[level-membership-for-pediatrics-category]Chapter 53
Congenital Lung Anomalies
Congenital lung anomalies refer to a heterogeneous group of pulmonary developmental disorders that affect the lung parenchyma, the arterial supply, and the venous drainage to the lung, or a combination of these entities. The reported incidence of congenital lung anomalies ranges from 1.2 : 10,000 to 1 : 35,000 pregnancies; however, these reports may be an underestimate of their true incidence.1–3 Although prenatal sonography (ultrasound), advances in postnatal imaging, and, more recently, fetal magnetic resonance imaging (MRI) have enhanced our understanding of congenital lung anomalies, substantial controversy continues regarding the nomenclature, classification, pathogenesis, description, and management of these lesions. In addition, considerable variability exists in their prenatal clinical presentation and outcome, ranging from in-utero involution to severe hydrops and fetal demise. Likewise, their postnatal clinical presentation also is variable, ranging from the completely asymptomatic newborn to the older child or young adult with recurrent pneumonias. In this chapter, the underlying etiology, clinical presentation, imaging findings, and management of various congenital lung anomalies encountered in the pediatric population are discussed.
Spectrum of Congenital Lung Anomalies
The classification of congenital lung anomalies is challenging and continuously controversial from embryologic, radiologic, pathologic, and clinical viewpoints. Several classifications and terminologies with their own advantages and disadvantages have been suggested.4–6 Some investigators have used embryology as a basis and have classified congenital lung anomalies according to the stage of intrauterine development in which the insult resulting in the malformation developed.4 Other investigators have categorized lesions based on their morphologic-radiologic features and divided them into two groups: whole lung malformations (e.g., lung hypoplasia) and focal malformations (e.g., bronchial atresia).5,7 Recently the Langston6 classification has become one of the most accepted classification systems for congenital lung anomalies, particularly from the pathological standpoint, although it is by no means the most widely used classification by all clinical groups. Langston categorizes the wide spectrum of respiratory tract malformations primarily as bronchial atresia, congenital pulmonary airway malformation (CPAM), extralobar bronchopulmonary sequestration (BPS), congenital lobar hyperinflation (CLH), and bronchogenic cysts. These five congenital lung anomalies comprise approximately 90% of the anomalies seen in clinical practice. However, this classification system is limited because other congenital lung anomalies (e.g., pulmonary arteriovenous malformation [AVM]) are not included.
For the purpose of relatively clear classification, easy differentiation on imaging studies, and preoperative assessment of surgical lesions, congenital lung anomalies discussed in this chapter are categorized according to their morphologic-radiologic-pathologic features. Such a classification system views congenital lung anomalies as a continuum ranging from predominantly parenchymal abnormalities (i.e., abnormal lung parenchyma, relatively normal vasculature, airway, and foregut derivatives, e.g., CPAM), to predominantly vascular abnormalities (i.e., normal lung parenchyma, normal airway, and no foregut abnormality but abnormal vasculature; e.g., AVM), to combined parenchymal and vascular abnormalities (e.g., pulmonary sequestration and scimitar syndrome) in which influencing factors (foregut and airway components) play an important role and the major abnormalities are intertwined (Fig. 53-1).
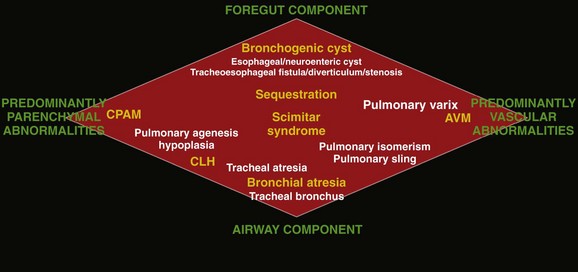
Figure 53-1 Diagram of the spectrum of congenital lung anomalies, including foregut and airway components.
The lesions in yellow denote those most commonly encountered in this entity, whereas the lesions in white represent additional lesions that can be considered part of the spectrum. AVM, Arteriovenous malformation; CPAM, congenital pulmonary airway malformation; CLH, congenital lobar hyperinflation. (Adapted from Newman B. Congenital bronchopulmonary foregut malformations: concepts and controversies. Pediatr Radiol. 2006;36:773-791.)
Given that specific terminology for these lesions may be controversial and occasionally confusing, we and other authors8–11 recommend that radiologists thoroughly describe all imaging findings of congenital lung anomalies rather than try to categorize the lesions by pathologic terminology. Specific imaging findings of congenital lung anomalies that need to be evaluated and described include (1) location of lesions, (2) associated anomalous vascular supply and drainage of the lesions, (3) internal components and the degree of aeration, (4) exclusion of communication with the gastrointestinal (GI) tract, (5) integrity of the diaphragm, and (6) an assessment of associated anomalies, such as vertebral anomalies.8,10
Predominantly Parenchymal Lesions
Etiology: Bronchial atresia refers to atresia of a lobar, segmental, or subsegmental bronchus at or near its origin resulting in a blind-ended atretic proximal bronchus. Bronchial atresia most frequently affects a segmental bronchus. The precise etiology of bronchial atresia remains unknown, but etiologies such as a vascular insult to the involved atretic or stenotic portion have been proposed.6,8 Several authors who used modern dissecting techniques found that bronchial atresia is more common than originally thought.6,12,13 Furthermore, bronchial atresia and BPSs coexist in nearly all cases,6,12,13 whereas it is found in nearly 70% of CPAM lesions.12 A malformation sequence resulting from airway obstruction during development has been proposed as the unifying element for such a wide spectrum of imaging appearances. Differences in degree, level, and timing of the bronchial obstruction are thought to be responsible for the association of bronchial atresia and other congenital lung anomalies.6,8,12 Bronchial atresia usually is diagnosed as an incidental finding on chest radiographs later in life in asymptomatic older children or adults.6,10 However, bronchial atresia increasingly is being diagnosed in utero, given the widespread use of prenatal imaging.6,9,10,12,13
Imaging: Prenatally, the involved portion of the lung appears hyperexpanded and shows increased homogenous echogenicity on ultrasound and high T2 signal on fetal MRI14 (Fig. 53-2). On occasion, it is possible to identify the centrally located, mucus-filled bronchocele/mucocele on prenatal ultrasound or MRI9,15 (e-Fig. 53-3).
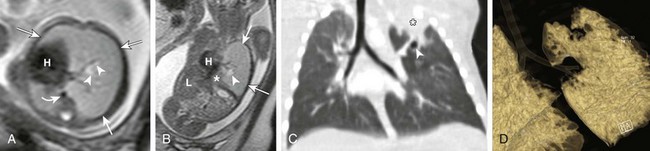
Figure 53-2 Bronchial atresia.
Axial (A) and coronal (B) T2-weighted fetal magnetic resonance images in a 22-week gestational age fetus demonstrate a large homogeneous lesion in the left upper lobe (arrows). Central fluid-filled bronchi are noted (arrowheads). The left lower lobe (asterisk) is compressed inferiorly and displaced medially adjacent to the fetal heart (H). The curved arrow denotes the aorta, which is also displaced to the right. L, Liver. C, A coronal multiplanar reconstruction computed tomography (CT) image shows partial opacification (asterisk) of the left upper lobe, presumably due to mucostasis related to bronchial atresia. In addition, a small air bubble is seen (arrowhead), reflecting a bronchocele. D, A volume-rendered reconstructed CT image could not delineate the left upper lobe bronchus.
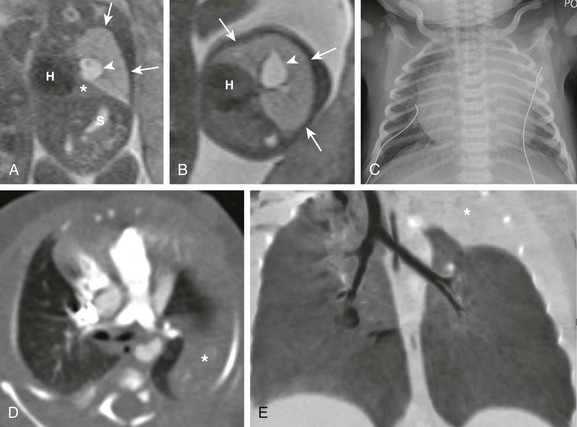
e-Figure 53-3 Bronchial atresia.
Coronal (A) and axial (B) T2-weighted fetal magnetic resonance images show a large hyperintense, homogeneous lesion in the left upper lobe (arrows) in a 21-week gestational age fetus. The left lower lobe (asterisk) is displaced and compressed inferiorly. The heart (H) is shifted to the right. Central fluid-filled bronchi are noted (arrowheads). S, Stomach. C, A frontal chest radiograph at birth shows complete opacification of the left upper lobe, presumably as a result of retained fetal fluid and mucostasis related to bronchial atresia. The fluid-filled central cystic structure reflects a large mucocele/bronchocele. Axial computed tomography (CT) angiography (D) and minimum intensity projection (E) images performed at day 2 of life show complete collapse of the left upper lobe (asterisk). In addition, the left upper lobe bronchus could not be delineated, a finding suggestive of bronchial atresia. Three-dimensional reconstructions are invaluable in showing the “interrupted connection” to the remainder of the airway.
Characteristically, the apicoposterior segmental bronchus of the left upper lobe is most commonly affected, followed by the segmental bronchi of the right upper, right middle, and lower lobes.16 In children, radiographic and computed tomography (CT) imaging features of bronchial atresia are characterized by a tubular or glove-shaped opacity representing mucus plugging in the region distal to the atretic bronchus, surrounding segmental hyperlucency due to air trapping, and decreased underlying vascularity15,17 (e-Fig. 53-4). However, in neonates, a portion of the lung distal to the atretic segment may remain atelectatic as a result of the in-utero mucostasis. Therefore some authors recommend avoiding immediate postnatal imaging evaluation because the under-aerated lung related to bronchial atresia may be difficult to differentiate from the normal lung with expected fetal fluid retention in the newborn period.10 Identification of the hallmark atretic bronchus and the “bronchocele/mucocele” by means of two-dimensional (2D) multiplanar or three-dimensional (3D) reconstructions either on prenatal or postnatal imaging may be helpful.10,18
Treatment and Follow-up: Management of bronchial atresia is somewhat varied. In general, surgical resection is primarily recommended in symptomatic pediatric patients because of recurrent infection.17,19 Although opinions vary, some centers advocate elective surgical resection of bronchial atresia even in asymptomatic pediatric patients because of potential future lung infection and increased association with CPAM.13,20
Bronchogenic Cysts
Etiology: Bronchogenic cysts result from abnormal tracheobronchial branching and presumably originate from an aberrant bud of the developing foregut, similar to other foregut duplication cysts. Bronchogenic cysts typically are unilocular, fluid-filled, or mucus-filled cysts lined by respiratory epithelium and are attached to but do not communicate with the tracheobronchial tree.6,17,19 Although most bronchogenic cysts are located within the mediastinum (predominantly near the carina), they may be encountered anywhere from the suprasternal area to the retroperitoneum. Bronchogenic cysts also may be found within the lung parenchyma, usually in the lower lobes.17,19 Such intrapulmonary bronchogenic cysts do not communicate with the airway unless superimposed infection with wall necrosis occurs,6 which may further predispose them to recurrent infections. The clinical symptomatology of affected pediatric patients primarily depends on the mass effect the lesion exerts on its neighboring structures including the airway, GI tract, and cardiovascular structures. Airway compression is usually mild, but it may be life threatening in some instances when large bronchogenic cysts are located near the carina regions.21
Imaging: A bronchogenic cyst typically presents as a round or oval-shaped cystic lesion located near the right paratracheal or subcarinal area within the middle mediastinum. Bronchogenic cysts are anechoic on prenatal ultrasound and show high signal intensity on T2-weighted prenatal MRI imaging14,19 (e-Fig. 53-5). On chest radiographs, a bronchogenic cyst manifests as a well-delineated round or oval-shaped middle mediastinal mass. On CT, approximately 50% of bronchogenic cysts demonstrate fluid attenuation value (~0 Hounsfield unit) (Fig. 53-6). The remaining bronchogenic cysts may have CT attenuation higher than water because of thick mucoid, milk-of-calcium, proteinaceous, or hemorrhagic contents. MRI, which can confirm the cystic nature of the bronchogenic cysts on T2-weighted images, is helpful for differentiating bronchogenic cysts with high attenuation value from a mildly enhancing solid mass on CT. Typically, no internal contrast enhancement is seen within the uncomplicated bronchogenic cysts on CT or MRI. The presence of an air-fluid level, thick wall enhancement, or surrounding inflammatory changes often is associated with superimposed infection.11,16,17
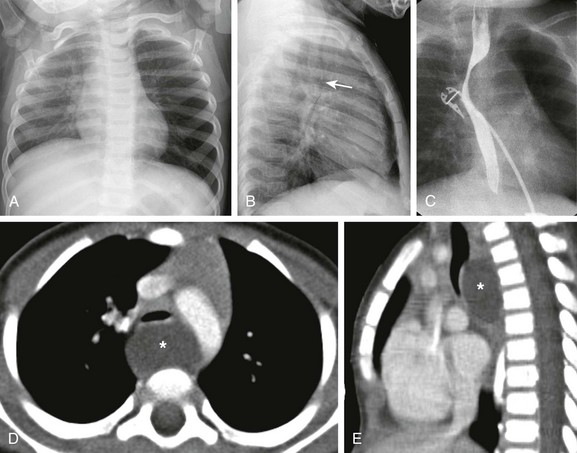
Figure 53-6 A bronchogenic cyst in a 15-month-old child with stridor.
The frontal chest radiograph (A) shows hyperinflation of the left lung. The lateral radiograph (B) reveals the effect of the mass and posterior displacement of the airway (arrow). C, An upper gastrointestinal image shows an esophageal displacement by a soft tissue density mass. Enhanced axial (D) and sagittal (E) computed tomography images show a fluid density lesion (asterisks) posterior to the lower trachea consistent with a bronchogenic cyst.
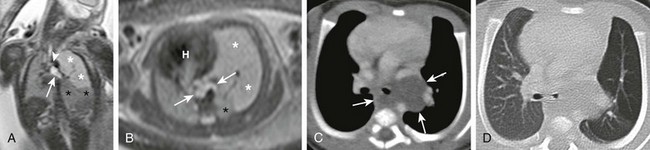
e-Figure 53-5 Bronchogenic cyst.
Coronal (A) and axial (B) T2-weighted fetal magnetic resonance images in a 26-week gestational age fetus show a cystic structure in the posterior mediastinum (arrows) apparently compressing the left mainstem bronchus (arrowhead) and resulting in hyperexpansion with increased fluid signal in the left upper lobe (white asterisks). The left lower lobe is compressed and displaced inferiorly (black asterisks). The fetal heart (H) and mediastinum have shifted to the right. Enhanced computed tomography images in mediastinal (C) and lung (D) windows show the fluid-filled cystic structure consistent with a bronchogenic cyst (arrows) centered at the left hilum and extending posteriorly and inferiorly. The cyst compresses the left mainstem bronchus and results in air trapping in the left upper lobe.
Treatment and Follow-up: Complete surgical resection is the current management of choice for bronchogenic cysts, particularly in symptomatic pediatric patients.22 Temporizing or palliative procedures such as transparietal, transbronchial, or mediastinal aspiration may be considered in symptomatic pediatric patients who are not surgical candidates.
