:48-53.
12. Hasso A.N., Broadwell R.A. Congenital anomalies. In: Som P.M., Bergeron R.T., editors. Head and Neck Imaging. St Louis: Mosby; 1991:960-966.
13. Lascaratos J., Assimakopoulos D. From the roots of otology: Diseases of the ear and their treatment in Byzantine times (324-1453 ad). Am J Otol. 1999;20:397-402.
14. Briau R. Chirurgie de Paul d’Egine. Paris: Masson; 1855.
15. Page J.R. Congenital bilateral microtia with total osseous atresia of the external auditory canals: Operation and report of cases. Trans Am Otol Soc. 1914;13:376-390.
16. Dean L.W., Gittens T.R. Report of a case of bilateral, congenital osseous atresia of the external auditory canal with an exceptionally good functional result following operation. Trans Am Laryngol Rhinol Otol Soc. 1917;23:296-309.
17. Ombredanne M. Chirurgie de la surdité: Fenestration dans les aplasies de l’oreille avec imperforation du conduit: Resultats. Otorhinolaryngol Int. 1947;31:229-236.
18. Pattee G.L. An operation to improve hearing in cases of congenital atresia of the external auditory meatus. Arch Otolaryngol Head Neck Surg. 1947;45:568-580.
19. Bellucci R.J. The problem of congenital auricular malformation, I: Construction of the external auditory canal. Trans Am Acad Ophthalmol Otolaryngol. 1960;64:840-852.
20. Meurman Y. Congenital microtia and meatal atresia. Arch Otolaryngol. 1957;66:443-463.
21. Nager G.T. Aural atresia: Anatomy and surgery. Postgrad Med. 1961;29:529-541.
22. Ombredanne M. Malformations des osselets dans les embryopathies de l’oreille. Acta Otorhinolaryngol (Belg). 1965;20:623-652.
23. Ruedi L. The surgical treatment of the atresia auris congenita: A clinical and histological report. Laryngoscope. 1954;64:666-684.
24. Scheer A.A. Correction of congenital middle ear deformities. Arch Otolaryngol. 1967;85:269-277.
25. Shambaugh G.E.Jr. Developmental anomalies of the sound conducting apparatus and their surgical correction. Ann Otol. 1952;74:873-887.
26. Woodman D.G. Congenital atresia of the auditory canal. Arch Otolaryngol. 1952;55:172-181.
27. Ombredanne M. Chirurgie des surdites congenitales par malformations ossiculaires. Acta Otorhinolaryngol Belg. 1971;25:837-869.
28. Crabtree J.A. Congenital atresia: Case selection, complications, and prevention. Otolaryngol Clin North Am. 1982;15:755-762.
29. Jahrsdoerfer R.A., Cole R.R., Gray L.E. Advances in congenital aural atresia. Adv Otolaryngol Head Neck Surg. 1991;5:1-15.
30. Jahrsdoerfer R.A. Clinical aspects of temporal bone anomalies. AJNR Am J Neuroradiol. 1992;13:821-825.
31. Marquet J. Homogreffes tympano-ossiculaires dans le traitement chirurgical de l’agenesie de woreille: Rapport preliminaire. Acta Otorhinolaryngol Belg. 1971;25:885-897.
32. Marquet J.E., Declau F., De Cock M., et al. Congenital middle ear malformations. Acta Otorhinolaryngol Belg. 1988;42:117-302.
33. Molony T.B., De la Cruz A. Surgical approaches to congenital atresia of the external auditory canal. Otolaryngol Head Neck Surg. 1990;103:991-1001.
34. Crabtree J.A. Tympanoplastic techniques in congenital atresia. Arch Otolaryngol. 1968;88:89-96.
35. Minatogawa T., Nishimura Y., Inamori T., Kumoi T. Results of tympanoplasty for congenital aural atresia and stenosis, with special reference to fascia and homograft as the graft material of the tympanic membrane. Laryngoscope. 1989;99:632-638.
36. Schuknecht H.F. Reconstructive procedures for congenital aural atresia. Arch Otolaryngol. 1975;101:170-172.
37. Bellucci R.J. Congenital auricular malformations: Indications, contraindications, and timing of middle ear surgery. Ann Otol Rhinol Laryngol. 1972;81:659-663.