Congenital Lobar Hyperinflation
Etiology: CLH, also referred to as infantile lobar emphysema or congenital lobar emphysema, presumably is caused by an intrinsic or extrinsic bronchial narrowing, resulting in subsequent air trapping. Intrinsic bronchial narrowing can be caused by weakness or absence of underlying bronchial cartilage, whereas extrinsic bronchial narrowing may be due to the compression from adjacent mediastinal masses or enlarged vessels. CLH clinically presents with respiratory distress in the newborn period14,17,19 in nearly half of the cases and by the age of 6 months in 80% of cases.23 There is a slight male predominance, and the upper lobes are affected more frequently than the lower lobes, with the left lung affected more often than the right.23
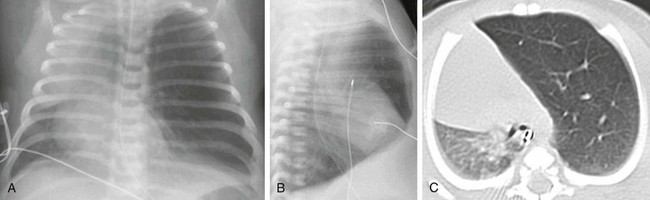
Imaging: On prenatal imaging, CLH manifests as a homogeneously hyperechogenic lesion on ultrasound or as a T2 hyperintense lesion on MRI without visible cysts.9,24 On prenatal imaging, CLH often is indistinguishable from other congenital lung anomalies, particularly bronchial atresia.9,24 CLH usually is diagnosed by its typical clinical presentation and characteristic radiographic features of progressive lobar hyperexpansion and hyperlucency, producing displacement or compression of adjacent structures. In the immediate postnatal period, CLH initially may appear as an area of increased opacity related to retained fetal lung fluid, which will clear on subsequent studies.6,17,19,23 Similar imaging findings are noted on CT, and the attenuated pulmonary vasculature is a helpful clue to distinguish CLH from a pneumothorax or other entities in cases of inconclusive chest radiographic findings17 (Fig. 53-7).
Congenital Pulmonary Airway Malformation
Etiology: CPAMs, formerly known as cystic adenomatoid malformations of the lung, were first described in the literature by Ch’In and Tang in 1949 as rare lung lesions occurring in premature or stillborn infants with significant hydrops.26,27 CPAMs are characterized by a heterogeneous group of congenital cystic and noncystic lung masses that communicate with an abnormal bronchial tree lacking supporting cartilage.8,17,28
In 1977, Stocker et al.28 classified these lesions based on their clinical and pathologic features, with subdivisions based on the size of the cysts (types I, II, and III) and according to the location of suspected development of the malformation along the airway. Type I CPAMs consist of cysts larger than 2 cm, with presumed bronchial/bronchiolar origin. Type II CPAMs consist of cysts smaller than 2 cm, with presumed bronchiolar origin. Type III CPAMs appear solid, with a presumed bronchiolar/alveolar origin. However, Stocker later expanded his CPAM classification into five types that included type 0 CPAMs, with presumed tracheobronchial origin, and type IV CPAMs, with presumed distal acinar origin. The term CPAM was now implemented instead of cystic adenomatoid malformations, because cystic changes were observed in only three of the aforementioned types (types, I, II, and IV), and adenomatoid change was observed only in type III.9,17,29,30 Increasing evidence indicates that type IV CPAM lesions and type I pleuropulmonary blastomas may represent the same entity.6,31 It is important to recognize that although Stocker’s classification is widely used, it is by no means universally accepted. For an example, Langston6 classifies CPAM lesions into two types and terms the Stocker type I CPAM as “large cyst type lesions” and the Stocker type II CPAMs as “small cyst type lesions” based on cyst size and pathologic criteria.6 Langston proposed that the type III CPAM actually represents a form of pulmonary hyperplasia and should be excluded from the CPAM classification.
Imaging: Prenatally, CPAMs are classified on the basis of cyst size as microcysts (<5 mm) and macrocysts (≥5 mm) on fetal ultrasound and MRI.32 Type 1 CPAMs may contain one, several, or multiple macrocysts, some of which are ≥5 mm in diameter9 (Fig. 53-8). Type II CPAMs have variable appearances, ranging from homogeneously hyperechoic or hyperintense lesions to microcystic lesions exhibiting multiple, uniform cysts that measure <5 mm9,14,19 (e-Figs. 53-9 and 53-10).
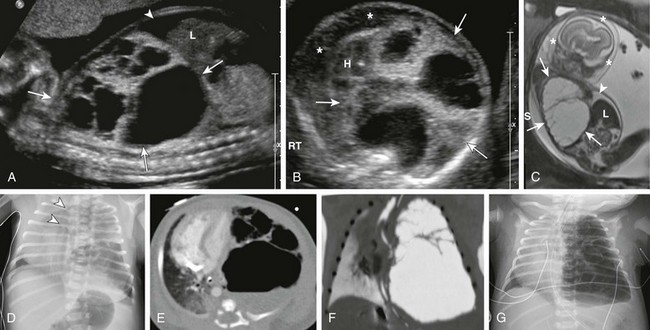
Figure 53-8 Congenital pulmonary airway malformation type I.
Sagittal ultrasound (A), axial ultrasound (B), and fetal T2-weighted magnetic resonance sagittal (C) images in a 22-week gestational age fetus show a large, heterogeneous, multicystic lesion occupying the vast majority of the left hemithorax (arrows) and resulting in inversion of the left hemidiaphragm. Note the ascites in the abdomen (arrowhead) and the skin thickening and edema (asterisks), which are reflective of hydrops. H, Heart; L, liver. D, A radiograph immediately after delivery shows the complex, partially aerated lesion resulting in significant mediastinal shift. The arrowheads denote the severe tracheal deviation. Axial computed tomography angiography (E) and minimum intensity projection (F) images obtained the following day show the large, heterogenous lesion with multiple macrocysts of varying sizes. Fluid is identified in some of the cysts. G, As fetal fluid is cleared from the lungs, the lesion appears more aerated and further mediastinal shift occurs.
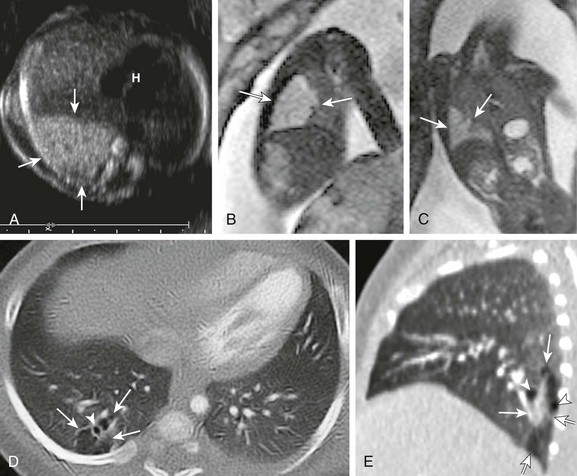
e-Figure 53-9 Congenital pulmonary airway malformation (CPAM) type II.
A, A transverse fetal ultrasound image through the chest in a 20-week gestational age fetus shows an echogenic right lower lobe lung lesion with tiny cysts only seen on high-resolution images (image not shown) consistent with a CPAM type II lesion (arrows). The heart (H) and mediastinum are only mildly shifted without cardiac compression. Sagittal (B) and coronal (C) fetal T2-weighted magnetic resonance images show a mildly heterogenous lesion (arrows) with slightly nodular contours but no visible cysts. Postnatal axial and sagittal computed tomography images (D and E) at 1 month of age show an ill-defined, relatively smaller lesion (arrows) in the right lower lobe with tiny, subcentimeter internal cysts (arrowheads). This lesion also demonstrated bronchial atresia components on histologic examination.
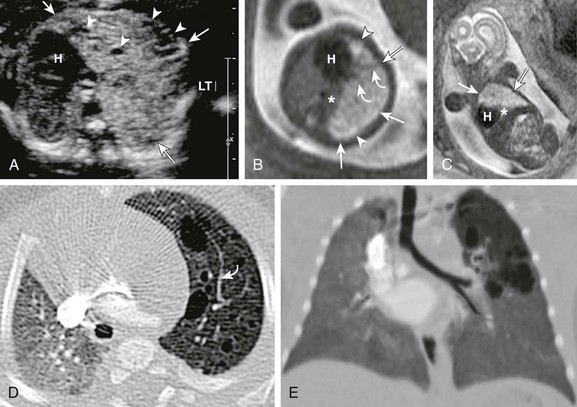
e-Figure 53-10 Congenital pulmonary airway malformation type II.
A, A transverse fetal ultrasound image through the chest in a 19-week gestational age fetus shows an echogenic left upper lobe lesion with several internal cysts (arrowheads) and findings consistent with a CPAM type 2 lesion (arrows). The heart (H) and mediastinum are moderately shifted to the right without cardiac compression. LT, left. Axial (B) and coronal (C) fetal T2-weighted magnetic resonance (MR) images show a heterogeneous lesion (arrows) with internal cysts (arrowheads); some of them even appear as “macrocysts.” Normal-appearing lung tissue is noted as being compressed posteriorly and inferiorly (asterisks). However, on postnatal axial (D) and minimum intensity projection (E) computed tomography (CT) images at 3 months of age, the lesion appears relatively smaller and exerts less mass effect and no mediastinal shift. All the cysts are aerated and measure less than 2.0 cm. Note that some slightly prominent, tortuous internal pulmonary vessels are seen within the lesion in both the fetal MR image (B) and the postnatal CT image (D) (curved arrows).
Postnatal imaging findings of CPAMs usually correlate with underlying histopathologic features.17 Large cyst type or type I CPAMs typically present with one or several larger air-filled cystic structures with intervening solid, unaerated lung parenchyma. The cysts of type I CPAMs are larger than 2 cm and may be accompanied by several microcysts, whereas small cyst type or type II CPAMs usually manifest as partially air-filled multicystic masses, with individual cysts smaller than 2 cm and with variable degrees of solid-appearing, unaerated lung tissue.10,17 Type 3 CPAMs typically appear as solid lesions with mild contrast enhancement because of microscopic cysts that can be identified only at histologic evaluation. Type IV CPAMs usually present as large cysts arising from the peripheral portion of the lung and can be radiographically indistinguishable from a predominantly cystic type 1 pleuropulmonary blastoma (see Chapter 55) (e-Fig. 53-11).
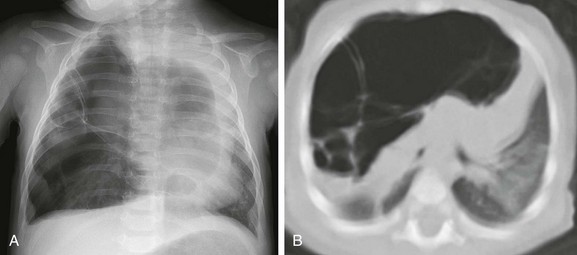
e-Figure 53-11 A cystic pleuropulmonary blastoma in a 6-month-old with increased work of breathing.
A, A frontal chest radiograph demonstrates a large radiolucent lesion with fine septations and multiple cysts within the right lung resulting in a mediastinal shift. B, An axial computed tomography image reveals multiple cysts of varying sizes with effect of the mass demonstrated on the mediastinum and contralateral left lung.
In their pure forms, CPAM blood supply is from the pulmonary artery and venous drainage is into the pulmonary veins. Although unilobar involvement of CPAM is far more common, multilobar and even bilateral lung involvement may occur.3,27,30 Although any lobe of the lung can be involved, predilection exists for the lower lobe.3 CPAMs that are complicated as a result of superimposed infection may have an imaging appearance similar to pneumonia or a lung abscess (Fig. 53-12).
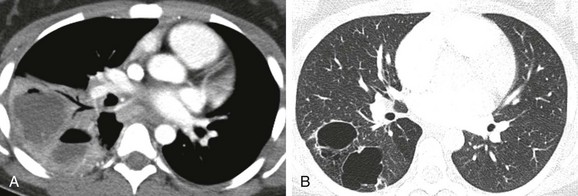
Figure 53-12 An infected congenital pulmonary airway malformation (CPAM) type I lesion in a 15-year-old girl.
A, An initial computed tomography (CT) enhanced image shows consolidation, with areas of low attenuation representing superimposed infection of the cystic components. B, A follow-up CT image obtained 7 months later shows resolution of the infection and visualization of CPAM cystic components.
Treatment and Follow-up: The generalized consensus is that symptomatic CPAMs should be resected, typically by lobectomy, regardless of the patient’s age at presentation.17,22,33 However, considerable controversy exists with regard to the management of prenatally diagnosed, asymptomatic, small CPAM lesions, and no consensus exists on the timing of34 or need for resection.35–39 Although some persons advocate a nonsurgical strategy with imaging follow-up, most medical centers advocate surgical resection before 1 year of age because of the potential risk of associated complications, such as infection, pneumothorax, and the small risk of malignant transformation, particularly in the case of CPAM type I lesions.3,21,22,33,40
Predominantly Vascular Lesions
Anomalies of the Pulmonary Artery
Pulmonary Agenesis, Aplasia, and Hypoplasia
Etiology: Pulmonary underdevelopment may be classified into three main types: (1) lung agenesis, consisting of the absence of the lung, bronchus, and pulmonary artery; (2) lung aplasia, that is, the presence of a rudimentary bronchus but the lack of lung tissue and pulmonary artery; and (3) lung hypoplasia, which consists of a hypoplastic bronchial tree and pulmonary artery with a variable amount of lung parenchyma.17,41
The etiology of lung agenesis or aplasia remains uncertain. Genetic, teratogenic, and mechanical factors may play a role.17 Pulmonary agenesis associated with ipsilateral radial ray defects or hemifacial microsomia may be the result of an abnormal development of the first and second arch derivatives or abnormal blood flow at this level inciting the developmental event, given the common association.42 On the other end of the spectrum, no identifiable cause has been found for lung hypoplasia.17
Persons with pulmonary agenesis, aplasia, and hypoplasia either are asymptomatic or present with variable degrees of respiratory distress, depending on the extent of lung underdevelopment. Associated congenital malformations may be seen in 50% to 80% of cases involving the heart, gastrointestinal tract, skeleton, and vascular and genitourinary systems.17,42–44
Imaging: On chest radiographs, affected pediatric patients may or may not present with a small, radiopaque hemithorax, depending on the degree of the abnormality. Ipsilateral displacement of mediastinal structures and elevation of the hemidiaphragm usually are present. The normal contralateral lung shows compensatory hyperinflation and herniation across the anterior midline, which is best seen on the lateral projections as a band of increased retrosternal lucency17 (Fig. 53-13). Left lung agenesis is more common than right lung agenesis. Multidetector CT with multiplanar 2D and 3D imaging capabilities can be used to distinguish among pulmonary agenesis, pulmonary aplasia, and pulmonary hypoplasia by clearly identifying the bronchial stump and/or the rudimentary bronchial tree17,45 (e-Figs. 53-14 and 53-15, Fig. 53-16, and e-Fig. 53-17).
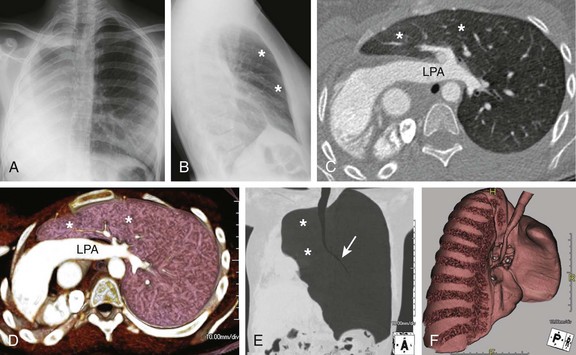
Figure 53-13 Pulmonary agenesis.
Frontal (A) and lateral (B) radiographs in a 15-year-old with worsening asthma demonstrate marked hyperinflation of the left lung that extends across the midline anteriorly and herniates toward the right, as evidenced by a band of retrosternal lucency on the lateral projection (asterisks). Associated dextroposition of the heart into the right hemithorax is seen. Enhanced axial computed tomography (C), axial volume-rendered (D), minimum intensity projection (E) and three-dimensional volume-rendered (F) images of the central airway and lungs demonstrate complete agenesis of the right bronchus and lung. Associated dextroposition of the heart and compensatory hyperexpansion is present, particularly of the left upper lobe (asterisks), which herniates into the right hemithorax. A normal left mainstem bronchus (arrow) is seen. LPA, Left pulmonary artery.
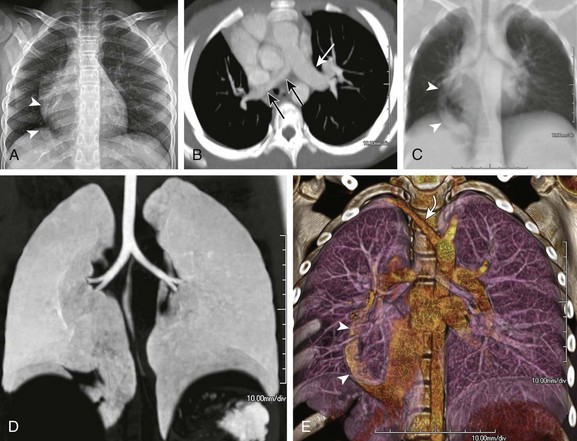
Figure 53-16 Hypoplastic right lung and scimitar syndrome in an 8-year-old.
A, A frontal chest radiograph shows asymmetric lung volumes with dextroposition of the heart and a vertically oriented curvilinear opacity (arrowheads) projecting over the right lower hemithorax. An axial maximum intensity projection computed tomography image (B) confirms mild dextroposition of the heart and hypoplastic right pulmonary artery (PA) (black arrows) relative to the normal left pulmonary artery (white arrow). Coronal thick multiplanar reconstruction (C), inverted minimum intensity projection (D), and three-dimensional volume rendered (E) images show hypoplasia of the right lung with absence of the right upper lobe bronchus and partial anomalous pulmonary venous return of a vast portion of the right lung via a scimitar vein (arrowheads) into the inferior vena cava. Incidentally noted is a right aberrant subclavian artery (curved arrow).
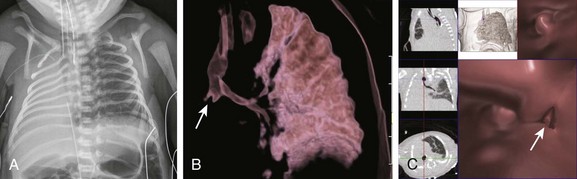
e-Figure 53-14 A 1-day-old with lung aplasia, esophageal atresia, and distal tracheoesophageal fistula.
A, A frontal chest radiograph shows complete opacification with volume loss in the right hemithorax. The airway and mediastinum are shifted to the right with associated dextroposition of the heart. A nasogastric tube terminates in the upper esophageal pouch consistent with esophageal atresia. The anterior view of a volume-rendered image of the central airway and lungs (B) and virtual bronchoscopic (C) images demonstrate a rudimentary right main stem bronchus (straight arrows) consistent with lung aplasia. No right pulmonary artery is identified (image not shown).
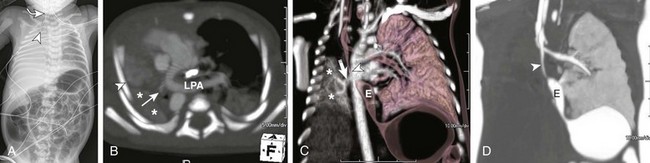
e-Figure 53-15 Ex-34 weeks premature with esophageal atresia, distal tracheoesophageal (TE) fistula, and imperforate anus.
A, A frontal radiograph of the chest and abdomen shows complete opacification with volume loss in the right hemithorax. The endotracheal tube (arrowhead) and mediastinum are shifted to the right with associated dextroposition of the heart. A nasogastric tube (curved arrow) terminates in the upper esophageal pouch consistent with esophageal atresia. Pronounced gaseous distention of numerous small and large bowel loops is seen that is consistent with imperforate anus and distal TE fistula. Note the dysplastic sacral elements. B, An axial maximum intensity projection computed tomography (CT) image shows an anomalous left pulmonary artery (LPA) arising from a tiny, hypoplastic right pulmonary artery (arrow) and coursing behind the mainstem bronchi in a slinglike configuration. The right lung is severely hypoplastic and collapsed (asterisks). C, A frontal volume-rendered CT image shows an aerated left lung and a collapsed, unaerated right lung (asterisks). Note the hypoplastic right pulmonary veins (arrow). The arrowhead denotes the distal TE fistulous communication. D, An inverted coronal minimum intensity projection CT image better demonstrates the distal TE fistulous communication (arrowhead). The right bronchi were fluid opacified and could not be depicted with these techniques. E, Esophagus.
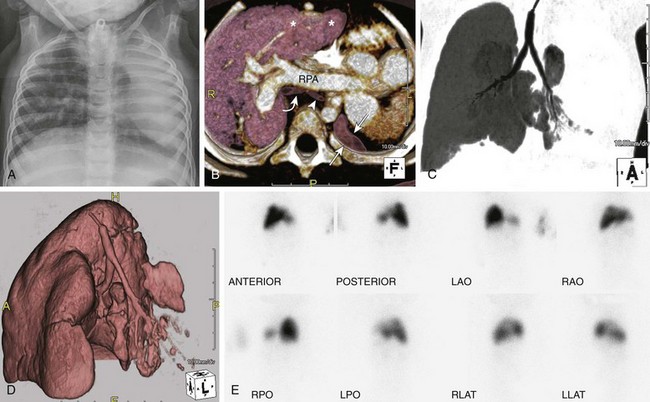
e-Figure 53-17 Left lung hypoplasia in a 2-year-old with multiple congenital anomalies.
A frontal chest radiograph (A) shows a small, hypoplastic left lung and leftward shift of the mediastinum. An axial volume-rendered computed tomography (CT) image (B) confirms leftward shift of the mediastinum and heart and shows a small, hypoplastic left lung (arrows), hypoplastic left bronchus (arrowhead) relative to the right (curved arrow) and compensatory hyperinflation with herniation of the right upper lobe (asterisks) into the left hemithorax. A secondary leftward mediastinal shift with levoposition of the heart is seen. Note the prominent right pulmonary artery (RPA) to the right lung. The left pulmonary artery was severely hypoplastic (image not shown). Coronal minimum intensity projection (C) and coronal oblique three-dimensional volume-rendered (D) CT images of the central airways and lungs show a hypoplastic left mainstem bronchus and left lung. Technetium-99m lung scan images (E) reveal significantly decreased perfusion of the left lung in comparison to the right lung. LAO, left anterior oblique; LLAT, left lateral; LPO, left posterior oblique; RAO, right anterior oblique; RLAT, right lateral; RPO, right posterior oblique.
Treatment and Follow-up: The prognosis of pediatric patients with pulmonary underdevelopment primarily depends on the extent of underdevelopment and the severity of other associated congenital malformations. Treatment usually is aimed at improving the respiratory status and symptoms related to concomitant congenital malformations.
Proximal Interruption of the Pulmonary Artery
Etiology: Proximal interruption of the pulmonary artery results from the abnormal involution of the proximal sixth aortic arch. Such involution causes the “absence” of the proximal pulmonary artery and a persistent connection of the hilar pulmonary artery to the distal sixth aortic arch, which ultimately becomes the ductus arteriosus. The hilar pulmonary artery supplying the ipsilateral affected lung continues to develop via the blood supply received from the ductus arteriosus (which originates either from the base of the right innominate artery or occasionally from an aberrant right subclavian artery).8,17 Progressive closure of the ductus arteriosus eventually results in loss of blood supply to the hilar pulmonary artery and the lung. Perfusion of the affected lung subsequently becomes dependent on collateral systemic vessels, mainly aortopulmonary and bronchial arteries but also transpleural branches of the intercostal, internal mammary, subclavian, and innominate arteries.17,46–48 Although asymptomatic pediatric patients with this anomaly may be detected incidentally, some children present with symptoms related to recurrent pulmonary infections, hemoptysis, and pulmonary hypertension.8,11,17
Imaging: The interrupted proximal pulmonary artery is characteristically located on the contralateral side of the aortic arch. Proximal interruption of the pulmonary artery is more commonly seen on the right side. Interruption of the proximal left pulmonary artery is less common and frequently is associated with congenital heart disease, typically tetralogy of Fallot and ventricular septal defect.8,17,45 On chest radiographs, the affected lung and hilum are smaller than those of the contralateral side (Fig. 53-18). Ipsilateral mediastinal shift, narrowed intercostal spaces, or rarely rib notching in the case of prominent intercostal collaterals also may be present.11,17,49 CT or MRI can definitely confirm and characterize this condition (e-Fig. 53-19). On CT, the interrupted pulmonary artery terminates within 1 cm of its origin from the main pulmonary artery. However, the hilar portion of the pulmonary artery and the intrapulmonary vascular network remain patent. A serrated pleural thickening and subpleural parenchymal bands also may be observed, which reflect the direct anastomosis of transpleural systemic collaterals with peripheral pulmonary arterial branches. Airway branching and pulmonary lobation anomalies are not uncommon. Additional imaging findings may include tiny peripheral cystic and possibly dysplastic lung changes, areas of mosaic attenuation, bronchiectasis, and an asymmetric thoracic cage.8,11,17,45,47,48
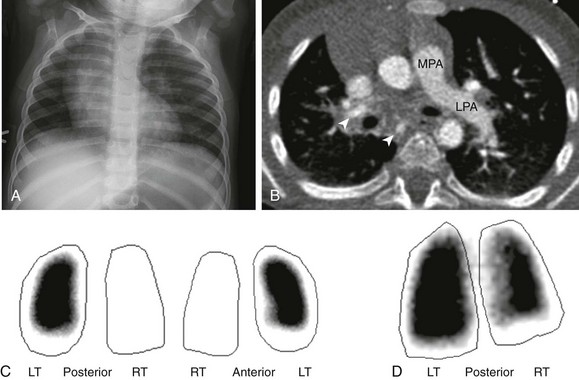
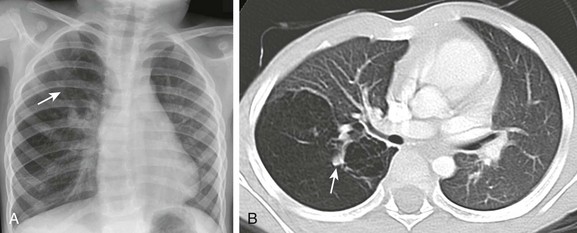
Figure 53-18 An interrupted proximal right pulmonary artery in a 16-month-old with a persistent abnormal chest radiograph following resolution of respiratory syncytial virus bronchiolitis.
A, A frontal chest radiograph shows dextroposition of the cardiomediastinal silhouette and mild asymmetry on pulmonary vascularity, decreased on the right. B, An axial computed tomography image reveals absence of the right pulmonary artery. Several collaterals to the right lung are present (arrowheads). Pulmonary blood flow is from the main pulmonary artery (MPA) to the left pulmonary artery (LPA). The aortic arch is left sided, and overall findings are consistent with proximal interruption of the right pulmonary artery. C, Techtetium-99m macroaggregated albumin lung perfusion images show absent blood flow to the right lung. D, A xenon-133 posterior planar image of the lungs shows decreased airflow to the right lung. LT, Left; RT, right.
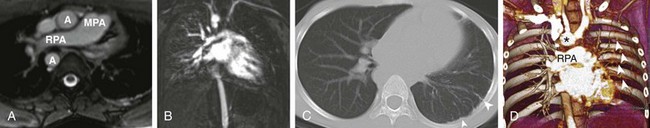
e-Figure 53-19 A 10-year-old with a history of proximal interruption of the left pulmonary artery.
A, An axial T2-weighted magnetic resonance (MR) image shows absence of the left pulmonary artery. Pulmonary blood flow is from the main pulmonary artery (MPA) to the right pulmonary artery (RPA). The aortic arch is right sided. A, aorta. B, An MR angiography image in the pulmonary arterial phase shows nearly no enhancement of the left lung. C, An axial computed tomography (CT) image shows a smaller left lung with slight mediastinal shift and serrated pleural thickening (arrowheads), reflective of the direct anastomosis of transpleural systemic collaterals with peripheral branches of the pulmonary arteries. D, A volume-rendered CT angiography image shows normal right pulmonary artery (RPA) arborization and poor vascularity in the left lung. Prominent left intercostal arteries (arrowheads) are providing collateral flow to the left lung. Note the right-sided aortic arch (asterisk).
Treatment and Follow-up: Treatment of this anomaly is aimed at prompt diagnosis and early surgical intervention, which may provide adequate blood supply to the affected lung, allowing improved pulmonary arterial and lung growth. Surveillance is indicated for patients with a late presentation who are considered unsuitable for intervention. Patients who present with recurrent hemoptysis or pulmonary hypertension may benefit from coil embolization of large systemic collaterals.8,17,50
Pulmonary Artery Sling
Etiology: Pulmonary artery sling (PAS) is a rare congenital anomaly in which the left pulmonary artery arises from the posterior aspect of the right pulmonary artery and courses between the trachea and esophagus to reach the left hilum. The anomalous left pulmonary artery forms a sling around the distal trachea and/or the proximal right mainstem bronchus.17,48,51,52 It is hypothesized that PAS develops as a result of proximal left sixth arch involution, and a secondary connection is acquired to the right sixth branchial arch via the embryonic peritracheal primitive mesenchymal vessels.48,51,52 The concurrent presence of a left ligamentum arteriosum connecting the main or right pulmonary artery and the left descending aorta results in a complete vascular ring that encircles the trachea but spares the esophagus.17,51
Two main types of PAS exist. In type I PAS, the position of the carina is normally situated at the T4-T5 level. In these instances, the airway is intrinsically normal or with an associated tracheal bronchus. The aberrant left pulmonary artery may compress the posterior wall of the distal trachea and/or the lateral aspect of the right mainstem bronchus, resulting in right lung air trapping.8,17,52 Type II PAS is more common52,53 and is associated with a more inferiorly located carina at the T6 level. Type II PAS often is associated with long-segment tracheal stenosis with complete cartilaginous rings and abnormal bronchial branching, including a T-shaped carina and a right-bridging bronchus. Other cardiovascular, GI, and right lung anomalies (e.g., lung hypoplasia, aplasia, agenesis, and scimitar syndrome) may coexist with PAS.8,17,52 Patients usually present as infants with respiratory symptoms, such as stridor, apneic spells, and/or hypoxia. The timing and severity of the respiratory symptoms depend on the severity of the accompanying airway abnormalities.8,11,52
Imaging: Imaging findings of PAS depend on its type and other coexisting congenital anomalies. On frontal chest radiographs, either hyperinflation or hypoinflation of the right lung as a result of partial or complete obstruction of the right mainstem bronchus may present in patients with type I PAS. In patients with type II PAS associated with long-segment tracheal stenosis, bilateral hyperinflation may be observed.8,52 On occasion, a small, rounded soft tissue density representing the anomalous left pulmonary artery may be seen between the midtrachea and esophagus on lateral chest radiographs in patients with both types of PAS (Fig. 53-20, A). Multidetector CT or MRI with 2D and 3D reconstructions are useful for evaluating the origin, size, and entire course of the anomalous vasculature (Fig. 53-20, A and B). CT is advantageous over MRI for evaluating associated central airway and lung abnormalities (Fig. 53-20, C). Inspiratory and expiratory imaging is particularly useful for assessing for associated tracheobronchomalacia.17,54
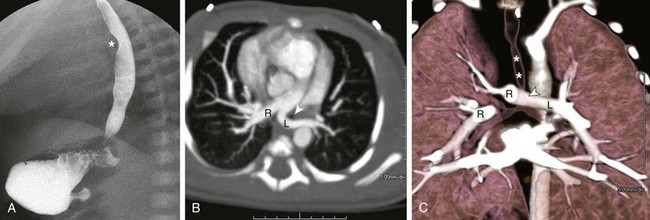
Figure 53-20 A neonate with respiratory distress and a left pulmonary artery (LPA) sling.
A, An esophagogram shows soft tissue density (asterisk) between the trachea and the esophagus, suggesting an LPA sling. B, An axial maximum intensity projection computed tomography angiography (CTA) image shows that the left pulmonary artery (L) originates from the proximal right pulmonary (R) before crossing behind the trachea (arrowhead) to feed the left lung. C, A coronal volume-rendered CTA image shows severe distal trachea narrowing (arrowhead) at the level of the anomalous course of the LPA, which originates off the right pulmonary artery (R) consistent with an LPA (L) sling. The distal trachea shows long segment narrowing (asterisks) from complete tracheal rings. Note the low T-shaped carina.