38. Linthicum F.H.Jr. Surgery of congenital deafness. Otolaryngol Clin North Am. 1971;4:401-409.
39. Patterson M.E., Linthicum F.H.Jr. Congenital hearing impairment. Otolaryngol Clin North Am. 1970;3:201-219.
40. Ruben R.J. Management and therapy of congenital malformations of the external and middle ears. In: Alberti P.W., Ruben R.J., editors. Otologic Medicine and Surgery. New York: Churchill Livingstone; 1988:1135-1154.
41. Jahrsdoerfer R.A., Garcia E.T., Yeakley J.W., Jacobson J.T. Surface contour three-dimensional imaging in congenital aural atresia. Arch Otolaryngol Head Neck Surg. 1993;119:95-99.
42. Ombredanne M. Absence congenitale de fenetre ronde dans certaines aplasies mineures. Ann Otolaryngol (Paris). 1968;85:369-378.
43. Aase J.M. Microtia: Clinical observations. Birth Defects. 1980;16:289-297.
44. Sando I., Shibahara Y., Takagi A., et al. Congenital middle and inner ear anomalies. Acta Otolaryngol (Stockh). Suppl 458, 1988, 76-78.
45. Van de Water T.R., Maderson P.F., Jaskoll T.F. The morphogenesis of the middle and external ear. Birth Defects. 1980;16:147-180.
46. Barrios-Montes J.M. Malformaciones auriculares. Acta Otorhinolaryngol Esp. 1976;27:17-46.
47. Savic D., Jasovic A., Djeric D. The relations of the mastoid segment of the facial canal to surrounding structures in congenital middle ear malformations. Int J Pediatr Otorhinolaryngol. 1989;18:13-19.
48. Tjellstrom A., Bergstrom K. Bone-anchored hearing aids and prostheses. In: Ars B., editor. Congenital External and Middle Ear Malformations: Management. Amsterdam: Kugler Publications; 1992:1-9.
49. Gill N.W. Congenital atresia of the ear. J Laryngol Otol. 1971;85:1251-1254.
50. Melnick M. The etiology of external ear malformations and its relation to abnormalities of the middle ear, inner ear, and other organ systems. Birth Defects. 1980;16:303-331.
51. Altmann F. Congenital atresia of the ear in man and animals. Ann Otol Rhinol Laryngol. 1955;64:824-858.
52. Bellucci R.J. Congenital aural malformations: Diagnosis and treatment. Otolaryngol Clin North Am. 1981;14:95-124.
53. Jahrsdoerfer R.A., Yeakley J.W., Aguilar E.A., et al. Grading system for the selection of patients with congenital aural atresia. Am J Otol. 1992;13:6-12.
54. Ishimoto S., Ito K., Karino S., et al. Hearing levels in patients with microtia: Correlation with temporal bone malformation. Laryngoscope. 2007;117:461-465.
55. Schuknecht H.F. Congenital aural atresia and congenital middle ear cholesteatoma. In: Nadol J.B.Jr., Schuknecht H.F., editors. Surgery of the Ear and Temporal Bone. New York: Raven Press; 1993:263-274.
56. Chiossone E. Surgical management of major congenital malformations of the ear. Am J Otol. 1985;6:237-242.
57. Sando I., Suehiro S., Wood R.P.II. Congenital anomalies of the external and middle ear. In: Bluestone C.D., Stool S.E., editors. Pediatric Otology. Philadelphia: Saunders; 1983:263-274.
58. Fernandez A.O., Ronis M.L. The Treacher Collins syndrome. Arch Otolaryngol. 1964;80:505-520.
59. Rapin I., Ruben R.J. Patterns of anomalies in children with malformed ears. Laryngoscope. 1976;86:1469-1502.
60. Ruben R.J., Toriyama M., Dische M.R., et al. External and middle ear malformations associated with mandibulo facial dysostosis and renal abnormalities: A case report. Ann Otol Rhinol Laryngol. 1969;78:605-624.
61. Sanchez-Corona J., Garcia-Cruz D., Ruenes R., Cantu J.M. A distinct dominant form of microtia and conductive hearing loss. Birth Defects. 1982;18:211-216.
62. Miyamoto R.T., Fairchild T.H., Daugherty H.S. Primary cholesteatoma in the congenitally atretic ear. Am J Otol. 1984;5:283-285.
63. Jahrsdoerfer R.A., Yeakley J.W., Hall J.W.III, et al. High-resolution CT scanning and auditory brain stem response in congenital aural atresia: Patient selection and surgical correlation. Otolaryngol Head Neck Surg. 1985;93:292-298.