Treatment and Follow-up: Asymptomatic pediatric patients with PAS may be followed up clinically. Symptomatic pediatric patients with PAS require surgical reimplantation of the anomalous left pulmonary artery to the main pulmonary artery or anterior translocation in conjunction with excision of the coexisting patent ductus arteriosus or ductal ligament. In patients with type II PAS, reimplantation or anterior translocation of the anomalous left pulmonary artery by itself may not result in respiratory improvement if the coexisting long-segment tracheal stenosis is not repaired, usually by slide tracheoplasty.52
Anomalies of the Pulmonary Veins
Partial Anomalous Pulmonary Venous Return
Etiology: In partial anomalous pulmonary venous return (PAPVR), one or several, but not all, pulmonary veins return anomalously to the systemic circulation. Such anomalous veins typically drain into the superior or inferior vena cava, the azygos vein, or the left innominate vein. The right cardiac chambers and pulmonary vasculature frequently become enlarged because of volume overload. In the pure forms of PAPVR, pulmonary hypertension and right heart failure subsequently may develop. Unlike patients with total anomalous pulmonary venous return, most patients with PAPVR are either mildly symptomatic or asymptomatic.
PAPVR usually is an isolated finding and is more frequent on the right. The most common type of PAPVR is right upper lobe pulmonary veins draining anomalously into the superior vena cava, with or without an associated sinus venosus defect. Other forms of PAPVR sometimes are associated with a secundum type of atrial septal defect or a patent foramen ovale. The second common type of PAPVR is an anomalous connection of the left upper pulmonary vein to the left innominate vein. This connection is followed by anomalous connections of pulmonary veins from the right lung to the inferior vena cava, which may be encountered in association with an intact atrial septum and bronchopulmonary sequestration. Unlike in total anomalous pulmonary venous return, obstruction of the anomalous venous drainage pathway is rare in PAPVR.45,51,55,56
Imaging: Imaging findings of PAPVR vary depending on the site of anomalous venous connection and the presence or lack of obstruction. On chest radiographs, mild to moderate increase in pulmonary blood flow with a pattern of overcirculation may be seen (e-Fig. 53-21). If overcirculation of blood is substantial and sufficient time has elapsed, mild to moderate right-sided cardiomegaly may be observed. CT and MRI can accurately depict the anomalous venous connections (see e-Fig 53-21 and Fig. 53-22). MRI provides the additional physiologic information necessary to determine whether surgical repair is warranted. The pulmonary to systemic (Qp/Qs) flow ratio can be calculated from phase-contrast velocity mapping of the right and left pulmonary arteries and the ascending aorta, obviating the need for invasive cardiac catheterization in most cases.55,57,58
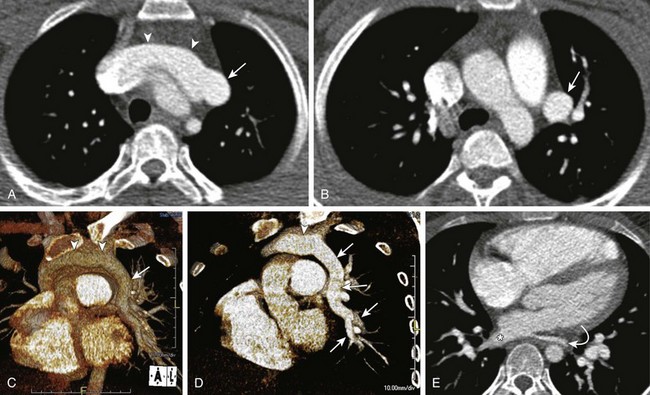
Figure 53-22 Partial anomalous pulmonary venous return to the left innominate vein.
Enhanced axial computed tomography images (A and B) show an anomalous left pulmonary vein (arrow) draining into the left innominate vein (arrowheads). Volume-rendered images (C and D) depict a large anomalous vein (arrows) draining a vast portion of the left lung into a prominent left innominate vein (arrowheads). E, An enhanced axial image shows a relative small size of the left inferior pulmonary vein (curved arrow) when compared with the right (asterisk). Only a portion of the left lower lobe drained into the left inferior pulmonary vein (curved arrow).
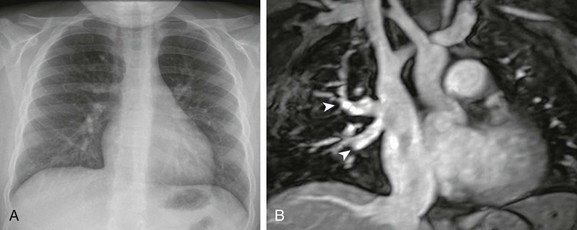
e-Figure 53-21 Partial anomalous pulmonary venous return with a sinus venosus defect in a 7-year-old girl.
A, A frontal chest radiograph demonstrates increased pulmonary blood flow. A sinus venosus defect was demonstrated on echocardiography (image not shown). B, A coronal T2-weighted magnetic resonance image shows at least two pulmonary veins (arrowheads) draining from the right upper lobe to the superior vena cava.
Treatment and Follow-up: In asymptomatic pediatric patients with PAPVR, particularly those who have a single anomalous draining vein that does not produce right ventricular volume overload, surgical management is not necessary. Although indications for surgical repair currently are not clearly established, surgical repair is recommended in symptomatic patients with a substantial volume-loaded right ventricle. The surgery type is determined by the location of the anomalous venous connection and generally consists of reconnection of the anomalous veins to the left atrium, either by direct anastomosis or through a baffle in most instances.59
Pulmonary Vein Atresia/Hypoplasia
Etiology: Pulmonary venous atresia is an uncommon anomaly associated with high morbidity and mortality. It is typically unilateral,8 and if bilateral and surgical repair is not performed on an emergency basis, the condition typically is fatal.57,60 The etiology is believed to represent the result of the unsuccessful incorporation of the common pulmonary vein into the left atrium, resulting in the lack of long segments of the central pulmonary veins.8 If unilateral, the condition may be asymptomatic or may manifest in infancy or childhood with recurrent episodes of pneumonia, hemoptysis (as a result of the systemic collateral supply to the affected lung), exercise intolerance, and pulmonary hypertension.8,61 Nearly half of the cases are associated with other forms of congenital heart disease.8,61,62
Imaging: Chest radiographs typically show a small affected hemithorax and hilum with ipsilateral mediastinal shift. The affected lung demonstrates circumferential pleural thickening, diffuse reticular opacities, and septal lines reminiscent of pulmonary edema, which are most pronounced in the lower lung zones.8,61 On enhanced CT examinations, the margins of the left atrium at the expected level of the pulmonary vein ostia appear smooth, and some adjacent enhancing soft tissue density may be present, reflecting collateral pulmonary-to-systemic venous channels. Additional collaterals may be seen in other portions of the mediastinum. The ipsilateral pulmonary artery appears small. On lung windows, diffuse ground-glass attenuation and smooth thickening of the interlobular septa and bronchovascular bundles may be observed.61 This constellation of findings is believed to represent prominent bronchial veins, dilated lymphatics, and patchy parenchymal fibrosis as a result of pulmonary infarcts61 (e-Fig. 53-23).
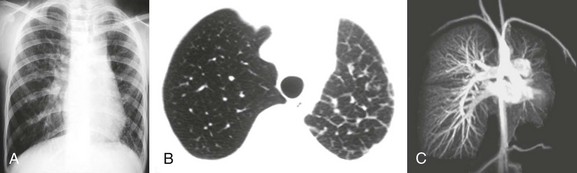
e-Figure 53-23 An asymptomatic 13-year-old girl with unilateral pulmonary vein atresia.
A, A frontal chest radiograph shows a smaller volume of the left lung, with mediastinal shift to the left. B, An axial computed tomography image shows thickened interlobular septal lines. C, A coronal magnetic resonance angiography maximum intensity projection image of the chest demonstrates decreased left pulmonary vascularity; however, a left pulmonary artery is present. The left pulmonary veins are not visualized. Note the normal arborization of the right pulmonary vascular bed and the presence of the right pulmonary veins. (Images reproduced with permission from Gasparetto TD, Daltro P, Marchiori E. Imaging findings of an asymptomatic child with pulmonary vein atresia. Pediatr Radiol. 2010;40:1458-1459, author reply 1460.)
Treatment and Follow-up: In cases of unilateral pulmonary venous atresia, surgical repair usually is not possible, depending on the age at diagnosis. Most patients present late in life once irreversible changes have occurred. Pneumonectomy may be necessary to prevent repeated pulmonary infections, to relieve the left-to-right shunt, and to remove the dead space contributing to exercise intolerance.62
Pulmonary Vein Stenosis
Etiology: Congenital pulmonary vein stenosis (PVS) is believed to be the result of an uninhibited myofibroblast-like proliferation causing endoluminal thickening and narrowing of the pulmonary veins.11,63 However, the term “primary” pulmonary vein stenosis would be more accurate, because increasing evidence exists that the disease is progressive and may not even be present at the time of delivery. Association with other congenital heart defects is high, ranging from 30% to 80%.11,64 Therefore echocardiographic evaluations of all forms of congenital heart defects should include assessment of the pulmonary veins.65 A strong association between PVS and prematurity has been recently reported, with a preponderance in premature newborns with cardiac shunt lesions.66 However, PVS also may occur in isolation, and in these cases, PVS usually progresses rapidly.64
Generally, the age at diagnosis and severity of symptoms are contingent on the number of pulmonary veins involved and the severity of pulmonary venous obstruction to individual pulmonary veins.57,64 Patients with more than three stenotic pulmonary veins have a poorer prognosis; their mortality rate approaches 85%, versus 0% in patients with one or two stenosed pulmonary veins.65 Most cases of PVS present in infancy with a history of worsening respiratory distress and recurrent pneumonias. With disease progression, pulmonary hypertension develops and becomes progressive. Therefore PVS always should be excluded in young patients with unexplained pulmonary hypertension. Hemoptysis may occur, particularly in older patients.64 Secondary PVS in pediatric patients typically occurs after anomalous pulmonary vein surgery. In approximately 10% of these patients, substantial stenosis develops either at the anastomotic site or within the central portions of the pulmonary veins.67,68
Imaging: Echocardiography usually can visualize all pulmonary veins in neonates and infants. Turbulent flow on color Doppler with flow velocities >1.6 m/s indicate hemodynamically significant pulmonary venous obstruction.69 Findings on chest radiographs include patchy reticular opacities and thickened septa, reflecting the impaired venous drainage in the affected lung. On CT, pulmonary vein thickening and narrowing can be seen. Although it typically affects the pulmonary venous-left atrial junction, it may involve more central and peripheral segments, resulting in long-segment narrowing, particularly in cases of advanced disease11,17 (Fig. 53-24). In advanced cases of PVS, lung and pleural findings may be indistinguishable from pulmonary venous atresia, with smooth septal thickening, reticular opacities, patchy ground-glass areas, and pleural thickening. MRI may show similar although lesser findings, because CT has better spatial resolution. However, MRI may provide additional physiologic information in patients.57
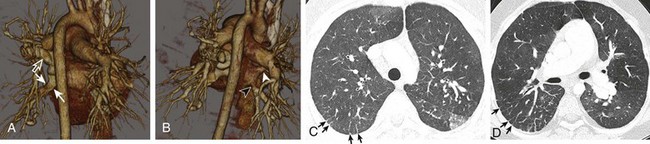
Figure 53-24 A 10-year-old boy with pulmonary venoocclusive disease.
Posterior oblique volume-rendered three-dimensional images (A and B) show severe right inferior pulmonary vein narrowing in the anteroposterior dimension resulting in flattening of the vein (solid arrows). Compensatory varicous enlargement of the right superior pulmonary vein is present (open arrow), likely because of the redirection of flow. The left inferior pulmonary vein shows approximately 50% stenosis (black arrowhead), whereas the left superior pulmonary vein (white arrowhead) remains patent. Axial computed tomography images on lung windows (C and D) show smooth interlobular septal thickening (arrows) and mild patchy areas of ground-glass opacity.
Treatment and Follow-up: PVS may be amenable to balloon dilatation occasionally followed by stent placement, although restenosis seems universal. Care should be taken, because stent implantation before surgery may limit the surgical approaches. The restenosis rate after surgery approaches 10%, despite advanced techniques reducing trauma to the veins to avoid any stimulus for regrowth of obstructive tissue. In severe cases with multiple vein involvement, lung transplantation may be necessary.64
Pulmonary Varix
Etiology: Congenital pulmonary varix is a rare congenital or acquired vascular anomaly resulting in focal aneurysmal dilatation of a pulmonary vein.11 Acquired congenital pulmonary varix typically is seen in pediatric patients who have underlying cardiac conditions resulting in pulmonary venous hypertension, such as mitral valve disease, aortic coarctation, and pulmonary vein stenosis.11 In most instances, pulmonary varices are incidental findings in otherwise asymptomatic patients. Rare complications such as rupture or thromboembolism may produce symptoms.11,17
Imaging: On chest radiographs, pulmonary varices usually present as well-defined pulmonary or mediastinal lesions in close proximity to the cardiac silhouette. They should be differentiated from other masses such as congenital lung anomaly, a neoplasm, or an infectious process. CT is particularly helpful in showing the characteristic imaging features of pulmonary varix, including contiguity between the pulmonary vein and the varix, simultaneous contrast enhancement within both structures, and lack of a feeding artery.11,17 Some authors advocate for the use of imaging modalities that can provide information regarding flow direction and pattern within the lesion, such as ultrasound, MRI, or conventional angiography to avoid confusion with a pulmonary AVM70 (e-Fig. 53-25).
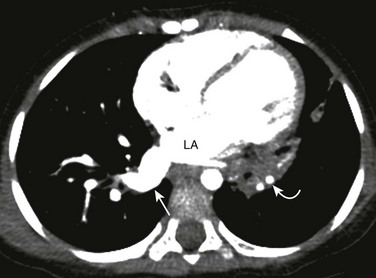
e-Figure 53-25 A pulmonary varix in a 27-month-old who underwent computed tomography (CT) study for evaluation of recurrent left lower lobe pneumonia.
An enhanced axial CT image shows an enlarged, incidentally found right inferior pulmonary vein (straight arrow) consistent with a pulmonary varix. Also noted is consolidation (curved arrow) involving the medial left lower lobe. LA, Left atrium.
Combined Anomalies of the Pulmonary Artery and Vein
Pulmonary Arteriovenous Malformation
Etiology: Pulmonary AVM is a vascular malformation due to an underlying direct connection between the pulmonary arteries and veins without an intervening capillary network.11 This bypass of the capillary network has two important physiologic consequences. First, the direct communication acts as a right-to-left shunt. Second, blood flowing through a pulmonary AVM circumvents the filter function of the normal pulmonary capillary bed, predisposing patients to paradoxical emboli.
Pulmonary AVMs can be congenital or acquired. The acquired form of pulmonary AVM usually is seen in patients with a prior history of congenital heart disease surgeries, chronic liver disease, or infections such as tuberculosis or actinomycosis.11,17 Congenital pulmonary AVMs may occur sporadically, although characteristically they are seen in 30% to 40% of family members with hereditary hemorrhagic telangiectasia (HHT), also known as Rendu-Osler-Weber syndrome. HHT is an autosomal-dominant condition, which is diagnosed clinically on the basis of Curaçao criteria (cerebral, pulmonary, or hepatic AVMs, epistaxis, family history of HHT, and telangiectasias).71 Given that each offspring of an affected person has a 50% chance of having inherited the condition, family members of patients with HHT should be screened for pulmonary AVMs.17,72
Pediatric patients with small pulmonary AVMs often are asymptomatic. Typical clinical symptoms of patients with larger or multiple pulmonary AVMs include dyspnea on exertion, cyanosis, chest pain, palpitations, and hemoptysis.73 Direct right-to-left shunting through larger or multiple pulmonary AVMs bypassing the pulmonary capillary bed can result in paradoxical emboli to the brain. Such paradoxical emboli may cause a stroke or brain abscess.
Imaging: On chest radiographs, pulmonary AVM may appear as a well-circumscribed serpiginous or lobulated opacity. Occasionally, curvilinear opacities directed toward the hilum representing the feeding artery or draining vein may be observed. Most pulmonary AVMs are located within the lower lobes. Small pulmonary AVMs or lesions in areas obscured by normal structures, such as the retrocardiac area or the pulmonary hila, may be overlooked easily.
In the past, pulmonary AVM traditionally was evaluated with conventional pulmonary angiography. Multidetector CT is now the preferred imaging modality for a complete assessment of pulmonary AVMs and can clearly show the often complex angioarchitecture of the pulmonary AVM with its feeding artery and draining vein. Reconstructed 2D and 3D images play an important role in the treatment of pulmonary AVMs by allowing preinterventional planning prior to embolization, which is of outmost importance when managing large or complex lesions17,73 (Fig. 53-26). MRA technology has narrowed the gap for the assessment of pulmonary AVMs.74
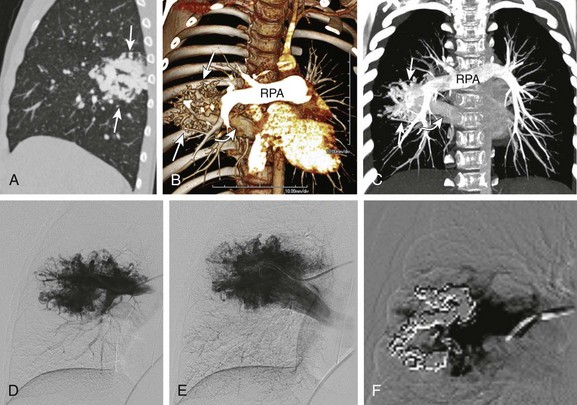
Figure 53-26 An 8-year-old with a family history of hereditary hemorrhagic telangiectasia and a positive echocardiogram for a pulmonary arteriovenous malformation (AVM).
A, An enhanced sagittal reformatted computed tomography (CT) image shows a large AVM (arrows) in the superior segment of the right lower lobe. Coronal-oblique volume-rendered (B) and coronal maximum intensity projection (C) CT angiography images show a large AVM (straight arrows) with a feeding artery from the right main pulmonary artery (RPA) and a large draining vein into an enlarged right inferior pulmonary vein (curved arrow). Pulmonary angiogram images (D and E) demonstrate a large AVM that correlates well with the CT angiography findings. A postembolization image (F) with platinum coils shows partial obliteration of a large portion of the lesion.
Pulmonary AVMs may be simple or complex. Approximately 80% to 90% of the simple angioarchitecture of pulmonary AVMs, consisting of single or multiple feeding arteries, all originate from one segmental artery and are connected directly to a single draining vein.11 Typically, both the artery and vein are dilated and are connected by the aneurysmal sac. The remaining 10% to 20% of cases involve complex architecture lesions, with two or more feeding arteries arising from at least two different segmental arteries and connecting with at least two draining veins.11 Nearly 5% of patients with HHT have multiple pulmonary AVMs.75
Treatment and Follow-up: As a general rule, treatment is offered for pulmonary AVMs with feeding arteries larger than 3 mm. However, symptomatic paradoxical emboli have been reported in patients with sub–3-mm feeding arteries. Consequently, many HHT centers currently are treating pulmonary AVMs with feeding arteries of less than 3 mm.76 The current management choice for pulmonary AVM is transvenous transcatheter embolotherapy, which can be performed using a variety of devices including coils, detachable balloons, and, most recently, with the Amplatzer vascular occluder (AGA Medical, Plymouth, MN). The latter, with the addition of at least one platinum coil, is believed to be the combination that most effectively prevents pulmonary AVM recanalization.73
Regarding pulmonary AVMs in pediatric patients, far less agreement exists with regard to who should be treated, particularly with children who are asymptomatic and younger than 12 years.75,77 Developing lungs in this setting may be at increased risk for reperfusion via pulmonary collaterals, which are even more difficult to treat. Symptomatic pulmonary AVMs should be treated regardless of the age of patients.73
Combined Parenchymal and Vascular Lesions
Hypogenetic Lung Syndrome (Scimitar Syndrome)
Etiology: Hypogenetic lung syndrome, also known as scimitar syndrome, refers to an anomalous connection of the right pulmonary veins to the inferior vena cava, in which an anomalous pulmonary vein drains part of or the entire right lung. The anomalous vein may on occasion drain into the hepatic veins, portal veins, azygos vein, coronary sinus, or right atrium. The anomalous vein often resembles a scimitar, a curved Turkish sword, hence the name “scimitar syndrome.” Hypogenetic lung syndrome frequently is associated with various degrees of right lung hypoplasia and abnormal lobation, along with heart dextroposition.45,78 Additional reported anomalies associated with hypogenetic lung syndrome include bronchogenic cyst, horseshoe lung, accessory diaphragm, hernia, and arterial supply of parts of the right lung by collateral arterial blood vessels, usually from the descending aorta.17,45 Affected infants may present with clinical signs and symptoms related to congestive heart failure from right-heart volume overload. Hypogenetic lung syndrome also may be seen as an incidental finding in older children or alternatively may manifest as recurrent right basilar pneumonia.17,79
Imaging: The classic vertically oriented curvilinear opacity, representing the scimitar vein, which projects over the right lower hemithorax in conjunction with a hypoplastic right lung, is usually seen on frontal chest radiographs. On lateral chest radiographs, a dense retrosternal band of variable width typically is observed, which is a result of the decrease in anteroposterior diameter of the hypoplastic lung, resulting in a lung–soft tissue interface.7,55 CT or MRI are the preferred imaging modalities for confirming and characterizing hypogenetic lung syndrome in pediatric patients (e-Figs. 53-27 and 53-28). Multidetector CT with 2D and 3D images have been reported as being particularly useful for displaying the entire course of the anomalous scimitar vein as a preprocedural or preoperative evaluation.11,45 In addition, they also are helpful noninvasive imaging tools for evaluating postoperative complications, including thrombosis or stenosis of a reimplanted anomalous vein. Furthermore, abnormal lung parenchymal changes, abnormal lung lobation, and anomalous bronchial branching patterns often are seen in patients with hypogenetic lung syndrome and also can be well evaluated with multidetector CT.11,45 The absence of an ipsilateral inferior pulmonary vein is a helpful finding that supports the diagnosis.
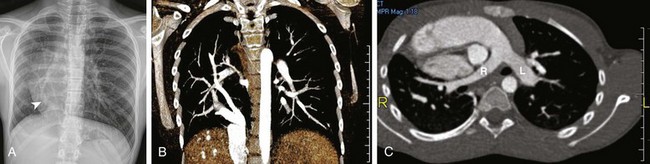
e-Figure 53-27 Hypogenetic lung syndrome in a 13-year-old with exercise intolerance.
A, A frontal chest radiograph shows a scimitar vein (arrowhead) extending to the medial right hemidiaphragm and heart dextroposition consistent with hypogenetic lung syndrome. B, A coronal volume-rendered computed tomography (CT) angiography image shows partial anomalous pulmonary venous drainage of the right lung to the inferior vena cava via two scimitar veins. A small portion of the right lung drains as expected into the left atrium. C, An axial maximum intensity projection CT image shows dextroposition of the heart and hypoplasia of the right pulmonary artery (R) relative to the left pulmonary artery (L).
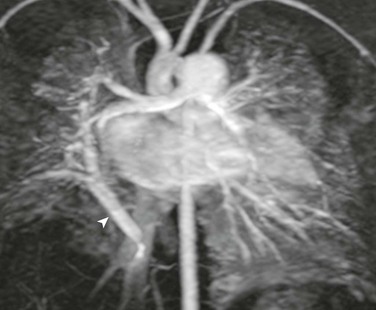
e-Figure 53-28 Hypogenetic lung syndrome in a 3-month-old with a history of an abnormal chest radiograph.
A coronal maximum intensity projection magnetic resonance image reveals anomalous pulmonary venous drainage of a vast portion of the right hypoplastic lung via a scimitar vein (arrowhead) into the inferior vena cava.
Treatment and Follow-up: For symptomatic pediatric patients with scimitar syndrome, several surgical techniques currently are available that aim to reconnect the anomalous vein to the left atrium with or without the creation of an intracardiac baffle. However, complications related to either restenosis or baffle obstruction are not uncommon.80 In addition, occlusion of the collateral arteries may be necessary in affected patients.59
Pulmonary Sequestration
Etiology: BPSs are congenital lung malformations that consist of nonfunctioning lung tissue that does not connect with the tracheobronchial tree. A BPS has a systemic arterial supply, usually from the aorta, although occasionally it may arise from branches of the celiac, splenic, intercostal, or subclavian arteries.
Sequestrations traditionally are classified as either extralobar (25%) or intralobar (75%). Extralobar sequestration (ELS) is defined as an isolated mass of lung tissue with its own pleural investment and aberrant systemic vascular supply. An ELS is believed to develop from a supernumerary lung bud that separates from the tracheobronchial tree and parasitizes its own vascular supply from the systemic circulation.6,81 Venous drainage is into the azygous or hemiazygous systems in most cases. However, an ELS may drain into the pulmonary veins or the systemic circulation, including the subclavian and intercostal veins, as well as the portal venous system.6,10,81 Although most ELSs are identified in isolation, occasionally an ELS may be associated with congenital heart defects, abnormal communications with the GI tract, pulmonary hypoplasia, ectopic pancreas, vertebral anomalies, and congenital diaphragmatic hernia, which is the most commonly associated anomaly.6,10,17,19,81 Associated microcystic maldevelopment or CPAM type 2 components have been described in many of these cases,10,82 an anomaly referred to by many investigators as hybrid lesions.83,84 Most patients with an ELS are asymptomatic.
Intralobar sequestration (ILS) lesions are defined as developmental malformations composed of isolated, nonfunctioning lung tissue without communication to the tracheobronchial tree and with an aberrant systemic vascular supply, typically embedded within a normal lobe.6,19,85 Unlike ELS lesions, ILS lesions do not have their own pleural coat, and venous drainage primarily occurs into the pulmonary veins.10,17,19,85 ILS has been considered an acquired lesion resulting from a chronic inflammatory process that recruited collateral flow from aortic branch arteries.85 Increasing reports of antenatal ILS detection, confirmed by postnatal resection, has challenged this previous concept. Although still uncertain, the hypothesis that ILS represents a developmental malformation rather than an acquired entity is most accepted.10 Pediatric patients with ILS often clinically present with a recurrent lung infection.
Imaging: Imaging findings of BPS may vary primarily on the basis of association with superimposed infection (e-Fig. 53-29), CPAM lesions (Fig. 53-30), or GI tract communication.10,17 In fetal imaging, BPS lesions present as echogenic lesions on ultrasound and as hyperintense lesions on T2-weighted images.24 Similar to CPAM and bronchial atresia, sequestrations can be detected as early as in the twelfth week of gestation,22 although most usually are diagnosed on routine ultrasound at 19 to 20 weeks. They exhibit a characteristic increase in volume from the twentieth to the twenty-sixth week and usually reach a plateau by the twenty-eighth week of gestation.3,9,22,24,40 These lesions usually decrease in size during the third trimester and seem to vanish in nearly half of the cases. However, they do not truly disappear. This phenomenon probably is partially related to the decrease in the size of the lesion, but technically it also is related to the relative increase in echogenicity of the adjacent normal lung parenchyma, which makes recognition challenging.24 Because complete regression is extremely unusual, follow-up cross-sectional postnatal imaging is recommended in all cases3,9,24 because these lesions may be overlooked on chest radiographs.86 In our experience, ultrasound duplex appears to be a highly sensitive modality for the depiction of the aberrant feeders. On fetal MRI, these feeders sometimes are difficult to identify and appear as low-signal linear structures extending from the aorta into the sequestration; they usually are best seen on coronal images.
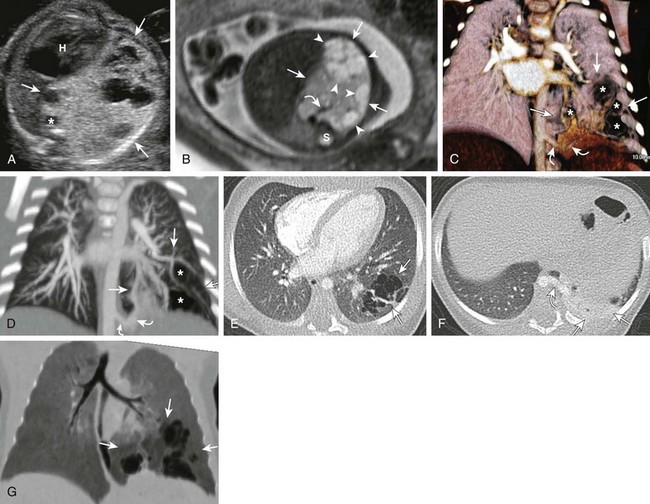
Figure 53-30 A hybrid lesion, intralobar sequestration, and microcystic maldevelopment (congenital pulmonary airway malformation [CPAM] type 2).
A, A transverse fetal ultrasound image shows a large left lower lesion (arrows) resulting in significant cardiac heart (H) and aortic (asterisk) displacement. Internal cystic components are noted mainly at the periphery of the lesion. B, An axial T2-weighted fetal magnetic resonance image shows the extensive hyperintense lesion within the left lower lobe. Cystic peripheral components consistent with CPAM are noted (arrowheads). An aortic feeder (curved arrow) is seen supplying the lesion. S, Spine. C-F, Postnatal imaging shows a complex, partially aerated left lower lesion supplied by an aberrant aortic vessel (curved arrows) with aerated, mainly peripheral cysts (asterisks) and overall findings consistent with a hybrid lesion (arrows). The lesion appears relatively smaller when compared with the prenatal images. Volume-rendered (C), maximum intensity projection (D), and axial computed tomography angiography (E and F) images show a systemic, aortic feeder (curved arrows) to the lesion (arrows). Internal cystic components are noted predominantly at the periphery (asterisks) of the lesion. A portion of unaerated lung is seen at the base of the lesion. G, A coronal reformatted minimum intensity projection image shows the internal cystic components of the CPAM (arrows) to better advantage.
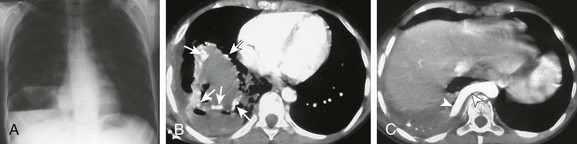
e-Figure 53-29 An infected hybrid lesion in a 12-year-old girl with a history of recurrent right lower lobe pneumonia.
A frontal chest radiograph (A) shows a large right lower lesion with an air fluid level. An axial computed tomography angiography (CTA) image (B) shows rounded, fluid-filled lesions with peripheral brisk, enhancing (arrows) margins. A small associated pleural effusion is present. An axial CTA image (C) shows a large aortic feeder (arrowheads) providing arterial systemic supply to the lesion.
On chest radiographs, ELS lesions typically present as a focal lung mass. ILS lesions often present as a focal lung mass and/or cyst but also may manifest as an area of consolidation or lung abscess, particularly in the setting of recurrent superimposed infection. Although ELSs may be encountered anywhere from the neck to below the diaphragm, they are found most commonly within the lower hemithorax, on the left side more often than the right side.6,10,81
On CT, ELS lesions characteristically appear as solid, unaerated lesions, although in nearly half of the cases, coexistent pathology with CPAM type 2 has been reported (Fig. 53-31). In these cases, internal cystic components may be identified.6,10,82 Because ILS lesions do not have a pleural investment, they typically manifest as aerated lesions, presumably from collateral air drift, if enough time has elapsed to adequately clear any retained fetal lung fluid10 (Fig. 53-32). Anomalous vascular components of BPS can be evaluated with either CT or MRI. When interpreting these studies, the real-time and interactive 2D and 3D imaging evaluation at the 2D/3D work stations can facilitate the accurate assessment of the anomalous vascular structures, which are better displayed in the z-plane.10,17,45
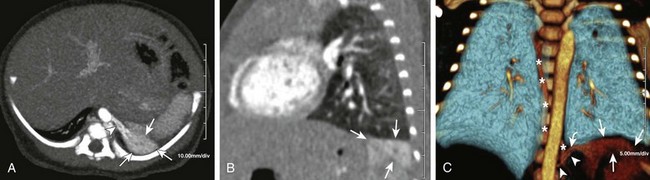
Figure 53-31 Extralobar bronchopulmonary sequestration in a 1-month-old infant.