64. Jahrsdoerfer R.A., Hall J.W. Congenital malformations of the ear. Am J Otol. 1986;7:267-269.
65. Brent B. The correction of microtia with autogenous cartilage grafts, I: The classic deformity. Plast Reconstr Surg. 1980;66:1-12.
66. Brent B. Auricular repair with autogenous rib cartilage grafts: Two decades of experience with 600 cases. Plast Reconstr Surg. 1995;90:355-374.
67. Gates G.A., Hough J.V., Gatti W.M., Bradley W.H. The safety and effectiveness of an implanted electromagnetic hearing device. Arch Otolaryngol. 1989;115:924-930.
68. Hakansson B., Liden G., Tjellstrom A., et al. Ten years of experience with the Swedish bone-anchored hearing system. Ann Otol Rhinol Laryngol Suppl. 1990;151:1-16.
69. Niparko J.K., Langman A.W., Cutler D.S., Carroll W.R. Tissue-integrated prostheses in the rehabilitation of auricular defects: Results with percutaneous mastoid implants. Am J Otol. 1993;14:343-348.
70. Naumann A. Plastic surgery to correct deformities of the ear. MMW Fortschr Med. 2005;147:28-31.
71. Romo T.3rd, Presti P.M., Yalamanchili H.R. Medpor alternative for microtia repair. Facial Plast Surg Clin North Am. 2006;14:129-136.
72. Kelley P.E., Scholes M.A. Microtia and congenital aural atresia. Otolaryngol Clin North Am. 2007;40:61-80.
73. Tjellstrom A., Hakansson B. The bone-anchored hearing aid: Design, principles, indications, and long-term clinical results. Otolaryngol Clin North Am. 1995;28:53-72.
74. van der Pouw K.T.M., Snik A.F.M., Cremers C.W.R.J. Audiometric results of bilateral bone-anchored hearing aid application in patients with bilateral congenital aural atresia. Laryngoscope. 1998;108:548-553.
75. Kunst S.J., Leijendeckers J.M., Mylanus E.A., et al. Bone-anchored hearing aid system application for unilateral congenital conductive hearing impairment: audiometric results. Otol Neurotol. 2008;29:2-7.
76. House J.W., Kutz J.W.Jr. Bone-anchored hearing aids: incidence and management of postoperative complications. Otol Neurotol. 2007;28:213-217.
77. Evans A.K., Kazahaya K. Canal atresia: “Surgery or implantable hearing devices? The expert’s question is revisited.”. Int J Pediatr Otorhinolaryngol. 2007;71:367-374.
78. Sortini A.J. Hearing aids for children with bilateral congenital ear canal atresia. Hear Instrument. 1981;6:20-23.
79. Zalzal G.H., Shott S.R., Towbin R., Cotton R.T. Value of CT scan in the diagnosis of temporal bone diseases in children. Laryngoscope. 1986;96:27-32.
80. Andrews J.C., Anzai Y., Mankovich N.J., et al. Three-dimensional CT scan reconstruction for the assessment of congenital aural atresia. Am J Otol. 1992;13:236-240.
81. Crabtree J.A. The facial nerve in congenital ear surgery. Otolaryngol Clin North Am. 1974;7:505-510.
82. Linstrom C.I., Meiteles L.Z. Facial nerve monitoring in surgery for congenital auricular atresia. Laryngoscope. 1993;103:406-415.
83. Chandrasekhar S.S., De La Cruz A., Lo W.W.M., Telischi F.J. Imaging of the facial nerve. In: Jackler R.A., Brackmann D.E., editors. Neurotology. St. Louis: Mosby–Year Book; 1993:341-359.
84. De la Cruz A., Teufert K.B. Congenital aural atresia surgery: Long-term results. Otolaryngol Head Neck Surg. 2003;129:121-127.
85. Teufert K.B., De la Cruz A. Advances in congenital aural atresia surgery: Effects on outcome. Otolaryngol Head Neck Surg. 2004;131:263-270.
86. Chole R.A. Meatoplasty using inferiorly based island pedicle flap for congenital aural atresia. Laryngoscope. 1983;93:954-955.