Axial maximum intensity projection (A) and a sagittal multiplanar reconstruction (B) computed tomography images show unaerated, enhancing lung tissue in the left costophrenic sulcus with a small aortic feeder (arrowhead), consistent with extralobar sequestration (ELS) (arrows). ELS in this location easily could be confused with atelectasis. A coronal volume-rendered three-dimensional image (C) demonstrates the relationship of the sequestration (arrows) to the left lung and depicts the course of the aortic feeder (arrowheads) and the draining vein (curved arrow) into the azygos system (asterisks).
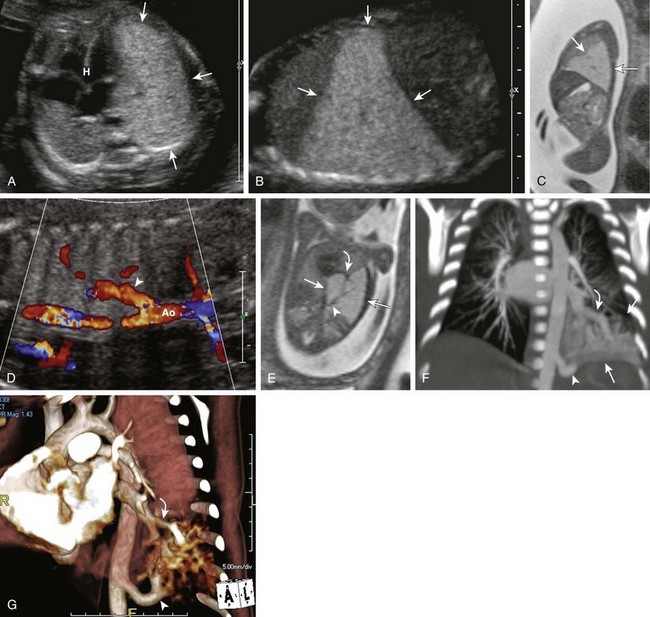
Figure 53-32 Intralobar sequestration.
A transverse ultrasound scan (A) through the chest shows a large, homogeneously hyperechoic lesion (arrows) in the left lower chest resulting in mild mediastinal shift and cardiac heart (H) displacement. Sagittal ultrasound (B) and sagittal T2-weighted fetal magnetic resonance (MR) (C) images show a homogeneous large left lower lesion without visible cysts (arrows). D, A coronal fetal ultrasound image shows an aortic feeder (arrowhead). Ao, Aorta. E, A coronal oblique T2-weighted fetal MR image shows a prominent aortic feeder (arrowhead) and a slightly prominent pulmonary vein (curved arrow) draining the large hyperintense lesion (arrows) within the left lower lobe. Coronal maximum intensity projection (F) and volume-rendered (G) computed tomography angiography images show a prominent aortic feeder (arrowhead) in a 7-day-old neonate. The lesion (straight arrows) is only partially aerated and drains into a prominent pulmonary vein (curved arrows) consistent with an intralobar bronchopulmonary sequestration. Note the relative decrease in the size of the lesion compared with prenatal imaging.
Treatment and Follow-up: Many authors support the elective surgical resection of ILSs because of the risk of complications, such as superimposed infection, pneumothorax, hemorrhage, sudden respiratory compromise, and the small risk of malignant transformation.21,22,33 Lobectomy performed via video-assisted thoracoscopic surgery is performed in many institutions, because segmentectomies may result in incomplete resection and air leaks.21,22,33
Management of ELSs is more controversial, because persons with an ELS appear to be at lower risk for the development of complications.21 Nonoperative, expectant management frequently is applied to an extrathoracic ELS, whereas an intrathoracic ELS usually is resected surgically. Their resection entails the ligation of the systemic vessels and the removal of the lesion. Arterial embolization has been reported as a successful alternative management, particularly for infants presenting with congestive heart failure,21 as evidenced by shrinkage of the lesion on follow-up imaging.87,88
 What the Clinician Needs to Know
What the Clinician Needs to Know
• Congenital lung anomalies refer to a heterogeneous group of pulmonary developmental disorders that affect the lung parenchyma, the arterial supply, and the venous drainage to the lung, or a combination of these entities.
• Despite the advent of prenatal imaging and advancement in postnatal imaging evaluation that has substantially enhanced our understanding of congenital lung anomalies, substantial controversy continues to exist regarding the nomenclature, classification, pathogenesis, description, and management of congenital lung anomalies.
• It is important to recognize that specific terminology for these lesions may be controversial and occasionally confusing. Therefore, careful evaluation and thorough understanding of all imaging findings are more important for practical care than trying to categorize the lesions by pathological terminology.
• Specific imaging findings of congenital lung anomalies that need to be evaluated include (1) location of lesions, (2) associated anomalous vascular supply and drainage of the lesions, (3) internal components and the degree of aeration, (4) exclusion of communication with the gastrointestinal tract, (5) integrity of the diaphragm, and (6) an assessment of associated anomalies, such as vertebral anomalies.
Epelman, M, Kreiger, PA, Servaes, S, et al. Current imaging of prenatally diagnosed congenital lung lesions. Semin Ultrasound CT MR. 2010;31(2):141–157.
Hellinger, JC, Daubert, M, Lee, EY, et al. Congenital thoracic vascular anomalies: evaluation with state-of-the-art MR imaging and MDCT. Radiol Clin North Am. 2011;49(5):969–996.
Langston, C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12:17–37.
Lee, EY, Dorkin, H, Vargas, SO. Congenital pulmonary malformations in pediatric patients: review and update on etiology, classification, and imaging findings. Radiol Clin North Am. 2011;49(5):921–948.
Lee, EY, Boiselle, PM, Cleveland, RH. Multidetector CT evaluation of congenital lung anomalies. Radiology. 2008;247(3):632–648.
Yikilmaz, A, Lee, EY. CT imaging of mass-like non-vascular pulmonary lesions in children. Pediatr Radiol. 2007;37(12):1253–1263.
References
1. Laberge, JM, Flageole, H, Pugash, D, et al. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: a Canadian experience. Fetal Diagn Ther. 2001;16:178–186.
2. Duncombe, GJ, Dickinson, JE, Kikiros, CS. Prenatal diagnosis and management of congenital cystic adenomatoid malformation of the lung. Am J Obstet Gynecol. 2002;187:950–954.
3. Azizkhan, RG, Crombleholme, TM. Congenital cystic lung disease: contemporary antenatal and postnatal management. Pediatr Surg Int. 2008;24:643–657.
4. Clements, BS, Warner, JO. Pulmonary sequestration and related congenital bronchopulmonary-vascular malformations: nomenclature and classification based on anatomical and embryological considerations. Thorax. 1987;42:401–408.
5. Mata, JM, Caceres, J. The dysmorphic lung: imaging findings. Eur Radiol. 1996;6:403–414.
6. Langston, C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12:17–37.
7. Mata, JM, Cáceres, J, Lucaya, J, et al. CT of congenital malformations of the lung. Radiographics. 1990;10:651–674.
8. Newman, B. Congenital bronchopulmonary foregut malformations: concepts and controversies. Pediatr Radiol. 2006;36:773–791.
9. Alamo, L, Gudinchet, F, Reinberg, O, et al. Prenatal diagnosis of congenital lung malformations. Pediatr Radiol. 2012;42(3):273–283.
10. Epelman, M, Kreiger, PA, Servaes, S, et al. Current imaging of prenatally diagnosed congenital lung lesions. Semin Ultrasound CT MR. 2010;31:141–157.
11. Lee, EY, Boiselle, PM, Cleveland, RH. Multidetector CT evaluation of congenital lung anomalies. Radiology. 2008;247:632–648.
12. Riedlinger, WF, Vargas, SO, Jennings, RW, et al. Bronchial atresia is common to extralobar sequestration, intralobar sequestration, congenital cystic adenomatoid malformation, and lobar emphysema. Pediatr Dev Pathol. 2006;9:361–373.
13. Kunisaki, SM, Fauza, DO, Nemes, LP, et al. Bronchial atresia: the hidden pathology within a spectrum of prenatally diagnosed lung masses. J Pediatr Surg. 2006;41:61–65.
14. Daltro, P, Werner, H, Gasparetto, TD, et al. Congenital chest malformations: a multimodality approach with emphasis on fetal MR imaging. Radiographics. 2010;30:385–395.
15. Talner, LB, Gmelich, JT, Liebow, AA, et al. The syndrome of bronchial mucocele and regional hyperinflation of the lung. Am J Roentgenol Radium Ther Nucl Med. 1970;110:675–686.
16. Zylak, CJ, Eyler, WR, Spizarny, DL, et al. Developmental lung anomalies in the adult: radiologic-pathologic correlation. Radiographics. 2002;22:S25–S43.
17. Lee, EY, Dorkin, H, Vargas, SO. Congenital pulmonary malformations in pediatric patients: review and update on etiology, classification, and imaging findings. Radiologic Clin North Am. 2011;49:921–948.
18. Abitayeh, G, Ruano, R, Martinovic, J, et al. Prenatal diagnosis of main stem bronchial atresia using 3-dimensional ultrasonographic technologies. J Ultrasound Med. 2010;29:633–638.
19. Biyyam, DR, Chapman, T, Ferguson, MR, et al. Congenital lung abnormalities: embryologic features, prenatal diagnosis, and postnatal radiologic-pathologic correlation. Radiographics. 2010;30:1721–1738.
20. Peranteau, WH, Merchant, AM, Hedrick, HL, et al. Prenatal course and postnatal management of peripheral bronchial atresia: association with congenital cystic adenomatoid malformation of the lung. Fetal Diagn Ther. 2008;24:190–196.
21. Eber, E. Antenatal diagnosis of congenital thoracic malformations: early surgery, late surgery, or no surgery? Semin Respir Crit Care Med. 2007;28:355–366.
22. Laje, P, Liechty, KW. Postnatal management and outcome of prenatally diagnosed lung lesions. Prenat Diagn. 2008;28:612–618.
23. Mani, H, Suarez, E, Stocker, JT. The morphologic spectrum of infantile lobar emphysema: a study of 33 cases. Paediatr Respir Rev. 2004;5(suppl A):S313–S320.
24. Bulas, D, Egloff, AM. Fetal chest ultrasound and magnetic resonance imaging: recent advances and current clinical applications. Radiol Clin North Am. 2011;49:805–823.
25. Ozcelik, U, Gocmen, A, Kiper, N, et al. Congenital lobar emphysema: evaluation and long-term follow-up of thirty cases at a single center. Pediatr Pulmonol. 2003;35:384–391.
26. Ch’In, KY, Tang, MY. Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol (Chic). 1949;48:221–229.
27. Rosado-de-Christenson, ML, Stocker, JT. Congenital cystic adenomatoid malformation. Radiographics. 1991;11:865–886.
28. Stocker, JT, Madewell, JE, Drake, RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8:155–171.
29. Stocker, JT. Congenital pulmonary airway malformation—a new name for an expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathology. 2002;41(suppl 2):424–431.
30. Stocker, JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol. 2009;28:155–184.
31. Priest, JR, Williams, GM, Hill, DA, et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009;44:14–30.
32. Adzick, NS, Harrison, MR, Glick, PL, et al. Fetal cystic adenomatoid malformation: prenatal diagnosis and natural history. J Pediatr Surg. 1985;20:483–488.
33. Laberge, JM, Puligandla, P, Flageole, H. Asymptomatic congenital lung malformations. Semin Pediatr Surg. 2005;14:16–33.
34. Colon, N, Schlegel, C, Pietsch, J, et al. Congenital lung anomalies: can we postpone resection? J Pediatr Surg. 2012;47:87–92.
35. Bush, A, Hogg, J, Chitty, LS. Cystic lung lesions—prenatal diagnosis and management. Prenat Diagn. 2008;28:604–611.
36. Chetcuti, PAJ, Crabbe, DCG. CAM lungs: the conservative approach. Arch Dis Childhood Fetal Neonatal Ed. 2006;91:F463–F464.
37. Jaffé, A, Chitty, LS. Congenital cystic adenomatoid malformations may not require surgical intervention. Arch Dis Childhood Fetal Neonatal Ed. 2006;91:F464.
38. Aziz, D, Langer, JC, Tuuha, SE, et al. Perinatally diagnosed asymptomatic congenital cystic adenomatoid malformation: to resect or not? J Pediatr Surg. 2004;39:329–334.
39. Fitzgerald, DA. Congenital cyst adenomatoid malformations: resect some and observe all? Paediatr Respir Rev. 2007;8:67–76.
40. Chuang, S, Sugo, E, Jaffe, A. A review of postnatal management of congenital pulmonary airway malformations. Fetal Maternal Med Rev. 2009;20:179–204.
41. Berrocal, T, Madrid, C, Novo, S, et al. Congenital anomalies of the tracheobronchial tree, lung, and mediastinum: embryology, radiology, and pathology. Radiographics. 2004;24:e17.
42. Cunningham, ML, Mann, N. Pulmonary agenesis: a predictor of ipsilateral malformations. Am J Med Genet. 1997;70:391–398.
43. Gilbert, EF, Opitz, JM. The pathology of some malformations and hereditary diseases of the respiratory tract. Birth Defects Orig Artic Ser. 1976;12:239–270.
44. Eroglu, A, Alper, F, Turkyilmaz, A, et al. Pulmonary agenesis associated with dextrocardia, sternal defects, and ectopic kidney. Pediatr Pulmonol. 2005;40:547–549.
45. Konen, E, Raviv-Zilka, L, Cohen, RA, et al. Congenital pulmonary venolobar syndrome: spectrum of helical CT findings with emphasis on computerized reformatting. Radiographics. 2003;23:1175–1184.
46. Trivedi, KR, Karamlou, T, Yoo, SJ, et al. Outcomes in 45 children with ductal origin of the distal pulmonary artery. Ann Thorac Surg. 2006;81:950–957.
47. Dillman, JR, Sanchez, R, Ladino-Torres, MF, et al. Expanding upon the unilateral hyperlucent hemithorax in children. Radiographics. 2011;31:723–741.
48. Castañer, E, Gallardo, X, Rimola, J, et al. Congenital and acquired pulmonary artery anomalies in the adult: radiologic overview. Radiographics. 2006;26:349–371.
49. Dillman, JR, Sanchez, R, Ladino-Torres, MF, et al. Expanding upon the unilateral hyperlucent hemithorax in children. Radiographics. 2011;31:723–741.
50. Apostolopoulou, SC, Kelekis, NL, Brountzos, EN, et al. “Absent” pulmonary artery in one adult and five pediatric patients: imaging, embryology, and therapeutic implications. Am J Roentgenol. 2002;179:1253–1260.
51. Hellinger, JC, Daubert, M, Lee, EY, et al. Congenital thoracic vascular anomalies: evaluation with state-of-the-art MR imaging and MDCT. Radiol Clin North Am. 2011;49:969–996.
52. Newman, B, Ya, Cho. Left pulmonary artery sling—anatomy and imaging. Semin Ultrasound CT MRI. 2010;31:158–170.
53. Lee, K-H, Yoon, CS, Choe, KO, et al. Use of imaging for assessing anatomical relationships of tracheobronchial anomalies associated with left pulmonary artery sling. Pediatr Radiol. 2001;31:269–278.
54. Lee, KS, Boiselle, PM. Update on multidetector computed tomography imaging of the airways. J Thorac Imaging. 2010;25:112–124.
55. Epelman, M, MacDonald, C. Partial and total anomalous pulmonary venous connections. In: Yoo S-J, MacDonald C, Babyn P, eds. Chest radiographic interpretation in pediatric cardiac patients. New York: Thieme Medical Publishers, 2010.
56. Keane, JF, Fyler, DC. Atrial septal defect. In Keane JF, Fyler DC, Lock JE, eds.: Nadas’ pediatric cardiology, 2nd ed, Philadelphia: WB Saunders, 2006.
57. Vyas, HV, Greenberg, SB, Krishnamurthy, R. MR Imaging and CT evaluation of congenital pulmonary vein abnormalities in neonates and infants. Radiographics. 2012;32:87–98.
58. Valsangiacomo Buechel, ER, Fogel, MA. Congenital cardiac defects and MR-guided planning of surgery. Magn Reson Imaging Clin N Am. 2011;19:823–840. [viii].
59. Webb, GDS, Smallhorn, JF, Therrien, J, et al. Congenital heart disease. In Bonow RO, Mann DL, Zipes DP, et al, eds.: Braunwald’s heart disease: a textbook of cardiovascular medicine, 9th ed, Philadelphia: WB Saunders, 2011.
60. Dominguez Garcia, O, Granados Ruiz, MA, Sanchez-Redondo, MD, et al. A difficult emergency surgical diagnosis: atresia of the common pulmonary vein. Pediatr Cardiol. 2009;30:989–991.
61. Heyneman, LE, Nolan, RL, Harrison, JK, et al. Congenital unilateral pulmonary vein atresia. Am J Roentgenol. 2001;177:681–685.
62. Pourmoghadam, KK, Moore, JW, Khan, M, et al. Congenital unilateral pulmonary venous atresia: definitive diagnosis and treatment. Pediatr Cardiol. 2003;24:73–79.
63. Riedlinger, WF, Juraszek, AL, Jenkins, KJ, et al. Pulmonary vein stenosis: expression of receptor tyrosine kinases by lesional cells. Cardiovasc Pathol. 2006;15:91–99.
64. Latson, LA, Prieto, LR. Congenital and acquired pulmonary vein stenosis. Circulation. 2007;115:103–108.
65. Breinholt, JP, Hawkins, JA, Minich, L, et al. Pulmonary vein stenosis with normal connection: associated cardiac abnormalities and variable outcome. Ann Thorac Surg. 1999;68:164–168.
66. Drossner, DM, Kim, DW, Maher, KO, et al. Pulmonary vein stenosis: prematurity and associated conditions. Pediatrics. 2008;122:e656–e661.
67. Hancock Friesen, CL, Zurakowski, D, Thiagarajan, RR, et al. Total anomalous pulmonary venous connection: an analysis of current management strategies in a single institution. Ann Thorac Surg. 2005;79:596–606.
68. Caldarone, CA, Najm, HK, Kadletz, M, et al. Relentless pulmonary vein stenosis after repair of total anomalous pulmonary venous drainage. Ann Thorac Surg. 1998;66:1514–1520.
69. Smallhorn, JF, Pauperio, H, Benson, L, et al. Pulsed Doppler assessment of pulmonary vein obstruction. Am Heart J. 1985;110:483–486.
70. Abujudeh, H. Pulmonary varix: blood flow is essential in the diagnosis. Pediatr Radiol. 2004;34:567–569.
71. Guttmacher, A, Pyeritz, R. Hereditary hemorrhagic telangiectasia. In Rimoin DL, Connor JM, Pyeritz RE, et al, eds.: Principles and practice of medical genetics, 5th ed, Philadelphia: Churchill Livingstone, 2007.
72. HHT Foundation International. Screening and treatment (website). http://hht.org/living-with-hht/screening-and-treatment/. [Accessed October 35, 2012].
73. Trerotola, SO, Pyeritz, RE. PAVM embolization: an update. Am J Roentgenol. 2010;195:837–845.
74. Schneider, G, Uder, M, Koehler, M, et al. MR angiography for detection of pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Am J Roentgenol. 2008;190:892–901.
75. Faughnan, ME, Thabet, A, Mei-Zahav, M, et al. Pulmonary arteriovenous malformations in children: outcomes of transcatheter embolotherapy. J Pediatr. 2004;145:826–831.
76. Faughnan, ME, Palda, VA, Garcia-Tsao, G, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48:73–87.
77. Pollak, JS, Saluja, S, Thabet, A, et al. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radiol. 2006;17:35–44. [quiz 45].
78. Godwin, JD, Tarver, RD. Scimitar syndrome: four new cases examined with CT. Radiology. 1986;159:15–20.
79. Lee, EY, Boiselle, PM, Cleveland, RH. Multidetector CT evaluation of congenital lung anomalies. Radiology. 2008;247:632–648.
80. Alsoufi, B, Cai, S, Van Arsdell, GS, et al. Outcomes after surgical treatment of children with partial anomalous pulmonary venous connection. Ann Thorac Surg. 2007;84:2020–2026.
81. Rosado-de-Christenson, ML, Frazier, AA, Stocker, JT, et al. From the archives of the AFIP. Extralobar sequestration: radiologic-pathologic correlation. Radiographics. 1993;13:425–441.
82. Conran, RM, Stocker, JT. Extralobar sequestration with frequently associated congenital cystic adenomatoid malformation, type 2: report of 50 cases. Pediatr Dev Pathol. 1999;2:454–463.
83. Cass, DL, Crombleholme, TM, Howell, LJ, et al. Cystic lung lesions with systemic arterial blood supply: a hybrid of congenital cystic adenomatoid malformation and bronchopulmonary sequestration. J Pediatr Surg. 1997;32:986–990.
84. Adzick, NS. Management of fetal lung lesions. Clin Perinatol. 2009;36:363–376.
85. Frazier, AA, Rosado de Christenson, ML, Stocker, JT, et al. Intralobar sequestration: radiologic-pathologic correlation. Radiographics. 1997;17:725–745.
86. Sauvat, F, Michel, JL, Benachi, A, et al. Management of asymptomatic neonatal cystic adenomatoid malformations. J Pediatr Surg. 2003;38:548–552.
87. Curros, F, Chigot, V, Emond, S, et al. Role of embolisation in the treatment of bronchopulmonary sequestration. Pediatr Radiol. 2000;30:769–773.
88. Lee, K-H, Sung, K-B, Yoon, H-K, et al. Transcatheter arterial embolization of pulmonary sequestration in neonates: long-term follow-up results. J Vasc Interv Radiol. 2003;14:363–367.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 53
Congenital Lung Anomalies
Congenital lung anomalies refer to a heterogeneous group of pulmonary developmental disorders that affect the lung parenchyma, the arterial supply, and the venous drainage to the lung, or a combination of these entities. The reported incidence of congenital lung anomalies ranges from 1.2 : 10,000 to 1 : 35,000 pregnancies; however, these reports may be an underestimate of their true incidence.1–3 Although prenatal sonography (ultrasound), advances in postnatal imaging, and, more recently, fetal magnetic resonance imaging (MRI) have enhanced our understanding of congenital lung anomalies, substantial controversy continues regarding the nomenclature, classification, pathogenesis, description, and management of these lesions. In addition, considerable variability exists in their prenatal clinical presentation and outcome, ranging from in-utero involution to severe hydrops and fetal demise. Likewise, their postnatal clinical presentation also is variable, ranging from the completely asymptomatic newborn to the older child or young adult with recurrent pneumonias. In this chapter, the underlying etiology, clinical presentation, imaging findings, and management of various congenital lung anomalies encountered in the pediatric population are discussed.
Spectrum of Congenital Lung Anomalies
The classification of congenital lung anomalies is challenging and continuously controversial from embryologic, radiologic, pathologic, and clinical viewpoints. Several classifications and terminologies with their own advantages and disadvantages have been suggested.4–6 Some investigators have used embryology as a basis and have classified congenital lung anomalies according to the stage of intrauterine development in which the insult resulting in the malformation developed.4 Other investigators have categorized lesions based on their morphologic-radiologic features and divided them into two groups: whole lung malformations (e.g., lung hypoplasia) and focal malformations (e.g., bronchial atresia).5,7 Recently the Langston6 classification has become one of the most accepted classification systems for congenital lung anomalies, particularly from the pathological standpoint, although it is by no means the most widely used classification by all clinical groups. Langston categorizes the wide spectrum of respiratory tract malformations primarily as bronchial atresia, congenital pulmonary airway malformation (CPAM), extralobar bronchopulmonary sequestration (BPS), congenital lobar hyperinflation (CLH), and bronchogenic cysts. These five congenital lung anomalies comprise approximately 90% of the anomalies seen in clinical practice. However, this classification system is limited because other congenital lung anomalies (e.g., pulmonary arteriovenous malformation [AVM]) are not included.
For the purpose of relatively clear classification, easy differentiation on imaging studies, and preoperative assessment of surgical lesions, congenital lung anomalies discussed in this chapter are categorized according to their morphologic-radiologic-pathologic features. Such a classification system views congenital lung anomalies as a continuum ranging from predominantly parenchymal abnormalities (i.e., abnormal lung parenchyma, relatively normal vasculature, airway, and foregut derivatives, e.g., CPAM), to predominantly vascular abnormalities (i.e., normal lung parenchyma, normal airway, and no foregut abnormality but abnormal vasculature; e.g., AVM), to combined parenchymal and vascular abnormalities (e.g., pulmonary sequestration and scimitar syndrome) in which influencing factors (foregut and airway components) play an important role and the major abnormalities are intertwined (Fig. 53-1).
Figure 53-1 Diagram of the spectrum of congenital lung anomalies, including foregut and airway components.
The lesions in yellow denote those most commonly encountered in this entity, whereas the lesions in white represent additional lesions that can be considered part of the spectrum. AVM, Arteriovenous malformation; CPAM, congenital pulmonary airway malformation; CLH, congenital lobar hyperinflation. (Adapted from Newman B. Congenital bronchopulmonary foregut malformations: concepts and controversies. Pediatr Radiol. 2006;36:773-791.)
Given that specific terminology for these lesions may be controversial and occasionally confusing, we and other authors8–11 recommend that radiologists thoroughly describe all imaging findings of congenital lung anomalies rather than try to categorize the lesions by pathologic terminology. Specific imaging findings of congenital lung anomalies that need to be evaluated and described include (1) location of lesions, (2) associated anomalous vascular supply and drainage of the lesions, (3) internal components and the degree of aeration, (4) exclusion of communication with the gastrointestinal (GI) tract, (5) integrity of the diaphragm, and (6) an assessment of associated anomalies, such as vertebral anomalies.8,10
Predominantly Parenchymal Lesions
Etiology: Bronchial atresia refers to atresia of a lobar, segmental, or subsegmental bronchus at or near its origin resulting in a blind-ended atretic proximal bronchus. Bronchial atresia most frequently affects a segmental bronchus. The precise etiology of bronchial atresia remains unknown, but etiologies such as a vascular insult to the involved atretic or stenotic portion have been proposed.6,8 Several authors who used modern dissecting techniques found that bronchial atresia is more common than originally thought.6,12,13 Furthermore, bronchial atresia and BPSs coexist in nearly all cases,6,12,13 whereas it is found in nearly 70% of CPAM lesions.12 A malformation sequence resulting from airway obstruction during development has been proposed as the unifying element for such a wide spectrum of imaging appearances. Differences in degree, level, and timing of the bronchial obstruction are thought to be responsible for the association of bronchial atresia and other congenital lung anomalies.6,8,12 Bronchial atresia usually is diagnosed as an incidental finding on chest radiographs later in life in asymptomatic older children or adults.6,10 However, bronchial atresia increasingly is being diagnosed in utero, given the widespread use of prenatal imaging.6,9,10,12,13
Imaging: Prenatally, the involved portion of the lung appears hyperexpanded and shows increased homogenous echogenicity on ultrasound and high T2 signal on fetal MRI14 (Fig. 53-2). On occasion, it is possible to identify the centrally located, mucus-filled bronchocele/mucocele on prenatal ultrasound or MRI9,15 (e-Fig. 53-3).
Figure 53-2 Bronchial atresia.
Axial (A) and coronal (B) T2-weighted fetal magnetic resonance images in a 22-week gestational age fetus demonstrate a large homogeneous lesion in the left upper lobe (arrows). Central fluid-filled bronchi are noted (arrowheads). The left lower lobe (asterisk) is compressed inferiorly and displaced medially adjacent to the fetal heart (H). The curved arrow denotes the aorta, which is also displaced to the right. L, Liver. C, A coronal multiplanar reconstruction computed tomography (CT) image shows partial opacification (asterisk) of the left upper lobe, presumably due to mucostasis related to bronchial atresia. In addition, a small air bubble is seen (arrowhead), reflecting a bronchocele. D, A volume-rendered reconstructed CT image could not delineate the left upper lobe bronchus.
e-Figure 53-3 Bronchial atresia.
Coronal (A) and axial (B) T2-weighted fetal magnetic resonance images show a large hyperintense, homogeneous lesion in the left upper lobe (arrows) in a 21-week gestational age fetus. The left lower lobe (asterisk) is displaced and compressed inferiorly. The heart (H) is shifted to the right. Central fluid-filled bronchi are noted (arrowheads). S, Stomach. C, A frontal chest radiograph at birth shows complete opacification of the left upper lobe, presumably as a result of retained fetal fluid and mucostasis related to bronchial atresia. The fluid-filled central cystic structure reflects a large mucocele/bronchocele. Axial computed tomography (CT) angiography (D) and minimum intensity projection (E) images performed at day 2 of life show complete collapse of the left upper lobe (asterisk). In addition, the left upper lobe bronchus could not be delineated, a finding suggestive of bronchial atresia. Three-dimensional reconstructions are invaluable in showing the “interrupted connection” to the remainder of the airway.
Characteristically, the apicoposterior segmental bronchus of the left upper lobe is most commonly affected, followed by the segmental bronchi of the right upper, right middle, and lower lobes.16 In children, radiographic and computed tomography (CT) imaging features of bronchial atresia are characterized by a tubular or glove-shaped opacity representing mucus plugging in the region distal to the atretic bronchus, surrounding segmental hyperlucency due to air trapping, and decreased underlying vascularity15,17 (e-Fig. 53-4). However, in neonates, a portion of the lung distal to the atretic segment may remain atelectatic as a result of the in-utero mucostasis. Therefore some authors recommend avoiding immediate postnatal imaging evaluation because the under-aerated lung related to bronchial atresia may be difficult to differentiate from the normal lung with expected fetal fluid retention in the newborn period.10 Identification of the hallmark atretic bronchus and the “bronchocele/mucocele” by means of two-dimensional (2D) multiplanar or three-dimensional (3D) reconstructions either on prenatal or postnatal imaging may be helpful.10,18
Treatment and Follow-up: Management of bronchial atresia is somewhat varied. In general, surgical resection is primarily recommended in symptomatic pediatric patients because of recurrent infection.17,19 Although opinions vary, some centers advocate elective surgical resection of bronchial atresia even in asymptomatic pediatric patients because of potential future lung infection and increased association with CPAM.13,20
Bronchogenic Cysts
Etiology: Bronchogenic cysts result from abnormal tracheobronchial branching and presumably originate from an aberrant bud of the developing foregut, similar to other foregut duplication cysts. Bronchogenic cysts typically are unilocular, fluid-filled, or mucus-filled cysts lined by respiratory epithelium and are attached to but do not communicate with the tracheobronchial tree.6,17,19 Although most bronchogenic cysts are located within the mediastinum (predominantly near the carina), they may be encountered anywhere from the suprasternal area to the retroperitoneum. Bronchogenic cysts also may be found within the lung parenchyma, usually in the lower lobes.17,19 Such intrapulmonary bronchogenic cysts do not communicate with the airway unless superimposed infection with wall necrosis occurs,6 which may further predispose them to recurrent infections. The clinical symptomatology of affected pediatric patients primarily depends on the mass effect the lesion exerts on its neighboring structures including the airway, GI tract, and cardiovascular structures. Airway compression is usually mild, but it may be life threatening in some instances when large bronchogenic cysts are located near the carina regions.21
Imaging: A bronchogenic cyst typically presents as a round or oval-shaped cystic lesion located near the right paratracheal or subcarinal area within the middle mediastinum. Bronchogenic cysts are anechoic on prenatal ultrasound and show high signal intensity on T2-weighted prenatal MRI imaging14,19 (e-Fig. 53-5). On chest radiographs, a bronchogenic cyst manifests as a well-delineated round or oval-shaped middle mediastinal mass. On CT, approximately 50% of bronchogenic cysts demonstrate fluid attenuation value (~0 Hounsfield unit) (Fig. 53-6). The remaining bronchogenic cysts may have CT attenuation higher than water because of thick mucoid, milk-of-calcium, proteinaceous, or hemorrhagic contents. MRI, which can confirm the cystic nature of the bronchogenic cysts on T2-weighted images, is helpful for differentiating bronchogenic cysts with high attenuation value from a mildly enhancing solid mass on CT. Typically, no internal contrast enhancement is seen within the uncomplicated bronchogenic cysts on CT or MRI. The presence of an air-fluid level, thick wall enhancement, or surrounding inflammatory changes often is associated with superimposed infection.11,16,17
Figure 53-6 A bronchogenic cyst in a 15-month-old child with stridor.