87. Colman B.H. Congenital malformations of the ear—aspects of management. J Otolaryngol Soc Austral. 1978;4:197-200.
88. Wigand M.E. Tympano-meatoplastie endurale pour les atresies congenitales severes de l’oreille. Rev Laryngol. 1978;99:14-28.
89. Ombredanne M. Transposition des osselets dans certaines “Aplasies Mineures.”. Ann Otolaryngol (Paris). 1966;83:273-280.
90. Weymuller E.A.Jr. Dressings for split-thickness skin graft donor sites. Laryngoscope. 1981;91:652-653.
91. Sheehy J.L. Surgery of chronic otitis media. In: English G.E., editor. Otolaryngology, Vol 1. Hagerstown, MD: Harper & Row, 1977.
92. Chandrasekhar S.S., De la Cruz A., Garrido E. Surgery of congenital aural atresia. Am J Otol. 1995;16:713-717.
93. Shih L., Crabtree J.A. Long-term surgical results for congenital aural atresia. Laryngoscope. 1993;103:1097-1102.
94. Digoy G.P., Cueva R.A. Congenital aural atresia: Review of short- and long-term surgical results. Otol Neurotol. 2007;28:54-60.
95. Trigg D.J., Applebaum E.L. Indications for the surgical repair of unilateral aural atresia in children. Am J Otol. 1998;19:679-684.
96. Schuknecht H.F. Congenital aural atresia. Laryngoscope. 1989;99:908-917.
97. Jahrsdoerfer R.A., Lambert P.R. Facial nerve injury in congenital aural atresia surgery. Am J Otol. 1998;19:283-287.
98. Lambert P.R. Congenital aural atresia: Stability of surgical results. Laryngoscope. 1998;108:1801-1805.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 5 Congenital Malformation of the External Auditory Canal and Middle Ear
Videos corresponding to this chapter are available online at www.expertconsult.com.
Congenital aural atresia is characterized by aplasia or hypoplasia of the external auditory canal (EAC), often associated with absence or deformity of the auricle (microtia) and the middle ear, with occasional inner ear abnormalities. Aural atresia occurs in 1 in 10,000 to 20,000 live births1–5; unilateral atresia is three times more common than bilateral atresia. This disorder occurs more commonly in males and on the right side.1 EAC atresia is more often bony rather than membranous, and bony atresia is regularly accompanied by malformation of the middle ear cavity and structures of the middle ear.6–9 More severe forms of congenital microtia are usually associated with EAC atresia; rarely, canal atresia may be seen in patients with a normal pinna.10 Generally, a more severe external deformity implies a more severe middle ear abnormality.11,12
Kiesselbach, in 1883, is often credited with the first deep operation attempting to correct this malformation.8 Lascaratos and Assimakopoulos13 noted that the Byzantine physician Paul of Aegina performed a surgical treatment of congenital aural atresia.14 The procedure done by Kiesselbach resulted in facial paralysis. Because of the lack of middle ear microsurgery and the high complication rate, congenital aural atresia surgery was considered dangerous and to be avoided for the most part. In 1914, Page15 reported hearing improvement after surgery in five of eight patients. This report was followed in 1917 by Dean and Gittens,16 who reported an excellent hearing result in a patient and reviewed the various types of operations that had been tried by other surgeons. The prevailing attitude toward surgical correction in these cases remained generally pessimistic, however, despite these and other occasional reports of successful operations, until 1947. That year, Ombredanne17 in France and Pattee18 in the United States each reported a series of patients successfully operated on to improve hearing. Pattee’s technique included removal of the incus to “mobilize” the stapes18; Ombredanne17 added fenestration of the lateral semicircular canal.
With the advent of tympanoplasty techniques in the 1950s, interest in atresiaplasty increased as the teachings of Wullstein and Zollner carried over into surgery of the congenital ear.8 Larger series with greater success rates were reported as surgeons attempted to improve their results, using ossiculoplasty, mastoidectomy, differing degrees of bone removal, and different types and techniques of graft placement.2,4,17,19–26 Ombredanne27 went on to report on more than 600 aplasia cases by 1971 and 1600 cases with major and minor malformations by 1976.8 Gill’s report of 83 cases in 19692 and Jahrsdoerfer’s 1978 article8 are considered landmarks. Crabtree,28 Jahrsdoerfer,29,30 Marquet,31,32 and De la Cruz6,33 and their colleagues all reported on large surgical series, with modifications of classification and operative techniques.