The frontal chest radiograph (A) shows hyperinflation of the left lung. The lateral radiograph (B) reveals the effect of the mass and posterior displacement of the airway (arrow). C, An upper gastrointestinal image shows an esophageal displacement by a soft tissue density mass. Enhanced axial (D) and sagittal (E) computed tomography images show a fluid density lesion (asterisks) posterior to the lower trachea consistent with a bronchogenic cyst.
e-Figure 53-5 Bronchogenic cyst.
Coronal (A) and axial (B) T2-weighted fetal magnetic resonance images in a 26-week gestational age fetus show a cystic structure in the posterior mediastinum (arrows) apparently compressing the left mainstem bronchus (arrowhead) and resulting in hyperexpansion with increased fluid signal in the left upper lobe (white asterisks). The left lower lobe is compressed and displaced inferiorly (black asterisks). The fetal heart (H) and mediastinum have shifted to the right. Enhanced computed tomography images in mediastinal (C) and lung (D) windows show the fluid-filled cystic structure consistent with a bronchogenic cyst (arrows) centered at the left hilum and extending posteriorly and inferiorly. The cyst compresses the left mainstem bronchus and results in air trapping in the left upper lobe.
Treatment and Follow-up: Complete surgical resection is the current management of choice for bronchogenic cysts, particularly in symptomatic pediatric patients.22 Temporizing or palliative procedures such as transparietal, transbronchial, or mediastinal aspiration may be considered in symptomatic pediatric patients who are not surgical candidates.
Congenital Lobar Hyperinflation
Etiology: CLH, also referred to as infantile lobar emphysema or congenital lobar emphysema, presumably is caused by an intrinsic or extrinsic bronchial narrowing, resulting in subsequent air trapping. Intrinsic bronchial narrowing can be caused by weakness or absence of underlying bronchial cartilage, whereas extrinsic bronchial narrowing may be due to the compression from adjacent mediastinal masses or enlarged vessels. CLH clinically presents with respiratory distress in the newborn period14,17,19 in nearly half of the cases and by the age of 6 months in 80% of cases.23 There is a slight male predominance, and the upper lobes are affected more frequently than the lower lobes, with the left lung affected more often than the right.23
Imaging: On prenatal imaging, CLH manifests as a homogeneously hyperechogenic lesion on ultrasound or as a T2 hyperintense lesion on MRI without visible cysts.9,24 On prenatal imaging, CLH often is indistinguishable from other congenital lung anomalies, particularly bronchial atresia.9,24 CLH usually is diagnosed by its typical clinical presentation and characteristic radiographic features of progressive lobar hyperexpansion and hyperlucency, producing displacement or compression of adjacent structures. In the immediate postnatal period, CLH initially may appear as an area of increased opacity related to retained fetal lung fluid, which will clear on subsequent studies.6,17,19,23 Similar imaging findings are noted on CT, and the attenuated pulmonary vasculature is a helpful clue to distinguish CLH from a pneumothorax or other entities in cases of inconclusive chest radiographic findings17 (Fig. 53-7).
Congenital Pulmonary Airway Malformation
Etiology: CPAMs, formerly known as cystic adenomatoid malformations of the lung, were first described in the literature by Ch’In and Tang in 1949 as rare lung lesions occurring in premature or stillborn infants with significant hydrops.26,27 CPAMs are characterized by a heterogeneous group of congenital cystic and noncystic lung masses that communicate with an abnormal bronchial tree lacking supporting cartilage.8,17,28
In 1977, Stocker et al.28 classified these lesions based on their clinical and pathologic features, with subdivisions based on the size of the cysts (types I, II, and III) and according to the location of suspected development of the malformation along the airway. Type I CPAMs consist of cysts larger than 2 cm, with presumed bronchial/bronchiolar origin. Type II CPAMs consist of cysts smaller than 2 cm, with presumed bronchiolar origin. Type III CPAMs appear solid, with a presumed bronchiolar/alveolar origin. However, Stocker later expanded his CPAM classification into five types that included type 0 CPAMs, with presumed tracheobronchial origin, and type IV CPAMs, with presumed distal acinar origin. The term CPAM was now implemented instead of cystic adenomatoid malformations, because cystic changes were observed in only three of the aforementioned types (types, I, II, and IV), and adenomatoid change was observed only in type III.9,17,29,30 Increasing evidence indicates that type IV CPAM lesions and type I pleuropulmonary blastomas may represent the same entity.6,31 It is important to recognize that although Stocker’s classification is widely used, it is by no means universally accepted. For an example, Langston6 classifies CPAM lesions into two types and terms the Stocker type I CPAM as “large cyst type lesions” and the Stocker type II CPAMs as “small cyst type lesions” based on cyst size and pathologic criteria.6 Langston proposed that the type III CPAM actually represents a form of pulmonary hyperplasia and should be excluded from the CPAM classification.
Imaging: Prenatally, CPAMs are classified on the basis of cyst size as microcysts (<5 mm) and macrocysts (≥5 mm) on fetal ultrasound and MRI.32 Type 1 CPAMs may contain one, several, or multiple macrocysts, some of which are ≥5 mm in diameter9 (Fig. 53-8). Type II CPAMs have variable appearances, ranging from homogeneously hyperechoic or hyperintense lesions to microcystic lesions exhibiting multiple, uniform cysts that measure <5 mm9,14,19 (e-Figs. 53-9 and 53-10).
Figure 53-8 Congenital pulmonary airway malformation type I.
Sagittal ultrasound (A), axial ultrasound (B), and fetal T2-weighted magnetic resonance sagittal (C) images in a 22-week gestational age fetus show a large, heterogeneous, multicystic lesion occupying the vast majority of the left hemithorax (arrows) and resulting in inversion of the left hemidiaphragm. Note the ascites in the abdomen (arrowhead) and the skin thickening and edema (asterisks), which are reflective of hydrops. H, Heart; L, liver. D, A radiograph immediately after delivery shows the complex, partially aerated lesion resulting in significant mediastinal shift. The arrowheads denote the severe tracheal deviation. Axial computed tomography angiography (E) and minimum intensity projection (F) images obtained the following day show the large, heterogenous lesion with multiple macrocysts of varying sizes. Fluid is identified in some of the cysts. G, As fetal fluid is cleared from the lungs, the lesion appears more aerated and further mediastinal shift occurs.
e-Figure 53-9 Congenital pulmonary airway malformation (CPAM) type II.
A, A transverse fetal ultrasound image through the chest in a 20-week gestational age fetus shows an echogenic right lower lobe lung lesion with tiny cysts only seen on high-resolution images (image not shown) consistent with a CPAM type II lesion (arrows). The heart (H) and mediastinum are only mildly shifted without cardiac compression. Sagittal (B) and coronal (C) fetal T2-weighted magnetic resonance images show a mildly heterogenous lesion (arrows) with slightly nodular contours but no visible cysts. Postnatal axial and sagittal computed tomography images (D and E) at 1 month of age show an ill-defined, relatively smaller lesion (arrows) in the right lower lobe with tiny, subcentimeter internal cysts (arrowheads). This lesion also demonstrated bronchial atresia components on histologic examination.
e-Figure 53-10 Congenital pulmonary airway malformation type II.
A, A transverse fetal ultrasound image through the chest in a 19-week gestational age fetus shows an echogenic left upper lobe lesion with several internal cysts (arrowheads) and findings consistent with a CPAM type 2 lesion (arrows). The heart (H) and mediastinum are moderately shifted to the right without cardiac compression. LT, left. Axial (B) and coronal (C) fetal T2-weighted magnetic resonance (MR) images show a heterogeneous lesion (arrows) with internal cysts (arrowheads); some of them even appear as “macrocysts.” Normal-appearing lung tissue is noted as being compressed posteriorly and inferiorly (asterisks). However, on postnatal axial (D) and minimum intensity projection (E) computed tomography (CT) images at 3 months of age, the lesion appears relatively smaller and exerts less mass effect and no mediastinal shift. All the cysts are aerated and measure less than 2.0 cm. Note that some slightly prominent, tortuous internal pulmonary vessels are seen within the lesion in both the fetal MR image (B) and the postnatal CT image (D) (curved arrows).
Postnatal imaging findings of CPAMs usually correlate with underlying histopathologic features.17 Large cyst type or type I CPAMs typically present with one or several larger air-filled cystic structures with intervening solid, unaerated lung parenchyma. The cysts of type I CPAMs are larger than 2 cm and may be accompanied by several microcysts, whereas small cyst type or type II CPAMs usually manifest as partially air-filled multicystic masses, with individual cysts smaller than 2 cm and with variable degrees of solid-appearing, unaerated lung tissue.10,17 Type 3 CPAMs typically appear as solid lesions with mild contrast enhancement because of microscopic cysts that can be identified only at histologic evaluation. Type IV CPAMs usually present as large cysts arising from the peripheral portion of the lung and can be radiographically indistinguishable from a predominantly cystic type 1 pleuropulmonary blastoma (see Chapter 55) (e-Fig. 53-11).
e-Figure 53-11 A cystic pleuropulmonary blastoma in a 6-month-old with increased work of breathing.
A, A frontal chest radiograph demonstrates a large radiolucent lesion with fine septations and multiple cysts within the right lung resulting in a mediastinal shift. B, An axial computed tomography image reveals multiple cysts of varying sizes with effect of the mass demonstrated on the mediastinum and contralateral left lung.
In their pure forms, CPAM blood supply is from the pulmonary artery and venous drainage is into the pulmonary veins. Although unilobar involvement of CPAM is far more common, multilobar and even bilateral lung involvement may occur.3,27,30 Although any lobe of the lung can be involved, predilection exists for the lower lobe.3 CPAMs that are complicated as a result of superimposed infection may have an imaging appearance similar to pneumonia or a lung abscess (Fig. 53-12).
Figure 53-12 An infected congenital pulmonary airway malformation (CPAM) type I lesion in a 15-year-old girl.
A, An initial computed tomography (CT) enhanced image shows consolidation, with areas of low attenuation representing superimposed infection of the cystic components. B, A follow-up CT image obtained 7 months later shows resolution of the infection and visualization of CPAM cystic components.
Treatment and Follow-up: The generalized consensus is that symptomatic CPAMs should be resected, typically by lobectomy, regardless of the patient’s age at presentation.17,22,33 However, considerable controversy exists with regard to the management of prenatally diagnosed, asymptomatic, small CPAM lesions, and no consensus exists on the timing of34 or need for resection.35–39 Although some persons advocate a nonsurgical strategy with imaging follow-up, most medical centers advocate surgical resection before 1 year of age because of the potential risk of associated complications, such as infection, pneumothorax, and the small risk of malignant transformation, particularly in the case of CPAM type I lesions.3,21,22,33,40
Predominantly Vascular Lesions
Anomalies of the Pulmonary Artery
Pulmonary Agenesis, Aplasia, and Hypoplasia
Etiology: Pulmonary underdevelopment may be classified into three main types: (1) lung agenesis, consisting of the absence of the lung, bronchus, and pulmonary artery; (2) lung aplasia, that is, the presence of a rudimentary bronchus but the lack of lung tissue and pulmonary artery; and (3) lung hypoplasia, which consists of a hypoplastic bronchial tree and pulmonary artery with a variable amount of lung parenchyma.17,41
The etiology of lung agenesis or aplasia remains uncertain. Genetic, teratogenic, and mechanical factors may play a role.17 Pulmonary agenesis associated with ipsilateral radial ray defects or hemifacial microsomia may be the result of an abnormal development of the first and second arch derivatives or abnormal blood flow at this level inciting the developmental event, given the common association.42 On the other end of the spectrum, no identifiable cause has been found for lung hypoplasia.17
Persons with pulmonary agenesis, aplasia, and hypoplasia either are asymptomatic or present with variable degrees of respiratory distress, depending on the extent of lung underdevelopment. Associated congenital malformations may be seen in 50% to 80% of cases involving the heart, gastrointestinal tract, skeleton, and vascular and genitourinary systems.17,42–44
Imaging: On chest radiographs, affected pediatric patients may or may not present with a small, radiopaque hemithorax, depending on the degree of the abnormality. Ipsilateral displacement of mediastinal structures and elevation of the hemidiaphragm usually are present. The normal contralateral lung shows compensatory hyperinflation and herniation across the anterior midline, which is best seen on the lateral projections as a band of increased retrosternal lucency17 (Fig. 53-13). Left lung agenesis is more common than right lung agenesis. Multidetector CT with multiplanar 2D and 3D imaging capabilities can be used to distinguish among pulmonary agenesis, pulmonary aplasia, and pulmonary hypoplasia by clearly identifying the bronchial stump and/or the rudimentary bronchial tree17,45 (e-Figs. 53-14 and 53-15, Fig. 53-16, and e-Fig. 53-17).
Figure 53-13 Pulmonary agenesis.
Frontal (A) and lateral (B) radiographs in a 15-year-old with worsening asthma demonstrate marked hyperinflation of the left lung that extends across the midline anteriorly and herniates toward the right, as evidenced by a band of retrosternal lucency on the lateral projection (asterisks). Associated dextroposition of the heart into the right hemithorax is seen. Enhanced axial computed tomography (C), axial volume-rendered (D), minimum intensity projection (E) and three-dimensional volume-rendered (F) images of the central airway and lungs demonstrate complete agenesis of the right bronchus and lung. Associated dextroposition of the heart and compensatory hyperexpansion is present, particularly of the left upper lobe (asterisks), which herniates into the right hemithorax. A normal left mainstem bronchus (arrow) is seen. LPA, Left pulmonary artery.
Figure 53-16 Hypoplastic right lung and scimitar syndrome in an 8-year-old.
A, A frontal chest radiograph shows asymmetric lung volumes with dextroposition of the heart and a vertically oriented curvilinear opacity (arrowheads) projecting over the right lower hemithorax. An axial maximum intensity projection computed tomography image (B) confirms mild dextroposition of the heart and hypoplastic right pulmonary artery (PA) (black arrows) relative to the normal left pulmonary artery (white arrow). Coronal thick multiplanar reconstruction (C), inverted minimum intensity projection (D), and three-dimensional volume rendered (E) images show hypoplasia of the right lung with absence of the right upper lobe bronchus and partial anomalous pulmonary venous return of a vast portion of the right lung via a scimitar vein (arrowheads) into the inferior vena cava. Incidentally noted is a right aberrant subclavian artery (curved arrow).
e-Figure 53-14 A 1-day-old with lung aplasia, esophageal atresia, and distal tracheoesophageal fistula.
A, A frontal chest radiograph shows complete opacification with volume loss in the right hemithorax. The airway and mediastinum are shifted to the right with associated dextroposition of the heart. A nasogastric tube terminates in the upper esophageal pouch consistent with esophageal atresia. The anterior view of a volume-rendered image of the central airway and lungs (B) and virtual bronchoscopic (C) images demonstrate a rudimentary right main stem bronchus (straight arrows) consistent with lung aplasia. No right pulmonary artery is identified (image not shown).
e-Figure 53-15 Ex-34 weeks premature with esophageal atresia, distal tracheoesophageal (TE) fistula, and imperforate anus.
A, A frontal radiograph of the chest and abdomen shows complete opacification with volume loss in the right hemithorax. The endotracheal tube (arrowhead) and mediastinum are shifted to the right with associated dextroposition of the heart. A nasogastric tube (curved arrow
[/not-level-membership-for-pediatrics-category]