Although techniques of canalplasty, meatoplasty, tympanoplasty, and ossiculoplasty have improved considerably, surgical correction of congenital aural atresia remains one of the most challenging operations performed by otologists. This is a complex surgical problem, requiring application of all tympanoplasty techniques and a thorough knowledge of the surgical anatomy of the facial nerve, oval window, and inner ear, and their congenital variants.1,6,8,17,21,26,27,29-31,33-40 The temporomandibular joint is displaced posteriorly by the lack of development of the EAC, narrowing the distance between the glenoid fossa and the anterior wall of the mastoid tip.19,41 Fusion of the incus and malleus is common, but because of its dual origin, the stapes footplate is usually normal.4,42
The surgical repair of aural atresia is recommended at age 6 years.6 The timing of repair must take into account any planned auricular reconstruction procedures. Criteria for patient selection must be stringent when attempting to achieve closure of the air-bone gap to within 20 to 30 dB. Preoperative counseling and several postoperative visits are essential for optimal results. In this chapter, we discuss these issues and provide guidelines for patient evaluation and selection, surgical techniques, and postoperative management.
EMBRYOLOGY
A review of the normal embryologic development of the ear aids in understanding the myriad of possible combinations of malformations encountered in congenital aural atresia. The inner ear, middle ear, and external ear develop independently and in such a way that deformity of one does not presuppose deformity of another.9,43 Most frequently, abnormalities of the outer and middle ear are encountered in combination with a normal inner ear.44,45
Microtia is a result of first and second branchial arch anomalies. Growth of mesenchymal tissue from the first and second branchial arches forms six hillocks around the primitive meatus that fuse to form the auricle (Table 5-1). By the end of the third month, the primitive auricle has been completed. The external auditory meatus develops from the first branchial groove. During the second month, a solid core of epithelium migrates inward from the rudimentary pinna toward the first branchial pouch. This core, the precursor of the EAC, starts to hollow out and take shape in the sixth month. It canalizes in the seventh month, causing the developing mastoid to become separated from the mandible. Its subsequent posterior and inferior development carries the middle ear and facial nerve to their normal positions.1,43,46–48 Some of the literature supports the notion that microtia grade can indicate the status of middle ear development in aural atresia. The better developed the external ear is, the better developed is the middle ear.
| First Branchial Arch | Second Branchial Arch |
|---|---|
| First hillock—tragus | Fourth hillock—antihelix |
| Second hillock—helical crus | Fifth hillock—antitragus |
| Third hillock—helix | Sixth hillock—lobule and lower helix |
The first branchial pouch grows outward to form the middle ear cleft. The plaque of tissue where this cleft meets the epithelium of the EAC forms the tympanic membrane. While the pouch is forming the eustachian tube, tympanic cavity, and mastoid air cells, Meckel’s cartilage (first branchial arch) is forming the neck and head of the malleus and incus body. Reichert’s cartilage (second branchial arch) forms the remainder of the first two ossicles’ long processes and the stapes superstructure. The footplate has a dual origin from the second arch and the otic capsule. The ossicles attain their final shape by the fourth month. By the end of the seventh to eighth month, the expanding middle ear cleft surrounds the ossicles and covers them with a mucous membrane.1,45,49
The facial nerve is the nerve of the second branchial arch. At 4.5 weeks, this developing nerve divides the blastema, which is the condensation of the second arch mesenchymal cells, into the stapes, the interhyale (stapedius muscle precursor), and the laterohyale (precursor of the posterior wall of the middle ear). The nerve’s intraosseous course is dependent on this bony expansion.45,47 The membranous portion of the inner ear develops during the third to sixth week from an auditory placode on the lateral surface of the hindbrain. The surrounding mesenchyme transforms into the bony otic capsule.50
Congenital aural atresia can range in severity from a thin membranous canal atresia to complete lack of tympanic bone, depending on the time of arrest of intrauterine development.1,51,52 The common finding of a normal inner ear is explained because the inner ear is formed by the time of external/middle ear development arrest. Facial nerve course abnormalities are often seen.
CLASSIFICATION SYSTEMS
Of historical significance is a classification in congenital aural atresia developed in 1955 by Altmann.51 In this system, atresias are categorized into three groups, as follows:
The De la Cruz classification includes surgical feasibility guidelines using only high-resolution computed tomography (CT), taking into consideration mastoid pneumatization, inner ear normality, and facial nerve and footplate relationship.6 The malformations are divided into minor and major malformations (Table 5-2). The clinical importance of this classification is that surgery in cases of minor malformations has a good possibility of yielding serviceable hearing, whereas cases of major malformations are frequently inoperable, but treatable with the bone-anchored hearing aid (BAHA) system.3
TABLE 5-2 De la Cruz Classification of Congenital Aural Atresia
| Minor Malformations |
| 1. Normal mastoid pneumatization |
| 2. Normal oval window/footplate |
| 3. Good facial nerve–footplate relationship |
| 4. Normal inner ear |
| Major Malformations |
| 1. Poor pneumatization |
| 2. Abnormal or absent oval window/footplate |
| 3. Abnormal course of facial nerve |
| 4. Abnormalities of inner ear |
From De la Cruz A, Linthicum FH Jr, Luxford WM: Congenital atresia of the external auditory canal. Laryngoscope 95:421-427, 1985.
Jahrsdoerfer and colleagues53 developed a widely used point grading system to guide surgeons in preoperative assessment of the best candidates for hearing improvement (Table 5-3). This system takes into account the parameters of mastoid pneumatization, presence of the oval and round windows, facial nerve course, status of the ossicles, and external appearance. Point allocation is based primarily on the findings on high-resolution CT. Jahrsdoerfer and colleagues53 proposed that when the preoperative evaluation of the patient is itemized into this grading system, the best results (>80% success) are achieved with a score of 8 or better. A score of 7 indicates the patient is a fair candidate, a score of 6 indicates the patient is a marginal candidate, and with a score less than 5 the patient becomes a poor candidate.
TABLE 5-3 Jahrsdoerfer’s Grading System of Candidacy for Surgery of Congenital Aural Atresia
| Parameter | Points |
|---|---|
| Stapes present | 2 |
| Oval window open | 1 |
| Middle ear space | 1 |
| Facial nerve normal | 1 |
| Malleus-incus complex present | 1 |
| Mastoid well pneumatized | 1 |
| Incus-stapes connection | 1 |
| Round window normal | 1 |
| Appearance of external ear | 1 |
| Total available points | 10 |
| Rating | Type of Candidate |
| 10 | Excellent |
| 9 | Very good |
| 8 | Good |
| 7 | Fair |
| 6 | Marginal |
| ≤5 | Poor |
From Jahrsdoerfer RA, Yeakley JW, Aguilar EA, et al: Grading system for the selection of patients with congenital aural atresia. Am J Otol 13:6-12, 1992.
Ishimoto and associates54 evaluated the relationship between hearing level and temporal bone abnormalities in patients with microtia, using Jahrsdoerfer’s CT scoring system and high-resolution CT scans of the temporal bone. They found that the hearing level in microtic ears correlated with the formation of oval/round windows and ossicular development, but not with the degree of middle ear aeration, facial nerve aberration, or severity of microtia.
Schuknecht’s55 system of classification of congenital aural atresia is based on a combination of clinical and surgical observations. Type A (meatal) atresia is limited to the fibrocartilaginous part of the EAC. Meatoplasty is the surgical procedure of choice, and when performed in a timely fashion prevents formation of canal cholesteatoma and conductive hearing loss. In type B (partial) atresia, there is narrowing of the fibrocartilaginous and bony EAC, but a patent dermal tract allows partial inspection of the tympanic membrane. The tympanic membrane is small and partly replaced by a bony septum. Minor ossicular malformations exist, and hearing loss may be mild to severe. Type C (total) atresia includes all cases with a totally atretic ear canal, but a well-pneumatized tympanic cavity. There is a partial or total bony atretic plate, the tympanic membrane is absent, the heads of the ossicles are fused, there may be no connection to a possibly malformed stapes, and the facial nerve is more likely to have an aberrant course over the oval window. Type D (hypopneumatic total) atresia is a total atresia with poor pneumatization, common in dysplasias such as Treacher Collins syndrome. There are abnormalities of the facial nerve canal and the bony labyrinth. These patients are poor candidates for hearing improvement surgery.
Chiossone’s56 classification is based primarily on the location of the glenoid fossa. In type I, the fossa is in the normal position; in type II, it is moderately displaced; in type III, the fossa overlaps the middle ear; and in type IV, in addition to the fossa overlapping the middle ear, there is lack of mastoid pneumatization. Patients with types I and II are ideal surgical candidates. Type III cases have a tendency toward graft lateralization. Patients with type IV are not surgical candidates. In atresiaplasty surgery, a classification scheme is useful for surgical planning, patient counseling, and comparison of outcomes.
INITIAL EVALUATION AND PATIENT SELECTION
When aural atresia is noted in a newborn, several issues must be addressed. Where one congenital abnormality is found, others must be sought. A high-risk registry for hearing loss is helpful in this regard.57 After the degree of aural deformity is assessed by physical examination, evaluation of auditory function in unilateral and bilateral atresia should be performed using auditory brainstem response audiometry within the first few days of life. An 11% to 47% incidence of inner ear abnormality is associated with congenital aural atresia.12 Occasionally, in unilateral cases, there is a total sensorineural hearing loss (SNHL) on the side of the normal-appearing ear, which might otherwise be missed.6,33
In bilateral cases, a bone-conduction hearing aid should be applied as soon as possible, ideally in the third or fourth week of life. In unilateral cases in which the opposite ear hears normally, a hearing aid is unnecessary. A child with aural atresia and associated cephalic abnormalities (e.g., hemifacial microsomia and Treacher Collins, Crouzon, or Pierre Robin syndrome) should be recognized.57–61 In this subset, surgical correction has poor results,55 and a long-term bone-conduction hearing aid or BAHA in these nonoperable bilateral congenital aural atresia situations is beneficial (see later).3
A patient with congenital aural atresia may present with an infected or draining ear or acute facial palsy; 14% have congenital cholesteatoma.6 The priority in these cases is removal of the cholesteatoma and resolution of the infection. Preoperative audiometry and high-resolution CT scanning may be necessary at an earlier age in children with repeated infections in the atretic ear.6,55,62
There are two requirements for planning surgery in congenital aural atresia: radiographic three-dimensional evaluation of the temporal bone and audiometric evidence of cochlear function.8,63 Other conditions mandating prompt surgical intervention are congenital cholesteatoma, a draining postoperative atretic ear, and acute facial palsy. The CT scan should always be reviewed for cholesteatoma, which necessitates surgery at any age.55,62,64 It is not included in any of the grading systems because these are used only for predicting hearing results in elective atresiaplasty surgery.
TIMING OF AURICULAR RECONSTRUCTION AND ATRESIAPLASTY
In bilateral or unilateral atresia, auricular reconstruction and atresiaplasty are recommended at 6 years of age. Before this age, there may be a tendency to form exostosis-like bony growth that may occlude the EAC, and there is less patient cooperation. By age 6, the costal cartilage has developed sufficiently to allow for reconstruction of the auricle, and the mastoid has become as pneumatized as possible. The microtia repair should be done first because the complex flaps and use of autologous rib graft demand excellent blood supply.65,66 Many surgeons do not perform cartilage microtia reconstruction on an ear with previous reconstruction attempts.64 The hearing restoration surgery is performed 2 months after the last step of the microtia repair. Rehabilitation of auricular defects can be done using an osseointegrated percutaneous mastoid implant prosthesis, with and without bone-conduction aids, such as BAHA.3,48,67–69 Alloplastic materials have been used for microtia repair in the past, but there is a high risk of extrusion associated with their use. Newer materials, such as Medpor, seem to be very well tolerated, however.70–72 As noted earlier, Medpor reconstruction can be done before or after atresiaplasty because intact blood supply is not indispensable.
In unilateral cases, atresiaplasty surgery may be indicated in a patient with “minor” unilateral atresia with normal middle ear, ossicles, and facial nerve, and excellent pneumatization. In such patients, atresiaplasty may be offered in childhood with the parents’ consent.33 We often see older adults with unilateral atresia who request surgery when their normal ear begins having high-frequency hearing loss (presbycusis).
Bone-Anchored Hearing Appliances
For patients with bilateral hearing loss, BICROS (Bilateral Contralateral Routing of Signal) hearing aid is an additional microphone that can be used as an accessory to an implanted BAHA fixture and sound processor to overcome the head shadow effect. The accessory microphone is placed in the contralateral ear to the BAHA fixture. The signal is routed from the accessory microphone to the BAHA sound processor via a wire worn behind the neck. It is intended to improve hearing by eliminating the head shadow effect, but it does so with little success and is not well accepted by patients.73
[/not-level-membership-for-surgery-category]
